
Exploring non-covalent interactions in excited states: beyond aromatic excimer models†
Ariel C.
Jones
 and
Lars
Goerigk
*
and
Lars
Goerigk
*
School of Chemistry, The University of Melbourne, Parkville, Australia. E-mail: lars.goerigk@unimelb.edu.au; Tel: +61-3-83446784
First published on 18th September 2024
Abstract
Time-dependent density functional theory (TD-DFT) offers a relatively accurate and inexpensive approach for excited state calculations. However, conventional TD-DFT may suffer from the same poor description of non-covalent interactions (NCIs) which is known from ground-state DFT. In this work we present a comprehensive benchmark study of TD-DFT for excited-state NCIs. This is achieved by calculating dissociation curves for excited complexes (‘exciplexes’), whose binding strength depends on excited-state NCIs including electrostatics, Pauli repulsion, charge-transfer, and London dispersion. Reference dissociation curves are calculated with the reasonably accurate wave function method SCS-CC2/CBS(3,4) which is used to benchmark a range of TD-DFT methods. Additionally, we test the effect of ground-state dispersion corrections, DFT-D3(BJ) and VV10, for exciplex binding. Overall, we find that TD-DFT methods generally under-bind exciplexes which can be explained by the missing dispersion forces. Underbinding errors reduce going up the rungs of Jacob's ladder. Further, the D3(BJ) dispersion correction is essential for good accuracy in most cases. Likewise, the VV10-type non-local kernel yields relatively low errors and has comparable performance in either its fully self-consistent implementation or as a post-SCF additive correction, but its impact is solely on ground-state energies and not on excitation energies. From our analysis, the most robust TD-DFT methods for exciplexes with localised excitations in their equilibrium and non-equilibrium geometries are the double hybrids B2GP-PLYP-D3(BJ) and B2PLYP-D3(BJ). Their range-separated versions ωB2(GP-)PLYP-D3(BJ) or the spin-opposite scaled, range-separated double hybrid SOS-ωB88PP86 can be recommended when charge transfer plays a role in the excitations. We also identify the need for a state-specific dispersion correction as the next step for improved TD-DFT performance.
1 Introduction
Linear-response time-dependent density functional theory (TD-DFT) in the adiabatic approximation has become the method of choice for excited-state computational chemistry.1,2 TD-DFT offers reasonable accuracy at a relatively low computational cost which makes it feasible to study chemical systems of interest (up to around 100 atoms). Within the adiabatic approximation, the exchange–correlation kernel is time-independent and is therefore known from ground state DFT.3,4 As such, it could be expected that TD-DFT calculations suffer from the same pitfalls as the underlying density functional approximation (DFA).It is well-established that common DFAs fail to correctly account for the London dispersion force5 which is a long-range electron correlation effect, whereas pure DFT correlation is (semi)-local.6–8 Comprehensive ground-state benchmark studies have demonstrated that properly accounting for non-covalent interactions (NCIs), particularly dispersion forces, is essential for good computational accuracy, including for barrier heights, reaction energies, and geometries.9–14 Nonetheless, the performance of TD-DFT methods for excited-state NCIs remains significantly undertested.15
TD-DFT benchmarking studies have primarily focused on single chromophores and their excited-state properties, including vertical transition energies,16–23 oscillator strengths,16,24 electronic circular dichroism,24–26 and UV-vis spectra,24,27 among others. The most robust methods generally belong to the time-dependent double hybrid density functional approximations (TD-DHDFAs).20,21,28,29 Generalised gradient approximation (GGA) and meta-GGA methods systematically redshift excitation energies and furthermore produce low-lying ‘ghost’ excitations which are not observed in experimental spectra.17,19,21,30,31 Hybrid methods, particularly with a large proportion of Fock exchange, reduce this redshift although can in turn lead to blueshifted excitations.18,19,26,30 Global TD-DFT methods fail to describe long-range excitations—Rydberg and charge-transfer (CT)—for which range-separation (RS) is required.20,31–35 TD-DHDFAs have been shown to describe exciton coupling in dimers.15,36
Excited-state NCIs have been investigated in the context of excited complexes known as ‘exciplexes’—or ‘excimers’ when both monomers are identical—which are more strongly bound when in their excited state compared to their ground state counterpart.37 Exciplex and excimer studies have been more thoroughly reviewed previously.15 These studies have generally tested a small number of TD-DFT methods and/or chemical systems.38–41 Typical studies that systematically tested ground-state dispersion corrections relied on the older DFT-D242 approach.40,41 Some authors have adjusted dispersion corrections specifically for excited-state complexes,41,43 again limited to a small number of DFAs and chemical systems. Nonetheless, improved performance is generally observed when either a ground-state or state-specific dispersion correction is applied.
Recently, Hancock and Goerigk performed a comprehensive benchmarking study of excited-state NCIs for aromatic excimers15 which was later44 expanded to test spin-component scaled45 (SCS) and spin-opposite scaled46 (SOS) TD-DHDFAs.29,47 It was shown that the smallest errors are achieved by spin-scaled RS double-hybrid methods. An additive ground-state dispersion correction generally reduced the error of TD-DFT methods although was inadequate to fully capture the excited-state dispersion forces in aromatic excimers. This strongly suggests the need to develop an excited-state dispersion correction.
However, before this endeavour can be undertaken, benchmark-quality data is needed to gain further insights into the current state-of-the art methods and allow for future training and cross-validation of any new excited-state specific dispersion corrections. Hence, this work aims to expand the currently limited benchmarking data on excited-state NCIs. We test a number of exciplexes—benzene–neon, benzene–argon, benzene–naphthalene, toluene–tetracyanoethylene (TCNE), and styrene–trimethylamine (TMA) (Fig. 1)—which range from dispersion-dominated exciplexes to those with a significant CT component.
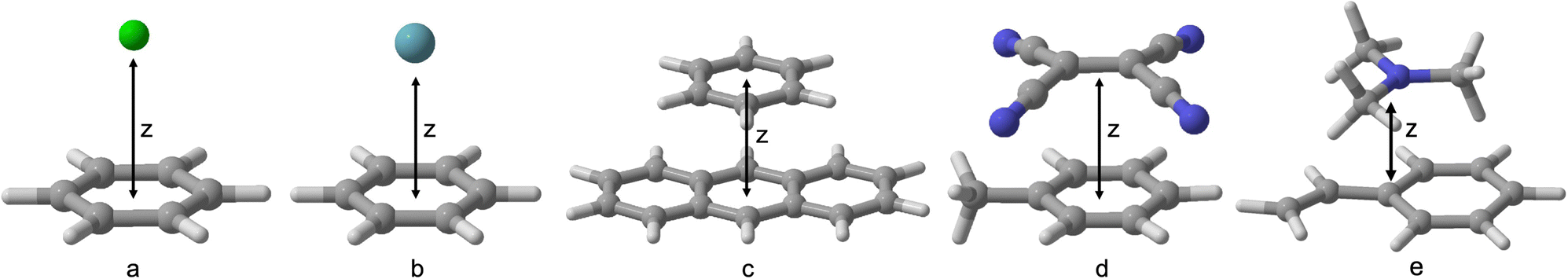 | ||
| Fig. 1 Benchmarked exciplexes with dissociation coordinate z: (a) benzene–neon; (b) benzene–argon; (c) benzene–naphthalene; (d) toluene–TCNE; (e) styrene–trimethylamine. | ||
Benzene-rare gas dimers have previously been explored by DFT in the ground state.8,48,49 However, as far as we are aware, this is the first time that TD-DFT methods have been applied to the benzene–neon and benzene–argon exciplexes. A small number of studies have used TD-DFT methods to investigate NCIs in benzene–naphthalene,39 toluene–TCNE,50 and styrene-TMA exciplexes.40 These studies employed a conservative number of DFAs and dispersion corrections either were not systematically tested or were neglected entirely. We build on these findings by testing a large number of TD-DFT methods, which include RS methods and double-hybrid DFAs, for five exciplexes.
Additionally, we present the first study of TD-SCS/SOS-DHDFAs for these exciplexes, namely with the 14 functionals presented by Casanova-Páez and Goerigk in 2021,29 which have shown promising performance for single chromophore excitations.29,51 Recently, these functionals were defined for their ground-state energy which has allowed their use for aromatic excimer interaction energies.44 In this study, we test these methods for new exciplexes beyond the aromatic dimers.
Furthermore, we systematically assess the performance of ground-state dispersion corrections when applied to exciplexes. This includes Grimme's additive dispersion correction DFT-D3(BJ)52,53 with Becke–Johnson damping. This correction adds the missing dispersion energy by an atom-pairwise summation:
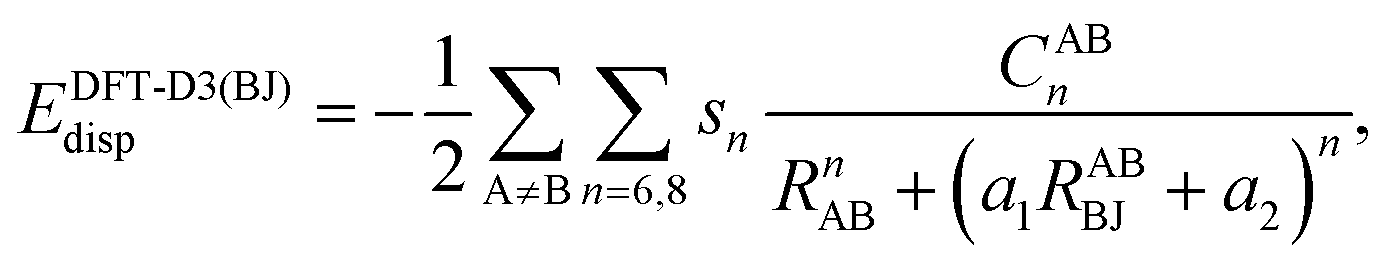 | (1) |
An alternative ground-state dispersion correction is van der Waals DFT (vdW-DFT) which incorporates non-local correlation into the DFA.55 As such, the dispersion force has an explicit dependence on electron density which is not the case for additive DFT-D approaches. Vydrov and Van Voorhis’ VV10 method56 is currently the most popular vdW-DFT approach which shows promising accuracy. Hujo and Grimme were the first to generalise non-local DFT (DFT-NL),57 in which the VV10 non-local correlation kernel can be used to augment an existing DFA as per eqn (2). DFT-NL requires the fitting of a single parameter b to ensure a seamless connection to the short-range DFA.
| EDFT-NLXC = EDFTX + EDFTC + EVV10C | (2) |
The DFT-NL approach is appealing in that it attempts to solve the dispersion issue at the DFA level. However, it has been shown that when VV10 non-local correlation is implemented in a post-SCF fashion, this yields highly comparable accuracy to the self-consistent implementation.57,58 We assess for the first time DFT-NL in the context of excited state NCIs, including both the self-consistent and post-SCF forms. We also assess other VV10-based vdW-DFT functionals that have shown good performance when tested for thermochemistry13 and on the extensive GMTKN55 test set.10,59
This study aims to expand the benchmarking data for excited-state NCIs. Specifically, we present the first comprehensive benchmarking study for exciplexes including those with significant CT character. We benchmark 38 TD-DFT methods, among which include some of the currently most accurate methods for excited state properties such as the recently developed TD-SCS/SOS-DHDFAs. Furthermore, we test the effect of the ground-state dispersion correction when applied to exciplex binding.
