
Leveraging cooperative photocatalysis for the concurrent production of solar fuels and value-added chemicals: mediated by a metal-free porphyrin-based polymeric framework†
Kirti
Dhingra
,
Neha
Saini
,
Amit
Kumar
and
Kamalakannan
Kailasam
 *
*
Advanced Functional Nanomaterials Group, Institute of Nano Science and Technology (INST), Knowledge City, Sector 81, SAS Nagar, Manauli PO, Mohali, 1140306, Punjab, India. E-mail: kamal@inst.ac.in; kkamal17@gmail.com
First published on 21st September 2024
Abstract
Limitations in the conventional energy-intensive anthraquinone oxidation process for H2O2 production have led researchers to develop an environmentally sustainable, energy-efficient, and cost-effective approach. The photocatalytic H2O2 generation from molecular oxygen has emerged as a leading edge in sustainable technology development, yet efficiency remains a key challenge. Various sacrificial agents are added to the reaction medium to improve efficiency, but their underutilization is the primary concern. To address this issue, we design a reaction system that considers the selective oxidation of the sacrificial agent along with the reduction of oxygen. Notably, we constructed a metal-free organic polymer Porp-Tz exhibiting broad visible light absorption and suitable band positions that consider the efficient reduction of O2 for the co-production of H2O2 with a remarkable generation rate of 25.13 mmol g−1 h−1 along with the synthesis of industrially important chemical N-benzylidenebenzylamine (AQY = 7.9% at 420 nm). In addition, the concurrent production of regioselective 3,4-dihydroisoquinolines (DHIQs) from tetrahydroisoquinolines (THIQs) alongside the H2O2 generation rate of 13.34 mmol g−1 h−1 was explored. Moreover, the photocatalytic reaction mechanism highlights the synergistic role of the reactive oxygen species (O2˙− and 1O2), h+, and proton donors, providing a comprehensive understanding of the photocatalytic process. This study emphasizes new insights into deploying the next-generation multifunctional polymeric framework for the photocatalytic co-production of solar fuel and the selective synthesis of fine value-added chemicals, broadening the scope of porous organic polymers for potential industrial interest.
Introduction
Energy crisis and environmental pollution have always been the reason for much solicitude across the globe. The two are often intertwined and have aroused alarming situations worldwide. A large share of energy demand has been resourced by non-renewable fossil fuels, namely coal, petroleum, natural gas, nuclear energy, and many more. Nevertheless, the production and utilization of these fuels release a substantial number of toxic gases and cause consequential environmental hazards.1 Consequently, to combat the situation of energy sustainability, security, and equity and to maintain an optimal balance between energy and the environment, there is an intense requisite for the transition of energy sources from non-renewable to more sustainable, cleaner, and economy-viable.Notably, harnessing inexhaustible solar energy for important photocatalytic chemical processes such as CO2 reduction, N2 fixation, H2 generation, organic transformations, degradation of environmental pollutants, and increasing solar-to-fuel conversion efficiency is one of the strategic goals towards sustainability. To imitate the artificial photosynthesis process, an effective photocatalyst is a prerequisite to facilitate the harvesting of solar energy and efficiently utilize it for chemical transformations.2 To develop a rational photocatalyst, semiconductors with optimal structure and excellent modularity are explored for better absorption of visible light and enhanced utility of photogenerated charge carriers with the least recombination of electron–hole pairs. Photocatalysts considered must have appropriate active sites to facilitate the photo-redox reactions and initiate the energy transfer process.3 Both inorganic (metal-based) and organic (metal-free) photocatalysts have been proposed, in which the metal-containing photocatalyst suffers from the challenges of low-solar absorption efficiency, vulnerable defects, wide bandgap, and low environment sustainability.4 However, the metal-free organic photocatalyst includes porous organic polymers (POPs) and possesses the features of reticular designing, tunability, better solar absorption efficiency, high porosity, excellent robustness, and tuneable band positions.5 Engineering of suitable band positions for the efficient separation of the charge carriers and the stability of the intermediates formed are crucial factors to work on while employing metal-free photocatalysts for better product efficiency and selectivity.6
Among the diverse array of structured POPs, many electron-donating organic linkers, such as pyrene, porphyrin, and phthalocyanine, have garnered widespread attention, especially the porphyrin moiety has been used so far owing to its excellent light absorption capacity, outstanding optoelectronic properties, ability to engineer the band positions by deliberately anchoring of the metal ion inside the ring and ease of substitution.7 Thus, porphyrin-based POPs exhibit excellent catalytic properties in photoconversion processes such as in water splitting, CO2 reduction, and organic transformations.8,9 Despite the wide range of photocatalytic applications of porphyrin-based POPs, there is a vast scope for future advancement. With this, we designed a metal-free porphyrin (Porp) and thiazole-based porous organic polymer through a Schiff-base condensation reaction. 4,4′-(thiazolo[5,4-d] thiazole-2,5-diyl) dibenzaldehyde (Tz) is employed as the monomer linker because it contains fused (bi)heterocyclic thiazolo[5,4-d] thiazole moieties that form a rigid planar structure with co-facial geometry, which facilitates the more photogenerated charge carrier mobility.10 The constructed donor–π–acceptor–π–donor (D–π–A–π–D) system enhances the charge separation efficiency through the extended electron delocalization across the polymeric network, leading to efficient visible-light absorption.11 The system can be further utilized to extend the lifetime of the charge carriers, suppressing charge carrier recombination, and hence efficient generation of reactive oxygen species (ROS) to carry out various organic transformations.12
Harnessing the efficient charge separation and effective ROS generation capabilities of Porp-Tz, we explored its improved oxidation and reduction abilities for cooperative photocatalysis. In this study, we stressed the judicious utilization of both the charge carriers and escalated the production rate of H2O2 by exploring specific proton donors as sacrificial agents, such as benzylamine and 1,2,3,4-tetrahydroisoquinoline, that undergo oxidation to yield high value-added products. The sacrificial agents improve the oxidation capacities of the holes by lowering the oxidation barrier.13 Therefore, the thoughtful selection of a sacrificial agent is crucial for the photocatalytic production of H2O2.
