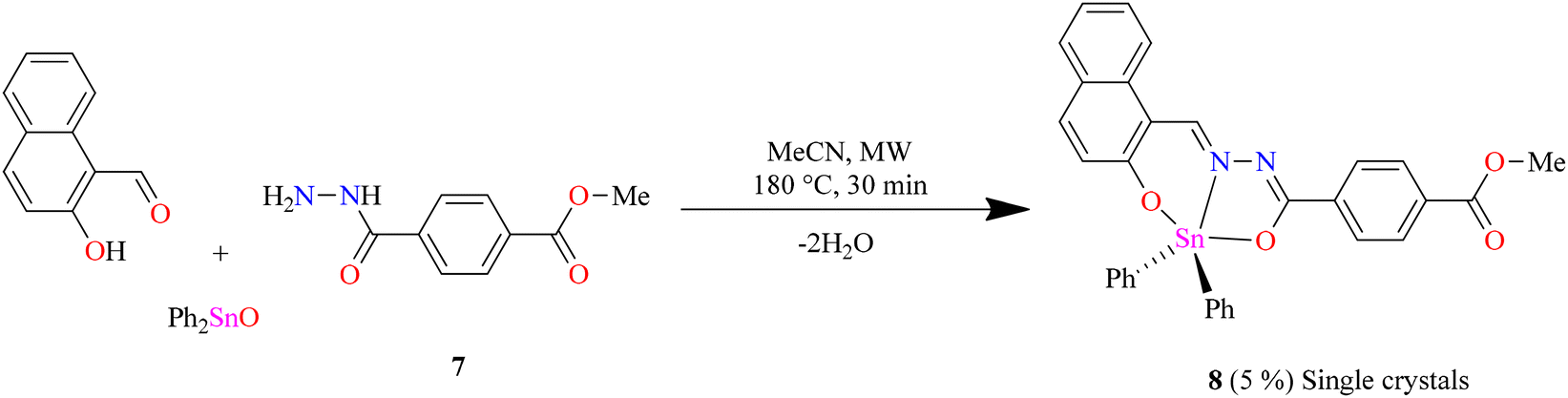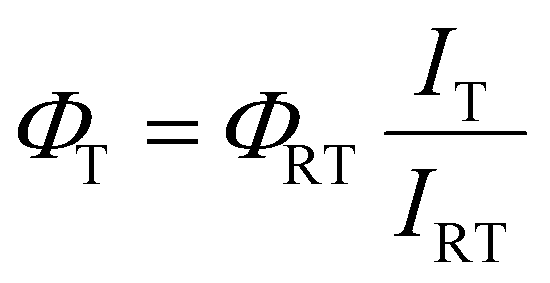DOI:
10.1039/D4DT01518E
(Paper)
Dalton Trans., 2024,
53, 15010-15031
Temperature-dependent photoluminescence down to 77 K of organotin molecular rotors: eco-friendly synthesis, photophysical characterization, X-ray structures, and DFT studies†
Received
24th May 2024
, Accepted 19th July 2024
First published on 6th August 2024
Abstract
Fluorescent organotin compounds are useful in sensing, optoelectronic devices, and in vitro bioimaging. Although in vitro fluorescence bioimaging shows low resolution at room temperature, a better resolution is possible at cryotemperatures. Therefore, the search for new cryoluminescent materials with potential application in high-resolution fluorescence bioimaging remains a great challenge. Herein, we report the cryoluminescence properties of two fluorescent bis-organotin compounds, namely, BisNTHySnBu2 (5) and BisNTHySnPh2 (6), synthesized via microwave irradiation. All compounds were fully characterized using 1H, 13C, and 119Sn NMR spectroscopy, Raman spectroscopy, IR spectroscopy, and HR-MS. The 119Sn δ and 3J(1H,119Sn) of 5 and 6 indicate that two Sn-ligands are chemically and electronically equivalent, as confirmed by cyclic voltammetry. The crystal structure of 6 showed pentacoordinate tin atoms with skeleton ligands. The study of self-assembled monolayers of both Sn-complexes via STM microscopy revealed a similar supramolecular packing in lamella-like patterns, adopting a face-on arrangement, where molecules stay flat lying on HOPG in accordance with the height profile of closely packed monolayers on graphite of about 0.33 nm thickness. However, only the Sn complex 6, which bears phenyls, covers large surface areas. The photophysical properties of bis-organotin compounds were also investigated in solution (room and low temperatures) and in the solid state. Good luminescence properties in solutions with fluorescence quantum yields (Φ) of approximately 35% and 50% were found. Despite this, Φ is quenched in the solid state because of aggregation, as supported by solvent/non solvent fluorescence studies, which is in agreement with STM and AFM investigation.
1. Introduction
Cryofluorochromism is the physical property of a material that exhibits fluorescence at low temperatures, below 120 K, and from room to low temperature, the fluorescence intensity increases due to a decrease in collisional quenching and non-radiative rate constant.1 This unique property has attracted considerable attention owing to its potential applications in optoelectronic devices,2 cryofluorescence microscopy,3,4 and nanospectroscopy.5 In the condensed phase, fluorescence properties such as lifetime, quantum yield, and emission maxima might be tuned by the pH, polarity, viscosity, and temperature. All these material properties are interesting in cellular biology. Fluorescence microscopy is becoming the most powerful and non-invasive diagnostic tools in cellular biology, where uncommon cellular changes are associated with diseases.6,7 Therefore, new fluorescent dyes with physicochemical and photophysical properties are required. However, some major problems in fluorescence microscopy at room temperature in some fluorophores are photobleaching and restricted resolution to 200 nm. A considerable improvement in fluorescence cryo-microscopy is avoiding photobleaching for high-resolution images.8,9 Therefore, the photophysical properties of fluorescent materials at low temperatures were investigated.
Dinuclear organotin Schiff bases have attracted significant attention in the field of coordination chemistry due to their unique structural features10,11 and potential biological applications.12–14 These compounds consist of two tin atoms connected by organic bridges, forming a central binuclear unit. The presence of two tin atoms in proximity creates a distinctive molecular environment that can influence both the electronic and luminescent properties of the compound. In recent years, the development of sustainable synthesis methods has become a critical focus in the field of main group chemistry, driven by the need for environmentally friendly and resource-efficient processes.15 Sustainable synthesis aims to minimize the use of hazardous reagents, reduce waste generation, and utilize renewable resources as starting materials. Applying the principles of sustainability to the synthesis of dinuclear organotin Schiff bases may offer significant advantages such as reducing the environmental impact and ensuring the long-term availability of raw materials.
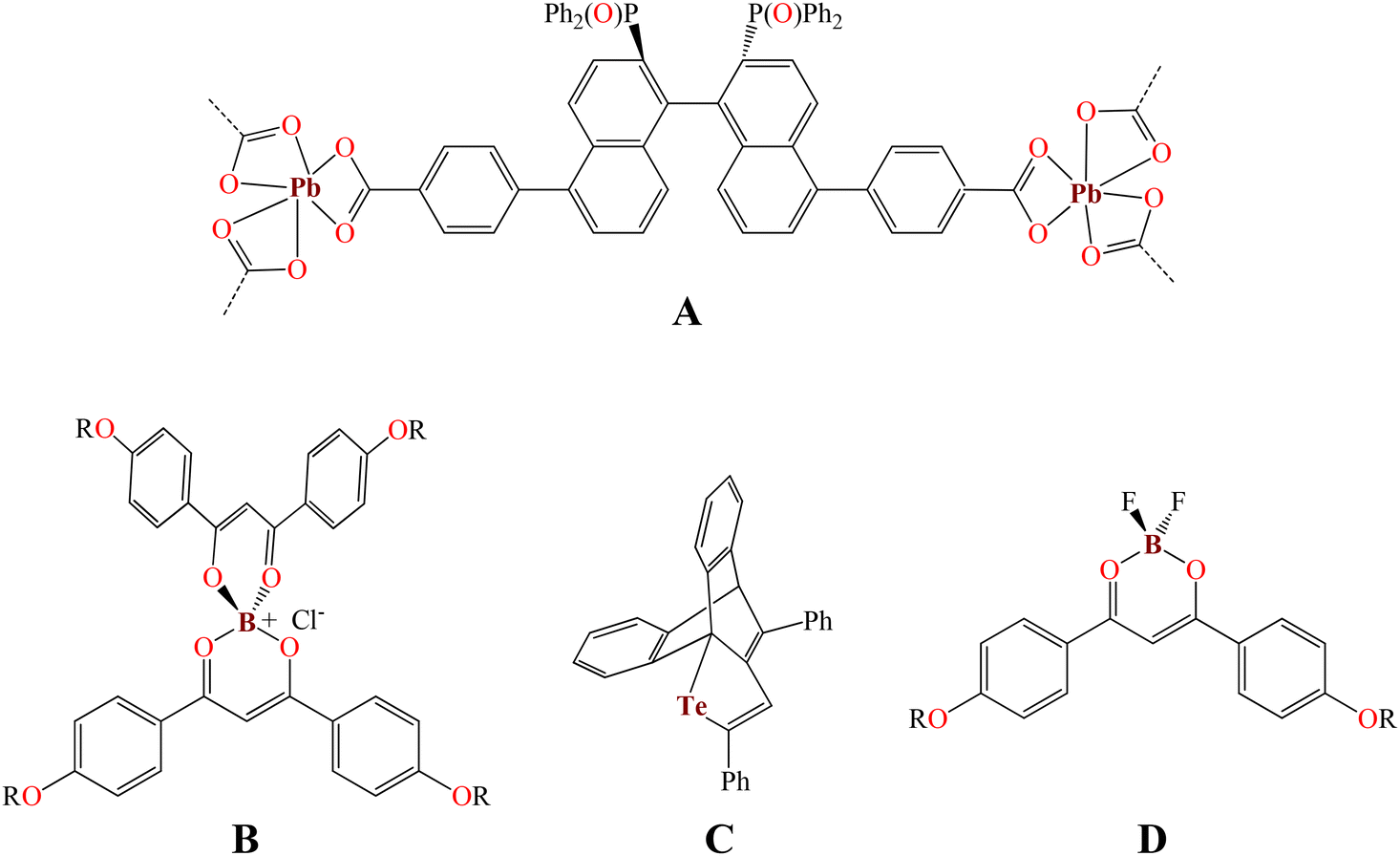
We have been interested in the green synthesis of fluorescent organotin complexes16,17 as well as their application in cell biology where these tin complexes are good cytoplasm,18,19 nucleolus, and cytoskeleton-like fluorescent probes observed by confocal microscopy.20 Furthermore, a plethora of reports on photophysical studies at low temperatures of transition metals21–24 or lanthanide complexes,25 QDs,26 and organic compounds27,28 as well as their review articles have been reported in the last two decades.29,30 However, photophysical studies at cryotemperatures with main group element complexes are still quite scarce. For example, Hey-Hawkins et al. reported a lead-based MOF (Fig. 1, A) where the emission spectrum at the cryotemperature (10 K) and at room temperature does not change in shape and position but shows a slight increase in the emission intensity.31 A short series of organoboronium bisdiketonates (Fig. 1, B) showed quenching effects at 77 K.32 Contrary to this, the telluride (C) reported by Ishii et al. displayed temperature-dependent fluorescence; it is fluorescent with a moderate quantum yield (ΦPL = 0.42) in 2-methyltetrahydrofuran at 77 K.33 Finally, difluoroboron β-diketonate fluorophores (D), in the solid state, showed a perfect linear relationship and red-shifted from ∼520 nm to ∼570 nm (380 K and 77 K, respectively) with a change in fluorescence from green to orange.34
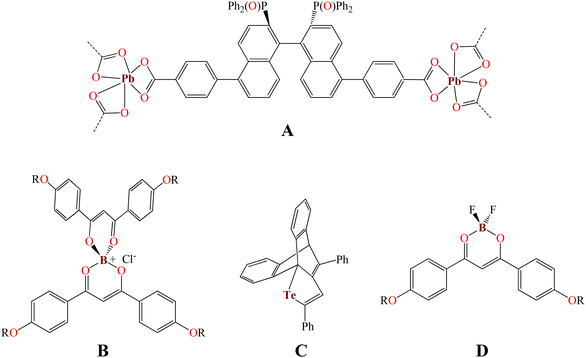 |
| | Fig. 1 Photophysical properties of main group element complexes at the cryotemperature. | |
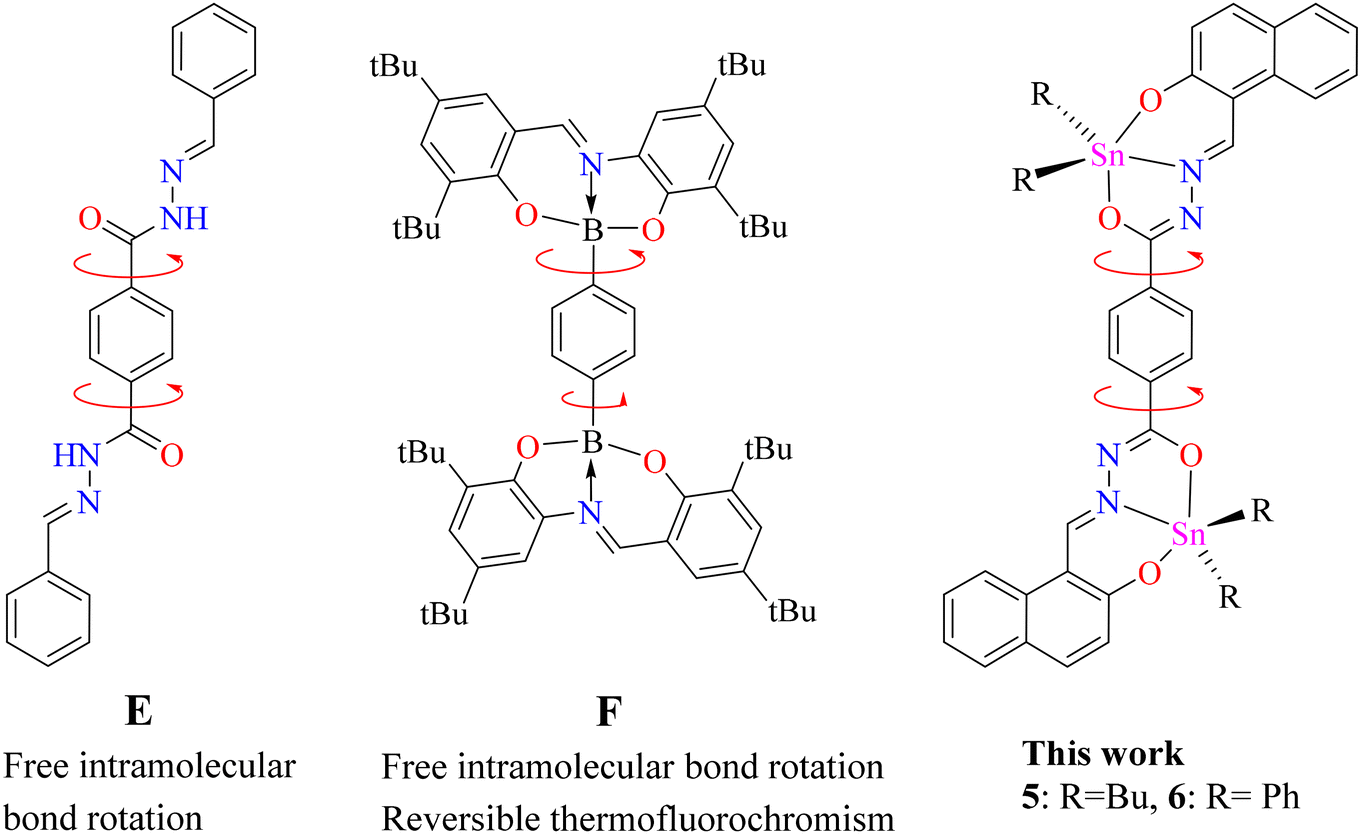
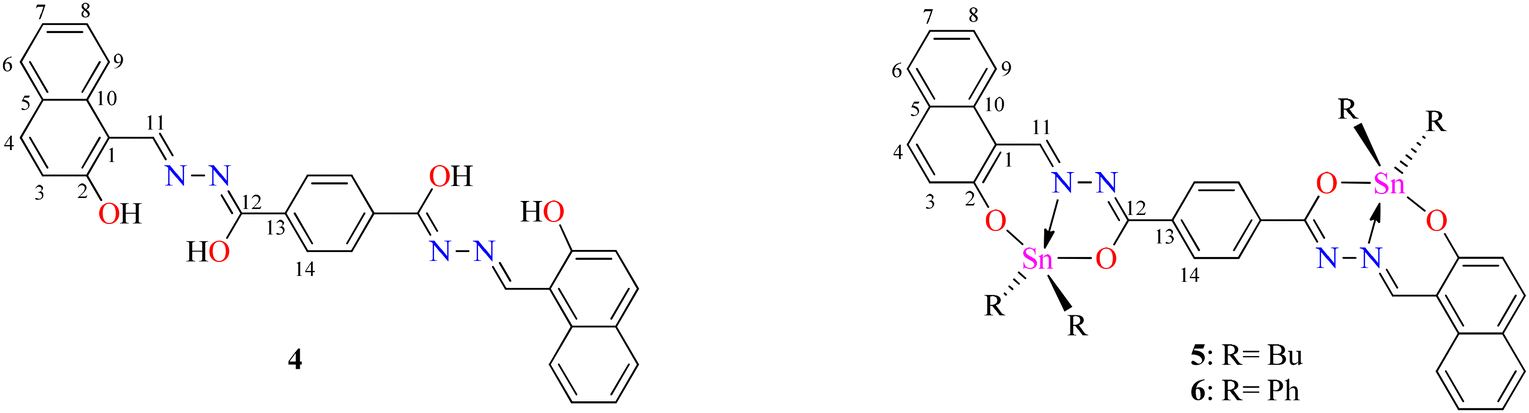
Bis-hydrazine (Fig. 2, E) has been considered as a non-emissive building unit due to free intramolecular bond rotation on the phenyl bridge. However, the fluorescence of this unit can be enhanced through restriction of intramolecular bond rotation (RIR).35 A couple of BOSCHIBAs (F) showed reversible thermochromism as well as viscochromism due to the phenylene bridge.36 Based on the these, a question arises, as to whether dinuclear organotin derived from terephthalohydrazide can display temperature-dependent fluorescence at a low temperature fixing the free rotation? (Fig. 2, 5 and 6). Therefore, herein we report the synthesis of dinuclear organotin Schiff bases, their photophysical properties at room and low temperatures and DFT calculations.
