
Effect of the protein environment on the excited state phenomena in a bacteriophytochrome†
Pradipta
Dey
 ,
Supriyo
Santra
and
Debashree
Ghosh
*
,
Supriyo
Santra
and
Debashree
Ghosh
*
School of Chemical Sciences, Indian Association for the Cultivation of Science, Jadavpur, Kolkata 700032, India. E-mail: pcdg@iacs.res.in
First published on 15th July 2024
Abstract
The excited state processes of a bacteriophytochrome are studied using high-level multireference methods. The various non-radiative channels of deactivation are identified for the chromophore. The effects of the protein environment and substituents are elucidated for these excited state processes. It is observed that while the excited states are completely delocalized in the Franck–Condon (FC) region, they acquire significant charge transfer character near the conical intersections. Earlier studies have emphasized the delocalized nature of the excited states in the FC region, which leads to absorption spectra with minimal Stokes shift [Rumyantsev et al., Sci. Rep., 2015, 5, 18348]. The effect of the protein environment on the vertical excitation energies was minimal, while that on the conical intersection (CI) energetics was significant. This may lead one to believe that it is charge transfer driven. However, energy decomposition analysis shows that it is the effect of the dispersion of nearby residues and the steric effect on the rings and substituents that lead to the large effect of proteins on the energetics of the CIs.
1 Introduction
Phytochromes are photoreceptors found in cyanobacteria,1 fungi,2 and non-photosynthetic bacteria in addition to plants.3 There are many examples such as biliverdin (BV; non-photosynthetic bacteria),4–6 phycocyanobilin (PCB; photosynthetic cyanobacteria),1 phytochromobilin (oat phytochrome),7etc. and they differ slightly in their chromophore structure and substituents. However, the core structure of the chromophore remains the same, consisting of four pyrrole rings connected by methine bridges. They regulate a wide range of physiological responses ranging from phototaxis in bacteria to seed germination, growth, organelle development, etc., in plants.8,9 They function as temperature sensors and are mainly involved in the photoactivity of plants and their growth. In bacteria and several aquatic species, they are involved in the absorption of light at low intensities.10,11As mentioned above, the chromophore of the phytochromes consists of a tetrapyrrole structure that has similarities to chlorophyll. The four pyrrole rings are denoted as A, B, C, and D rings, starting from the pyrrole that is connected to the cysteine residue (shown in Fig. 1). The pyrroles in phytochromes are linearly arranged, while in chlorophyll they are circularly organized, and this shifts the spectral maxima quite significantly.12,13 Phytochromes show photoactivity in the red and far red regions of the visible spectra. They have two main photo-interconvertible forms, called Pr and Pfr, denoting red (700 nm) and far red (754 nm) absorbances, respectively (shown in Fig. 1).14,15
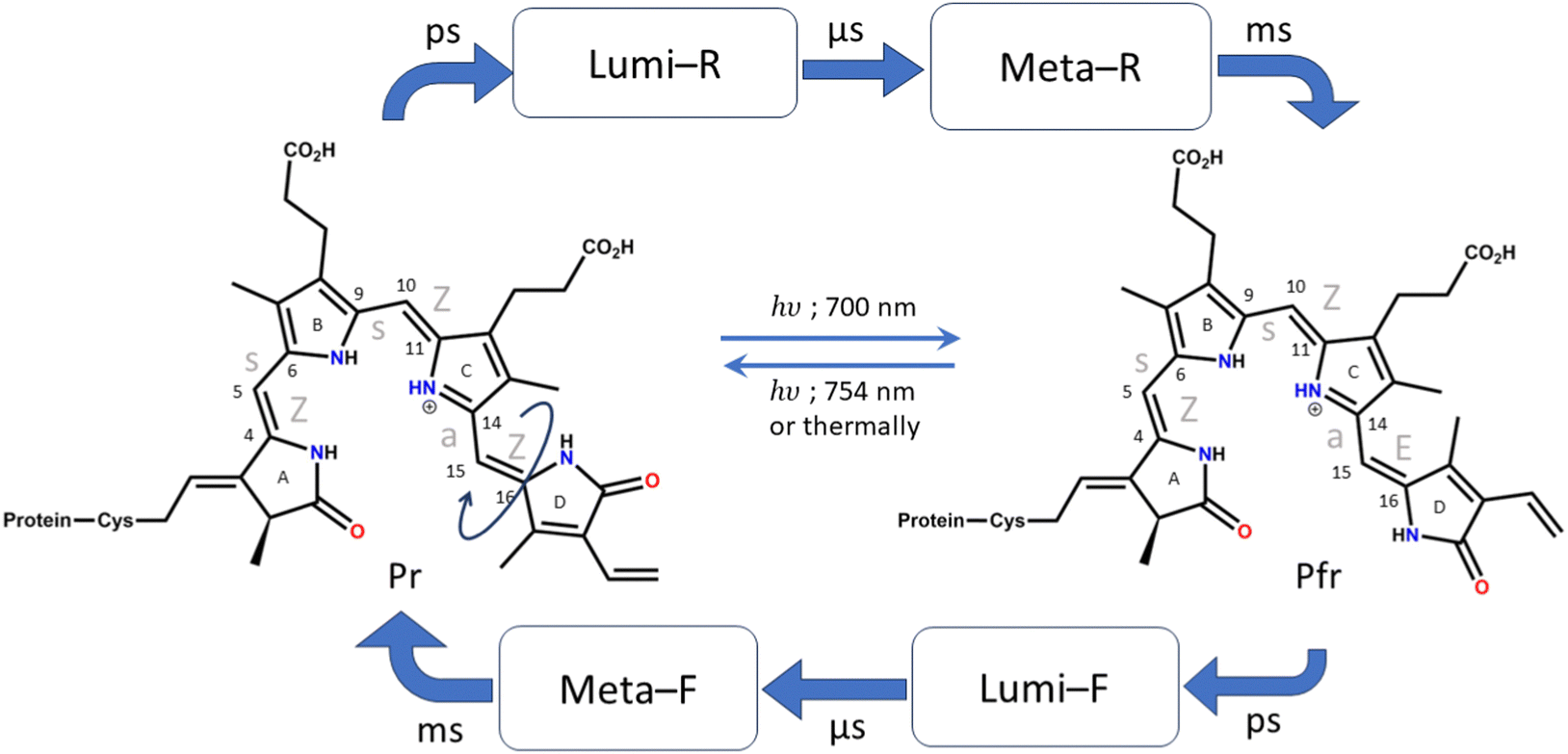 | ||
| Fig. 1 Photocycle of Pr to Pfr photoconversion and Pfr to Pr photoconversion. The names of the intermediates are shown. Typical timescales are given above the arrows for the formation of those intermediates. The chromophore and conformation of the Pr and Pfr forms are shown, and the atoms in the methine bridge are numbered. | ||
Photoisomerization across double bonds, i.e., cis–trans isomerization initiated by photoactivation, is a common process in several biological species, such as rhodopsins,16 carotenoids,17etc. In phytochromes, several such photoisomerization sites are present across all methine bridges. However, biologically observed isomerization occurs only at the last methine bridge (between the C and D rings) to form the Pfr structure.4 It is, therefore, crucial to understand the cause behind this process and its specificity. Furthermore, it is known that despite efficient absorption of light by the phytochromes, they are non-fluorescent.18,19 Therefore, ultrafast nonradiative decay channels are expected.
