
Atomic structure and water arrangement on K-feldspar microcline (001)†
Tobias
Dickbreder‡
 *a,
Franziska
Sabath‡
*a,
Bernhard
Reischl
b,
Rasmus V. E.
Nilsson
b,
Adam S.
Foster
cd,
Ralf
Bechstein
a and
Angelika
Kühnle
a
*a,
Franziska
Sabath‡
*a,
Bernhard
Reischl
b,
Rasmus V. E.
Nilsson
b,
Adam S.
Foster
cd,
Ralf
Bechstein
a and
Angelika
Kühnle
a
aFaculty of Chemistry, Physical Chemistry I, Bielefeld University, 33615 Bielefeld, Germany. E-mail: dickbreder@uni-bielefeld.de; franziska.sabath@uni-bielefeld.de
bInstitute for Atmospheric and Earth System Research (INAR)/Physics, Faculty of Science, University of Helsinki, 00014 Helsinki, Finland
cDepartment of Applied Physics, Aalto University, Finland
dNano Life Science Institute (WPI-NanoLSI), Kanazawa University, Kanazawa 920-1192, Japan
First published on 19th December 2023
Abstract
The properties of clouds, such as their reflectivity or their likelihood to precipitate, depend on whether the cloud droplets are liquid or frozen. Thus, understanding the ice nucleation mechanisms is essential for the development of reliable climate models. Most ice nucleation in the atmosphere is heterogeneous, i.e., caused by ice nucleating particles such as mineral dusts or organic aerosols. In this regard, K-feldspar minerals have attracted great interest recently as they have been identified as one of the most important ice nucleating particles under mixed-phase cloud conditions. The mechanism by which feldspar minerals facilitate ice nucleation remains, however, elusive. Here, we present atomic force microscopy (AFM) experiments on microcline (001) performed in an ultrahigh vacuum and at the solid–water interface together with density functional theory (DFT) and molecular dynamics (MD) calculations. Our ultrahigh vacuum data reveal features consistent with a hydroxyl-terminated surface. This finding suggests that water in the residual gas readily reacts with the surface. Indeed, the corresponding DFT calculations confirm a dissociative water adsorption. Three-dimensional AFM measurements performed at the mineral–water interface unravel a layered hydration structure with two features per surface unit cell. A comparison with MD calculations suggests that the structure observed in AFM corresponds to the second hydration layer rather than the first water layer. In agreement with previous computation results, no ice-like structure is seen, questioning an explanation of the ice nucleation ability by lattice match. Our results provide an atomic-scale benchmark for the clean and water-covered microcline (001) plane, which is mandatory for understanding the ice nucleation mechanism on feldspar minerals.
1. Introduction
The aggregate state of water in clouds crucially influences their properties,1,2 and thus the formation of ice in clouds plays a fundamental role in weather and the global climate.3,4 For instance, most continental rain precipitates from the ice phase rather than the liquid phase.4,5 Moreover, the existence of ice in clouds significantly influences the Earth's albedo by changing the optical properties of clouds.1–3 Consequently, a fundamental understanding of ice nucleation is crucial for the development of more reliable climate models.Most ice nucleation in the atmosphere occurs heterogeneously initiated by so-called ice nucleating particles such as biological particles,6 organic aerosols7 and mineral dusts.1,2 In this regard, feldspar minerals have gained great research interest because they have been identified as one of the most important atmospheric ice nucleating particles.8 It has been shown based on a combination of droplet-freezing experiments and atmospheric modelling that K-rich feldspar minerals dominate ice nucleation by mineral dusts under mixed-phase cloud conditions.8 This is the case despite feldspar being a minor component of mineral dusts due to the higher ice nucleation efficiency of feldspar minerals compared to other components of mineral dusts, such as clay minerals.8 To understand the origin of the exceptional ice nucleation activity of feldspar minerals and elucidate the corresponding mechanism, feldspar minerals have been studied intensively. Ice nucleation experiments have been conducted using feldspar particles immersed in water8–14 and aqueous solution.15–17 Moreover, the deposition of ice crystals from the gas phase onto the natural (001) and (010) cleavage planes of feldspar crystals has been investigated.14,18–21 To identify ice nucleation active sites (INAS) on the feldspar surface, these experiments were combined with environmental scanning electron microscopy (ESEM),10–13,20–22 optical microscopy,10,13,14,18 and atomic force microscopy (AFM) under ambient conditions.10,18,21 Immersion-mode ice nucleation experiments with feldspar samples of varying composition and crystal structure consistently reveal a higher ice nucleation efficiency of alkali feldspars compared to plagioclase feldspar minerals.8,9,11–13 It has been speculated that this higher ice nucleation efficiency is due to a perthitic microtexture associated with ex-solution of alkali feldspars into K-rich and Na-rich domains.13 Moreover, it has been reported that the ice nucleation efficiency of alkali feldspars correlates with the crystallographic order, making microcline the most active ice nucleator and sanidine the least.11
Concerning the identification of INAS on K-rich feldspars, recent ESEM measurements by Kiselev et al. have revealed that ice crystals forming on the (001) and (010) faces of microcline predominantly share the same crystal orientation.20 This finding has been explained by an epitaxy of ice crystals with an unstable (100) crystal face, which is expected to be exposed in cracks or cavities, and thus acts as INAS.20 Furthermore, it has been shown that ice formation on K-feldspar in immersion and deposition modes starts at steps18 and pores,10,21 highlighting the role of surface topography in ice nucleation. However, the pores observed in these studies are on the micrometre scale while the features relevant for ice nucleation are expected to be nanometre sized.10,21 Moreover, optical microscopy experiments show that the INAS on feldspar minerals can differ depending on the conditions, i.e., different sites are relevant for ice nucleation in immersion and deposition modes.14
In addition to experimental studies, the surfaces of K-feldspar minerals have been investigated theoretically based on density functional theory (DFT)23 and molecular dynamics (MD) calculations.20,24–26 DFT calculations by Pedevilla et al. have revealed an ice-like second water layer on microcline (001), despite the first layer having a non-ice-like structure. Moreover, Kiselev et al. have reported based on MD simulations that ice is more stable on the (100) plane than on the (001) and (010) cleavage planes of microcline.20 However, two recent MD simulations by Soni and Patey and Kumar et al. have not found any indication for ice nucleation on the (001), (010) or (100) surfaces of K-rich feldspar. Hence, the atomistic mechanism and surface structures causing the exceptional ice nucleation efficiency of K-rich feldspar minerals remain elusive. Despite this, experimental studies on the atomic-scale structure of both the pristine feldspar surface and the feldspar–water interface are still sparse. Pioneering work on the surface structure of alkali feldspars and plagioclase has been presented by Drake and Hellmann and Hellmann et al. using AFM and transmission electron microscopy, but these studies mainly focus on the alkaline or acidic alteration of feldspar minerals.27–29 Furthermore, Fenter et al. have studied the mineral–water interface of the less ice nucleation active orthoclase feldspar with X-ray reflectivity (XRR).30,31 Albite feldspar has been investigated in a recent publication presenting the hydration structure on the (001) and (010) surfaces.32 However, to the best of our knowledge, experimental studies on the atomic-scale surface and hydration structure of the most ice nucleation active K-feldspar microcline have not been presented so far.
