
Light switching for product selectivity control in photocatalysis
Bayan G. D.
Peelikuburage
,
Wayde N.
Martens
 and
Eric R.
Waclawik
*
and
Eric R.
Waclawik
*
Centre of Materials Science & School of Chemistry and Physics, Faculty of Science, Queensland University of Technology (QUT), 2 George Street, Brisbane, Queensland 4000, Australia. E-mail: e.waclawik@qut.edu.au
First published on 29th April 2024
Abstract
Artificial switchable catalysis is a new, rapidly expanding field that offers great potential advantages for both homogeneous and heterogeneous catalytic systems. Light irradiation is widely accepted as the best stimulus to artificial switchable chemical systems. In recent years, tremendous progress has been made in the synthesis and application of photo-switchable catalysts that can control when and where bond formation and dissociation take place in reactant molecules. Photo-switchable catalysis is a niche area in current catalysis, on which systematic analysis and reviews are still lacking in the scientific literature, yet it offers many intriguing and versatile applications, particularly in organic synthesis. This review aims to highlight the recent advances in photo-switchable catalyst systems that can result in two different chemical product outcomes and thus achieve a degree of control over organic synthetic reactions. Furthermore, this review evaluates different approaches that have been employed to achieve dynamic control over both the catalytic function and the selectivity of several different types of synthesis reactions, along with the remaining challenges and potential opportunities. Owing to the great diversity of the types of reactions and conditions adopted, a quantitative comparison of efficiencies between considered systems is not the focus of this review, instead the review showcases how insights from successful adopted strategies can help better harness and channel the power of photoswitchability in this new and promising area of catalysis research.
 Bayan G. D. Peelikuburage | Bayan received his bachelor's degree (Honours) in chemistry from the University of Sri Jayewardenepura, Sri Lanka, in 2018. He is also a graduate chemist with a Graduateship in Chemistry Honours degree from the Institute of Chemistry Ceylon. Bayan is currently a PhD student at the Queensland University of Technology studying photoswitchable product selectivity control in organic synthesis processes. His research interests are the synthesis and characterization of nanomaterials and the optimization of nanophotocatalysts for optimum yields and selectivity control. |
 Wayde N. Martens | Wayde Martens earned his doctorate in 2004 and has since garnered over $5 million in research grants. He collaborates with global research institutions, specializing in analytical and industrial chemistry. Dr Martens focuses on synthesizing and analyzing inorganic materials, including clays and minerals, with applications in catalysis, photocatalysis, batteries, and sorbents. His expertise spans various analytical techniques such as vibrational spectroscopy, atomic spectroscopy, electron microscopy, and chemometrics. Currently, Wayde applies his expertise to commercial projects, designing and constructing customized analysis systems for HPA production, membrane systems, and water purification. |
 Eric R. Waclawik | Eric Waclawik studied chemistry at Flinders University and received his doctorate in Physical Chemistry in 1997. He continued studies in the field of high-resolution molecular spectroscopy as a postdoctoral fellow at the University of Toronto and the University of Exeter. In 2003, Eric joined the Queensland University of Technology, where he is Professor of Physical Chemistry and leads a research program involving the controlled growth and deposition of inorganic semiconductor nanocrystals, quantum dots and their applications. His research bridges fields of materials science, spectroscopy and photonics, with an application focus of harnessing plasmonic nanoparticles and semiconductor nanostructures for photocatalysis. |
1. Introduction
Catalysis research, particularly at the industrial level, often involves transformations that yield valuable chemicals, versatile drug candidates, biomolecules or polymers. More than 90% of such chemical manufacturing processes, throughout the world, incorporate catalysts in one form or another.1 Given the scale of the products generated in terms of tonnes of product by chemical industries annually, two scientific challenges need to be overcome in order to achieve a sustainable ‘future Earth’: (i) continuous development and advances in terms of performance while (ii) minimizing adverse impacts on the environment, human and planetary wellbeing.2–4 Chemical transformations have recently experienced a surge in ‘green’ chemical syntheses, with a plethora of achievements made in the fields of ‘green chemical synthesis’ and ‘green engineering’, which lead to better energy efficiency and atom economy with the help of renewable energy sources.5 The optimization of catalytic systems to achieve higher efficiency; better product yields and/or higher chemo-, regio- or stereoselectivity has frequently been a focus of studies in the field of green chemical synthesis.6,7 Photo-switches can potentially alter catalytic activity and selectivity in many chemical processes and thus improve reactions and applications, ranging from relatively straightforward chemical syntheses to the fabrication of more sophisticated polymer microstructures, data storage systems and light-driven molecular motors.8–11 This is well demonstrated through the contributions of Jean-Pierre Sauvage, Fraser Stoddart and Ben Feringa to chemistry through the design and development of molecular machines for which they were awarded the Nobel Prize in 2016.12–14Man-made catalytic systems that take advantage of photo-switchable pathways can potentially draw inspiration from the powerful precedents and mechanisms that developed through natural selection and are present in natural systems. Nature regulates a host of essential functions in living organisms that are controlled by specific molecular units that can respond and trigger changes based on the reception of specific external stimuli. Natural photo responsive processes such as photosynthesis15 and vision16 clearly demonstrate the capacity for using electromagnetic radiation to run complex biological and molecular processes with exquisite levels of proficiency. In typical chemistry batch synthetic processes in contrast, the properties of the catalyst must be changed to achieve a different outcome or to direct a different chemical transformation, and the outcome of such optimized systems is limited to a specific reaction rate and/or selectivity, once the conditions are set.17 Hence, the concept of modulating the level of catalytic activity in response to a simple external stimulus is an appealing concept that would eliminate the need for extra, expensive and sophisticated synthetic steps, which could substantially alter and impact the way catalysis could progress in the future.
Recently, efforts have been directed to exploiting a variety of external stimuli including light,18 temperature,19 pH,20 redox states,21 solvents,22 mechanical forces23 and the presence of cations or anions24,25 that can influence and modulate the intrinsic catalytic performances of catalysts. Many of these stimuli such as mechanical forces, temperature and pH suffer from poor temporal and spatial resolutions, and offer little parallelization capability.26 The best possible spatial resolution for inducing nano scale chemical reactions can be provided using a scanning tunneling microscope (STM) tip.27 However, there are trade-offs that are associated with the STM spatial resolution advantage, and the STM has certain well-known limitations such as poor scalability, low speed and serial processing that are all impediments to using this technique. Light is generally accepted to be a most convenient, attractive and non-invasive stimulus to stimulate reactions, offering the potential to channel reactants along reaction pathways towards particular products with minimal energy input and minimal waste formation. Therefore, light-assistance has great potential for the devising of synthetic organic processes that are green and eco-friendly.5 Light could be an ideal stimulus in some situations, due to its ability to deliver information into chemical systems with very high precision in space and time resolutions. The spatial resolution of light however can be broadened by diffraction, a problem that can sometimes be significantly improved using two-photon processes28 and super resolution techniques.29 The choice of a defined wavelength of light and intensity also allows us to selectively excite and trigger specific photo reactions in a highly controlled manner, with no permanent contaminations of the samples under interest.30,31
Current photocatalytic methods provide a basic understanding of how catalytic performances may be controlled using light as a stimulus, with the achievement of chemo-, regio-, and stereoselectivity into only one desired reaction pathway, while suppressing others.32–34 Taking a step further, the development of catalytic structures with photo-switchable characteristics requires the development of methodologies to fine-tune the surface, electronic or steric features of the active catalyst upon irradiation and to regulate its interaction with substrates.
1.1 Different approaches to harvesting light in catalysis
Essentially three conceptually different approaches are used to achieve artificial, photo responsive catalysis, namely, photocatalysis, photo-activated catalysis and photo-switchable catalysis. Photocatalysis utilizes a pre-catalyst that is in its inactive state, which gets activated via light irradiation to generate a catalytically active photo-excited state. The high-energy excited state that is typically formed in this way is unstable and it subsequently reacts with a substrate while dissipating its excess energy.35 In the second approach, a photo-activated or photocaged catalyst, generally comprising photo-acid/photo-base generators and metal complexes, gets activated following the dissociation of photo-labile ligands. These complexes initially exist in a catalytically inactive state that becomes catalytically active upon light irradiation.36 A photo-switchable catalyst has the capacity to toggle between two or more catalytically active states using reversible photochemical transformations, while in the initial state, the catalyst may be active or inactive.37 These photoswitchable transformations mainly result in two outcomes, either with a difference in the rate at which the catalyst facilitates a given reaction (A → B: k1 ≠ k2), or it may allow the catalyst to switch reaction intermediate pathways, promoting a completely different reaction (C → D) (Fig. 1). This review highlights examples from the latter category of photo-switchable transformations. It is evident that photo-switching is a distinct and different approach, compared to photocatalysis or photo-activated catalysis, which offers advantages through possibilities for control of the catalyst during the process of the reaction, in addition to sustaining the initial catalytic pathway. | ||
| Fig. 1 Mode of operation of a photoswitchable catalyst. | ||
This review on photoswitchable catalysis stands out due to its sole focus on reactions yielding two distinct product outcomes upon photoactivation, offering insights into mechanisms and synthetic chemistry applications. This review comprehensively covers both homogeneous and heterogeneous catalytic systems, catering to a broader audience and providing a holistic view of the field. We have analyzed selectivity differences among catalyst types, elucidating factors influencing selectivity and guiding catalyst design. A focus on mechanistic aspects offers detailed insights into molecular-level interactions and reaction pathways, enhancing theoretical understanding and catalyst development strategies. This review also includes merits and limitations sections, critically assessing the field's current state, highlighting the advantages and challenges of photoswitchable catalysis. By focusing on dual-product reactions, covering diverse catalytic systems, analyzing selectivity, providing mechanistic insights, and offering a critical evaluation, the review advances the understanding of photoswitchable catalysis and its applications, contributing significantly to the field.
1.2 Selectivity control in heterogeneous catalysis
Controlling the features of a macroscopic system through an alteration of microscopic, molecular configurations, stimulated by artificial external stimuli, is an interesting catalytic strategy. This has become the most common means by which photo-switching has been achieved with heterogeneous catalytic pathways. Smart macroscopic catalytic systems could provide many new opportunities in the field of heterogeneous catalysts, in addition to using traditional heterogeneous catalyst dispersions; nonetheless, the literature on such systems is still somewhat scarce and limited.38,39 Nanoparticles have become a popular choice for the fabrication of differential-rate and of different chemical-output photo-switch systems and are highly efficient due to the enormous surface area they provide for the catalytic reactions to take place.40 However, this inherent feature of high surface area/volume ratio also endows nanoparticles with a higher surface energy and they often tend to aggregate due to their instability. Therefore, bare unsupported NPs often need to be passivated with surface structures, or molecule layers, in order to prevent aggregation.41 Most ligand types that are attached to NP surfaces are rigidly bound and have no capability to change their conformation. However, the attachment of some reconfigurable molecular structures, such as azobenzenes when incorporated into the ligand structure on the nanoparticle solid surfaces, can be artificially manipulated. Different configurations of the attached ligand can be optimised to favour different surface reactions. Such examples are almost always associated with ON/OFF switching reactions and alteration of the rate of a specific reaction rather than resulting in two distinct chemical outcomes.42–44 Therefore, over the past 3 decades heterogeneous catalysts have most often utilized photo responsive moieties in order to achieve photo-switching in organic synthetic reactions with respect to change in rates and ON/OFF switching mechanisms. This approach to tuning catalysed organic reaction profiles is underexplored. Besides the utilization of photochromic moieties, other surface phenomena can occur on heterogeneous surfaces such as surface adsorption, plasmonic resonance and photoelectron generation based on light as a stimulus, which appeared to be more popular mechanisms by which the selectivity of a photo-switchable reaction is switched from one product output to another.1.3 Selectivity control in homogeneous catalysis
The focus of most of the reported studies among ON/OFF and differential rate photo-switchable reactions has been directed at homogeneous catalysts, where the goal has usually been to control reaction selectivity via E/Z isomerization of the molecular groups, often using light-sensitive azobenzenes.45–50 However, azobenzenes are seldom employed in homogeneous photo-switchable reactions where two different product outcomes are delivered. Reactions reported generally rely on the E/Z isomerization of non-azo olefins and other modes of photochromism of functional molecules such as the change of enantio configuration of rotary motors and different conformations adapted by molecular helices.51–56 Feringa's research on molecular motors, for which he was awarded the Nobel Prize in Chemistry in 2016, ties-in with the idea of selectivity control.14 The development of molecular motors involves the design of molecules that can undergo controlled movements, often unidirectional rotation. This controlled movement at the molecular level reflects a high degree of selectivity in response to external stimuli, such as light in the case of Feringa's molecular motor. Such systems showcase the potential for achieving precise control over molecular transformations, highlighting the importance of selectivity in catalysis.Photochromism refers to a reversible transformation of a chemical species under light irradiation between two isomeric forms that have different absorption spectra.42 This photoisomerization leads to a change in the properties of the molecule, including its geometry and hence spatial-steric properties and molecule rigidity that can all influence reactivity, changed electronic properties, basicity, etc.57–61 Some remarkable progress has been made via the incorporation of photo responsive units within the structure of the active catalyst. For these catalyst systems, an external stimulus can change their intrinsic catalytic activity, which is subsequently translated to an alteration of the rate of a specific reaction, or to switch it ON/OFF, or generate a completely different product outcome. In a few reported photo-switchable pathways, a change in reaction type results, yielding different product molecules. Interesting and novel mechanisms are described, different from the conventional dependence on the presence of geometric isomerizable moieties. Some of these approaches make use of photoexcitation of homogeneous catalyst species where a variable light-factor, typically the wavelength (photon energy) or intensity (photon number), is used to achieve the switchability.31,62,63
2. Photo-switchable organic reactions
2.1. Heterogeneous catalysis
2.1.1.1. Semiconductor catalysts. Besides dual wavelength light switching (DWLS) effects, titania-based systems can yield different product distributions with a single-wavelength light source, based on the temperature at which the reaction is performed. The evaluation of different product distributions upon varying reaction temperatures shows that thermal effects can strongly influence photo catalytic reactions when there is a competition between thermal desorption and secondary photochemical reactions, for example.64 Thermodynamics determines the rapid conversion of tert-butanol into isobutene by water elimination via a bimolecular elimination (E2) mechanism. However, another reaction pathway for tert-butanol with the reduced TiO2(110) surface was observed by Walenta et al., giving a different product selectivity under photochemical conditions.65 It was observed that when the crystal facet (110) of a reduced n-type TiO2 (r-TiO2) was illuminated with UV light in the presence of tert-butanol at 100 K, the photo generated holes on r-TiO2 induced C–C bond cleavage with the ejection of a methyl radical, to yield acetone (Fig. 2). This observation has been reported with several carbonyls, but has not been observed with alcoholic compounds.66 Therefore, this novel reaction approach represents a deviation from the usual α-H abstraction process, which is commonly shown by alcohols under photo chemical conditions.
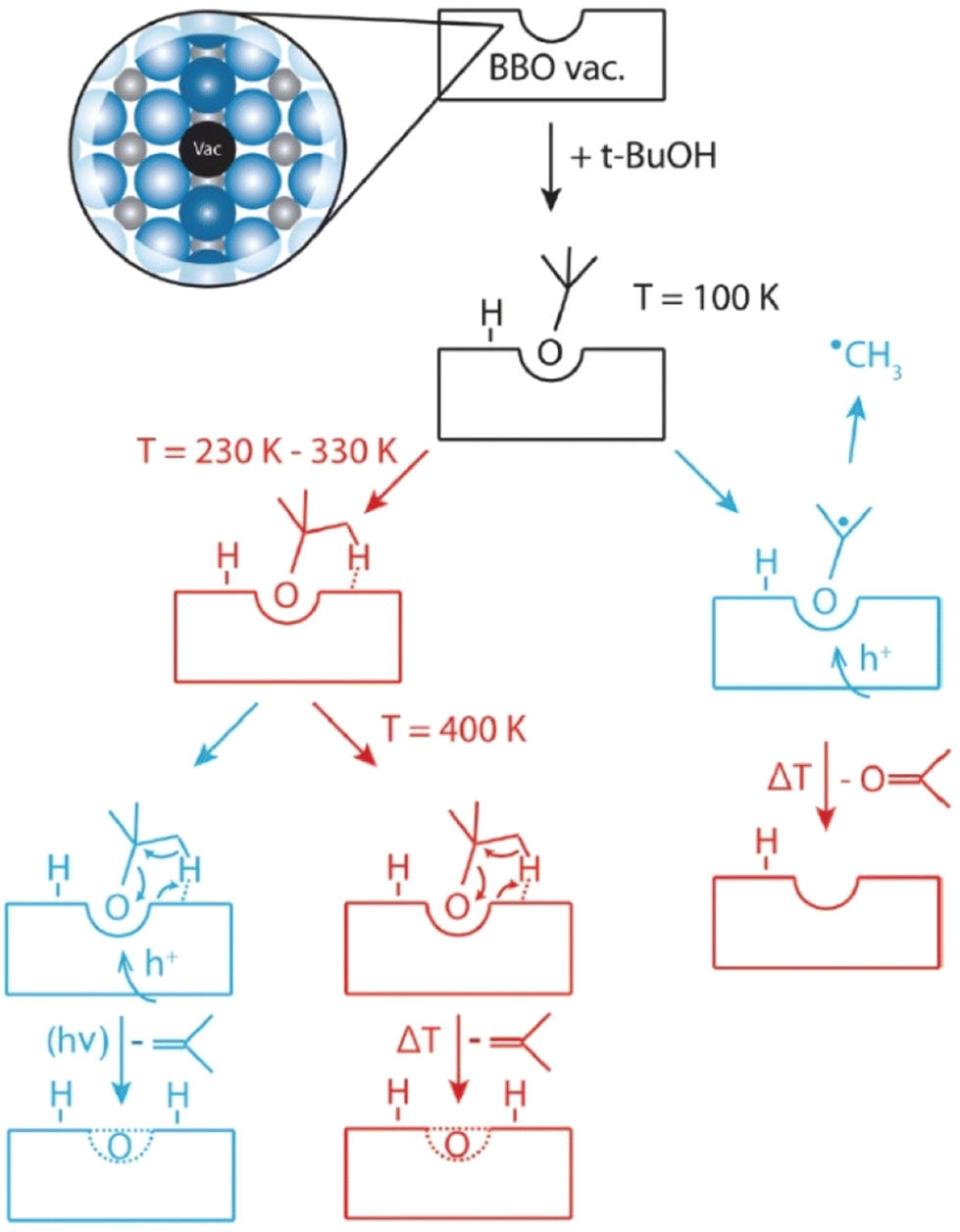 | ||
| Fig. 2 Reaction mechanism that occurs on the r-TiO2(110) surface. Photochemical pathways are indicated in blue and thermocatalytic steps in red. “Reproduced from ref. 65 with permission from the Royal Society of Chemistry, copyright 2018”. | ||
Efficient oxidation catalysis of organics by titania is widely used for the decomposition of organic pollutants.67–69 The TiO2 rutile (110) surface is frequently used for the oxidation of primary and secondary alcohols such as methanol,70 ethanol71 and 2-propanol,72 which occurs via α-H abstraction of their corresponding alkoxy species. However, tertiary alcohols such as tert-butanol have been somewhat neglected in this area because they are usually resistant towards oxidation.73 Previous findings confirm that alkoxy species act as photo active elements in photo reactions of alcohols and are generated mostly on defects.74,75 In accordance with this, scanning tunnelling microscopy (STM) revealed that the formation of butoxy radicals occur mainly at bridge-bonding oxygen (BBO) defects in the r-TiO2 lattice. Even though the ejection of a methyl radical is observed accompanying the formation of acetone as the only photo reaction pathway under cryogenic conditions (100 K), the thermal reaction at the same temperature exclusively produces isobutene as the reaction product. When the photo chemical reaction is carried out at 293 K instead of 100 K, the selectivity of the reaction changes from pure acetone to a mixture of acetone and isobutene. Even if the ejection of methyl radicals to give acetone under UV irradiation represents a photo induced reaction, the efficiency of acetone formation is greatly enhanced at 293 K compared to 100 K, implying that a thermally regulated reaction step might also be involved in the mechanism at a higher temperature. Data obtained at 239 K give a nearly equal composition of acetone and isobutene. When the temperature is increased from 239 K to 293 K, the percentage yield of acetone rises sharply with temperature (from nearly 50% at 239 K to over 80% at 294 K), while isobutene production shows only a slight rate increase (Fig. 3a and b).
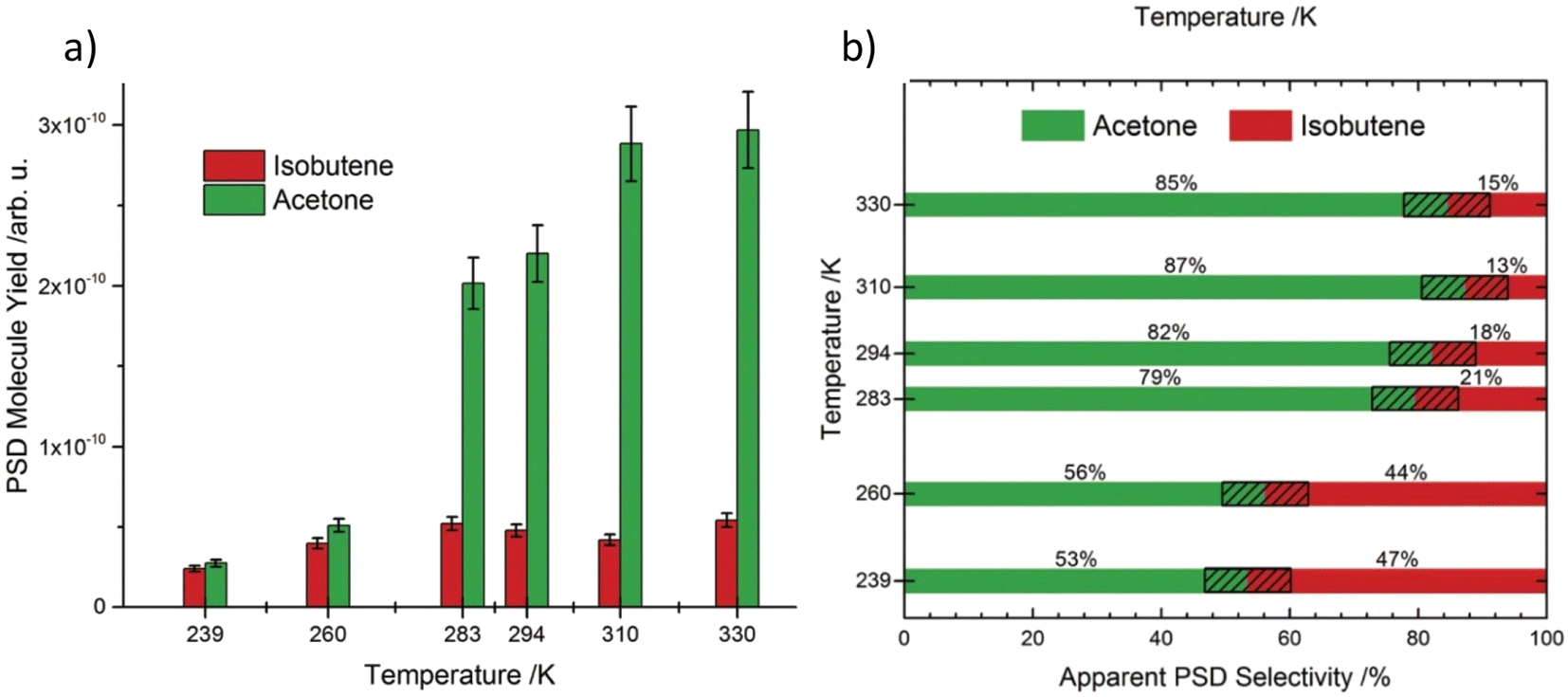 | ||
| Fig. 3 (a) Integrated molecule yields indicating a higher overall yield at higher temperatures. (b) Selectivity of the products obtained at different temperatures. “Reproduced from ref. 65 with permission from Royal Society of Chemistry, copyright 2018”. | ||
Isobutene desorption is thought to readily take place after its formation, but the involvement of a cyclic transition state with a BBO-defect site makes the reaction dependent on temperature. Under UV illuminated conditions, the charge redistribution that takes place makes the reaction feasible, making isobutene one of the products obtained under photo chemical conditions at elevated temperatures. Hence, it is quite evident that both dehydration and dehydrogenation reaction pathways take place in a simple TiO2 catalytic system, displaying rather complex thermal and photo chemical effects that govern the ultimate product selectivity of the chemical transformation. For the same photochemical reaction wavelength, the product distribution can therefore be shifted from dehydration to dehydrogenation using a simple change of temperature.