The crystal structures of the bacterial4–6 and cyanobacterial1 phytochromes show that the A-ring of the chromophore binds to the protein via a thioether bond of the cysteine residue, while the D ring is free at the far end. The orientation of the adjacent pyrrole rings is Z, Z, Z with respect to the double bond and syn, syn, anti with respect to the single bond of the methine bridge. Due to this reason, the Pr form of the chromophore (which is the resting state) is also denoted as ZZZssa. The protein interacts with the chromophore in such a way that allows only the C15![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C16 bond isomerization.9 Therefore, upon excitation by red light, when the photoconversion occurs and the Pfr structure is formed, the configuration changes to ZZEssa. The photocycle of conversion between these forms is shown in detail in Fig. 1.18
C16 bond isomerization.9 Therefore, upon excitation by red light, when the photoconversion occurs and the Pfr structure is formed, the configuration changes to ZZEssa. The photocycle of conversion between these forms is shown in detail in Fig. 1.18
NMR,20 time-resolved mid-IR,21,22 resonance Raman (RR),23–25 and time-resolved femtosecond Raman spectral studies26 have elucidated the first intermediate Lumi-R structure as the primary photoproduct formed during Pr to Pfr photoconversion. The Lumi-R formation takes around 30 ps.26
The protonation state of the chromophore has been studied theoretically27,28 and experimentally29–31 and the fully protonated structure is expected to be in the Pr form. However, some studies have also proposed protein-aided proton release and uptake mechanisms during photoconversion.32
There have been several theoretical studies to characterize the primary photoconversion step. The absorption spectra have been studied, and the effect of different isomers on the spectral intensities has been elucidated.33–35 Durbeej and co-workers have observed isomerization about the C9–C10 and C10–C11 bonds using the TDDFT method (rotation around the central methine bridge).36,37 Since this is not a biologically relevant isomerization step, other levels of theory have been employed to understand the C15–C16 isomerization. However, due to the steep computational cost of multireference methods, either a truncated model has been used or semi-empirical methods have been resorted to. From a truncated model consisting of only two pyrrole rings, the effects of clockwise and anti-clockwise rotations and substitution have been studied.38 Here, it has been observed that one bond flip becomes a preferred mechanism over hula twist upon carbonyl substitution. Minimal effects of alkyl group substitution were also observed. On the other hand, the study that considered the full chromophore while resorting to semi-empirical methods (OM2/MRCI) shows that isomerization across the C9–C10 and C10–C11 bonds becomes predominant.28 This result was similar to that obtained from earlier TDDFT observations.36,37
Garavelli and co-workers39 have done CIS/CASPT2 calculations and observed C9–C10/C10–C11 and C15–C16 bond isomerization pathways. Slavov et al.40 have shown the tendency of the D ring of the chromophore to undergo counterclockwise rotation through QM/MM computations. QM/MM dynamics of the dimer protein were used to observe the evolution of the C15–C16 bond rotation, and they observed a similar counterclockwise rotation41 to that reported in a previous study. Mennucci and co-workers have shown the effect of a protein on the dynamics of photoisomerization using the FOMO-CASCI method.42 They have observed C15–C16 isomerization in the protein environment. From all these observations, it is clear that multireference methods are crucial to appropriately describe the relative energetics of different bond rotations in the phytochrome.
In this study, we have investigated the initial step of photoisomerization of the BV chromophore in the gas phase and used multireference methods with sufficient active space to appropriately describe the potential energy surface (PES). We have worked with the fully protonated chromophore in the Pr form. The effect of a protein on the PES is also studied at both CASPT2 and TDDFT levels of theory to ascertain the most important interactions that modulate the energetics of the pathways.
2 Computational details
