
Trigonal bipyramidal or square planar? Density functional theory calculations of iron bis(dithiolene) N-heterocyclic carbene complexes†
Katherine
Merkel
,
Alyssa V. B.
Santos
 and
Scott
Simpson
*
and
Scott
Simpson
*
Department of Chemistry, St Bonaventure University, St Bonaventure, NY 14778, USA. E-mail: ssimpson@sbu.edu
First published on 31st October 2024
Abstract
Density functional theory (DFT) calculations of 57 iron bis(dithiolene)-N-heterocyclic carbene adducts were conducted to determine what parameters predict, and possibly influence, the coordination of these aforementioned adducts. The parameters considered herein include three different types of nuclear magnetic resonance (C-NMR, Se-NMR, and P-NMR) isotropic chemical shifts, the Tolman Electronic Parameter (TEP), the Huynh Electronic Parameter (HEP), and the percent buried volume (%Vbur) of the different N-heterocyclic carbenes (NHCs) calculated from DFT. These parameters were selected based upon prior literature connection to σ-donor ability, π-acidity, and steric effects. The computed values of the properties were compared via multivariable linear regression models to see which properties best predict the Addison τ parameter—a measure of whether the complex would assume the square pyramidal or trigonal bipyramidal geometry. It was determined that a combination of TEP and %Vbur are the best predictors of the τ value, for the parameters considered herein. The inclusion of additional parameters yields mild improvement to the statistical models for the prediction of the τ value.
Introduction
N-heterocyclic carbenes (NHCs) are a class of molecules with interesting electronic properties that have application in many areas of chemistry–most dominantly in organometallic chemistry, materials chemistry, and catalysis.1–3 These ligands have many applications because they are highly modular, meaning they are extremely diverse in terms of both structure and stereo-electronic diversity.1,4 This modular behavior is enhanced by the large libraries of NHCs that have been prepared.2,4–6Though there are many different structures of NHCs, this work will focus on the simple, imidazole-2-ylidenes (shown below in Fig. 1).2,7 All imidazole-2-ylidenes NHCs contain a pentagonal carbon/nitrogen structure, including a lone pair of electrons on the head carbon. The nucleophilic carbene is stabilized by the donation of π-electron density from the two adjacent nitrogen heteroatoms to the carbon and by the σ-withdrawing nature of the nitrogen atoms.2,5,8
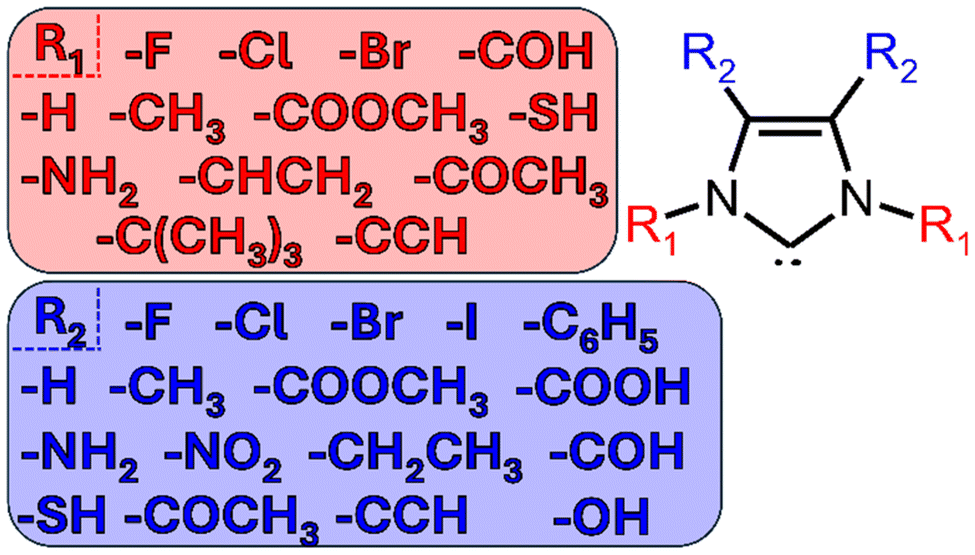 | ||
| Fig. 1 Skeletal structures of the N-heterocyclic carbenes (NHCs) considered in this study. | ||
In an experimental sense, NHCs are fairly easy to prepare when paired with a transition metal, and contain a strong bond between the metal and the NHCs. NHCs play an important role in transition metal complexes because of their ability to act as strong σ-donor ligands while behaving as π-acceptors, making the interaction tunable.2,6,8–10 The NHC will donate electron density from an occupied orbital to the metal while simultaneously accepting electron density from the metal into an unoccupied molecular orbital. This bonding mechanism results in a strong bond between the metal center and the NHC ligand.8 Examples of different techniques/parameters that have been considered to deconvolute the NHC-metal center bond can be found in Fig. 2.