2.1.1.2. Metal–semiconductor catalysts. In 1901, Ullmann described the first reported carbon–carbon coupling of aryl halides to form symmetrical biaryl compounds.76 The importance of the Ullmann reaction is reflected in the many pharmacological systems that rely upon it as a synthetic step, particularly in the synthesis of non-symmetrical biaryl compounds, due to their high synthetic value. In this direction, Marina, Lanterna and Scaiano described a hybrid Pd@TiO2 heterogeneous photo-switchable system that imparted wavelength-selective reactivity over product formation in the Ullmann synthesis by irradiation with two different color light sources, simultaneously.77 The excitation of Pd@TiO2 with UV radiation catalyzes the Ullmann coupling between two 4-iodoanisole molecules, while the irradiation with a combination of both UV and blue light induces the formation of a cross-coupled product between 4-iodoanisole and 4-iodobenzoate. This hybrid system harvests visible radiation with Pd NPs and UV radiation through the TiO2 semiconductor structures, respectively. Hence, a combination of both materials promotes different reactivities, based on the applied incident wavelength of irradiation. Both UV and visible radiation are required to achieve improvements in cross-coupling yields (100% conversion in 1 h). UV light alone initiates the cross-coupling reaction, but visible light component is required when wanting to achieve better selectivity towards the cross-coupled product. It is interesting to note that the same dose of blue light alone yields only a small amount of cross-coupled product, even after 24 hours of irradiation, even though plasmonic light absorption by Pd can occur for this wavelength.
After extensive screening experiments, the Pd@TiO2 catalyst reaction was optimised using a Cs2CO3 base, THF solvent and Ar atmosphere to achieve the best yields. Cs2CO3 appeared to favor the formation of coupling products with only a little formation of the reduced product 5 (Fig. 4). Tetrahydrofuran (THF) also played a crucial role, both as a solvent and as a participant in the catalyzed reaction. No reaction occurred without it. The reaction is believed to be initiated by electron–hole pair formation upon UV irradiation of TiO2, where holes generated in the valence band are initially trapped by THF, giving THF˙ and a proton which is scavenged by the base (Cs2CO3). THF˙ then transfers its high energy electron to the aryl halide, producing an aryl radical anion that then loses the iodide and associates with the Pd@TiO2 surface. The oxidative addition of the second aryl halide follows, with the help of THF˙, and the biaryl product forms via reductive elimination.
 | ||
| Fig. 4 Activity of the Pd@TiO2 catalyst towards Ullmann coupling under UV irradiation and cross coupling under a combination of UV and blue light. “Reproduced from ref. 77 with permission from the American Chemical Society, copyright 2018”. | ||
Zhu, Wu and co-workers have demonstrated from previous work the capacity of Pd NPs to induce hot-electron processes under light irradiation.78 Photons carrying higher energy than the energy of the lowest unoccupied molecular orbitals (LUMO orbitals) of reactants adsorbed to the photocatalyst surface can initiate photoreactions, and hence, there exists a photon energy threshold for each photocatalytic reaction. This energy threshold depends on factors such as wavelength, intensity, reactant molecule structure and temperature. In these processes, the characteristics of the photocatalyst along with their band structures also play key roles in the catalytic efficiency. For metals that can support a localised surface plasmon resonance (LSPR) with the incident light wavelength, a large number of hot electrons are generated compared to non-LSPR wavelengths (even high energy and short wavelengths).79 In photocatalytic reactions, hot electrons generated through LSPR absorption can efficiently drive the reaction when the reactant's LUMO energy is low. This results in a close alignment between the action spectra of plasmonic metal nanoparticles and light absorption spectra, with higher AQE values at LSPR wavelengths.
The parameter quantum yield (QY) can be expressed for any deactivation process such as fluorescence, phosphorescence or a chemical reaction that has taken place as a result of a molecular excitation. Quantum yield is expressed as a fraction of the number molecules that have undergone a specific deactivation process relative to the total number of ground of state molecules that have been excited upon shining of light (eqn (1)).79 The denominator of the quantum yield equation, which is the number of molecules excited can be replaced by the number of photons absorbed considering the Stark Einstein law of photochemical equivalence. According to this law which is also known as the principle of quantum activation, in a primary photochemical process each molecule is activated by the absorption of one quantum of radiation (one photon).80 In other words, one molecule requires one photon.
 | (1) |
In practical terms when light is incident on a substance, only a fraction of the incident light will be absorbed. A part of light will be reflected and transmitted and will not play any role in absorption or in the chemical reaction under interest. It should be noted that in a photochemical reaction the actual quantum efficiency should be the internal quantum efficiency, which is calculated from the number of molecules reacted divided by the number of absorbed photons. However it is difficult to determine the accurate amount of photons absorbed by a molecule or a photo-promoter in a dispersed medium because of light scattering and losses. Therefore, an assumption has to be made as to the total number of incident photons that are irradiated will be equal to the number absorbed by the photocatalyst, the reactant molecules, or both. Therefore, the obtained quantum yield is approximate and referred to as the ‘apparent quantum yield’ (AQY) at a specific monochromatic wavelength. Therefore,
 | (2) |
Y (light) and Y(dark) represents the amount of reactants converted into products under light (at a specific wavelength) and dark conditions respectively.
When the reactant has a higher LUMO energy, most hot electrons lack the energy to activate the reaction, leading to a lower AQE at longer wavelengths. In high-energy threshold reactions, shorter-wavelength light is more effective, as many LSPR-generated electrons fall short of the energy needed. In contrast, in low-energy threshold reactions, the AQE at the LSPR peak wavelength dominates due to the surplus of high-energy hot electrons. Unlike plasmonic metals, higher quantum yields for non-plasmonic metals such as Pd, for which the LSPR wavelength lies in the UV region, are always achieved using short-wavelength irradiation and the efficiency of a reaction catalysed by a non-plasmonic metal drops drastically at lower energy, visible light wavelengths.81 Hence, different wavelength irradiations result in different effects and efficiencies in the transfer of photo-generated hot electrons to LUMO orbitals of substrates and for the specific reaction of Ullmann coupling, blue irradiation gives the highest efficiency. The developed catalytic system was reversible and even after 4 reaction cycles, it was observed that the Pd@TiO2 catalyst retained 100% conversion capability and excellent selectivity.
In 2016, Luo et al. were able to simplify the depolymerisation of β-O-4 alcohol functionalities to a one-pot photocatalytic system using a catalyst of Pd nanoparticles supported on both ZnIn2S4 and TiO2 that employed tandem oxidation and hydrogenolysis steps with two light irradiation wavelengths.62 Initially, the hydrogenolysis of the C–O bond in β-O-4 ketone 1b in Fig. 5 was tested at 365 nm with anatase TiO2 (40 nm) and sodium acetate (NaOAc) dissolved in ethanol, in an inert (N2) atmosphere. Ethanol played a dual role, acting as the solvent and as the hydrogen donor, while the NaOAc base enhanced the catalytic activity, playing a vital role in the H+ ion transfer step from ethanol to the adsorbed −OPh species. The (101) crystal facet of anatase TiO2 was determined to be the most exposed and most active crystal facet in the hydrogenolysis.
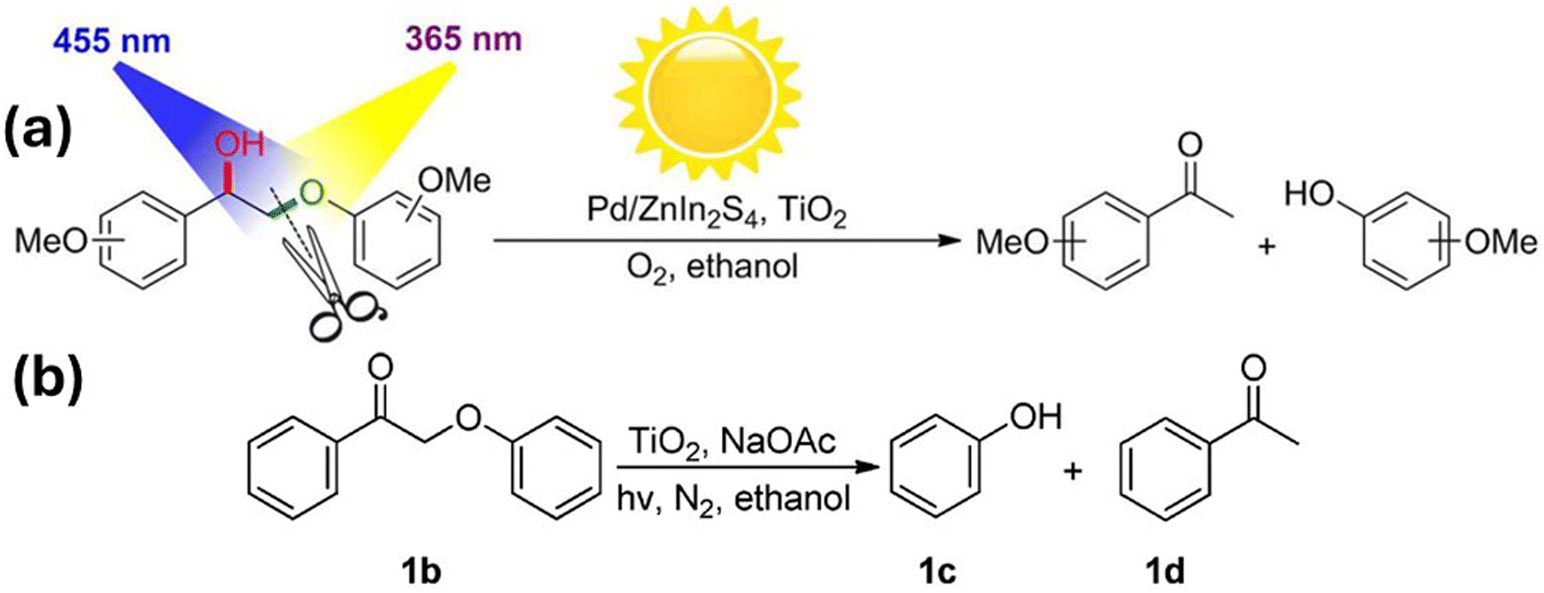 | ||
| Fig. 5 (a) Oxidation–hydrogenolysis reaction via dual-light wavelength switching to cleave β-O-4 alcohols into phenols and ketones. (b) Photocatalytic hydrogenolysis of 1b. “Reproduced from ref. 62 with permission from the American Chemical Society, copyright 2016”. | ||
In the analysis of the mechanistic pathway, protons (H+), electrons (e−) and alkoxy radicals were identified as potentially active species under the adopted reaction conditions. Different additives such as K2S2O7 (electron scavenger), NaC2O4 (h+ trapping agent) and BuOH (˙OH or ˙OEt scavenger) were used to gain insights into individual steps in the reaction mechanism. Through these studies, it was discovered that ˙OEt plays a vital role in the hydrogenolysis pathway and that the reduction is not directly caused by photo generated electrons on the TiO2 surface. It was postulated that 365 nm irradiation photogenerated electrons in TiO2 initially converted Ti4+ to Ti3+in situ and that the electrons were then transferred from Ti3+ to 1b, weakening the C–O bond and causing its cleavage. The authors further showed that the oxidation of α-C-OH to a ketone group can be achieved photocatalytically using irradiation of 455 nm LED on the Pd/ZnIn2S4 catalyst.
Lignin occupies a key place among renewable aromatic sources because it is considered to be a highly abundant, natural-product aromatic polymer.82 In order to depolymerize lignin, many synthetic pathways have been developed, among which the oxidation of α-C-OH in β-O-4 alcohols to α-C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O followed by the cleavage of C–O bond through hydrogenolysis has gained great interest. To explore the possibility of one-pot transformation of lignin, two photo catalytic systems (Pd/ZnIn2S4 and TiO2-NaOAc) were combined with no change in the solvent (ethanol) or the reaction atmosphere (O2). Interestingly, it was found that switching the excitation light wavelength from 455 nm to 365 nm converted the initial α-C-OH and C–O functionalities to produce ketones and phenols while leaving all other reaction conditions (ethanol, NaOAc, and O2 atmosphere) unchanged. Upon illumination with 365 nm light, holes and electrons are simultaneously generated and the holes oxidize ethanol to ˙OEt, while the electrons reduce Ti4+ to Ti3+. The initial oxidation step of the alcohol to a carbonyl reduces the bond energy of the nearby C–O group by nearly 13 kcal mol−1, facilitating its easy dissociation.83 Interaction between 1b onto the in situ-generated Ti3+ weakens the C–O bond, causing the cleavage of 1b. Therefore, the first step in the process appears to be the oxidation of β-O-4 alcohols to β-O-4-ketones on Pd/ZnIn2S4 and the second step, the hydrogenolysis cleavage of C–O bond over TiO2, can be triggered by switching the initial 455 nm light to 365 nm (dual-light wavelength switching – DLWS) or with simultaneous irradiation of both 455 nm and 365 nm radiations (dual-light wavelength co-existence). Even though the discovered photo synthetic protocol has some practical limitations when applied to actual lignin depolymerisations in batch reactors, the immobilization of the catalyst on a flow reactor could be a way to overcome the drawbacks.
O followed by the cleavage of C–O bond through hydrogenolysis has gained great interest. To explore the possibility of one-pot transformation of lignin, two photo catalytic systems (Pd/ZnIn2S4 and TiO2-NaOAc) were combined with no change in the solvent (ethanol) or the reaction atmosphere (O2). Interestingly, it was found that switching the excitation light wavelength from 455 nm to 365 nm converted the initial α-C-OH and C–O functionalities to produce ketones and phenols while leaving all other reaction conditions (ethanol, NaOAc, and O2 atmosphere) unchanged. Upon illumination with 365 nm light, holes and electrons are simultaneously generated and the holes oxidize ethanol to ˙OEt, while the electrons reduce Ti4+ to Ti3+. The initial oxidation step of the alcohol to a carbonyl reduces the bond energy of the nearby C–O group by nearly 13 kcal mol−1, facilitating its easy dissociation.83 Interaction between 1b onto the in situ-generated Ti3+ weakens the C–O bond, causing the cleavage of 1b. Therefore, the first step in the process appears to be the oxidation of β-O-4 alcohols to β-O-4-ketones on Pd/ZnIn2S4 and the second step, the hydrogenolysis cleavage of C–O bond over TiO2, can be triggered by switching the initial 455 nm light to 365 nm (dual-light wavelength switching – DLWS) or with simultaneous irradiation of both 455 nm and 365 nm radiations (dual-light wavelength co-existence). Even though the discovered photo synthetic protocol has some practical limitations when applied to actual lignin depolymerisations in batch reactors, the immobilization of the catalyst on a flow reactor could be a way to overcome the drawbacks.
2.1.2.1. Plasmonic metal catalysts. While the manipulation of plasmonic resonance in metal NPs such as gold is a well-established field, achieving dynamic tuning of plasmonic coloration is constrained by existing methodologies. Addressing this limitation, Li et al. explored a novel approach – the wavelength-switched photoredox method – enabling bidirectional control of LSPR in crystalline gold nanoparticles.84 This method involves reversible surface-plasmon-resonance-based colour changes, which presents new avenues for photonic applications. The reversible control of plasmonic resonance was realized by alternately illuminating nanoparticles with UV and near-UV-Vis light. This induced photoreduction of gold ions and then photooxidation of gold nanorods. The capacity to adjust the plasmon resonance peak of gold nanorods between 630 and 660 nm makes this a versatile technique, as demonstrated through reversible colour pattern creation via mask illumination of a gold nanorod solution. The bidirectional colour tuning process hinges on the photoreduction of gold ions, giving rise to coloured gold nanorods (AuNRs), and the subsequent photooxidation of AuNRs to yield colourless gold ions. This wavelength-dependent process was meticulously controlled, with incident light switching between photooxidation and photoreduction conditions, thereby dictating the colouration process.
Gold nanorods have two oscillation modes: a transverse surface plasmon resonance peak (TSPRP) and a longitudinal surface plasmon resonance peak (LSPRP). The LSPRP is key to altering the frequency of LSPR and achieving diverse colours, and can be adjusted from the visible to the infrared range by manipulating the aspect ratio of AuNRs. Specific photoredox conditions facilitate bidirectional tuning of the LSPRP, involving photoreduction reactions inducing electron donors and elongation of AuNRs. Subsequently, photooxidation occurs when the exposure wavelength changes, leading to the shortening of AuNRs. The process of tuning the LSPRP involves red-shifts via photoreduction and blue-shifts through a photooxidation-based etching method. Photooxidation, reliant on strong coupling between AuNRs and light at the SPR frequency, is achieved by exposing the AuNR solution to visible light of low intensity (Fig. 6). This work emphasizes the indispensable role of visible light in initiating photooxidation and the significance of surface plasmon resonance excitation by visible light in the overall process. The approach also illuminates the transformative potential of reversible tuning of LSPR in nanoscale gold particles, offering insights that can shape the future of photonic technologies such as active-substrate-based surface-enhanced Raman spectroscopy, erasable optical data storage, and dynamic laser colour printing (Fig. 7).
 | ||
| Fig. 6 Photooxidation-induced modulation of SPR by varying the exposure time with gold nanorod (AuNR) solutions under the light of wavelength λ2. (a) Extinction spectra recorded at different irradiation times: 0 min (curve 1), 90 min (curve 2), and 180 min (curve 3). The observed blue-shift of the longitudinal surface plasmon resonance peak (LSPRP) and the reduction in intensity are indicative of the oxidation process leading to the shortening of AuNRs. Transmission electron microscopy (TEM) images of AuNRs captured before oxidation (b) and after 180 min of oxidation (c). Distribution histograms of AuNRs corresponding to (b) and (c) are presented in (d) and (e). For (d), the mean length (ML) and mean diameter (MD) of AuNRs are 33.8 ± 3.3 nm and 9.5 ± 1.1 nm, respectively. In contrast, for (e), the ML and MD are 23.7 ± 3.5 nm and 9.4 ± 1.4 nm, respectively. Scale bars in the insets of (b) and (c) indicate 10 nm. “Reproduced from ref. 84 with permission from the Royal Society of Chemistry, copyright 2018”. | ||
 | ||
| Fig. 7 Periodically adjusting the longitudinal surface plasmon resonance peak (LSPRP) of gold nanorods (AuNRs) by altering the wavelengths of the irradiating light between λ1 and λ2. (a) Cyclic tuning of LSPRP between two distinct values: LSPRP = 630 nm (states “A”, “C”, and “E”) and LSPRP = 660 nm (states “B” and “D”). (b and c) Transmission electron microscopy (TEM) images corresponding to states “B” and “C”. (d and e) Distribution histograms of AuNRs for these states. The mean length (ML) of AuNRs for (b) is 51.7 ± 9.7 nm, and for (c), it is 45.3 ± 8.5 nm. Scale bars in both (b) and (c) indicate 100 nm. (f) Optical field distribution of AuNRs for states “A”, “B”, “C”, and “D.” The aspect ratios (ARs) of AuNRs for LSPRP in states “B” and “C” are 2.5 and 2.2, respectively. The scale bar represents 10 nm. (g) Extinction spectra corresponding to states “B”, “C”, and “D.” Experimental extinction spectra (black curves) are peak-differentiated to fit Gaussian curves (red curves for LSPRP and green curves for transverse SPR). The transverse SPR (TSPRP) and LSPRP for states “B”, “C”, and “D” are 530 nm and 660 nm, 527 nm and 630 nm, and 530 nm and 660 nm, respectively. “Reproduced from ref. 84 with permission from the Royal Society of Chemistry, copyright 2018”. | ||
Much recently, Xiao and co-workers reported a photo-switchable, solvent-free system that could tune reaction product selectivity.85 A composite catalyst consisting of Au nanorod-decorated TiO2(B) nanofibers catalyzed the direct oxidation of benzylamine to benzaldoxime under dark conditions with over 50% selectivity. Upon irradiation, the reaction pathway switched to oxidative coupling, forming imine via the generation of superoxide radical species (O2˙−). The LSPR light absorption peak around 525 nm of isolated gold nanoparticles (Au NPs) in conventional solvents has only slight wavelength variations based on particle size. Gold nanorods (Au NRs) have two absorption peak maxima: the transversal LSPR peak around 525 nm and a stronger longitudinal LSPR peak at longer wavelengths that depends on the nanorod's aspect ratio. The aspect ratio strongly influences the longitudinal plasmon absorption, allowing easy adjustment from visible to near-infrared light ranges. Au nanorods with longitudinal absorption in the visible-near IR light range were fabricated by Xiao and coworkers and loaded onto TiO2(B) nanofibers by an electrostatic interaction-annealing method.
In the experiments using TiO2(B) nanofiber supports with benzylamine, the yield of imine with Au NRs/TiO2(B) was slightly higher than that with TiO2(B) under visible light, but significantly greater under visible-near IR light. The supported Au NRs exhibited higher imine selectivity than TiO2(B) under visible-near IR light, while the turnover frequency based on the Au surface area for Au NRs/TiO2(B) was notably higher than that for Au nanoparticles on TiO2(B) under both visible and visible-near IR light. The selectivity of the coupling product for Au nanorods was linked to their extended light absorption observed in the UV-vis spectra and the influence of light on selectivity was evident from control experiments, with increased yield and selectivity of benzylidenebenzylamine occurring when the light was on, and an increase in the yield and selectivity of benzaldoxime when the light was off. The reaction pathway was found to shift from direct oxidation to oxidative coupling with benzylamine, depending on the presence or absence of light.