Here, we focus on the atomic-scale structure of the most easily cleaved (001) surface of K-feldspar microcline,30 because a solid understanding of the kinetically-favoured crystal face is crucial for the investigation of more complex systems. We present AFM experiments on microcline (001) performed in an ultrahigh vacuum (UHV) and at the solid–water interface in combination with DFT and MD calculations. Our UHV data reveal features that can be explained by protruding hydroxyl groups exposed on the α-termination. This finding suggests that water in the residual gas readily reacts with the dangling bonds of the as-cleaved surface, resulting in a hydroxyl-terminated surface. The corresponding DFT calculations confirm that water indeed adsorbs dissociatively with an energy gain of about 3 eV. Three-dimensional AFM measurements performed at the mineral–water interface unravel a hydration structure with two features within the unit cell per layer. MD calculations indicate that the AFM measurements correspond to the second hydration layer and that the first layer cannot be penetrated by the AFM tip. In the second layer, the water molecules appear to be rather mobile and do not reside localized on well-defined sites. In particular, no ice-like structure is seen, questioning a simple explanation of the ice nucleation ability by lattice match. The latter finding is in perfect agreement with previous computation results. Our results provide an atomic-scale benchmark for the clean and water-covered microcline (001) plane, which is a prerequisite for unravelling the ice nucleating mechanism of microcline and feldspar minerals in general.
2. Results and discussion
2.1 Bulk structure of microcline
To understand the surface structure and characteristics of K-feldspar microcline, a basic understanding of the bulk structure of feldspar minerals is required. Hence, we give a brief description of the bulk crystal structure of feldspar minerals on the example of microcline before turning to the structure of the (001) cleavage plane. The crystal structure of K-feldspar microcline with the chemical formula KAlSi3O8 is shown in Fig. 1.33Fig. 1 shows projections of a single microcline unit cell along the [100] and [010] directions in (a) and (c), respectively, and on the (001) lattice plane in (b).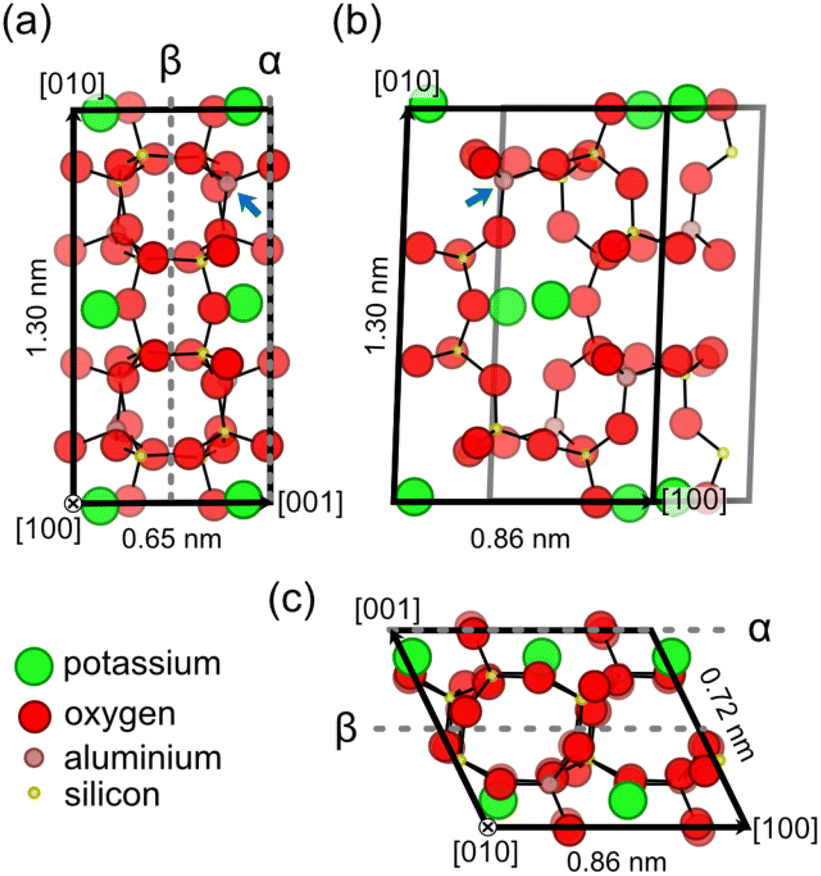 | ||
| Fig. 1 Bulk structure of K-feldspar microcline. (a) and (c) Side views on the (001) crystal plane along the [100] and [010] directions, respectively. (b) A top view on the (001) crystal plane. In all the three views, all atoms contained within the unit cell of monoclinic feldspar are displayed.§ Moreover, atoms are drawn with decreasing opacity the further they are in the back of the structure. The α- and β-terminations for the microcline (001) surface are indicated in (a) and (c) by dashed lines. The blue arrows indicate one of the T1(o) tetrahedral sites, where the aluminium atoms in the microcline structure are concentrated. The structure shown here is based on the XRD results obtained for the microcline used in this work (see the ESI†). All dimensions agree with the parameters presented in the literature for maximum microcline (ref. 34 and 37). | ||
Here, we show the C-centred unit cell, which contains four formula units, because this unit cell is commonly used in literature.23,30,34 For triclinic feldspar varieties (space group C![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) such as microcline, the unit cell is a parallelepiped with a parallelogram as a base, while monoclinic feldspar varieties (space group C2/m) such as sanidine and orthoclase have a rectangular base.34 As for all feldspar minerals, microcline is a framework silicate consisting of (Al,Si)O4 tetrahedra linked by shared oxygen atoms.34,35 These tetrahedra create a three-dimensional network of four- and eight-membered rings as shown in Fig. 1. These rings of tetrahedra are especially visible in the projections normal to the [100] and [010] directions as shown in Fig. 1(a) and (c). The negative charge associated with the replacement of silicon by aluminium atoms in the aluminosilicate tetrahedra is compensated by potassium ions located in cavities inside the framework.34 The counter ions form a centred lattice in the (001) lattice plane as shown in the top view in Fig. 1(b). The potassium ions can be gradually replaced by sodium or calcium ions creating a ternary mixing series between the end members of orthoclase (K-feldspar), albite (Na-feldspar) and anorthite (Ca-feldspar).34,35 Note that the replacement of alkali metal ions (potassium and sodium) is accompanied by an increase in the aluminium content of feldspar minerals, where the calcium end member has the chemical formula CaAl2Si2O8.34
) such as microcline, the unit cell is a parallelepiped with a parallelogram as a base, while monoclinic feldspar varieties (space group C2/m) such as sanidine and orthoclase have a rectangular base.34 As for all feldspar minerals, microcline is a framework silicate consisting of (Al,Si)O4 tetrahedra linked by shared oxygen atoms.34,35 These tetrahedra create a three-dimensional network of four- and eight-membered rings as shown in Fig. 1. These rings of tetrahedra are especially visible in the projections normal to the [100] and [010] directions as shown in Fig. 1(a) and (c). The negative charge associated with the replacement of silicon by aluminium atoms in the aluminosilicate tetrahedra is compensated by potassium ions located in cavities inside the framework.34 The counter ions form a centred lattice in the (001) lattice plane as shown in the top view in Fig. 1(b). The potassium ions can be gradually replaced by sodium or calcium ions creating a ternary mixing series between the end members of orthoclase (K-feldspar), albite (Na-feldspar) and anorthite (Ca-feldspar).34,35 Note that the replacement of alkali metal ions (potassium and sodium) is accompanied by an increase in the aluminium content of feldspar minerals, where the calcium end member has the chemical formula CaAl2Si2O8.34