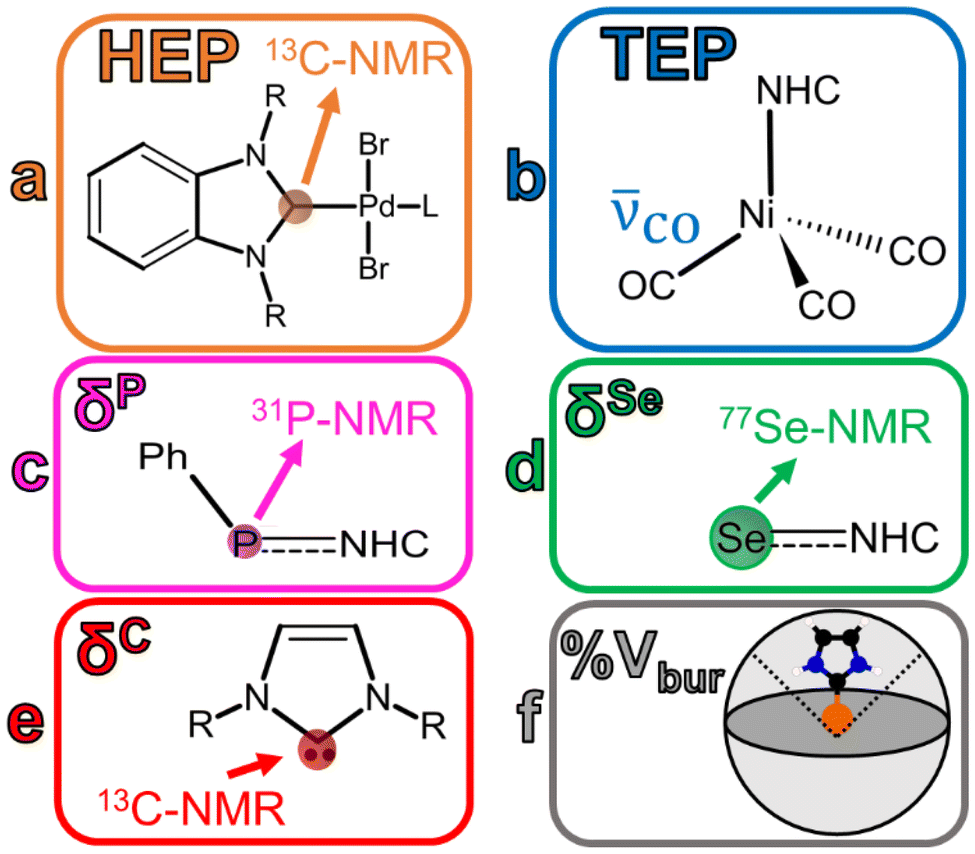 | ||
| Fig. 2 Schematic representations of (a/orange) the Huynh Electronic Parameter (HEP), (b/blue) the Tolman electronic parameter (TEP), (c/pink) phosphorous isotropic chemical shift (δP), (d/green) selenium isotropic chemical shift (δSe), (e/red) free-carbene carbon isotropic chemical shift (δC), and (f/grey) percent buried volume (%Vbur). | ||
The Tolman Electronic Parameter (TEP) is determined by measuring the symmetric CO stretch in nickel-containing complexes. Originally this technique was applied to carbonyl complexes with phosphorous ligands11 but has since been applied to NHC complexes.5 While the previously mentioned NMR techniques can be utilized to parse out σ/π contributions, TEP has been associated with only net bonding impact and cannot be utilized to determine σ/π donating/accepting character. A stronger net electron donor should increase electron density at the Ni atom, strengthening the metal–carbon bond but weaken the CO bond order, yielding a lower CO frequency value. A stronger net acceptor would do the opposite–ligands competing for backbonding from the metal center's electron density result in a higher CO stretching frequency.