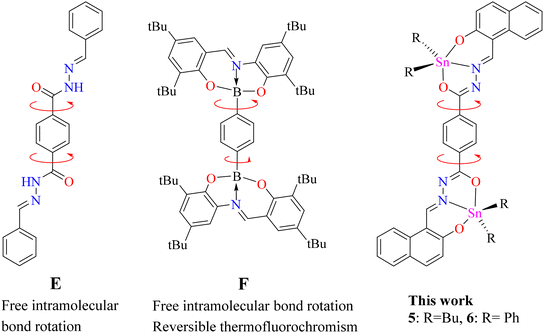 |
| | Fig. 2 Molecular rotors with a phenylene bridge. | |
2. Results and discussion
2.1. Syntheses
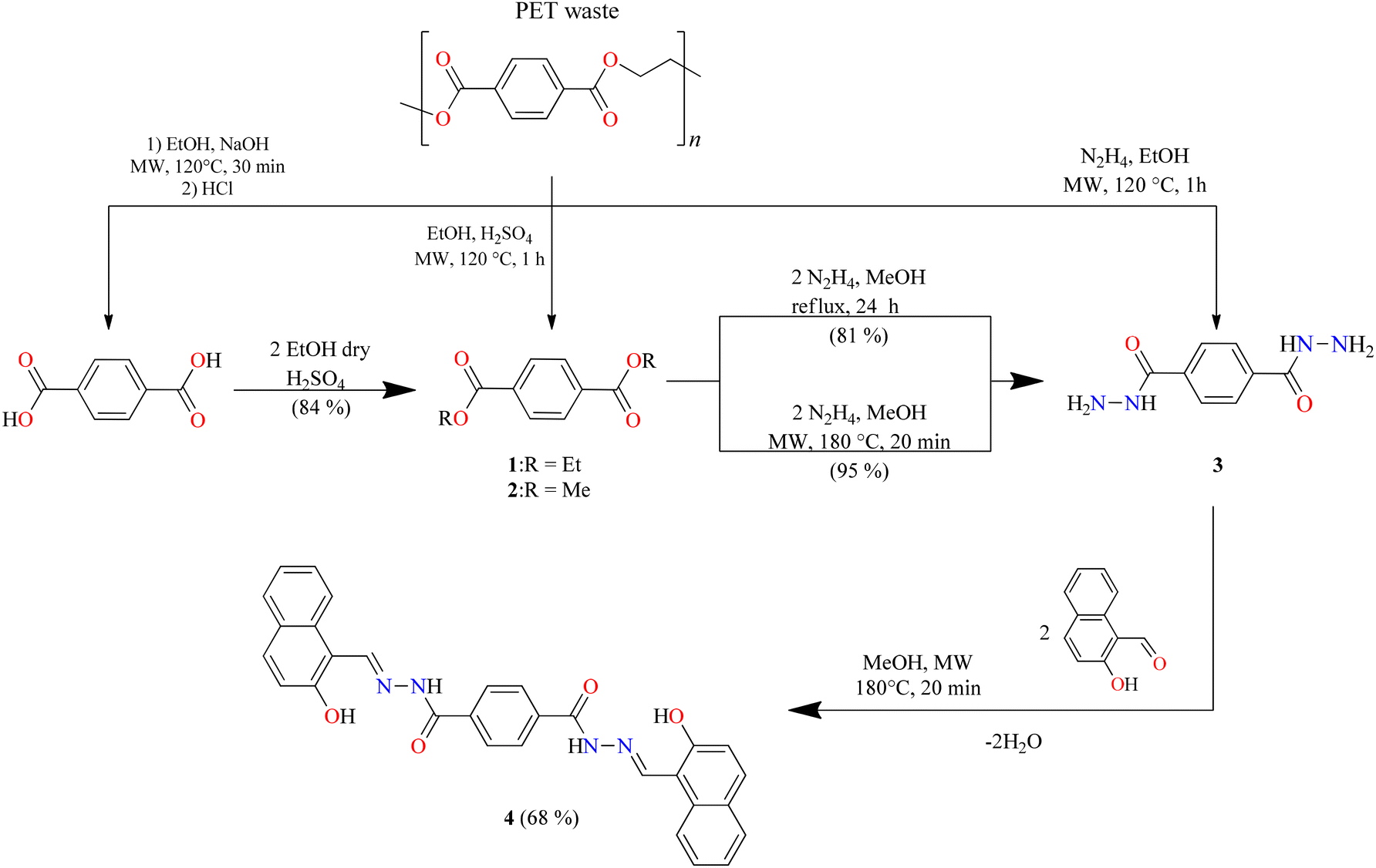
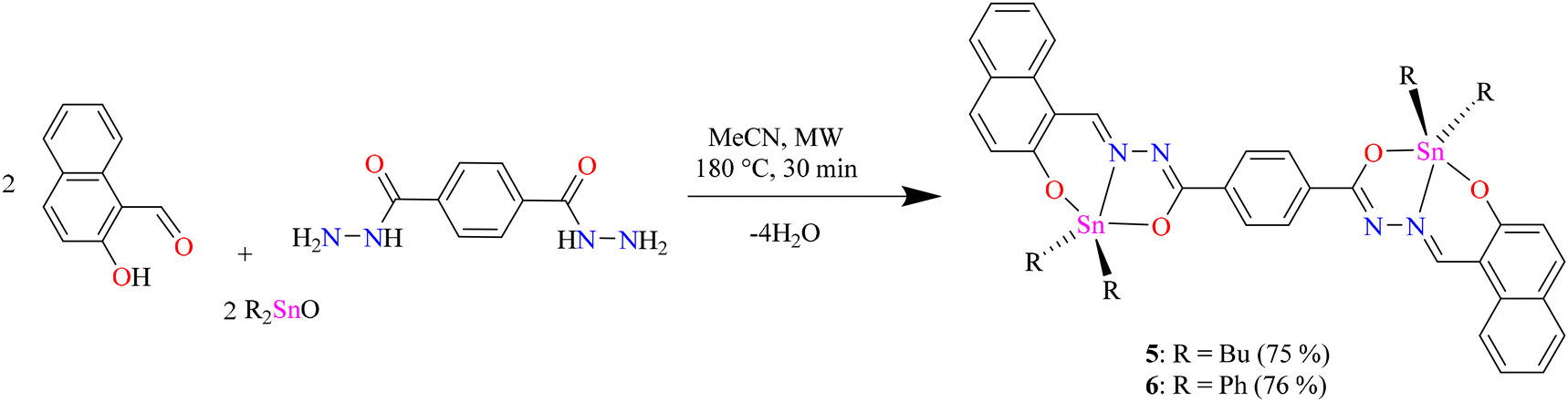
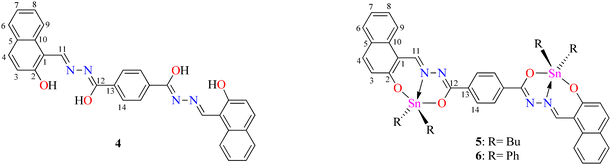
To obtain the centrosymmetric and hexadentate ligand 4 (Scheme 1), three synthetic steps were carried out. After the Brønsted acid-catalyzed esterification of terephthalic acid in ethanol, hydrazinolysis of 1 or 2 by conventional heating or microwave (MW)-assisted synthesis afforded compound 3 in good yields. It is worth mentioning that, using microwave irradiation, the reaction time was improved over 70-fold by comparison with conventional heating. Finally, after further 20 minutes of using microwave irradiation and via condensation reaction with 2 equivalents of 2-hydroxynaphtalehyde, the ligand 4 was isolated as a pale-yellow powder after work-up. It is important to mention that terephthalic acid, diethyl terephthalate, and terephthalohydrazide were synthesized by microwave irradiation in less than one hour and in good yields from polyethylene terephthalate plastic waste (see ESI, Fig. S1–S9†). Organotin compounds 5 and 6 were synthesized via multicomponent reactions with 2-hydroxynaphtalehyde, terephthalohydrazide, and dibutyltin/diphenyltin oxide in stoichiometric proportion (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2) with reaction yields of 75 and 76%, respectively (Scheme 2). The compounds were soluble in common organic solvents such as dichloromethane, chloroform, and tetrahydrofuran, while ligand 4 just in dimethyl sulfoxide. The unexpected compound 8 (Scheme 3) was isolated in low yield (5%) due to the uncompleted hydrazinolysis of 2 under microwave (MW) irradiation. The compound 8 was crystallized from ethanol/hexane (2:9) as red single crystals (vide infra).
:2:2) with reaction yields of 75 and 76%, respectively (Scheme 2). The compounds were soluble in common organic solvents such as dichloromethane, chloroform, and tetrahydrofuran, while ligand 4 just in dimethyl sulfoxide. The unexpected compound 8 (Scheme 3) was isolated in low yield (5%) due to the uncompleted hydrazinolysis of 2 under microwave (MW) irradiation. The compound 8 was crystallized from ethanol/hexane (2:9) as red single crystals (vide infra).
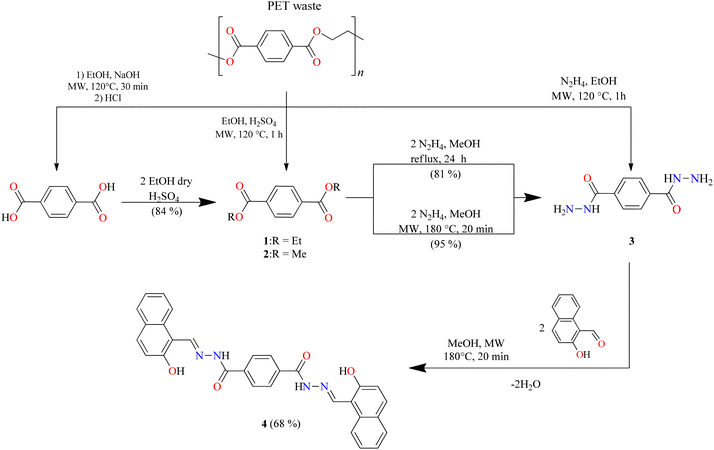 |
| | Scheme 1 Synthetic route of ligand 4. | |
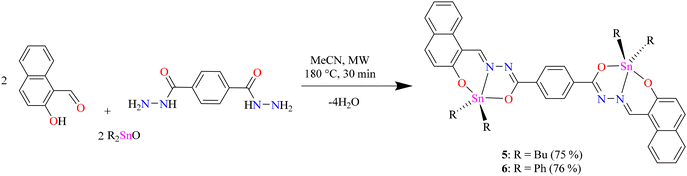 |
| | Scheme 2 Microwave-assisted synthesis via multicomponent reactions of 5 and 6. | |
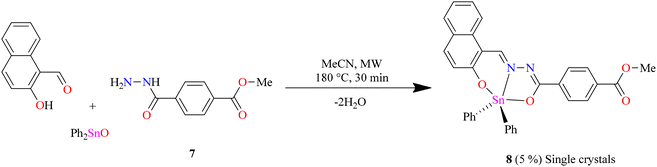 |
| | Scheme 3 Unexpected compounds 7 and 8. | |
2.2. Spectroscopic characterization
The 1H NMR spectra of compounds 5 and 6 exhibit one signal from 9.73 to 9.78 ppm with coupling constants 3J(1H,119Sn) of 45 and 56 Hz, respectively, which are indicative of the new N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond formation and the N → Sn coordination (Fig. S11 and S16†). The 1H signals of ortho protons with [3J(1H,119Sn) = 84 Hz] for 6 show a de-shielding effect due to their intramolecular interaction with the oxygen atoms, which was corroborated by X-ray diffraction analysis (vide infra) (C(17)–Ho⋯O(1) 2.629(3) Å; ∠C–H⋯O 120.52°), (C(21)–Ho⋯O(2) 2.624(3) Å; ∠C–H⋯O 119.73°), and the measured values suggest that the phenyl groups linked to the tin centre contributed to the stability of the complex in dissolution.37 For the unambiguous assignment of the proton and carbon resonant signals, it was essential to realize HETCOR and COSY experiments (Fig. S12, S14, S16 and S19†). The 119Sn NMR spectra of the complexes 5 and 6 in a CDCl3 solution exhibit a single resonance signal in the characteristic range for five-coordinated (Fig. S15 and S20†) diphenyltin structure for complex 6, as later confirmed by X-ray diffraction. For the dibutyltin complex derivatives, the 119Sn NMR spectrum of 5 shows a single resonance at −189 ppm (Table 1), while the diphenyl tin complex 6 at −328 ppm. In both complexes, there are characteristics of pentacoordinate tin atoms, where the tin atoms reside in a trigonal bipyramidal molecular geometry.38,39 In the 13C NMR spectra of both organotin compounds (Fig. S13 and S18†), the signals of the imine carbon, C11, appear in the range from 157.26 to 157.88 ppm for 5 and 6, which are deshielded with respect to the free ligand, due to the N → Sn coordination bond.
C bond formation and the N → Sn coordination (Fig. S11 and S16†). The 1H signals of ortho protons with [3J(1H,119Sn) = 84 Hz] for 6 show a de-shielding effect due to their intramolecular interaction with the oxygen atoms, which was corroborated by X-ray diffraction analysis (vide infra) (C(17)–Ho⋯O(1) 2.629(3) Å; ∠C–H⋯O 120.52°), (C(21)–Ho⋯O(2) 2.624(3) Å; ∠C–H⋯O 119.73°), and the measured values suggest that the phenyl groups linked to the tin centre contributed to the stability of the complex in dissolution.37 For the unambiguous assignment of the proton and carbon resonant signals, it was essential to realize HETCOR and COSY experiments (Fig. S12, S14, S16 and S19†). The 119Sn NMR spectra of the complexes 5 and 6 in a CDCl3 solution exhibit a single resonance signal in the characteristic range for five-coordinated (Fig. S15 and S20†) diphenyltin structure for complex 6, as later confirmed by X-ray diffraction. For the dibutyltin complex derivatives, the 119Sn NMR spectrum of 5 shows a single resonance at −189 ppm (Table 1), while the diphenyl tin complex 6 at −328 ppm. In both complexes, there are characteristics of pentacoordinate tin atoms, where the tin atoms reside in a trigonal bipyramidal molecular geometry.38,39 In the 13C NMR spectra of both organotin compounds (Fig. S13 and S18†), the signals of the imine carbon, C11, appear in the range from 157.26 to 157.88 ppm for 5 and 6, which are deshielded with respect to the free ligand, due to the N → Sn coordination bond.
Table 1 Chemical shifts of 1H, 13C, and 119Sn and constant coupling [nJ(13C, 119Sn)]
|
|
| Comp. |
1H |
13C |
119Sn |
IR CN |
| H-11 |
H-β/CH-o |
C-1 |
C-2 |
C-12 |
C-14 |
C-α C-i |
C-β |
C-γ |
C-δ |
| C-o |
C-m |
C-p |
|
4
|
12.29 |
|
|
|
|
|
|
|
|
|
|
1622 |
|
5
|
9.73 [ref. 45] |
1.67 |
107.0 |
169.5 |
167.9 |
127.3 |
21.8 |
26.6 |
26.2 |
13.4 |
−189.2 |
1602 |
|
6
|
9.78 [ref. 56] |
7.91 |
107.5 |
169.7 |
167.7 |
127.5 |
137.4 |
136.3 |
129.1 |
130.7 |
−328.1 |
1605 |
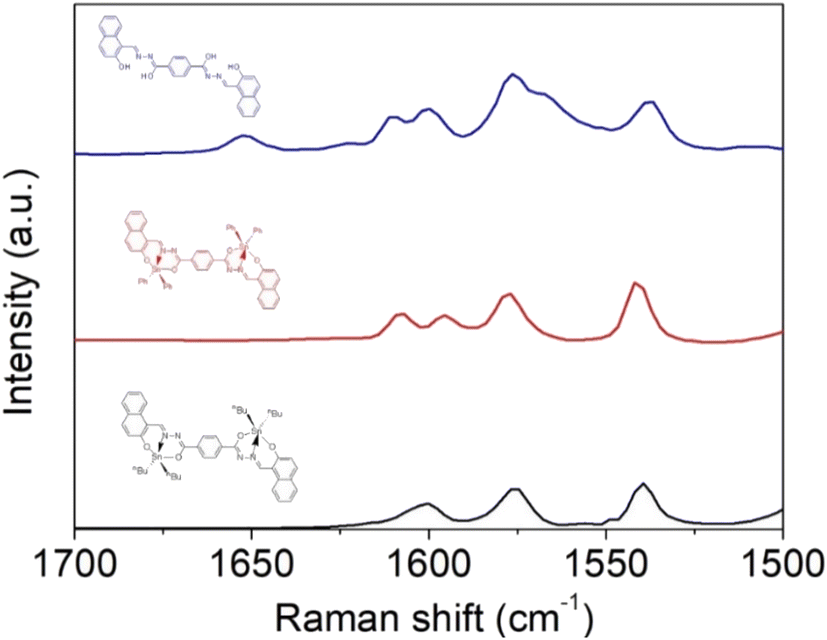
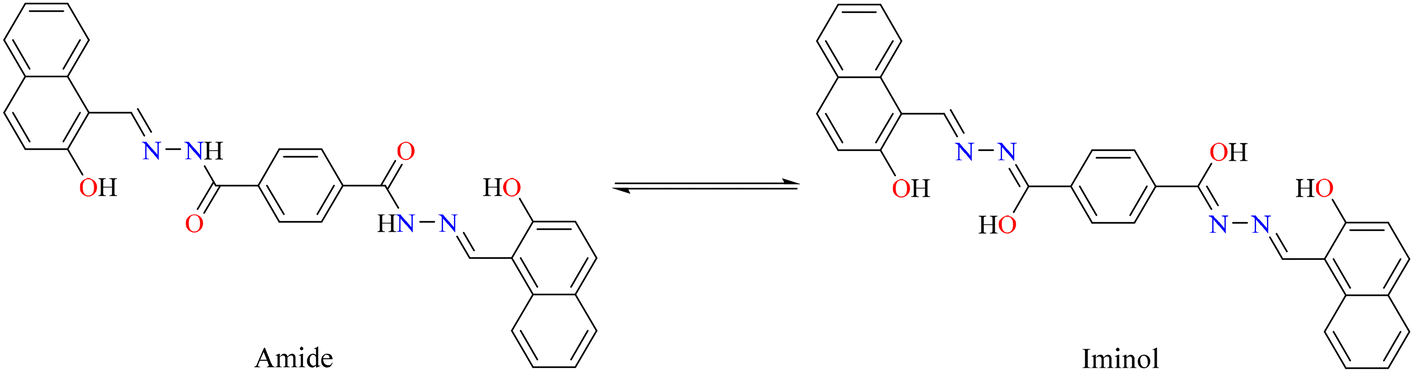
IR spectral analysis of ligand 4 showed stretching bands at 3318–3107, 3105–3000 and 1642 cm−1, which correspond to the vibrations of the N–H, O–H, and CO bonds, respectively. It is well known that benzohydrazine might show tautomerism for amide-iminol (Scheme 4). However, the above-mentioned vibrations exhibit a dominant amide at solid state.40 These bands disappear when the tin atom is coordinated with the ligand, and the ν(CN) stretching vibration that appears at 1622 cm−1 for ligand presents a displacement to lower frequencies (5: 1602; 6: 1605 cm−1), indicating the coordination of the imino with the diorganotin moiety (CN → Sn).41 Moreover, a signal at 1598–1580 cm−1 is observed in the complexes and assigned to the CN–NC group42 (Fig. S24–S26†). This was confirmed by Raman spectroscopy. Fig. 3 reports the Raman spectra of the tin complexes and their reference compound, in the region between 1700 and 1500 cm−1, where the most characteristic vibrations appear. In the free ligand, there exists the possibility of tautomerism between the amide and oxime isomers. This was evidenced in the IR spectra where both the O–H and N–H stretching vibrations were observed. Accordingly, the amide vibration is observed in the Raman spectra at 1652 cm−1, while amide II (N–H vibration) gives the strong signal at 1575 cm−1. The bands at 1600 cm−1 can be attributed to the imine CN vibrations, overlapping with the CC aromatic vibrations. After complexation, the CO band disappears, indicating the complete stabilization of the imine form, for which CN vibration is at ∼1600 cm−1. The strong signal at 1585 cm−1 is now attributed to the conjugated CN–NC stretching vibration; except for the CC vibration of the phenyl ligand, which is absent in the n-butyl complex, no significant changes were observed in the Raman bands of the two complexes.
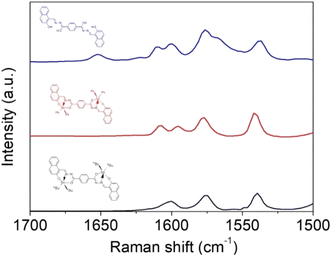 |
| | Fig. 3 Raman spectra of tin complexes 5 and 6 and their ligand 4. | |
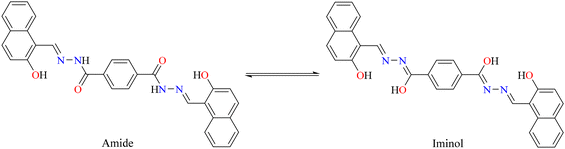 |
| | Scheme 4 Tautomerism of benzohydrazine. | |
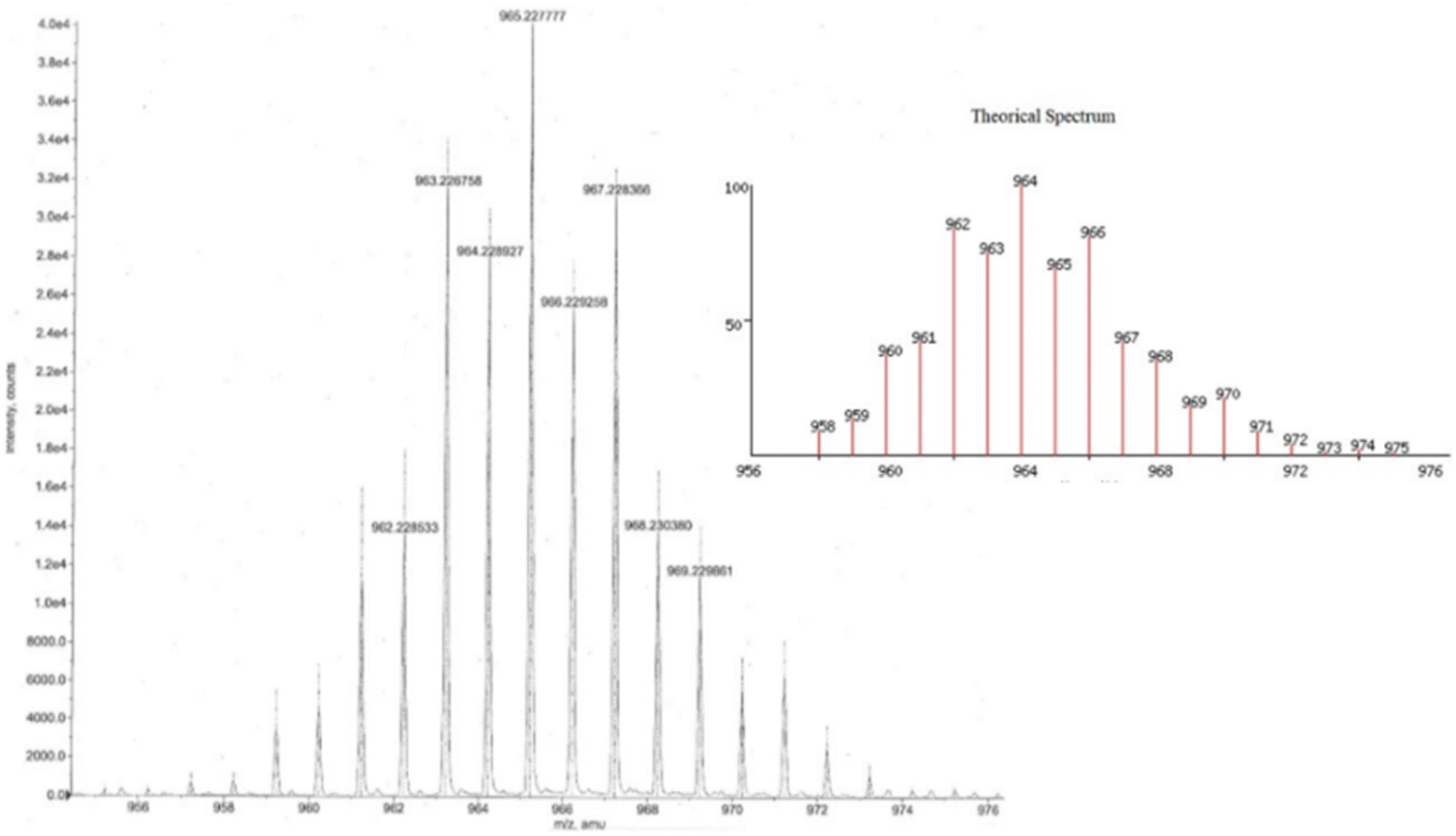
The high-resolution mass spectra of organotin compounds confirmed that the expected molecular ion peak for 5 is 967.2261 and 6 is 1047.1009 u m a (Fig. S27–S28†). Both organotin compounds showed the loss of the butyl and phenyl substituents, respectively, as well as the tin atoms (Scheme 5b and c). In addition, the isotopic pattern observed in the mass spectrum indicates that the molecules have two tin atoms in their skeleton (Fig. 4, compound 5 as example).43 In compound 4, the loss of water and naphthol group showed the possible formation of benzoindazole and oxadiazole, respectively (Scheme 5a).