2 Computational details
Geometry optimisations were performed for all exciplexes in their lowest-lying singlet excited state, calculated at the second-order approximate coupled-cluster (CC2) level of theory60 in its SCS form61 and with the def2-TZVP62 triple-ζ atomic orbital (AO) basis set. Dissociation curves were generated by freezing the internal coordinates and translating monomers along the z-coordinate shown in Fig. 1. 65 geometries were obtained per dimer, with z ranging from 2 Å to 16 Å and with the greatest density of geometries about the equilibrium distance. Interaction energies were defined relative to the energy at 16 Å, where the monomers are sufficiently non-interacting. At the dissociation limit only one monomer is excited while the other remains in its electronic ground state.Reference dissociation curves were calculated for each exciplex in its lowest-lying singlet excited state. These calculations were performed at the SCS-CC2 level of theory with a triple-ζ quadruple-ζ (TZ-QZ) complete basis set (CBS) extrapolation. The CBS extrapolation procedure has been previously described in ref. 15, see ESI† for details. The authors additionally showed that this method provides suitably accurate reference data for excimer binding of small to moderately sized systems. We ran additional tests to confirm its suitability and refer the reader to Section 1 of the ESI.†
Benchmarking dissociation curves were calculated for the TD-DFT methods shown in Table 1, also for the lowest-lying singlet excitation. Where the D3(BJ) correction has been parameterised for a given method, D3(BJ) dispersion energies were calculated using the DFTD3 V3.1 standalone program.63 All TD-DFT benchmarking used the def2-QZVP basis set, except for the benzene–neon exciplex which required the decontracted def2-QZVP basis set to obtain the correct dissociation curves in ORCA 5.0.3.64,65 Testing revealed that decontracted def2-QZVP resulted in the same well-depth and position of the minimum compared to this basis set in its contracted form; however, the larger decontracted basis set was needed to produce smooth curves that otherwise suffered from numerical noise.
| ORCA 5.0.3 | TURBOMOLE 7.4.1 | QCHEM 6.0.1 |
|---|---|---|
| a D3(BJ) correction has been parameterised for this method. b The D3(BJ) correction replaces the VV10 kernel of the ωB97X-V functional.58 c Not to be confused with SOS-RSX-QIDH or SOS1-RSX-QIDH as presented later in ref. 92 and 93. | ||
| GGA | ||
BLYPa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 53,67–69 53,67–69 |
||
| PBEa53,70 |
||
| BP86a53,67,71 |
||
| meta-GGA | ||
| B97M-V72 | ||
| Hybrid | ||
| Global | ||
| B3LYPa53,73,74 |
||
| BHLYPa14,75 |
||
| PBE0a53,76,77 |
||
| Range-separated | ||
| LC-BLYP78 | CAM-B3LYPa14,79 |
ωB97X-V80 |
| LC-PBE81 | ωB97X82 | ωB97M-V83 |
| ωB97X-D3(BJ)ab58,80 |
||
| Double hybrid | ||
| Global | ||
| B2GP-PLYPa14,84 |
||
| SCS-B2GP-PLYPa29,44 |
||
| SOS-B2GP-PLYPa29,44 |
||
| B2PLYPa14,85 |
||
| SOS-B2PLYPa29,44 |
||
| PBE-QIDHa86,87 |
||
| SCS-PBE-QIDHa29,44 |
||
| SOS-PBE-QIDHa29,44 |
||
| PBE0-DHa88,89 |
||
| Range-separated | ||
| RSX-0DHa59,90 |
||
| RSX-QIDHa59,91 |
||
| SCS-RSX-QIDHa29,44 |
||
| SOS-RSX-QIDHac29,44 |
||
| ωB2GP-PLYPa20,59 |
||
| SCS-ωB2GP-PLYPa29,44 |
||
| SOS-ωB2GP-PLYPa29,44 |
||
| ωB2PLYPa20,59 |
||
| SOS-ωB2PLYPa29,44 |
||
| ωB88PP86a29,44 |
||
| SCS-ωB88PP86a29,44 |
||
| SOS-ωB88PP86a29,44 |
||
| ωPBEPP86a29,44 |
||
| SCS-ωPBEPP86a29,44 |
||
| SOS-ωPBEPP86a29,44 |
||
SCS-CC2 calculations were performed with TURBOMOLE 7.4.194 and TD-DFT calculations were performed using TURBOMOLE 7.4.1, ORCA 5.0.3,64,65 or QCHEM 6.0.195 as per Table 1. To obtain total energies for TD-SCS/SOS-DHDFAs, additional keywords had to be added to the ORCA inputs, as outlined in ref. 44. All calculations in TURBOMOLE and ORCA used the resolution of the identity (RI) approximation with the appropriate auxiliary basis sets.62,96 In ORCA, calculations additionally used the COSX approximation for exchange integrals.97 For all double-hybrid calculations, the RI approximation98,99 was applied to the (SCS/SOS-)MP2 ground state term and excited-state (SCS/SOS-) configuration interaction singles with perturbative doubles [CIS(D)] terms.100,101 The same approximation was also used in all SCS-CC2 calculations.
The RI approximation was not used in QCHEM TD-DFT calculations due to a technical issue with its parallel implementation on the National Computational Infrastructure. Nonetheless, it was found that the RI approximation resulted in essentially no change to the calculated interaction energy.
Additionally, the frozen core approximation was applied throughout, for which the chemical-core definition was chosen. The SCF convergence threshold for all calculations was 10−7Eh, and in QCHEM THRESH was set to 10−12Eh. Calculations used the grid options ‘defgrid2’ for ORCA and gridsize 7 for TURBOMOLE. QCHEM calculations required a large unpruned integration grid containing 99 radial points and 590 angular points. The default SG-1 grid was used for VV10 non-local correlation in QCHEM where applicable.
Dissociation curves were analysed in terms of their minimum values. A cubic interpolation was used between each point on the dissociation curve, resulting in a finely interpolated curve containing 14000 points from which the minimum was located with a resolution of 0.001 Å. The dissociation energy (De) is defined as the negative interaction energy at the minimum. The equilibrium distance (re) is the distance between monomers at the minimum, in terms of the coordinate z (Fig. 1). Where the TD-DFT method predicts an entirely repulsive dissociation curve (i.e. all positive interaction energies with no minimum), De has been estimated as the negative interaction energy at the reference re distance. In these cases, no re for the method is reported and the De is negative.
3 Results and discussion