Previously, the electronic structure of NHCs has been examined on the basis of gauging their π-acidity.12 This is typically done via Se-NMR and/or P-NMR.5,13–15 Additionally, the σ-donating capabilities of these NHCs have also been judged utilizing the Huynh electronic parameter (HEP) which uses 13C-NMR to gauge the σ-donor strength of a ligand. The isotropic chemical shift indicates more (upfield) or less (downfield) shielding on the carbene carbon atom of the 1,3-diisopropylbenzimidazolin-2-ylidene ligand, attached to a Pd atom complex. An upfield signal indicates a weaker σ-donor, while a downfield signal represents a stronger σ-donor. Together, HEP and Se-/P-NMR probe techniques can be utilized to understand σ-donor/π-acceptor trends and net impacts on bonding for NHCs.13,14
The percent buried volume (%Vbur) calculation defines the percentage of a coordination sphere around a metal–ligand complex that is occupied by the NHC ligand, thereby relating %Vbur to steric hinderance/bulk.8,9,16 The center is the metal contained in the complex and the sphere has a defined radius (usually 3.50 Å, but can be user defined).9,17 A higher %Vbur value should indicate a bulkier ligand and has been connected to steric hinderance. Clavier et al. have demonstrated a correlation between the Tolman cone angle (θ) and %Vbur for a variety of different phosphine ligands contained in gold(I) chloride complexes.9
In terms of physical structure, the geometry of NHC-metal complexes can be understood utilizing the Addison τ parameter.18–20 This parameter reveals information such as the coordination number of complex and is relevant to the catalytic ability of the complex.19,21,22 The value of τ is calculated as
| τ = (β − α)/60 | (1) |
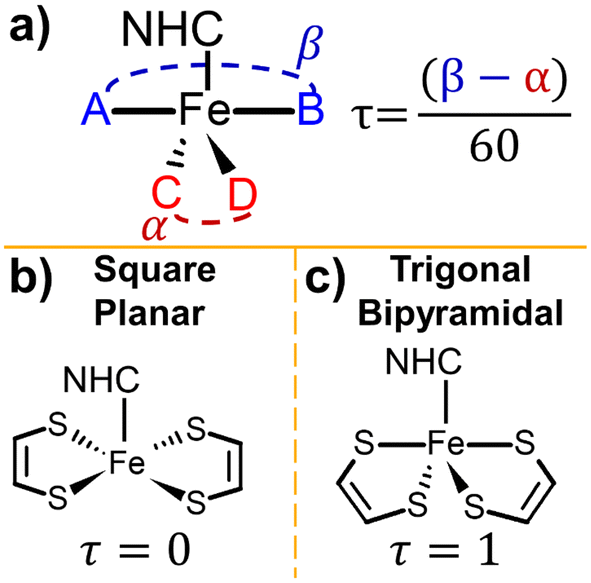 | ||
| Fig. 3 (a) A visual description of the Addison τ parameter. Examples of square planar geometry (b) and trigonal bipyramidal geometry (c). For the square planar geometry, α = β = 180°, τ = 0 and for trigonal bipyramidal geometry α ≈ 120°, β ≈ 180°, τ ≈ 1.18,20 | ||
Understanding the relationship between electronic structure, physical structure, and the effect of different R groups on the τ parameter is imperative to our understanding of how NHCs function and how they might be useful for future applications, especially with regards catalysis or how these molecules interact with surfaces as a possible route to safe hydrogen storage.23,24
Computational details
With regards to experimental design: previous studies have outlined the basic structure of many different varieties of these molecules, and the imidazolyidene form was selected for investigation. All structures were constructed then optimized using the UFF force field in the program Avogadro.25 All density functional theory (DFT) calculations were conducted using TURBOMOLE,26 the BP86 functional,27 and the def2-TZVPD28 basis set. We, and others, have found the BP86 functional to be sufficient for obtaining the correct spin-states for iron bis(dithiolene) complexes.19,29Nuclear Magnetic Resonance (NMR) shielding constant calculations were completed with the previously described basis functions and functional for all systems other than our Huynh Electronic Parameter (HEP).5,30 In calculations of the HEP, the dhf-TZVP basis was used for each of the lighter elements, and the dhf-ecp was utilized for Pd atoms. HEP and free NHC 13C chemical shifts (δC) were calculated utilizing eqn (2):
| δ = σreference − σprobe | (2) |
| δP = σreference − σprobe − δreference | (3) |
For Tolman Electronic Parameter (TEP) calculations, nickel containing complexes were analysed via a vibrational frequency calculation that predicted the CO stretch. Vibrational frequencies were visualized using the TMoleX19 application.