For the photo-oxidation reactions involving the co-production of H2O2 along with the homocoupling of benzylamine, metal-based photocatalysts, such as CdIn2S4 (ref. 14) and ZrS1−yS2−x,15 have been explored so far, but they face certain limitations, such as toxicity and low solar absorption, in the visible region, resulting in a comparatively lower H2O2 production rate. Metal-free photocatalysts, such as DCM-HCPs16 and H3LP-HCPs,17 have also been investigated but resulted in lower yields of H2O2 than Porp-Tz. For THIQ dehydrogenative oxidation, there is no such report on the synergistic production of H2O2 along the oxidation pathway for metal-free photocatalysis. Notably, as per our knowledge, this is the first report of a metal-free porous organic polymer in which both benzylamine and THIQs are deployed as sacrificial agents for their simultaneous oxidation alongside the production of H2O2 in the visible region of the spectrum with enhanced photocatalytic activity when compared to the previous reports [Tables S1 and S2†].
The oxidative coupling of benzylamine is a challenging reaction from the synthesis viewpoint, as it is difficult to control the selectivity of the coupled product.18 Moreover, the imines produced are employed as the key intermediates in synthesizing numerous nitrogen-containing heterocyclic compounds used in the production of many pharmaceutical drugs, such as alkaloids, polymers, and various chemicals.19 Moreover, the semi-dehydrogenation of THIQs produces DHIQ derivatives, which also act as valuable intermediates in the synthesis of drugs, such as antifungal drugs and antitumor drugs, and in many important organic reactions.20,21
Hence, this report proposes a sustainable approach to deploy a porous organic polymer for synchronously carrying out the cooperative production of H2O2 and the industrially significant fine chemicals.
Results and discussions
The as-synthesized Porp-Tz consists of a polymeric network connected through the imine linkage formed via a Schiff-base condensation between a porphyrin-based amine and a thiazole-based aldehyde monomeric unit under the solvothermal reaction conditions (Scheme 1), as discussed in ESI 1.3.† The chemical structure of the POP was first scrutinized by solid-state 13C CP/MAS SSNMR analysis, which was further supported by Fourier-transform infrared spectroscopy (FTIR). Successful condensation was demonstrated in the presence of imine linkages, as demonstrated by the peak at 138 ppm. Signals located at 168 ppm and 152 ppm correspond to the C atoms of the thiazole ring and peaks from 100 to 150 ppm were assigned to the carbon atoms of the phenyl and porphyrin ring [Fig. 1(a)], indicating the existence of both the monomeric units in the polymeric network.10,22 FTIR measurements also supported the effective polymerization by exhibiting a characteristic signal of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching at 1670 cm−1 along with the disappearance of both aniline –N–H stretching bands and –CO stretching signal [Fig. 1(b) and S1†]. From the powder X-ray diffraction pattern (PXRD), a sharp peak at 2.34° was observed together with three weak peaks at 3.43°, 7.59°, and 12.54° and a broad peak at 20°–30° ascertaining the partial crystalline nature of the Porp-Tz framework [Fig. S2†]. Further, to study the type of linkages and gain more insights into the surface of the framework, X-ray photoelectron spectroscopy (XPS) was employed. The XPS survey scan interpreted the elemental composition of C, N, and S as present in the Porp-Tz. Fig. S3(a)† shows the typical chemical states for C, N, and S. For N 1s high-resolution XPS, three deconvoluted peaks corresponding to the protonated pyrrolic N of the porphyrin (–NH–, 399.9 eV), non-protonated pyrrolic N of porphyrin, and –S–CN– of the thiazole ring (–NC–, 397.2 eV),22 and N of the imine bond (–CN–, 398.6 eV) [Fig. S3(b)†].23 These investigations indicate the successful synthesis of the polymer Porp-Tz.