An isolated BV chromophore was taken from a PDB ID-2O9C,5 and the fully protonated form of the chromophore was used. Fig. 2a shows the protein and the chromophore structure. The unsubstituted (alkyl groups are removed) small chromophore is shown in Fig. 2b. This small chromophore form is used for the most detailed study to calculate the PES and nonradiative decay pathways. The ground state geometry is optimized at the B3LYP/6-31G(d) level of theory. VEEs for the Franck–Condon (FC) region are calculated at various levels of theory – TDDFT43,44 with B3LYP and CAM-B3LYP functionals and 6-31G(d) and 6-311++G(d,p) basis sets, EOM-CCSD/6-31G(d),45 CASSCF46 and CASPT247 with (6o,6e) active space and the 6-31G(d) basis set. State-specific CASPT2 calculations with a level shift of 0.25 a.u. are used to avoid the problem of intruder states.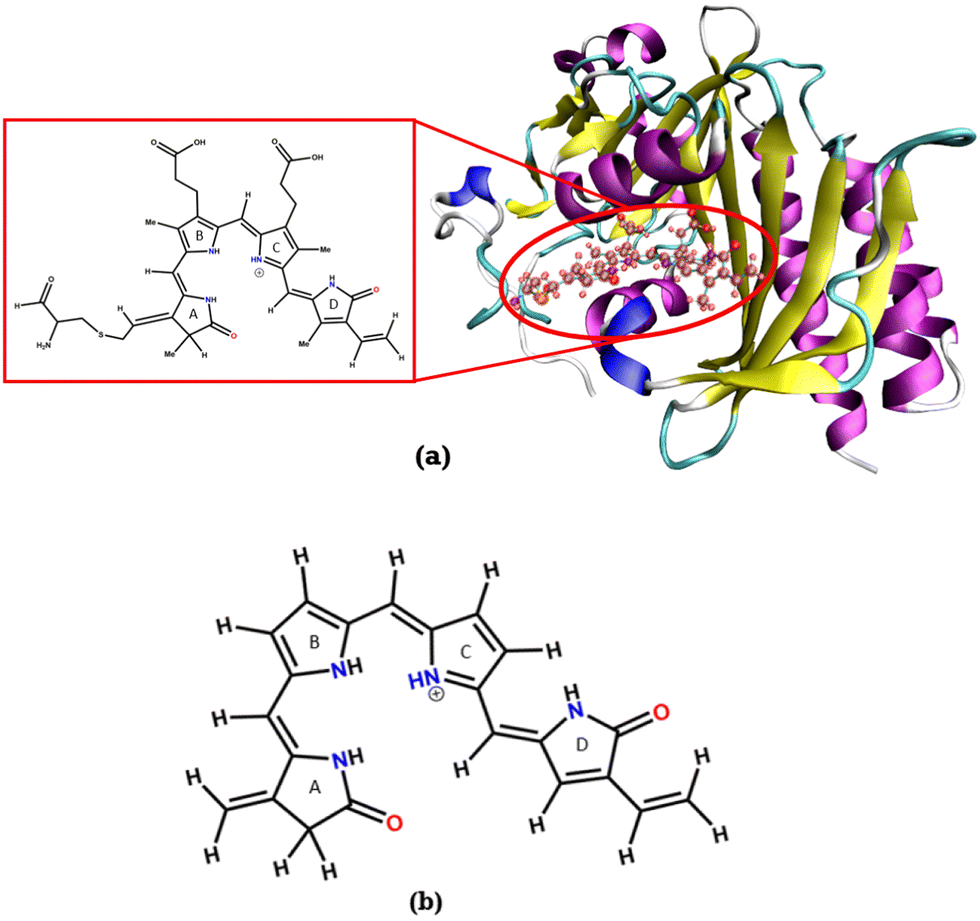 | ||
| Fig. 2 (a) Structure of the bacteriophytochrome BV in a protein, the large chromophore considered for the chromophore in protein calculations (shown in the inset) and (b) the chromophore without the substituents (Chr-sm). | ||
CASSCF with (6o,6e) and TDDFT have been used to calculate the stationary points of the excited state surface, including the S1-Min and conical intersections. The minimum energy pathways (MEPs) between FC, S1-Min, and the CIs are constructed to study the non-radiative decay mechanism. Then, single-point calculations were done on each geometry at CASPT2/6-31G(d) level of theory to create the surface.
To study the fate of the molecule after reaching the CIs, the 3D surfaces around the CIs are created using four topological parameters of the system,48 namely
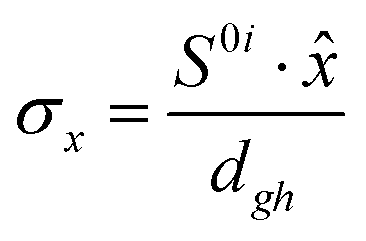
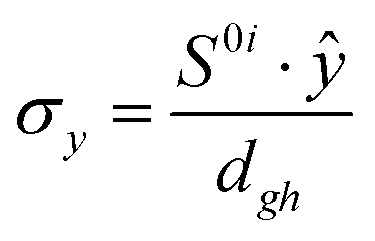
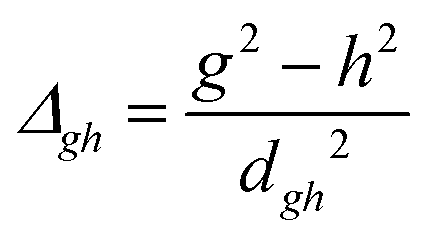
where S0i is the gradient sum vector with i representing the state with which the ground state has a CI (here i is 1) and
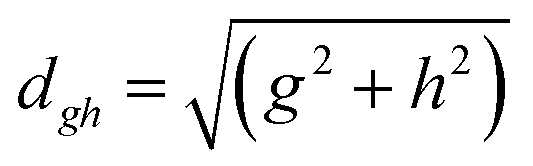
![[x with combining circumflex]](https://www.rsc.org/images/entities/i_char_0078_0302.gif) and ŷ are the unit vectors based on the Schmidt-orthogonalised energy gradient difference vector (g0i) and the derivative coupling vector (h0i), which are given as
and ŷ are the unit vectors based on the Schmidt-orthogonalised energy gradient difference vector (g0i) and the derivative coupling vector (h0i), which are given as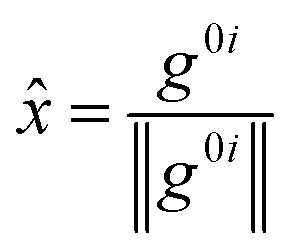
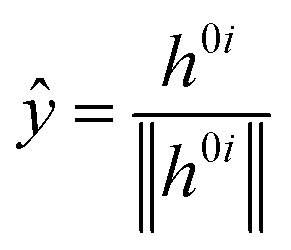
The parameters σx and σy give the tilt of the cone and dgh and Δgh denote the pitch and divergence of the cone. These parameters help to understand the fate of the molecule after reaching the CI.
The SA-CASSCF and state-specific CASPT2 calculations were done using Molpro-2015 software.49 The CASPT2/MM calculations were done using Molpro-2024 software.50 All TDDFT single-point calculations, excited state optimizations, EOM-CCSD excitation energies and the TDDFT/MM calculations were done using Q-Chem 5.1 software.51
The configurations around the methine bridges are shown by the dihedral angles denoted as – τA–B, τB–A between A and B rings, τB–C, τC–B between B and C rings, and τC–D, τD–C between C and D rings. Dihedral NA–C4C5–C6, i.e., the N of the A ring and the methine bridge C atoms between A and B rings, is referred to as τA–B, while the same for the N of the B ring is referred to as τB–A and so on.
For the chromophore in protein calculations, the Amber force field (ff14SB)52 was used and solvated in TIP3P water.53 For the cysteine attached BV chromophore the parameters were generated using the parmchk2 program and ff14SB force field and Amber atom types. The solvated protein crystal structure was then optimized. With this optimized structure, excited state QM/MM calculations were performed with TDDFT and CASPT2 as the QM method and point charges of the Amber force field for protein residues. The TIP3P charges on nearby water molecules were also included in the excited state QM/MM calculations.
The FC and CI geometries of the gas phase were embedded in the protein. The gas phase (Chr-sm) geometries were first placed in the protein cavity by translation and rotation of these geometries with respect to the crystal structure of the protein and its cavity. The extra propionate and cysteine groups were added to Chr-sm and therefore a large chromophore structure was formed. The part of the chromophore denoted as Chr-sm was kept fixed and the propionate groups and the protein around it were relaxed at the MM level of theory. This protocol was followed for FC and CI geometries. Then QM/MM calculations were done to determine the effective energies of those optimized geometries in the protein environment. This protocol preserves the structure of the CI as obtained in the gas phase while studying the effect of the protein environment and substituents on the energetics of the excited state PES. However, as artifacts of this protocol it should be mentioned that the CIs in the protein will be approximate in nature.