According to the analysis, upon illumination, plasmonic-generated hot electrons transferred from the Au nanorods (AuNRs) to the TiO2 support, activating O2 molecules to form O2˙− radical species. The Au nano rods that are left electro positive could generate benzylamine radical cations via abstraction of an electron from a benzylamine molecule. The O2˙− present and the benzylamine radical cation then reacted to form a benzaldimine intermediate, which subsequently reacted with another benzylamine molecule to give the homo-coupled imine. White-light irradiation of AuNR-loaded TiO2(B) nanofibers and AuNP-loaded TiO2(B) nanofibers for similar noble metal loadings revealed that light could switch the reaction pathway from benzylamine-to-benzaldoxime (direct oxidation) to benzylamine-to-benzylidenebenzylamine (oxidative coupling) in both cases (Fig. 8).
 | ||
| Fig. 8 Switch in the selectivity for oxidation products of benzylamine for benzaldoxime in the dark and imine under light irradiation. “Reproduced from ref. 85 with permission from Elsevier, copyright 2020”. | ||
Glaser homocoupling using a copper catalyst was discovered in 1869.86 The co-addition Pd to Cu species to promote the cross coupling of acetylenes and vinyl or aryl halides was uncovered by Sonogashira and Hagihara in 1975.87 To date, using a single catalyst system by which these two distinct reaction types can be effectively controlled, with high yield, is limited. Metal catalysts with Pd and Au have been extensively studied for the Sonogashira reaction, while the other inexpensive transition metals such as Fe, Cu and Ni have gained interest in the field.88 Zhao and co-workers reported the first efficient heterogeneous catalytic system for controlling the Glaser and Sonogashira coupling processes with CuxO nanoparticles on carbon nanotubes with high yield and selectivity.89 The supported CuxO NPs catalysed Sonogashira cross coupling between phenylacetalene and isobenzene under visible light irradiation, while the Glaser homocoupling product of phenylacetalene predominated under dark conditions, thus demonstrating a remote switching of the reaction pathway with light as an external stimulus.
CuxO nanoparticles synthesised via the impregnation method with a particle size distribution of 1–3 nm were used. Reactions were conducted in DMF with a K2CO3 base and under H2/Ar atmosphere, because O2 was found to inhibit the yield of Sonogashira product over the CuxO/CNT catalyst. The CNTs are visible light absorbers, which were thought to facilitate the reaction. When semi-conductors such as TiO2 with similar size distribution to the CNTs were employed with the active Cu catalyst, the reaction rate was much reduced. Under visible light irradiation, aromatic iodides adsorbed well on the surface of CuxO nanoparticles of the CuxO/CNT catalyst and hence the adsorbed aryl iodide together with the light-excited copper(I) phenylacetylide intermediate gave high Sonogashira product selectivity (Fig. 9). Under dark conditions, a dimer complex based on Cu(II) and Cu(I) acetylides was proposed and the synergistic effect between the dimer complexes was shown to prefer the Glaser homo diyne formation.
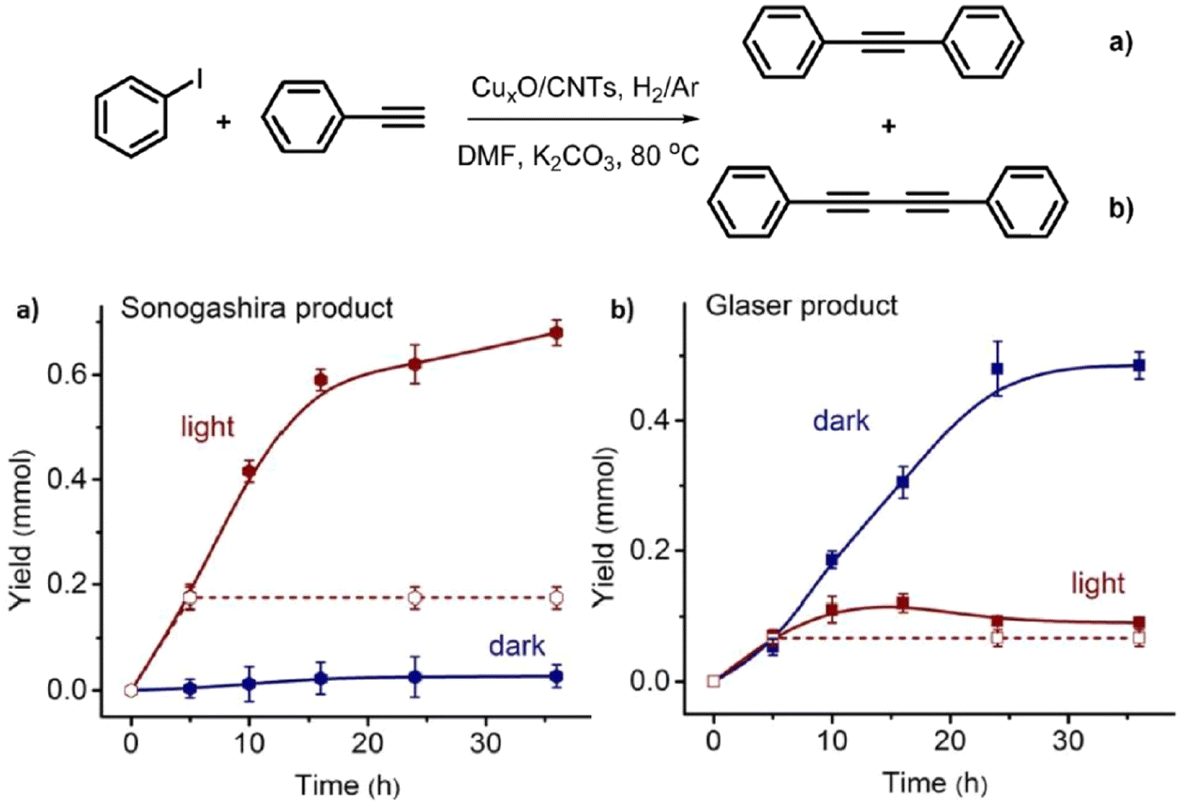 | ||
| Fig. 9 Yield vs. time graphs of (a) Sonogashira and (b) Glaser reactions under visible light irradiation and dark conditions. 1.0 mmol phenylacetylene, 1.5 mmol iodobenzene, 1.5 mmol K2CO3, 2 mL DMF, 100 mg CuxO/CNTs, 0.75 W cm−2, 80 °C, H2/Ar. “Reproduced from ref. 89 with permission from Elsevier, copyright 2022”. | ||
The presence of base (K2CO3) is responsible for the formation of copper(I) phenylacetylide and Cu(II)-CuS dimeric acetylide intermediates and found to have no any influence on the subsequent reaction steps of Sonogashira coupling under visible light or the Glaser coupling under dark conditions. As shown in Fig. 10 under visible light irradiation, Cu(I) phenylacetylide was photo-excited via LMCT to give an electron deficient acetylene, which reacted with iodobenzene on CuxO through its phenyl ring and C–I bond. The Sonogashira product formed with the addition of the aryl iodide intermediate and the excited Cu(I) phenylacetylide under light irradiation. Under dark conditions, the Glaser product was generated with the reaction of the Cu(II)–Cu(I) dimeric complex with a second phenylacetylene. The formation of HI was observed under illumination but was not observed in the darkness. Different aryl iodides carrying para-substituted electron-donating groups were found to give similar yields as non-substituted substrates in the Sonogashira and Glaser coupling reactions. The Sonogashira cross-coupling was favoured with electron-withdrawing groups such as NO2 because the abstraction of the I atom from aryl iodide over CuxO was facilitated by the deactivation of the aromatic ring.
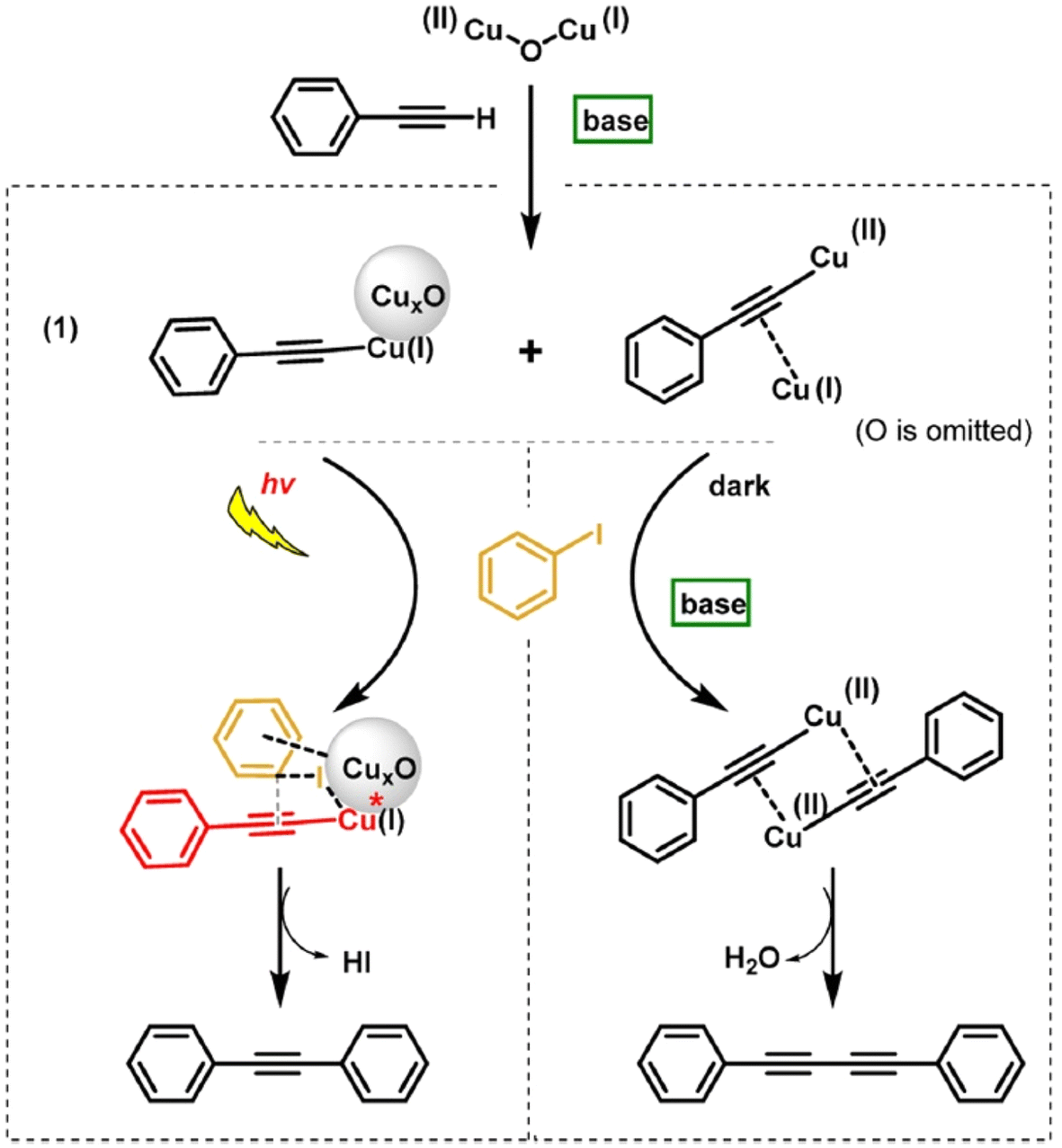 | ||
| Fig. 10 Sonogashira and Glaser coupling reaction pathways over the CuxO/CNT catalyst under light irradiation and dark conditions respectively. “Reproduced from ref. 89 with permission from Elsevier, copyright 2022”. | ||
LSPR-based photocatalysis switching is not exclusively observed in supported plasmonic metal alloy systems. Much recently, we have reported a novel, efficient and base-free approach to switch the Sonogashira and Glaser coupling reaction pathways through wavelength-dependent transmetallation (TM) between Cu and Pd surfaces.90 Based on previous work, it was anticipated that irradiation of this bimetallic system using different wavelength light sources might activate a different reaction pathway for a coupling product through the LSPR absorption of light by plasmonic Cu NPs and the strong light absorption properties of the cuprous phenylacetylide (Ph–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Cu) intermediate. The reaction system was efficiently driven on a mixed catalyst of Pd/Al2O3 and Cu NPs under very mild conditions of 40 °C, without added base or additives to promote the Glaser homo-coupling diyne product at shorter wavelengths (400–500 nm) and the Sonogashira cross-coupled product at longer wavelengths (500–940 nm) with iodobenzene and phenylacetylene as reactants (Fig. 11b).
C–Cu) intermediate. The reaction system was efficiently driven on a mixed catalyst of Pd/Al2O3 and Cu NPs under very mild conditions of 40 °C, without added base or additives to promote the Glaser homo-coupling diyne product at shorter wavelengths (400–500 nm) and the Sonogashira cross-coupled product at longer wavelengths (500–940 nm) with iodobenzene and phenylacetylene as reactants (Fig. 11b).
 | ||
| Fig. 11 Dependence of selectivity of the reaction for Glaser homo coupling and Sonogashira cross coupling products on the irradiation wavelength (a) with the Cu NP catalyst and (b) with the mixed Cu/Pd NP catalyst. (c) Repeated product selectivity switching cycles with blue (445 ± 5 nm) and red (850 ± 5 nm) irradiations. (d) Observed photo-enhanced reactant conversions on irradiation wavelength on Cu Nps and Cu/Pd mixed catalysts (thermal conversion figures of phenylacetylene subtracted). “Reproduced from ref. 90 with permission from John Wiley and Sons, copyright 2022”. | ||
Commercial Cu NPs were thermally treated in a H2 atmosphere at 350 °C for 4 hours to ensure that Cu was in its metallic state and then they were mechanically mixed with Pd/Al2O3 to form the composite catalyst with a Cu/Pd molar ratio of 37/1 to achieve the optimum product outcome. The mixed Cu/Pd NP catalyst was illuminated by white light to yield 61% cross coupling selectivity and 39% homo-coupled product selectivity. Upon irradiation with wavelengths longer than 730 nm, Sonogashira cross-coupled product (diphenylacetylene) occurred with over 80% selectivity. Using light-sources with wavelengths shorter than 455 nm, almost 90% of the Glaser homo-coupled product (diyne) was obtained. This switching pathway was not observed with other metal nanoparticles mixed with Cu NPs, and hence, the switching was selective specifically for the Pd-mixed Cu NP system. The reaction had much lower conversion rates with the mixed Cu/Pd catalyst in the absence of light, a much higher selectivity of 91% for the Glaser homo-coupled product with only Cu NPs and no photocatalytic or thermal activity when only Pd/Al2O3 was employed as the catalyst.
Molecular O2 facilitates the cleavage of the acidic, acetylenic C–H bond and coupling to the surface of Cu, and thereby, phenylacetaldehyde can react with Cu NPs via oxidative addition to give the intermediate Ph–CC–Cu. This intermediate strongly absorbs short-wavelength light (λmax – 465 nm) whereupon this organic Ph–CC– species transmetallates between Pd and Cu surfaces, depending on the wavelength of irradiation.91 When system was irradiated with short wavelengths, a higher local concentration of Ph–CC– species was generated, which suppressed adsorption of iodobenzene at the surface of Pd NPs. This resulted in a higher conversion of phenylacetylene over the iodobenzene, favouring the Glaser homocoupled diyne formation. The activation of aniline on Pd surfaces is believed to take place through the formation of a Ph–Pd–I intermediate. The initially formed Ph–CC–C– of cuprous phenylacetylide shifts to the Pd surface and reacts with Ph–Pd–I via transmetallation to form the complex Ph–CC–Pd, which subsequently reacts with another Ph–CC–Pd to yield the homo diyne Ph–CC–CC–Ph via reductive elimination. The iodine present in the Ph–Pd–I intermediate complex can react also with Ph–CC–Cu to remove iodine in the form of CuI, and this pathway gives rise to the cross coupled Ph–CC–Ph product (Fig. 12).
 | ||
| Fig. 12 Proposed mechanism of the coupling reaction with Cu/Pd mixed NPs. Blue circle depicts the mechanism under short-wavelength irradiation and red circle, under long-wavelength irradiation. “Reproduced from ref. 90 with permission from John Wiley and Sons, copyright 2022”. | ||
A key aspect in this mixed catalytic system is that the highest yield of the cross-coupled Sonogashira product was observed at 560 nm (the LSPR wavelength of the CuNPs), presumably by producing the intermediate cuprous phenylacetylide in higher quantities.92 This intermediate transferred to the Pd surface through transmetallation, to form the cross coupled product, as the coupling of Ph–CC– with Ph– happens more readily at the Pd NP surface than at the Cu NP surface. Upon irradiation with shorter wavelengths, the higher quantity of cuprous phenylacetylide that formed at the Pd NPs suppressed the adsorption of iodobenzene and promotes the formation of the homo coupled product rather than the cross-coupled product. At longer wavelengths (>730 nm) only very low amounts of cuprous phenylacetylide was generated, so a greater proportion of Sonogashira cross-coupling product was observed compared to the Glaser homo-coupling product. This novel catalytic protocol addresses the limitations of Sonogashira coupling such as the use of additives and inert atmospheres and creates opportunities in the field of selective catalysis to fine-tune the fundamental steps in organic reaction mechanisms along different chemical pathways which are switchable by the wavelength of light.
Another switchable, bifunctional heterogeneous catalytic system introduced in 2019 by Pratihar and co-workers employed Cu-doped ZnO. This photocatalyst system used a single active site to drive two distinct catalytic transformations.93 The catalyst was synthesised via exchange between Cu(II) and Na(I) of the Zn–Na-acetate complex followed by hydrothermal treatment in the presence of hydrazine hydrate. Copper-loaded ZnO is responsive to stimuli O2/light and toggles reversibly between its oxidised state of ZnO–CuO (S-2) and the reduced Zn–Cu(0) (S-1) state. The oxidised (S-2) state drives the oxidation of alkylarenes/heteroarenes to aldehydes/ketones with a 7-fold higher rate than S-1, while the reduced (S-1) state promotes the reduction of nitro aromatics to amines with a 6-fold higher rate than that of the S-2, in different reaction environments (Fig. 13). In this comparative study, the oxidation of toluene to benzaldehyde was chosen as the model reaction for the oxidised (S-2) catalytic state, while the reduction of 4-nitrophenol (NP) to 4-aminophenol (AP) was used for the reduced (S-1) state.
 | ||
| Fig. 13 Activity of the switchable bistate catalyst with ZnO-Cu(0) catalysing the oxidation reaction and ZnO–CuO catalysing the reduction reaction. “Reproduced from ref. 93 with permission from the American Chemical Society, copyright 2018”. | ||
Doping ZnO with CuO in S-2 broadened the light absorption response of ZnO, extending it into the visible range while reducing the electron hole pair recombination process. Upon exposure to visible light, the oxidised (S-2) state was reduced to the (S-1) state, facilitated by the self-reduction of CuO to Cu(0), using photo-generated electrons of the adsorbed hydrogen on the catalytic surface. Copper also played a role in the reoxidation of the reduced S-1, via the formation of reactive superoxo (O2−) species from O2, thereby, allowing the catalyst to be reversibly switched between two states, depending on the external stimuli (light/O2) applied.
Reaction samples were collected at different time intervals for the characterization of the catalyst with PXRD, and the results indicated that the transformation of Cu(0) in ZnO-Cu(0) (S-1) into ZnO–CuO (S-2) occurred via the intermediate Cu2O form. This phenomenon was not observed in an inert N2 atmosphere, where the reaction was completely quenched. Insights into the mechanism by which reactive oxygen species were generated in an O2 atmosphere for the reaction were gained through tests with different scavenger molecules. These tests were carried out using selected scavengers, benzoquinone (superoxide radical scavenger) and ammonium oxalate/EDTA (hole scavengers), which caused the yield of the model reaction in acetonitrile to decrease sharply. This indicated that superoxide radicals and photogenerated holes were the key species that drove the selective oxidation of toluene into benzaldehyde, in acetonitrile solvent. In the antagonistic reduction model reaction, the presence of light and an inert (N2) atmosphere were key requirements for the transformation of 4-nitrophenol into 4-aminophenol, where no reaction was observed in the darkness or in the presence of O2 as the reaction atmosphere. Notably, in this catalytic procedure, the intrinsic reactivity of the two states S-2 and S-1 stayed the same irrespective of the sequential order of oxidation or reduction reactions conducted with the catalyst. The generated in situ reactivity of S-1 and S-2 states upon switching the catalyst via an external stimulus essentially resembled the reactivity observed with ex situ generated S-1 and S-2 states, which is also a key factor to note in the proposed catalytic model.
This bistate system was extended towards assisted tandem catalysis involving both oxidation and reduction reactions in a single pot, where the aldehydes generated from the S-1 state undergoes a condensation reaction with the generated amino aromatics through the S-2 state to give the corresponding imine compounds, which eventually get reduced to amines under the experimental conditions employed, giving N-benzylaniline derivatives in good to moderate yields (Fig. 14). In the substrate screening experiments with the oxidation of substituted toluenes to aldehydes, many substituents such as Cl, Br, F, NO2, CHO, CO2Me2, CO2H and OH displayed good functional group tolerance, whereas with groups such as NH2, NMe2, Co2H and several pyridine groups, an inhibiting effect was observed, where no aldehyde was generated as a result. Similarly, the reduction reaction also had good tolerance with Cl, Br, F, I, CO2H, OH and CO2Me groups, whereas the presence of aldehyde and ketone substituents resulted in failed reactions.
 | ||
| Fig. 14 One-pot mechanism for assisted tandem catalysis of S-2 and S-1 states to give N-benzylaniline derivatives with different substituent groups attached. “Reproduced from ref. 93 with permission from the American Chemical Society, copyright 2018”. | ||
Plasmonic metal nano particles with their characteristic LSPR light absorption properties are catalytically active and capable of initiating reactions via the injection of generated hot charges into the LUMO of molecules adsorbed to their surfaces.94,95 Recently, rhodium NPs supported on Al2O3 (Rh/Al2O3) were found to demonstrate a plasmonic switching behaviour toward CO2 reduction giving methane as the exclusive product under mild illumination with blue or ultra violet LEDs, whereas a mixture of methane and CO in equal proportion was produced under dark conditions (Fig. 15).96 In the photo catalytic pathway, blue LEDs showed a selectivity of >86% for CH4, whereas UV LED gave as high as >98% conversion. Both illumination sources produced CH4 at nearly twice the rate as the corresponding thermocatalytic reaction at 350 °C. On heating (thermocatalytic) Rd/Al2O3, CO and CH4 formed at a very low reaction rate in nearly equal proportions. The illumination of unheated catalyst resulted in only a slight increase in CO production.
 | ||
| Fig. 15 Rates of production of CH4 (green) and CO (black) at 623 K on Rh/Al2O3 (solid lines) and Al2O3 (dotted lines) under UV illumination and dark conditions (light intensity – 3 W cm−2). CH4 production and selectively is greatly enhanced by ultraviolet light on Rh/Al2O3. Rates of production of CO (black) and CH4 (green) at 623 K on Au/Al2O3 visible illumination and under dark conditions (light intensity – 3 W cm−2). “Reproduced from ref. 96 with permission from Springer Nature, copyright 2017”. | ||
A comparable and similar experiment performed with a gold (Au) photo catalyst gave a different selectivity with the formation of CO as the main product under illumination (white and ultraviolet) and dark conditions. This shows that the plasmonic effect or the irradiated wavelength do not govern the observed changes in selectivity and that a specific property related to Rd metal and/or Rd metal–adsorbate interactions play a role in the selectivity switch. CO2 is known to dissociatively adsorb on Rd catalysts to give oxygen (O) and adsorbed CO and the latter can be hydrogenated to generate CHO. The dissociation of the CH–O bond can result in a CH fragment, which undergoes further hydrogenation to form the final product CH4. It therefore appears that the competition between the desorption of CO from the Rd surface and the bond dissociation of CH–O to give a CH fragment governs the ultimate selectivity of product formation in the catalytic process. When the desorption of CO is favoured over the CH–O bond cleavage, CO is generated with a higher selectivity over CH4 and vice versa.