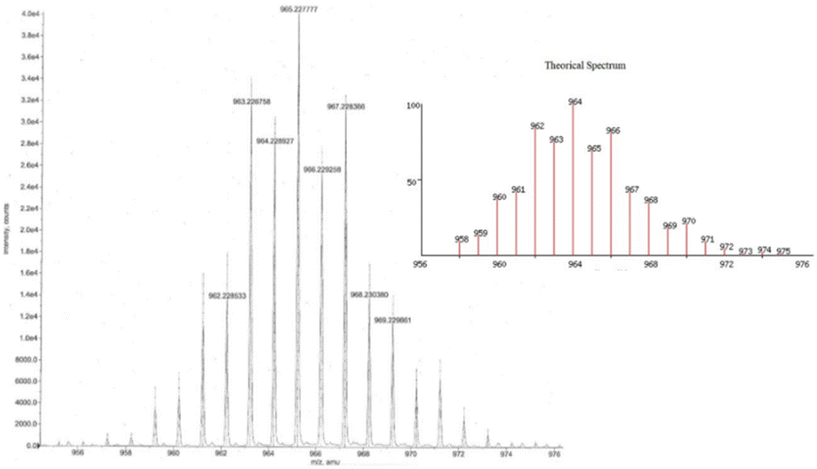 |
| | Fig. 4 Isotopic pattern of compound 5. | |
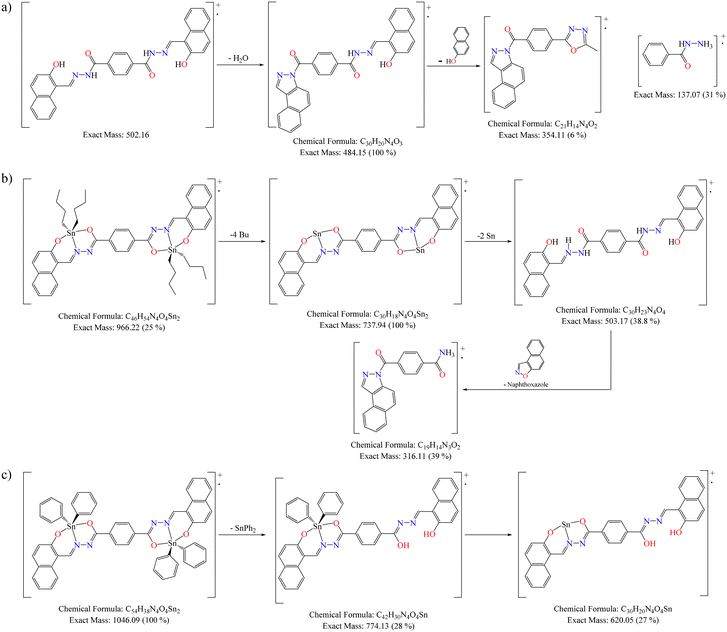 |
| | Scheme 5 Proposed fragmentation for compounds 4 (a), 5 (b), and 6 (c). | |
2.3. X-ray analysis
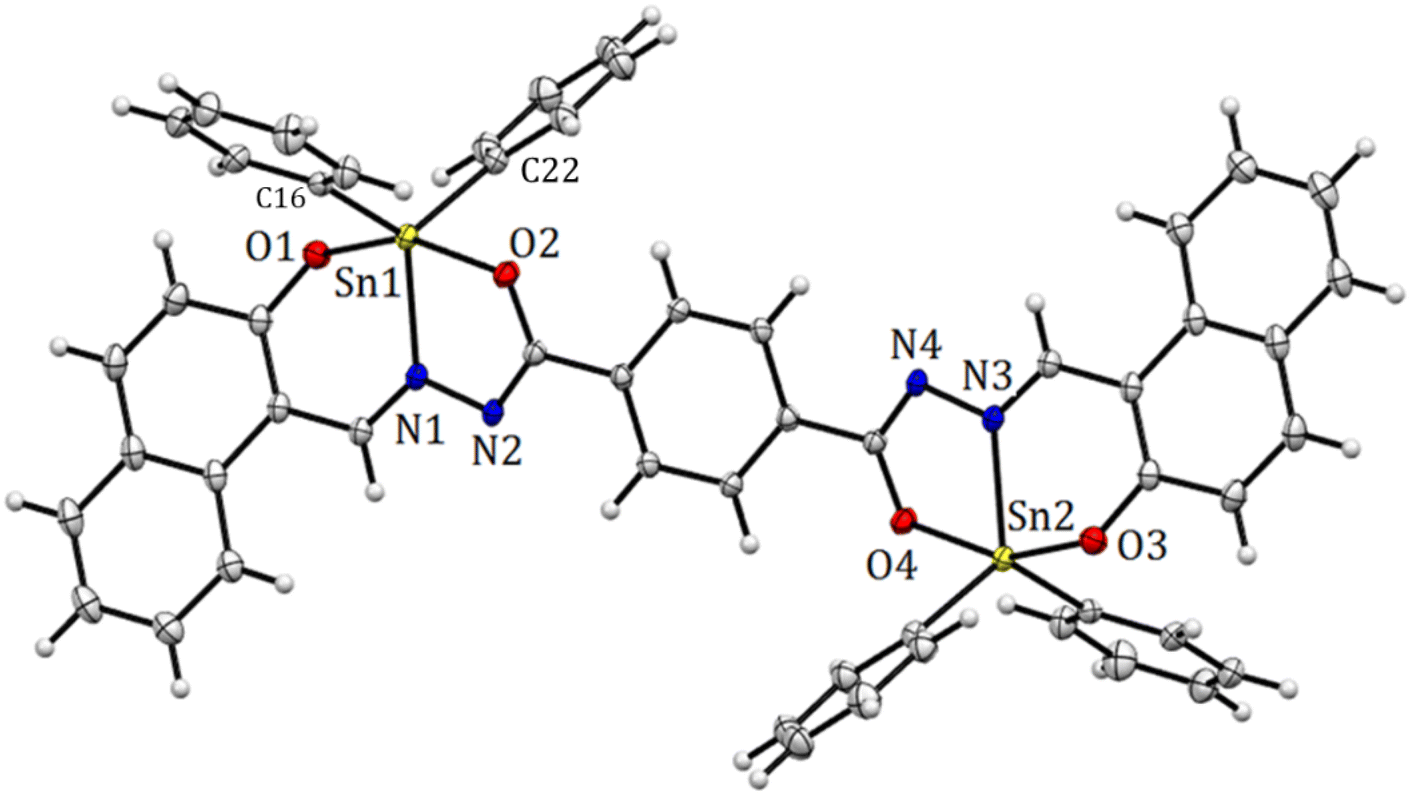
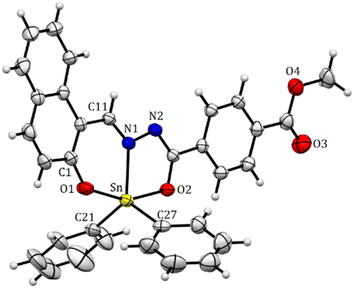
The structure of compound 6 is represented by the ellipsoid plot in Fig. 5. Crystal data were determined by single-crystal X-ray diffraction analysis (Table 2). Selected bond length and angles are listed in Table 3. The crystal structure corresponds to a binuclear and centrosymmetric molecule with a planar array containing two pentacoordinated tin atoms with a distorted trigonal bipyramidal molecular geometry (O–Sn–O range angle 156.00°, C–Sn–C from 129.84°), which mainly results from the strained five-membered Sn–NN–C–O chelate ring. The binuclear compound presents a N → Sn coordination length of 2.1409(13) Å. Similarly, the axial positions are occupied by oxygen atoms 6: Sn–O1 2.0688(11), Sn–O2 2.1205(11) Å and the equatorial positions are occupied by nitrogen and Cipso atoms (6: Sn–C(16) 2.109 (15) Sn–C(22) 2.121(15)). Additionally, the X-ray structure provides the molecular constitution with a planar system and shows π-stacking as a dimer with a π–π distance of 3.47 Å (Fig. 6 and 7).
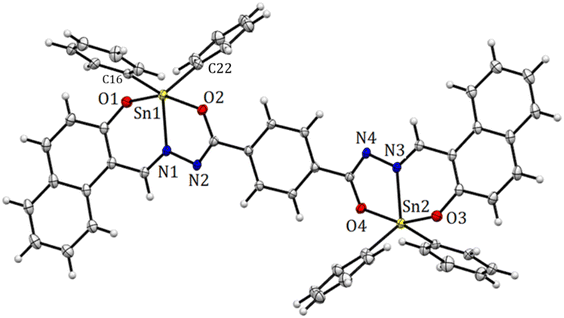 |
| | Fig. 5 Molecular structure deduced by X-ray crystallography for organotin compound 6. Selected bond distances (Å), Sn(1)–N(1) 2.1409(13), Sn(1)–O(1) 2.0688(11), Sn(1)–O(2) 2.0688(11), 2.1205(11), Sn(1)–C(16) 2.1090(15), and Sn(1)–C(22) 2.1213(15). Selected angles (°): O(1)–Sn(1)–O(2) 156.0(5), C(16)–Sn(1)–C(22) 123.84(5), C(16)–Sn(1)–N(1) 117.92 (5), and C(22)–Sn(1)–N(1) 117.71(6). | |
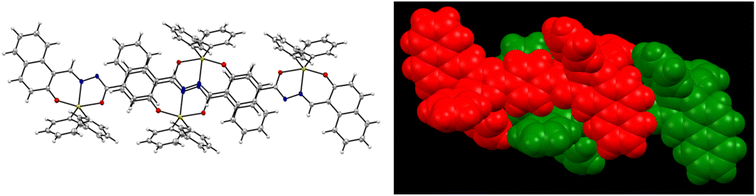 |
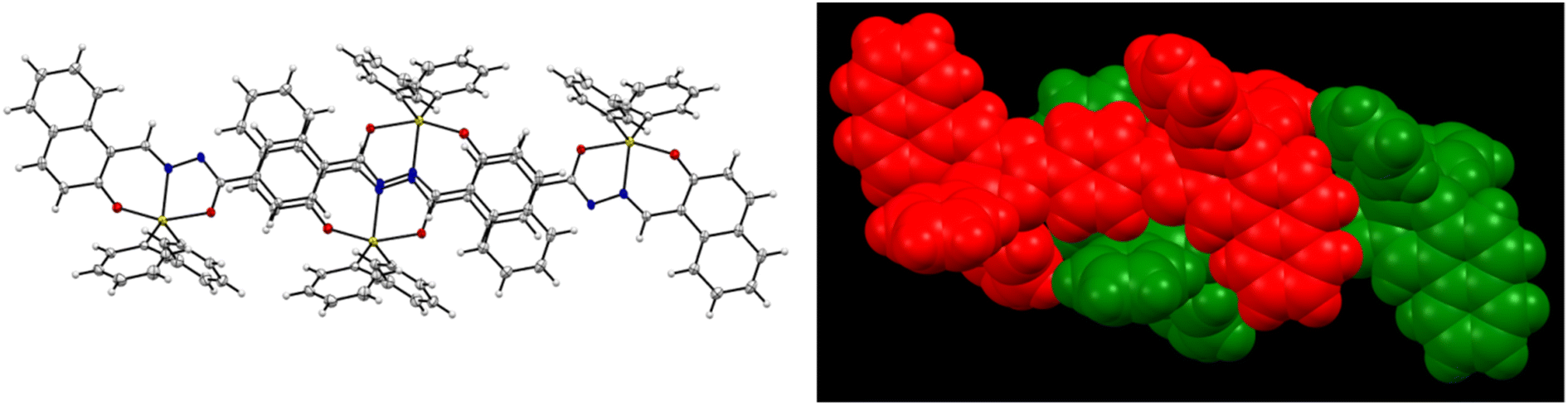
| | Fig. 6 Crystal structure of 6 showing intermolecular interactions. | |
 |
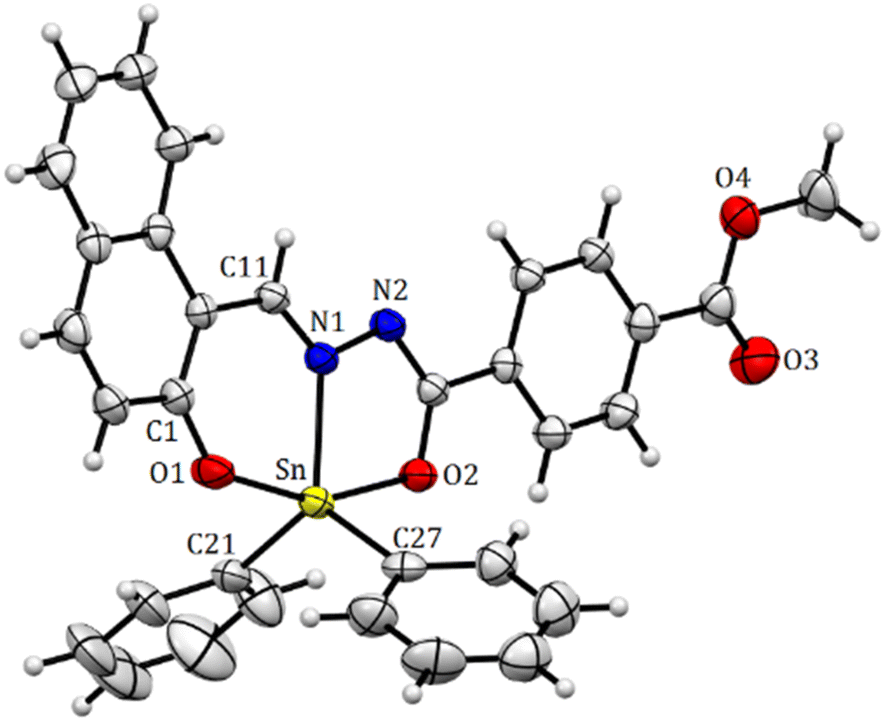
| | Fig. 7 Molecular structure of organotin compound 8. Selected bond distances (Å), Sn(1)–N(1) 2.147(2), Sn(1)–O(1) 2.087(2), Sn(1)–O(2) 2.1068(19), 2.1205(11), Sn(1)–C(27) 2.113(3), and Sn(1)–C(20) 2.122(3). Selected angles (°): O(1)–Sn(1)–O(2) 156.47(7), C(27)–Sn(1)–C(21) 121.70(11), C(27)–Sn(1)–N(1) 118.99 (5), and C(21)–Sn(1)–N(1) 119.03(9). | |
Table 2 Crystal data of compounds 6 and 8
| |
6
|
8
|
| Empirical formula |
C54H38N4O4Sn2 |
C32H24N2O4Sn |
| Formula weight |
1044.26 |
619.2 |
| Temperature, K |
100(2) |
298(2) |
| Wavelength Å |
0.71073 |
0.71073 |
| Crystal system |
Triclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P2(1)/c |
|
a, Å |
9.1150(8) |
14.1091(10) |
|
b, Å |
10.5354(9) |
21.6813(14) |
|
c, Å |
12.3958(11) |
9.2066(6) |
|
α
|
80.286(2)° |
90.00° |
|
β
|
72.486(2)° |
101.065(2)° |
|
γ
|
68.711(2)° |
90.00° |
|
V, Å3 |
1055.35(16) |
2764.0(3) |
|
Z
|
1 |
2 |
|
ρ
calc,mg cm−3
|
1.643 |
1.488 |
|
μ, mm−1 |
1.239 |
0.963 |
|
F(000) |
522.0 |
1248.0 |
| Crystal size/mm3 |
0.15 × 0.14 × 0.06 |
0.35 × 0.28 × 0.1 |
| Radiation |
MoKα (λ = 0.71073) |
MoKα (λ = 0.71073) |
| 2θ range for data collection |
6.752 to 61.018° |
2.386–29.209° |
| Index ranges |
−12 ≤ h ≤ 13 |
−29 ≤ k ≤ 29 |
| −19 ≤ h ≤ 19 |
−12 ≤ l ≤ 12 |
| −21 ≤ k ≤ 21 |
−15 ≤ k ≤ 15 |
| −12 ≤ l ≤ 12 |
−17 ≤ l ≤ 17 |
| No. of reflns collected |
14656 |
157026 |
| No. of indep reflns |
6385 |
5649 |
| [Rint] |
0.0185 |
0.0443 |
| Goodness of fit |
1.057 |
1.108 |
|
R
1, wR2 (I > 2σ(I)) |
0.0216/0.0540 |
0.0353/0.0826 |
| 0.1318 |
0.1283 |
|
R
1, wR2 (all data) |
0.0239/0.0550 |
0.0453/0.0885 |
| 0.1387 |
0.1207 |
| Largest diff. peak/hole/e Å−3 |
0.69/−0.75 |
0.80/−0.68 |
| 0.1387 |
0.1207 |
| CCDC number |
1871586
|
2293069
|
Table 3 Thermal data (°C) of organotin derivatives 5 and 6
| Compound |
T
o
|
T
d5
|
T
max
|
T
m
|
T
f
|
|
5
|
281 |
329 |
382 |
172 |
540 |
|
6
|
299 |
356 |
386 |
336 |
429 |
2.4. Scanning tunneling microscopy (STM)
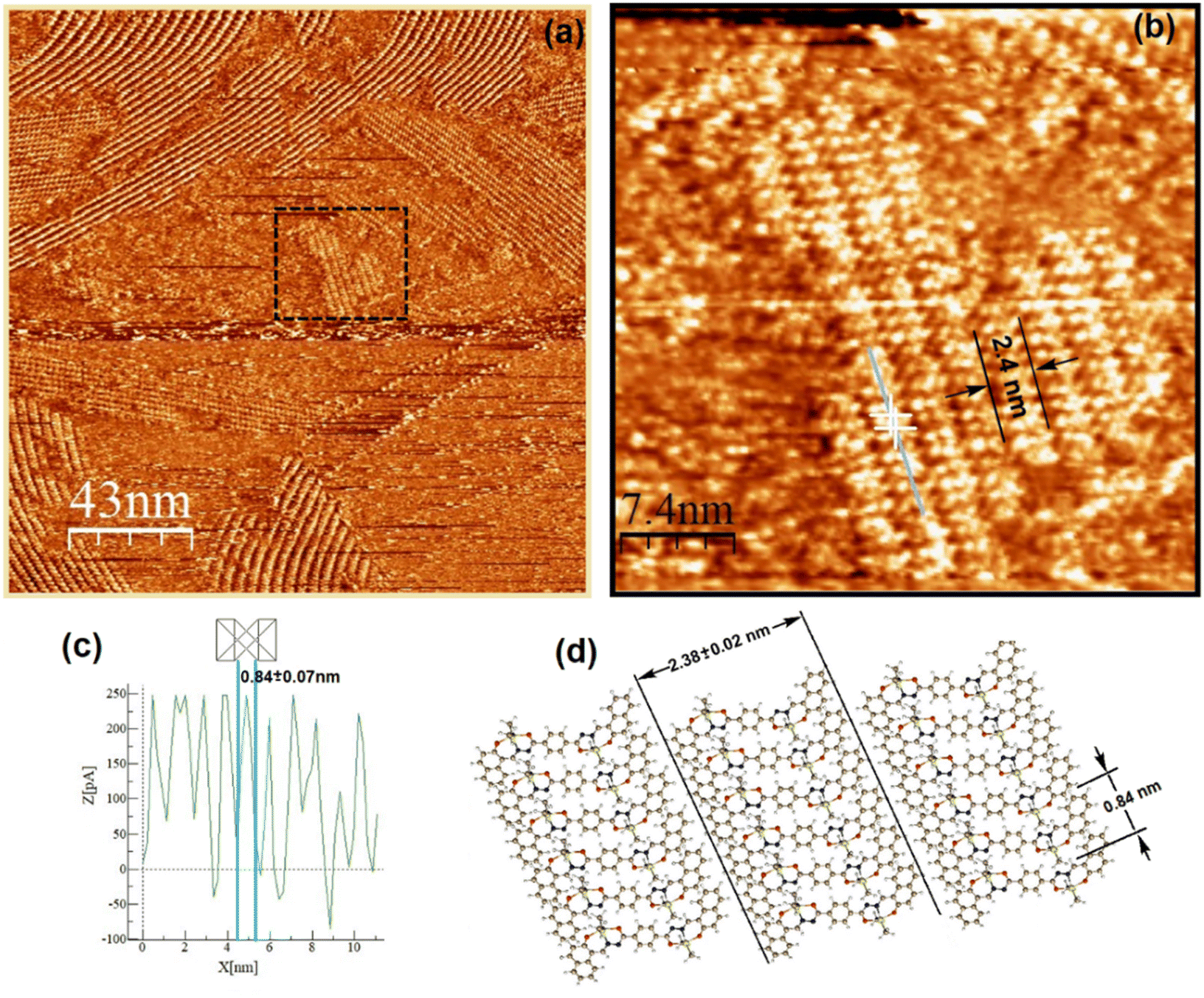
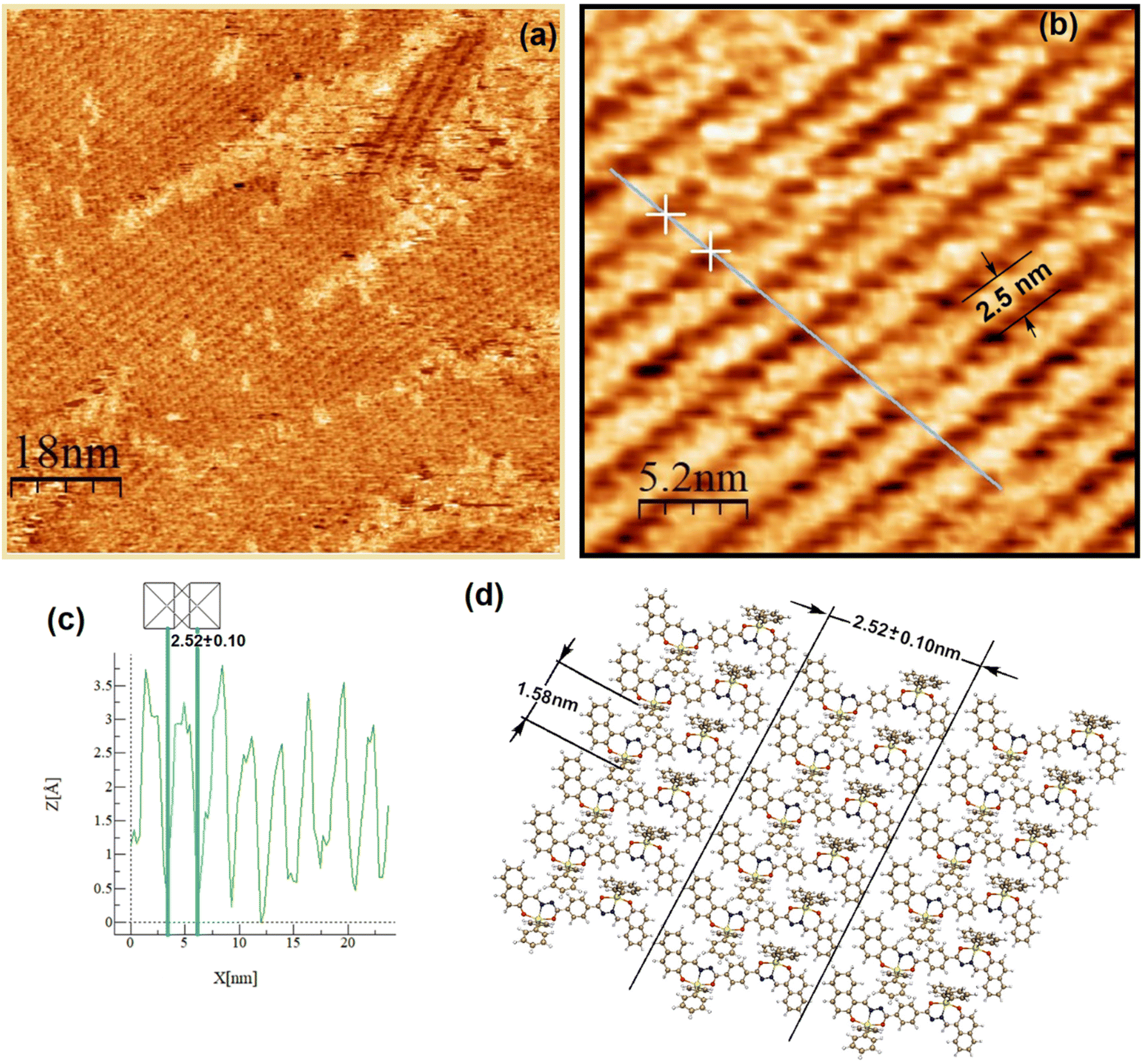
Both complexes were investigated at the atomic level by STM. After deposition of a drop of a 2.8 mM solution on highly oriented pyrolytic graphite (HOPG), a pulse of 2–6 V was immediately applied in order to induce the self-assembled monolayer (SAM) at the HOPG/TCB interface. Fig. 8a and 9a show the mesoscopic STM images, while those obtained at high resolution are shown in Fig. 8b and 9b, respectively. It can be seen, that on a mesoscopic scale, Sn-complex 5 does not cover large surface areas as Sn-complex 6 does. In fact, 5 self-organizes in randomly oriented microdomains, while 6 spontaneously arranges into a well-defined molecular layer. However, both complexes pack tightly on the surface, forming long lamellar patterns, with differences in the lamellar stacking because of the change in the ligand (butyl or phenyl) but always adopting a face-on arrangement,44 where the conjugated backbones are flat-lying on the HOPG surface, probably due to the π–π interaction of the aryls with the HOPG. The average lamella distance (Fig. 8a and b) is 2.38 ± 0.02 nm for complex 5 and 2.52 ± 0.10 nm for complex 6 (Fig. 9a and b). These values fit well with their theoretical molecular length in their most extended conformation (∼2.69 nm), which was obtained for the equilibrium geometries through density functional theory calculations. Within the lamellas, a spacing of 0.84 ± 0.07 nm for 5 and a spacing of 1.58 ± 0.15 nm for 6 were determined according to their profiles (Fig. 8c and Fig. S41 and S42†). In this case, the difference with the theoretical value (i.e., 1.37 nm corresponding to the alkyl-alkyl spacing for 5 and 1.46 nm to the phenyl-phenyl distance for 6) is more significant, suggesting that the molecules stack with the butyl or the phenyl ligands interdigitated as sketched in Fig. 8d and 9d, and certainly complex 6 presents a more extended separation because of the major steric volume of the phenyl with respect to the butyl ligands of complex 5.
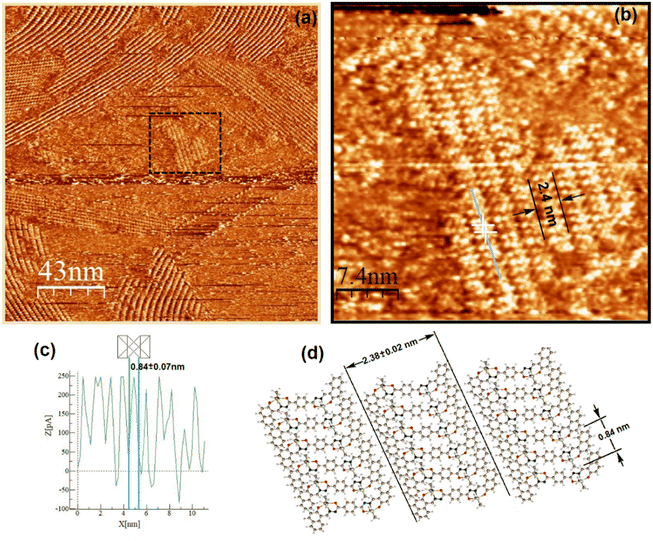 |
| | Fig. 8 Two-dimensional STM lamellar images of the HOPG/TCB interface of 5: (a) pronounced drift due to the instability and irregularity of the crystals on the HOPG taken at Vbias = −740 mV and It = 90 pA, (b) high-resolution STM image of 5 at Vbias = −740 mV and It = 90 pA, (c) current profile showing an individual molecule length packed within the lamellas and (d) molecular sketch depicting the lamella length that corresponds to the dimension of 5, and molecule dimensions were determined from the optimized molecular geometry. | |
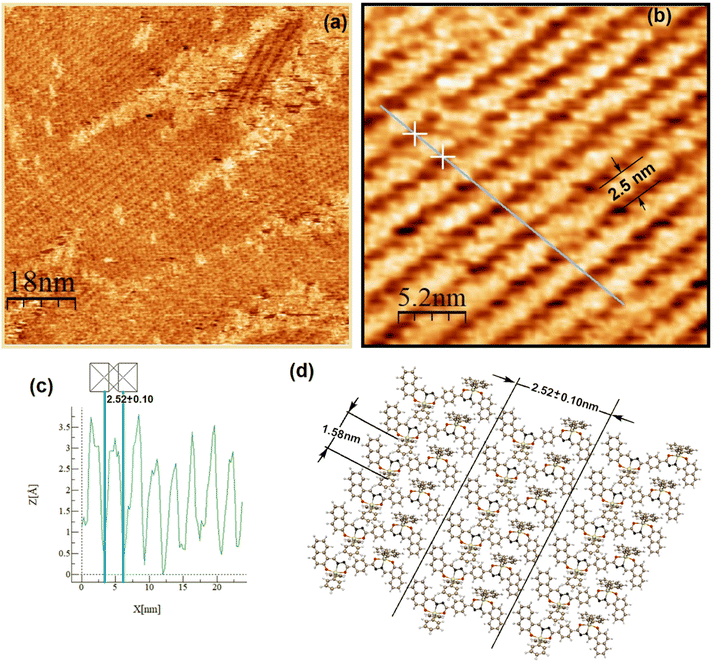 |
| | Fig. 9 Two-dimensional STM lamellar images of the HOPG/TCB interface of 6: (a) image taken at Vbias = −830 mV and It = 110 pA, (b) high-resolution STM image of 6 at Vbias = −830 mV and It = 110 pA, (c) current profile showing an individual molecule length packed into the lamellas and (d) molecular sketch depicting the lamella length that corresponds to the dimension of 6, and molecule dimensions were determined from the optimized molecular geometry. | |