A comparison of the theoretical spectroscopic values and experimental values for different NHCs considered herein is provided in the ESI.†
For the buried volume calculation (%Vbur), the web application SambVca 2.117,33 was utilized which calculated the percent buried volume of the NHC in the NHC-iron bis(dithiolene) complexes.
All statistics and regression models were calculated with the statistics software package R, version 4.3.0.34 Max-min normalization was applied to each variable when constructing regression models, following eqn (4):
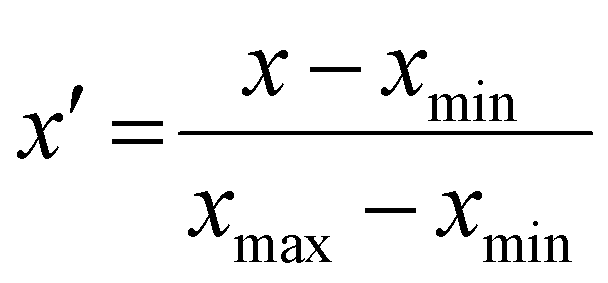 | (4) |
Results and discussion
In order to gain more clarity regarding the impact of the electronic structure of the NHCs on the complexes, the different parameters were calculated (HEP, TEP, δP, δSe, and δC). The Addison τ values and %Vbur were extracted from the geometry optimizations of the iron bis(dithiolene) adducts. HEP values were extracted from vibrational frequency calculations of optimized HEP complexes. TEP values were extracted from vibrational frequency calculations of optimized Ni(CO)3-NHC complexes. Se isotropic chemical shifts (δSe) were calculated from NMR calculations of optimized Se-NHC adducts. P isotropic chemical shifts (δP) were calculated from NMR calculations of optimized phosphinidene-NHC adducts. Carbene carbon isotropic chemical shifts (δC) were calculated from optimized free NHC geometries. A schematic representation of the aforementioned parameters can be seen in Fig. 2. These values were normalized in preparation for the regression analysis.A pairwise plot (also often referred to as a scatter plot matrix) and heatmap of the previously mentioned parameters can be found in Fig. 4. Fig. 4a illustrates relationship between two variables within the considered parameters while Fig. 4b provides the associated Pearson correlation coefficient between each variable. A value of +1 signifies a total positive correlation, a value of zero indicates no correlation, and a value of −1 signifies a negative linear correlation. Selenium isotropic chemical shift seems have positive correlations with δP, δC, & %Vbur. This is unsurprising as these previously mentioned chemical shifts are a metric of π-acidity. The most negatively correlated parameters were HEP and %Vbur. The most positively correlated parameter with τ was %Vbur while TEP was the most negatively correlated.
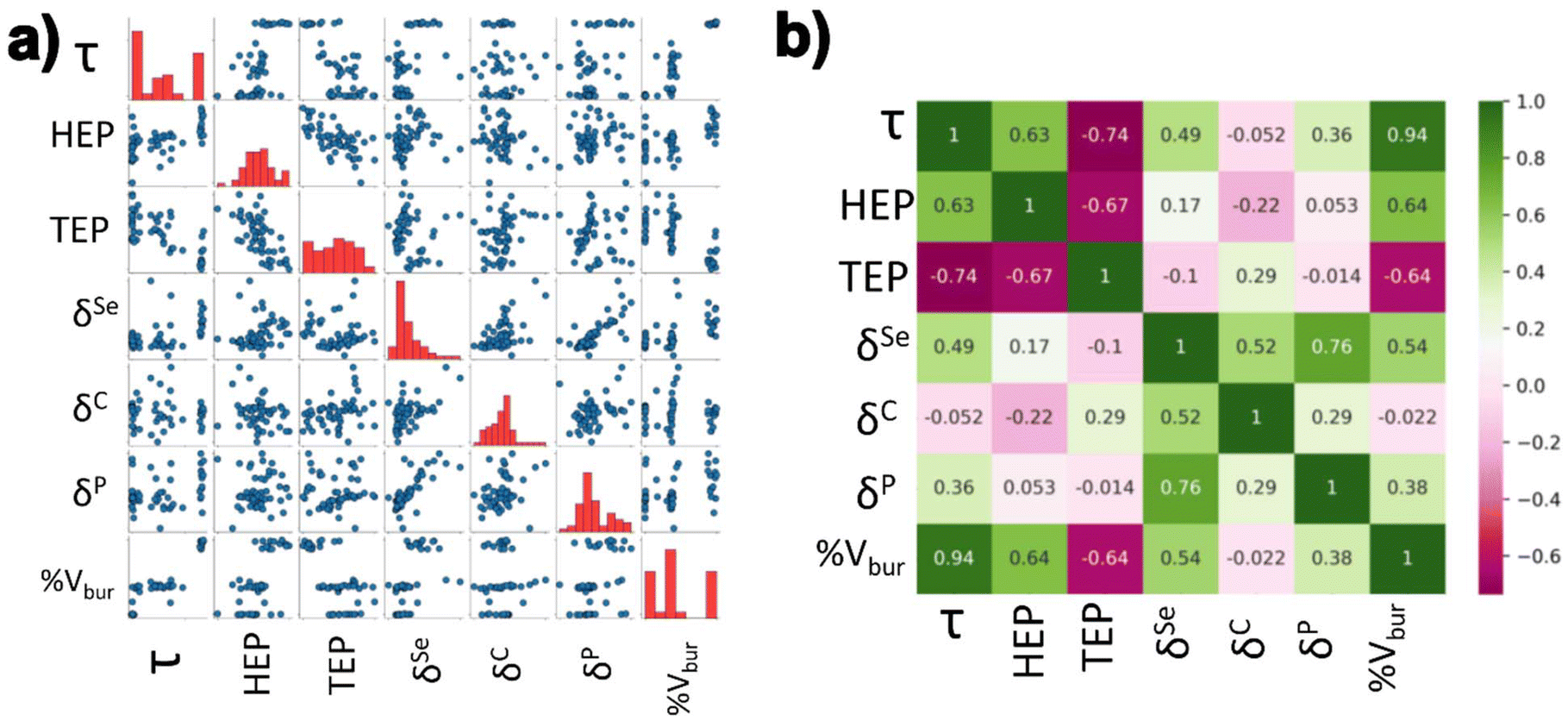 | ||