The energy of adsorption of oxygen (Eads,O) also plays a role in the thermocatalytic processes where high Eads,O provides energy for the dissociation of the CH–O bond, hence favouring the formation of CH4 over CO. The comparison of Eads,O figures for Rh (5.22 eV) vs. Au (3.25 eV) explains the slight preference of Rh surface towards CH4 production over CO, while the Au surface to exclusively produce CO. The mechanism of the reaction was shown to be mediated via the generation of hot electrons on the plasmonic Rh surface because the dependence of the rate of photo reaction with light intensity was observed to shift from linear to super linear beyond ∼1 W cm−2 light intensity. Therefore, under photocatalytic conditions, the energy of hot electrons and that of antibonding orbitals of the reaction intermediates critically affect the specific electron pathways chosen and this, in turn, is observed to give the described switch in the product selectivity with the Rh/Al2O3 catalyst in its photo chemical vs. thermocatalytic routes.
The photothermal effect is also a fundamental phenomenon in nanoparticle photocatalysis, playing a critical role in driving chemical transformations via the conversion of absorbed light energy into thermal energy.97 In plasmonic nanostructures, such as metal nanoparticles, this process is particularly pronounced due to their unique optical properties, which result in enhanced light absorption and strong electromagnetic field confinement. The coherent electron oscillation resulting from plasmon excitation exhibits a brief lifetime of approximately 5–100 femtoseconds (fs), during which it undergoes three primary decay mechanisms.98 These mechanisms include elastic radiative re-emission of photons, nonradiative Landau damping leading to the excitation of energetic electrons and holes, and chemical interface damping (CID), involving direct electron injection into unpopulated adsorbate acceptor states.99 While all three processes can transfer energy to adsorbates, they operate via distinct mechanisms. Landau damping, occurring approximately 10 fs after plasmon excitation, converts photon energy into single electron/hole pair excitations, which subsequently interact with other electrons via coulombic inelastic scattering, leading to a cascading process of energy spreading. Concurrently, low-energy electrons couple with phonon modes, heating the metal lattice over a timescale of approximately 1 picosecond (ps), followed by dissipation of this heat to the surrounding environment within 10–100 ps.100
Plasmon-mediated nanostructure heating has gained significant interest in the field of photocatalysis due to its potential to facilitate chemical reactions by providing additional thermal energy. The photothermal effect can influence reaction kinetics by lowering activation barriers, thereby accelerating the rate of chemical transformations through the Arrhenius dependence of rate on surface temperature. Additionally, it may affect reaction selectivity by altering the energetics of different reaction pathways. This influence on selectivity arises from changes in the distribution of energetic charge carriers and their interactions with adsorbates. Studies have demonstrated a linear relationship between changes in the surface temperature of plasmonic nanoparticles and illumination intensity.101 Higher light intensities result in increased contributions from the photothermal effect, leading to more significant temperature increases within the nanoparticle system. However, it is crucial to note that the efficacy of the photothermal effect in inducing chemical transformations is contingent upon various factors, including environmental conditions, such as temperature and pressure, as well as the specific reaction mechanism.
In a recent investigation involving the conversion of CO2 and H2 over Au/ZnO catalysts, researchers observed a notable correlation between the rate of CO production and the intensity of laser illumination.102 This correlation exhibited an exponential trend, aligning closely with expectations for a thermally driven process. Notably, ZnO, serving as a semiconductor in this context with a bandgap of approximately 3.3 eV, facilitated visible light absorption primarily through the Au nanostructures utilized. It was observed that a laser intensity of 250 W cm−2 was necessary to initiate any CO2 conversion, which significantly exceeded the typical intensity of solar irradiance by over three orders of magnitude. Hence, under low-intensity illumination, the photothermal effect may have limited efficacy in driving chemical reactions, particularly in temperature-regulated environments. Plasmon-mediated nanostructure heating, even under solar intensity illumination, may result in transient temperature increases of negligible magnitude for driving chemical reactions. This limitation suggests that the utilization of the photothermal effect to induce reactions may require high photon flux, making it less applicable under low-intensity illumination conditions. Moreover, the plasmon heating mechanism does not offer a distinct pathway to control reaction selectivity, resembling external heating methods in its effects on reaction outcomes.100 Furthermore, the interaction between photothermal effects and illumination intensity is complex and multifaceted. While higher light intensities lead to increased contributions from the photothermal effect and subsequent temperature increases within the nanoparticle system, the efficacy of this effect in inducing chemical transformations depends on factors such as the availability of reactants, presence of catalysts, and the overall reaction environment.
In essence, the interaction between light and metal nanoparticles leads to a dual influence: the surface chemistry affects the optical properties of the nanoparticles, while the optical properties, in turn, impact the surface chemistry and heterogeneous chemical reactions occurring on the nanoparticle surface. Understanding the intricate interplay between light exposure, surface charge distribution, and catalytic activity is crucial for elucidating the diverse reaction pathways observed under light and dark conditions.103 We have explained that the disparities in reaction paths stem from variations in reactant adsorption states and the existence forms of active sites on the catalyst surface, which are intrinsically linked to the electronic states of the catalyst. The way reactants bind to the catalyst is strongly influenced by the electronic setup of the catalyst, which can be altered by exposure to light.
Moreover, when light transforms the active sites on the catalyst, it also changes how charges are distributed across the catalyst's surface. These variations in surface charge distribution significantly impact the energetics of reactions including adsorption, desorption, and the actual chemical transformations. The changes in the electronic structure of the nanoparticle induced by light exposure can alter its surface chemistry and charge distribution.104 For example, light-induced transitions between different electronic states or surface defects can modulate the binding affinity of molecules to the nanoparticle surface and hence impact its charge distribution.105 Additionally, the presence of light can influence the chemical environment surrounding the nanoparticle, leading to modifications in surface charge distribution. Photochemical reactions initiated by light can often result in the formation of reactive intermediates or changes in the pH of the solution, which, in turn, can affect the surface charge of the nanoparticle. Hence, variations in light conditions can significantly influence the surface charge distribution on nanoparticles, with implications for their reactivity, stability, and interactions with surrounding environments.
2.1.2.2. Plasmonic metal-alloy catalysts. A report on photo catalytic activity involving plasmonic Au NPs supported on ZrO2 investigated how the hydroamination reaction between alkynes and aniline can give cross-coupled imines.106 Taking this a step further, our group devised a photo-switchable system by incorporating metallic Co into the plasmonic metal NP system as Au2Co bimetallic alloy NPs/ZrO2, suitable for the alkyne hydroamination reaction with aniline.107 The product selectivity of the reaction between phenyl acetylene and aniline was made switchable with a bimetallic alloy plasmonic NP, utilizing light as the external stimulus, giving cross-coupled imine under visible light and homo-coupled 1,4-diphenylbutadiyne under dark conditions.
It has been proposed that the visible light irradiation alters the concentrations of alkyne and aniline at the solid-solution interface, and this change is subsequently translated towards a switch in the reaction pathway (Fig. 16). In the absence of light, alkyne molecules were found to adsorb with twice the surface abundance of aniline on the catalyst surface, which subsequently gave the homo-coupled diyne as the major product. Light irradiation, however, destabilizes the adsorption of alkynes on the catalyst surface, while promoting the adsorption of aniline, giving rise to a high imine yield under illuminated conditions (Fig. 17). The photo-switchable hydroamination was not observed with monometallic Co or Au nanoparticles deposited on ZrO2 and an accelerated hydroamination reaction rate was observed with Au2Co/ZrO2 compared to the rate over unalloyed Au/ZrO2. Au2Co alloy NPs of mean particle size 7 nm used in these experiments adsorbed visible light over a very broad wavelength range, peaking at 520 nm, similar to the absorption spectrum of Au/ZrO2 particles. It was proposed that the selective attraction of aniline to the surface of Au2Co/ZrO2 occurs due to the presence of cobalt in the alloy, in concert with large electromagnetic (EM) fields generated in close proximity to the NPs at plasmonic hotspots, mimicking an optical trap-type force. It was confirmed that the EM hot-spots resulted in an enhanced SERS signal when aniline was adsorbed on Au2Co/ZrO2, much higher than the aniline SERS signal observed on either Au/ZrO2 or ZrO2 alone (Fig. 16b). Therefore, by incorporating Co into the plasmonic alloy nanoparticle, a reaction switch was introduced through an exaggerated, light-induced change in the reactant surface concentrations at the catalyst.
 | ||
| Fig. 16 (a) Adsorption patterns of phenyl acetylene and aniline on the Au2Co/ZrO2 catalyst under irradiation and dark conditions (b) Surface-enhanced Raman spectra of aniline molecules adsorbed on monometallic gold (Au) NPs and gold and cobalt alloy NPs (Au2Co). The vibrations specific to aniline are indicated with asterisks (“*”). (c) Visible-light irradiation induced changes in the reactant concentration on Au2Co NPs, Au NPs and Co NPs in light-on and light-off cycles. “Reproduced from ref. 107 with permission from John Wiley and Sons, copyright 2019”. | ||
 | ||
| Fig. 17 Hydroamination reaction and homo diyne formation with alkyne and aniline. (A) Reactions carried out with Au2Co/ZrO2 under visible light irradiation and (B) in the dark. (C) The effect of temperature under visible light and (D) in the dark. “Reproduced from ref. 107 with permission from John Wiley and Sons, copyright 2019”. | ||
In a similar vein, Peiris et al. have recently used visible light to direct a model phenylene hydrogenation reaction towards a homocoupling path using an alloy AgPd plasmonic NP system supported on ZrO2.108 The mechanism by which this reaction switch was achieved was by plasmon-enhanced H2 desorption, which had the effect of decreasing the concentration of H-species at the heterogeneous catalyst surface.109 The amount of Pd in the alloy NPs needed to be quite low (0.7% molar ratio), to maintain LSPR activity. In the absence of Pd, the reaction did not proceed, and so it was assumed that Pd must have been present at the surface of the alloy NPs in the photocatalyst to make it an active reaction switch. Isopropanol was the solvent system used, a solvent frequently used in proof-of-concept studies of this type, because it is a suitable scavenger of hot-holes.110 In the absence of Ag, or under dark conditions, the hydrogenation of phenylacetylene occurred, producing styrene and ethylbenzene at 70 °C (Fig. 18). Under light conditions, the LSPR component influences effectively ejected surface adsorbed H-atoms on the catalyst as H2, which removed the catalyst's ability to cause hydrogenation, leaving the homocoupling pathway open to produce 1,4-diphenylbutadiyne.
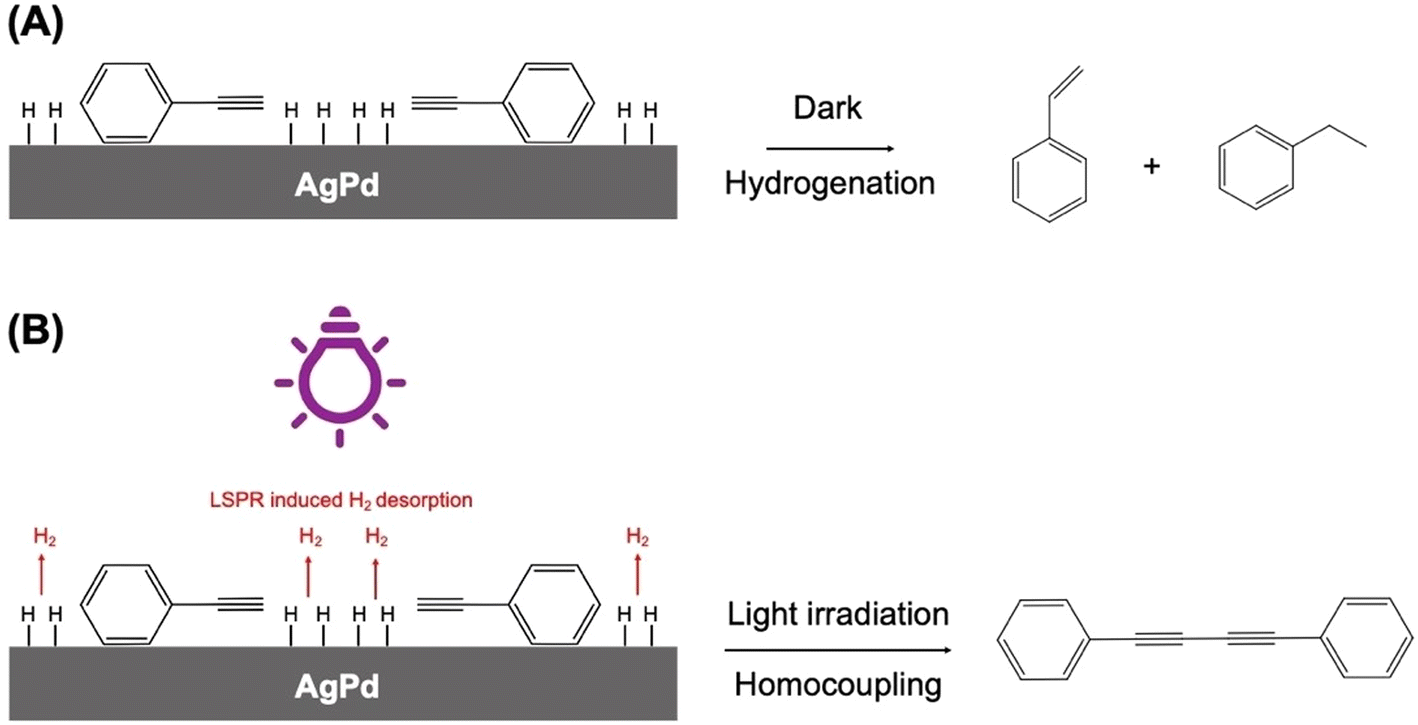 | ||
| Fig. 18 Proposed mechanisms for phenylacetylene conversion by AgPd/ZrO2 NPs. (A) Under dark conditions, conversion follows the hydrogenation pathway in which adsorbed H species are added to surface species, leading to the formation of styrene and ethylbenzene as the main products. (B) Under light irradiation, LSPR hot carriers drive H desorption from the surface, depleting the concentration of H surface species. This changes the reaction pathway to homocoupling and leads to the formation of 1,4-diphenylbutadiyne as the main product. “Reproduced from ref. 108 with permission from John Wiley and Sons, copyright 2023”. | ||
2.2. Homogeneous catalysis
The florescent dye Rh-6G undergoes photo excitation to give Rh-6G*, which has a reduction potential of −0.8 V vs. SCE. In the presence of the e− donor N,N-diisopropylethylamine (DIPEA), under green light irradiation, the excited state Rh-6G* transforms to stable Rd-6G˙− radical anion with a slightly enhanced potential of −1.0 V with respect to SCE (Fig. 19). This radical anion can again be excited upon blue light irradiation to give the excited radical anion (Rd-6G˙−*) that carries an even higher reduction potential value of −2.4 V vs. SCE. This photo catalytic system therefore allows sequential and selective activation of aromatic C–Br bonds towards C–H arylations.
 | ||
| Fig. 19 Chromoselective one-fold (left) and two-fold (right) substitution reactions. “Reproduced from ref. 63 with permission from John Wiley and Sons, copyright 2016”. | ||
When 1,3,5-tribromobenzene was irradiated with a green LED (530 nm), a radical anion formed which subsequently lost its Br− substituent to give an aryl radical. When N-methylpyrrole was introduced to this system, a rapid re-aromatization took place, resulting in mono-substituted products. The reduction potential of Rh-6G (−1.0 V vs. SCE) is insufficient to activate the remaining C–Br bond/s in the substrate, and hence, only single-substituted derivatives were obtained. When the same substrate was irradiated with a blue LED (455 nm), Rh-6G was converted into Rd-6G˙−*, which has a reduction potential of −2.4 V vs. SCE. The high reduction potential of the excited photo redox catalyst led to substitution of the second C–Br bond, forming bi-substituted products. One-pot synthesis of two-fold substitution products was also found to be feasible upon irradiation of blue colour LED from the very start of the reaction. The scope of the reaction could be further extended by introducing two different substituents to the substrate. The scope of the reaction could be extended by introducing two substituents to the substrate, before the wavelength was switched from green-to-blue. Extending the substrate scope further, aryl bromide substrates bearing electron-withdrawing groups were used to substitute allylic substrates with biologically important pyrroles and styrene derivatives.63
The Tsuji–Trost reaction is a powerful and commonly used way to alkylate allylic substrates to obtain the E-isomer of the alkylated product.111 Photocatalytic arylation of hetero-aromatic rings such as pyrroles have been reported and widely studied, but allylations of hetero-aromatics have not been a central focus.112–114 In an attempt to fill this gap in knowledge, a novel photocatalytic chromoselective approach to allylation that used a variety of nucleophiles, such as heteroarenes, amines, and alcohols and which is also compatible with biologically important indole and pyrrole derivatives was reported in 2019.115 The catalytic protocol was run under mild conditions with the merit of being able to switch between the allylated E-isomer under visible irradiation and the Z-allylated product under UV irradiation. The latter had been previously inaccessible (Fig. 20).
 | ||
| Fig. 20 Photocatalytic allylation reaction and Isomerization studies of E-1a with photocatalysts 3e and 3g under different irradiation wavelengths. “Reproduced from ref. 115 with permission from Springer Nature, copyright 2019”. | ||
Different transition metal catalysts and several photoorganocatalysts were used to screen allylation reactions with pyrrole and allylic acetate derivatives, and out of the catalysts tested, only 10-phenyl-10H-phenothiazine (PTH) produced the Z-allylated pyrrole, under light irradiation. CH3CN was identified to be the best solvent for the reaction with two pyrrole equivalents and the optimum wavelength of irradiation was 365 nm, which coincides with the λmax value of the PTH photocatalyst. With the optimized conditions and expansion of the substrate scope, N-methyl pyrrole was found to be a better substrate, giving higher yields than pyrrole with excellent selectivity to the Z-isomer (Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E selectivity up to 96:4).
:E selectivity up to 96:4).
Unsubstituted indoles at the nitrogen atom were also found to give better yields than pyrroles, maintaining a higher selectivity for the Z-isomer. The reaction was feasible with many leaving groups present at the allylic position of the substrate, such as benzoate, carbonate or carbonates. Unreactive hydroxyl groups with very high reduction potentials were not active towards allylation. With the excellent selectivity obtained for the Z-isomer with 10-phenyl-10H-phenothiazine, it was suggested that the irradiation of initial substrate E-1a with visible light could avoid E → Z isomerization. Hence, when phenoxazine (3g), which has a higher reduction potential and absorbs in the visible region, was employed in the reaction, it selectively produced the E-allylated product in good yields under 420 nm irradiation, with minimal E → Z isomerization.
With the phenoxazine photocatalyst, N-substituted pyrroles maintained the same selectivity with good yields. Following a similar pattern to the previous (3e) photo catalyst, unsubstituted as well as substituted enols with methyl, bromo and several electron-donating and -withdrawing groups afforded even better yields and excellent selectivity (up to >98:2) than pyrroles with the phenoxazine (3g) photocatalyst.
It was observed that the metal complex photocatalyst Irsppy, upon blue colour photon irradiation, can either act as a sensitizer in some reactions by a triple–triplet energy transfer mechanism, or as a photoredox catalyst that facilitates one-electron reduction processes. The Irppy triplet state has an energy of 2.65 eV and an oxidation potential of 1.01 V relative to the normal hydrogen electrode (NHE). Upon irradiation of Irsppy with 447 nm light, operating at an intensity of 1 W, two photons can be simultaneously absorbed by the system with the help of a simple inexpensive lens (Fig. 21). The lens collimates the beam to a spot (smaller than 1 mm2) with a much higher light power density and this concentrated beam of light makes it possible to deliver an extra photon to the photoactive catalyst within the lifetime of the first photonic excitation, causing biphotonic (two-photon) ionization.
 | ||
| Fig. 21 Generation of 3Irsppy from Irsppy under one-photon natural laser irradiation (left) and the formation of high-energy eaq˙+ upon two-photon collimated laser beam (right). “Reproduced from ref. 31 with permission from the Royal Society of Chemistry, copyright 2019”. | ||
Interestingly, when a second photon was absorbed by Irsppy during the life time of its triplet excited state, high energy electrons (eaq˙−) with super reducing abilities were generated. These hydrated electrons (eaq˙−) are very aggressive, with almost double the available redox energy when compared with single photon excited 3Irsppy (−2.9 V relative to NHE) and hence they have greater potential and likelihood to cause reduction reactions than the usual triplet–triplet energy transfer mechanisms known for 3Irsppy. Thus, the same substrate is able to undergo completely different one-photon (3Irsppy) and two-photon (eaq˙−) pathways depending on the intensity per unit area of irradiation applied opening novel and fundamentally different photo redox selectivity control mechanisms.
In the first reaction studied, the substrate 4-bromo-2-chloro-5-fluorobenzoic acid and the catalyst Irsppy were used to test the selectivity control approach with triethanolamine (TEOA), which was used both as an electron donor and as a H atom donor in the reaction (Fig. 22).
 | ||
| Fig. 22 Photoswitchable dehalogenations. Selective debromination and subsequent dechlorination carried out with (a) 4-bromo-2-chloro-5-fluorobenzoic acid using the blue-light driven photocatalytic system in Ar-saturated solutions. (b) Selective debromination with one-photon conditions. (c) Debromination followed by dechlorination upon two-photon conditions. “Reproduced from ref. 31 with permission from the Royal Society of Chemistry, copyright 2019”. | ||
From a thermodynamic point of view, C–Cl bonds are more difficult to reduce than C–Br bonds. When the substrate was irradiated with a 447 nm laser for 30 h in an aqueous medium with Irsppy and TEOA, the debromination product (2-chloro-5-fluorobenzoic acid) was observed under 1-photon conditions (without lens). With all other parameters kept constant, when the lens was placed between the lamp and the reaction solution giving two photo conditions, 3-fluorobenzoic acid was observed upon complete conversion of the substrate within 6 h. This shows that the generated eaq˙− under two-photon conditions is powerful enough to initiate both reductive debromination and dechlorination processes, and as a super reductant with very high energy, it can facilitate the reaction over short reaction times.
The same approach of light intensity-dependent selectivity was applied to control cis–trans isomerization and photoreduction of trans-3-fluorocinnamate. The F atom in the molecule allowed convenient detection of the cis isomer as well as the hydrogenated product through 19F NMR spectroscopy. The substrate trans-3-fluorocinnamate underwent trans → cis isomerism upon irradiation with 1-photon absorption by an unaltered laser beam (447 nm), giving 75% of cis and 25% of the trans isomer at the PSS. With the lens in place and under two-photon absorption conditions, the hydrogenation of the olefinic group in the substrate was observed. The hydrogenation was thought to occur by an initial one electron reduction of cinnamate ion by eaq˙− to form a radical dianion, followed by protonation of the dianion and ultimate H atom abstraction of the protonated radical anion from TEOA to form the reduced olefin. The irradiation of a similar system with the fumaric acid (trans) substrate did not undergo trans → cis isomerization under one-photon conditions initially, but with longer reaction times and higher catalyst loadings, a reasonable yield of maleic acid (cis) was obtained. The slow isomerism was attributed to the endothermic TTET transition, as fumarate has a higher triplet energy than 3Irsppy. Two-photon photo reduction was very efficient, with fumaric acid being converted into its reduced substrate in 3 h.