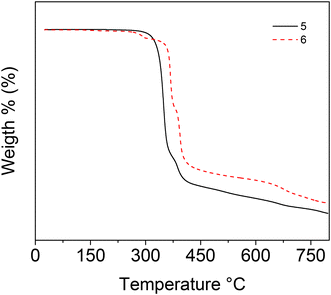
2.5. Thermogravimetric analysis
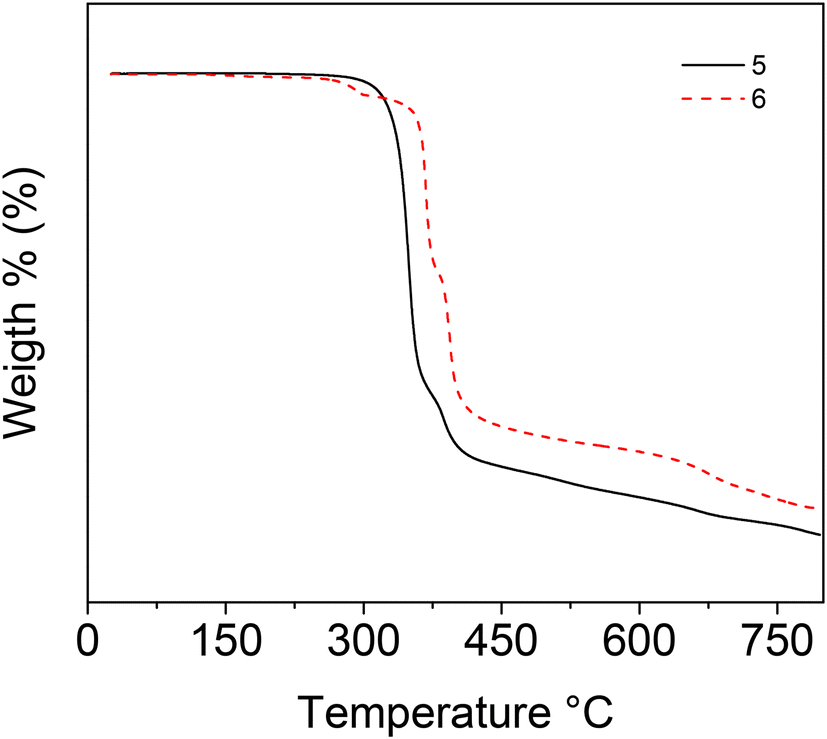
The TGA curves (Fig. 10, Fig. S31 and S32†) indicate that both bis-diorganotin complexes 5 and 6 begin to decompose at 329 and 356 °C respectively. The melting transition (Tm), decomposition temperatures (Td5), and other relevant thermal date are summarized in Table 3. The decomposition is a multistep process with four steps for 5 and 6, respectively; a similar behaviour has been reported for other diorganotin complexes.45
 |
| | Fig. 10 Thermogravimetric curves corresponding to organotin compounds 5 and 6. | |
For 5, the first step is at 298–345 °C corresponding to a mass loss of 44.4% that is associated with the release of C27H32N2O2Sn (cal. 44.5%); the second step at 370–452 °C corresponds to a weight loss of 12.0% due to C20H28O2Sn (cal. 12.0%). The third one at 457–740 °C with a mass loss of 7.27% can be originated from the loss of C16H22OSn (cal. 7.24%). For 6, the first step occurs at 240–302 °C (Fig. S32†), exhibiting a mass loss of 3.29% and corresponding to C51H38N4O4Sn2 (cal. 3.42%). The second step is at 380–408 °C, with a mass loss of 24.69% corresponding to C43H36O4Sn8 (cal. 24.85%). The last step appears at 410–430 °C with a loss of 22.06% attributed to C28H20O4Sn2 (cal. 21.50%). The next step appears at 618–711 °C with a loss of 9.58% attributed to C23H18O4Sn2 (cal. 8.66%).
2.6. Theoretical calculations
The geometry of both complexes was optimized in chloroform at the B3LYP/ZORA-def2-TZVP level of theory. The optimized ground-state structures of both complexes are depicted in Fig. 11, while the structural parameters with their corresponding experimental data are included in Table 5.
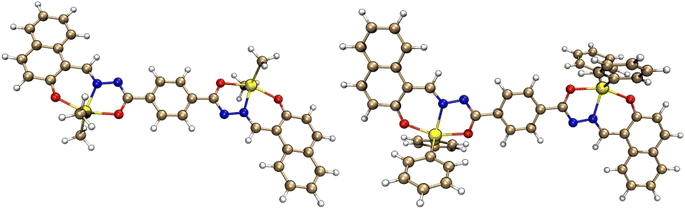 |
| | Fig. 11 Optimized molecular structures of 5 and 6 calculated at the B3LYP/ZORA-def2-TZVP level of theory. | |
As can be seen from Table 4, both complexes present slight differences with regard to the structural parameters between the theoretical and the experimental (X-ray) values because the optimization was carried out in solvents. While the distance between tin and the substituents’ carbon is in good agreement with X-ray data, the most prominent discrepancy is observed for the bond between tin and oxygen, which is higher, in the DFT optimization, for about 0.045 Å for both compounds. Concerning the bond angles, their values differ very little from the experimental ones. Complex 6 presents the biggest error for the angle between oxygen, tin and carbon, which is about 3.4°. In spite of the aforementioned differences with experimental data, the TDDFT simulation fits quite well the electronic transitions for both complexes in chloroform.
Table 4 Structural parameters of complexes 5 and 6, as determined using X-rays and DFT B3LYP/ZORA-def2-TZVP in chloroform
| |
X-Ray |
Chloroform |
X-Ray |
Chloroform |
| Complex 5 |
Complex 6 |
|
Bond length (Å)
|
| Sn(1)–N(1) |
2.152(5) |
2.180 |
2.1409(13) |
2.171 |
| Sn(1)–O(1) |
2.091(4) |
2.131 |
2.0688(11) |
2.113 |
| Sn(1)–O(2) |
2.127(4) |
2.170 |
2.1205(11) |
2.155 |
| Sn(1)–C(16) |
2.108(8) |
2.135 |
2.1090(15) |
2.133 |
| Sn(1)–C(22) |
2.130(8) |
2.135 |
2.1213(15) |
2.133 |
| |
|
Bond angles (°)
|
| O1–Sn–C16 |
94.40(3) |
94.65 |
97.86(5) |
95.29 |
| O1–Sn–C20 |
93.20(3) |
95.77 |
96.05(5) |
94.97 |
| C16–Sn–C20/C22 |
126.80(3) |
128.80 |
123.84(5) |
128.41 |
| O1–Sn–O2 |
155.30(2) |
154.97 |
156.00(5) |
156.23 |
| O1–Sn–N1 |
82.20(2) |
82.03 |
82.63(5) |
82.77 |
| C16–Sn–N1 |
117.20(3) |
116.201 |
117.92 (5) |
115.15 |
| N1–Sn–C20/C22 |
116.00(3) |
114.86 |
117.71(6) |
116.26 |
| O2–Sn–C16 |
96.709(2) |
95.16 |
96.61(5) |
95.29 |
| O2–Sn–C20 |
97.70(3) |
95.90 |
91.64(5) |
95.02 |
| O2–Sn–N1 |
73.20(2) |
72.98 |
73.69(4) |
73.47 |
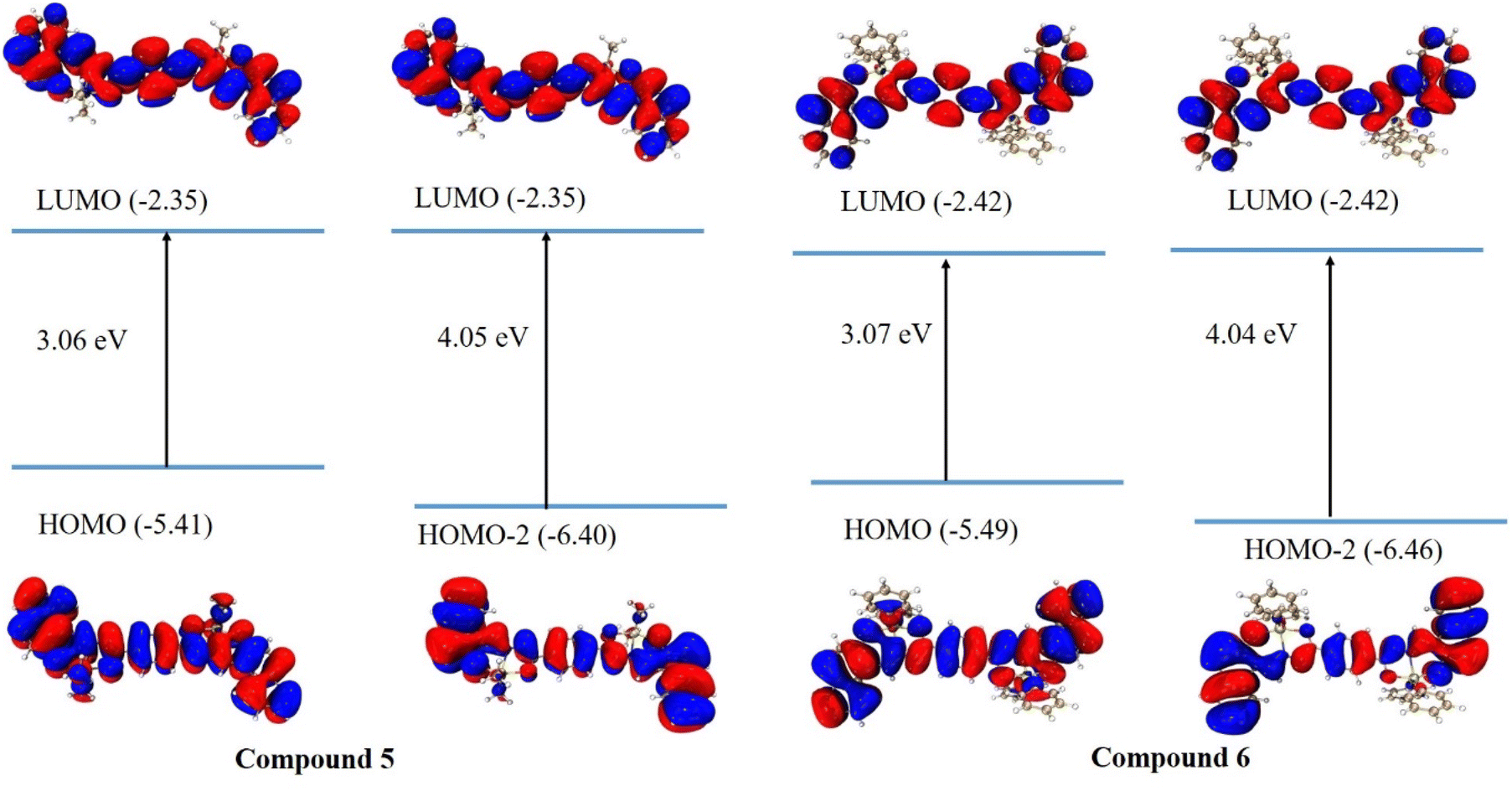
Fig. 12 shows the frontier molecular orbitals (FMOs) calculated from the ground-state optimized geometry in chloroform. The values of HOMO/LUMO of both compounds are very similar and do not depend on the tin's ligands: phenyl or butyl. The HOMO and LUMO orbitals show the π-type molecular orbital features. It is worth noting that both HOMO−2 and LUMO present the distribution all over the conjugated backbone including am imine group and without any participation of Sn atom for both compounds. The feature of HOMO and HOMO−2 orbitals may explain the similar results obtained for the excited states of both compounds. Naphthalene moieties principally contribute to the electron transition without any participation of the ligand substituent. The TDDFT procedure was used to get first 30 excited states. The calculated values of absorption, oscillator strength, and HOMO–LUMO transitions are listed in Table 5. In general, the calculated wavelengths are similar to the experimental values (vide infra). All the calculations are reported in Table S1, ESI.†
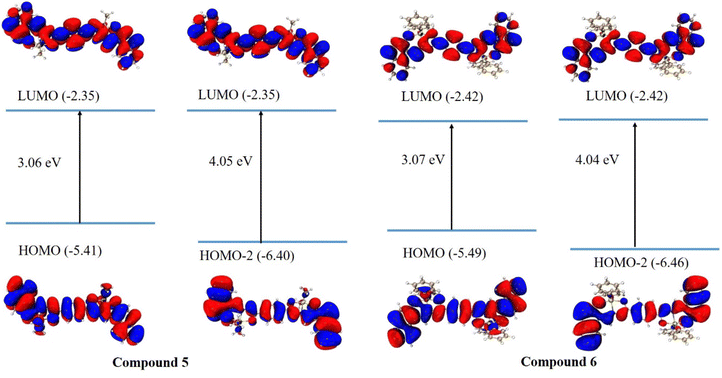 |
| | Fig. 12 Calculated frontier molecular orbitals (FMOs) for ground-state optimized compounds 5 and 6. Isosurface value = 0.025. | |
Table 5 Calculated optical properties of compounds 5 and 6 in CHCl3
| Compound |
λ
abs (nm) |
Oscillator strength f |
Electronic transitions |
|
5
butyl
|
336 |
0.23 |
HOMO−2 → LUMO (82%) |
| 464 |
1.40 |
HOMO → LUMO (94%) |
|
6
phenyl
|
339 |
0.24 |
HOMO−2 → LUMO (83%) |
| 460 |
1.46 |
HOMO → LUMO (94%) |
The calculated electrostatic potential map (EPM) of ligand 4 and Sn-complexes 5, 6 is shown in Fig. 13. The ligand exhibits an inhomogeneous electrostatic distribution map (in green) all along the structure. The greatest surface of electronegative charge (in red) susceptible to redox process is mainly located on both –OH groups of the naphthalene and on the nitrogen atoms of the azine group (N–N); in contrast, the lowest electronegative charge (in blue) is mainly found on the hydrogen atoms of the aryls. Interestingly, when ligand 4 is complexed with Sn, either with butyls or with phenyls, the EPM images show that the greatest surface of negative or electron-rich region is distributed on the central phenyl, at the –O and N– coordination centre, while the lowest electrostatic potential surface is located around the –Sn– coordination centre, meaning that their redox behaviour and contribution are lower with respect to the entire conjugation segment, probably because Sn imparts an electron donor character. As a term of comparison, the analogue Sn-mononuclear complex I shows that the greatest surface of negative charge is concentrated around the –C–O-phenyl, –C–O-naphthyl and on one of the coordination nitrogen Sn ← –NN–, while a slight EPM is distributed (yellow) on the conjugated aryls.