In this section, we first present the calculated dissociation curves for each exciplex categorised into those with a localised or CT excitation. The following discussion primarily concerns the calculated De which is of greater chemical relevance than re. For simplicity and given the large number of methods tested, complete dissociation curves for TD-SCS/SOS-DHDFAs are provided in the ESI† and discussed generally in Section 3.3. Likewise for the D3(BJ) dispersion-corrected methods which are discussed in Section 3.4.1. Additionally, all values for De and re are provided in the ESI.† We then discuss the physical origins for exciplex binding and verify the nature of the excitation (localised or CT) based on orbital analysis and an Energy Decomposition Analysis (EDA) in Section 3.5. Finally, the averaged results for all DFAs are presented in Section 3.6.3.1 Benchmarking exciplexes with localised excitations
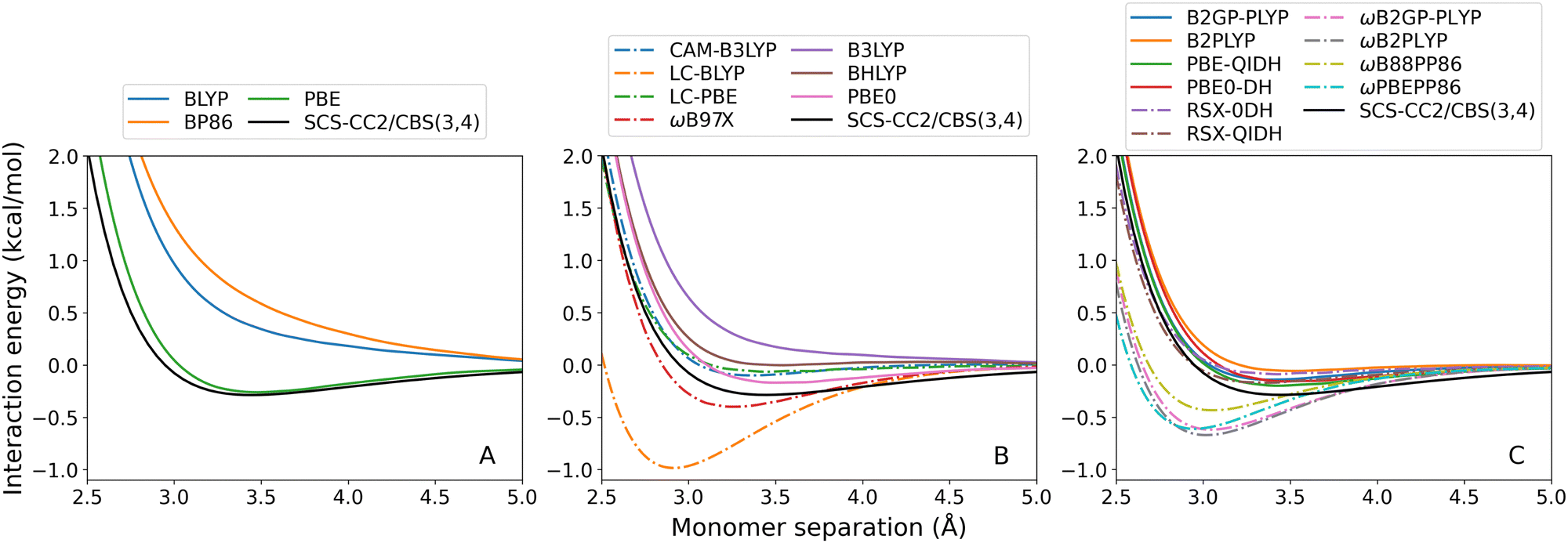 | ||
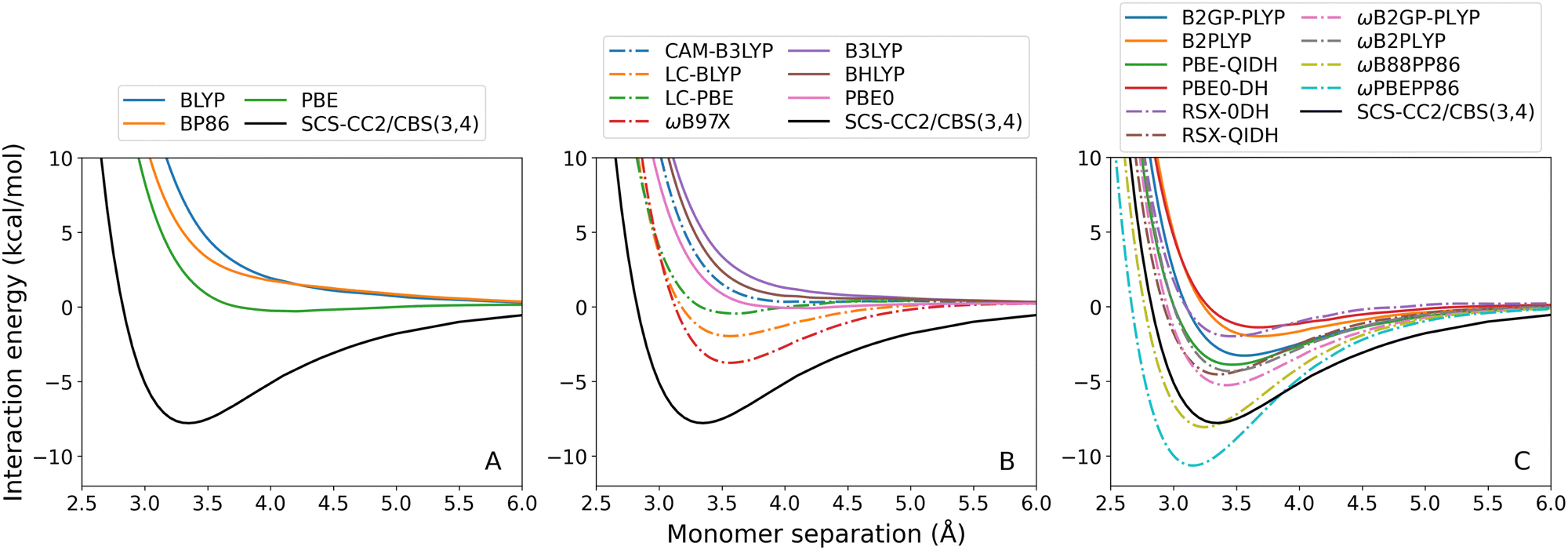
| Fig. 2 Benzene–neon TD-DFT dissociation curves, calculated for the lowest-lying singlet excited state. From left to right: (A) GGA; (B) hybrid; and (C) double-hybrid DFAs. Range-separated methods are indicated by dash-dotted lines. | ||
 | ||
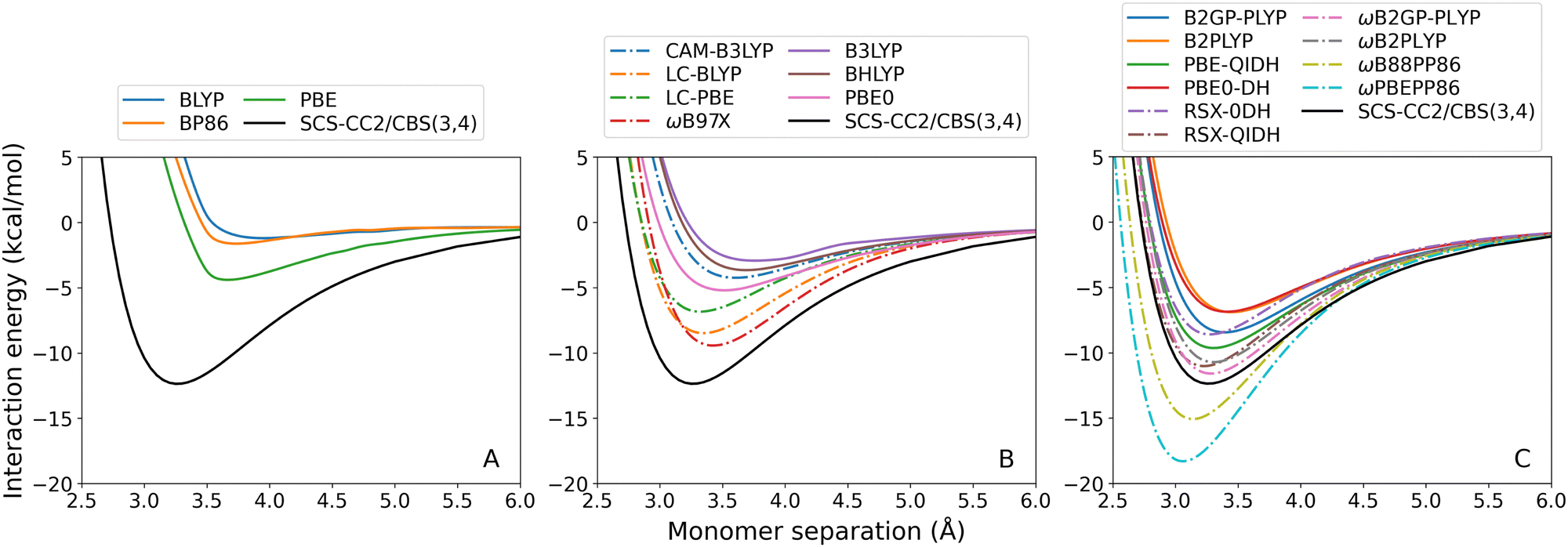
| Fig. 3 Benzene–argon TD-DFT dissociation curves, calculated for the lowest-lying singlet excited state. From left to right: (A) GGA; (B) hybrid; and (C) double-hybrid DFAs. Range-separated methods are indicated by dash-dotted lines. | ||
For both exciplexes, GGA (panel A in the two figures) and global hybrid methods (both panels B, continuous lines) tend to underestimate De. Some of these DFAs show entirely repulsive dissociation curves and fail to predict exciplex formation, which are BLYP, BP86, and B3LYP. Underbinding can be attributed to the known limitation of GGA and hybrid methods for describing dispersion forces. Of the GGA methods tested, only PBE correctly predicts an attractive De for both the benzene-rare gas exciplexes. This aligns with the established trend that PBE shows partially attractive behaviour when describing ground-state NCIs.42,102–104
The observed underbinding is improved with the inclusion of range-separation (panels B and C in both figures, dash-dotted lines): however; this results in overbinding for a number of dispersion-uncorrected RS methods (LC-BLYP, ωB97X, ωB2PLYP, ωB2GP-PLYP, ωB88PP86, and ωPBEPP86). CAM-B3LYP is barely attractive with De = 0.08 kcal mol−1 for benzene–argon and De = 0.10 kcal mol−1 for benzene–neon. While the global double-hybrid DFAs show the most consistent performance, interestingly these do not comprise the best methods for the benzene-rare gas exciplexes. Surprisingly, dispersion-uncorrected PBE is the best-performing method for benzene–neon in terms of the De error which is only 0.03 kcal mol−1. PBE-QIDH has the next best result for this exciplex, with 0.09 kcal mol−1 error. For benzene–argon, three DFAs have errors below the 0.1 kcal mol−1 chemical accuracy threshold for NCIs. These are: LC-BLYP (0.04 kcal mol−1); ωB2PLYP (0.07 kcal mol−1); and ωB97X (0.09 kcal mol−1). PBE does not perform as well for the argon system as for the neon one.
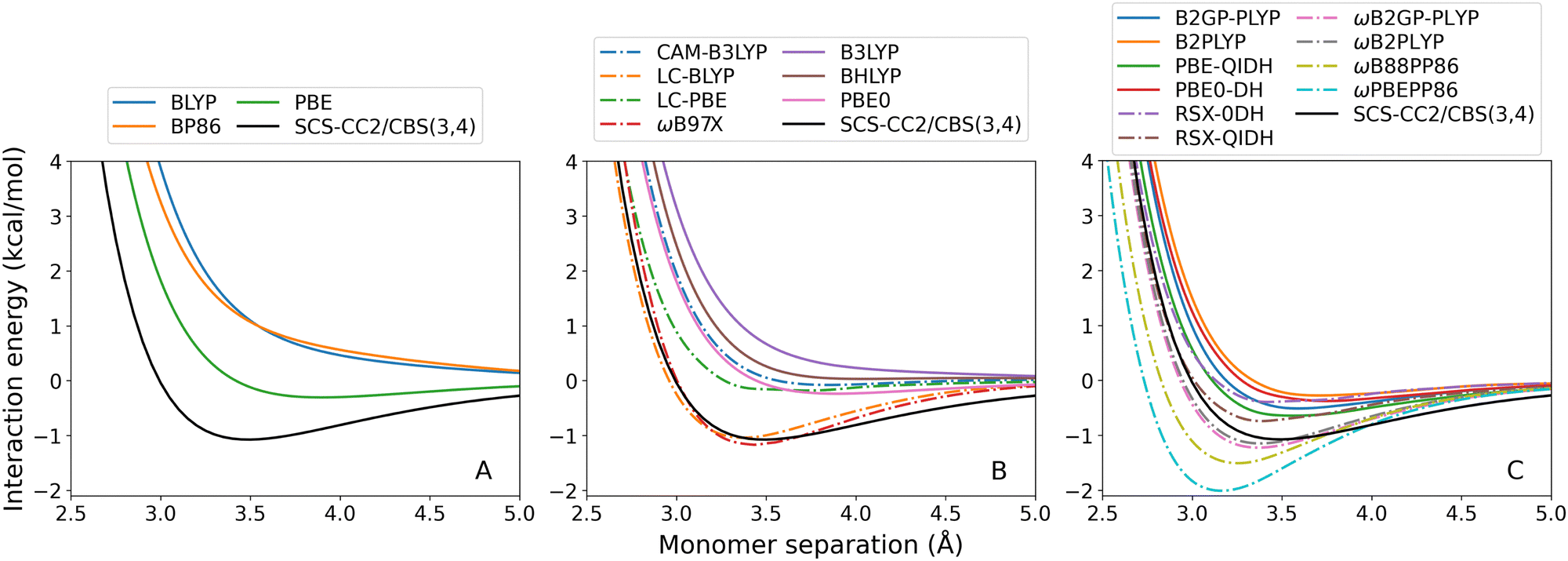
 | ||
| Fig. 4 Benzene–anthracene TD-DFT dissociation curves, calculated for the lowest-lying singlet excited state. From left to right: (A) GGA; (B) hybrid; and (C) double-hybrid DFAs. Range-separated methods are indicated by dash-dotted lines. | ||
As previously observed for the benzene-rare gas exciplexes, GGA (Fig. 4(A)) and global hybrid (Fig. 4(B), continuous lines) DFAs systematically underbind the benzene–anthracene exciplex. Both BLYP and BP86 predict entirely repulsive dissociation curves, and the De error for PBE is large (7.51 kcal mol−1). Of the global hybrids, only PBE0 predicts a positive De however also with a large error of 7.70 kcal mol−1. The benzene–anthracene exciplex is also under-bound by the RS hybrids (Fig. 4(B), dash-dotted lines), global double-hybrids (Fig. 4(C), continuous lines), and most of the RS double-hybrids (Fig. 4(C), dash-dotted lines), which was not the case for the benzene-rare gas exciplexes. CAM-B3LYP yields a purely repulsive curve with an unusual inflection point at 4.91 Å.
As before, ωB88PP86 and ωPBEPP86 overestimate De for this exciplex. Nonetheless, ωB88PP86 has the smallest absolute error for benzene–anthracene which is 0.27 kcal mol−1 compared to the reference. Of the methods that do not over-bind this exciplex, ωB2GP-PLYP has the smallest error which is 2.54 kcal mol−1. None of the dispersion-uncorrected DFAs tested are able to reproduce De within the 0.1 kcal mol−1 chemical accuracy threshold. However, for ωB88PP86 the error comprises only 3% of the total binding energy.
 | ||
| Fig. 5 Toluene–TCNE TD-DFT dissociation curves for the second singlet excited state 3A′. From left to right: (A) GGA; (B) hybrid; and (C) double-hybrid DFAs. Range-separated methods are indicated by dash-dotted lines. | ||
For toluene–TCNE 3A′, the dispersion-uncorrected methods again systematically underbind this exciplex with the exception of ωB88PP86 and ωPBEPP86 which over-bind every tested exciplex. Unlike for previous exciplexes, every GGA and global hybrid method correctly predicts a positive (attractive) binding strength for toluene–TCNE 3A′. Nonetheless, these methods have large errors ranging from 8.00 to 11.17 kcal mol−1 for the GGAs (Fig. 5(A)) and from 7.16 to 9.43 kcal mol−1 for the global hybrid methods (Fig. 5(B), continuous lines). An unusual feature of this system is that the GGA methods fail to capture the correct underlying shape of the dissociation curves, instead appearing with a sharp elbow about the calculated minima.
Underbinding persists for the RS methods (dash-dotted lines in Fig. 5(B)) and double-hybrid methods (Fig. 5(C)), although errors in De are generally reduced. For toluene-TCNE 3A′, the DFA with the closest agreement for De is ωB2GP-PLYP with 0.78 kcal mol−1 error. While this is not within the chemical accuracy limit, this represents a reasonably small 6% error.