The reduction of benzylhalides proceeds via the formation of benzyl radicals, and this provides an opportunity to utilize the switching capacity of the Irsppy catalyst to produce either toluene by reductive dechlorination or the dimer dibenzyl upon dimerization. Benzyl bromides tend to easily hydrolyse under aqueous conditions, and therefore, 4-chloromethylbenzoate was chosen as a suitable substrate which additionally provided water solubility and stability in basic media. With single-photon irradiation, the conversion of the substrate to the reduced product was very slow and insufficient due to difficulty in the reduction of benzyl chlorides, particularly when compared to their corresponding bromides. In contrast, the two-photon experiment, with a collimated beam, was significantly different and complete conversion of the substrate was achieved after 2 h with a selectivity of 54% for the dibenzyl product, accompanying an overall enhancement of rate by a factor of 3. The findings in this work on selectivity control of a single substrate to give different products by varying the light intensity per area might well encourage more photo-switchable applications in the field of photo-redox catalysis.
Visible light-driven artificial molecular motors have been designed that can both catalyse the activity and control the enantioselectivity of chiral transformations via configurational changes accompanied in a 360° rotary cycle of the motor.10 Seminal work done by Nobel Laureate, Feringa and co-workers on the dynamic control of chirality clearly demonstrates how a photochemically/thermally driven unidirectional rotary cycle be used to achieve different tasks at the molecular level. Feringa's pivotal contribution came in the form of the first molecular motor with an ingenious design of a rotor blade powered by light, and he achieved controlled molecular-scale movement. A significant challenge in the field of enantioselective catalysis is to achieve each enantiomer of the asymmetric product using a single enantiomer of a ligand through switching (dual control), eliminating the need for renewed ligand synthesis.134 However, conventional chiral ligands do not switch their chirality upon thermal or photochemical treatments. Light-driven artificial molecular motors that can act as multi-stage chiral switches, between two pseudoenantiomeric forms, provide a potential platform to achieve this goal.135,136
As shown in Fig. 23b, a typical rotary catalytic system comprises a light/heat-driven molecular motor integrated with a bifunctional organocatalyst.10 In one promising strategy, the motor features overcrowded olefin with a naphthalene or substituted phenyl chromophore, which is linked through a central olefin bridge to an identical (first-generation molecular motor) or a different (second-generation molecular motor) aromatic chromophore.137 The chromophore acts as the rotor, whereas the olefin serves as the axle of the motor. Each of the fragments of the bifunctional organocatalyst, designated A and B, connect to the chromophore and the chromophore is free to rotate along the axle (with respect to the stator) under thermally or photochemically induced steps. This rotation provides different relative helical orientations of the components of bifunctional organocatalysts (A and B), generating sequential chiral catalytic configurations, which dictate the overall activity and stereoselectivity of the system.
 | ||
| Fig. 23 (a) Schematic diagram of the unidirectional molecular motor with A and B representing bifunctional organocatalysts. (b) Structure of the molecule (2R,2′R)-(P,P)-trans-1. “Reproduced from ref. 10 with permission from the American Association for the Advancement of Science, copyright 2011”. | ||
In 2011, Feringa and Wang described a molecular motor decorated with dimethylaminopyridine (DMAP) as the Brønsted base (A) and thiourea as the Brønsted base (B) to catalyse the enantioselective sulfa-Michael addition reaction between 2-methoxythiophenol and cyclohexanone (Fig. 24a).10 In Fig. 24, the molecule motor is a single R enantioisomer, where R indicates the absolute chiral configuration at the chiral methyl substituted indanyl centre, M and P the helicity of the pair of indane groups, whereas trans and cis the relative configuration of the A and B organocatalytic fragments to each other. trans orientation between A and B groups occurs when they lie remotely distant to each other, while cis occurs when both groups are in close proximity. These cis–trans isomers can additionally adopt either M or P helicity; thus, cooperative effects that operate with A and B groups, upon the rotation of the motor, are able to induce a preferred specific chiral sense in the product of the asymmetric addition reaction. The rate of sulfa-Michael addition with (P,P)-trans-1 isomer was very low with only 7% product yield after 15 hours, and resulted in a racemic thiol adduct with no enantioselectivity to either of the enantiomers. This low activity of the trans motor was attributed to the distantly oriented thiourea and DMAP functionalities, which prevented a strong cooperative bifunctional catalysis effect.
 | ||
| Fig. 24 (a) Sulfa-Michael addition reaction. (b) Four-step isomerization stages of the 360° rotary motor.10 “Reproduced from ref. 10 with permission from the American Association for the Advancement of Science, copyright 2011”. | ||
Molecule (P,P)-trans-1 undergoes E–Z photoisomerism to (M,M)-cis-1 isomer upon 312 nm wavelength irradiation at 20 °C, bringing the two catalytic moieties of the bifunctional catalyst to close proximity along with an inversion of the helicity from P to M. This generated an efficient cooperative effect and the sulfa-Michael addition occurred at a faster rate, with enhanced yields (50%). Here, it is noteworthy that (M,M-cis-1) isomer facilitates the selective formation of the S enantiomer of the thiol adduct with an enantiomeric ratio (er) of 75/25 (S/R). A thermal inversion of the helix at 70 °C transforms (M,M)-cis-1 isomer to (P,P)-cis-1, which accelerates the reaction up to 83% yield favouring the opposite enantiomer (R) with an er of 23/77 (S/R). A further photochemical step with 312 nm at −60 °C gives an (M,M)-trans-1 isomer with very low thermal stability, and is followed by thermal isomerization at −10 °C to reset the motor in its original (P,P)-trans-1 configuration. Therefore, with (2R,2′R)-trans-1 as the starting configuration, a rotary cycle that operates clockwise sequentially generates racemic (R,S)-, (S)- and (R)-enantiomers upon completion of its UV and thermal triggered 4-step full rotary cycle. All of these individual steps of the 360° rotation were analysed by 1H NMR and UV-Vis spectroscopy, circular dichroism (CD) and chiral HPLC.
Following this discovery, the same catalyst 1 (Fig. 23b) was examined for the reactions involving nitroalkanes and nitroalkenes such as the Henry reaction.138 However, catalyst 1 was not found to be effective for this class of reactions in terms of both its activity and enantioselectivity. Nonetheless, the same research group attempted to develop a similar catalyst retaining the same motor design but removed the phenyl spacers between the bifunctional catalytic fragments. The removal of spacer groups generated a stronger cooperative effect between the bifunctional active moieties (DMAP and thiourea). This new molecule motor greatly enhanced catalysis of the Henry reaction between nitromethane and ααα-trifluoroketones.138 Catalyst 2 (Fig. 25a) has a photostationary state with a ratio of 92/8 in favour of the cis isomer in the transformation between (R,R)-(P,P)-trans-2 → (R,R)-(M,M)-cis-2. Similar to catalyst 1, the trans isomer of catalyst 2 was not catalytically effective at controlling the enantioselectivity in the Henry reaction between nitromethane and α,α,α-trifluoroketones. The two cis isomers, namely, (M,M)-cis-2 and (P,P)-cis-2 behaved similarly to catalyst 1, but with higher yields (93% for R and 92% for S). This also provides a switchable enantiomeric product composition with an er of 86/14 (S/R) for (M,M)-cis-2 and 29:71 (S/R) for (P,P)-cis-2 isomer at −25 °C in toluene. Therefore, as expected, the same 4-step unidirectional rotary motor was again effective in the control of handedness of the product obtained in an asymmetric chemical transformation.
 | ||
| Fig. 25 (a) Design of catalyst 2 and its enhanced catalytic activity. (b) Henry reaction between nitromethane and α,α,α-trifluoroketones. “Reproduced from ref. 138 with permission from the Royal Society of Chemistry, copyright 2014”. | ||
Bisphosphine ligands bearing a C2 symmetry have also attracted huge attention since the same conceptual model of an unidirectional rotary motor, based on overcrowded alkenes, was extended to palladium-catalysed asymmetric transformations with phosphenes acting as photoresponsive ligands in this system.139 The catalyst (shown in Fig. 26) comprised a motor core, with outer-chiral bisphosphine ligands attached to a crowded alkene, via amide linkers and the structure undergoes the 360° rotary cycle with (P,P)-trans, (M,M)-cis, (P,P)-cis and (M,M)-trans configurations. This generates helical structures with varying spatial distances between pendant phosphine groups that control the chirality of the catalysed reaction at each step of the rotary cycle.
 | ||
| Fig. 26 Schematic diagram of thermally and photochemically induced four isomerization stages of bisphosphine-based molecular rotary motor. “Reproduced from ref. 139 with permission from Springer Nature, copyright 2015”. | ||
An extension of the biphosphine ligand catalyst has also used two different ligands L1 and L2, carrying the same motor design (Fig. 27) but subtly differing through the attachment direction of the amide linker, which has been tested as a potential photoswitchable phosphine ligand system. Photoisomerization of L1 was very weak at λmax = 312 nm (THF, −15 °C), giving only 7% conversion of (P,P)-transL1 to (M,M)-trans-L1 at PSS, while L2 carrying a reversed amide bond gave 93% (M,M)-cis and was found to function as an effective 4 stage molecular motor.
 | ||
| Fig. 27 (a) Photoresponsive phosphine ligands L1 and L2 with different amide linkers. (b) Enantioselective synthesis of (3S,4R)-(3R,4S) oxazolidinones. “Reproduced from ref. 139 with permission from Springer Nature, copyright 2015”. | ||
Out of the available four isomers, only (P,P)-trans-L2, (M,M)-cis-L2 and (P,P)-cisL2 were employed in the model Pd-catalysed desymmetrization of meso-cyclopent-2-en-1,4-diol bis(carbamate) in toluene with Pd2(dba)3 (2.5 mol%) and (R,R)(P,P)-cis-L2 (7.5 mol%). Mimicking the previous examples by Feringa et al., the trans isomer (P,P)-trans-L2 was incapable of the desymmetrization process, but upon photoisomerization at 312 nm to (M,M)-cis-L2, the reaction resulted in 89% yield of oxazolidone giving an er of 11/89 (3S,4R/3R,4S). N,N-Diisopropylethylamine was found to be the best at enhancing the er, up to 6/94 (3S,4R/3R,4S). For optimum conditions (P,P)-trans-L2 resulted in a 65% yield giving a near mixture with an er of 53/47 (3R,4S,/3S,4R). Following the conversion of (P,P)-trans-L2 to (M,M)-cis-L2 at −15 °C, the Pd-complex of L2 was successful at reaching an excellent stereoselectivity with an er of 93/7 (3R,4S/3S,4R). In comparison, upon isomerization to (P,P)-cis-L2, the opposite enantiomer was observed with an excellent er of 6/94 (3R,4S/3S,4R), showing a near ideal product switching behaviour between two helical cis configurations. This clearly demonstrates that the photo responsive chiral phosphine ligands with its photochemical cis–trans isomerization and thermal helix inversions from (M,M)-cis to (P,P)-cis have a huge potential to control the chirality of the Pd-catalysed product formation from almost racemic to nearly pure enantiomers.
Control of the reaction selectivity of a catalyst to different functional groups in a molecule is a challenge in synthetic organic chemistry. The control of selectivity of a chemical reagent for the same type of functional groups is even more challenging.140 Thus, a catalyst that can selectively react with one of two similar functional groups, with only small differences in their chemical environments, and can switch the selectivity through an external stimulus is noteworthy. One of the first examples of this phenomenon reported a photo-switchable, site-selective esterification reaction. This novel photo-switch was demonstrated for in situ, site-selective, acetylation of partially protected sugars used in peptide catalysts.141 An oligopeptide catalyst integrated with a photo isomerizable azobenzene moiety was used to govern the size and shape of the active catalytic pocket. This unit switched the selectivity of the acylation reaction to two different sites of the same functional group within the same substrate.
The sugar molecule (4,6-O-benzylidene) methyl-α-D-pyranoside acted as the substrate for the acetylation with π-methyl-L-histidine (Pmh), the active moiety in the peptide catalytic pocket that transfers the electrophile. Irradiation of a sample with catalyst 6, as shown in Fig. 28b (R1 = Me, R2 = Cy), using continuous irradiation of 365 nm induced E → Z isomerization, with 94% of the Z isomer at PSS. Thermal isomerization of the Z isomer back to E was favoured with the increase in temperature, giving a half-life of 48 days at 0 °C and just 1 hour at 50 °C. This isomerization process of Z → E occurred readily under photochemical conditions probed with pulsed irradiation of blue light (λ = 448 nm). In the E-6 confirmation, the Pmh catalytic active site lies to one side of the azo functional group and hence provides a well-defined and exposed binding site to the substrate, whereas in Z-6, the folded arrangement buries the active Pmh moiety inside the fold. In addition to the shape and steric factors, other interactions such as H bonding, polar and dispersion forces also vary in the two isomeric forms (Fig. 29).
 | ||
| Fig. 28 (A) Different axial and equatorial orientation of the diol group in (4,6-O-benzylidene) methyl-α-D-pyranoside. (B) Different acetylation catalysts. (C) cis and trans photoisomerization of the azo group in catalyst 6. “Reproduced from ref. 141 with permission from the American Chemical Society, copyright 2019”. | ||
 | ||
| Fig. 29 Different acetylated products of (4,6-O-benzylidene) methyl-α-D-pyranoside. “Reproduced from ref. 141 with permission from the American Chemical Society, copyright 2019”. | ||
In the acetylation of substrates 1–3a, the catalyst 4 did not result in any site selective acetylation, while catalysts 5–7 displayed noticeable site preferences, particularly in the substrate 3a to give axial-2-acetylated product (14a) with the E isomer and equatorial-3-acetylated (15a) product with the Z isomer of the catalyst. However, diacetylated product (16a) was also observed as one of the products and the attempts to lower or eliminate its formation was not successful (Fig. 30). It was found that the selectivity for the 3-acetylated product with the Z isomer could be enhanced lowering the reaction temperature (0 °C) though the corresponding E isomer displayed a reduction of selectivity towards 2-acetylation under similar cryogenic conditions upon 448 nm irradiation.
 | ||
| Fig. 30 Acetylation of different substituted (4,6-O-benzylidene) methyl-α-D-pyranosides. “Reproduced from ref. 141 with permission from the American Chemical Society, copyright 2019”. | ||
In several experiments conducted with 3a and methyl-α-D-mannopyranosides protected by groups such as napthylidene, methylidene and acetonide (3b–d) at 0 °C, the size and orientation-dependent catalysis was observed. Groups substituted at the R1 position of 3a–d resulted in a selectivity variation of H < Nap ≪ Ph while that at R2, where substituents larger than H are substituted, gave reduced selectivity but enhanced reactivity. Regardless of the protecting group used in 3a–d, 2-acetylation was favoured by the E-6 catalyst, while 3-acetylation by Z-6. Thus, the selectivity of the reaction was not governed by substitution or steric demands of the protecting groups, but was due to catalyst–substrate binding features. The substrate (3a) was placed inside E-6 catalytic pocket, resulting in an inside catalytic event for acetylation to take place at the 2nd position to give the 2-acetylated derivative, whereas the substrate interacted with the outside of the ball-shaped Z-6 catalyst, resulting in an outside catalytic event to yield the 3-acetylated product. Accordingly, the analyzed structures of various transition states confirm that the reaction's natural ability to selectively respond to light and target a specific site have been successfully attained. This achievement is attributed to the substrate adopting varied orientations in relation to the active Pmh moiety, leading to alterations in how the substrate undergoes nucleophilic attack in the reaction. This synthetic strategy was extended to test site-selective acetylation of the natural product quercetin (which carries 5 hydroxy groups), with successful photo-switchable product-selectivity outcomes.
Phototropism in vascular plants,142 phototaxis in prokaryotes143 and vision in animals:144 are responses to light stimulus shown by different levels of organisms. These responses are generally created by a two-component system comprising a protein that can adapt multiple conformations and a chromophore, capable of harvesting light. The combination of these two functions; the protein scaffold and the chromophore, either covalently or non-covalently bound to each other, provides a photo-switchable foldamer that regulates biochemical processes with light.145 The process of human vision is modulated via cis–trans isomerization of the chromophore retinal, attached to protein opsin, where the structural change triggered by the geometric isomerism propagates through the protein structure. This reorganization in the conformation is finally transformed to an electrical signal, which is sent to the brain via the optic nerve.
Mimicking these natural mechanisms, foldamers represent a synthetic analogue of biomolecules for which a keen interest has been drawn in recent years, often involving conjugation of photo-switchable chromophores such as azobenzenes, to make them photo responsive.146 With the discovery of the first photo-switchable foldamer by Hecht and coworkers in 2006,147 azobenzenes with their firmly established photochemistry, ease of synthesis, photostability, recyclability and ability to generate high quantum yields have been the dominant choice among available chromophores for incorporating into foldamers such as peptides.148–150 Fumaramide/maleamide units containing C□C unsaturation with a photochemically isomerizable compact dicarboximide structure, but as a substitute for azobenzenes, have been less exploited.151 Upon inclusion of a distant terminal chiral residue in an achiral helical foldamer, remote chiral induction of chemical transformations up to 4 nm can be achieved resembling remote communication devices.152 In order to observe this phenomenon, the Moretto group described an approach where a fumaramide group was attached to the N-terminus of a range of helical foldamers comprising Aib oligomers and the other side of the fumaramide to a chiral group capable of inducing a specific screw sense preference in the Aib oligomer.153 This chiral residue can influence a specific screw sense conformation adapted by the Aib oligomer chain. These fumaramide/maleamide linkages may be coupled to Aib peptide foldamers to allow the formation of a hydrogen bond between the moieties present on either side of the unsaturation in the Z-maleamide geometry.
Hydrogen bond formation was not observed in trans (E) fumaramide, whereas upon UV irradiation (λ = 254 nm) in MeOH, photo isomerization of fumaramide into cis (Z) maleamide restored the H-bonding and, therefore, the conformational communication mechanism between two amide groups across the point of unsaturation and along the Aib peptide foldamer. The changes accompanied by the formation of the hydrogen bond is communicated along the peptide foldamer by means of a specific screw-sense conformational preference in the maleamide-linked foldamer, whereas this effect is absent in fumaramides, since the E-geometric configuration does not support the formation of such H bond (Fig. 31). Thus, the fumaramide/maleamide linker behaves as a photodiode that relays electronic information upon the presence or absence of an external UV light stimulus and the interconversion between the maleamide geometry that permits conformational conduction and fumaramide that inhibits any conduction allows selective control of the stereochemical aspects with light as an ON/OFF switch.
 | ||
| Fig. 31 Conversion of fumaramide 2a–f derivatives to maleamide isomers via photoisomerization. “Reproduced from ref. 153 with permission from the American Chemical Society, copyright 2016”. | ||
It is understood that Aib helical foldamers adopt a 310 helical form in non-polar media and exist as rapidly interconverting isoenergetic screw-sense forms. This interconverting, bi-stable system can be biased to preferred one screw-form, upon attachment of a chiral controller. L-(α-Me)Val was identified as the chiral residue that imparts the strongest preference to a single screw sense (right handed). A range of different, functionalized fumaramide Aib oligomers 2a–f were synthesized, out of which 2a,b have glycinamide residues at C termini and 2c–f consist of Aib homopeptides carrying methyl esters with increasing chain lengths from 1, 2, 4, or 8 Aib units. These fumaramides (2a–f) were isomerized in MeOH to corresponding maleamide derivatives upon λ = 254 nm irradiation to obtain 3a–f in 65–97% selectivity. NOESY, 1H and 13C NMR spectra of 2a and 3a suggested that the Aib peptide segment exists in a helical arrangement in both isomeric E–Z forms. The 1D 1H NMR spectra indicated different NH chemical shifts for L-(αMe)Val and Aib(2) units and the sharp difference in signals of Aib(2) between the two isomers (2a and 3a) shows that the newly formed H bond involves the NH group on Aib(2).
The change of geometrical configuration in the double bond in fumaramide (E) to maleamide (Z) has a huge influence on the –(Aib4)– peptide portion of the foldamer. The 1H NMR spectra obtained for 2a gives a singlet for 2H of the glycine methylene group, whereas upon isomerization to 3a, the peak splits into an AB system having a separation of 218 ppb. Hence, the shift in chemical properties, transitioning from no change (0 ppb) in the E isomer to a significant shift (218 ppb) in the Z isomer, serves as an indicator of the degree of alteration in the helical structure along the oligomer chain. This change likely signifies the process of deracemization in the foldamer chain, possibly influenced by the introduction of a new hydrogen bond.
The isomerization of E → Z brings the chiral L-(αMe)Val residue in close contact with the Aib peptide domain in the Z-isomer, inducing a specific screw configuration. This induced preference in the Z isomer towards a distinct chemical output has been utilized in a chain extension reaction with peptide oxazolones, where the diastereoisomeric ratio can be altered (Fig. 32).