| Fig. 4 (a) A pairwise plot and (b) heat map of the Pearson coefficients for the parameters summarized in Fig. 2 & 3. For the pairwise plot, the blue circles in (a) represent the calculated properties of each individual NHC, while the red bars indicate the distribution of data. For the heatmap in (b), green values show a positive Pearson correlation while pink indicates a negative correlation between the different parameters. | ||
Utilizing least-squares-fits, single-/multi-variable regression models were constructed from the normalized aforementioned parameters to predict the τ parameter of 57 iron bis(dithiolene)-NHC adducts. The structure of the considered NHCs can be found in Fig. 1. A total of 63 different linear-regression models were created that predicted τ values. These models contained varying degrees of freedom (DOF) ranging from 3 DOF to 8 DOF. Models including only 1 independent variable were found to have poor performance in the predictability of τ values. A full summary of all models can be found in the ESI.† In order to determine the optimal model, a series of statistical parameters were considered, including residual standard error (S), adjusted R2 values, and Akaike Information Criterion (AIC)35 values, and Bayesian information criterion (BIC)36 values. See S2 in ESI† for full statistical analysis, but for brevity only the 5 most accurate models, as seen in Table 1, are discussed herein.
| Model # | Model equation | Adj-R2 | S | AIC | BIC |
|---|---|---|---|---|---|
| 1 | τ = −1.927 × 10−2 (TEP) − 7.625 × 10−4 (δP) + 6.956 × 10−2 (%Vbur) + 38.019 | 0.9077 | 0.1577 | −42.11 | −31.98 |
| 2 | τ = −1.732 × 10−2 (TEP) + 7.362 × 10−2 (%Vbur) + 33.716 | 0.9091 | 0.1595 | −41.79 | −33.68 |
| 3 | τ = −1.925 × 10−2 (TEP) − 1.353 × 10−4 (δSe) + 6.876 × 10−2 (%Vbur) + 37.987 | 0.9067 | 0.1586 | −41.41 | −31.29 |
| 4 | τ = −1.861 × 10−2 (TEP) − 1.116 × 10−3 (δC) + 7.265 × 10−2 (%Vbur) + 36.341 | 0.9052 | 0.1599 | −40.58 | −30.45 |
| 5 | τ = −2.043 × 10−2 (TEP) − 1.966 × 10−2 (HEP) − 7.127 × 10−4 (δP) + 7.113 × 10−2 (%Vbur) + 40.369 | 0.9066 | 0.1587 | −40.58 | −28.42 |
Model 1 (see Table 1) had the 2nd largest adjusted R2 value (0.9077) of the tested models, indicating a “linear” fit of the data to τ values. This model also has the lowest S, and AIC value, and the 2nd lowest BIC model indicating the model is more suitable to predict τ values compared to the other models. AIC and BIC are utilized to balance model fit with complexity of the model. It is well known that AIC emphasizes prediction accuracy while BIC is better in the identification of the “true” model in smaller sample sizes.