3.2 Benchmarking exciplexes with CT transitions
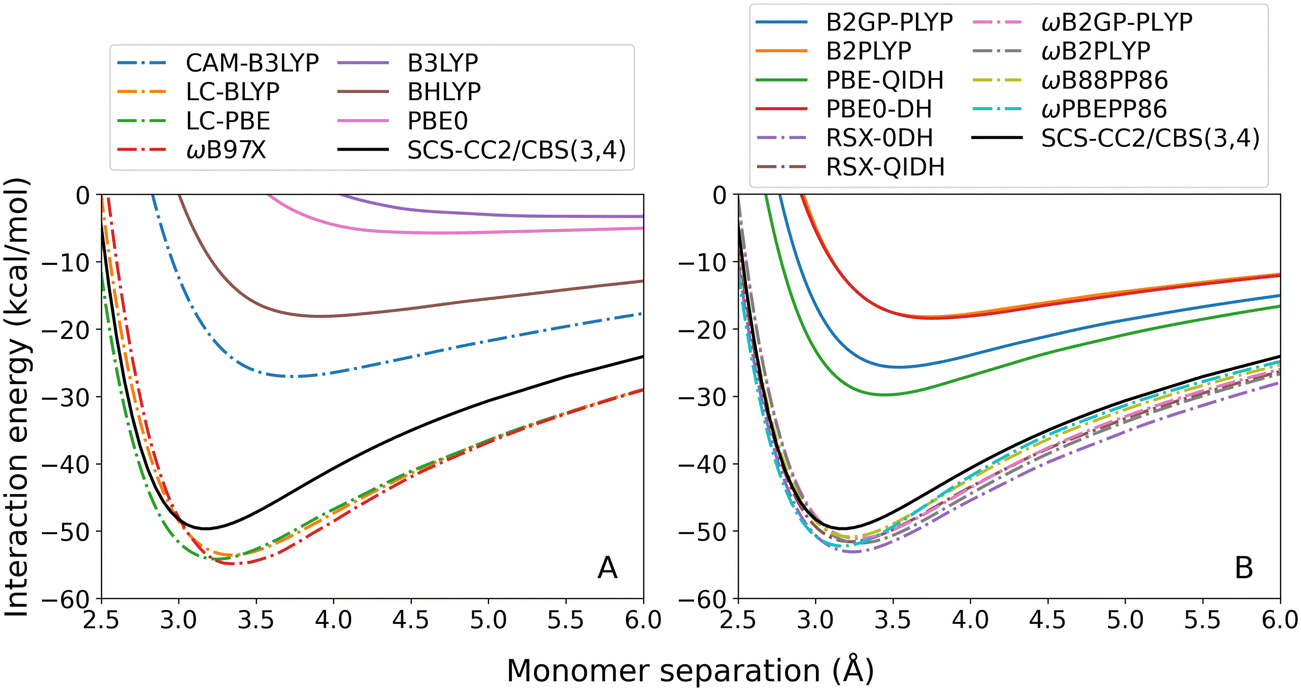 | ||
| Fig. 6 Toluene–TCNE TD-DFT dissociation curves for the first singlet excited state 2A′. From left to right: (A) hybrid; and (B) double-hybrid DFAs. Range-separated methods are indicated by dash-dotted lines. | ||
None of the GGA methods were able to calculate correct dissociation curves for the 2A′ state. This reflects the inherent limitation of GGA methods, which contain only semi-local DFT exchange, for calculating long-range excitations.31 Of the global hybrids (panel A, continuous lines) only BHLYP correctly predicts dimer formation however shows severe under-binding. Correspondingly, the calculated re is far too large with an error of 0.74 Å (23%). Notably, the performance of the hybrid functionals directly correlates with the proportion of Fock exchange they contain: 20% for B3LYP, which is the worst-performing global hybrid; 25% for PBE0; and 50% for BHLYP, which is the best-performing global hybrid. The global double-hybrid methods (panel B, continuous lines) again reduce exciplex underbinding although with particularly large errors in De ranging from 19.87 to 31.46 kcal mol−1.
For both the hybrid and double-hybrid methods, RS (dash-dotted lines) greatly improves the calculated De and re which is unsurprising given the importance of long-range Fock exchange for describing CT excitations. However, with the exception of CAM-B3LYP, this results in an over-correction that leads to over-binding. Nonetheless, the lowest absolute error is given by ωB88PP86 which over-estimates De by 1.20 kcal mol−1 (2% error).
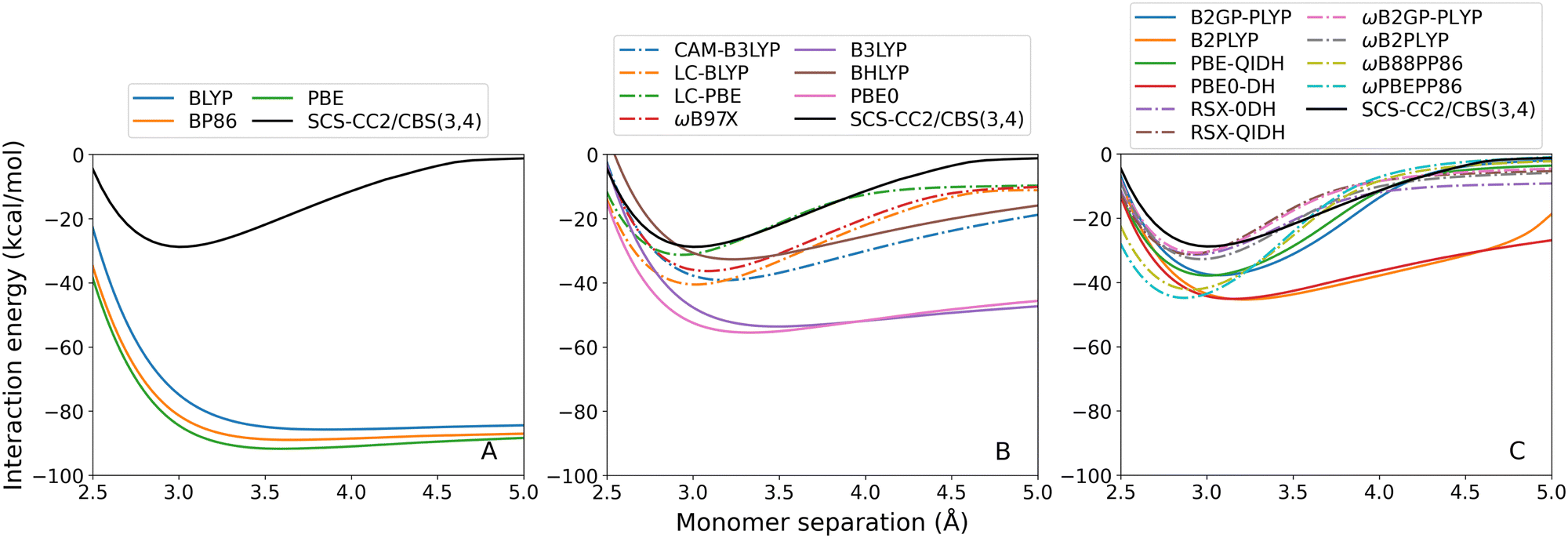 | ||
| Fig. 7 Styrene-TMA TD-DFT dissociation curves, calculated for the lowest-lying singlet excited state. From left to right: (A) GGA; (B) hybrid; and (C) double-hybrid DFAs. Range-separated methods are indicated by dash-dotted lines. | ||
The styrene-TMA dissociation curves show the opposite trend in that all TD-DFT methods over-bind this exciplex. This over-binding can be attributed to strongly redshifted excitations at the minimum which is a CT excitation, whereas this redshift is less pronounced or becomes a blueshift at the endpoint which has a localised excitation. This imbalance leads to an artificially lowered minimum in the interaction energy curves due to the total energies of the minimum structures being too low and the interaction energies being the differences between total energies. Examples for select DFAs are shown in Table 2. GGA methods, which show the most pronounced over-binding and the largest redshifts near their respective minima at 3 Å, underestimate SCS-CC2/def2-QZVP energies by about 2 eV. This redshift is reduced with the inclusion of Fock exchange, and in particular for RS methods. Excitation energies for all assessed DFAs are shown in the ESI† (Section 4.2).
| DFA | Deviation (eV) | |
|---|---|---|
| 3 Å | 16 Å | |
| BP86 | −2.10 | 0.58 |
| PBE | −2.13 | 0.58 |
| BLYP | −2.03 | 0.54 |
| B3LYP | −1.26 | 0.04 |
| BHLYP | −0.07 | 0.42 |
| CAM-B3LYP | −0.41 | 0.31 |
| ωB97X | 0.02 | 0.43 |
| SOS-ωB2PLYP | 0.10 | 0.00 |
The GGA methods (Fig. 7(A)) have particularly large errors in De ranging from 57.00 to 62.94 kcal mol−1. The double-hybrid and RS methods show improvements compared to the other methods. In particular, RS is needed for a more accurate treatment of this CT excitation. The RS double-hybrid methods (7C, dash-dotted lines) generally show the most robust performance, with the exception of ωB88PP86 and ωPBEPP86. The best performance is achieved by ωB2GP-PLYP with 1.92 kcal mol−1 absolute error in De.
3.3 Spin-scaled double-hybrid methods
Fourteen TD-SCS/SOS-DHDFAs have been recently developed for the calculation of excitation energies.29 They have then been extended to also define their ground-state total energies, which allows the calculation of interaction energies for excited states.44 These methods were tested for the exciplexes in the current study. For brevity, the complete dissociation curves are given in the ESI† and examples of typical SCS/SOS results are shown in Fig. 8 for the toluene–TCNE localised 3A′ excitation.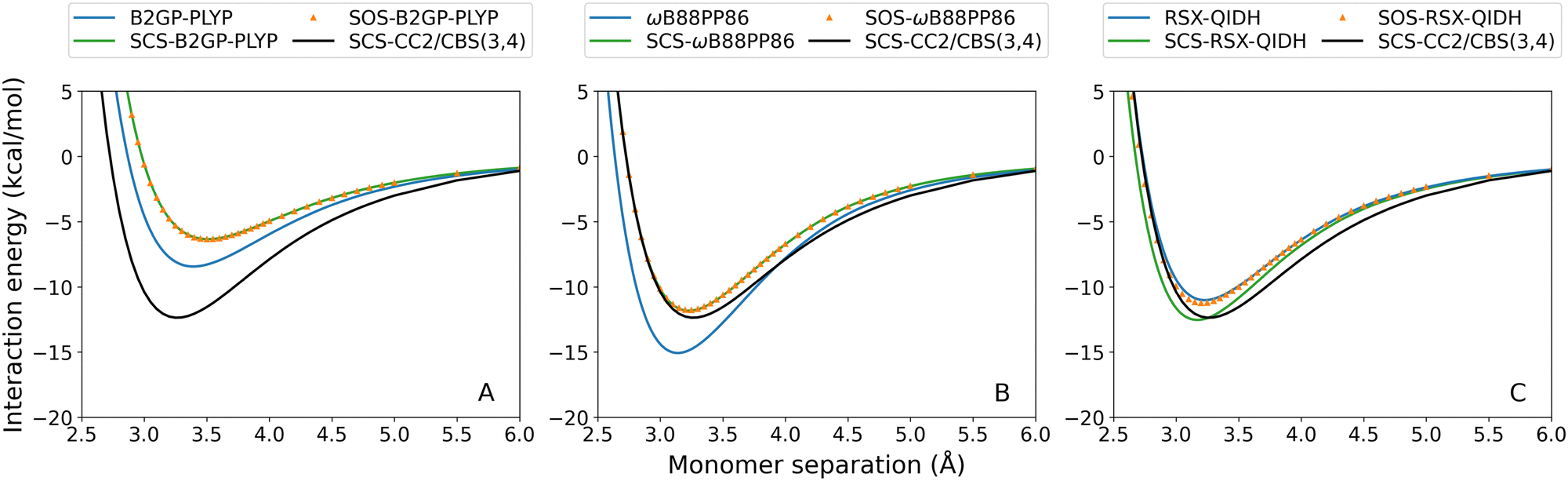 | ||
| Fig. 8 Toluene–TCNE 3A′ dissociation curves for representative TD-DHDFAs and their SCS/SOS variants: (A) B2GP-PLYP; (B) ωB88PP86; (C) RSX-QIDH. | ||
With the exception of SCS-RSX-QIDH, spin-scaling (both SCS and SOS) consistently reduces the binding strength predicted for all exciplexes. This is unsurprising given the small scaling parameters fitted for the SCS and SOS methods, which results in reduced non-local correlation compared to the unscaled methods.29,44 Of the spin-scaled methods, SOS methods generally predict weaker binding although in some cases the SCS and SOS methods calculate virtually identical results. This can again be rationalised by the similar parameterisation between SCS and SOS variants. For example, parameter fitting for both B2PLYP and ωB2PLYP leads to SCS results that are identical to the SOS fit (i.e. no same-spin contribution and identical opposite-spin scaling). Indeed, all of the SCS methods have small or negligible contributions to their same-spin term with the only exception being SCS-RSX-QIDH. As a result, SCS-RSX-QIDH shows the reverse trend in which this method calculates stronger exciplex binding compared to unscaled RSX-QIDH.
The overall effect of TD-SCS/SOS-DHDFAs is both method- and system-dependent. Where the unscaled functional already demonstrates under-binding, this is exacerbated by spin-scaling (e.g.Fig. 8(A)). However for methods that overestimate De, the SCS and SOS methods yield improved performance (e.g.Fig. 8(B)). The only exception is for SCS-RSX-QIDH which shows the opposite trend in that SCS improves under-binding (e.g.Fig. 8(C)) or exacerbates over-binding. Generally, ωB88PP86 and ωPBEPP86 show the greatest improvements by spin-scaled methods. These methods have a large MP2 contribution and consistently over-bind the tested exciplexes, and as such their improved behaviour with SCS/SOS is unsurprising.