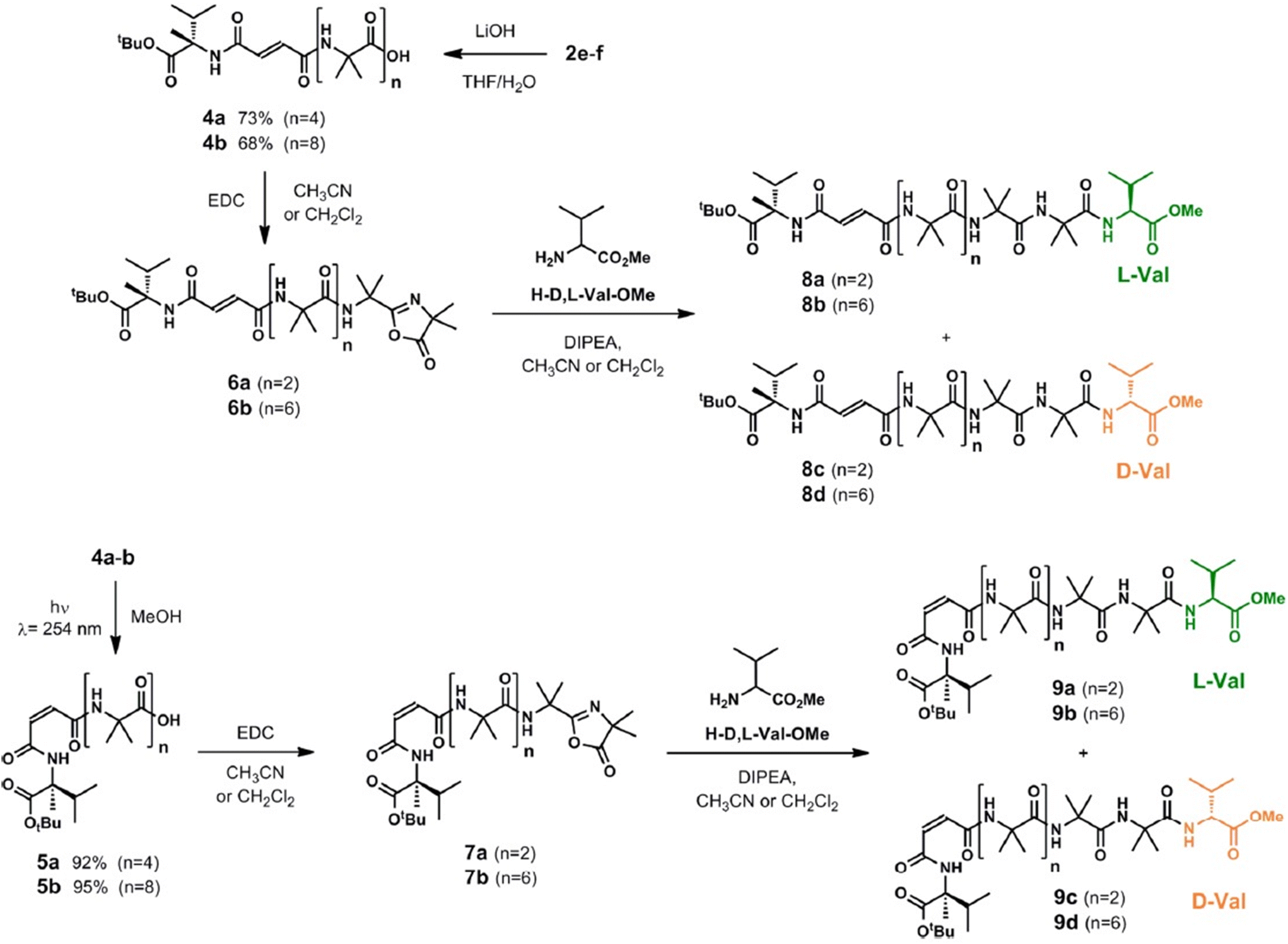 | ||
| Fig. 32 Synthesis of peptide oxazolones (6a,b and 7a,b) and their involvement in diastereomeric chain extension reactions with H-D,L-Val-OMe. “Reproduced from ref. 153 with permission from the American Chemical Society, copyright 2016”. | ||
The peptide oxazolones 6a,b (E) and 7a,b (Z) were synthesized and separately reacted with 8 equivalents of H-D,L-Val-OMe at temperatures of 20 °C and 35 °C in CH2Cl2. This resulted in no observed stereoisomerism with 6a,b (E) and short chain 7a (Z) structures giving corresponding diastereomeric pairs in 50/50 ratio, while long-chain 7b (Z) carrying a (Aib)7 peptide chain showed stereoselectivity with the preferential incorporation of L-Val-OMe in the chain extension reaction, up to a diastereoisomeric ratio of 74/26 (9b/9D) at 20 °C. Similar reactions carried out in acetonitrile at 20, 40, and 70 °C failed to induce any stereoselectivity while that conducted in CH2Cl2 at 35 °C gave a lower diastereoisomeric ratio of 62/38 (9b/9d). The number of intramolecular H-bonds that can be formed in the helix increases with the chain length, showing greater effects with the longer 7b (Z) than shorter 7a (Z) in the experiment with CH2Cl2 at 20 °C. Thus, for a low polarity solvent, lower temperature and longer main-chain length of the peptide were observed to favour the stereoselectivity in the reaction studied.
Helix-to-helix conformational communication between helical domains has also been identified with fumaramide/maleamide as an OFF/ON switch modulated by light. In compound 12, synthesised via the coupling of 10 and 11, the photo-switchable unsaturated linker acts as a bridge that allows conduction or insulation of the induced conformational changes between two Aib4 peptide helices that are linked head-to-head at their N-termini. Chiral L-Ala-OtBu and glycinamide were attached at each of the C-termini of two Aib peptide foldamers with a high preference of the esterified L-Ala to the left-handed helical screw sense conformation. Fumaramide derivative 12 carrying E configuration resulted in a singlet 1H NMR peak for glycinamide methylene group, indicating the lack of preference for any specific screw sense at the glycinamide unit. However, after isomerization of 12 to 13 (E-fumaramide → Z-maleamide) upon 254 nm irradiation, the same peak appeared as a doublet showing that the Z conformation permits communication from the helix bearing L-Ala to a more distant helical domain carrying the Gly residue via the point of unsaturation (Fig. 33). Therefore, the ability of fumaramide/maleamide linkage has also been successfully applied to design helical foldamers capable of helix-to-helix communication.
 | ||
| Fig. 33 Synthesis of helix 12 and isomerization to 13 under UV irradiation. “Reproduced from ref. 153 with permission from the American Chemical Society, copyright 2016”. | ||
Natural proteins have the ability to retain similar tertiary structures and catalyse non-identical catalytic processes despite making substantial alternations in their primary structure. This characteristic has been key to evolution via random mutations from the very beginning of life.154 In comparison, small modifications made in foldamers determine significant structural changes.155 Some foldamers have the ability to induce different stereochemical outcomes of a chemical reaction upon switching of the conformational structure, which can be triggered by external factors such as light and heat. One example has described how even a change in either insertion or removal of a single hydrogen (H) bond regulated by light or pH as a switch can lead to complete inversion of the helical conformation of the foldamer.156
Oligomers comprising more than three Aib residues, particularly in non-polar media, are essentially 310 helical and exist as two different conformations with opposite screw senses.157 When the oligomer is entirely achiral, each interconverting screw sense conformation becomes enantiomeric. These conformers made of Aib oligomers are made dissymmetrical upon ligation of a chiral terminus, and this leads to a difference in the population of two interconverting conformers. The ability of the ligated terminus to cause a specific preference in the screw sense is related to the organization of the final β turn of the 310 helix structure, and this phenomenon can be exploited towards the simple addition or the elimination of a hydrogen bond causing a pronounced alternation in the structure, hence modifying its conformation.156
The helical oligomer 4 features an alanine ligated to 5-bromo-7-nitroindoline (Bni) in its C-terminus and an isotopically labelled 13C methyl group at the N-terminus to facilitate 13C NMR studies. Oligomer 4 adopts a left-handed (M) screw sense in THF room temperature with a preference of 72:28 (left:right). Upon irradiation with 360 nm for 1 h in the presence of excess isopropylamine, a photo chemically induced switch from left (M) to right (P) helix occurs with a 99/1 helical ratio (Fig. 34). The refolding of the helical structure (inversion) is presumed to take place due to the transformation of the tertiary amide group in the C-terminus to a secondary amide, generating a novel H-bond at the C terminus. This introduces newly formed β turn, thus a switch in the screw sense preference.
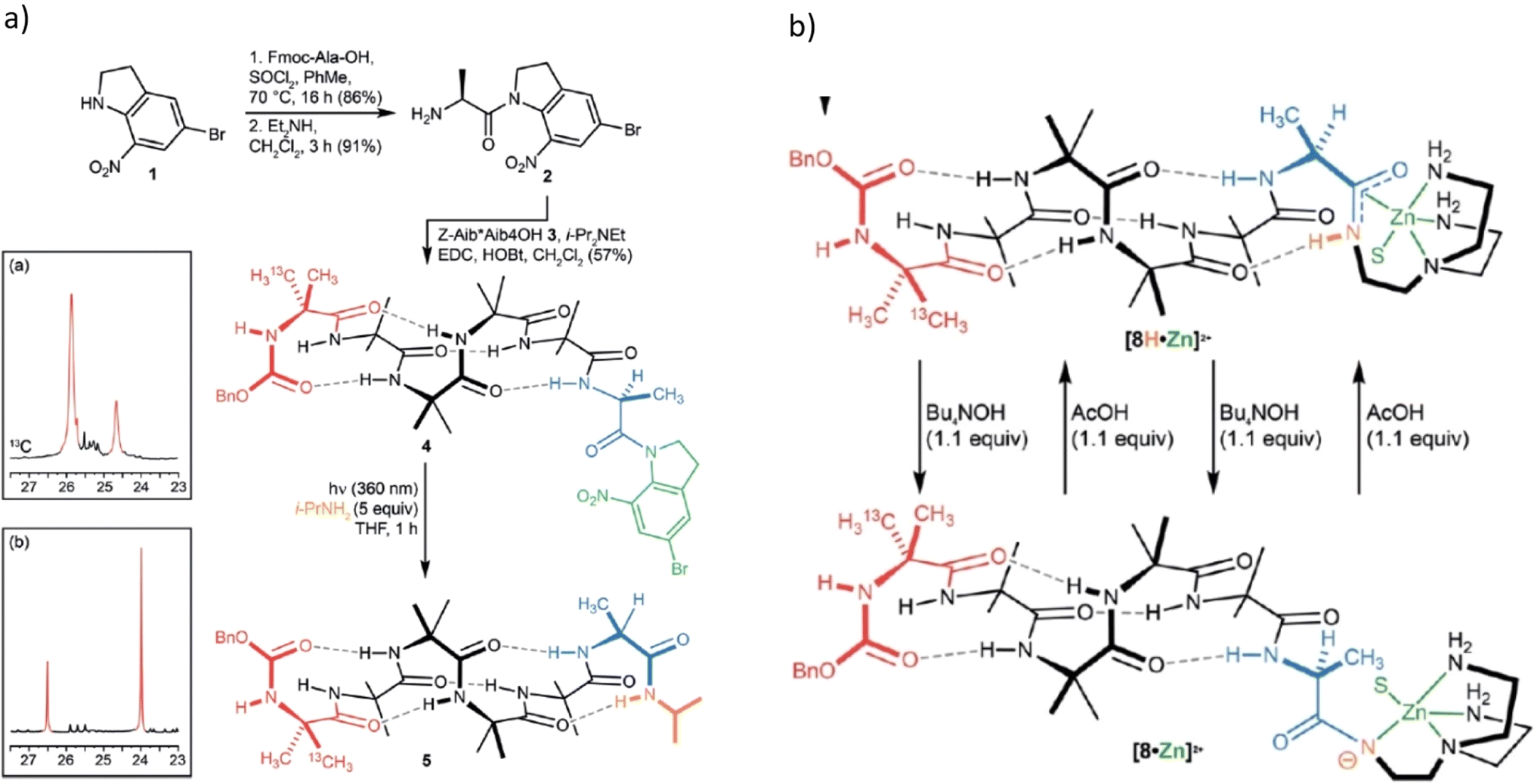 | ||
| Fig. 34 (a) Synthesis of left-handed 4 helix and the photoisomerization to right-handed 5 helix. (b) pH-Induced interconversion of screw sense preference of 8H·Zn2+ and 8·Zn2+. “Reproduced from ref. 156 with permission from John Wiley and Sons, copyright 2016”. | ||
This reversible H bond inclusion and deletion can also be achieved with the simple addition or removal of a proton (H+) with the help of an acid or base. The amide functionality present in peptides can bind to metals through amide N atoms, and in this way, a metal complex containing a Zn core was ligated with 1:1 metal binding stoichiometry to the amide linkage at the C-terminus of the helical foldamer. The downfield shift of the peak for amide NH fragment in the 13C NMR spectrum suggested the higher acidity of the NH group through binding of the terminal amide group with Zn in the 8H·Zn2+ structure. Treatment of the metal complex-bound helical foldamer with 1.1 equivalent of Bu4NOH base caused the removal of the H atom bonded to amide nitrogen, which was coordinated with the Zn complex (8·Zn2+). The loss of H atom removed the H-bond in the helix, causing a switch in the screw sense from P in 8H·Zn2+ to M in 8·Zn2+. The lost H-bond could be reintroduced to the helical foldamer when the added base was neutralised by acetic acid (1.1 equivalent) giving a complete restoration of the original P screw sense in 8H·Zn2+. The repeatability of this pH-regulated switching was confirmed with the second addition of Bu4NOH and successful P →M refolding process.
Structures 4 and 8 were modified to convert the conformational change into a detectable chemical output by ligating a series of achiral catalytic moieties to the N terminus of the oligomer (foldamers 9a–f). These foldamers carried no local chirality in their catalytic sites; however, their conformation could be made chiral through a distant stereogenic centre of 13–16 bonds away. Foldamers 9a–f were used to catalyse the addition of diethyl/dimethyl malonate to β-nitrostyrene and out of the series of catalytic foldamers used, only less sterically crowded 9e and 9f were effective in the reaction (Fig. 35). Even though the source of asymmetry and the catalytic site are well spatially separated, the foldamer catalysed the addition reaction enantioselectively to yield preferably the R isomer (R-10) with enantiomeric ratios of 75/25 for diethyl malonate and 82/18 for dimethyl malonate, as the substrates. A control reaction where the catalytic site and the point of asymmetry located in two separate molecules essentially gave a racemic mixture of 10 show no enantioselectivity.
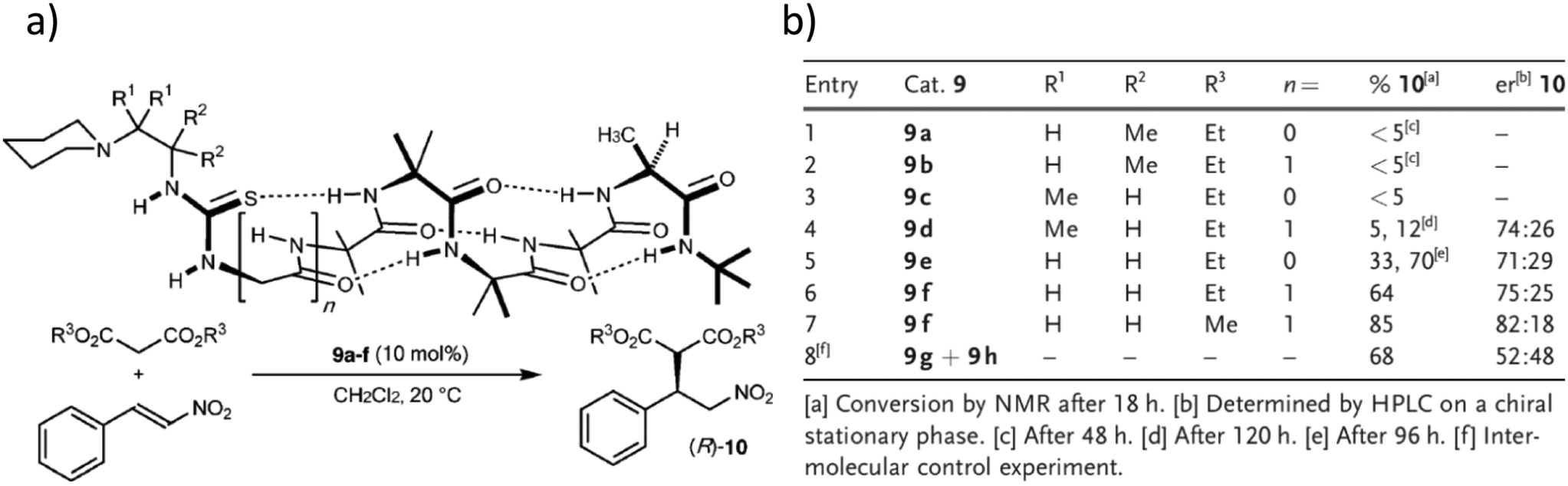 | ||
| Fig. 35 (a) Reaction between malonate and β-nitrostyrene catalysed by foldamers 9a–f. (b) Optimization of the catalytic foldamers 9a–f. “Reproduced from ref. 156 with permission from John Wiley and Sons, copyright 2016”. | ||
The catalyst 4 bearing a AlaBni group at its C-terminus was modified with the incorporation of the active catalytic site of 9f at its N-terminus to give the modified catalytic foldamer 11. This catalyst in the addition reaction of dimethyl malonate and nitrostyrene resulted in the S-10 product with an enantiomeric ratio of 37/63 in 18 h in CH2Cl2 (Fig. 36). Upon irradiation of 11 with 360 nm irradiation in the presence of 5 equivalents of i-PrNH2, a right-handed (P) helix was converted upon refolding to the left-handed (M) oligomer. The same reaction with the resulted M conformer gave a 78% yield of R-10 isomer (er 77/23) in 72 h in CH2Cl2 showing the potential of the foldamer to induce asymmetric organocatalysis upon switching of its screw sense preference with light as the external stimulus. The synthetic protocol also clearly demonstrates how the insertion or deletion of a single H bond located distant to the active catalytic site can induce a switch in the overall helical screw sense that relays its influence along the helical chain to cause a significant and a detectable enantioselective chemical output in an addition reaction.
 | ||
| Fig. 36 Induction of enantioselecctivity in the reaction between malonate and β-nitrostyrene for preferential S and R isomers upon photoisomerization of the catalytic foldamer. “Reproduced from ref. 156 with permission from John Wiley and Sons, copyright 2016”. | ||
3. Remarks and future outlook
3.1. Merits
Nature, through the process of natural selection, creates the most efficient catalytic systems known and biology therefore teems with examples of incredibly efficient and selective synthesis, with control of chirality, in living systems. The inherent homochirality in nature allows access only to a specific enantiomer while almost completely suppressing the other. In comparison, most artificial catalysts developed to date use elements that fabricate higher-order, complex structures with photo-switchable chirality that generate both possible enantiomers of a compound through switching of the catalyst upon orthogonal external stimuli. These applications can exceed the capabilities of nature and may therefore have enormous potential.The scope of ‘remote controls’ or artificial stimuli to influence reaction switching is currently in development and experimental, proof-of-concept stages.133,158 The capacity for reaction switching by the modes described in this review can potentially achieve an excellent temporal resolution in the switching process, thus paving the way to exactly time individual reaction steps of a multistep catalytic pathway. These ideas offer great potential to synthesize complex molecules, even biological and macromolecules, with access to well-programmed sequences of exact blocks of interest. This realizes the possibility to attain complete control over the length, composition of monomers as well as the manner by which the pendant groups are arranged in the main chain (tacticity) in the synthesis of macromolecules such as polymers. The photomodulated rotational motion of molecular motors reflects the conversion of light energy into kinetic or potential energy and the approach could be useful in solar power devices and related smart materials. Molecular motors when arranged and aligned in a desired manner could be used to harness work while the incorporation of such molecular motors into hard matter (such as MOFs) may deliver an array of future devices related to conductivity and magnetism.159 The bistable molecular switches can be used and employed in data storage devices; however, molecular motors that show multistate switching can serve as better candidates in data storage applications over the bistable analogues as they can significantly improve the bit density owing to the availability of multiple states.160 Hence, it is clear that these improved designs will be extremely powerful in synthetic applications and have a tremendous impact on new material development.161 Further, the ability of a single catalyst to be product selective and the switch catalyst reaction pathway of chosen substrates eliminate the need for multiple catalysts and varying reaction conditions making the catalysed processes more economically friendly.
Photoswitchable catalysts represent a burgeoning field within organic chemistry, offering unprecedented control over reaction selectivity and efficiency. Through the modulation of catalyst structure or activity upon illumination, these catalysts enable the synthesis of intricate molecules with heightened precision. This capability holds promise for applications ranging from drug discovery to materials science, where the ability to precisely control reaction outcomes is paramount. Recent advancements in photoswitchable catalysis have demonstrated dynamic reaction optimization for on-demand adjustments of reaction conditions, to enhance yields and minimize side reactions. Furthermore, the integration of photoswitchable catalysts into continuous-flow systems has streamlined reaction processes, leading to improved scalability and safety in chemical synthesis.
The spatial control afforded by photoswitchable catalysts opens new avenues for catalysis, enabling precise manipulation of chemical transformations in defined regions of a reaction environment. This spatial precision mimics natural processes where spatial organization plays a crucial role, facilitating the synthesis of complex molecules with specific spatial arrangements. Additionally, the integration of photoswitchable reactions with artificial intelligence (AI) algorithms has enabled automated design and optimization of reaction conditions, accelerating the discovery of novel catalysts and predicting their behavior with high accuracy. Such advancements have implications not only in traditional organic synthesis but also in bioorthogonal catalysis, where photoswitchable catalysts operate under biocompatible conditions, allowing for the precise manipulation of biological processes with high spatiotemporal control.
Beyond traditional synthetic chemistry applications, photoswitchable reactions hold promise in environmental remediation, energy storage, and emerging materials. By harnessing light energy to drive chemical transformations, these reactions contribute to the development of sustainable technologies for pollutant degradation and energy conversion/storage. Moreover, the exploration of new reaction mechanisms enabled by photoswitchable catalysts provides insights into fundamental chemical processes and may lead to the discovery of novel transformations. In materials science, photoswitchable reactions pave the way for the development of dynamic materials with applications in responsive coatings, optical devices, and molecular machines, offering unprecedented opportunities for advanced materials design and engineering.
3.2. Limitations
Each class of photo switches inevitably has demerits in certain contexts that can limit their applicability. No one system can be a universal catalyst! Azobenzenes have comparatively low thermal stability although they are widely used as a photoswitches. Most photoswitches involving azobenzene use UV irradiation to achieve the photoisomerization. In the dark, at equilibrium, azobenzene exists in the more stable trans conformation and the cis isomer is obtained by irradiating the system with UV light, which reverts back to its trans isomer thermally or upon blue light irradiation. Hence, most of the azobenzene-modified biomolecules targeting proteins and nucleic acids depend on UV light for the isomerization process, limiting the applicability of any such molecular switch in vivo.150,162,163 This is due to the fact that high-energy UV light is strongly scattering that makes the penetration to the cells difficult, while it can also cause apoptotic events and other undesired responses.164,165 To overcome this issue, azobenzene derivatives that show photoisomerization entirely within the visible region have been developed that can be utilized in in vivo applications via different approaches such as substitution of azo molecules or coupling to upconversion nanophosphors.166,167 When involving photochromic molecules, the switching activity of such catalytic switches between their different states is not perfect and the photostationary state (PSS) composition is governed by the difference in population of the two states of a catalyst. Therefore, to achieve a complete photoswitching behavior and thereby a quantitative photo conversion, the absorption bands for the two states have to be well separated to facilitate selective excitation. The quantum yield is also a key challenge, while the chemo and stereoselectivity of the obtained products also have to be further optimized in many of the reported examples. Further, the switching behaviour of photochromic molecules once attached to a solid surface such as a MOF is not as reversible as it is in solutions, and hence, this factor needs to be considered and overcome in future solid catalytic fabrications incorporating photochromic moieties.168,169 Homogeneous catalysts are comparatively less stable and immobilized catalysts usually do not achieve satisfactory turnovers, and hence, along with the cost incurred in immobilizing such catalysts, it is quite ambiguous whether immobilization will be of interest for industrial use in terms of efficiency and cost-effectiveness.170To attain a higher reversibility of the chemical reactions associated with photochromism, a higher fatigue resistance of the catalyst should be maintained with the elimination of any competing side reactions.171,172 A significant progress in terms of effective designs for fatigue resistance in switches has been made in applications such as data storage, but the extension of this key criterion to academic research in this new field has not occurred. Molecules at their nano scale are subjected to Brownian motion as well as diffusion processes, which pose a challenge towards the proper control of molecular assembly lines. In these setups, the substrate molecules need to shift from a specific catalytic site to the other and also the formed product must leave away from the “machines” to prevent product inhibition. These barriers may be minimized with the suitable strategies to achieve a smooth, fast and more efficient utility of molecular machines.
The commercialization of switchable catalysts presents several challenges rooted in their intrinsic properties and the associated manufacturing requirements. One significant drawback is the poor stability of the catalysts themselves, which can compromise their performance over extended operational periods. This instability may stem from factors such as catalyst degradation or deactivation under reaction conditions, limiting the catalyst's lifespan and necessitating frequent replacement or regeneration. Additionally, switchable catalysts often exhibit unsatisfactory turnover rates, indicating their inefficiency in catalyzing reactions or their limited capacity to convert reactants into products within a given timeframe. These suboptimal turnover rates can hinder their practical application in industrial processes where high throughput and productivity are priorities.