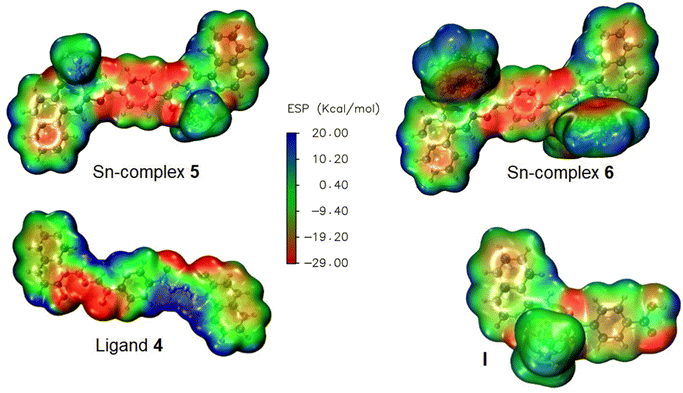 |
| | Fig. 13 Electrostatic potential map (EPM) of binuclear Sn-complexes 5 and 6 and their corresponding ligand 4. The EPM of mononuclear Sn complex I ([N-(2-oxido-1-naphthaldehyde)-4-hydroxybenzyhydrazidato]di-nbutyltin) is given for comparison purposes. | |
2.7. Electrochemistry
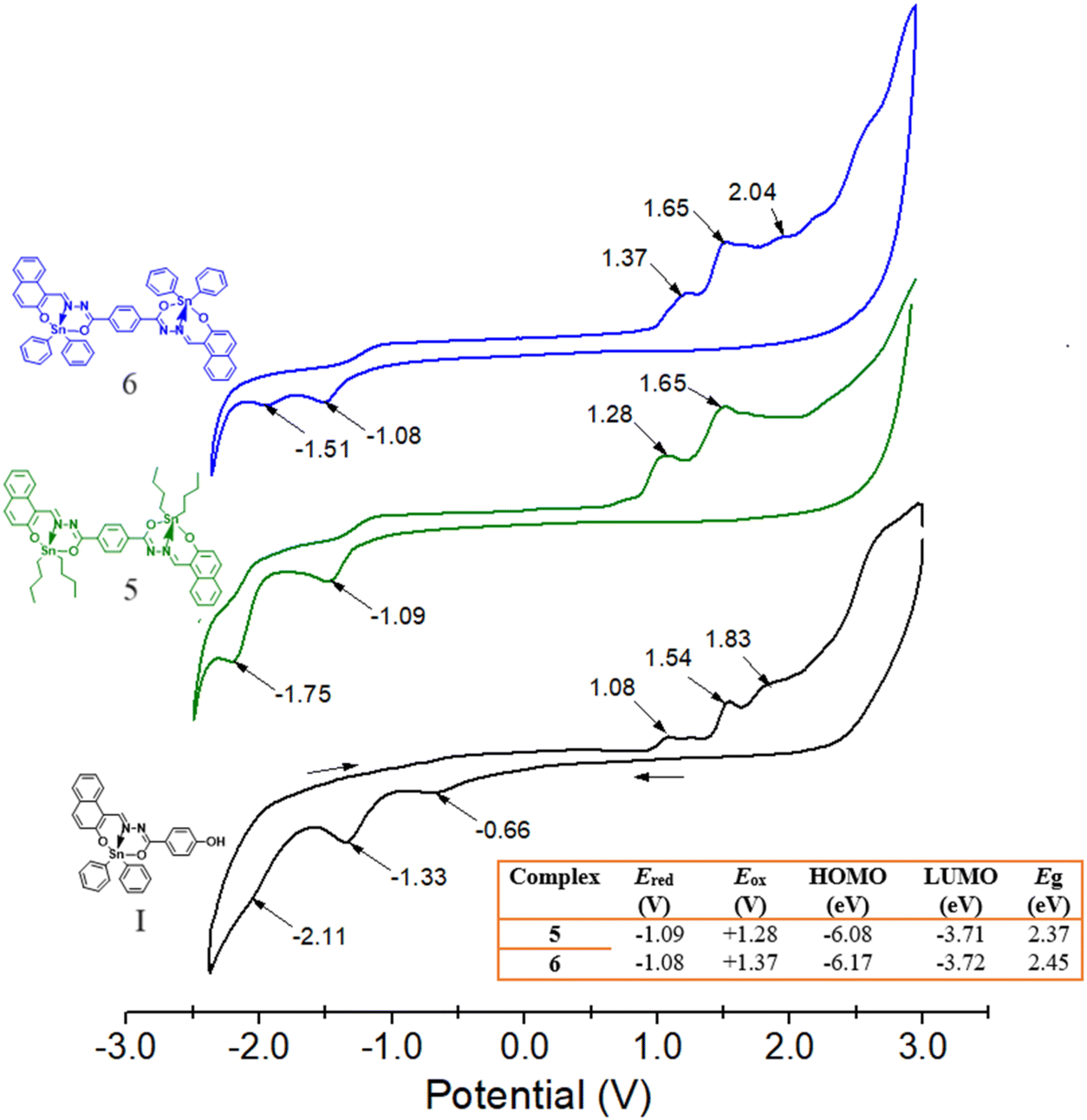
Both complexes 5 and 6 are electroactive in methylene chloride in either forward (from −3 to +3 V) or backward (+3 to −3 V) direction but not when separately analysed (0 to +3 V and 0 to −3 V) (Fig. S43 and S44†). In order to better understand the electrochemical behaviour of these complexes, Fig. 14 shows the voltammogram for 5 and 6, together with the mononuclear complex I as term of comparison. We have found that the Sn-mononuclear complex I exhibits at least four distinguishable oxidation peaks, whereas three reduction peaks are well defined; both processes are irreversible. From the molecular structures and EPM images, we may consider the –C–O-aryl and N–N aryl functional groups to be susceptible to redox processes, while the metal ion and its coordination with the azine group could rather be associated with the oxidation processes. In Fig. 14, the first peak at +1.08 V was associated with the loss of electrons of both the Sn and N–N, since we have previously found that the oxidation of the azine group appears for the free ligand at +0.9 V.46 Similarly, in the reduction process, the first peak of the reduction processes at −0.66 V is associated with the reduction of both, the Sn and its coordination with the azine because the corresponding free ligand reduction of the azine to (–NH–NH–) is found around −0.9 V.
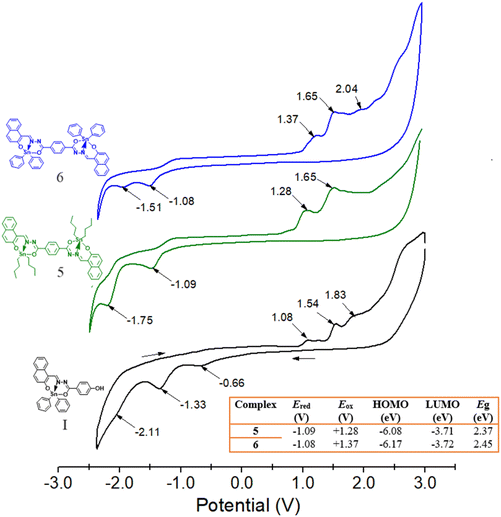 |
| | Fig. 14 Cyclic voltammograms of binuclear Sn complexes 5 and 6 and of mononuclear Sn complex I ([N-(2-oxido-1-naphthaldehyde)-4-hydroxybenzyhydrazidato]di-butyltin) as a term of comparison in 0.1 mmol CH2Cl2 at a scan rate of 50 mV s−1, V vs. Ag/AgCl and using GC as the working electrode. The inserted table summarises the electrochemical parameters determined for both binuclear complexes. | |
In contrast, voltammograms of complexes 5 and 6 disclose slight significant changes in both Ered potentials (1.08 V), as is observed in the inserted table of Fig. 14. This behaviour is in accordance with what was observed by theoretical calculations and UV/Vis spectroscopy (see further), i.e. there is no difference in the LUMO orbital distribution (3.71 eV), and a slight difference in the HOMO orbitals for the complexes by changing the butyls (6.08 eV) per phenyls (6.17 eV). The voltammograms show one weak oxidation peak around +1.37 V for 6 and 1.28 V for 5, which are assigned to the Sn oxidation; however this peak also overlaps with the peak associated with the loss of electrons of N–N group, as aforementioned for I. The most current intensity peaks are found at +1.65 and +2.04 V, associated with the conjugated aryls that become more difficult either to oxidize or to reduce. The presence of only one peak in the voltammograms for Sn suggests that the two atoms are chemically equivalent (as observed by NMR) and are not electronically interacting. The electrochemical band gap of Eg = 2.37 eV for 5 and of 2.46 eV for 6 are quite consistent with those obtained by UV-Vis spectroscopy of Eg = 2.45 and 2.49 eV, considering chloroform in both determinations.
2.8. Photophysical properties
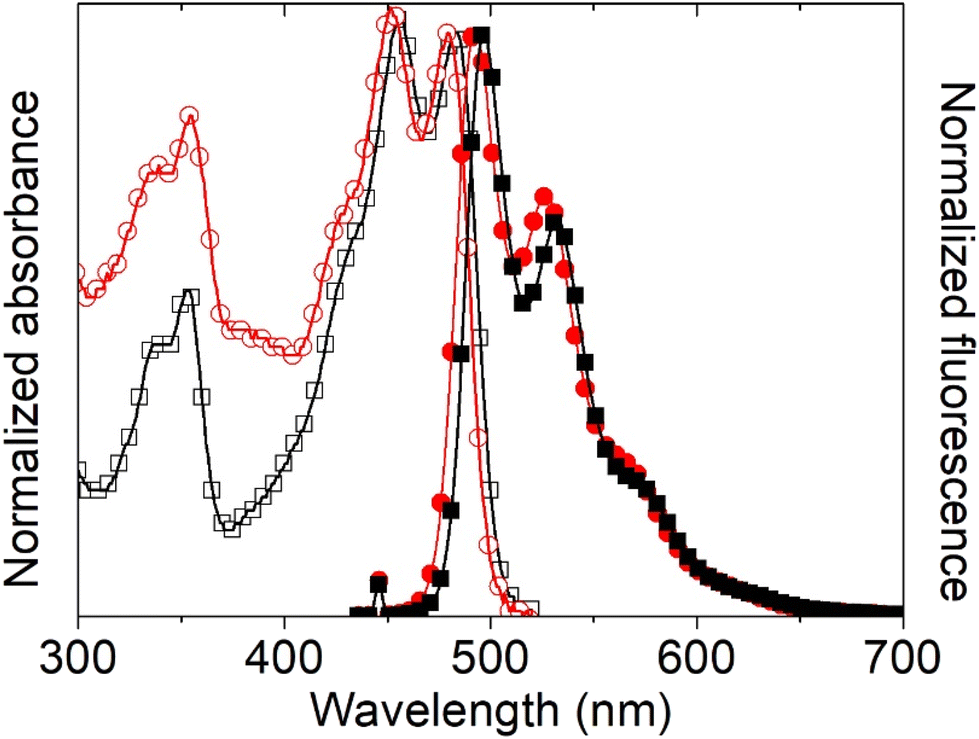
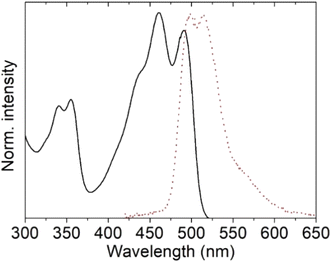
First, the intrinsic photophysical properties of complexes 5 and 6 were studied in chloroform, and are presented in Table 6. Fig. 15 shows the absorption and (inserted) fluorescence spectra of both tin complexes in chloroform. Note that ligand 4 is poorly soluble in chloroform, so its photophysical properties in this solvent were not investigated. Both compounds present two UV peaks at 337 nm and at 353 nm, and two peaks in the visible region at 456/453 nm and 484/481 nm, which are ascribed to HOMO−2 → LUMO (S0 → S5) and to HOMO → LUMO (S0 → S1) electronic transitions, respectively, according to the theoretical calculations. The peaks in the visible region exhibit excitonic features with a spacing between the excitonic peak and the first replica of 28 nm, and do not present a significant shift between the two complexes, indicating that the phenyl and butyl ligands do not participate in the electronic delocalization of the HOMO and LUMO orbitals, as previously found for other Sn complexes9 and according to the theoretical calculations. The optical band gap was almost equivalent, ∼2.5 eV, which corresponds to semiconducting materials.
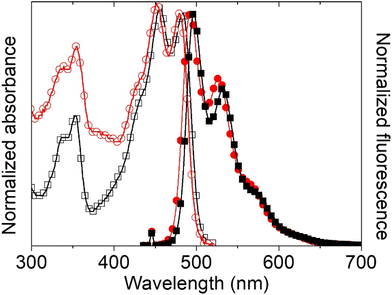 |
| | Fig. 15 Normalized UV-vis and fluorescence spectra of organotin compounds 5 (black squares) and 6 (red circles) in chloroform. | |
Table 6 Photophysical properties of organotin derivatives 5 and 6 and ligand 4 in chloroform, thin films and powders
| Comp. |
|
λ
abs (nm) |
ε
|
E
g (eV) |
λ
em (nm) |
Δν (cm−1) |
ϕ (%) |
τ (ns) |
|
Calculated on the bold marked wavelength.
|
|
4
|
Powder |
374, 395 |
— |
2.51 |
482 |
4570 |
— |
— |
|
5
|
CHCl3 |
337, 353, 456, 483 |
7.9 × 104 M−1 cm−1 |
2.45 |
496 |
543 |
35.0 |
1.76 |
| 533 |
1.77 |
| 571 |
1.78 |
| Thin film |
341, 355, 460, 491 |
3.9 × 10−3 nm−1 |
2.42 |
499 |
326 |
0.43 |
— |
| Powder |
460, 493 |
— |
2.16 |
557 |
2330 |
— |
— |
|
6
|
CHCl3 |
337, 353, 452, 479 |
4.3 × 104 M−1 cm−1 |
2.49 |
492 |
552 |
49.5 |
1.95 |
| 527 |
1.98 |
| 569 |
2.00 |
| Powder |
455, 490 |
— |
2.01 |
553 |
2324 |
— |
— |
The fluorescence spectra are quite mirror-like with respect to the visible part of the absorption spectra, presenting two main peaks: a maximum at 496/491 nm and a first replica at 533/527 nm being the spacing equal to 37/36 nm, similar to the values found in the absorption spectra. A shoulder is visible at ∼570 nm, attributed to the second vibronic replica and the corresponding excitation spectra are identical for the three emission features and match well with the absorption spectra. Moreover, the fluorescence spectra do not change with the excitation wavelength (Fig. S33†). This means that, even at the excitation of the naphthalene moiety (HOMO−2 → LUMO excitation), it corresponds to a higher energy excited state and the emission of naphthalene is not observed because it de-activates fast to the first excited state that corresponds to the extended π conjugated system. Moreover, the fluorescence spectra do not change with the excitation wavelength (Fig. S33†). From all these results, it is deduced that there is only one emitting state. In general, the optical properties are similar to those previously reported for mononuclear Sn complexes derived from [N-(2-oxido-1-naphthaldehyde)-4-hydroxybenzyhydrazidate]47 but with red-shifts in the absorption and emission maxima, indicating an increase in the effective conjugation length. The fluorescence quantum yields are also in the same range, with even shorter Stokes shifts Δ, and the fluorescence decays can be well fitted (χ2 ≤ 1.2) by a monoexponential curve with a lifetime (τ) of ∼2 ns at any of the different emission features (Table 9) and regardless of the excitation wavelength (Fig. S34 and Table S2, ESI†). This confirms that the emission peaks at 496/492, 533/527 and 571/569 (shoulder) are all related to the same excited state, and there is one de-activation pathway. The extremely small Δ values reveal that the geometry is maintained after excitation. The deviation from the unity for the fluorescence quantum yield is thus expected to be mainly due to competitive processes, different from internal conversion. Intersystem crossing has been found for mononuclear analogues.48 Theoretically, the intersystem crossing process is favoured when two conditions are met: a high spin–orbit coupling (SOC) value, i.e. in the range of 0.3–5 cm−1 and a small energy difference between the first singlet and triplet states (ES1–Tn).49 For the present complexes, the first triplet state T1 was calculated at 2.06 eV and 2.07 eV respectively for 5 and 6, giving a quite large energy barrier with the first singlet excited state (ΔES1–T1 ∼ 0.6 eV). Moreover, the SOC for S1–Tn (n = 1, 2 or 3) has very low values (0.80, 0.87, and 0.81 cm−1 for 5 and 0.10, 0.81, and 0.08 cm−1 for 6, respectively). Nevertheless, it is evident from Fig. 15 that there is a large overlapping between the absorption and emission spectra, which causes the emitted light to be re-absorbed. Conversely, another possible cause is non-radiative loss induced by intramolecular rotation, as found for other Schiff base complexes.50 To support this hypothesis, fluorescence spectra were recorded in the temperature range −10 to 40 °C. Fig. 16 shows the spectral evolution for 5 (Fig. S35,† for 6), as an example, where a gradual increase in the emission intensity by lowering the temperature can be observed, confirming this not-radiative loss.