3 Results and discussion
The B3LYP/6-31G(d) optimized structure of the ground state is given in the ESI† (S11) and also shown in Fig. 3. The optimized ground state geometry of the chromophore is somewhat out of plane with a dihedral angle of 16° between A and B rings, 10.50° between the B and C rings, and about 14° between the C and D rings.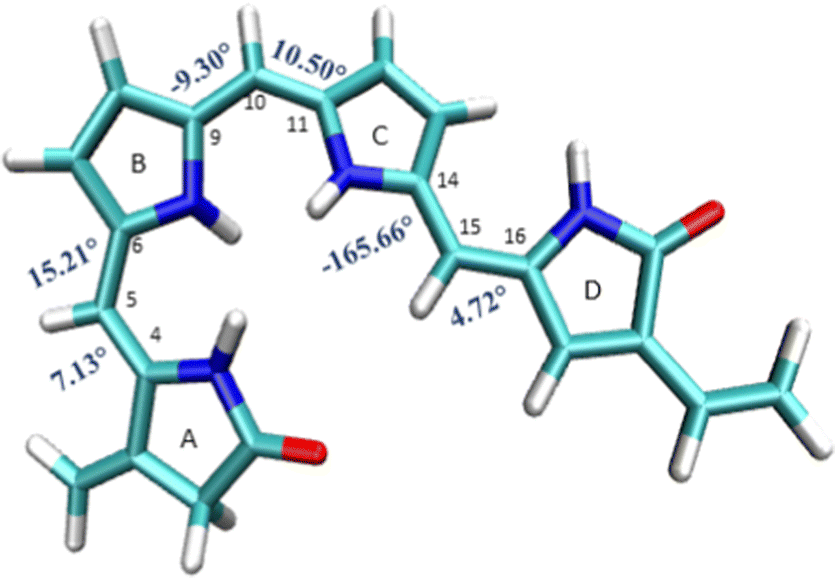 | ||
| Fig. 3 Geometrical parameters of FC: (a) dihedral angles τA–B, τB–A, τB–C, τC–B, τC–D and τD–C (in degrees). | ||
3.1 Vertical excitation energy and absorption spectra
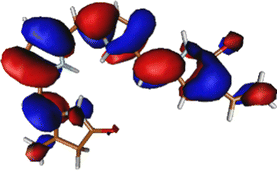
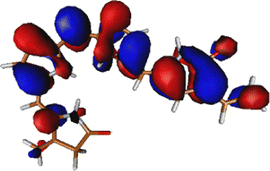
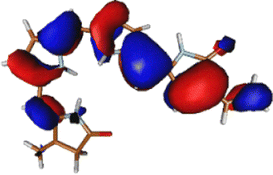
As noted in the earlier literature, there are significant differences in the absorption spectra of the phytochrome chromophore14 and porphyrin.54–57 In this work, since we are interested in the excited state processes at the lowest optically bright state, we target only two excited states in our calculations. Both these excited states are π–π* in nature. However, it is only the first excited state S1 that is optically bright. The S2 state is mostly optically dark.Table 1 shows the excitation energies of these states calculated using TDDFT (B3LYP and CAM-B3LYP), EOM-EE-CCSD, and CASPT2(6,6) with the 6-31G(d) basis set. The oscillator strengths are mentioned in brackets. The effect of the basis set is also checked at the TDDFT level with the 6-311++G(d,p) basis set. It shows very little effect of the basis set on the excitation energy (given in Table S1, ESI†). The reason for the low effect of the basis set can be understood from the orbitals involved in the excitations (also shown in Table 1). Since both the excitations are purely π and π* in nature, it is unsurprising that the effect of the basis set is minimal.
| Excited state | B3LYP | CAM-B3LYP | EOM-EE-CCSD | CASPT2 | Orbitals from | To |
|---|---|---|---|---|---|---|
| S1 | 1.99 (0.95) | 2.10 (1.04) | 2.53 (1.33) | 1.65 (1.45) |
|
|
| S2 | 2.48 (0.07) | 3.25 (0.27) | 3.63 (0.04) | 2.26 (0.17) |
|
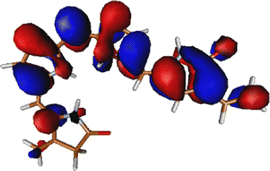
|
The excitation is completely delocalized over the chromophore, with no charge transfer component. The Mulliken charge analysis done on different parts of the optimized Chr-sm has a qualitative similarity to those of a recent study,58 yet we did not observe any charge transfer as evident from the attachment detachment density analysis shown in ESI† (Fig. S4). This is the reason why the absorption spectra remain at the same position irrespective of mutations.35,59 This is also the reason behind high oscillator strength and the ability of the phytochrome to absorb strongly in this red region. The orbitals involved in the (6o,6e) active space are given in Fig. S1 (ESI†). Since between the first two excitations, the only orbitals involved are the HOMO, HOMO−1, and LUMO, the (6o,6e) active space is adequate.39 In ref. 39, several active spaces have been tested, and it was observed that the size of active space has very little effect on the nature and energy of the S1 state.
3.2 Excited state potential energy surface
The optimized geometry for the S1 minimum is given in Fig. S5 (ESI†). The S1 minimum is near the ground state geometry. This is expected since there is no charge transfer component in the excited state, and therefore, the S1 has a similar electron density to the ground state.Two conical intersections could be obtained for the molecule between the S1 and S0 states (shown in Fig. 4a and b). CI1 (Fig. 4a) consists of an anticlockwise rotation of the D-ring. CI2 (Fig. 4b), on the other hand, has the rotation of the A and B rings, or it can be envisaged as a C and D ring rotation. Since the S1 state is an excitation state delocalized over the chromophore, the CIs can be obtained by reducing the conjugation length. The rotation of the different rings out of the molecular plane leads to a reduction in the effective conjugation length and, therefore, causes the near degeneracy between the S0 and S1 states. It is also unsurprising that the CD ring rotation causes a greater reduction in the conjugation length and, therefore, leads to a lower energy or easier-to-access CI. The effective reduction in the conjugation can be noticed from the dihedral angles and bond lengths of the methine bridge C–C bond lengths (shown in Table 2). The dihedral angles at both the CIs show a complete perpendicular geometry between C–D or B–C rings, respectively (the values of dihedral angles are tabulated in Table S2, ESI†). The alternating double and single bonds in the methine bridges are also disturbed at those locations.