In addition to the chemical composition, the crystallographic order of aluminium and silicon atoms, the Al/Si order, is crucial for the structure of feldspar minerals. Monoclinic feldspar varieties exhibit two distinct tetrahedral sites T1 and T2, where each centred unit cell contains eight sites of each type.34 The aluminium atoms in the feldspar structure can either be distributed randomly onto these sites (disordered) or they can be concentrated to specific tetrahedral sites (ordered). With increased Al/Si order, however, previously identical tetrahedral sites become different, and the crystal structure changes from the monoclinic (space group C2/m) to a triclinic symmetry (space group C).34 Due to the lower symmetry, triclinic feldspar varieties exhibit four different tetrahedral sites, where each centred unit cell contains four sites of each type (for naming of tetrahedral sites, see Fig. S1†). For K-rich feldspar varieties, the fully disordered variety sanidine and the intermediately ordered orthoclase have a monoclinic crystal structure.34 The fully ordered K-feldspar variety microcline is triclinic with aluminium atoms predominantly occupying one type of tetrahedral site referred to as T1(o) (see the blue arrow in Fig. 1).34 Note that while all microcline samples are ordered with respect to the positions of aluminium atoms, the specific degree of Al/Si order, and thus the crystal structure, varies depending on the sample. Fully ordered microcline samples (also referred to as maximum microcline) have crystal structure parameters close to a = 0.859 nm, b = 1.297 nm, 0.722 nm, α = 90.6°, β = 115.9° and γ = 87.7°.34,36,37 Microcline samples with an intermediate ordering, however, can have structural parameters much closer to those of orthoclase such as a = 0.857 nm, b = 1.296 nm, 0.721 nm, α = 90.3°, β = 116.0° and γ = 89.1°.33
Feldspar minerals have two natural cleavage planes, the (001) and (010) surfaces, which exhibit perfect and good cleavage, respectively.34,35 The third low-index plane, the (100) surface, on the other hand, is not observed in natural feldspar samples.34 Here, we are interested in the (001) cleavage plane as it is the most easily cleaved plane of K-feldspar microcline.31,34 For the (001) surface, the possibility of two different surface terminations, the α- and β-terminations, has been discussed in the literature.30 In Fig. 1(a) and (c), the cleavage planes corresponding to these two terminations are indicated by dashed grey lines. One of the key differences between both surface terminations is that the α-termination exclusively exposes T1 tetrahedral sites, while the β-termination only exposes T2 sites. Moreover, the creation of a β-terminated surface requires breaking eight bonds per unit cell compared to four for the α-termination. Hence, a cleavage along the α plane is favoured. This expectation has been confirmed by XRR measurements by Fenter et al., which have found an α-terminated surface at the orthoclase (001)–water interface.30 Following this theoretical explanation and experimental evidence, we assume that the (001) surfaces prepared in our experiments exhibit an α-terminated surface. A top view of a bulk cut of the α-terminated surface (001) is shown in Fig. 1(b). The surface exhibits a centred lattice of potassium ions with unit cell dimensions of 0.86 × 1.30 nm2 and an angle of 88.0° for maximum microcline. The calculated height of monoatomic step edges is 0.65 nm.33,34 The potassium ions are located in cavities formed by the surrounding aluminosilicate framework of the feldspar. From this framework, four oxygen atoms per unit cell protrude out of the surface plane.30 These oxygen atoms are located on the cleaving plane, and thus they are shared by the displayed unit cell and the unit cell one layer above (see Fig. 1(c)). Upon cleavage, stoichiometry and charge neutrality are preserved, so two of these oxygen atoms remain with each of the two created surfaces. Hence, the as-cleaved α-terminated (001) surface exhibits two protruding oxygen atoms and two oxygen vacancies per surface unit cell.
2.2 Surface structure in ultrahigh vacuum
To evaluate the surface structure expected in our UHV AFM measurements, we performed DFT calculations of the as-cleaved microcline (001) surface. These calculations are based on the bulk structure of maximum microcline as the samples used in our UHV AFM experiments have a crystal structure close to maximum microcline with an aluminium occupation of 0.85 in the T1(o) sites (see the X-ray diffraction (XRD) results in the ESI†). The initial surface structure is created from the bulk structure by cleavage along the α-plane such that only Al–O bonds (length 0.174 nm) are broken as these are significantly longer, and thus weaker than the corresponding Si–O bonds (length 0.160 nm, see XRD in the ESI†). This procedure creates an initial surface structure, where the oxygen vacancies are distributed periodically following the highly ordered arrangement of aluminium atoms in the microcline structure. As all our experimental data (see below and the ESI†) reveal no indication of a reconstruction, but rather an unreconstructed surface unit cell, we start our DFT calculation with the unreconstructed surface structure. The results of our DFT calculation are shown in Fig. 2(a). Here, we show features in the surface plane in grayscale and those protruding from the surface in colour to highlight the depth of the surface features. The relaxed surface structure exhibits a centred unit cell with dimensions of 0.86 × 1.31 nm2 and an angle of 87.6°, which agrees with the unit cell dimensions of the bulk-terminated structure within the accuracy of DFT. Moreover, Fig. 2(a) shows two protruding oxygen atoms (red circles) and two oxygen vacancies (dashed red circles) per unit cell. These oxygen vacancies correspond to dangling bonds of aluminium atoms in the surface structure below. Both protruding oxygen atoms and vacancies follow the same centred surface structure as observed for the potassium ions (dark grey circles). For our AFM measurements, we thus expect to observe two different types of features for the protruding oxygens and vacancies, respectively. Alternatively, the tip could be sensitive to only one of these features. In this case, we would expect two identical features per unit cell.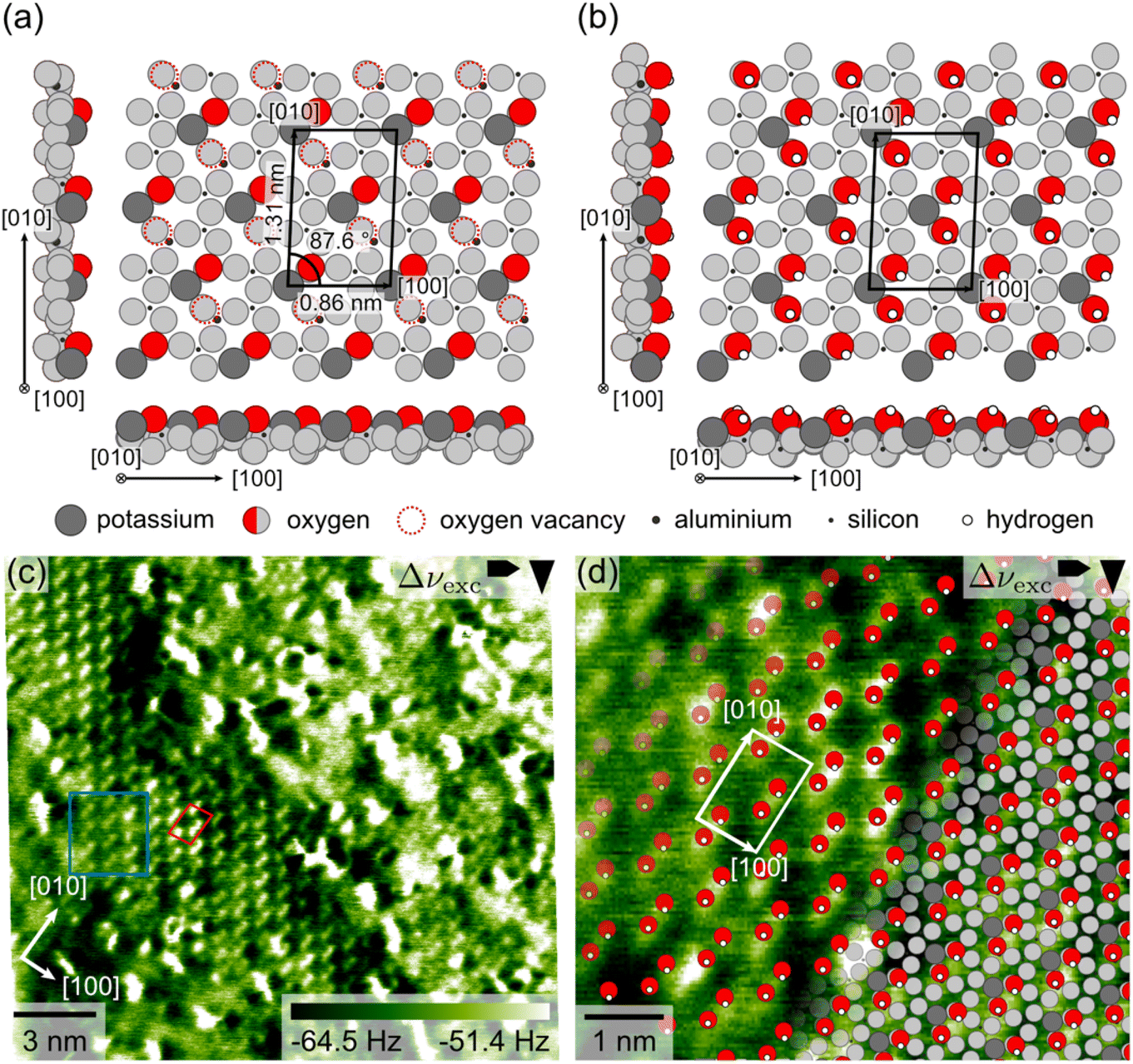 | ||
| Fig. 2 Theoretical and experimental structures of the microcline (001) surface. (a) and (b) DFT structures of the as-cleaved and hydroxyl-terminated microcline (001) surfaces, respectively. In the DFT structures, the surface plane is presented in grayscale while the protruding features are drawn in colour. (c) An experimental AFM image of the microcline (001) cleavage plane measured under UHV conditions at room temperature. The experimental AFM image was calibrated and corrected for linear drift. (d) DFT structure of the hydroxyl-terminated surface (b) superimposed onto a cut-out of the experimental AFM image shown in (c). For the overlay image, the experimental unit cell parameters are adjusted to match the lattice parameters of the DFT structure. | ||