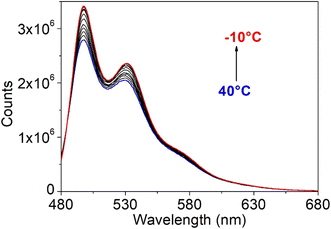 |
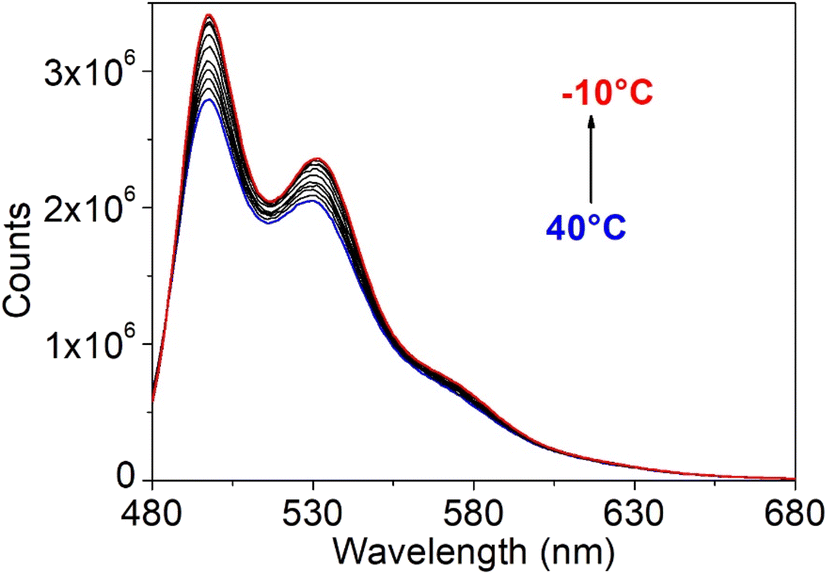
| | Fig. 16 Fluorescence spectra of organotin compound 5 in chloroform at different temperatures. | |
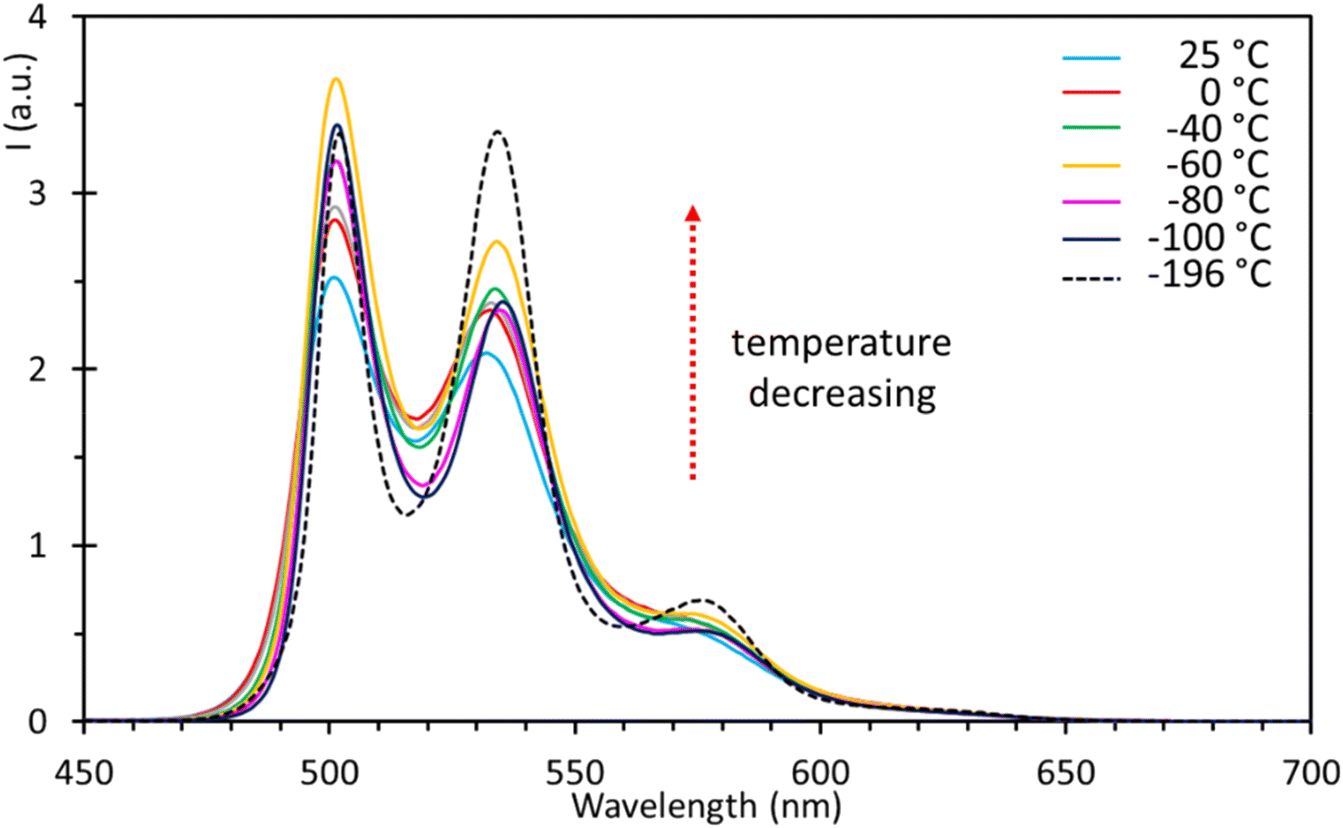
To further prove this hypothesis, several fluorescence measurements in a temperature range from 25 to −196 °C have been performed using 2-methyl tetrahydrofuran (Me-THF) as the solvent. This allowed us to achieve good solubility for both complexes 5 and 6, and for the poorly soluble ligand 4. For complex 5, two main emission maxima were observed (Fig. 17), respectively at 502 and 534 nm, slightly red-shifted with respect to the chloroform solution but with the same spectral features, i.e. excitonic-like, with the first one usually being the most intense. An additional shoulder was detected at 577 nm, gradually increasing in intensity with the decrease in temperature and becoming relatively maximum at −196 °C. According to lifetime measurements (vide infra), where no notable changes have been observed on lowering the temperature, this behaviour was not ascribed to additional radiative processes (i.e., phosphorescence), but was simply due to the appearance of the vibronic structure at low temperatures. At −196 °C, the intensity of the maximum at 534 nm became very similar to that of the maximum at 502 nm. The general trend observed was an increase in emission intensity with the decrease in temperature (Fig. 17). More in detail, there is a net increase from 25 to −60 °C, followed by a decrease at −80 °C, after which an increase can be observed again, reaching the maximum intensity at −196 °C. Complex 6 showed instead two emission maxima at 496 and 530 nm (Fig. S38†). All the observations reported for 5 can be applied to complex 6 as well, validating that there is no marked difference in the electronic properties of the complexes inferred by the ligand bound to tin (phenyl or butyl); moreover, a slight hypsochromic shift of ca. 6 nm of the maxima was observed for complex 6 at −196 °C.
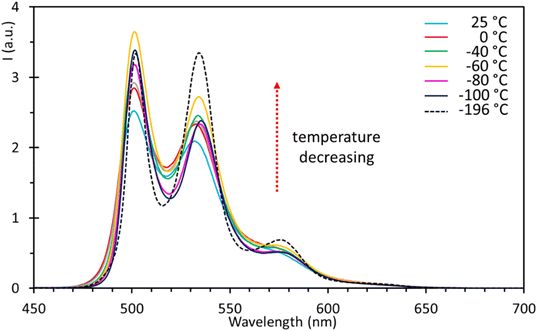 |
| | Fig. 17 Fluorescence spectra of organotin compound 5 in 2-methyl tetrahydrofuran (5 × 10−5 M) at different temperatures. | |
Interestingly, using 2-methyl tetrahydrofuran, it was also possible to study the fluorescence properties at low temperatures of compound 4 (Fig. S36†). The ligand exhibited the same trend described for the relative complexes, with a gradual increase in emission intensity by lowering the temperature. The maximum emission at 488 nm was recorded, with a small shoulder at 509 nm, which progressively became more pronounced upon cooling. However, compound 4 displayed lesser emissive properties all along the temperature range compared to the corresponding complexes 5 and 6, as demonstrated by quantum yield measurements. Indeed, absolute fluorescence quantum yields in 2-methyl tetrahydrofuran were first recorded for compounds 4, 5 and 6, at room temperature (Table 7). The ligand displayed a very low quantum yield of 5%, whereas the organotin compounds reached values similar to those analysed in the chloroform solution (28 and 43% respectively for 5 and 6). Then, starting from the corresponding intensity of the emission, fluorescence quantum yields at low temperatures in 2-methyl tetrahydrofuran were estimated indirectly by comparison with the results at room temperature (see eqn (1) in the Experimental section). As expected, the quantum yields of both compounds 5 and 6 increased upon lowering the temperature, thus confirming that the deviation from the unity of quantum yields is probably due to non-radiative losses caused by rotational relaxation, prevented at lower temperatures. Moreover, the highest fluorescent quantum yields of the complexes with respect to ligand 4 are due to the more rigidity of the whole system imposed upon coordination to the metal centre.
Table 7 Estimated quantum yields of compounds 4, 5, and 6 at different temperatures
| Comp. |
25 °C |
0 °C |
−10 °C |
−40 °C |
−60 °C |
−80 °C |
−100 °C |
−196 °C |
Improved Φ |
|
Experimental values recorded in 2-methyl tetrahydrofuran (5 × 10−5 M).
|
| 4 |
0.05a |
0.07 |
0.09 |
0.10 |
0.18 |
0.12 |
0.13 |
0.24 |
4.8-fold |
| 5 |
0.28a |
0.32 |
0.32 |
0.35 |
0.40 |
0.35 |
0.38 |
0.37 |
1.3-fold |
| 6 |
0.43a |
0.53 |
0.60 |
0.60 |
0.68 |
0.59 |
0.67 |
0.67 |
1.5-fold |
Finally, lifetime data were collected for both complexes 5 and 6 at different temperatures and at all emission maxima (Tables 8 and 9). Complex 5 showed bi-exponential decay curves (Table 8) in the whole series, most probably due to the possible different radiative decay pathways generated by the high mobility of butyl substituents. The τ1 value is in the range of 1.40–1.99 ns (at 502 nm), 1.29–1.84 ns (at 534) and 1.29–1.70 ns (at 577 nm), whereas the second lifetime (τ2) is distributed between 3.14 and 3.67 ns (at 502 nm), 3.01–3.53 ns (at 534 nm) and 3.06–3.47 ns (at 577 nm). Upon lowering the temperature, the relative percentages of the two lifetimes change marginally. As a result, the average weighted lifetime values (Table S3†) slightly increase upon lowering the temperature, a trend which occurred at all maxima. It is noteworthy that the differences between the lifetime values recorded at different maxima are very negligible (Table S3†). According to the values reported in Table S3,† complex 5 always showed average weighted lifetimes comprised between 2.34 and 2.84 ns (at 502 nm), 2.34 and 2.86 ns (at 534 nm) and 2.40 and 2.88 ns (at 577 nm). At a fixed temperature, the differences between lifetimes recorded at different maxima are always less than 0.11 ns, so within experimental errors. The same considerations can be done for complex 6 (Table 9), where again the lifetimes increase at low temperatures at both maxima. In this case, a mono-exponential fit is observed, due to the lesser mobility of phenyl groups than that of butyl. The only exceptions are represented by decays measured at −100 °C and −196 °C, where the curves are described by a bi-exponential fit: reasonably, at these temperatures, some heterogeneity in the complex surrounding is responsible for an additional decay pathway. Such a bi-exponential fit is somehow intrinsic in the ligand behaviour and attributed to possible different radiative decay pathways generated by free rotation around C–C and N–N bonds (Table S4†). The lifetime values are lower than those observed in the complexes (averaged weighted values: τ = 0.97–2.02 ns at 488 nm; τ = 0.96–1.95 ns at 509). In conclusion, no significant variations are observed in all the series of lifetimes collected regardless of the temperature, thus excluding distinct emission processes (like phosphorescence) at different temperatures.
Table 8 Fluorescence lifetimes of compound 5, at different temperatures, recorded in 2-methyl tetrahydrofuran (5 × 10−5 M)
|
T (°C) |
τ (ns) at 502 nm |
Goodness of fit (χ2) |
τ (ns) at 534 nm |
Goodness of fit (χ2) |
τ (ns) at 577 nm |
Goodness of fit (χ2) |
| 25 |
1.40 (45.80%) |
1.109 |
1.29 (38.93%) |
1.036 |
1.29 (37.30%) |
1.195 |
| 3.14 (54.20%) |
3.01 (61.07%) |
3.06 (62.70%) |
| 0 |
1.53 (40.52%) |
1.173 |
1.45 (39.06%) |
0.867 |
1.40 (35.13%) |
0.909 |
| 3.16 (59.48%) |
3.19 (60.94%) |
3.10 (64.87%) |
| −10 |
1.48 (44.76%) |
1.190 |
1.48 (43.46%) |
1.150 |
1.37 (36.62%) |
1.073 |
| 3.19 (55.24%) |
3.19 (56.54%) |
3.18 (63.38%) |
| −40 |
1.45 (36.99%) |
1.065 |
1.31 (29.99%) |
1.106 |
1.34 (28.77%) |
0.902 |
| 3.21 (63.01%) |
3.12 (70.01%) |
3.13 (71.23%) |
| −60 |
1.65 (39.44%) |
1.047 |
1.62 (33.42%) |
1.198 |
1.32 (26.03%) |
1.039 |
| 3.41 (60.56%) |
3.36 (66.58%) |
3.24 (73.97%) |
| −80 |
1.19 (23.08%) |
1.091 |
1.36 (22.30%) |
0.934 |
1.65 (30.03%) |
1.098 |
|
|
3.24 (76.92%) |
|
3.26 (77.70%) |
|
3.41 (69.97%) |
|
| −100 |
1.99 (49.32%) |
1.177 |
1.84 (39.37%) |
0.982 |
1.67 (32.31%) |
1.160 |
| 3.67 (50.68%) |
3.53 (60.63%) |
3.45 (67.69%) |
| −196 |
1.77 (42.72%) |
1.100 |
1.64 (35.57%) |
1.100 |
1.70 (41.99%) |
1.028 |
| 3.40 (57.28%) |
3.32 (64.43%) |
3.47 (58.01%) |
Table 9 Fluorescence lifetimes of compound 6 at different temperatures recorded in 2-methyl tetrahydrofuran (5 × 10−5 M)
|
T (°C) |
τ (ns) at 496 nm |
Goodness of fit (χ2) |
Average τ (ns) at 496 nm |
τ (ns) at 530 nm |
Goodness of fit (χ2) |
Average τ (ns) at 530 nm |
| 25 |
2.04 |
1.018 |
— |
2.09 |
1.003 |
— |
| 0 |
2.01 |
0.867 |
— |
2.01 |
1.001 |
— |
| −10 |
2.00 |
1.182 |
— |
2.14 |
1.013 |
— |
| −40 |
2.04 |
1.120 |
— |
2.05 |
1.190 |
— |
| −60 |
1.97 |
0.937 |
— |
2.08 |
1.175 |
— |
| −80 |
1.98 |
1.090 |
— |
2.02 |
1.109 |
— |
| −100 |
1.51 (21.69%) |
1.066 |
2.97 |
1.48 (20.12%) |
1.001 |
2.97 |
| 3.38 (71.31%) |
3.35 (79.88%) |
| −196 |
1.67 (40.97%) |
1.118 |
2.73 |
1.65 (42.70%) |
1.103 |
2.72 |
| 3.46 (59.03%) |
3.52 (57.30%) |
Due to the good quantum yields and previous results reported for other Sn complexes,51,52 the feasibility of obtaining good optical quality films for optoelectronic applications was investigated. Despite covering a large area surface in the monolayer, complex 6 unfortunately could not form homogeneous thin films of the thickness that is required for light-emitting diodes or solar cells (100–150 nm) as its solubility is limited to 5 g L−1. On the contrary, complex 5 can be dissolved in concentrated solutions (5–30 g L−1) from different solvents (chloroform, dichloromethane, THF, and chlorobenzene) and could be deposited by spin-coating. Based on the thickness and roughness values, 10 g L−1 chlorobenzene solution was used for the preparation of films. However, the AFM study (Fig. S39†) shows the coexistence of a granular morphology that is typical of aggregated materials, with big irregular structures, which persist even in small areas, and can be probably associated with the microdomains found at the atomic level, in the STM study. The UV-Vis spectra in films (Fig. 18) are similar to those studied in solutions, but the maxima are slightly red shifted, which is due to intermolecular interactions occurring in the solid state.
 |
| | Fig. 18 UV-vis (solid line) and fluorescence (dotted line) spectra of a spun film of organotin compound 5. | |
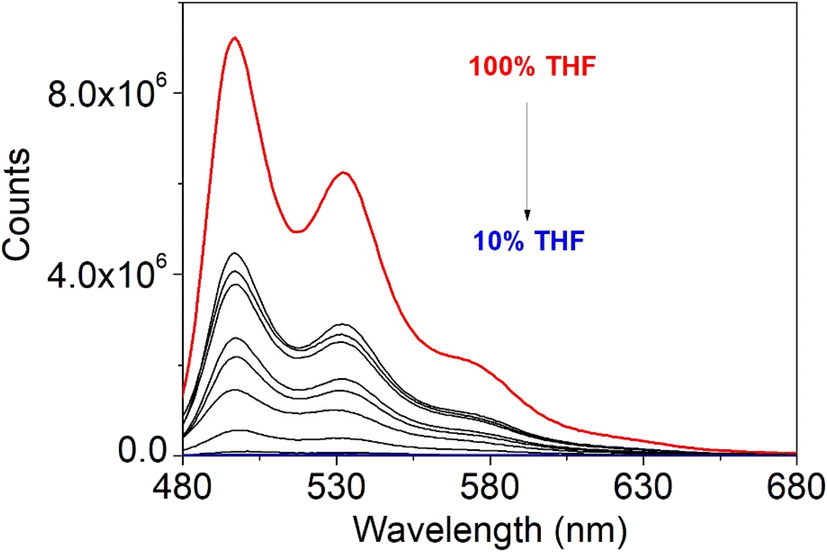
As found in the solution, the Stokes shift value is very small suggesting very minor influence of internal conversion but possible self-absorption. The fluorescence quantum yield is ∼500-fold quenched with respect to the solution. As the fluorescence quenching is very large, and aggregates were observed in the AFM study, aggregation studies were carried out in THF/water mixtures (5 and 6 are soluble in THF but insoluble in water), increasing the water content from 0 to 90% with a 10% volume percent increase. Fig. 19 reports the study on complex 5 as example; the fluorescence intensity decreases with the increase in water content to 90%. The aggregation-caused quenching (ACQ)53 is consistent with the strong π–π stacking interactions observed in the crystallographic study for the single crystal, which are expected to increase as the non-solvent content increases.