Of the variables studied in this work that most correctly predicted the τ values, it can be seen that there is a trend in which the five best models, which all utilize TEP, and %Vbur as independent variables. This indicates that electronic parameters, such as TEP, and steric parameters, such as %Vbur, must be considered when predicting the τ value. δC also appears in the top 4 models, another electronic parameter. Interestingly, HEP and δSe, highly regarded and useful metrics to better understand the electronic structure of NHCs, were found to only appear in 2 of the 5 top performing models. What can be inferred from all of these top performing statistical models is that both electronic effects and steric interactions must be taken into account when exploring the impact of modifying the NHC on the geometry of the transition metal complex.10,15 Sole consideration of only one or a few of these properties will not provide a wholistic picture of the bonding mechanisms occurring in these systems. Therefore, those looking to make correlations with these aforementioned properties should be cautious to make those correlations with limited data.
Conclusions
DFT calculations at the BP86/def2-TZVPD level of theory of iron bis(dithiolene)-N-heterocyclic carbene adducts were conducted to investigate what factors influence the coordination in these iron-containing complexes. Several parameters were connected to the coordination based on their documented associations with σ-donor ability, π-acidity, and steric effects. Using multivariable linear regression models, these parameters were compared to determine their correlation with the Addison τ parameter, which indicates whether the complex adopts a square pyramidal or trigonal bipyramidal geometry. The analysis revealed that a combination of TEP and %Vbur together are the most effective predictors of the τ value among the parameters considered in this study. Adding more parameters leads to only slight improvements in the statistical models for predicting the τ value.Author contributions
K. M.: investigation, data curation, methodology, project administration, writing – original draft, writing – review & editing, formal analysis, visualization; A. V. B. S.: writing – review & editing; S. S.: conceptualization, resources, supervision, funding acquisition, investigation, data curation, methodology, project administration, writing – original draft, writing – review & editing, software, formal analysis, validation, visualization.Data availability
All optimized coordinates, statistical information, R scripts, and additional data is included in the ESI† of this article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the useful comments from Dr Matthew Hanson and Dr Katsu Ogawa in relation to this work. The authors acknowledge the National Science Foundation (Award #2142874) for support of this research. The authors acknowledge support from the Camille & Henry Dreyfus Foundation (Award # TH-23-033).References
- H. V. Huynh, T. T. Lam and H. T. T. Luong, Anion Influences on Reactivity and NMR Spectroscopic Features of NHC Precursors, RSC Adv., 2018, 8(61), 34960–34966, 10.1039/C8RA05839C.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, An Overview of N-Heterocyclic Carbenes, Nature, 2014, 510(7506), 485–496, DOI:10.1038/nature13384.
- C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, N -Heterocyclic Carbenes in Materials Chemistry, Chem. Rev., 2019, 119(8), 4986–5056, DOI:10.1021/acs.chemrev.8b00514.
- O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Beyond Conventional N -Heterocyclic Carbenes: Abnormal, Remote, and Other Classes of NHC Ligands with Reduced Heteroatom Stabilization, Chem. Rev., 2009, 109(8), 3445–3478, DOI:10.1021/cr8005087.
- H. V. Huynh, Electronic Properties of N-Heterocyclic Carbenes and Their Experimental Determination, Chem. Rev., 2018, 118(19), 9457–9492, DOI:10.1021/acs.chemrev.8b00067.
- A. A. Danopoulos, T. Simler and P. Braunstein, N-Heterocyclic, Carbene Complexes of Copper, Nickel, and Cobalt, Chem. Rev., 2019, 119(6), 3730–3961, DOI:10.1021/acs.chemrev.8b00505.