Under environmental conditions, however, the as-cleaved microcline (001) surface is expected to react with water to create a hydroxyl-terminated surface structure.30,35 As water also constitutes an important component of the residual gas in our experimental UHV setup, we investigated the adsorption of water on the microcline surface. To this end, we calculated the adsorption of two water molecules per unit cell on the pristine surface in Fig. 2(a). Our DFT results confirm the expected dissociative adsorption of water molecules on the microcline (001) surface. We found a very high adsorption energy of −3.03 eV per water molecule, which indicates a very high affinity of the surface to adsorb water molecules. The water adsorption creates a hydroxyl-terminated surface as shown in Fig. 2(b). Here, all oxygen vacancies of the as-cleaved surface are saturated with hydroxyl groups. Moreover, the remaining hydrogen atoms from the adsorbed water react with the protruding oxygen atoms to form hydroxyl groups as well. Hence, all dangling bonds of the pristine surface are saturated, and the surface is fully covered with hydroxyl groups. Our results are in perfect agreement with previous DFT and experimental studies.23,30 Whether the clean or hydroxyl-terminated surface is present in our experiments should be possible to be distinguished by our UHV AFM data. The pristine surface exhibits two different types of features (two protruding oxygen atoms and two oxygen vacancies) per unit cell, while the hydroxyl-terminated surfaces have only one type of feature (four protruding hydroxyl groups).
Fig. 2(c) shows the experimental AFM data of the atomic-scale structure of microcline (001). The AFM image presented here was acquired on a microcline sample kept under UHV conditions at room temperature. The atomic-scale contrast is especially pronounced in a band from the top left to the bottom right corner of the image. Here, the surface exhibits bright, elongated features, which we refer to as sticks. The sticks are arranged in a regular pattern, where the corresponding unit cell contains two sticks. In this image, the unit cell has dimensions of (0.90 ± 0.03) × (1.31 ± 0.02) nm2 and an angle of (80 ± 2)° (see 2D Fourier transform in the ESI†), and the orientation of the [100] and [010] directions agrees well with the directions expected due to the orientation of the sample.¶ In addition to the very pronounced contrast shown in Fig. 2(c), we observed two slightly different contrasts shown in the ESI.† Note that we can rule out an artificial origin of the observed stick contrast caused by the tip shape as also smaller features are imaged, showing a single feature (see in Fig. 2(c)). The average unit cell determined from all our measurements has dimensions of (0.90 ± 0.03) × (1.29 ± 0.04) nm2 and an angle of (82 ± 3)° as determined from 72 images. Hence, the experimentally determined unit cell dimensions exhibit a small, but significant deviation from the bulk-truncated and DFT structures. This discrepancy could either originate from a real distortion of the surface structure or measurement artefacts that are not included in our error estimation. Regarding the former possibility, perthitic feldspar minerals are known to exhibit strain at the interfaces between the K-rich and Na-rich phases.34 It seems likely that this strain could cause a distortion of the surface structure. However, as we do not know whether our microcline sample exhibits a perthitic structure or internal strain, we cannot fully assess this possibility. In terms of potential measurement artefacts, we cannot fully rule out a scan angle dependence of the calibration parameters, which is why the stated error might be too small. In this case, the measured unit cell dimensions would be in accordance with the literature values for bulk maximum microcline.33,34
Next, we discuss the shape and internal structure of the sticks and compare them to the DFT structures. The sticks are elongated along the [010] direction and each stick consists of two smaller round features imaged together. This is especially visible for the sticks highlighted with a blue box in Fig. 2(c). Between the sticks, the AFM image does not show any additional surface features. Thus, we do not observe any indication for a second type of surface feature as expected for the as-cleaved microcline surface. Instead, we see a close match between the AFM image and the structure of the hydroxyl-terminated surface in Fig. 2(b). The experimental image exhibits four identical features per unit cell (two sticks containing two smaller features each), which correspond to the four hydroxyl groups in Fig. 2(b). To illustrate this interpretation, we superimposed the DFT structure of hydroxyl-terminated microcline (001) on a cut-out of the experimental AFM image in Fig. 2(d). As we are interested in comparing the structure within the unit cell, the unit cell parameters of the AFM image presented in the overlay image in Fig. 2(d) are adjusted to match those from the DFT calculations. In the overlay image, the superimposed positions of the hydroxyl groups agree perfectly with the positions of the sticks, where each stick is comprised of two surface hydroxyl groups. Hence, the experimental data provides evidence that microcline (001) is terminated by hydroxyl groups even under UHV conditions. This finding suggests that the dangling bonds of the as-cleaved microcline surface readily react with residual water in the UHV setup. To check whether the amount of water available in the UHV setup is sufficient to create a fully hydroxylated surface, we calculated the required water pressure from the exposure time and impingement rate based on kinetic gas theory. During preparation, our microcline sample was exposed to the chamber atmosphere for approximately 20 h. Assuming that each water molecule colliding with the surface reacts, this means that a water pressure of 7 × 10−12 mbar would be required to provide enough water molecules to saturate all vacancies. This is a reasonable water pressure for our system, especially during sample annealing. Hence, we expect that our samples were exposed to enough water during the annealing procedure. The finding that small amounts of water as available in a UHV setup are sufficient to hydroxylate the microcline surface stresses the fact that the hydroxyl-terminated surface structure is the relevant surface termination for microcline (001).