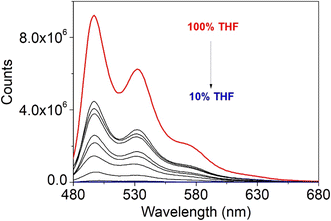 |
| | Fig. 19 Emission spectra of 5 in THF/water mixtures with different water fractions. From top to bottom: 100%, 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, and 10% THF. | |
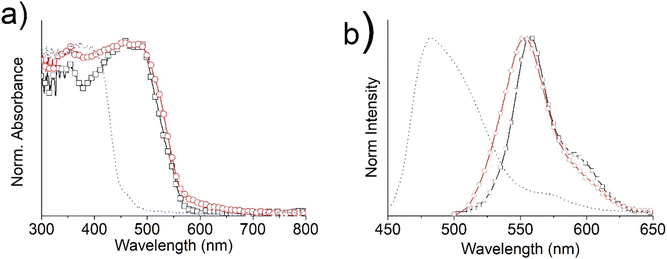
In order to investigate if this behaviour was retained in bulk, powders were also analysed. Fig. 20 presents the UV-Vis absorption (a) and emission (b) spectra of complexes 5 and 6 as bulk. The optical properties of the ligand 4 are also shown, for comparison.
 |
| | Fig. 20 Powder (a) UV-vis and (b) fluorescence spectra of organotin compounds 5 (squares), 6 (circles) and the ligand 4 (dotted lines). | |
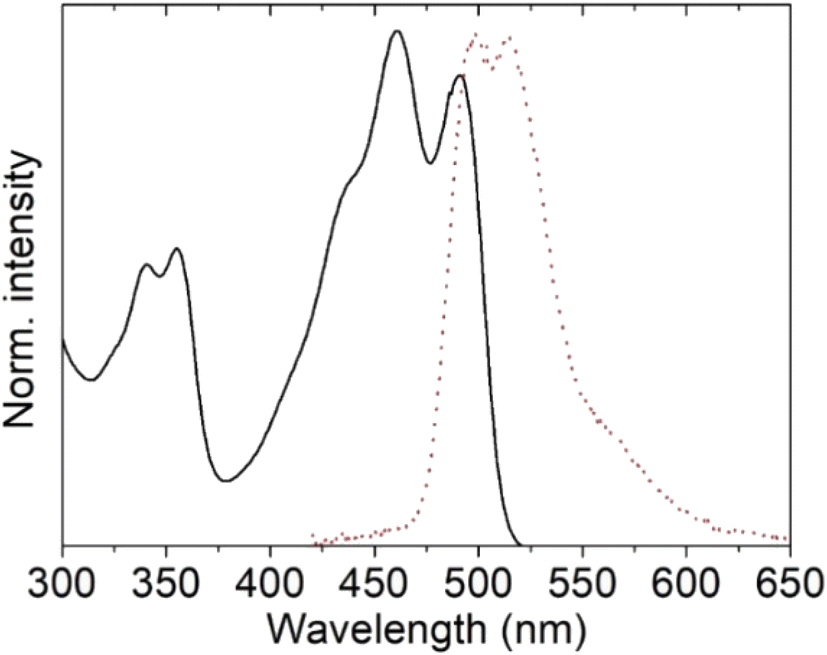
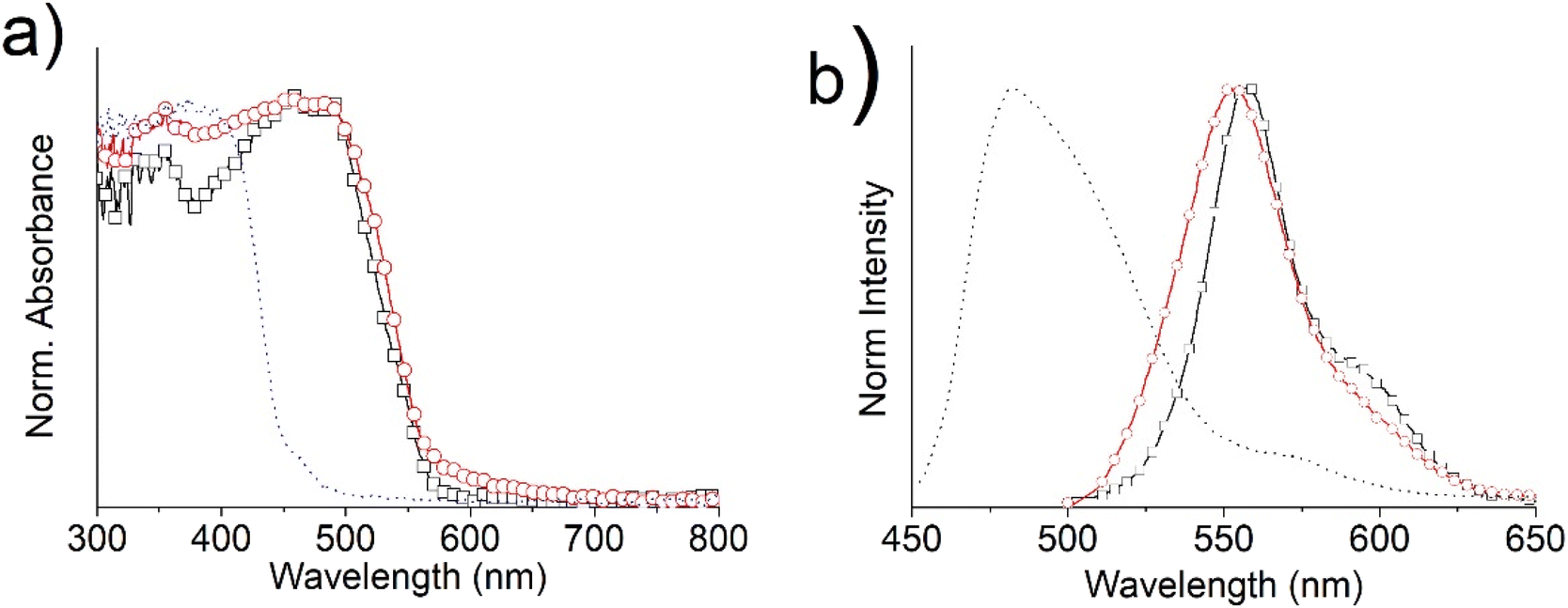
Both the absorption and emission spectra of the tin complexes in powders maintain spectral features similar to those of the solutions, but red-shifted almost 10 nm for the UV-Vis and 50 nm for the fluorescence maxima, consistent with intermolecular interactions. The UV-Vis spectrum of the ligand 4 presents a broad band at around 400 nm, due to the π–π* electronic transitions of the conjugated Schiff base, which gives rise to a very broad emission band at 482 nm.
3. Conclusion
In summary, dinuclear organotin Schiff bases represent a fascinating class of compounds that exhibit cryoluminescence properties. The sustainable synthesis of these compounds is of paramount importance in achieving environmentally friendly and resource-efficient processes. Understanding and harnessing the cryoluminescence properties of dinuclear organotin Schiff bases will not only advance the field of coordination chemistry but also pave the way for the development of novel luminescent applications at cryogenic temperatures. Waste polyethylene terephthalate (PET) is a suitable precursor for the synthesis of fluorescent organic ligands and organotin complexes. The compounds exhibited good luminescence properties in solutions with quantum yields of 35.0 and 49.5%, which decrease in films or solid state because of non-radiative losses and aggregation-caused quenching, as found by a fluorescence study in solutions with temperature (−10 to 40 °C) and in solvent/non solvent mixtures.
The supramolecular organization was studied in self-assembled monolayers at the atomic level on HOPG of both Sn-complexes. From the STM study, we conclude that the alkyls or phenyls in the Sn-complex essentially drive the 2D assembly of the structures, and if both Sn-complexes well form lamella-like patterns, it was the phenyl 6 that covered the entire surface of the HOPG. The lamella dimensions correspond to the length of the conjugated structures with an average value of 2.38 nm for 5 and of 2.52 nm for 6; the values that are consistent for Sn-complexes that self-assemble with the alkyl and phenyl chains tightly interdigitated. The NMR, photophysical, electrochemical and theoretical studies including the electrostatic potential map (EPM) reveal that the two Sn-nuclei do not electronically interact.
4. Experimental section
4.1. Materials and measurements
All starting materials such as Bu2SnO, Ph2SnO, dimethylterephthalate, hydrazine monohydrate and organic solvent were purchased from Aldrich Chemical Company. 2-Methyltetrahydrofuran (stabilized with BHT) was purchased from TCI chemicals. Solvents were used without further purification. The synthesis of all complexes was performed using an Anton Paar Monowave 300 microwave reactor. Melting points were carried out using an Electrothermal Mel-Temp apparatus and are uncorrected. Infrared spectra were recorded using a Bruker Tensor 27 FT-IR spectrophotometer equipped with a Pike Miracle™ ATR accessory with a single-reflection ZnSe ATR crystal. Raman spectra were obtained using Horiba Xplora equipment, focusing the sample as powders on a microscopic slide with a 10X objective. The excitation wavelength was 785 nm and the Nanoled power was 10% of total laser power (25 mW). Spectra were acquired with 10 s acquisition time and 10 accumulations, at a spectral resolution of 2 cm−1. High-resolution mass spectra were acquired by LC/MSD TOF using an Agilent Technologies instrument with APCI as the ionization source. 1H, 13C and 119Sn-NMR spectra were recorded in CDCl3 and (CD3)2SO using a Bruker advance DPX 400. Chemical shifts (ppm) are relative to (CH3)4Si for 1H and 13C and to (CH3)4Sn for 119Sn. The thermal stability of compounds (vacuum dried samples) was determined using a thermogravimetric analyser (TGA 951 from DuPont Instruments) connected to a N2 vector gas, heating from 25 to 600 °C at 10 °C min−1.
4.2. Theoretical calculations
All theoretical calculations were performed using Orca, version 5.0.1.54 Geometry optimizations in chloroform via LR-CPCM55 under equilibrium conditions were performed with the B3LYP functional, including D3BJ dispersion correction with Becke–Johnson damping56 and considering scalar relativistic zeroth-order regular approximation (ZORA).57 For light elements such as H, C, O and N, Hamiltonian-specific recontractions of all-electron nonrelativistic Karlsruhe basis sets58 were used, while for Sn specially designed segmented all-electron relativistically contracted (SARC) basis set59 was employed for both geometry optimization and TDDFT calculations in combination with the corresponding auxiliary SARC/J and def2-TZVP/C basis sets. Analytical frequencies were computed for all optimized structures and the absence of imaginary modes confirmed that true minima were obtained in all cases. The excited state analysis was performed using Multiwfn 3.8.60 Electrostatic potential maps (MEPs) were plotted for the optimized geometries using the VMD program.61 The blue colour represents positive electrostatic potential while the red one corresponds to a negative electrostatic potential and the green is attributed to zero potential. All calculations were made on cluster Thubat Kaal II of National Supercomputing Center-IPICYT.
4.3. Photophysical characterization
For the determination of the intrinsic photophysical properties, dilute solutions (∼10−6 M) in freshly distilled spectroscopic grade CHCl3 (Sigma-Aldrich) were studied as prepared, to avoid any solvolysis or photodegradation effect.62 The UV-Vis absorption spectra were recorded using a Shimadzu 2401PC spectrophotometer. The extinction coefficient was obtained from the slope of the absorbance vs. molar concentration for four solutions. The optical band gap (Eg) corresponds to the value where the X axis (energy) crosses the tangent drawn at absorbance of 0.1 of the normalized absorption spectra. The excitation and emission spectra were recorded using a Horiba PTI Quantamaster QM-8450-22-c spectrofluorimeter, equipped with an integrating sphere for the determination of the quantum yield and a Peltier accessory for the temperature studies (with a refrigeration liquid for a temperature range between −10 and 40 °C). All the spectra were recorded using a background correction. The excitation wavelength was 10 nm below the main absorption peak and the absorbance at that wavelength was adjusted to be lower than 0.1. Three solutions were analysed for each complex and the quantum yield was averaged. The Stokes shift values were calculated from the absorption and fluorescence maxima. Fluorescence lifetimes were obtained at room temperature by the time-correlated single-photon counting (TCSPC) technique using the kinetic module of the same spectrofluorimeter with a nanoLED laser (1.2 ns of pulse, 370 nm or 455 nm). Fits were performed using the Felix software of the instrument. Aggregation studies were carried out in THF/water mixtures increasing the water content from 0 to 90% with a 10% volume increase.
The emission spectra at a lower temperature of 25 to −196 °C in 2-methyl tetrahydrofuran (Me-THF) were recorded using a Photon Technology International (PTI) QuantaMaster QM-40 spectrometer equipped with an Xe arc lamp (70 W), using a quartz cold finger Dewar accessory. The corresponding lifetime measurements were performed using an Edinburgh Instruments FS5 fluorimeter equipped with an EPLED-320 (Edinburgh Instruments) as the pulsed source. The analysis of the lifetime decay curve was done using the Fluoracle® Software package (Ver. 1.9.1), which runs the FS5 instrument.
Fluorescence quantum yields at low temperatures in 2-methyl tetrahydrofuran were estimated indirectly using the following equation:
| | 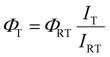 | (1) |
where
ΦT is the fluorescence quantum yield calculated at a given temperature,
ΦRT is the fluorescence quantum yield obtained experimentally at room temperature,
IT is the maximum intensity of the emission spectrum at a given temperature and
IRT is the maximum intensity of the emission spectrum recorded at room temperature. The correction depending on the refractive index has been neglected due to lack of experimental data regarding the refractive indexes of 2-methyl tetrahydrofuran at all the temperatures; for this reason, the calculated fluorescence quantum yields have to be intended as estimates and not as absolute values.
For the film characterization, samples were prepared on quartz slides (Spi. Inc.) by spin coating using a WS-400B-6NPP-LITE spin processor from Laurell Technologies. A 30 mg mL−1 C6H5Cl solution was spun with a first step of 25 s at 1000 rpm speed and a second step of 20 s at 2500 rpm. These parameters were optimized after different assays to have a good homogeneity and thickness around 100 nm, as found by AFM, using a Veeco Digital Instruments 3100 microscope in the tapping mode at a scanning rate of 0.2 Hz. UV-Vis powder spectra were recorded in the diffuse reflectance mode, with the same spectrophotometer used for the solutions, coupled with an integrating sphere.
4.4. X-ray crystallography
The crystal of 6 was covered with a layer of hydrocarbon oil that was selected and mounted with paratone-N oil on a cryo-loop, and immediately placed in the low-temperature nitrogen stream at 100(2) K. The data for 6 was recorded using a Bruker SMART APEX CCD area detector system equipped with an Oxford Cryosystems 700 Series Cryostream cooler, a graphite monochromator, and a Mo Kα fine-focus sealed tube (λ = 0.71073 Å). The structure was solved by direct methods using SHELXS-9763 and refined against F2 on all data by full-matrix least-squares with SHELXL-97.64 All of the software manipulations were done under the WIN-GX environment program set.65 All the heavier atoms were found by Fourier map difference and refined anisotropically. Some hydrogen atoms were found by Fourier map differences and refined isotropically. The remaining hydrogen atoms were geometrically modelled and not refined. The XRD measurements for the samples were carried out at 2θ in the range of 0–90° with a step size of 0.01° and a counting time of 4.5 s for each step. The CCDC numbers are 1871586 and 2293069 for 6 and 8, respectively. These files contain supplementary crystallographic data.
4.5. Electrochemistry
Cyclic voltammetry was performed in a C3 Stand cell from Basi, coupled to an ACM Gill AC potentiostat/galvanostat. The system consisted of a conventional three-electrode cell: a glassy carbon electrode as the working electrode (polished with alumina after each run), a Pt wire as the counter electrode, Ag/AgCl as the reference electrode and ferrocene/ferrocenium as the internal reference (at the maximum potential: Eox = 0.590 V, Ered = 0.510 V); a value of −4.8 eV below the vacuum level was considered. Voltammetric measurements were performed at room temperature in acetonitrile containing Bu4NPF6 (0.1 M) as the supporting electrolyte. Prior to recording the voltammograms, all the solutions (∼0.1 mmol) were deoxygenated by bubbling nitrogen at least for 15 min. The experiments were carried out in a nitrogen atmosphere at a scan rate of 50 mV s−1.
4.6. STM characterization
It was performed between ∼23 and 25 °C using an AA5000 Scanning Probe Microscope (Angstrom Advanced Inc., Braintree, MA USA). The scanner tube calibration was performed by means of atomic resolution images obtained using commercial HOPG substrates from Ted Pella (Redding, CA USA) and SPI supplies (grade SPI-3). Samples were exfoliated before each measurement by the adhesive tape method. Pt/Ir tips were prepared by mechanically cutting a 0.25 mm diameter 80/20 wire from Nanoscience, Switzerland. 1,2,4-Tricholorobenzene (TCB) (Sigma Aldrich, ≥99%) was used as received. The Sn-complexes 5 and 6 were dissolved in TCB (2.8 mM), sonicated and heated to 60 °C for one hour. A droplet of the solution, ∼3 μL, was placed using a micro-syringe between the substrate and the tip, and then a pulse of 2–6 V was applied to obtain a monolayer. The STM images were taken in the constant-current mode at the solid–liquid interface. The bias and current values were constantly adjusted during each scan. Details on the experiment bias and current set-point are given below each image. The raw and FFT images were processed using the WSxM 5.0 software with the aim to reduce noise and normally observed drift.66
4.7. General procedure for the synthesis of precursors
4.7.1. Synthesis of terephthalate acid from PET waste.
Sodium hydroxide (200 mg) was dissolved in 10 mL ethanol and PET waste (500 mg) was added. The reaction mixture was synthesized by irradiation of microwave for 30 min. The product precipitated from the mixture. After cooling to room temperature, the precipitate was filtered, washed thoroughly with ethanol and dried completely to give a solid white powder. Then, the product was dissolved in water and 4–5 drops of concentrated HCl was added to precipitate terephthalate acid. The precipitate was filtered, washed thoroughly with ethanol and dried completely to give a solid white powder. Yield: 417 mg (96%). m.p.: 300 °C. IRνmax (ATR): 3063 (C–H, ArH), 2817 (OH), 1678 (CO), 728 (C–H ArH). UV/vis (DMSO) λabs (nm): 287 [0.44]. Fluorescence (DMSO): λem (nm): 363. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.06 (s, 4H, ArH), 13.30 (s, 2H, OH) ppm. 13C NMR (100.61 MHz, DMSO-d6, 298 K): δ = 129.6 (C3), 134.6 (C2), 166.8 (C1) ppm.
4.7.2. Synthesis of diethyl terephthalate (1): route 1.
Terephthalic acid (1.6 g) in ethanol dry (60 mL) containing 2–3 drops of concentrated H2SO4 (AR) was refluxed till it dissolves. Then, the reaction mixture was poured onto ice cold water, and immediately a solid started separating from the clear solution. Sodium bicarbonate was used to neutralize the acid excess. The ester was filtered, washed several times with water, and dried in air. Yield: 84% (1.79 g). m.p.: 44 °C (Lit.67 m. p. 44.5 °C). 1H NMR (400.13 MHz, CDCl3, 298 K): δ = 1.42 (t, J = 7.0 Hz, 6H; CH3), 4.40 (q, J = 7.7 Hz, 4H; CH2), 8.10 (s, 4H, aryl) ppm.
4.7.3. Synthesis of diethyl terephthalate (1): route 2 from PET waste.
PET waste (500 mg) was added to 10 mL ethanol containing concentrated H2SO4 (2 mL). The reaction mixture was synthesized by irradiation of microwave for 60 min. The product precipitated from the mixture. After cooling to room temperature, the precipitate was filtered, washed thoroughly with ethanol and dried completely to give a solid white powder. Yield: 262 mg (82%). m.p.: 45 °C. IRνmax (ATR): 2968 (C–H, ArH), 1717 (CO), 1250 (C–O), 725 (C–H ArH). UV/vis (CHCl3) λabs (nm): 299 [0.07]. Fluorescence (CHCl3): λem (nm): 347. 1H NMR (400.13 MHz, CDCl3, 298 K): δ = 8.10 (s, 4H, ArH), 4.69 (q, 4H, CH2), 0.07 (t, 6H, CH3) ppm. 13C NMR (100.61 MHz, CDCl3, 298 K): δ = 129.6 (C3), 134.6 (C2), 166.8 (C1) ppm.
4.7.4. Synthesis of dihydrazide (3): route 1.
A mixture of diethyl terephthalate (0.5 g, 2.25 mmol) was dissolved in 50 mL ethanol and hydrazine monohydrate (1 mL) was added. The reaction mixture was refluxed for 24 h. The product precipitated from the mixture. After cooling to room temperature, the precipitate was filtered, washed thoroughly with water and dried completely to give a solid white powder. Yield: 4.27 g (81%). m.p. >300 °C (Lit.68 m.p. >300 °C). 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.89 (s, 2H), 7.87 (s, 4H), 4.59 (s, 4H J = 5.0 Hz) ppm.
4.7.5. Synthesis of dihydrazide (3): route 2.
Preparation via route 2 was accomplished following the same procedure of route 1 with 50 mL methanol. Yield: 4.27 g (81%). m.p. >300 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.89 (s, 2H), 7.87 (s, 4H), 4.59 (s, 4H J = 5.0 Hz). (Lit.69 m.p. >300 °C).
4.7.6. Synthesis of dihydrazide (3) route 3.
A mixture of dimethyl terephthalate (0.2 g, 1.03 mmol) was dissolved in 5 mL methanol and hydrazine monohydrate (0.45 mL) was added. The reaction mixture was synthesized by irradiation of microwave for 20 min. The product precipitated from the mixture. After cooling to room temperature, the precipitate was filtered, washed thoroughly with water and methanol and dried completely to give a solid white powder. Yield: 0.19 g (95%). m.p. >300 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.87 (s, 2H), 7.86 (s, 4H), 4.54 (s, 4H) ppm.
4.7.7. Synthesis of dihydrazide (3) route 4 from PET waste.
PET waste (500 mg) was added to 10 mL ethanol and hydrazine monohydrate (1.25 mL) was added. The reaction mixture was synthesized by irradiation of microwave for 60 min. The product precipitated from the mixture. After cooling to room temperature, the precipitate was filtered, washed thoroughly with ethanol and dried completely to give a solid white powder. Yield: 409 mg (81%). m.p.: 370 °C. IRνmax (ATR): 3313 (NH), 3029 (C–H, ArH), 1602 (CO), 1100 (C–N), 736 (C–H ArH). UV/vis (DMSO) λabs (nm) [εmax × 104 (M−1 cm−1)]: 312 [0.21]. Fluorescence (DMSO): λem (nm): 383. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 4.57 (s, 4H, NH2), 7.86 (s, 4H, ArH), 9.89 (s, 2H, NH) ppm. 13C NMR (100.61 MHz, DMSO-d6, 298 K): δ = 127.0 (C3), 135.5 (C2), 165.2 (C1) ppm. HRMS (TOF) C8H11N4O2. [M+] found: 195.087652, ppm error: −0.892786.