3.4 Effect of ground-state dispersion correction
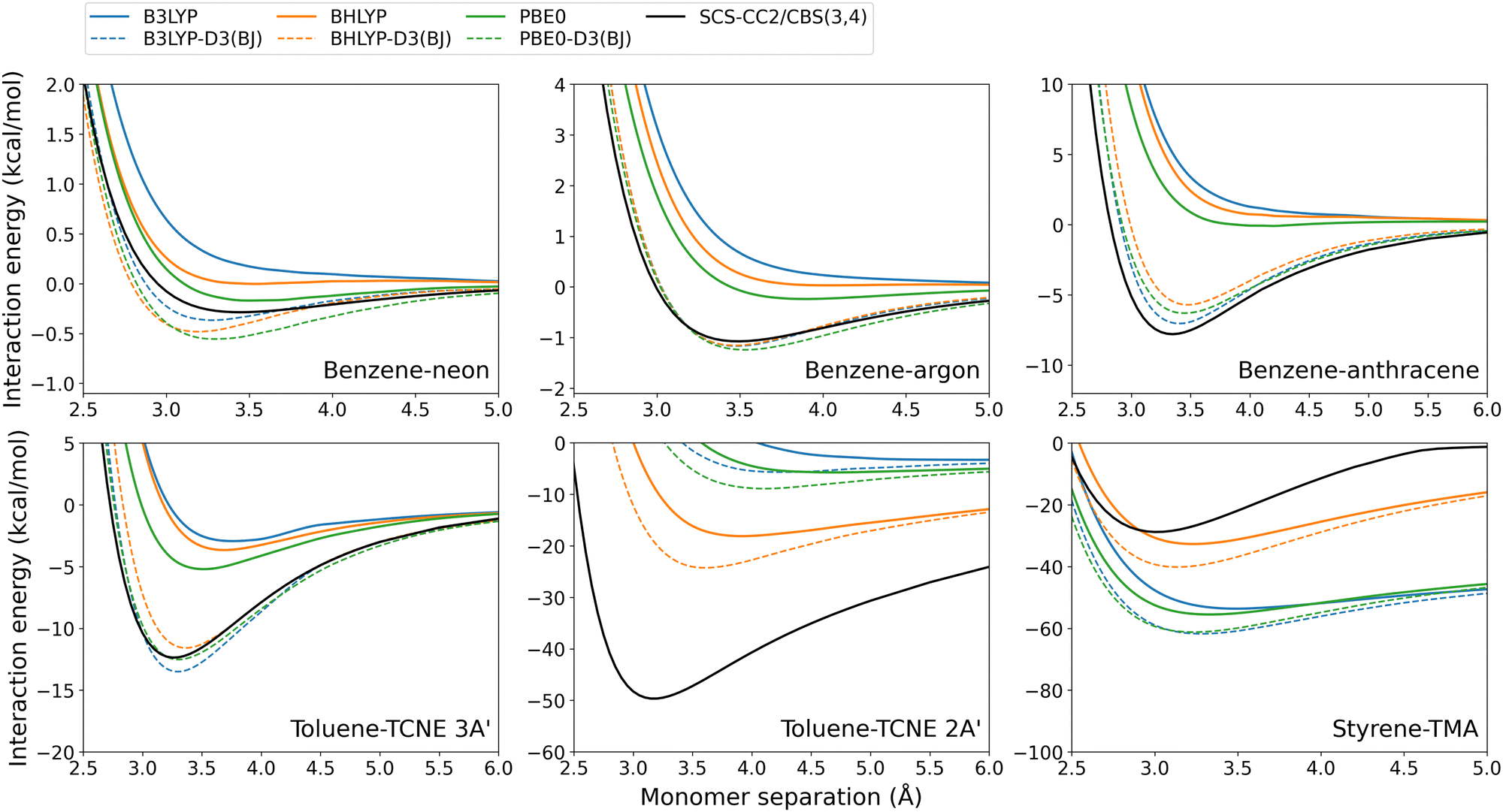 | ||
| Fig. 9 Exciplex dissociation curves for global hybrid DFAs with and without the D3(BJ) dispersion correction. | ||
Unsurprisingly, the D3(BJ) correction was found to increase the binding strength predicted by almost every DFA which results in a larger De and shorter re. In some cases, the dispersion-uncorrected DFA failed entirely to predict exciplex formation (for example, the benzene-rare gas dimers calculated with B3LYP shown in Fig. 9) which is generally fixed by the D3(BJ) correction. A notable exception is that D3(BJ) makes little or negligible difference to the binding strength predicted by RS TD-DHDFAs, including their SCS/SOS variants. This has been previously noted for excimer binding15,44 and on the GMTKN55 test set.59,107 The physical reason for this remains unclear however is unlikely to be a result of the fit set used for the damping parameters. A recent re-fit was performed for the D3(BJ) damping parameters on a larger fit set, for the RS double hybrid methods ωB2GP-PLYP, ωB2PLYP, and RSX-QIDH.44 This refit made no difference when tested for exciplex binding (see Fig. S14, ESI†) as was also the case in the original excimer study.44
In general, D3(BJ) greatly improves the results for the DFAs and exciplexes tested herein, as is discussed in Section 3.6. Nonetheless, the performance of this correction depends on the method and system tested. The benzene-rare gas exciplexes, which have particularly weak binding, show reasonably good agreement for some dispersion-uncorrected DFAs. As such, D3(BJ) can result in an overcorrection with a predicted De that is larger than the reference, which is particularly so for benzene–neon. Despite this, D3(BJ) generally results in reduced absolute errors for these exciplexes. On the other hand, the larger benzene–anthracene and toluene–TCNE 3A′ exciplexes generally exhibit underbinding for the dispersion-uncorrected methods. Applying the D3(BJ)-correction leads to good agreement with the reference and greatly reduces the error for each DFA tested (not including the RS TD-DHDFAs as discussed above).
The CT exciplexes again proved challenging to model with dispersion-corrected methods. For toluene–TCNE 2A′, the global hybrid and global double-hybrid DFAs showed severe underbinding which could not be entirely compensated for by D3(BJ). The calculated De values remain significantly underestimated by up to 44 kcal mol−1 in the case of B3LYP-D3(BJ). In contrast, the RS methods show much better agreement to the reference for toluene–TCNE 2A′. As such, these methods can result in over-binding when the D3(BJ) correction is added.
Styrene-TMA is the only exciplex which is systematically over-bound by the dispersion-uncorrected DFAs. As previously discussed in Section 3.2.2, this is due to the CT excitation at the equilibrium distance being strongly redshifted while at the dissociation limit this redshift decreases or becomes a blueshift. This imbalance causes an artificial over-binding from dispersion-uncorrected DFAs. As such, adding the D3(BJ) dispersion correction, which is by definition attractive, exacerbates this over-binding. This reflects a limitation of the underlying DFA rather than the dispersion correction.
The exception to this is for spin-scaled TD-DHDFAs, specifically SOS-ωB2GP-PLYP, SCS-ωB2GP-PLYP, and SOS-ωB2PLYP. For these methods, spin-scaling counteracts the over-binding predicted by the plain DFA and as such the D3(BJ) correction can improve De (see Fig. S6, ESI†).
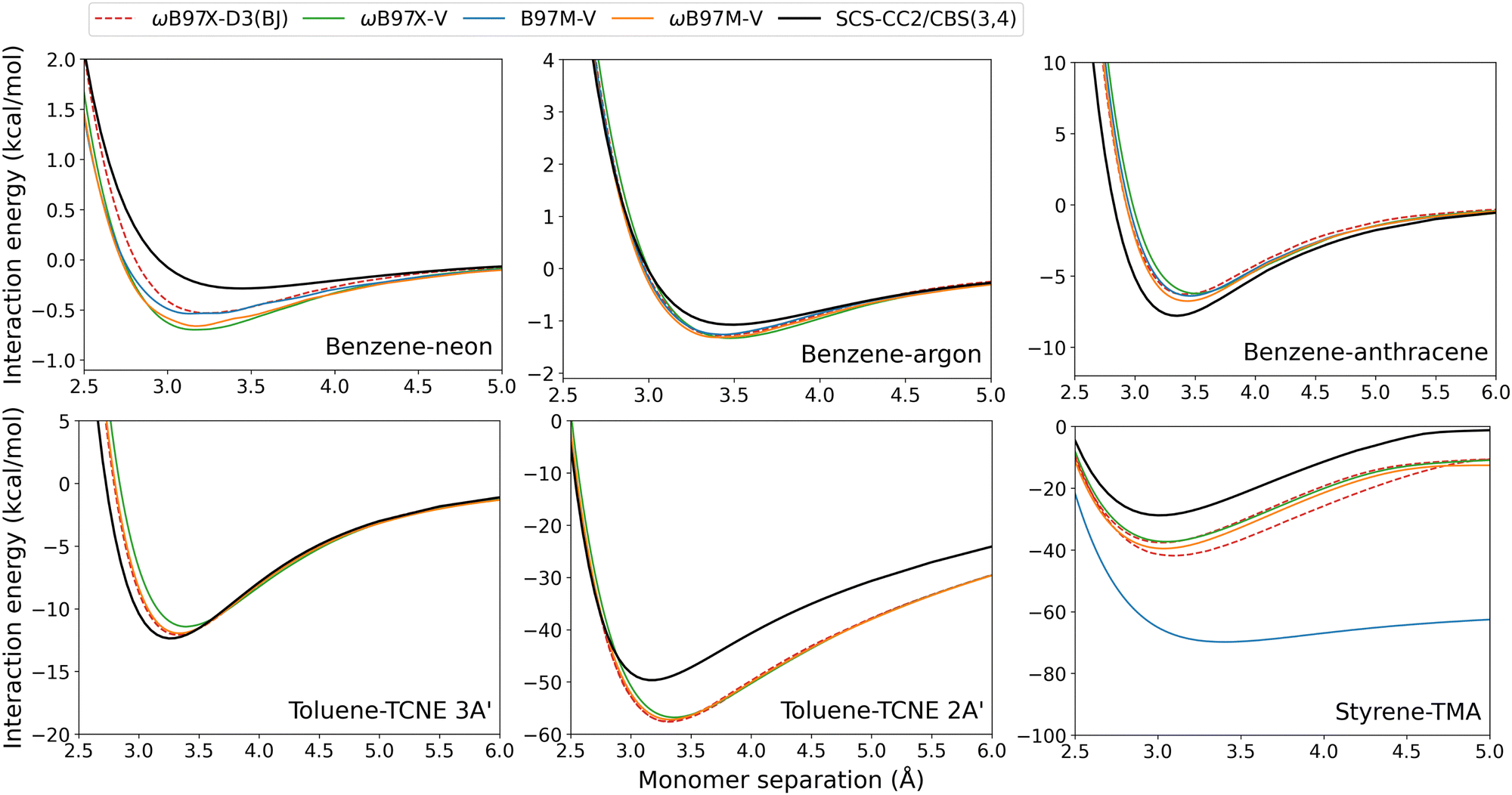 | ||
| Fig. 10 Exciplex dissociation curves for combinatorially-optimised functionals with the VV10 dispersion correction. The ωB97X-D3(BJ) results are also shown for comparison. | ||
These VV10-corrected methods overbind both of the benzene-rare gas exciplexes and underbind benzene–anthracene and toluene–TCNE 3A′. This mirrors our observations made for the D3(BJ) correction. For these four exciplexes with localised excitations, De errors are reasonably small and range from: 0.19–1.40 kcal mol−1 for B97M-V; 0.26–1.56 kcal mol−1 for ωB97X-V; and 0.24–1.03 kcal mol−1 for ωB97M-V. Interestingly, ωB97X-D3(BJ) slightly outperforms the original ωB97X-V functional for each of these four exciplexes.
The VV10-corrected methods show the greatest errors for the CT exciplexes. Toluene–TCNE 2A′ is over-bound by both ωB97X-V and ωB97M-V. In Section 3.2.1, it was shown that dispersion-uncorrected RS hybrid methods yield close agreement to the reference dissociation curve for toluene–TCNE 2A′. As such, this overbinding can be explained as an over-correction from VV10 dispersion. Styrene-TMA again shows overbinding by all of the VV10-corrected methods, especially for B97M-V which contains no Fock exchange. This overbinding is caused by a redshift in the CT excitation energy as described previously. The ωB97X-D3(BJ) and ωB97X-V functionals again show similar performance, although for these exciplexes ωB97X-V has slightly reduced errors.