2.3 Water structure at the solid–water interface
Besides pristine microcline (001), the interface of this surface with bulk water is of interest to shed light on the ice nucleating ability of this mineral. To this end, we performed MD simulations and compared them to two- and three-dimensional AFM experiments.We have carried out atomistic molecular dynamics simulations of a fully hydroxylated, α-terminated microcline (001)–water interface using the model system shown in Fig. 3(a) and full details of the simulation protocol are provided in the Methods section. On a 10 ns timescale, K+ ions on the surface can go fully into solution or return from solution to vacant sites. On average, about 3/4 surface ion sites were found to be occupied, with vacant sites being temporarily occupied by water molecules. These ion dynamics at the surface convolute the hydration layer structure and thus require averaging interfacial properties over sufficiently long simulation trajectories. Our analyses were performed on a 105 ns trajectory following 35 ns of equilibration (see Fig. S6 in the ESI†). The water density profile along the z direction, perpendicular to the interface, exhibits two peaks before levelling off to the bulk water density around 1 nm from the surface, as shown in Fig. 3(b). The full 3D water density map reveals that the first hydration layer contains contributions from multiple maxima at different lateral (x,y) and z positions, as illustrated in Fig. 3, panels (c)–(g). The second hydration layer exhibits less lateral order than the first one, as shown in Fig. 3(h). The first hydration layer consists of highly mobile water molecules coordinating the hydroxyl groups and K+ ions at the mineral surface, as shown in Fig. 3(c). Water exchange around Si hydroxyl groups and surface ions occurs on a sub-100 ps timescale and the exchange around Al hydroxyls is slightly slower. On average, the first layer contains 9.7 water molecules per surface unit cell. The average numbers of water molecules in the first coordination shells of surface K+ ions, silicon hydroxyl groups, and aluminium hydroxyl groups are 1.8, 2.0, and 2.5, respectively (see Fig. S7 and S8 in the ESI†). Molecules coordinating the K+ ions are oriented in a way that their hydrogen atoms point away from the ion while molecules coordinating hydroxyls can do so both by donating or accepting hydrogen bonds. The distribution of water molecule positions and orientations in the hydration layer arises from the coordination of surface species as well as the hydrogen bond network between water molecules (see Fig. S9 in the ESI†). Hydrogen bonding between water molecules in the first hydration layer and the second hydration layer is comparatively weaker. We note that the solubility of surface ions depends strongly on the atomistic interactions in the model and that the interactions in the CLAYFF forcefield are not specifically fitted to reproduce the structural and thermodynamic properties of the microcline (001)–water interface. However, the solvation free energy of an isolated K+ ion in SPC/Fw water obtained from free energy perturbation, −269 kJ mol−1, is in very good agreement with the experimental values of −269.9 kJ mol−1 (ref. 38) or −295.0 kJ mol−1.39 Similar surface ion dynamics were also observed in previous simulations of the orthoclase (001)–water interface,24 while in the microcline–water interface simulations of Soni and Patey,26 all atoms in the mineral were kept at fixed positions, except for surface hydrogens. We conclude the exchange of ions on the surface observed in the simulations is a realistic feature of the system, but we have also performed simulations where surface ions were constrained to their equilibrium positions by a harmonic potential acting along the z axis. In these simulations, we observed similar equilibrium hydration layer structures, which confirms that our sampling in the simulation with unconstrained ions is adequate and indicates that the transient perturbation of the hydration layer structure, as ions dissolve from or re-adsorb onto the surface, is quite minor (see Fig. S10†). For subsequent comparison with AFM experiments, we have further averaged water density data over equivalent positions within the six surface unit cells of our model interface.
 | ||
| Fig. 3 Equilibrium structure of the microcline (001)–water interface from MD simulations. (a) Snapshot of the atomistic model. Atoms in the mineral surface are shown as spheres: silicon, yellow; aluminium, pink; oxygen, red; hydrogen, white; potassium, green. Water molecules are shown as red and white sticks. The (001) surfaces are exposed along the z axis of the simulation box. (b) Density profiles along the [001] direction of different atom types present at the interface. The z distance range shown in the graph is also indicated by the dashed black rectangle in panel (a). The water oxygen density profile exhibits two pronounced peaks at the interface, corresponding to two hydration layers marked HL1 and HL2. (c) Top view of the interface with the positions of water molecule oxygen atoms in HL1, sampled every 100 ps, marked by orange, magenta, and green dots when they coordinate silicon hydroxyls, aluminium hydroxyls, or surface K+ ions, respectively. (d)–(h) Water density maps in (x,y) planes parallel to the (001) surface at different z positions, indicated by the arrows in panel (b). The positions of K+ ions and hydroxyl groups in the bulk-terminated surface are shown by spheres and the surface unit cell is indicated by red lines. Additional visualizations of the hydration layer structure are provided in the ESI.† | ||
To compare these theoretical results with experiments, a microcline crystal was cleaved along the (001) plane under ambient conditions and a droplet of ultrapure water was positioned on top of the surface before performing two- and three-dimensional AFM experiments. In Fig. 4(a), a 5 × 5 nm2 drift-corrected40 two-dimensional AFM image of the (001) microcline–water interface is shown. Therein, a periodic structure with a unit cell of 0.85 × 1.31 nm2 and an angle of 85.7° can be seen as marked by the red quadrangle. Within the measurement accuracy of our AFM (0.025 nm and 2°),40 the determined unit cell agrees excellently with the unit cell of maximum microcline. The averaged unit cell determined from 162 AFM images at the (001) microcline–water interface has dimensions of (0.83 ± 0.01) × (1.31 ± 0.03) nm2 and (86.7 ± 2.2)°, again in excellent agreement with the unit cell of maximum microcline. The unit cell contains two bright features. The shown periodic structure in Fig. 4(a) of the (001) microcline–water interface appears blurred. This blurred structure might originate from the fact that the water molecules at this interface are rather mobile. Two-dimensional AFM experiments do not allow for identifying in which hydration layer the AFM images are acquired and whether one or more hydration layers are involved. To answer these questions, three-dimensional AFM is required.
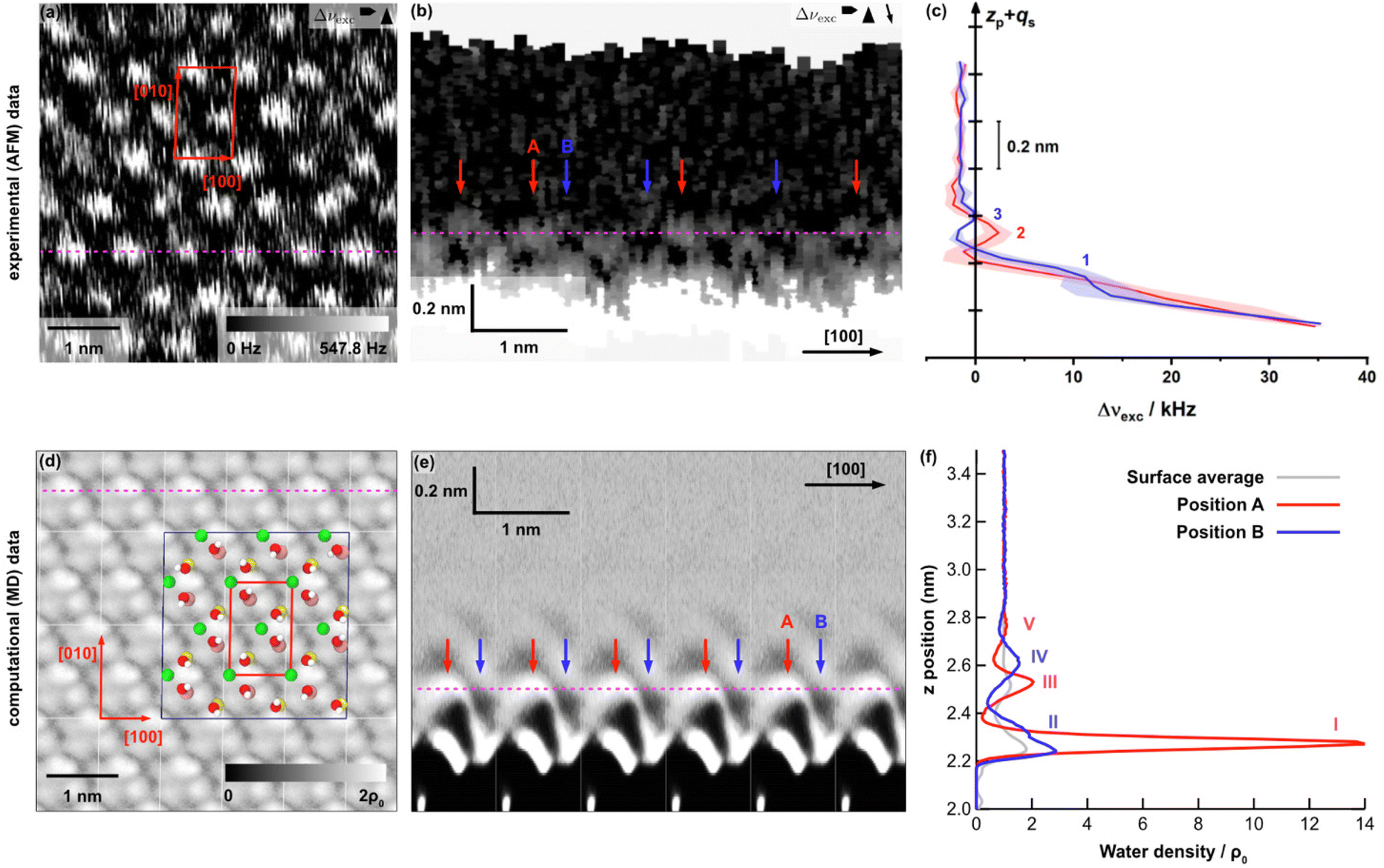 | ||