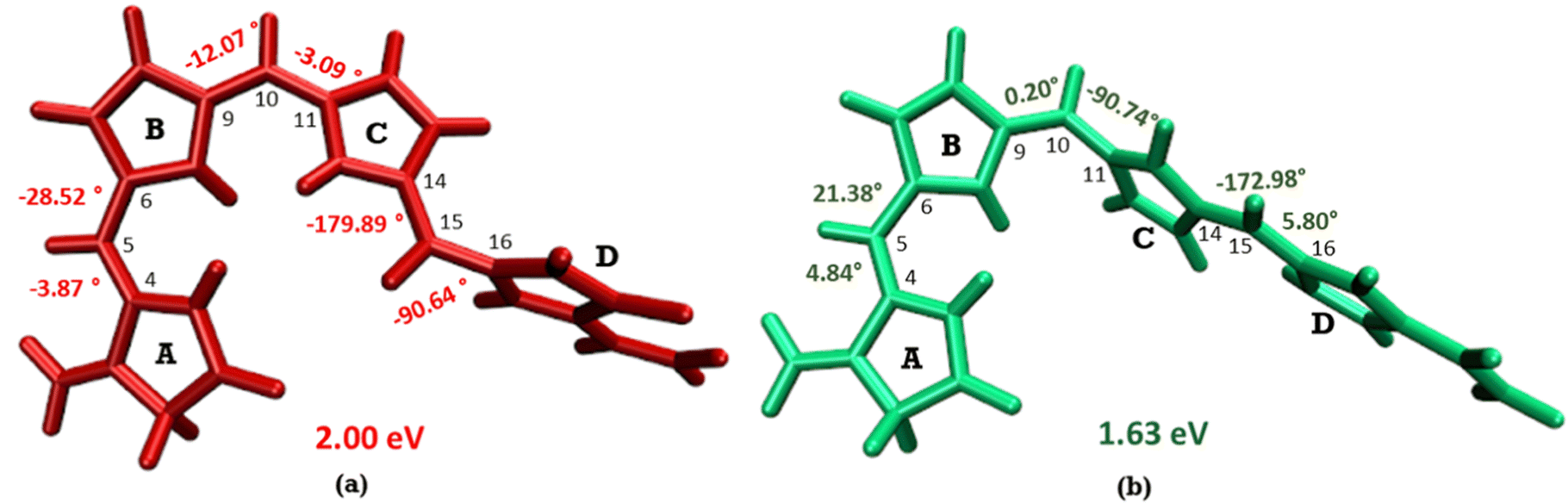 | ||
| Fig. 4 (a) CI1, D ring rotated CI and (b) CI2, C–D ring rotated CI with their respective dihedral angle values. | ||
| Geometry | C4–C5 | C5–C6 | C9–C10 | C10–C11 | C14–C15 | C15–C16 |
|---|---|---|---|---|---|---|
| FC | 1.37 | 1.42 | 1.40 | 1.39 | 1.42 | 1.37 |
| S1-Min | 1.39 | 1.40 | 1.36 | 1.44 | 1.43 | 1.37 |
| CI1 | 1.33 | 1.46 | 1.42 | 1.36 | 1.34 | 1.45 |
| CI2 | 1.35 | 1.44 | 1.36 | 1.46 | 1.40 | 1.36 |
Fig. 5 shows the minimum energy pathways from the FC/S1 minimum to the two CIs. At the CASPT2 level of theory, the CI1 is much higher in energy than the S1 state at the FC region (>8 kcal mol−1), while an energy barrier of 2 kcal mol−1 is observed to reach C–D ring rotated CI2. Therefore, the CI2 is energetically easier to approach in the gas phase than CI1. This gas phase observation is contrary to the observation of the Lumi-R structure in proteins, which is closer to the CI1 structure, i.e., involves D ring rotation. Therefore, large effects of the protein environment are expected.
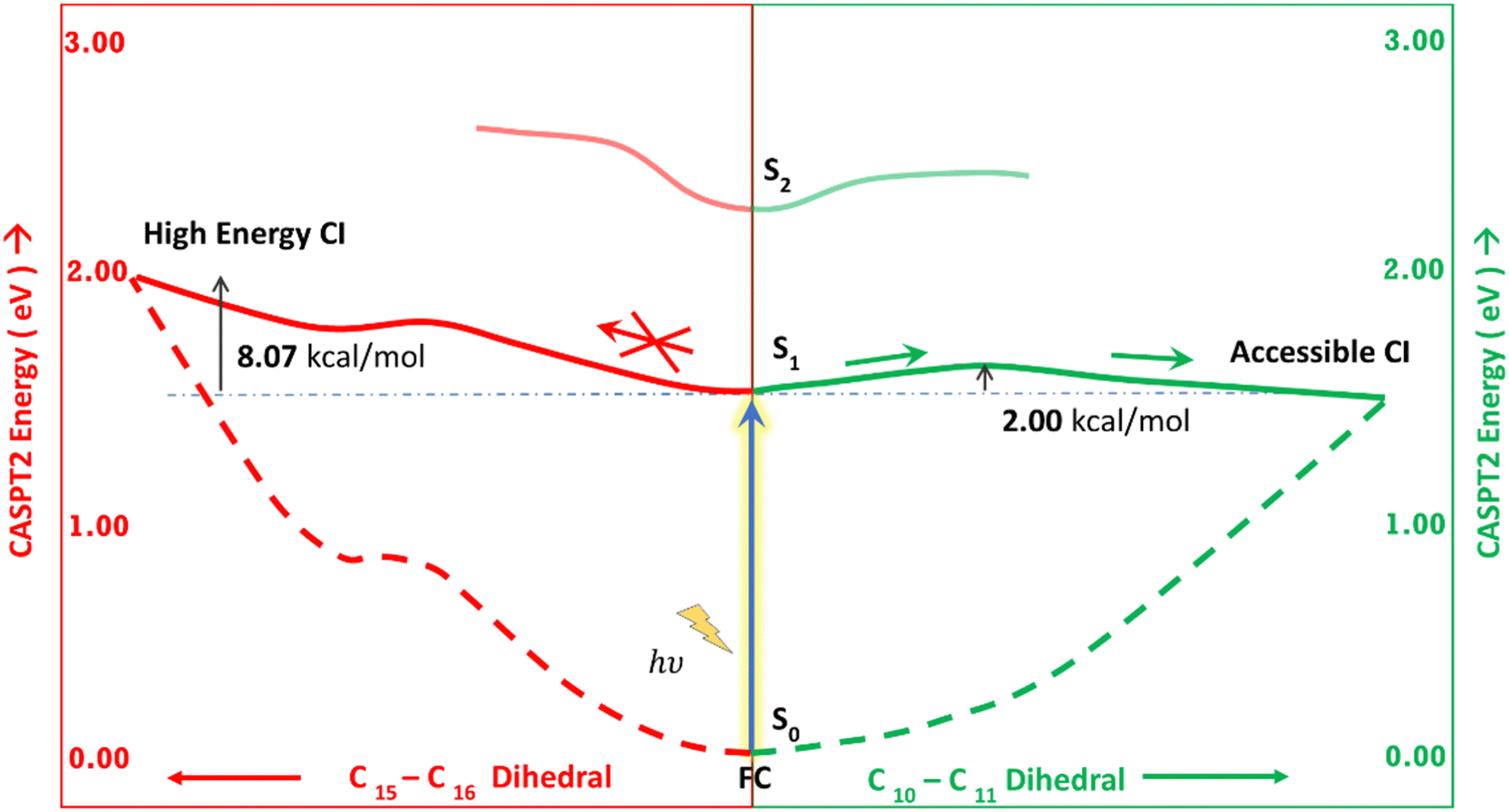 | ||
| Fig. 5 Schematic representation of the two MEPs. The left side refers to the pathway from FC to CI1 (red). The right side refers to the pathway from FC to CI2 (green). MEPs are calculated at the CASPT2/6-31g(d) level of theory. | ||
The orbitals involved in the S1 excitation near the CI are shown in Fig. S6 in the ESI.† It shows large changes in electron density at these geometries. The CI1 shows the charge transfer component from the B and C rings to the D ring. The CI2 shows the charge transfer component from C and D rings to A and B rings.