Previous studies have shown that incorporating VV10 non-local correlation in a post-SCF fashion yields accuracies highly similar to self-consistent VV10.57,58 Furthermore, the effect of VV10 on TD-DFT excitation energies was previously shown to be small. For example, the excitation energies of single chromophores were calculated with and without VV10 for the functionals B97M-V, ωB97X-V, and ωB97M-V and the VV10 correction resulted in less than 0.01 eV change to the mean signed error.18
We tested two GGA functionals, BLYP and PBE, which were augmented with VV10 as per standard DFT-NL (eqn (2)) with values for b that were respectively 4.0057 and 6.40.108 The VV10 correction was implemented in three ways:
1. Self-consistent VV10 for the ground-state energy and VV10 included in the exchange–correlation kernel of the TD-DFT calculation.
2. Self-consistent VV10 for the ground-state energy and VV10 not included in the exchange–correlation kernel of the TD-DFT calculation.
3. Post-SCF VV10 correction for the ground-state energy and VV10 not included in the exchange–correlation kernel of the TD-DFT calculation.
These methods were tested for the benzene–argon exciplex as a case study and the results for De and re are shown in Table 3.
| Method | D e (kcal mol−1) | r e (Å) |
|---|---|---|
| Reference | 1.07 | 3.49 |
| BLYP | ||
| A | 1.13 | 3.44 |
| B | 1.15 | 3.46 |
| C | 1.09 | 3.48 |
| D3(BJ) | 1.10 | 3.49 |
| PBE | ||
| A | 1.41 | 3.48 |
| B | 1.42 | 3.48 |
| C | 1.40 | 3.46 |
| D3(BJ) | 1.27 | 3.49 |
The calculated De and re show minimal change depending on how VV10 non-local correlation is implemented. For BLYP-NL, the difference in calculated binding energy is 0.04 kcal mol−1 and for PBE-NL is only 0.01 kcal mol−1 for post-SCF (C) versus fully self-consistent VV10 (A). The effect of VV10 on the calculated excitation energies is also small. This is reflected by the minimal change to De when VV10 is included fully self-consistently (A) compared to when only the ground-state energy is treated self-consistently and VV10 is not applied to the excitation energy (B).
For both BLYP-NL and PBE-NL, post-SCF VV10 (C) results in a smaller binding strength compared to when VV10 is included self-consistently (A). As a result, both of the post-SCF DFT-NL methods have the best agreement to the reference due to a reduction in over-binding. PBE-D3(BJ) outperforms any of its VV10 counterparts. For BLYP, the best-performing method is BLYP-NL with post-SCF VV10 (C), although this is closely followed by BLYP-D3(BJ) which outperforms the self-consistent VV10 methods.
Overall, this case study supports that VV10 non-local correlation can be used post-SCF as an additive dispersion correction without loss of accuracy and at a lower computational cost.57,58 Additionally, VV10 makes little difference to calculated excitation energies as previously reported by Head-Gordon and coworkers.18 We further recommend that D3(BJ) can be used as an inexpensive dispersion correction which shows comparable accuracy to VV10.
3.5 Origin of exciplex binding strength
In this section, we provide an analysis of the physical origins for binding strength of the herein assessed dimers and verify the localised or CT nature of the excitations. An EDA scheme for exciplexes has been developed by Ge et al. which uses absolutely localised molecular orbitals (ALMO-EDA).109 Exciplex interaction energy is decomposed into a frozen component, containing electrostatics and Pauli repulsion, energy from polarisation, and that from charge transfer (CT). The current scheme suffers from known limitations in that dispersion corrections contaminate the Pauli term and furthermore that the polarisation contribution is dependent on basis-set size.109 Nonetheless, ALMO-EDA calculations have been performed for all exciplexes to qualitatively measure contributions to their binding strengths. These calculations were performed with ωB97M-V which has shown sufficiently accurate performance for each exciplex. The def2-TZVP basis set was used to balance the basis set superposition error with the polarisation issue known for ALMO-EDA. The ALMO-EDA results are shown in Fig. 11.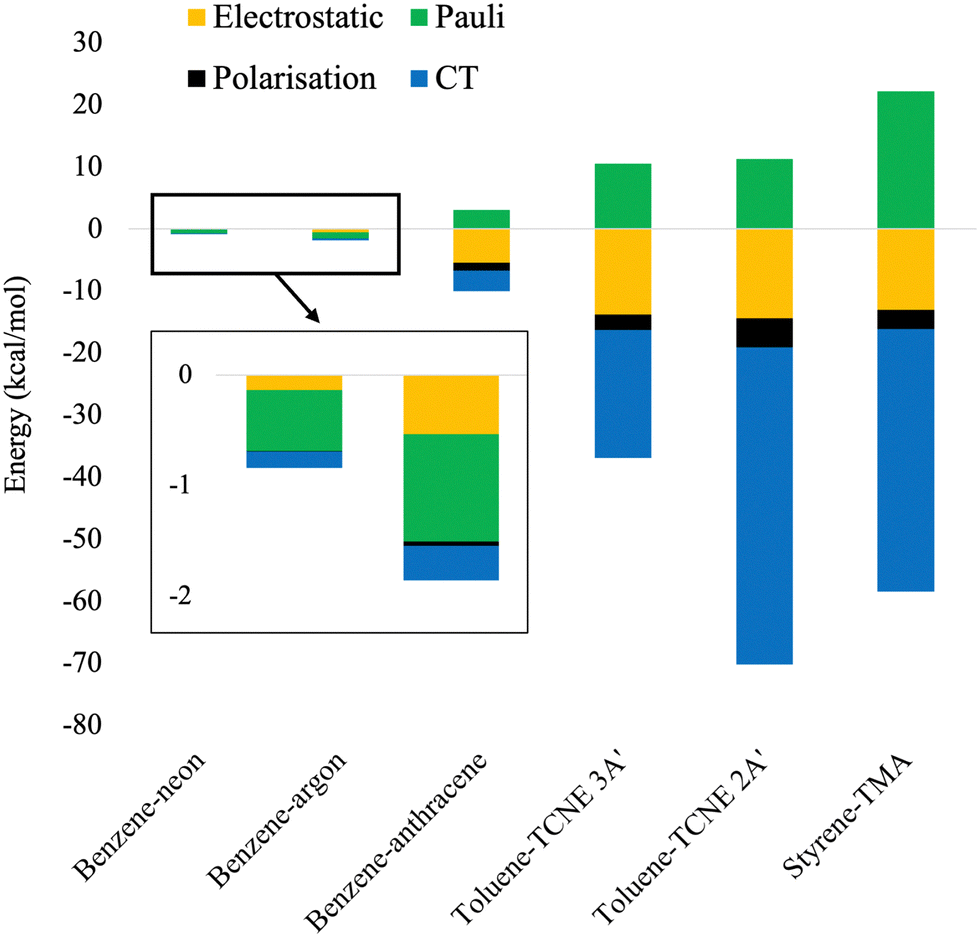 | ||
| Fig. 11 Stacked energy diagram of ALMO-EDA energies for exciplexes calculated with ωB97M-V/def2-TZVP. | ||
From Fig. 11, the ALMO-EDA calculations reproduce the correct qualitative trend for exciplex binding strength. The benzene-rare gas exciplexes have small dispersion-dominated interaction energies. For these exciplexes, their unphysically attractive Pauli energy is an artefact of ALMO-EDA wherein dispersion is included in this term. This contamination of the Pauli term has been noted in ref. 109 and which we further verified, see ESI† for details. Benzene–anthracene shows an overall larger binding strength which is significantly due to electrostatics and some CT nature. Toluene–TCNE 3A′ again has stronger binding and contains significant electrostatic and CT contributions. Finally, toluene–TCNE 2A′ and styrene-TMA have the largest binding strengths which are a result of their large CT components.
A different scheme for quantifying the CT nature of an excitation was developed by Plasser and Lischka, which uses the one-electron transition density matrix to calculate CT metrics.110 In this scheme, the calculated CT number varies from 0 for entirely localised excitations to 1 for entirely charge-separated excitations. The calculated CT numbers, and additional CT metrics, are given in the ESI.† These calculated CT numbers generally complement the ALMO-EDA findings. The benzene-rare gas exciplexes have no CT character (CT number = 0) and the excitation is entirely localised on benzene. There is some CT quality for the benzene–anthracene exciplex (CT number = 0.04), although this is minimal and the excitation is primarily localised on anthracene. For toluene–TCNE, the 3A′ state has a small CT number of 0.04. This does not reflect the more significant CT component as predicted by ALMO-EDA although is consistent with the visualised molecular orbitals (MOs) for this transition ((HOMO−2)–LUMO excitation, Fig. S15, ESI†) which shows this excitation localised on the TCNE fragment. The CT number is 0.92 for toluene–TCNE 2A′ and is 0.83 for styrene-TMA, which confirms that these exciplexes have predominantly CT excitations. This is consistent with the MOs for these excitations (e.g. HOMO–LUMO transition for toluene–TCNE 2A′, see Fig. S15, ESI†) and agrees with the ALMO-EDA results.
In conclusion, the analyses presented in this section complement the system-specific trends observed in the previous sections.
3.6 Averaged functional performance
To assess the overall performance of a TD-DFT method across all exciplexes, a mean absolute error (MAE) can be calculated as: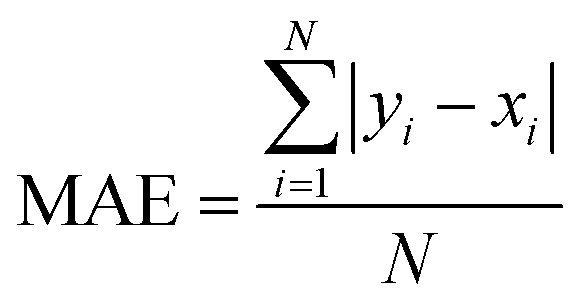 | (3) |
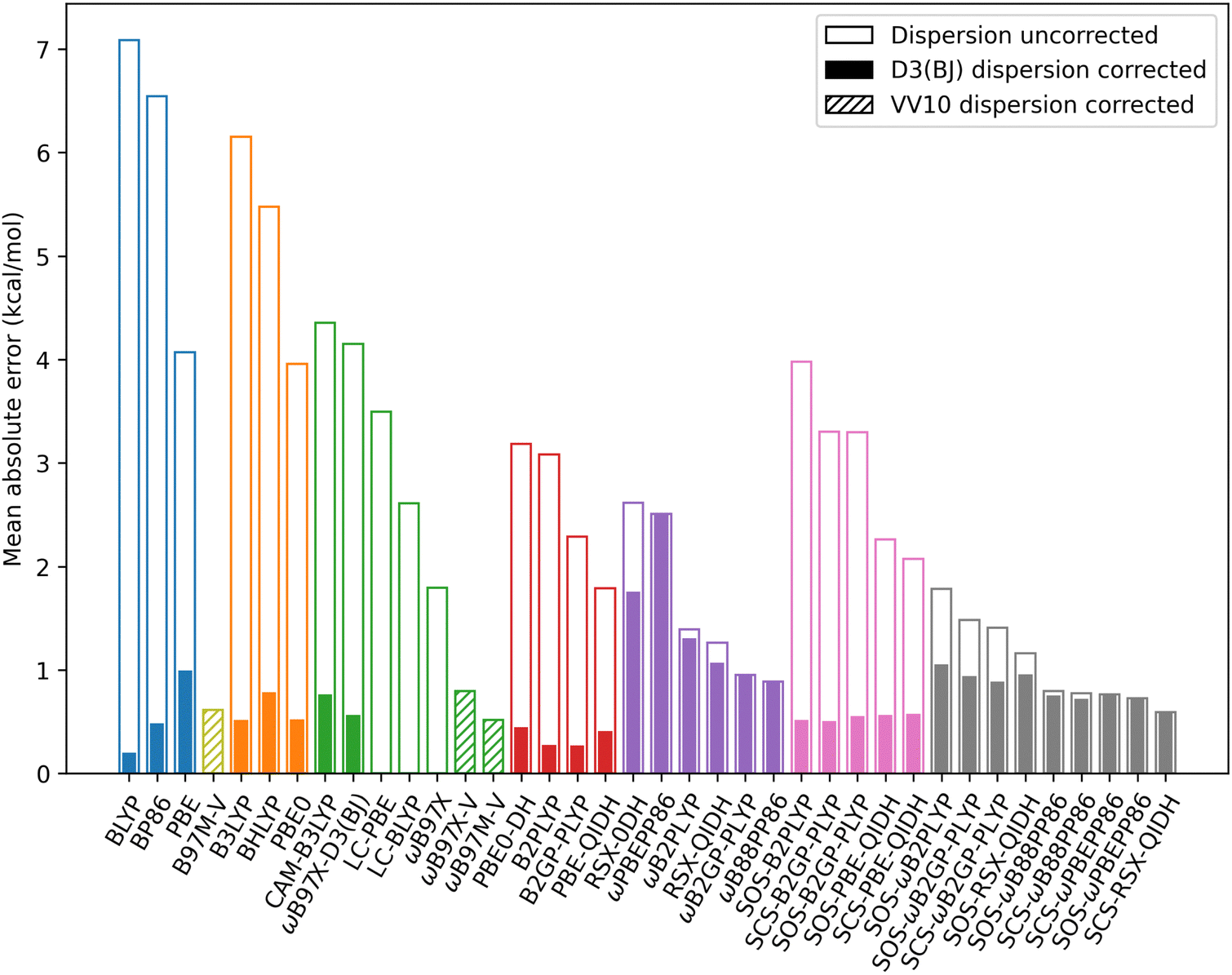 | ||
| Fig. 12 MAEs for exciplexes with a localised excitation which are benzene–neon, benzene–argon, benzene–anthracene, and toluene–TCNE 3A′. MAEs are for De in kcal mol−1. | ||
RS reduces the MAEs for hybrid methods from an average of 5.19 kcal mol−1 to 3.28 kcal mol−1 Similarly, for the unscaled double-hybrids the averaged MAE reduces from 2.59 kcal mol−1 to only 1.60 kcal mol−1 with RS. Spin-scaling has a variable effect on the MAE for TD-DHDFAs. Each global double-hybrid shows an increased MAE when either SCS or SOS is applied. However, three of the RS double-hybrids are on average improved by spin-scaling (both SCS and SOS variants). These are RSX-QIDH, ωB88PP86, and ωPBEPP86. This is unsurprising for the latter two DFAs since spin-scaling was shown to counter the systematic overbinding for these methods.