| Fig. 4 Results for the (001) microcline–water interface from two- and three-dimensional AFM experiments (a–c) and MD simulations (d–f). (a) Drift-corrected two-dimensional 5 × 5 nm2 AFM image. The determined unit cell (shown in red) has dimensions of 0.85 × 1.31 nm2 and an angle of 85.7°. (b) Slice normal to the microcline (001) surface extracted from a 3D AFM map taken along the [100] direction. The y position at which the slice normal to the surface where extracted corresponds to the dashed magenta line in the 2D AFM image in (a), which correlates with the lateral slice on peak position 2 (see Fig. S4(b)†). Two different positions A and B (highlighted by red and blue arrows) can be identified. The dashed magenta line corresponds to the z position where the 2D AFM image in (a) was taken. (c) Averaged Δνexcvs. zp + qs profiles taken at the red and blue positions in (b). Three peaks labelled (1)–(3) can be identified. (d) Water density map parallel to the (001) microcline–water interface within the second hydration layer between z = 2.50 and 2.55 nm from MD simulations. The positions of K+ ions and hydroxyl groups in the bulk-terminated surface are overlaid and the surface unit cell is shown in red. The dark blue marked box displays the original simulation box with a size of 2 × 3 unit cells in [010] and [100] directions. (e) Vertical slice through the water density extracted along the [100]-direction at the y position indicated by the dashed magenta line in panel (d). The dashed magenta line indicates the z position of the horizontal slice shown in panel (d). As in the 3D AFM data, two different positions A and B can be identified, marked by red and blue arrows. (f) Water density profiles extracted at the two positions A and B, averaged over an area of 0.1 × 0.1 nm2 in each of the six surface unit cells. Additionally, the average water density profile over the entire surface (grey) is shown. Five peaks can be identified in total, labelled (I)–(V). The reference bulk water density is ρ0 = 33.7 nm−3. | ||
Hence, we collected three dimensional maps with a size of 10 nm × 10 nm × 1.2 nm. The [100] direction of the microcline crystal was aligned with the fast scan direction of the AFM. Fig. 4(b) shows a frequency shift slice normal to the microcline (001) surface along the [100] direction. At the lower part of the vertical slice, i.e., close to the microcline surface, the frequency shift is the largest due to the repulsive interactions between the tip and the sample. Above this, an oscillatory structure can be observed normal to the surface up to a height of 0.5 nm. Further away from the microcline (001) surface, the water was found to be unstructured as in bulk water. According to the solvent tip approximation (STA),41 we assigned bright parts in the frequency shift image to the high water density at the (001) microcline–water surface.
Besides the structure normal to the surface, a faint lateral structure is seen as well. In accordance with the blurred structure observed in the two-dimensional image shown in Fig. 4(a), the local maxima and minima are, however, sometimes not clearly separated.
The local maxima and minima within a layer are shifted with respect to the maxima and minima within the next layer. Consequently, there are two distinguishable positions A and B normal to the surface, highlighted by red and blue arrows in Fig. 4(b). At the highlighted positions, Δνexcvs. zp + qs profiles were extracted and averaged depending on the position. Fig. 4(c) shows the averaged profiles at the two positions. Note that the left scale bar coincides with the height of the vertical slice shown in Fig. 4(b) to facilitate the comparison. As expected from the results shown in the vertical slice, both profiles show a significant increase near the surface due to the repulsive interactions between the tip and the (001) microcline surface. In addition, the profile extracted at position B shows a shoulder (1) in this slope. This shoulder is referred to as the first water peak. Further from the surface, two more local maxima can be identified. One of which is seen in the profile extracted at position A (2) and the other in the profile extracted at position B (3). The distance between (1) and (3) is 0.29 nm, which fits in size with the dimensions of a water molecule. The peaks (2) and (3) are close together, with only 0.07 nm between them. This can be understood from Fig. 4(b), showing the water layers shifted into each other. From the 3D maps, we can extract two-dimensional images parallel to the surface (see the ESI†). Very close to the surface, i.e., at the height of peak (1), no contrast can be obtained in the images. Further away from the surface, e.g., at position (2), the images reveal a periodic structure that closely resemble the structure shown in the two-dimensional image given in Fig. 4(a).
We now compare the AFM data with the MD calculations. Fig. 4(d) shows a calculated lateral slice of the water density parallel to the microcline (001) surface. The best match with the experiments was obtained when extracting the parallel slice at a height of z = 2.50–2.55 nm, about 0.4 nm above the surface ions. In this layer, the microcline unit cell can be identified with two bright features per unit cell as in the experiments.
Fig. 4(e) shows the vertical slice extracted along the [100] direction. As in the experimental data, an oscillating water density is observed up to a height of about 0.8 nm away from the surface. We can again identify two positions A and B exhibiting a distinct profile normal to the surface marked by red and blue arrows, respectively. In Fig. 4(f), the calculated water density profiles extracted at these two positions are presented. The shown water density profiles were obtained by averaging over six unit cells and an area of 0.1 × 0.1 nm2 around the given positions. In addition, the water density averaged over the entire surface, consisting of six surface unit cells, is shown (grey). The number density of the local maxima decreases with increasing distance to the microcline (001) surface. The two water density profiles exhibit five peaks in total, i.e., two more than in the experimental data. The MD calculations show a significantly increased water density near the microcline (001) surface at position A (peak I) and, somewhat less pronounced, at position B (peak II). The water peaks (I) and (II) are superimposed. Subsequently, the water density profile at position A shows two more local maxima, labelled (III) and (V). The distances between the water layers (I), (III) and (V) are 0.24 nm each. In addition, the water density profile at position B contains another local maximum (IV) close to the water layer (III) determined at position A. The distance between the water layers (III) and (IV) is 0.08 nm, which is much smaller than the size of a water molecule. The distance between the two water layers identified in the water density profile at position B is 0.32 nm. Triggered by the fact that the 2D images parallel to the surface are similar in experiment and theory when extracting the images at peak (2) in experiment and peak (III) in theory, we speculate that the first large peak (I) in the MD data is not seen in the experiment, while peak (II) is identified as the shoulder (1) in the repulsive part. This assignment is consistent with the picture that the first clear peaks in the MD data correspond to the first hydration layer. Similar to what has been observed before for other systems,42,43 we expect that we are not able to penetrate this first water layer by AFM. Close to the bound layer, a strong repulsive force is detected, which prevents imaging. Moreover, the weak peak (V) in theory might not be resolved in AFM, i.e., we arrive at three peaks in AFM instead of five peaks as in theory.