Furthermore, the immobilization of catalysts for photo-switchable systems could impose a considerable cost on commercialization. The integration of these catalysts into functional devices requires specialized immobilization techniques such as surface anchoring or encapsulation to ensure their stability and activity under operational conditions. These immobilization processes can be labour-intensive and resource-demanding, adding to the overall production costs and potentially limiting scalability for large-scale industrial applications. Moreover, the implementation of industrial photo-switchable systems requires integration of additional equipment for light modulation. Light intensity adjustment devices or specialized optical components are essential for controlling the wavelength and intensity of light exposure, which are crucial for activating the switchable functionalities of the catalysts. However, the incorporation of such equipment adds complexity to the production and requires additional investment in infrastructure and technology.
Addressing these challenges requires concerted efforts in catalyst design, process optimization, and technology innovation. Improving the stability and efficiency of switchable catalysts through advanced materials design and synthesis strategies is essential for enhancing their performance and longevity. Furthermore, streamlining immobilization processes and developing cost-effective manufacturing approaches can help reduce production costs and enhance scalability for industrial deployment. Ultimately, overcoming these obstacles will be critical for realizing the full commercial potential of switchable catalysts and facilitating their widespread adoption in various industrial sectors.
One of the other main limitations of photoswitchable catalysts is that they rarely have the ability to run more than a few ON–OFF or different product formation cycles.46 However, a large number of cycles is generally a requirement in practical applications. One strategy to achieve this requirement is by choosing reactions such that the substrates and products do not compete with switchable ligands for the catalytic active sites of NPs, which ensures a higher catalytic efficiency over repeated cycles. Most photochromic entities undergo photoisomerization under UV light and such systems are utilized in many in vitro experimental set-ups. To expand the scope of these photoisomerization catalysts to in vivo experiments such as in human tissues and other cellular models, photoswitches that operate using long-wavelength visible radiation (red or orange) or in the near-IR region are more suitable, owing to greater light penetration into living tissues at longer wavelengths.
Molecular machines continue to provide the opportunity to expand the chemical space beyond the capabilities of both biological systems and conventional chemistry. The growth of molecular machines along with the progress achieved in metal, organo, organometallic and supramolecular catalysis will help realize new generations of robust, multifunctional photoresponsive catalysts and incorporated synthetic tools that can perform complex sequences of tasks, having command-adaptive behaviour. Hence, these advancements can be expected to benefit numerous novel applications that are tunable with light making the photoswitchable catalytic transformations a broader field than current perception.118
To achieve smooth, fast, and efficient utilization of molecular machines in photochromic reactions and catalysis, several constructive suggestions can be implemented. Catalysts should be designed with enhanced fatigue resistance, drawing insights from materials science and catalysis to incorporate durable materials or surface modifications that mitigate degradation over repeated cycles of photochromic reactions. Strategies from organic synthesis and reaction engineering should be employed to minimize competing side reactions that decrease the reversibility of photochromic reactions, involving the optimization of reaction conditions, catalyst selection, or design of reaction pathways favouring desired product formation. Effective designs for fatigue resistance demonstrated in applications like data storage could be adapted, including the development of robust molecular architectures or protective coatings to enhance the stability and longevity of photochromic switches.
Furthermore, principles of molecular self-assembly and supramolecular chemistry can be utilized to control assembly processes of molecular machines, designing molecular scaffolds or templates to guide precise positioning of substrate molecules and facilitate efficient catalytic reactions. Additionally, challenges posed by Brownian motion and diffusion processes at the nanoscale should be addressed by developing strategies such as confinement effects, surface patterning techniques, or microfluidic devices to control movement and interactions of molecules within the catalytic environment. The optimization of molecular machine design to ensure efficient access to catalytic sites by substrate molecules is crucial, requiring engineering of dynamic molecular architectures or responsive materials that facilitate rapid and selective substrate binding. Molecular machines should be designed with enhanced capabilities for product diffusion and removal from catalytic sites, potentially incorporating molecular channels or pores to facilitate efficient transport of products away from reaction sites, thus preventing product inhibition and improving overall reaction efficiency. The integration of these suggestions and leveraging scientific knowledge across various disciplines can aid researchers in overcoming barriers and advancing the utility of molecular machines in photochromic reactions and catalysis.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Bayan would like to acknowledge QUT for a PhD scholarship. We gratefully acknowledge financial support from the Australian Research Council Discovery (DP210103357) and Linkage Projects schemes (LP210301424). ERW thanks A. Prof Sarina Sarina for helpful discussions and assistance regarding this work.References
-
J. M. Thomas and W. J. Thomas, Principles and practice of heterogeneous catalysis, John Wiley & Sons, 2014 Search PubMed
.
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Designing for a green chemistry future, Science, 2020, 367(6476), 397–400 CrossRef CAS PubMed
- T. Collins, Toward sustainable chemistry, Science, 2001, 291(5501), 48–49 CrossRef CAS PubMed
- P. Marion, B. Bernela, A. Piccirilli, B. Estrine, N. Patouillard, J. Guilbot and F. Jérôme, Sustainable chemistry: how to produce better and more from less?, Green Chem., 2017, 19(21), 4973–4989 RSC
- S. Peiris, J. McMurtrie and H.-Y. Zhu, Metal nanoparticle photocatalysts: emerging processes for green organic synthesis, Catal. Sci. Technol., 2016, 6(2), 320–338 RSC
- Z. Li, Y. Yan, M. Liu, Z. Qu, Y. Yue, T. Mao, S. Zhao, M. Liu and Z. Lin, Robust ring-opening reaction via asymmetrically coordinated Fe single atoms scaffolded by spoke-like mesoporous carbon nanospheres, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(14), e2218261120 CrossRef CAS PubMed
- B. Wang, X. Zhu, X. Pei, W. Liu, Y. Leng, X. Yu, C. Wang, L. Hu, Q. Su and C. Wu, Room-temperature laser planting of high-loading single-atom catalysts for high-efficiency electrocatalytic hydrogen evolution, J. Am. Chem. Soc., 2023, 145(25), 13788–13795 CrossRef CAS PubMed
- F. Eisenreich, M. Kathan, A. Dallmann, S. P. Ihrig, T. Schwaar, B. M. Schmidt and S. Hecht, A photoswitchable catalyst system for remote-controlled (co) polymerization in situ, Nat. Catal., 2018, 1(7), 516–522 CrossRef CAS
- X. Yao, T. Li, J. Wang, X. Ma and H. Tian, Recent progress in photoswitchable supramolecular self–assembling systems, Adv. Opt. Mater., 2016, 4(9), 1322–1349 CrossRef CAS
- J. Wang and B. L. Feringa, Dynamic control of chiral space in a catalytic asymmetric reaction using a molecular motor, Science, 2011, 331(6023), 1429–1432 CrossRef CAS PubMed
- M. M. Russew and S. Hecht, Photoswitches: from molecules to materials, Adv. Mater., 2010, 22(31), 3348–3360 CrossRef CAS PubMed
- J. P. Sauvage, From chemical topology to molecular machines (Nobel lecture), Angew. Chem., Int. Ed., 2017, 56(37), 11080–11093 CrossRef CAS PubMed
- J. F. Stoddart, Mechanically interlocked molecules (MIMs)—Molecular shuttles, switches, and machines (Nobel Lecture), Angew. Chem., Int. Ed., 2017, 56(37), 11094–11125 CrossRef CAS PubMed
- B. L. Feringa, The art of building small: From molecular switches to motors (Nobel lecture), Angew. Chem., Int. Ed., 2017, 56(37), 11060–11078 CrossRef CAS PubMed
- J. Barber, Photosynthetic energy conversion: natural and artificial, Chem. Soc. Rev., 2009, 38(1), 185–196 RSC
- D. F. O'Brien, The chemistry of vision, Science, 1982, 218(4576), 961–966 CrossRef PubMed
- V. Blanco, D. A. Leigh and V. Marcos, Artificial switchable catalysts, Chem. Soc. Rev., 2015, 44(15), 5341–5370 RSC
- R. Göstl, A. Senf and S. Hecht, Remote-controlling chemical reactions by light: Towards chemistry with high spatio-temporal resolution, Chem. Soc. Rev., 2014, 43(6), 1982–1996 RSC
- L. Tzounis, M. Doña, J. M. Lopez-Romero, A. Fery and R. Contreras-Caceres, interfaces, Temperature-controlled catalysis by core–shell–satellite AuAg@ pNIPAM@ Ag hybrid microgels: a highly efficient catalytic thermoresponsive nanoreactor, ACS Appl. Mater. Interfaces, 2019, 11(32), 29360–29372 CrossRef CAS PubMed
- S. Semwal and J. Choudhury, Switch in Catalyst State: Single Bifunctional Bi–state
Catalyst for Two Different Reactions, Angew. Chem., Int. Ed., 2017, 56(20), 5556–5560 CrossRef CAS PubMed
- J. Wei and P. L. Diaconescu, Redox-switchable ring-opening polymerization with ferrocene derivatives, Acc. Chem. Res., 2019, 52(2), 415–424 CrossRef CAS PubMed
- R. J. Chew, X. R. Li, Y. Li, S. A. Pullarkat and P. H. Leung, Pd–Catalyzed Enantiodivergent and Regiospecific phospha–Michael Addition of Diphenylphosphine to 4-oxo-Enamides: Efficient Access to Chiral Phosphinocarboxamides and Their Analogues, Chem. – Eur. J., 2015, 21(12), 4800–4804 CrossRef CAS PubMed
- J. C. Robertson, M. L. Coote and A. C. Bissember, Synthetic applications of light, electricity, mechanical force and flow, Nat. Rev. Chem., 2019, 3(5), 290–304 CrossRef
- I. Vassalini and I. Alessandri, Switchable stimuli-responsive heterogeneous catalysis, Catalysts, 2018, 8(12), 569 CrossRef
- N. Mittal, S. Pramanik, I. Paul, S. De and M. Schmittel, Networking nanoswitches for ON/OFF control of catalysis, J. Am. Chem. Soc., 2017, 139(12), 4270–4273 CrossRef CAS PubMed
- R. S. Stoll and S. Hecht, Artificial light–gated catalyst systems, Angew. Chem., Int. Ed., 2010, 49(30), 5054–5075 CrossRef CAS PubMed
- W. Ho, Single-molecule chemistry, J. Chem. Phys., 2002, 117(24), 11033–11061 CrossRef CAS
- D. Chen, W. Qin, H. Fang, L. Wang, B. Peng, L. Li and W. Huang, Recent progress in two-photon small molecule fluorescent probes for enzymes, Chin. Chem. Lett., 2019, 30(10), 1738–1744 CrossRef CAS
- B. Huang, H. Babcock and X. Zhuang, Breaking the diffraction barrier: super-resolution imaging of cells, Cell, 2010, 143(7), 1047–1058 CrossRef CAS PubMed
- X. Ke, S. Sarina, J. Zhao, X. Zhang, J. Chang and H. Zhu, Tuning the reduction power of supported gold nanoparticle photocatalysts for selective reductions by manipulating the wavelength of visible light irradiation, Chem. Commun., 2012, 48(29), 3509–3511 RSC
- C. Kerzig and O. S. Wenger, Reactivity control of a photocatalytic system by changing the light intensity, Chem. Sci., 2019, 10(48), 11023–11029 RSC
- L. Guo, F. Song, S. Zhu, H. Li and L. Chu, syn-Selective alkylarylation of terminal alkynes via the combination of photoredox and nickel catalysis, Nat. Commun., 2018, 9(1), 1–8 Search PubMed
- X. Q. Chu, D. Ge, M. L. Wang, W. Rao, T. P. Loh and Z. L. Shen, Chemo–and Regioselective Ring Construction Driven by Visible–Light Photoredox Catalysis: an Access to Fluoroalkylated Oxazolidines Featuring an All–Substituted Carbon Stereocenter, Adv. Synth. Catal., 2019, 361(17), 4082–4090 CrossRef CAS
- P. Ji, Y. Zhang, Y. Wei, H. Huang, W. Hu, P. A. Mariano and W. Wang, Visible-Light-Mediated, Chemo-and Stereoselective Radical Process for the Synthesis of C-Glycoamino Acids, Org. Lett., 2019, 21(9), 3086–3092 CrossRef CAS PubMed
- S. Zhu and D. Wang, Photocatalysis: basic principles, diverse forms of implementations and emerging scientific opportunities, Adv. Energy Mater., 2017, 7(23), 1700841 CrossRef
- P. Li, Y. Song and C.-M. Dong, Hyperbranched polypeptides synthesized from phototriggered ROP of a photocaged N ε-[1-(2-nitrophenyl) ethoxycarbonyl]-l-lysine-N-carboxyanhydride: microstructures and effects of irradiation intensity and nitrogen flow rate, Polym. Chem., 2018, 9(29), 3974–3986 RSC
- E. Peiris, S. Sarina, E. R. Waclawik, G. A. Ayoko, P. Han, J. Jia and H. Y. Zhu, Plasmonic Switching of the Reaction Pathway: Visible–Light Irradiation Varies the Reactant Concentration at the Solid–Solution Interface of a Gold–Cobalt Catalyst, Angew. Chem., 2019, 131(35), 12160–12164 CrossRef
- B. A. Grzybowski and W. T. Huck, The nanotechnology of life-inspired systems, Nat. Nanotechnol., 2016, 11(7), 585–592 CrossRef CAS PubMed
- L. Zhai, Stimuli-responsive polymer films, Chem. Soc. Rev., 2013, 42(17), 7148–7160 RSC
- I. Khan, K. Saeed and I. Khan, Nanoparticles: Properties, applications and toxicities, Arabian J. Chem., 2019, 12(7), 908–931 CrossRef CAS
- C. M. Phan and H. M. Nguyen, Role of capping agent in wet synthesis of nanoparticles, J. Phys. Chem. A, 2017, 121(17), 3213–3219 CrossRef CAS PubMed
- L. Wang and Q. Li, Photochromism into nanosystems: towards lighting up the future nanoworld, Chem. Soc. Rev., 2018, 47(3), 1044–1097 RSC
- R. S. Stoll and S. Hecht, Immobilization of a photoswitchable piperidine base, Org. Lett., 2009, 11(21), 4790–4793 CrossRef CAS PubMed
- A. B. Kanj, K. Müller and L. Heinke, Stimuli–responsive metal–organic frameworks with photoswitchable azobenzene side groups, Macromol. Rapid Commun., 2018, 39(1), 1700239 CrossRef PubMed
- M. Zhu and H. Zhou, Azobenzene-based small molecular photoswitches for protein modulation, Org. Biomol. Chem., 2018, 16(44), 8434–8445 RSC
- R. Liu, X. Zhang, F. Xia and Y. Dai, Azobenzene-based photoswitchable catalysts: State of the art and perspectives, J. Catal., 2022, 409, 33–40 CrossRef CAS
- M. Szewczyk, G. Sobczak and V. Sashuk, Photoswitchable catalysis by a small swinging molecule confined on the surface of a colloidal particle, ACS Catal., 2018, 8(4), 2810–2814 CrossRef CAS
- W. T. Yao, Z. Q. Liu, A. M. Zheng, J. Q. Li, X. F. Feng, L. F. Ma, C. S. Yan, M. B. Luo and F. Luo, Photoswitching storage of guest molecules in metal–organic framework for photoswitchable catalysis: exceptional product, ultrahigh photocontrol, and photomodulated size selectivity, J. Mater. Chem. A, 2017, 5(17), 7961–7967 RSC
- S. Neri, S. Garcia Martin, C. Pezzato and L. J. Prins, Photoswitchable catalysis by a nanozyme mediated by a light-sensitive cofactor, J. Am. Chem. Soc., 2017, 139(5), 1794–1797 CrossRef CAS PubMed
- Y. Wei, S. Han, J. Kim, S. Soh and B. A. Grzybowski, Photoswitchable catalysis mediated by dynamic aggregation of nanoparticles, J. Am. Chem. Soc., 2010, 132(32), 11018–11020 CrossRef CAS PubMed
- R. Dorel and B. L. Feringa, Photoswitchable catalysis based on the isomerisation of double bonds, Chem. Commun., 2019, 55(46), 6477–6486 RSC
- J. Ludwig, J. Helberg, H. Zipse and R. Herges, Azo-dimethylaminopyridine-functionalized Ni(II)-porphyrin as a photoswitchable nucleophilic catalyst, Beilstein J. Org. Chem., 2020, 16(1), 2119–2126 CrossRef CAS PubMed
- D. Cameron and S. Eisler, Photoswitchable double bonds: Synthetic strategies for tunability and versatility, J. Phys. Org. Chem., 2018, 31(10), e3858 CrossRef
- T. Arif, C. Cazorla, N. Bogliotti, N. Saleh, F. Blanchard, V. Gandon, R. Métivier, J. Xie, A. Voituriez and A. Marinetti, Bimetallic gold(I) complexes of photoswitchable phosphines: synthesis and uses in cooperative catalysis, Catal. Sci. Technol., 2018, 8(3), 710–715 RSC
- A. Ahmed, H. Ullah, Z. Ullah, M. Tariq and K. Ayub, External stimulus controlled recombination of hydrogen in photochromic dithienylethene frustrated lewis pairs, Int. J. Hydrogen Energy, 2019, 44(59), 31141–31152 CrossRef
- N. Kumagai and M. Shibasaki, Catalytic chemical transformations with conformationally dynamic catalytic systems, Catal. Sci. Technol., 2013, 3(1), 41–57 RSC
- M. V. Peters, R. S. Stoll, A. Kühn and S. Hecht, Photoswitching
of basicity, Angew. Chem., Int. Ed., 2008, 47(32), 5968–5972 CrossRef CAS PubMed
- R. Göstl, B. Kobin, L. Grubert, M. Pätzel and S. Hecht, Sterically crowding the bridge of dithienylcyclopentenes for enhanced photoswitching performance, Chem. – Eur. J., 2012, 18(45), 14282–14285 CrossRef
- P. R. Huddleston, V. V. Volkov and C. C. Perry, The structural and electronic properties of 3, 3′-azothiophene photo-switching systems, Phys. Chem. Chem. Phys., 2019, 21(3), 1344–1353 RSC
- Y. Kim, Photoswitching Molecular Junctions: Platforms and Electrical Properties, ChemPhysChem, 2020, 21(21), 2368–2383 CrossRef CAS
- N. Grebenovsky, L. Luma, P. Müller and A. Heckel, Introducing LNAzo: More Rigidity for Improved Photocontrol of Oligonucleotide Hybridization, Chem. – Eur. J., 2019, 25(53), 12298–12302 CrossRef CAS PubMed
- N. Luo, M. Wang, H. Li, J. Zhang, H. Liu and F. Wang, Photocatalytic oxidation–hydrogenolysis of lignin β-O-4 models via a dual light wavelength switching strategy, ACS Catal., 2016, 6(11), 7716–7721 CrossRef CAS
- I. Ghosh and B. König, Chromoselective photocatalysis: controlled bond activation through light–color regulation of redox potentials, Angew. Chem., Int. Ed., 2016, 55(27), 7676–7679 CrossRef CAS PubMed
- C. Courtois, C. A. Walenta, M. Tschurl, U. Heiz and C. Friend, Regulating Photochemical Selectivity with Temperature: Isobutanol on TiO2 (110), J. Am. Chem. Soc., 2020, 142(30), 13072–13080 CrossRef CAS PubMed
- C. A. Walenta, S. L. Kollmannsberger, C. Courtois, M. Tschurl and U. Heiz, Photocatalytic selectivity switch to C–C scission: α-methyl ejection of tert-butanol on TiO 2 (110), Phys. Chem. Chem. Phys., 2018, 20(10), 7105–7111 RSC
- N. G. Petrik, M. A. Henderson and G. A. Kimmel, Insights into acetone photochemistry on rutile TiO2 (110). 2. New photodesorption channel with CH3 ejection along the surface normal, J. Phys. Chem. C, 2015, 119(22), 12273–12282 CrossRef CAS
- B. Szczepanik, Photocatalytic degradation of organic contaminants over clay-TiO2 nanocomposites: A review, Appl. Clay Sci., 2017, 141, 227–239 CrossRef CAS
- Y. Hunge, A. Yadav, S. Khan, K. Takagi, N. Suzuki, K. Teshima, C. Terashima and A. Fujishima, Photocatalytic degradation of bisphenol A using titanium dioxide@ nanodiamond composites under UV light illumination, J. Colloid Interface Sci., 2021, 582, 1058–1066 CrossRef CAS
- L. Han, B. Li, H. Wen, Y. Guo and Z. Lin, Photocatalytic degradation of mixed pollutants in aqueous wastewater using mesoporous 2D/2D TiO2 (B)-BiOBr heterojunction, J. Mater. Sci. Technol., 2021, 70, 176–184 CrossRef CAS
- K. R. Phillips, S. C. Jensen, M. Baron, S.-C. Li and C. M. Friend, Sequential photo-oxidation of methanol to methyl formate on TiO2 (110), J. Am. Chem. Soc., 2013, 135(2), 574–577 CrossRef CAS PubMed
- C. A. Walenta, S. L. Kollmannsberger, J. Kiermaier, A. Winbauer, M. Tschurl and U. Heiz, Ethanol photocatalysis on rutile TiO 2 (110): the role of defects and water, Phys. Chem. Chem. Phys., 2015, 17(35), 22809–22814 RSC