- C. Heinemann, T. Müller, Y. Apeloig and H. Schwarz, On the Question of Stability, Conjugation, and “Aromaticity” in Imidazol-2-Ylidenes and Their Silicon Analogs, J. Am. Chem. Soc., 1996, 118(8), 2023–2038, DOI:10.1021/ja9523294.
- D. J. Nelson and S. P. Nolan, Quantifying and Understanding the Electronic Properties of N-Heterocyclic Carbenes, Chem. Soc. Rev., 2013, 42(16), 6723, 10.1039/c3cs60146c.
- H. Clavier and S. P. Nolan, Percent Buried Volume for Phosphine and N-Heterocyclic Carbene Ligands: Steric Properties in Organometallic Chemistry, Chem. Commun., 2010, 46(6), 841, 10.1039/b922984a.
- C. A. Gaggioli, G. Bistoni, G. Ciancaleoni, F. Tarantelli, L. Belpassi and P. Belanzoni, Modulating the Bonding Properties of N–Heterocyclic Carbenes (NHCs): A Systematic Charge–Displacement Analysis, Chem. – Eur. J., 2017, 23(31), 7558–7569, DOI:10.1002/chem.201700638.
- C. A. Tolman, Steric Effects of Phosphorus Ligands in Organometallic Chemistry and Homogeneous Catalysis, Chem. Rev., 1977, 77(3), 313–348, DOI:10.1021/cr60307a002.
- O. Back, M. Henry-Ellinger, C. D. Martin, D. Martin and G. Bertrand, 31 P NMR Chemical Shifts of Carbene–Phosphinidene Adducts as an Indicator of the π–Accepting Properties of Carbenes, Angew. Chem., Int. Ed., 2013, 52(10), 2939–2943, DOI:10.1002/anie.201209109.
- C. Barnett, J. B. Harper and M. L. Cole, Correlating Electronic Properties of N –Heterocyclic Carbenes with Structure, and the Implications of Using Different Probes, ChemistrySelect, 2022, 7(2), e202104348, DOI:10.1002/slct.202104348.
- C. Barnett, M. L. Cole and J. B. Harper, A Dual NMR Probe Approach to Understanding the Electronic Properties of N –Heterocyclic Carbenes, Chem.: Methods, 2021, 1(8), 374–381, DOI:10.1002/cmtd.202100043.
- C. Barnett, M. L. Cole and J. B. Harper, Steric Properties of N –Heterocyclic Carbenes Affect the Performance of Electronic Probes, Eur. J. Inorg. Chem., 2021, 2021(47), 4954–4958, DOI:10.1002/ejic.202100796.
- Y. Kim, Y. Kim, M. Y. Hur and E. Lee, Efficient Synthesis of Bulky N-Heterocyclic Carbene Ligands for Coinage Metal Complexes, J. Organomet. Chem., 2016, 820, 1–7, DOI:10.1016/j.jorganchem.2016.07.023.
- SambVca 2.1 A web application to characterize catalytic pockets. https://www.molnac.unisa.it/OMtools/sambvca2.1/index.html (accessed 2023-10-27).
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, Synthesis, Structure, and Spectroscopic Properties of Copper(II) Compounds Containing Nitrogen–Sulphur Donor Ligands; the Crystal and Molecular Structure of Aqua[1,7-Bis(N-Methylbenzimidazol-2′-Yl)-2,6-Dithiaheptane]Copper(II) Perchlorate, J. Chem. Soc., Dalton Trans., 1984,(7), 1349–1356, 10.1039/DT9840001349.
- J. Selvakumar, S. M. Simpson, E. Zurek and K. Arumugam, An Electrochemically Controlled Release of NHCs Using Iron Bis(Dithiolene) N-Heterocyclic Carbene Complexes, Inorg. Chem. Front., 2021, 8(1), 59–71, 10.1039/D0QI00638F.
- I. Sánchez-Lombardo, S. Alvarez, C. C. McLauchlan and D. C. Crans, Evaluating Transition State Structures of Vanadium–Phosphatase Protein Complexes Using Shape Analysis, J. Inorg. Biochem., 2015, 147, 153–164, DOI:10.1016/j.jinorgbio.2015.04.005.