3. Conclusions
In conclusion, we have investigated the surface structure and water arrangement on the most stable surface plane of a K-rich, fully ordered feldspar, namely maximum microcline (001). The UHV AFM results reveal a surface termination that is consistent with the α-termination. Residual water in the UHV chamber appears to readily react with the dangling bonds of the as-cleaved surface, resulting in a hydroxyl-terminated surface. This finding is corroborated by DFT calculations, indicating that water adsorbs dissociatively with an energy gain of about 3 eV. MD simulations at the microcline (001)–water interface reveal a well-defined, but complex, lateral structure in the first water layer and a less ordered second water layer. Based on the sub-nanosecond timescale for water exchange both within the first hydration layer and between the first and second hydration layers and the bulk, we conclude that water molecules at the microcline (001)–water interface are highly mobile, compared to other mineral–water interfaces.44,45 Furthermore, we observed that K+ surface ions can go into solution and return to vacant surface sites on a timescale of tens of nanoseconds, which causes surprisingly minor perturbations within the first hydration layer. Based on these simulation results, we do not expect AFM experiments to be able to resolve the ion dynamics at the interface. AFM experiments carried out at the mineral–water interface reveal a periodic structure with two features per surface unit cell. A comparison with MD calculations suggests that the AFM does not penetrate into the first water layer. Instead, the AFM contrast is likely due to the structuring of the water in the second water layer, having less well-defined water positions. This interpretation is consistent with the comparatively weak contrast both in two- and three-dimensional experiments. Based on these results, no indication was found for a favoured lattice match with an ice-like structure, in good agreement with previous literature results.25,26 The molecular-level insights into the microcline (001)–water interface presented in this work constitute a prerequisite to understand not only ice nucleation, but also reactions on feldspar surfaces in general.4. Methods
4.1 Experimental methods
Oriented microcline single crystals were purchased from SurfaceNet, Germany. X-ray diffraction (XRD) data presenting the unit cell dimensions, potassium and sodium contents as well as aluminium occupation for the crystals used here are given in the ESI,† confirming maximum microcline. The crystals were brought into vacuum and degassed at 700 K for several hours. The crystals were cleaved in situ with a tungsten carbide blade to expose a pristine surface parallel to the (001) lattice plane. The samples were annealed for 20 h at 450 K to remove residual surface charges.
For 3D mapping, the z-piezo signal was modulated with a triangular waveform.42 The threshold for the frequency shift was set to 40 kHz and a constant value of 1 Å was used for the oscillation amplitude. After 3D mapping, the deflection sensitivity of the cantilever was determined by recording the deflection while the unexcited cantilever was moved into the sample.
MI-UHV-4b. K and Na share the same position with a ratio of 93
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7. Crystal data for AlK0.935Na0.065O8Si3 (M = 277.30 g mol−1): triclinic, space group P (no. 2), a = 7.2227(13) Å, b = 7.6478(14) Å, c = 7.9029(14) Å, α = 113.221(17)°, β = 104.213(15)°, γ = 103.675(15)°, V = 360.67(12) Å3, Z = 2, T = 100.0(1) K, μ(Mo Kα) = 1.335 mm−1, Dcalc = 2.551 g cm−3, 7178 reflections measured (6.062° ≤ 2Θ ≤ 60.064°), 2542 unique (Rint = 0.0429, Rsigma = 0.0803) which were used in all calculations. The final R1 was 0.0607 for 1990 reflections with I > 2σ(I) and wR2 was 0.1791 for all data.
:7. Crystal data for AlK0.935Na0.065O8Si3 (M = 277.30 g mol−1): triclinic, space group P (no. 2), a = 7.2227(13) Å, b = 7.6478(14) Å, c = 7.9029(14) Å, α = 113.221(17)°, β = 104.213(15)°, γ = 103.675(15)°, V = 360.67(12) Å3, Z = 2, T = 100.0(1) K, μ(Mo Kα) = 1.335 mm−1, Dcalc = 2.551 g cm−3, 7178 reflections measured (6.062° ≤ 2Θ ≤ 60.064°), 2542 unique (Rint = 0.0429, Rsigma = 0.0803) which were used in all calculations. The final R1 was 0.0607 for 1990 reflections with I > 2σ(I) and wR2 was 0.1791 for all data.
Mi-I. K and Na share the same position with a ratio of 93
:7. Crystal data for AlK0.93Na0.07O8Si3 (M = 277.22 g mol−1): triclinic, space group P (no. 2), a = 7.2098(8) Å, b = 7.7022(14) Å, c = 7.8231(15) Å, α = 113.060(18)°, β = 104.062(12)°, γ = 103.900(12)°, V = 359.32(11) Å3, Z = 2, T = 293.70(11) K, μ(Mo Kα) = 1.338 mm−1, Dcalc = 2.562 g cm−3, 6152 reflections measured (6.112° ≤ 2Θ ≤ 60.056°), 2195 unique (Rint = 0.0323, Rsigma = 0.0532) which were used in all calculations. The final R1 was 0.0642 for 1841 reflections with I > 2σ(I) and wR2 was 0.1704 for all data.
4.2 Theoretical methods
Author contributions
T. D. carried out the UHV experiments. F. S. performed the experiments at the mineral–water interface. Both analysed the experimental data with contributions of R. B. and A. K. The DFT calculations were done by A. S. F. MD data were collected by R. V. E. N. and B. R. The first version of the manuscript was written by T. D., F. S., B. R., A. S. F. and A. K. All authors commented on and contributed to the final manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Hans-Georg Stammler for the XRD measurements and for his help in interpreting the XRD data. We gratefully acknowledge the financial support from the DFG through grant KU 1980/18-1. B. R. and R. V. E. N. acknowledge funding from the Academy of Finland Centers of Excellence Program (Grant No. 346368 VILMA). A. S. F. acknowledges funding from the Academy of Finland's Flagship Programme under project no. 318890 and 318891 (Competence Center for Materials Bioeconomy, FinnCERES) and project no. 314862. Computational resources were provided by CSC-IT Centre for Science Ltd, Espoo, Finland and the Aalto Science-IT project.References
- C. Hoose and O. Möhler, Atmos. Chem. Phys., 2012, 12, 9817–9854 CrossRef CAS
.