For all DFAs tested, the ground-state D3(BJ) correction considerably reduces the MAE except for those RS double-hybrids for which the dispersion correction has no impact. Overall, the dispersion-corrected method with the lowest MAE is BLYP-D3(BJ) with 0.20 kcal mol−1 error for De. This is followed closely by B2GP-PLYP-D3(BJ) (0.27 kcal mol−1 MAE) and B2PLYP-D3(BJ) (0.28 kcal mol−1 MAE). These MAEs outperform all of the dispersion-uncorrected methods. The strong performance of BLYP-D3(BJ) mirrors findings that this functional is competitive with double-hybrid accuracy for ground-state NCIs when tested on the S22 and S66 datasets.10
The VV10 dispersion-corrected methods generally have MAE's comparable, or slightly worse, compared to the D3(BJ)-corrected methods. B97M-V, which belongs to the meta-GGA rung on Jacob's ladder, is out-performed by two D3(BJ)-corrected GGAs but has a smaller MAE than PBE-D3(BJ). For the RS hybrid methods tested, MAEs are similar for VV10-corrected methods (ranging from 0.52 to 0.80 kcal mol−1) compared to D3(BJ)-corrected methods (0.57 to 0.76 kcal mol−1). Notably, ωB97X-D3(BJ) outperforms the original ωB97X-V functional by 0.23 kcal mol−1. Of the VV10-corrected methods, ωB97M-V has the best performance with a MAE of 0.52 kcal mol−1.
In terms of the equilibrium geometry re, trends to the MAEs are generally consistent with the observed trends for De. Going up Jacob's ladder and incorporating RS tend to decrease the MAE for re. However, MAEs are particularly large for the RS double-hybrid methods which have shown exciplex over-binding: ωB2GP-PLYP, ωB2PLYP, ωB88PP86, and ωPBEPP86. These methods predict a distance re which is considerably shorter than the reference. As before, the D3(BJ) correction greatly improves the calculated re except for the RS double-hybrids. When considering re, the best performing method is PBE-QIDH-D3(BJ) and the MAE is 0.03 Å. More details on the statistics for re can be found in the ESI.†
Finally, DFAs were assessed in terms of their performance for non-equilibrium geometries. In the spirit of the S66X8 data set,11 8 geometries were selected by scaling the equilibrium distance by multipliers which are: 0.90, 0.95, 1.00, 1.05, 1.10, 1.25, 1.50, and 2.00. That is, the dissociation curves were sampled at two compressed geometries, at the equilibrium geometry, and at 5 stretched geometries. The equilibrium distances were taken from the minima of the SCS-CC2/CBS(3,4) reference curves. The MAEs averaged over all non-equilibrium distances are given in the ESI.† Mean absolute percentage errors (MAPEs) were also calculated as per the original S66X8 analysis11 and are also shown in the ESI.† From this analysis, B2PLYP-D3(BJ) has the smallest MAE which is 0.24 kcal mol−1. The MAPEs generally yield similar trends, however the best method becomes PBE-QIDH-D3(BJ) (10% MAPE).
Exciplexes with a localised excitation showed near-universal improvement when a dispersion correction was included. This is not the case for the CT exciplexes. For styrene-TMA, the poor performance of the dispersion-corrected methods was shown to be a limitation of the underlying DFAs which over-bind this exciplex due to a redshifted CT excitation (see Section 3.2.2). The dispersion correction exacerbates overbinding for this exciplex. On the other hand, toluene–TCNE 2A′ shows improved performance when a dispersion correction is applied. However, the dispersion correction was insufficient to recover the true well-depth. The inconsistent behaviour of ground-state dispersion corrections for these exciplexes suggests that a state-specific dispersion correction is needed for robust performance with TD-DFT methods. These exciplexes further highlight the inherent limitations of current TD-DFT methods for long-range excitations.
4 Conclusions
In this study we have investigated excited-state non-covalent interactions (NCIs) in a series of dimers that consist of two different monomers with both older popular and more modern TD-DFT approaches. Accurate dissociation curves were calculated at the SCS-CC2/CBS(3,4) level of theory for five dimers with a total of six excited states. This reference method was used to benchmark a large number of DFAs which span the commonly-used rungs of Jacob's ladder and include RS methods and TD-SCS/SOS-DHDFAs. Additionally, we evaluated the performance of two ground-state dispersion corrections which are the DFT-D3(BJ) additive dispersion correction and the VV10 non-local kernel. Overall, the DFAs tested generally showed improved accuracy going up rungs of Jacob's ladder. GGA methods should be avoided in TD-DFT as these resulted in large errors, ghost excited states, and significant redshifts for CT excitation energies. For exciplexes with a localised excited state, GGA methods showed considerable under-binding and in some cases failed entirely to predict exciplex formation. Hybrid and double-hybrid DFAs reduced exciplex under-binding as did the RS methods.Exciplexes with CT character proved particularly challenging for many of the assessed TD-DFT methods. For styrene-TMA, TD-DFT methods showed artificial over-binding due to redshifted excitation energies. The CT exciplexes required methods with a large proportion of non-local Fock exchange. Specifically, RS methods resulted in greatly improved accuracy for these exciplexes. CT exciplexes also benefitted from TD-SCS/SOS-DHDFAs which were found to reduce exciplex binding compared to their unscaled counterparts.
In general, the ground-state DFT-D3(BJ) dispersion correction significantly improved the accuracy for all DFAs tested, except for RS TD-(SCS/SOS)-DHDFAs where D3(BJ) made little or no difference to the total energy. The other exception is styrene-TMA, which was already artificially over-bound and as such was not improved by a dispersion correction. The VV10 correction was also tested for the first time in the context of excited state NCIs and gave reasonably small errors comparable to D3(BJ)-corrected methods. The VV10 correction was also tested for its post-SCF implementation in its ground state, which resulted in no loss of accuracy and a reduced cost compared to self-consistent VV10. In fact, incorporating VV10 in the actual calculation of the excitation energies resulted to be negligible mirroring results previously reported in single-chromophore benchmarking.
Overall, the best performing TD-DFT methods for exciplex binding when only local excitations were involved were BLYP-D3(BJ), B2GP-PLYP-D3(BJ), and B2PLYP-D3(BJ). In particular, B2PLYP-D3(BJ) showed the best performance when non-equilibrium geometries were included. The latter two methods have previously shown excellent performance for excimer binding15 and can be recommended for exciplexes. Despite its strong performance for localised excitations, BLYP-D3(BJ) is not recommended for TD-DFT due to its demonstrated inability to capture CT excitations and problems with ghost states. For the two CT exciplexes, dispersion-uncorrected SOS-ωB88PP86 gave the smallest errors, but ωB2(GP-)PLYP and a series of other RS TD-SCS/SOS-DHDFAs can also be recommended.
Finally, this study has reinforced that dispersion corrections are essential for good accuracy in (TD)-DFT calculations. While the D3(BJ) and VV10 dispersion corrections showed reduced errors, these did not reach chemical accuracy. In particular, the performance of the D3(BJ) correction was system- and method-dependent. We strongly recommend the development of a dispersion correction specifically for excited-state TD-DFT.
Author contributions
ACJ: conceptualization (equal), formal analysis (lead), investigation (lead), methodology (equal), validation (lead), visualization (lead), data curation (lead), writing – original draft (lead), writing – review & editing (equal). LG: conceptualization (equal), methodology (equal), formal analysis (supporting), investigation (supporting), funding acquisition (lead), project administration (lead), resources (lead), supervision (lead), writing – review & editing (equal).Data availability
The data supporting this article have been included as part of the ESI.† Any raw data can be obtained from the corresponding author upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
ACJ acknowledges the Australian Commonwealth Government and the University of Melbourne for a Research Training Program Scholarship. The authors are thankful for the allocation of computing resources by the National Computational Infrastructure (NCI) National Facility within the National Computational Merit Allocation Scheme (project no. fk5). This research was additionally supported by the Research Computing Services NCI Access scheme at The University of Melbourne.References
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- F. London, Z. Phys., 1930, 63, 245–279 CrossRef CAS.
- S. Kristyán and P. Pulay, Chem. Phys. Lett., 1994, 229, 175–180 CrossRef.
- J. M. Pérez-Jordá and A. D. Becke, Chem. Phys. Lett., 1995, 233, 134–137 CrossRef.
- P. Hobza, J. Šponer and T. Reschel, J. Comput. Chem., 1995, 16, 1315–1325 CrossRef CAS.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
- L. Goerigk, A. Hansen, C. Bauer, S. Ehrlich, A. Najibi and S. Grimme, Phys. Chem. Chem. Phys., 2017, 19, 32184–32215 RSC.
- J. Řezáč, K. E. Riley and P. Hobza, J. Chem. Theory Comput., 2011, 7, 2427–2438 CrossRef.
- J. Řezáč, K. E. Riley and P. Hobza, J. Chem. Theory Comput., 2011, 7, 3466–3470 CrossRef.
- N. Mardirossian and M. Head-Gordon, Mol. Phys., 2017, 115, 2315–2372 CrossRef.
- L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- A. C. Hancock and L. Goerigk, RSC Adv., 2023, 13, 35964–35984 RSC.
- M. R. Silva-Junior, M. Schreiber, S. P. A. Sauer and W. Thiel, J. Chem. Phys., 2008, 129, 104103 CrossRef.
- D. Jacquemin, V. Wathelet, E. A. Perpète and C. Adamo, J. Chem. Theory Comput., 2009, 5, 2420–2435 CrossRef PubMed.
- J. Liang, X. Feng, D. Hait and M. Head-Gordon, J. Chem. Theory Comput., 2022, 18, 3460–3473 CrossRef PubMed.