- S. Escayola, N. Bahri-Laleh and A. Poater, % VBur Index and Steric Maps: From Predictive Catalysis to Machine Learning, Chem. Soc. Rev., 2024, 53(2), 853–882, 10.1039/D3CS00725A.
- A. Gorczyński, M. Zaranek, S. Witomska, A. Bocian, A. R. Stefankiewicz, M. Kubicki, V. Patroniak and P. Pawluć, The Cobalt(II) Complex of a New Tridentate Schiff-Base Ligand as a Catalyst for Hydrosilylation of Olefins, Catal. Commun., 2016, 78, 71–74, DOI:10.1016/j.catcom.2016.02.009.
- S. Simpson, The Search for Molecular Corks beyond Carbon Monoxide: A Quantum Mechanical Study of N-Heterocyclic Carbene Adsorption on Pd/Cu(111) and Pt/Cu(111) Single Atom Alloys, JCIS Open, 2021, 3, 100013, DOI:10.1016/j.jciso.2021.100013.
- M. D. Hanson and S. M. Simpson, Geometric and Electronic Effects in the Binding Affinity of Imidazole-Based N-Heterocyclic Carbenes to Cu(100)- and Ag(100)-Based Pd and Pt Single-Atom Alloy Surfaces, ACS Omega, 2023, 8(40), 37402–37412, DOI:10.1021/acsomega.3c05376.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform, J. Cheminf., 2012, 4(1), 17, DOI:10.1186/1758-2946-4-17.
- S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding, C. Hättig, A. Hellweg, B. Helmich-Paris, C. Holzer, U. Huniar, M. Kaupp, A. Marefat Khah, S. Karbalaei Khani, T. Müller, F. Mack, B. D. Nguyen, S. M. Parker, E. Perlt, D. Rappoport, K. Reiter, S. Roy, M. Rückert, G. Schmitz, M. Sierka, E. Tapavicza, D. P. Tew, C. Van Wüllen, V. K. Voora, F. Weigend, A. Wodyński and J. M. Yu, TURBOMOLE: Modular Program Suite for Ab Initio Quantum-Chemical and Condensed-Matter Simulations, J. Chem. Phys., 2020, 152(18), 184107, DOI:10.1063/5.0004635.
- A. D. Becke, Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior, Phys. Rev. A, 1988, 38(6), 3098–3100, DOI:10.1103/PhysRevA.38.3098.
- A. Schäfer, C. Huber and R. Ahlrichs, Fully Optimized Contracted Gaussian Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr, J. Chem. Phys., 1994, 100(8), 5829–5835, DOI:10.1063/1.467146.
- H. Jacobsen and J. P. Donahue, Computational Study of Iron Bis(Dithiolene) Complexes: Redox Non-Innocent Ligands and Antiferromagnetic Coupling, Inorg. Chem., 2008, 47(21), 10037–10045, DOI:10.1021/ic801277r.
- H. V. Huynh, Recent Applications of the Huynh Electronic Parameter (HEP), Chem. Lett., 2021, 50(10), 1831–1841, DOI:10.1246/cl.210435.
- B. Maryasin and H. Zipse, Theoretical Studies of 31P NMR Spectral Properties of Phosphanes and Related Compounds in Solution, Phys. Chem. Chem. Phys., 2011, 13(11), 5150, 10.1039/c0cp02653k.
- P.-A. Payard, L. A. Perego, L. Grimaud and I. Ciofini, A DFT Protocol for the Prediction of 31 P NMR Chemical Shifts of Phosphine Ligands in First-Row Transition-Metal Complexes, Organometallics, 2020, 39(17), 3121–3130, DOI:10.1021/acs.organomet.0c00309.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Towards the Online Computer-Aided Design of Catalytic Pockets, Nat. Chem., 2019, 11(10), 872–879, DOI:10.1038/s41557-019-0319-5.
- R Core Team R: A Language and Environment for Statistical Computing, 2023. https://www.R-project.org/(accessed 2024-03-06).
- H. Akaike, A New Look at the Statistical Model Identification, IEEE Trans. Autom. Control, 1974, 19(6), 716–723, DOI:10.1109/TAC.1974.1100705.
- M. Stone, Comments on Model Selection Criteria of Akaike and Schwarz, J. R. Stat. Soc. Ser. B Methodol., 1979, 41(2), 276–278, DOI:10.1111/j.2517-6161.1979.tb01084.x.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt02650k |
| This journal is © The Royal Society of Chemistry 2024 |