- D. Brinkley and T. Engel, Photocatalytic dehydrogenation of 2-propanol on TiO2 (110), J. Phys. Chem. B, 1998, 102(39), 7596–7605 CrossRef CAS
- C. Courtois, M. Eder, K. Schnabl, C. A. Walenta, M. Tschurl and U. Heiz, Reactions in the photocatalytic conversion of tertiary alcohols on rutile TiO2 (110), Angew. Chem., Int. Ed., 2019, 58(40), 14255–14259 CrossRef CAS PubMed
- M. Shen and M. A. Henderson, Identification of the active species in photochemical hole scavenging reactions of methanol on TiO2, J. Phys. Chem. Lett., 2011, 2(21), 2707–2710 CrossRef CAS
- C. A. Walenta, M. Tschurl and U. Heiz, Introducing catalysis in photocatalysis: what can be understood from surface science studies of alcohol photoreforming on TiO2, J. Phys.: Condens. Matter, 2019, 31(47), 473002 CrossRef CAS
- F. Ullmann and J. Bielecki, Ueber synthesen in der biphenylreihe, Ber. Dtsch. Chem. Ges., 1901, 34(2), 2174–2185 CrossRef CAS
- N. Marina, A. E. Lanterna and J. C. Scaiano, Expanding the Color Space in the Two-Color Heterogeneous Photocatalysis of Ullmann C–C Coupling Reactions, ACS Catal., 2018, 8(8), 7593–7597 CrossRef CAS
- S. Sarina, H. Y. Zhu, Q. Xiao, E. Jaatinen, J. Jia, Y. Huang, Z. Zheng and H. Wu, Viable photocatalysts under solar–spectrum irradiation: nonplasmonic metal nanoparticles, Angew. Chem., Int. Ed., 2014, 126(11), 2979–2984 CrossRef
- S. Sarina, E. Jaatinen, Q. Xiao, Y. M. Huang, P. Christopher, J. C. Zhao and H. Y. Zhu, Photon energy threshold in direct photocatalysis with metal nanoparticles: key evidence from the action spectrum of the reaction, J. Phys. Chem. Lett., 2017, 8(11), 2526–2534 CrossRef CAS PubMed
- J. Galego, F. J. Garcia-Vidal and J. Feist, Many-molecule reaction triggered by a single photon in polaritonic chemistry, Phys. Rev. Lett., 2017, 119(13), 136001 CrossRef PubMed
- J. A. Creighton and D. G. Eadon, Ultraviolet–visible absorption spectra of the colloidal metallic elements, J. Chem. Soc., Faraday Trans., 1991, 87(24), 3881–3891 RSC
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. Bruijnincx and B. M. Weckhuysen, Paving the way for lignin valorisation: recent advances in bioengineering, biorefining and catalysis, Angew. Chem., Int. Ed., 2016, 55(29), 8164–8215 CrossRef CAS PubMed
- J. D. Nguyen, B. S. Matsuura and C. R. Stephenson, A photochemical strategy for lignin degradation at room temperature, J. Am. Chem. Soc., 2014, 136(4), 1218–1221 CrossRef CAS PubMed
- W. Li, J. Xu, Q. Zhou, S. Wang, Z. Feng, D. Hu, X. Li and Y. J. N. Cao, Bidirectional plasmonic coloration with gold nanoparticles by wavelength-switched photoredox reaction, Nanoscale, 2018, 10(46), 21910–21917 RSC
- Y. Xiao, Y. Huang, S. Xue and J. Zhao, Light switching of amine oxidation products from oximes to imines: Superior activity of plasmonic gold nanorods-loaded TiO2 (B) nanofibers under visible-near IR light, Appl. Catal., B, 2020, 265, 118596 CrossRef CAS
- C. Glaser, Beiträge zur kenntniss des acetenylbenzols, Ber. Dtsch. Chem. Ges., 1869, 2(1), 422–424 CrossRef
- K. Sonogashira, Y. Tohda and N. Hagihara, A convenient synthesis of acetylenes: catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines, Tetrahedron Lett., 1975, 16(50), 4467–4470 CrossRef
- M. B. Thathagar, J. Beckers and G. Rothenberg, Palladium-free and ligand-free Sonogashira cross-coupling, Green Chem., 2004, 6(4), 215–218 RSC
- Y. Zhou and J. Zhao, Glaser coupling-and Sonogashira coupling-control over CuxO nanoparticles/carbon nanotube by switching visible-light off and on, Appl. Catal., B, 2022, 300, 120721 CrossRef CAS
- X. Liu, Y. Shi, Y. Jin, T. Tana, E. Peiris, X. Zhang, F. Xu, E. R. Waclawik, S. E. Bottle and H. Zhu, Surface–Plasmon–Enhanced Transmetalation between Copper and Palladium Nanoparticle Catalyst, Angew. Chem., Int. Ed., 2022, e202203158 CAS
- H. Y. Jiang, P. Zhou, Y. Wang, R. Duan, C. Chen, W. Song and J. Zhao, Copper–Based Coordination Polymer Nanostructure for Visible Light Photocatalysis, Adv. Mater., 2016, 28(44), 9776–9781 CrossRef CAS PubMed
- Y. Huang, Z. Liu, G. Gao, G. Xiao, A. Du, S. Bottle, S. Sarina and H. Zhu, Stable copper nanoparticle photocatalysts for selective epoxidation of alkenes with visible light, ACS Catal., 2017, 7(8), 4975–4985 CrossRef CAS
- K. Sarmah, S. Mukhopadhyay, T. K. Maji and S. Pratihar, Switchable Bifunctional Bistate Reusable ZnO–Cu for Selective Oxidation and Reduction Reaction, ACS Catal., 2019, 9(1), 732–745 CrossRef CAS
- Z. Zhang, C. Zhang, H. Zheng and H. Xu, Plasmon-driven catalysis on molecules and nanomaterials, Acc. Chem. Res., 2019, 52(9), 2506–2515 CrossRef CAS PubMed
- P. Han, W. Martens, E. R. Waclawik, S. Sarina and H. Zhu, Metal nanoparticle photocatalysts: synthesis, characterization, and application, Part. Part. Syst. Charact., 2018, 35(6), 1700489 CrossRef
- X. Zhang, X. Li, D. Zhang, N. Q. Su, W. Yang, H. O. Everitt and J. Liu, Product selectivity in plasmonic photocatalysis for carbon dioxide hydrogenation, Nat. Commun., 2017, 8(1), 1–9 CrossRef
- X. Bian, Y. Zhao, G. I. Waterhouse, Y. Miao, C. Zhou, L. Z. Wu and T. Zhang, Quantifying the Contribution of Hot Electrons in Photothermal Catalysis: A Case Study of Ammonia Synthesis over Carbon–supported Ru Catalyst, Angew. Chem., Int. Ed., 2023, 62, e202304452 CrossRef CAS PubMed
- S. Link and M. A. El-Sayed, Size and temperature dependence of the plasmon absorption of colloidal gold nanoparticles, J. Phys. Chem. B, 1999, 103(21), 4212–4217 CrossRef CAS
- K. Watanabe, D. Menzel, N. Nilius and H.-J. Freund, Photochemistry on metal nanoparticles, Chem. Rev., 2006, 106(10), 4301–4320 CrossRef CAS PubMed
- M. J. Kale, T. Avanesian and P. Christopher, Direct photocatalysis by plasmonic nanostructures, ACS Catal., 2014, 4(1), 116–128 CrossRef CAS
- H. H. Richardson, M. T. Carlson, P. J. Tandler, P. Hernandez and A. O. Govorov, Experimental and theoretical studies of light-to-heat conversion and collective heating effects in metal nanoparticle solutions, Nano Lett., 2009, 9(3), 1139–1146 CrossRef CAS PubMed
- C. Wang, O. Ranasingha, S. Natesakhawat, P. R. Ohodnicki, M. Andio, J. P. Lewis and C. Matranga, Visible light plasmonic heating of Au–ZnO for the catalytic reduction of CO2, Nanoscale, 2013, 5(15), 6968–6974 RSC
- S. Bai, W. Jiang, Z. Li and Y. Xiong, Surface and interface engineering in photocatalysis, ChemNanoMat, 2015, 1(4), 223–239 CrossRef CAS
- Y. Gao, W. Nie, Q. Zhu, X. Wang, S. Wang, F. Fan and C. Li, The polarization effect in surface–plasmon–induced photocatalysis on Au/TiO2 nanoparticles, Angew. Chem., Int. Ed., 2020, 59, 18218–18223 CrossRef CAS PubMed
- S. Bai, J. Ge, L. Wang, M. Gong, M. Deng, Q. Kong, L. Song, J. Jiang, Q. Zhang, Y. Luo, Y. Xie and Y. Xiong, A Unique Semiconductor–Metal–Graphene Stack Design to Harness Charge Flow for Photocatalysis, Adv. Mater., 2014, 26, 5689–5695 CrossRef CAS PubMed
- J. Zhao, Z. Zheng, S. Bottle, A. Chou, S. Sarina and H. Zhu, Highly efficient and selective photocatalytic hydroamination of alkynes by supported gold nanoparticles using visible light at ambient temperature, Chem. Commun., 2013, 49(26), 2676–2678 RSC
- E. Peiris, S. Sarina, E. R. Waclawik, G. A. Ayoko, P. Han, J. Jia and H. Y. Zhu, Plasmonic Switching of the Reaction Pathway: Visible–Light Irradiation Varies the Reactant Concentration at the Solid–Solution Interface of a Gold–Cobalt Catalyst, Angew. Chem., Int. Ed., 2019, 58(35), 12032–12036 CrossRef CAS PubMed
- E. Peiris, S. Hanauer, T. Le, J. Wang, T. Salavati-fard, P. Brasseur, E. V. Formo, B. Wang and P. H. Camargo, Controlling Selectivity in Plasmonic Catalysis: Switching Reaction Pathway from Hydrogenation to Homocoupling Under Visible–Light Irradiation, Angew. Chem., Int. Ed., 2023, 62(4), e202216398 CrossRef CAS PubMed
- D. F. Swearer, H. Zhao, L. Zhou, C. Zhang, H. Robatjazi, J. M. P. Martirez, C. M. Krauter, S. Yazdi, M. J. McClain and E. Ringe, Heterometallic antenna− reactor complexes for photocatalysis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(32), 8916–8920 CrossRef CAS PubMed
- S. Yu, A. J. Wilson, J. Heo and P. K. Jain, Plasmonic control of multi-electron transfer and C–C coupling in visible-light-driven CO2 reduction on Au nanoparticles, Nano Lett., 2018, 18(4), 2189–2194 CrossRef CAS PubMed
- B. M. Trost and D. L. Van Vranken, Asymmetric transition metal-catalyzed allylic alkylations, Chem. Rev., 1996, 96(1), 395–422 CrossRef CAS
- I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Visible light mediated photoredox catalytic arylation reactions, Acc. Chem. Res., 2016, 49(8), 1566–1577 CrossRef CAS PubMed
- L. Marzo, I. Ghosh, F. Esteban and B. König, Metal-free photocatalyzed cross coupling of bromoheteroarenes with pyrroles, ACS Catal., 2016, 6(10), 6780–6784 CrossRef CAS
- J. Zoller, D. C. Fabry and M. Rueping, Unexpected dual role of titanium dioxide in the visible light heterogeneous catalyzed C–H arylation of heteroarenes, ACS Catal., 2015, 5(6), 3900–3904 CrossRef CAS
- A. M. Martínez-Gualda, R. Cano, L. Marzo, R. Pérez-Ruiz, J. Luis-Barrera, R. Mas-Ballesté, A. Fraile, A. Víctor and J. Aleman, Chromoselective access to Z-or E-allylated amines and heterocycles by a photocatalytic allylation reaction, Nat. Commun., 2019, 10(1), 1–10 CrossRef PubMed
- S. Protti, D. Ravelli and M. Fagnoni, Wavelength dependence and wavelength selectivity in photochemical reactions, Photochem. Photobiol. Sci., 2019, 18(9), 2094–2101 CrossRef CAS PubMed
- F. Glaser, C. Kerzig and O. S. Wenger, Multi–Photon Excitation in Photoredox Catalysis: Concepts, Applications, Methods, Angew. Chem., Int. Ed., 2020, 59(26), 10266–10284 CrossRef CAS PubMed
- T. van Leeuwen, A. S. Lubbe, P. Štacko, S. J. Wezenberg and B. L. Feringa, Dynamic control of function by light-driven molecular motors, Nat. Rev. Chem., 2017, 1(12), 0096 CrossRef CAS
- M. Schliwa and G. Woehlke, Molecular motors, Nature, 2003, 422(6933), 759–765 CrossRef CAS PubMed
- K. Barlan, M. J. Rossow and V. I. Gelfand, The journey of the organelle: teamwork and regulation in intracellular transport, Curr. Opin. Cell Biol., 2013, 25(4), 483–488 CrossRef CAS PubMed
- Y. Feng, M. Ovalle, J. S. Seale, C. K. Lee, D. J. Kim, R. D. Astumian and J. F. Stoddart, Molecular Pumps and Motors, J. Am. Chem. Soc., 2021, 143(15), 5569–5591 CrossRef CAS PubMed
- H. Itoh, A. Takahashi, K. Adachi, H. Noji, R. Yasuda, M. Yoshida and K. Kinosita, Mechanically driven ATP synthesis by F 1-ATPase, Nature, 2004, 427(6973), 465–468 CrossRef CAS PubMed
- S. Sengupta, M. M. Spiering, K. K. Dey, W. Duan, D. Patra, P. J. Butler, R. D. Astumian, S. J. Benkovic and A. Sen, DNA polymerase as a molecular motor and pump, ACS Nano, 2014, 8(3), 2410–2418 CrossRef CAS PubMed
- J. M. Scholey, I. Brust-Mascher and A. Mogilner, Cell division, Nature, 2003, 422(6933), 746–752 CrossRef CAS PubMed
- F. Lancia, A. Ryabchun and N. Katsonis, Life-like motion driven by artificial molecular machines, Nat. Rev. Chem., 2019, 3(9), 536–551 CrossRef CAS
- V. García-López, D. Liu and J. M. Tour, Light-activated organic molecular motors and their applications, Chem. Rev., 2019, 120(1), 79–124 CrossRef PubMed
- M. Baroncini, S. Silvi and A. Credi, Photo-and redox-driven artificial molecular motors, Chem. Rev., 2019, 120(1), 200–268 CrossRef PubMed
- E. R. Kay and D. A. Leigh, Rise of the molecular machines, Angew. Chem., Int. Ed., 2015, 54(35), 10080–10088 CrossRef CAS PubMed
- G. Saper and H. Hess, Synthetic systems powered by biological molecular motors, Chem. Rev., 2019, 120(1), 288–309 CrossRef PubMed
- D. Roke, M. Sen, W. Danowski, S. J. Wezenberg and B. L. Feringa, Visible-light-driven tunable molecular motors based on oxindole, J. Am. Chem. Soc., 2019, 141(18), 7622–7627 CrossRef CAS PubMed
- W. Danowski, F. Castiglioni, A. S. Sardjan, S. Krause, L. Pfeifer, D. Roke, A. Comotti, W. R. Browne and B. L. Feringa, Visible-light-driven rotation of molecular motors in a dual-function metal–organic framework enabled by energy transfer, J. Am. Chem. Soc., 2020, 142(19), 9048–9056 CrossRef CAS PubMed
- Z.-T. Shi, Y.-X. Hu, Z. Hu, Q. Zhang, S.-Y. Chen, M. Chen, J.-J. Yu, G.-Q. Yin, H. Sun and L. Xu, Visible-light-driven rotation of molecular motors in discrete supramolecular metallacycles, J. Am. Chem. Soc., 2020, 143(1), 442–452 CrossRef PubMed
- M. Suda, Y. Thathong, V. Promarak, H. Kojima, M. Nakamura, T. Shiraogawa, M. Ehara and H. M. Yamamoto, Light-driven molecular switch for reconfigurable spin filters, Nat. Commun., 2019, 10(1), 2455 CrossRef PubMed
- J. Escorihuela, M. I. Burguete and S. V. Luis, New advances in dual stereocontrol for asymmetric reactions, Chem. Soc. Rev., 2013, 42(12), 5595–5617 RSC
- N. Koumura, R. W. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Light-driven monodirectional molecular rotor, Nature, 1999, 401(6749), 152–155 CrossRef CAS PubMed
- B. L. Feringa, The art of building small: from molecular switches to molecular motors, J. Org. Chem., 2007, 72(18), 6635–6652 CrossRef CAS PubMed
- M. M. Pollard, A. Meetsma and B. L. Feringa, A redesign of light-driven rotary molecular motors, Org. Biomol. Chem., 2008, 6(3), 507–512 RSC
- M. Vlatković, L. Bernardi, E. Otten and B. L. Feringa, Dual stereocontrol over the Henry reaction using a light-and heat-triggered organocatalyst, Chem. Commun., 2014, 50(58), 7773–7775 RSC
- D. Zhao, T. M. Neubauer and B. L. Feringa, Dynamic control of chirality in phosphine ligands for enantioselective catalysis, Nat. Commun., 2015, 6(1), 1–7 Search PubMed
- Z. Huang and G. Dong, Site-selectivity control in organic reactions: A quest to differentiate reactivity among the same kind of functional groups, Acc. Chem. Res., 2017, 50(3), 465–471 CrossRef CAS PubMed
- D. Niedek, F. R. Erb, C. Topp, A. Seitz, R. C. Wende, A. K. Eckhardt, J. Kind, D. Herold, C. M. Thiele and P. R. Schreiner, In Situ Switching of Site-Selectivity with Light in the Acetylation of Sugars with Azopeptide Catalysts, J. Org. Chem., 2019, 85(4), 1835–1846 CrossRef PubMed
- M. Ahmad, J. A. Jarillo, O. Smirnova and A. R. Cashmore, Cryptochrome blue-light photoreceptors of Arabidopsis implicated in phototropism, Nature, 1998, 392(6677), 720–723 CrossRef CAS PubMed
- E. B. Purcell and S. Crosson, Photoregulation in prokaryotes, Curr. Opin. Microbiol., 2008, 11(2), 168–178 CrossRef CAS PubMed
- R. Feuda, S. C. Hamilton, J. O. McInerney and D. Pisani, Metazoan opsin evolution reveals a simple route to animal vision, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(46), 18868–18872 CrossRef CAS
- X. E. Zhou, K. Melcher and H. E. Xu, Structure and activation of rhodopsin, Acta Pharmacol. Sin., 2012, 33(3), 291–299 CrossRef CAS PubMed
- Z. Yu and S. Hecht, Remote control over folding by light, Chem. Commun., 2016, 52(40), 6639–6653 RSC
- A. Khan, C. Kaiser and S. Hecht, Prototype of a photoswitchable foldamer, Angew. Chem., Int. Ed., 2006, 45(12), 1878–1881 CrossRef CAS PubMed
- Z. Mahimwalla, K. G. Yager, J.-I. Mamiya, A. Shishido, A. Priimagi and C. J. Barrett, Azobenzene photomechanics: prospects and potential applications, Polym. Bull., 2012, 69(8), 967–1006 CrossRef CAS
- R. J. Mart and R. K. Allemann, Azobenzene photocontrol of peptides and proteins, Chem. Commun., 2016, 52(83), 12262–12277 RSC
- A. A. Beharry and G. A. Woolley, Azobenzene photoswitches for biomolecules, Chem. Soc. Rev., 2011, 40(8), 4422–4437 RSC
- V. Peddie and A. D. Abell, Photocontrol of peptide secondary structure through non-azobenzene photoswitches, J. Photochem. Photobiol., C, 2019, 40, 1–20 CrossRef CAS
- R. A. Brown, V. Diemer, S. J. Webb and J. Clayden, End-to-end conformational communication through a synthetic purinergic receptor by ligand-induced helicity switching, Nat. Chem., 2013, 5(10), 853–860 CrossRef CAS PubMed
- D. Mazzier, M. Crisma, M. De Poli, G. Marafon, C. Peggion, J. Clayden and A. Moretto, Helical foldamers incorporating photoswitchable residues for light-mediated modulation of conformational preference, J. Am. Chem. Soc., 2016, 138(25), 8007–8018 CrossRef CAS PubMed
- M. A. Nowak, M. C. Boerlijst, J. Cooke and J. M. Smith, Evolution of genetic redundancy, Nature, 1997, 388(6638), 167–171 CrossRef CAS PubMed
- A. Altmayer–Henzien, V. Declerck, J. Farjon, D. Merlet, R. Guillot and D. Aitken, Fine Tuning of β–Peptide Foldamers: a Single Atom Replacement Holds Back the Switch from an 8-Helix to a 12-Helix, Angew. Chem., 2015, 127(37), 10957–10960 CrossRef
- B. A. Le Bailly, L. Byrne and J. Clayden, Refoldable foldamers: Global conformational switching by deletion or insertion of a single hydrogen bond, Angew. Chem., 2016, 128(6), 2172–2176 CrossRef
- J. Venkatraman, S. C. Shankaramma and P. Balaram, Design of folded peptides, Chem. Rev., 2001, 101(10), 3131–3152 CrossRef CAS PubMed
- E. Moulin, L. Faour, C. C. Carmona-Vargas and N. Giuseppone, From molecular machines to stimuli–responsive materials, Adv. Mater., 2020, 32(20), 1906036 CrossRef CAS
- K. Zhu, C. A. O′keefe, V. N. Vukotic, R. W. Schurko and S. J. Loeb, A molecular shuttle that operates inside a metal–organic framework, Nat. Chem., 2015, 7(6), 514–519 CrossRef CAS PubMed
- J. E. Green, J. Wook Choi, A. Boukai, Y. Bunimovich, E. Johnston-Halperin, E. DeIonno, Y. Luo, B. A. Sheriff, K. Xu, Y. Shik Shin, H.-R. Tseng, J. Fraser Stoddart and J. R. Heath, A 160-kilobit molecular electronic memory patterned at 1011 bits per square centimetre, Nature, 2007, 445(7126), 414–417 CrossRef CAS PubMed
- I. Aprahamian, The future of molecular machines, ACS Cent. Sci., 2020, 6(3), 347–358 CrossRef CAS PubMed
- P. Gorostiza and E. Y. Isacoff, Optical switches for remote and noninvasive control of cell signaling, Science, 2008, 322(5900), 395–399 CrossRef CAS PubMed
- C. Hoppmann, P. Schmieder, P. Domaing, G. Vogelreiter, J. Eichhorst, B. Wiesner, I. Morano, K. Rück-Braun and M. Beyermann, Photocontrol of contracting muscle fibers, Angew. Chem., Int. Ed., 2011, 50(33), 7699–7702 CrossRef CAS PubMed
- S. Cooper, J. Ranger-Moore and T. G. Bowden, Differential inhibition of UVB–induced AP–1 and NF–κB transactivation by components of the jun bZIP domain, Mol. Carcinog., 2005, 43(2), 108–116 CrossRef CAS PubMed
- W.-F. Cheong, S. A. Prahl and A. J. Welch, A review of the optical properties of biological tissues, IEEE J. Quantum Electron., 1990, 26(12), 2166–2185 CrossRef
- W. Wu, L. Yao, T. Yang, R. Yin, F. Li and Y. Yu, NIR-light-induced deformation of cross-linked liquid-crystal polymers using upconversion nanophosphors, J. Am. Chem. Soc., 2011, 133(40), 15810–15813 CrossRef CAS PubMed
- A. A. Beharry, O. Sadovski and G. A. Woolley, Azobenzene photoswitching without ultraviolet light, J. Am. Chem. Soc., 2011, 133(49), 19684–19687 CrossRef CAS PubMed
- S. Hübner, J. G. de Vries and V. Farina, Catalysis, Why does industry not use immobilized transition metal complexes as catalysts?, Adv. Synth. Catal., 2016, 358(1), 3–25 CrossRef
- Z. Wang, L. Heinke, J. Jelic, M. Cakici, M. Dommaschk, R. J. Maurer, H. Oberhofer, S. Grosjean, R. Herges and S. Bräse, Photoswitching in nanoporous, crystalline solids: an experimental and theoretical study for azobenzene linkers incorporated in MOFs, Phys. Chem. Chem. Phys., 2015, 17(22), 14582–14587 RSC
- S. Hübner, J. G. de Vries and V. Farina, Why does industry not use immobilized transition metal complexes as catalysts?, Adv. Synth. Catal., 2016, 358(1), 3–25 CrossRef
- K. Higashiguchi, K. Matsuda, S. Kobatake, T. Yamada, T. Kawai and M. Irie, Fatigue mechanism of photochromic 1, 2-bis (2, 5-dimethyl-3-thienyl) perfluorocyclopentene, Bull. Chem. Soc. Jpn., 2000, 73(10), 2389–2394 CrossRef CAS
- M. Herder, B. M. Schmidt, L. Grubert, M. Pätzel, J. Schwarz and S. Hecht, Improving the fatigue resistance of diarylethene switches, J. Am. Chem. Soc., 2015, 137(7), 2738–2747 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2024 |