- L. Goerigk and S. Grimme, J. Chem. Phys., 2010, 132, 184103 CrossRef.
- M. Casanova-Páez, M. B. Dardis and L. Goerigk, J. Chem. Theory Comput., 2019, 15, 4735–4744 CrossRef PubMed.
- L. Goerigk, J. Moellmann and S. Grimme, Phys. Chem. Chem. Phys., 2009, 11, 4611–4620 RSC.
- M. Véril, A. Scemama, M. Caffarel, F. Lipparini, M. Boggio-Pasqua, D. Jacquemin and P.-F. Loos, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, 1517 Search PubMed.
- R. Grotjahn and M. Kaupp, J. Chem. Phys., 2021, 155, 124108 CrossRef PubMed.
- A. D. Laurent and D. Jacquemin, Int. J. Quantum Chem., 2013, 113, 2019–2039 CrossRef.
- C. Diedrich and S. Grimme, J. Phys. Chem. A, 2003, 107, 2524–2539 CrossRef.
- L. Goerigk and S. Grimme, J. Phys. Chem. A, 2009, 113, 767–776 CrossRef CAS.
- K. Batra, S. Zahn and T. Heine, Adv. Theory Simul., 2020, 3, 1900192 CrossRef CAS.
- S. Grimme and F. Neese, J. Chem. Phys., 2007, 127, 154116 CrossRef PubMed.
- M. Casanova-Páez and L. Goerigk, J. Chem. Theory Comput., 2021, 17, 5165–5186 CrossRef.
- L. Goerigk and M. Casanova-Páez, Aust. J. Chem., 2021, 74, 3–15 CrossRef CAS.
- A. Dreuw, J. L. Weisman and M. Head-Gordon, J. Chem. Phys., 2003, 119, 2943–2946 CrossRef CAS.
- D. J. Tozer, R. D. Amos, N. C. Handy, B. O. Roos and L. Serrano-Andres, Mol. Phys., 1999, 97, 859–868 CrossRef.
- D. J. Tozer, J. Chem. Phys., 2003, 119, 12697–12699 CrossRef.
- A. Dreuw and M. Head-Gordon, J. Am. Chem. Soc., 2004, 126, 4007–4016 CrossRef PubMed.
- M. Casanova-Páez and L. Goerigk, J. Comput. Chem., 2021, 42, 528–533 CrossRef.
- L. Goerigk, H. Kruse and S. Grimme, Comprehensive Chiroptical Spectroscopy, Wiley-Blackwell, 2012, pp. 643–673 Search PubMed.
- T. Förster, Angew. Chem., Int. Ed. Engl., 1969, 8, 333–343 CrossRef.
- M. Kołaski, C. R. Arunkumar and K. S. Kim, J. Chem. Theory Comput., 2013, 9, 847–856 CrossRef PubMed.
- R. A. Krueger and G. Blanquart, J. Phys. Chem. A, 2019, 123, 1796–1806 CrossRef CAS PubMed.
- R. Huenerbein and S. Grimme, Chem. Phys., 2008, 343, 362–371 CrossRef CAS.
- E. A. Briggs and N. A. Besley, Phys. Chem. Chem. Phys., 2014, 16, 14455–14462 RSC.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef PubMed.
- Y. Ikabata and H. Nakai, J. Chem. Phys., 2012, 137, 124106 CrossRef.
- A. C. Hancock, E. Giudici and L. Goerigk, J. Comput. Chem., 2024, 45, 1667–1681 CrossRef.
- S. Grimme, J. Chem. Phys., 2003, 118, 9095–9102 CrossRef.
- Y. Jung, R. C. Lochan, A. D. Dutoi and M. Head-Gordon, J. Chem. Phys., 2004, 121, 9793–9802 CrossRef.
- T. Schwabe and L. Goerigk, J. Chem. Theory Comput., 2017, 13, 4307–4323 CrossRef CAS PubMed.
- B. G. Janesko, T. M. Henderson and G. E. Scuseria, J. Chem. Phys., 2009, 131, 034110 CrossRef PubMed.
- O. A. Vydrov, Q. Wu and T. van Voorhis, J. Chem. Phys., 2008, 129, 014106 CrossRef.
- P. Mach, v Budzák, M. Medved’ and O. Kysel’, Theor. Chem. Acc., 2012, 131, 1268 Search PubMed.
- J. Van Dijk, M. Casanova-Páez and L. Goerigk, ACS Phys. Chem. Au, 2022, 2, 407–416 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- L. Goerigk and S. Grimme, J. Chem. Theory Comput., 2011, 7, 291–309 CrossRef CAS.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- O. A. Vydrov and T. Van Voorhis, J. Chem. Phys., 2010, 133, 244103 CrossRef.
- W. Hujo and S. Grimme, J. Chem. Theory Comput., 2011, 7, 3866–3871 CrossRef CAS PubMed.
- A. Najibi and L. Goerigk, J. Chem. Theory Comput., 2018, 14, 5725–5738 CrossRef CAS.
- A. Najibi, M. Casanova-Páez and L. Goerigk, J. Phys. Chem. A, 2021, 125, 4026–4035 CrossRef CAS PubMed.
- O. Christiansen, H. Koch and P. Jørgensen, Chem. Phys. Lett., 1995, 243, 409–418 CrossRef CAS.
- A. Hellweg, S. A. Grün and C. Hättig, Phys. Chem. Chem. Phys., 2008, 10, 4119–4127 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- DFT-D3 V3.1, S. Grimme, University of Bonn, 2014 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- J. P. Perdew and K. Schmidt, AIP Conf. Proc., 2001, 577, 1–20 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef PubMed.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- N. Mardirossian and M. Head-Gordon, J. Chem. Phys., 2015, 142, 074111 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- Y. Tawada, T. Tsuneda, S. Yanagisawa, T. Yanai and K. Hirao, J. Chem. Phys., 2004, 120, 8425–8433 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- N. Mardirossian and M. Head-Gordon, Phys. Chem. Chem. Phys., 2014, 16, 9904–9924 RSC.
- H. Iikura, T. Tsuneda, T. Yanai and K. Hirao, J. Chem. Phys., 2001, 115, 3540–3544 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef.
- N. Mardirossian and M. Head-Gordon, J. Chem. Phys., 2016, 144, 214110 Search PubMed.
- A. Karton, A. Tarnopolsky, J.-F. Lamère, G. C. Schatz and J. M. L. Martin, J. Phys. Chem. A, 2008, 112, 12868–12886 CrossRef CAS PubMed.
- S. Grimme, J. Chem. Phys., 2006, 124, 034108 CrossRef PubMed.
- E. Brémond, J. C. Sancho-García, A. J. Pérez-Jiménez and C. Adamo, J. Chem. Phys., 2014, 141, 031101 CrossRef.
- J. C. Sancho-García, E. Brémond, M. Savarese, A. J. Pérez-Jímenez and C. Adamo, Phys. Chem. Chem. Phys., 2017, 19, 13481–13487 RSC.
- E. Brémond and C. Adamo, J. Chem. Phys., 2011, 135, 024106 CrossRef.
- L. Goerigk and S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 576–600 Search PubMed.
- E. Brémond, A. J. Pérez-Jiménez, J. C. Sancho-García and C. Adamo, J. Chem. Phys., 2019, 150, 201102 CrossRef.
- E. Brémond, M. Savarese, A. J. Pérez-Jiménez, J. C. Sancho-García and C. Adamo, J. Chem. Theory Comput., 2018, 14, 4052–4062 CrossRef PubMed.
- M. Alipour and N. Karimi, J. Chem. Theory Comput., 2021, 17, 4077–4091 CrossRef PubMed.
- E. Brémond, A. J. Pérez-Jiménez, J. C. Sancho-García and C. Adamo, J. Chem. Phys., 2023, 159, 234104 CrossRef.
- S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding, C. Hättig, A. Hellweg, B. Helmich-Paris, C. Holzer, U. Huniar, M. Kaupp, A. Marefat Khah, S. Karbalaei Khani, T. Müller, F. Mack, B. D. Nguyen, S. M. Parker, E. Perlt, D. Rappoport, K. Reiter, S. Roy, M. Rückert, G. Schmitz, M. Sierka, E. Tapavicza, D. P. Tew, C. van Wüllen, V. K. Voora, F. Weigend, A. Wodyński and J. M. Yu, J. Chem. Phys., 2020, 152, 184107 CrossRef PubMed.
- E. Epifanovsky, A. T. B. Gilbert, X. Feng, J. Lee, Y. Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons, A. L. Dempwolff, Z. Gan, D. Hait, P. R. Horn, L. D. Jacobson, I. Kaliman, J. Kussmann, A. W. Lange, K. U. Lao, D. S. Levine, J. Liu, S. C. McKenzie, A. F. Morrison, K. D. Nanda, F. Plasser, D. R. Rehn, M. L. Vidal, Z.-Q. You, Y. Zhu, B. Alam, B. J. Albrecht, A. Aldossary, E. Alguire, J. H. Andersen, V. Athavale, D. Barton, K. Begam, A. Behn, N. Bellonzi, Y. A. Bernard, E. J. Berquist, H. G. A. Burton, A. Carreras, K. Carter-Fenk, R. Chakraborty, A. D. Chien, K. D. Closser, V. Cofer-Shabica, S. Dasgupta, M. de Wergifosse, J. Deng, M. Diedenhofen, H. Do, S. Ehlert, P.-T. Fang, S. Fatehi, Q. Feng, T. Friedhoff, J. Gayvert, Q. Ge, G. Gidofalvi, M. Goldey, J. Gomes, C. E. González-Espinoza, S. Gulania, A. O. Gunina, M. W. D. Hanson-Heine, P. H. P. Harbach, A. Hauser, M. F. Herbst, M. Hernández Vera, M. Hodecker, Z. C. Holden, S. Houck, X. Huang, K. Hui, B. C. Huynh, M. Ivanov, A. Jász, H. Ji, H. Jiang, B. Kaduk, S. Kähler, K. Khistyaev, J. Kim, G. Kis, P. Klunzinger, Z. Koczor-Benda, J. H. Koh, D. Kosenkov, L. Koulias, T. Kowalczyk, C. M. Krauter, K. Kue, A. Kunitsa, T. Kus, I. Ladjánszki, A. Landau, K. V. Lawler, D. Lefrancois and S. Lehtola, et al. , J. Chem. Phys., 2021, 155, 084801 CrossRef CAS PubMed.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 242, 652–660 CrossRef CAS.
- R. Izsák and F. Neese, J. Chem. Phys., 2011, 135, 144105 CrossRef.
- F. Weigend and M. Häser, Theor. Chem. Acc., 1997, 97, 331–340 Search PubMed.
- F. Weigend, M. Häser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143–152 CrossRef CAS.
- M. Head-Gordon, R. J. Rico, M. Oumi and T. J. Lee, Chem. Phys. Lett., 1994, 219, 21–29 CrossRef.
- Y. M. Rhee and M. Head-Gordon, J. Phys. Chem. A, 2007, 111, 5314–5326 CrossRef.
- S. Grimme, A. Hansen, J. G. Brandenburg and C. Bannwarth, Chem. Rev., 2016, 116, 5105–5154 CrossRef.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef.
- M. Friede, S. Ehlert, S. Grimme and J.-M. Mewes, J. Chem. Theory Comput., 2023, 19, 8097–8107 CrossRef PubMed.
- E. Caldeweyher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2017, 147, 034112 CrossRef.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- N. Mehta, M. Casanova-Páez and L. Goerigk, Phys. Chem. Chem. Phys., 2018, 20, 23175–23194 RSC.
- T. Risthaus and S. Grimme, J. Chem. Theory Comput., 2013, 9, 1580–1591 CrossRef CAS.
- Q. Ge, Y. Mao and M. Head-Gordon, J. Chem. Phys., 2018, 148, 064105 CrossRef PubMed.
- F. Plasser and H. Lischka, J. Chem. Theory Comput., 2012, 8, 2777–2789 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details for CBS extrapolation, values for De and re, dissociation curves, styrene-TMA excitation energies, more details on ALMO-EDA, CT statistics, mean absolute errors, optimised geometries and geometries used to generate dissociation curves. See DOI: https://doi.org/10.1039/d4cp03214d |
| This journal is © the Owner Societies 2024 |