We have calculated the parameters associated with the topology of the CIs and have re-created the linearly interpolated structure of the CIs. For the high energy CI1, the tilt of the cone (Fig. 6a) is towards the positive of the ![[h with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0068_20d1.gif) vector and negative of the
vector and negative of the ![[g with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0067_20d1.gif) vector (the vectors are shown in Fig. S8, ESI†). This observation shows that there is an equal probability of the molecule undergoing photo-product formation and photo-protection (returning to the Franck–Condon region) for this CI. The vector corresponds to out of plane bending motion of the C14–C15 and C15–C16 bonds. It also shows asymmetric stretching of C9–C10 and C10–C11 bonds. The photoproduct will form by the torsion of τC–D and τD–C dihedrals as it involves out-of-plane rotation.
vector (the vectors are shown in Fig. S8, ESI†). This observation shows that there is an equal probability of the molecule undergoing photo-product formation and photo-protection (returning to the Franck–Condon region) for this CI. The vector corresponds to out of plane bending motion of the C14–C15 and C15–C16 bonds. It also shows asymmetric stretching of C9–C10 and C10–C11 bonds. The photoproduct will form by the torsion of τC–D and τD–C dihedrals as it involves out-of-plane rotation.
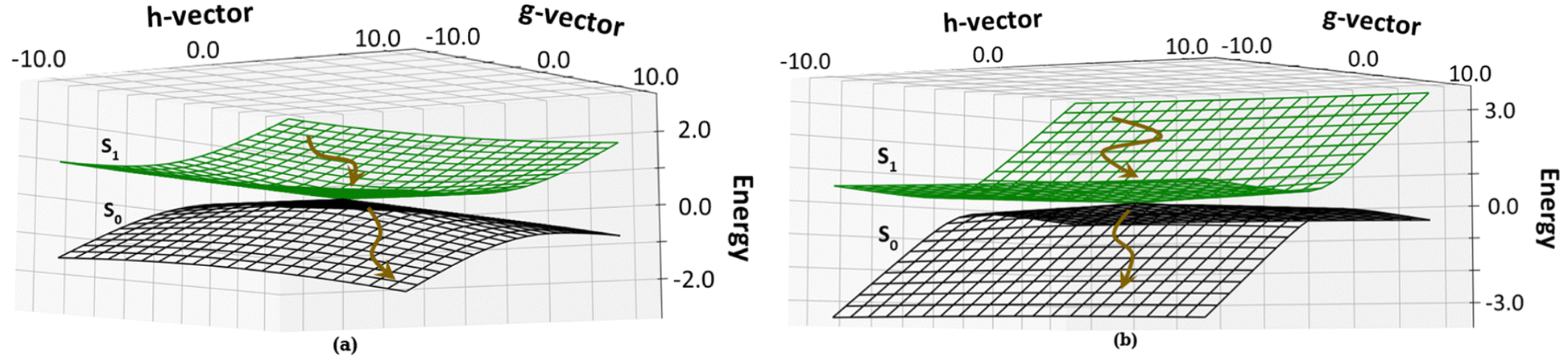 | ||
| Fig. 6 Topology around the Cis: (a) CI1, D ring rotated CI and (b) CI2, C–D ring rotated CI. The deactivation of the chromphore from the S1 state to the S0 state is shown by brown arrows. | ||
The energetically accessible CI2 describes simultaneous torsion of τB–C and τC–B dihedral angles. The topology of CI2 (Fig. 6b) has a tilt towards the negative of the vector and is symmetric about the vector. This refers to a greater probability of the molecule undergoing photoprotection than forming a photoproduct. The vector corresponds to the out of plane motion of C9–C10 bonds and C10–C11 bonds (shown in Fig. S8, ESI†). From this information, it is inferred that if photoproduct formation occurs, it must involve the isomerization of the bridge connecting the B and C pyrrole rings.
3.3 Effect of substituents and the protein environment
To understand the effect of substituents on the excited state surface, the S0 and S1 energies were calculated with the large chromophore at the FC and CI geometries. The substituents and added atoms were relaxed for all these calculations. No large effects were observed in this case, proving that the substituents, especially the alkyl groups, do not alter the PES significantly.To understand the effect of the protein environment, the chromophore with the protein was taken. The protein structure was relaxed while keeping the chromophore geometry fixed at the FC or CI geometries. With this geometry, further QM/MM computations were performed at the B3LYP/MM and CASPT2/MM levels of theory.
Table 3 shows the energy of the FC, CI1 and CI2 of the gas phase and in the protein system calculated at the TDDFT/MM level of theory. It is observed that while in the gas phase, CI2, i.e., rotation of both C and D rings (across the central methine bridge), is lower in energy, the protein environment destabilizes this CI. On the other hand, the CI1, i.e., rotation of only the D ring, is significantly stabilized in the protein environment. This leads to the facile D ring rotation in the protein en route to the Lumi-R structure. A similar effect is seen for CASPT2, where due to the extra stability of the protein environment, the CI1 g.s. is stabilized by 4.38 kcal mol−1 (0.19 eV), while CI2 g.s. is de-stabilized by 44.04 kcal mol−1. Steric effects and lack of favourable dispersion interactions primarily drive the large de-stabilization of CI2.
| System | CI1 | FC | CI2 |
|---|---|---|---|
| Chr-min | 2.04 | 1.99 | 1.48 |
| Large chromophore | 1.92 | 1.91 | 1.67 |
| Chromophore in protein | 1.27 | 1.62 | 1.92 |
Here, it should also be noted that the embedding of gas phase geometries in the protein is an approximation due to which the exact geometry of the CIs gets significantly affected as noticed from the somewhat lack of degeneracy between the ground and excited states at the gas phase CI geometries with the protein environment.