4.8. General procedure for the synthesis of ligands, organotin compounds 1–2 and Schiff bases
4.8.1. Bis(2-hydroxy-1-naphthaldehyde)terephthalohydrazone (4).
A mixture of terephthalohydrazide 0.10 g (0.51 mmol) and 2-hydroxy-1-naphthaldehyde 0.20 g (0.10 mmol) was refluxed in methanol for 24 h. A yellow solid formed was filtered, washed with methanol and vacuum dried. Yield: 0.17 g (68%). m.p.: 345 °C. IRνmax (ATR): 3189 (s, N–H), 3003 (s, O–H), 1640 (s, CO), 1622 (m, CN). 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 12.84 (s, 2H, OH), 12.29 (s, 2H, NH), 9.57 (s, 2H, CH), 8.20 (d, J = 8.0 Hz, 2H, Np), 7.59 (t, J = 7.6 Hz, 2H, Np), 7.40 (t, J = 7.6 Hz, 2H, Np) 7.21 (d, J = 8.8 Hz, 2H, Np) ppm. HRMS (APCI/TOF-Q) m/z: 484.1503 [M+] found: 502.1641 PPM error: −0.1445.
4.8.2. Synthesis of bis(2-hydroxy-1-naphthaldehyde)terephthalohydrazone di-n-butyltin(IV) (5).
A microwave tube was placed with a magnetic shuffler, and terephthalohydrazide 0.05 g (0.25 mmol) and 2-hydroxy-1-naphthaldehyde 0.08 g (0.51 mmol), and more dibutyltin oxide 0.12 g (0.51 mmol) in acetonitrile 5 mL were added into the tube and heated to 180 °C for 20 min. After this time, the solution was cooled, and the solvent evaporated under reduced pressure and washed with hexane and acetonitrile. The precipitate was obtained by filtration and crystalized with dichloromethane/methanol (1:1) to afford 0.20 g (0.19 mmol, 76% yield) of 5 as an orange-yellow solid. m. p. 172 °C. IRνmax (ATR): 3057 (C–H ArH), 2326 (O–CN) 1602 (CN), 1581 (CN–NC), 715 (C–H ArH). UV/vis (CHCl3) λabs (nm) [εmax × 104 (M−1 cm−1)]: 456 [0.08]. Fluorescence (CHCl3): λem (nm): 495. 1H NMR (400.13 MHz, CDCl3, 298 K): δ = 9.73 [s, 2H, 3J(1H–119Sn) = 45.08 Hz, H-11], 8.14 (s, 4H, ArH, H-14), 8.09–8.11 (d, J = 12 Hz, 2H, H-9), 7.76–7.78 (d, J = 8 Hz, 2H, H-4), 7.69–7.71 (d, J = 8.0 Hz, 2H, H-6), 7.53 (t, J = 16 Hz, 2H, H-8), 7.33 (t, J = 8 Hz, 2H, H-7), 6.95–6.97 (d, J = 8.0 Hz, 2H, H-3), 1.40 (t, 6H, 3J = 7.2 Hz, CH3-γ), 0.85–0.89 (m, 4H, CH2- δ), 1.55–1.59 (t, 4H, 3J = 7.2 Hz, CH2-α), 1.65–1.71 (m, 4H, 3J = 7.2 Hz, CH2-β). 13C NMR (100.61 MHz, CDCl3, 298 K): δ = 13.41 (δ, CH3), 26.27 [Cγ, 3J(13C–119Sn) = 83.86 Hz], 26.66, [Cβ, 2J(13C–119Sn) = 33.75 Hz], 21.87 [Cα, 1J(13C–119Sn) = 595/568 Hz], 107.24 (C1), 169.24 (C2), 119.31 (C3), 136.81 (C4), 127.31 (C5), 128.18 (C6), 123.15 (C7), 124.52 (C8), 129.30 (C9), 135.97 (C10), 157.26 (C11), 168.17 (C12), 135.83 (C13), 127.51 (C14) ppm. 119Sn NMR (128.0 MHz, C6D6, 298 K): δ = −189.28 ppm. HRMS (APCI/TOF-Q) m/z: 966.2261 [M+] found: 967.226181, ppm error: 0.1445.
4.8.3. Synthesis of bis(2-hydroxy-1-naphthaldehyde)terephthalohydrazone diphenyltin(IV) (6).
Preparation of 6 was accomplished following the same procedure used for preparing 4 from terephthalohydrazide (0.05 g, 0.25 mmol), 2-hydroxy-1-naphthaldehyde (0.08 g, 0.51 mmol) and more diphenyltin oxide (0.14 mg, 0.51 mmol) at 180 °C for 30 min. Yield: 0.23 g (0.52 mmol, 75%). Yellow solid. m.p.: 336 °C. IRνmax (ATR): 3065 (C–H ArH), 2326 (O–CN), 1580 (C–N–N–C), 1596 (CN), 1582 (CN–NC), 715 (C–H ArH). UV/vis (CHCl3) λabs (nm) [εmax × 104 (M−1 cm−1)]: 453 [0.07]. Fluorescence (CHCl3): λem (nm): 491. 1H NMR (400 MHz, CDCl3, 298 K): δ = 9.78 [s, 2H, 3J(1H–119Sn) = 56 Hz, H-11], 8.39 (s, 4H, ArH, H-14), 8.09–8.12 (d, J = 12 Hz, 2H, H-9), 7.91–7.93 (d, J = 8 Hz, 2H, H-4), 7.73–7.78 (d, J = 8.0 Hz, 2H, H-6), 7.54 (t, J = 16 Hz, 2H, H-8), 7.46 (t, J = 8 Hz 2H, H-7), 7.42–7.43 (d, 3J = 8.0 Hz, 2H, H-3), 7.91 (s, 8H, 3J = 7.2 Hz, H-o), 7.43 (s, 8H, 3J = 8.0 Hz, H-m), 7.45 (s, 4H, 3J = 8.0 Hz, H-p). 13C NMR (100.61 MHz, CDCl3, 298 K): δ = 136.39 (H-o), 129.15 (H-m), 130.76 (H-p), 137.40 (C-i) 107.58 (C1), 169.78 (C2), 119.60 (C3), 141.43 (C4), 127.62 (C5), 129.35 (C6), 123.55 (C7), 124.64 (C8), 129.54 (C9), 134.00 (C10), 157.88 (C11), 167.72 (C12), 139.13 (C13), 127.51 (C14). 119Sn NMR (100.7 MHz, C6D6, 298 K): δ = −328.69 ppm. HRMS (APCI/TOF-Q) m/z: calcd For C37H37NO2Sn [M+] 1046.0900 found: 1047.1009 u m a (error: −0.8524 ppm).
Author contributions
Arelly M. Cantón-Díaz, investigation and formal analysis; Blanca M. Muñoz-Flores, interpretation, and formal analysis; Luis F. Macías-Gamboa, investigation and formal analysis; Ivana Moggio, interpretation, formal analysis; Eduardo Arias, interpretation and formal analysis; Gleb Turlakov, DFT studies, H. V. Rasika Dias, X-ray diffraction analysis; Gioele Colombo, cryoluminescence properties and analysis, Stefano Brenna, conceptualization, project administration, and funding acquisition; Víctor M. Jiménez-Pérez, conceptualization, project administration, and funding acquisition.
Data availability
The data supporting this article have been included as part of the ESI.†
Crystallographic data for compounds 6 and 8 have been deposited at the CCDC under 1871586 (6) and 2293069 (8), respectively, and can be obtained from https://doi.org/10.1039/D4DT01518E.
Conflicts of interest
Universidad Autónoma de Nuevo León has applied for a patent on this work. V. M. J. P. and B. M. M. F. are founders of a company related to this work.
Acknowledgements
The authors thank the PAICYT (Grant: CE-1675-21 and 252-CE-2022) and the Facultad de Ciencias Químicas-UANL for the financial support. A. M. C. D. thank the CONACYT for the Ph.D. scholarships. Authors acknowledge the National Supercomputing Center-IPICYT for the computational resources supported in this research through grant TKII.
References
- Y. Xie, Y. Ge, Q. Peng, C. Li, Q. Li and Z. Li, Adv. Mater., 2017, 29, 1606829 CrossRef PubMed.
- H.-J. Chuang, X. Tan, N. J. Ghimire, M. M. Perera, B. Chamlagain, M. M.-C. Cheng, J. Yan, D. Mandrus, D. Tománek and Z. Zhou, Nano Lett., 2014, 14(6), 3594–3601 CrossRef CAS PubMed.
- R. Kaufmann, C. Hagen and K. Grünewald, Curr. Opin. Chem. Biol., 2014, 20, 86–91 CrossRef CAS PubMed.
- P. D. Dahlberg and W. E. Moerner, Annu. Rev. Phys. Chem., 2021, 20, 253–278 CrossRef PubMed.
- J. Huebinger, H. Grecco, M. E. Masip, J. Christmann, G. R. Fuhr and P. I. H. Bastiaens, Sci. Adv., 2021, 7, 1–12 Search PubMed.
- J. Yin, L. Huang, L. Wu, J. Li, T. D. James and W. Lin, Chem. Soc. Rev., 2021, 50, 12098 RSC.
- P. Gao, W. Pan, N. Li and B. Tang, Chem. Sci., 2019, 10, 6035–6071 RSC.
- R. Kaufmann, C. Hagen and K. Grünewald, Curr. Opin. Chem. Biol., 2014, 20, 86–91 CrossRef CAS PubMed.
- C. L. Schwartz, V. I. Sarbash, F. I. Ataullakhanov, J. R. McIntosh and D. Nicastro, J. Microsc., 2007, 227, 98–109 CrossRef PubMed.
- I. Rojas-León, H. Alnasr, K. Jurkschat, M. G. Vasquez-Ríos, G. Gómez-Jaimes, H. Höpfl, I. F. Hernández-Ahuactzí and R. Santillan, Organometallics, 2019, 38, 2443–2460 CrossRef.
- A. González-Hernández and V. Barba, Inorg. Chim. Acta, 2018, 483, 284–292 CrossRef.
- J. Wang, H. Chen, Q. Song, X. Liu, C. Li, H. Wang, C. Li and M. Hong, J. Inorg. Biochem., 2022, 236, 111983 CrossRef CAS PubMed.
- M. Yousefi, T. Sedaghat, J. Simpson, H. Motamedi and M. Reza Dayer, Polyhedron, 2018, 155, 153–162 CrossRef CAS.
- H. Zafarian, T. Sedaghat, H. Motamedi and H. A. Rudbari, J. Organomet. Chem., 2016, 825, 25–32 CrossRef.
- J. Berrones-Reyes, C. C. Vidyasagar, B. M. Muñoz Flores and V. M. Jiménez Pérez, J. Lumin., 2018, 195, 290–313 CrossRef CAS.
- A. Cantón-Diaz, B. M. Muñoz-Flores, J. Berrones-Reyes, I. Moggio, E. Arias, G. Turlakov, R. Santillán and V. M. Jiménez-Pérez, J. Organomet. Chem., 2021, 954–955, 122111 CrossRef.
- M. López-Espejel, A. Gómez-Treviño, B. M. Muñoz-Flores, M. Treto-Suarez, E. Schott, D. Paez-Hernández, X. Zarate and V. M. Jiménez-Pérez, J. Mater. Chem. B, 2021, 9, 7698–7712 RSC.
- J. C. Berrones-Reyes, B. M. Muñoz-Flores, A. Uscanga-Palomeque, R. Santillán, C. Del Angel-Mosqueda, D. Nobis, M. A. Cochrane, S. W. Magennis and V. M. Jiménez-Pérez, ChemistrySelect, 2020, 5, 1623–1627 CrossRef CAS.
- V. M. Jiménez-Pérez, M. C. García-López, B. M. Muñoz-Flores, R. Chan-Navarro, J. Berrones-Reyes, I. Moggio, E. Arias, H. V. Rasika Dias, J. A. Serrano and A. Chávez-Reyes, J. Mater. Chem. B, 2015, 3, 5731–5745 RSC.
- B. M. Muñoz, J. Cabrera, A. Chávez-Reyes, H. V. Rasika Dias, C. Viñas, R. Nuñez and V. M. Jiménez-Pérez, Chem. – Eur. J., 2018, 24, 5601–5612 CrossRef PubMed.
- H. V. Rasika Dias, H. V. K. Diyabalanage, M. G. Eldabaja, O. Elbjeirami, M. A. Rawashdeh-Omary and M. A. Omary, J. Am. Chem. Soc., 2005, 127, 7489–7501 CrossRef PubMed.
- D. Cauzzi, R. Pattacini, M. Delferro, F. Dini, C. Di Natale, R. Paolesse, S. Bonacchi, M. Montalti, N. Zaccheroni, M. Calvaresi, F. Zerbetto and L. Prodi, Angew. Chem., Int. Ed., 2012, 51, 9662–9665 CrossRef CAS PubMed.
- S.-Z. Zhan, X. Jiang, J. Zheng, X.-D. Huang, G.-H. Chen and D. Li, Dalton Trans., 2018, 47, 3679–3683 RSC.
- M. S. Deshmukh, A. Yadav, R. Pant and R. Boomishankar, Inorg. Chem., 2015, 54, 1337–1345 CrossRef CAS PubMed.
- A. Ansari, A. K. Parchur, M. K. Nazeeruddin and M. M. Tavakoli, Coord. Chem. Rev., 2021, 444, 214040 CrossRef CAS.
- L. J. Mohammed and K. M. Omer, Nanoscale Res. Lett., 2020, 15, 182 CrossRef PubMed.
- K. Tanaka, D. Sakamaki and H. Fujiwara, Chem. – Eur. J., 2021, 27, 4430–4438 CrossRef CAS PubMed.
- T. Nishiuchi, K. Tanaka, Y. Kuwatani, J. Sung, T. Nishinaga, D. Kim and M. Iyoda, Chem. – Eur. J., 2013, 19, 4110–4116 CrossRef CAS PubMed.
- X.-D. Wang, O. S. Wolfbeis and R. J. Meier, Chem. Soc. Rev., 2013, 42, 7834 RSC.
- M. M. Ogle, A. D. S. McWilliams, B. Jiang and A. A. Martí, ChemPhotoChem, 2020, 4, 1–17 CrossRef.
- W. W. Lestari, P. Lönnecke, H. C. Streit, M. Handke, C. Wickleder and E. Hey-Hawkins, Eur. J. Inorg. Chem., 2014, 1775–1782 CrossRef CAS.
- X. Chen, X. Zhang and G. Zhang, Chem. Commun., 2015, 51, 161–163 RSC.
- T. Annaka, N. Nakata and A. Ishii, Organometallics, 2015, 34, 1272–1278 CrossRef CAS.
- J. Wang, Y. Yu, L. Niu, B. Zou, K. Wang and Q. Yang, Chem. Commun., 2020, 56, 6269–6272 RSC.
- X. Li, Q. Gao, J. Wang, Y. Chen, Z.-H. Chen, H.-S. Xu, W. Tang, K. Leng, G.-H. Ning, J. Wu, Q.-H. Xu, S. Y. Quek, Y. Lu and K. P. Loh, Nat. Commun., 2018, 9, 2335 CrossRef PubMed.
- M. Ibarra-Rodríguez, B. M. Muñoz-Flores, H. V. Rasika Dias, M. Sánchez, A. Gómez, R. Santillán, N. Farfán and V. M. Jiménez-Pérez, J. Org. Chem., 2017, 82, 2375–2385 CrossRef PubMed.
- V. M. Jiménez-Pérez, H. Nöth, A. Ariza-Castolo, A. Flores-Parra and R. Contreras, J. Organomet. Chem., 2006, 691, 1584–1589 CrossRef.
- J. Holecek, M. Nadvornik, K. Handlir and A. Lycka, J. Organomet. Chem., 1986, 315, 299–308 CrossRef CAS.
- F. P. Pruchnik, M. Banbula, Z. Ciunik, M. Latocha, B. Skop and T. Wilczok, Inorg. Chim. Acta, 2003, 356, 62–68 CrossRef CAS.
- N. Ramesh Babu, S. Subashchandrabose, M. Syed Ali Padusha, H. Saleem, V. Manivannan and Y. Erdoǧdu, J. Mol. Struct., 2014, 25, 84–93 CrossRef.
- L. Tian, X. Liu, X. Zheng, Y. Sun, D. Yan and L. Tu, Appl. Organomet. Chem., 2011, 25, 298–304 CrossRef CAS.
- H. D. Yin, M. Hong and D. Q. Wang, J. Organomet. Chem., 2005, 690, 3714–3719 CrossRef CAS.
- Scientific instrument service, INC (SIS), https://www.sisweb.com/mstools/isotope.htm (accesed Oct. 4, 2018).
- H. Yang, L. Wang, J. Zhang, X. Yu, Y. Geng and Y. Han, Macromol. Chem. Phys., 2014, 215, 405–411 CrossRef CAS.
-
(a) C. Ma, G. Tian and R. Zhang, Inorg. Chem. Commun., 2006, 9, 882–886 CrossRef CAS;
(b) C. L. Ma, M. Yang, R. Zhang and J. Li, Inorg. Chem. Commun., 2007, 10, 1330–1334 CrossRef CAS.
- R. Chan-Navarro, B. M. Muñoz-Flores, V. M. Jimenez-Pérez, I. Moggio, E. Arias, G. Ramos-Ortíz, M. C. García-López, V. M. Rosas-García, P. Elizondo and M. Rodríguez, Int. J. Innov. Res. Sci. Eng. Technol., 2014, 1, 462–475 Search PubMed.
- B. M. Muñoz, J. Cabrera, A. Chávez-Reyes, H. V. Rasika Dias, C. Viñas, R. Nuñez and V. M. Jiménez-Pérez, Chem. – Eur. J., 2018, 24, 5601–5612 CrossRef PubMed.
- A. M. Cantón-Díaz, B. M. Muñoz-Flores, I. Moggio, E. Arias, G. Turlakov, C. Del Angel-Mosqueda, P. I. Ramírez-Montes and V. M. Jiménez-Pérez, J. Mol. Struct., 2019, 1180, 642–650 CrossRef.
- M. Klessinger, Theor. Comput. Chem., 1998, 5, 581–610 CAS.
- A. Molina-Paredes, V. M. Jiménez-Pérez, J. Lara-Cerón, I. Moggio, E. Arias, R. Santillán, M. Sánchez, A. Suacedo-Yañez and B. M. Muñoz-Flores, Appl. Organomet. Chem., 2019, 33, e4609 CrossRef.
- M. C. García-López, B. M. Muñoz-Flores, V. M. Jiménez-Pérez, I. Moggio, E. Arias, R. Chan-Navarro and R. Santillán, Dyes Pigm., 2014, 106, 188–196 CrossRef.
- A. M. Cantón-Díaz, B. M. Muñoz, I. Moggio, E. Arias, A. De León, M. C. García-López, R. Santillán, M. E. Ochoa and V. M. Jiménez-Pérez, New J. Chem., 2018, 42, 14586–14596 RSC.
-
(a) M. Yang, D. Xu, W. Xi, L. Wang, J. Zheng, J. Huang, J. Zhang, H. Zhou, J. Wu and Y. Tian, J. Org. Chem., 2013, 78, 10344–10359 CrossRef CAS PubMed;
(b) M. Xiao-Feng, S. Rui, C. Jinghui, L. Jiaoyan, G. Fei, X. Haifeng and Z. Xiangge, J. Chem. Educ., 2016, 93, 345–350 CrossRef.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- R. Cammi, B. Mennucci and J. Tomasi, J. Phys. Chem. A, 2000, 104, 5631 CrossRef CAS.
-
(a) S. Grimmea, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed;
(b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
-
(a) E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597 CrossRef CAS;
(b) E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783 CrossRef CAS;
(c) C. van Wüllen, J. Chem. Phys., 1998, 109, 392 CrossRef.
- F. Weigenda and R. Ahlrich, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. D. Rolfes, F. Neese and D. A. Pantazis, J. Comput. Chem., 2020, 41, 1842–1849 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- A. Felouat, A. D'Aléo and F. Fages, J. Org. Chem., 2013, 78, 4446–4455 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
-
G. M. Sheldrick, SHELX-97: Program for the solution and refinement of crystal structures, Universität Göttingen, Göttingen, Germany, 1997 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
- H. Hosseini-Monfared, N. Asghari-Lalami, A. Pazio, K. Wozniak and C. Janiak, Inorg. Chim. Acta, 2013, 406, 241–250 CrossRef CAS.
-
(a) B. K. Lakshmi, N. Shivananda, G. A. Prakash, A. M. Isloor and K. N. Mahendra, Bull. Korean Chem. Soc., 2012, 33, 673–482 CrossRef;
(b) P. G. Avaji, C. H. Vinod, A. Sangamesh, K. N. Shivananda and C. Bagaraju, Eur. J. Med. Chem., 2009, 44, 3552–3559 CrossRef CAS PubMed.
- X. Zhang, B. Tang, P. Zhang, M. Li, M. Jing and W. Tian, J. Mol. Struct., 2007, 846, 55–64 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Ivana
Moggio
a,
Ivana
Moggio