- B. J. Murray, D. O'Sullivan, J. D. Atkinson and M. E. Webb, Chem. Soc. Rev., 2012, 41, 6519–6554 RSC
- M. B. Baker, Science, 1997, 276, 1072–1078 CrossRef CAS
- M. B. Baker and T. Peter, Nature, 2008, 451, 299–300 CrossRef CAS
- J. Mülmenstädt, O. Sourdeval, J. Delanoë and J. Quaas, Geophys. Res. Lett., 2015, 42, 6502–6509 CrossRef
- L. Eickhoff, M. Bayer-Giraldi, N. Reicher, Y. Rudich and T. Koop, Biogeosciences, 2023, 20, 1–14 CrossRef CAS
- N. Hiranuma, O. Möhler, K. Yamashita, T. Tajiri, A. Saito, A. Kiselev, N. Hoffmann, C. Hoose, E. Jantsch, T. Koop and M. Murakami, Nat. Geosci., 2015, 8, 273–277 CrossRef CAS
- J. D. Atkinson, B. J. Murray, M. T. Woodhouse, T. F. Whale, K. J. Baustian, K. S. Carslaw, S. Dobbie, D. O'Sullivan and T. L. Malkin, Nature, 2013, 498, 355–358 CrossRef CAS PubMed
- A. D. Harrison, T. F. Whale, M. A. Carpenter, M. A. Holden, L. Neve, D. O'Sullivan, J. Vergara Temprado and B. J. Murray, Atmos. Chem. Phys., 2016, 16, 10927–10940 CrossRef CAS
- M. A. Holden, T. F. Whale, M. D. Tarn, D. O'Sullivan, R. D. Walshaw, B. J. Murray, F. C. Meldrum and H. K. Christenson, Sci. Adv., 2019, 5, eaav4316 CrossRef PubMed
- A. Welti, U. Lohmann and Z. A. Kanji, Atmos. Chem. Phys., 2019, 19, 10901–10918 CrossRef CAS
- T. Zolles, J. Burkart, T. Häusler, B. Pummer, R. Hitzenberger and H. Grothe, J. Phys. Chem. A, 2015, 119, 2692–2700 CrossRef CAS PubMed
- T. F. Whale, M. A. Holden, A. N. Kulak, Y.-Y. Kim, F. C. Meldrum, H. K. Christenson and B. J. Murray, Phys. Chem. Chem. Phys., 2017, 19, 31186–31193 RSC
- M. A. Holden, J. M. Campbell, F. C. Meldrum, B. J. Murray and H. K. Christenson, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2022859118 CrossRef CAS PubMed
- T. F. Whale, M. A. Holden, T. W. Wilson, D. O'Sullivan and B. J. Murray, Chem. Sci., 2018, 9, 4142–4151 RSC
- J. Yun, N. Link, A. Kumar, A. Shchukarev, J. Davidson, A. Lam, C. Walters, Y. Xi, J.-F. Boily and A. K. Bertram, ACS Earth Space Chem., 2020, 4, 873–881 CrossRef CAS
- A. Kumar, C. Marcolli, B. Luo and T. Peter, Atmos. Chem. Phys., 2018, 18, 7057–7079 CrossRef CAS
- R. W. Friddle and K. Thürmer, Nanoscale, 2019, 11, 21147–21154 RSC
- J. D. Yakobi-Hancock, L. A. Ladino and J. P. D. Abbatt, Atmos. Chem. Phys., 2013, 13, 11175–11185 CrossRef CAS
- A. Kiselev, F. Bachmann, P. Pedevilla, S. J. Cox, A. Michaelides, D. Gerthsen and T. Leisner, Science, 2017, 355, 367–371 CrossRef CAS PubMed
- E. Pach and A. Verdaguer, J. Phys. Chem. C, 2019, 123, 20998–21004 CrossRef CAS
- A. Peckhaus, A. Kiselev, T. Hiron, M. Ebert and T. Leisner, Atmos. Chem. Phys., 2016, 16, 11477–11496 CrossRef CAS
- P. Pedevilla, S. J. Cox, B. Slater and A. Michaelides, J. Phys. Chem. C, 2016, 120, 6704–6713 CrossRef CAS
- S. Kerisit, C. Liu and E. S. Ilton, Geochim. Cosmochim. Acta, 2008, 72, 1481–1497 CrossRef CAS
- A. Kumar, A. K. Bertram and G. N. Patey, ACS Earth Space Chem., 2021, 5, 2169–2183 CrossRef CAS
- A. Soni and G. N. Patey, J. Chem. Phys., 2019, 150, 214501 CrossRef
- B. Drake and R. Hellmann, Am. Mineral., 1991, 76, 1773–1776 CAS
- R. Hellmann, J. M. Penisson, R. L. Hervig, J. H. Thomassin and M. F. Abrioux, Phys. Chem. Miner., 2003, 30, 192–197 CrossRef CAS
- R. Hellmann, Y. Zhai, E. Robin, N. Findling, S. Mayanna, R. Wirth, A. Schreiber, M. Cabié, Q. Zeng, S. Liu and J. Liu, Chem. Geol., 2021, 569, 120133 CrossRef CAS
- P. Fenter, H. Teng, P. Geissbühler, J. M. Hanchar, K. L. Nagy and N. C. Sturchio, Geochim. Cosmochim. Acta, 2000, 64, 3663–3673 CrossRef CAS
- P. Fenter, L. Cheng, C. Park, Z. Zhang and N. C. Sturchio, Geochim. Cosmochim. Acta, 2003, 67, 4267–4275 CrossRef CAS
- K. Umeda, K. Kobayashi, T. Minato and H. Yamada, Langmuir, 2018, 34, 9114–9121 CrossRef CAS PubMed
- S. W. Bailey, Am. Mineral., 1969, 54, 1540–1545 CAS
-
W. A. Deer, R. A. Howie and J. Zussman, An Introduction to the Rock-Forming Minerals, in MineralogicalSociety of Great Britain and Ireland, 2013 Search PubMed
-
I. Parsons, Feldspars and their Reactions, Springer Science & Business Media, 2012 Search PubMed
- A. Blasi, C. De Pol Blasi and P. F. Zanazzi, Can. Mineral., 1987, 25, 527–537 CAS
- J. J. Finney and S. W. Bailey, Z. Kristallogr. - Cryst. Mater., 1964, 119, 413–436 CrossRef CAS
- F. David, V. Vokhmin and G. Ionova, J. Mol. Liq., 2001, 90, 45–62 CrossRef CAS
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1991, 87, 2995–2999 RSC
-
T. Dickbreder, F. Sabath, L. Höltkemeier, R. Bechstein and A. Kühnle, Beilstein J. Nanotechnol., 2023, vol. 14, pp. 1225–1237 Search PubMed
- M. Watkins and B. Reischl, J. Chem. Phys., 2013, 138, 154703 CrossRef PubMed
- H. Söngen, M. Nalbach, H. Adam and A. Kühnle, Rev. Sci. Instrum., 2016, 87, 063704 CrossRef PubMed
- H. Söngen, A. Silvestri, T. Roshni, S. Klassen, R. Bechstein, P. Raiteri, J. D. Gale and A. Kühnle, J. Phys. Chem. C, 2021, 125, 21670–21677 CrossRef
- B. Reischl, P. Raiteri, J. D. Gale and A. L. Rohl, J. Phys. Chem. C, 2019, 123, 14985–14992 CrossRef CAS
- H. Söngen, C. Marutschke, P. Spijker, E. Holmgren, I. Hermes, R. Bechstein, S. Klassen, J. Tracey, A. S. Foster and A. Kühnle, Langmuir, 2017, 33, 125–129 CrossRef PubMed
- H. Adam, S. Rode, M. Schreiber, K. Kobayashi, H. Yamada and A. Kühnle, Rev. Sci. Instrum., 2014, 85, 023703 CrossRef PubMed
- S. Rode, R. Stark, J. Lübbe, L. Tröger, J. Schütte, K. Umeda, K. Kobayashi, H. Yamada and A. Kühnle, Rev. Sci. Instrum., 2011, 82, 073703 CrossRef CAS PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed
- T. Bučko, S. Lebègue, J. Hafner and J. G. Ángyán, J. Chem. Theory Comput., 2013, 9, 4293–4299 CrossRef
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed
- T. Bučko, S. Lebègue, J. G. Ángyán and J. Hafner, J. Chem. Phys., 2014, 141, 034114 CrossRef PubMed
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed
- R. T. Cygan, J.-J. Liang and A. G. Kalinichev, J. Phys. Chem. B, 2004, 108, 1255–1266 CrossRef CAS
- Y. Wu, H. L. Tepper and G. A. Voth, J. Chem. Phys., 2006, 124, 024503 CrossRef PubMed
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: XRD data, further AFM data obtained under UHV and at the mineral–water interface, and further MD calculations. See DOI: https://doi.org/10.1039/d3nr05585j |
| ‡ These authors contributed equally to this work. |
| § For monoclinic feldspar varieties such as sanidine or orthoclase, the atoms shown here are part of the unit cell. For triclinic varieties, the atom positions change slightly, so the centres of some atoms which were located on the faces of the unit cell are now outside of the unit cell. However, we think that it is easier to recognize some of the patterns in the feldspar structure if these atoms are drawn. Hence, we decided to show them in the structure of triclinic microcline. |
| ¶ The absolute orientation of the [010] direction in the coordinate system of our AFM instrument is (175 ± 2)°. This value deviates slightly from the orientation of (180 ± 2)° specified by the manufacturer. The discrepancy of 1° between the error ranges of both values can be explained with an additional error caused by the alignment of the crystal in the sample holder and the sample holder in the AFM stage. |
| This journal is © The Royal Society of Chemistry 2024 |