To decompose the effect of the nearby residues and ascertain the main cause of the extra stabilization of CI1 in the protein, EDA analysis is performed with HIS290, HIS260, TYR263, and PHE203. The details of the EDA analysis are included in Table S3 in the ESI.† Quite surprisingly, it was noticed that TYR263 and PHE203 showed significant stabilization effects on the CI1 structure, and this was mainly due to the dispersion interaction between the D ring and the amino acids. In CI2, both these interactions were largely absent due to large distances and mismatch of ring faces. Furthermore, it is noteworthy that HIS290, which is near the D ring, does not show much differential effect on the stability of the CIs. This is true even though the excited states in the CIs show a significant charge transfer component. The reason could be the relatively large distance of the HIS290 from the ring itself. On the other hand, HIS260, which remains stacked in FC geometry between the B and C rings, offers preferential stability to the CI1. This is because the B and C rings remain unchanged in the CI1 and FC geometry. On the other hand, in CI2 due to rotation around the central methine bridge, this stacking interaction is largely diminished, thereby preferentially destabilizing the CI2 conformer. The tethering effect of the alkyl carboxylic acid substituents could also result in the destabilization of the CI2 geometry. Essentially, the tethers retain the B and C rings in the original FC geometry and deter rotation around the methine bridge between these rings.
To surmise the effect of substituents, carboxylic acid groups and the protein environment cause the CI1 to be preferred. The modulation by these factors is mainly driven by steric and dispersion interactions rather than specific H-bond or electrostatic interactions. This observation corroborates with the earlier study conducted by Durbeej and co-workers.60
4 Conclusion
In summary, we have studied the bacteriophytochrome chromophore's excited state potential energy surface using high-level multireference methods. We have noticed that the excited states are delocalized and do not involve a charge transfer component. The lowest excited state is optically bright, characterized by high oscillator strength (absorption coefficient). Two low-energy conical intersections between the S1 and ground states are found. The CIs involve rotation across methine bridges – either the central methine bridge or the one between the C and D rings. While in the gas phase, the CI along central methine bridge rotation is lower in energy, in the protein environment, this trend is reversed. This observation also proves the reason behind the varying results obtained in different theoretical studies.Furthermore, the origin of a specific protein interaction that leads to preferential D ring rotation is ascertained. It is observed that the dispersion and steric interactions in proteins play a crucial role.
Author contributions
D. G. formulated the problem and computational approaches. P. D. and S. S. performed the calculations. D. G. and S. S. wrote the manuscript.Data availability
All geometries (coordinates) and software versions used have been included/mentioned in the ESI† and main manuscript. No other data are required to reproduce the study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. D. thanks the UGC for a junior research fellowship. S. S. thanks DST-INSPIRE for a senior research fellowship. D. G. thanks generous funding from SERB (SPF/2021/000194 and CRG/2023/001806). All the authors thank computational facilities provided by IACS.Notes and references
- L.-O. Essen, J. Mailliet and J. Hughes, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14709–14714 CrossRef CAS PubMed
.
- A. C. Froehlich, B. Noh, R. D. Vierstra, J. Loros and J. C. Dunlap, Eukaryotic Cell, 2005, 4, 2140–2152 CrossRef CAS PubMed
- P. H. Quail, M. T. Boylan, B. M. Parks, T. W. Short, Y. Xu and D. Wagner, Science, 1995, 268, 675–680 CrossRef CAS PubMed
- J. R. Wagner, J. S. Brunzelle, K. T. Forest and R. D. Vierstra, Nature, 2005, 438, 325–331 CrossRef CAS PubMed
- J. R. Wagner, J. Zhang, J. S. Brunzelle, R. D. Vierstra and K. T. Forest, J. Biol. Chem., 2007, 282, 12298–12309 CrossRef CAS PubMed
- X. Yang, E. A. Stojković, J. Kuk and K. Moffat, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12571–12576 CrossRef CAS PubMed
- M. J. Terry, M. T. McDowell and J. C. Lagarias, J. Biol. Chem., 1995, 270, 11111–11118 CrossRef CAS PubMed
- N. C. Rockwell, Y.-S. Su and J. C. Lagarias, Annu. Rev. Plant Biol., 2006, 57, 837–858 CrossRef CAS PubMed
- N. C. Rockwell and J. C. Lagarias, Plant Cell, 2006, 18, 4–14 CrossRef CAS PubMed
-
J. Mach, Phytochromes in Diatoms: Sensing Far-Red Light in the Deep Blue Sea, 2016 Search PubMed
- M. Legris, Y. Ç. Ince and C. Fankhauser, Nat. Commun., 2019, 10, 5219 CrossRef CAS PubMed
- C. French, J. Brown and M. Lawrence, Plant Physiol., 1972, 49, 421–429 CrossRef CAS PubMed
- G. Cinque, R. Croce and R. Bassi, Photosynth. Res., 2000, 64, 233–242 CrossRef CAS PubMed
- R. Tasler, T. Moises and N. Frankenberg-Dinkel, FEBS J., 2005, 272, 1927–1936 CrossRef CAS PubMed
- E. S. Burgie, J. Zhang and R. D. Vierstra, Structure, 2016, 24, 448–457 CrossRef CAS PubMed
- J. von Lintig, P. D. Kiser, M. Golczak and K. Palczewski, Trends Biochem. Sci., 2010, 35, 400–410 CrossRef CAS PubMed
- H.-E. Khoo, K. N. Prasad, K.-W. Kong, Y. Jiang and A. Ismail, Molecules, 2011, 16, 1710–1738 CrossRef CAS PubMed
- D. Buhrke, F. Velazquez Escobar, L. Sauthof, S. Wilkening, N. Herder, N. N. Tavraz, M. Willoweit, A. Keidel, T. Utesch and M.-A. Mroginski,
et al.
, Sci. Rep., 2016, 6, 28444 CrossRef CAS PubMed
- K. A. Rumyantsev, D. M. Shcherbakova, N. I. Zakharova, A. V. Emelyanov, K. K. Turoverov and V. V. Verkhusha, Sci. Rep., 2016, 5, 18348 CrossRef PubMed
- T. Rohmer, C. Lang, J. Hughes, L.-O. Essen, W. Gärtner and J. Matysik, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15229–15234 CrossRef CAS PubMed
- J. J. van Thor, K. L. Ronayne and M. Towrie, J. Am. Chem. Soc., 2007, 129, 126–132 CrossRef CAS PubMed
- C. Schumann, R. Groß, N. Michael, T. Lamparter and R. Diller, ChemPhysChem, 2007, 8, 1657–1663 CrossRef CAS PubMed
- F. Andel, J. C. Lagarias and R. A. Mathies, Biochemistry, 1996, 35, 15997–16008 CrossRef CAS PubMed
- C. Kneip, P. Hildebrandt, W. Schlamann, S. E. Braslavsky, F. Mark and K. Schaffner, Biochemistry, 1999, 38, 15185–15192 CrossRef CAS PubMed
- F. Andel, J. T. Murphy, J. A. Haas, M. T. McDowell, I. van der Hoef, J. Lugtenburg, J. C. Lagarias and R. A. Mathies, Biochemistry, 2000, 39, 2667–2676 CrossRef CAS PubMed
- J. Dasgupta, R. R. Frontiera, K. C. Taylor, J. C. Lagarias and R. A. Mathies, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1784–1789 CrossRef CAS PubMed
- V. Modi, S. Donnini, G. Groenhof and D. Morozov, J. Phys. Chem. B, 2019, 123, 2325–2334 CrossRef CAS PubMed
- H. Huang, C. Xu, K. Lin, J. Peng, F. L. Gu and Z. Lan, Chin. Chem. Lett., 2023, 34, 107850 CrossRef CAS
- T. Rohmer, H. Strauss, J. Hughes, H. de Groot, W. Gaertner, P. Schmieder and J. Matysik, J. Phys. Chem. B, 2006, 110, 20580–20585 CrossRef CAS PubMed
- D. Von Stetten, M. Günther, P. Scheerer, D. H. Murgida, M. A. Mroginski, N. Krauß, T. Lamparter, J. Zhang, D. M. Anstrom and R. D. Vierstra,
et al.
, Angew. Chem., Int. Ed., 2008, 47, 4753–4755 CrossRef CAS PubMed
- J. A. Rumfeldt, H. Takala, A. Liukkonen and J. A. Ihalainen, Photochem. Photobiol., 2019, 95, 969–979 CrossRef CAS PubMed
- J. J. van Thor, B. Borucki, W. Crielaard, H. Otto, T. Lamparter, J. Hughes, K. J. Hellingwerf and M. P. Heyn, Biochemistry, 2001, 40, 11460–11471 CrossRef CAS PubMed
- R. A. Matute, R. Contreras, G. Pérez-Hernández and L. González, J. Phys. Chem. B, 2008, 112, 16253–16256 CrossRef CAS PubMed
- R. A. Matute, R. Contreras and L. González, J. Phys. Chem. Lett., 2010, 1, 796–801 CrossRef CAS
- S. Santra, R. N. Manna, S. Chakrabarty and D. Ghosh, J. Phys. Chem. B, 2024, 128, 3614–3620 CrossRef CAS PubMed
- B. Durbeej, Phys. Chem. Chem. Phys., 2009, 11, 1354–1361 RSC
- A. Strambi and B. Durbeej, Photochem. Photobiol. Sci., 2011, 10, 569–579 CrossRef CAS PubMed
- A. G. Rao and I. Schapiro, Phys. Chem. Chem. Phys., 2022, 24, 29393–29405 RSC
- P. Altoe, T. Climent, G. C. De Fusco, M. Stenta, A. Bottoni, L. Serrano-Andrés, M. Merchán, G. Orlandi and M. Garavelli, J. Phys. Chem. B, 2009, 113, 15067–15073 CrossRef CAS PubMed
- C. Slavov, T. Fischer, A. Barnoy, H. Shin, A. G. Rao, C. Wiebeler, X. Zeng, Y. Sun, Q. Xu and A. Gutt,
et al.
, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 16356–16362 CrossRef CAS PubMed
- D. Morozov, V. Modi, V. Mironov and G. Groenhof, J. Phys. Chem. Lett., 2022, 13, 4538–4542 CrossRef CAS PubMed
- G. Salvadori, V. Macaluso, G. Pellicci, L. Cupellini, G. Granucci and B. Mennucci, Nat. Commun., 2022, 13, 6838 CrossRef CAS PubMed
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS
- J. F. Stanton and R. J. Bartlett, J. Chem. Phys., 1993, 98, 7029–7039 CrossRef CAS
- B. O. Roos, Ab Initio Methods in Quantum Chemistry Part 2, Adv. Chem. Phys., 1987, 69, 399–445 CrossRef CAS
- J. Finley, P.-Å. Malmqvist, B. O. Roos and L. Serrano-Andrés, Chem. Phys. Lett., 1998, 288, 299–306 CrossRef CAS
- M. Barbatti, A. J. Aquino and H. Lischka, J. Phys. Chem. A, 2005, 109, 5168–5175 CrossRef CAS PubMed
-
H. Werner, P. Knowles, G. Knizia, F. Manby, M. Schütz, P. Celani, W. Györffy, D. Kats, T. Korona and R. Lindh, et al., MOLPRO, version 2015.1, a package of ab initio programs, 2015 Search PubMed
- H.-J. Werner, P. J. Knowles, F. R. Manby, J. A. Black, K. Doll, A. Heßelmann, D. Kats, A. Köhn, T. Korona and D. A. Kreplin,
et al.
, J. Chem. Phys., 2020, 152, 144107 CrossRef CAS PubMed
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng and X. Feng,
et al.
, Mol. Phys., 2015, 113, 184–215 CrossRef CAS
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed
- P. Mark and L. Nilsson, J. Phys. Chem. A, 2001, 105, 9954–9960 CrossRef CAS
- M. Aydin and D. L. Akins, Comput. Theor. Chem., 2018, 1132, 12–22 CrossRef CAS
- C. Weiss, J. Mol. Spectrosc., 1971, 44, 37–80 CrossRef
- J. Lin and D. Shi, Appl. Phys. Rev., 2021, 8, 011302 CAS
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS
- E. Maximowitsch and T. Domratcheva, ChemRxiv, 2020, preprint DOI:10.26434/chemrxiv.12278780.v1.
- J. R. Wagner, J. Zhang, D. Von Stetten, M. Günther, D. H. Murgida, M. A. Mroginski, J. M. Walker, K. T. Forest, P. Hildebrandt and R. D. Vierstra, J. Biol. Chem., 2008, 283, 12212–12226 CrossRef CAS PubMed
- O. Falklöf and B. Durbeej, ChemPhysChem, 2016, 17, 954–957 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02112f |
| This journal is © the Owner Societies 2024 |