N stretching at 1670 cm−1 along with the disappearance of both aniline –N–H stretching bands and –CO stretching signal [Fig. 1(b) and S1†]. From the powder X-ray diffraction pattern (PXRD), a sharp peak at 2.34° was observed together with three weak peaks at 3.43°, 7.59°, and 12.54° and a broad peak at 20°–30° ascertaining the partial crystalline nature of the Porp-Tz framework [Fig. S2†]. Further, to study the type of linkages and gain more insights into the surface of the framework, X-ray photoelectron spectroscopy (XPS) was employed. The XPS survey scan interpreted the elemental composition of C, N, and S as present in the Porp-Tz. Fig. S3(a)† shows the typical chemical states for C, N, and S. For N 1s high-resolution XPS, three deconvoluted peaks corresponding to the protonated pyrrolic N of the porphyrin (–NH–, 399.9 eV), non-protonated pyrrolic N of porphyrin, and –S–CN– of the thiazole ring (–NC–, 397.2 eV),22 and N of the imine bond (–CN–, 398.6 eV) [Fig. S3(b)†].23 These investigations indicate the successful synthesis of the polymer Porp-Tz.
 | ||
| Scheme 1 Synthesis of Porp-Tz. | ||
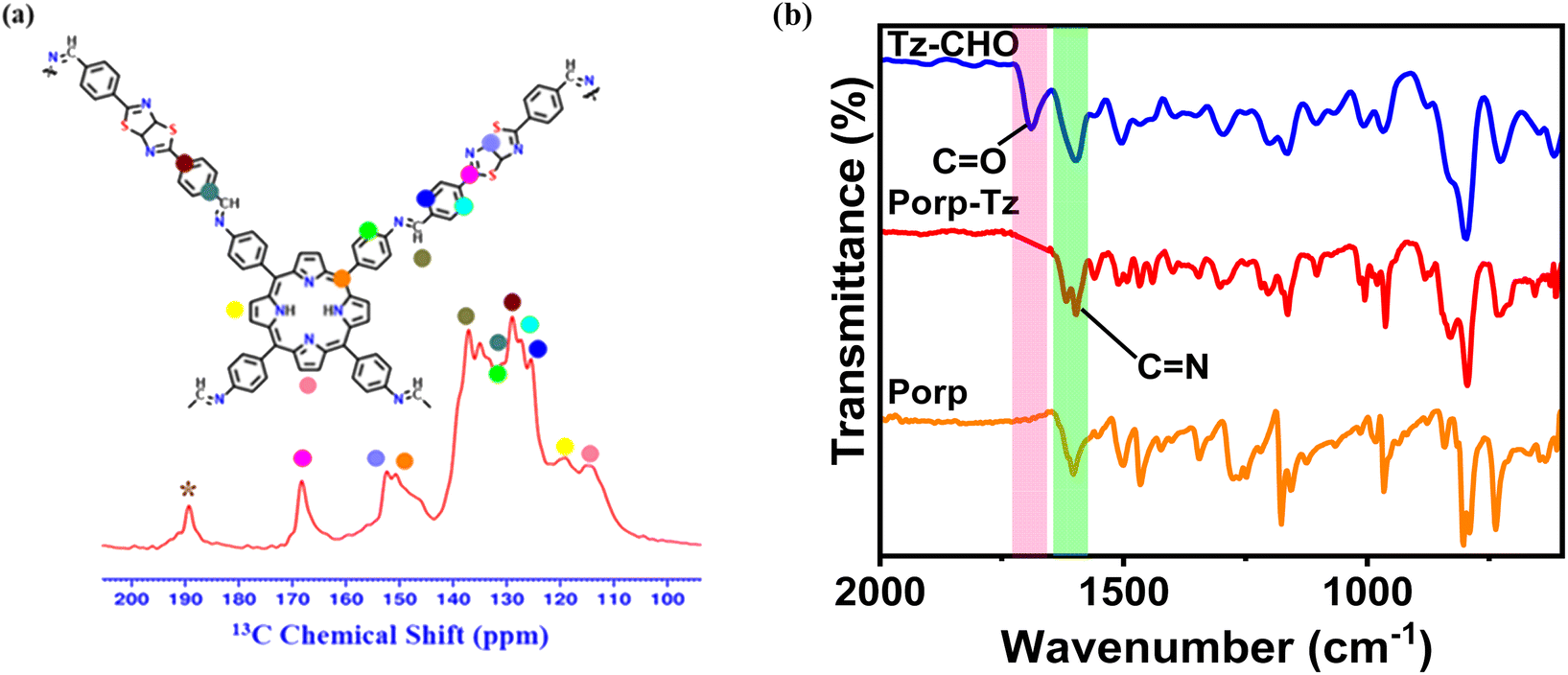 | ||
| Fig. 1 (a) 13C CP/MAS solid-state NMR spectra and (b) FTIR spectra. | ||
From the FESEM and TEM images, the rod-shaped morphology of the Porp-Tz can be visualized [Fig. S4†]. The POP exhibited excellent porosity by manifesting a characteristic of the Type-IV sorption isotherm with a Brunauer–Emmet–Teller (BET) specific surface area of 186 m2 g−1 [Fig. S5(a)†]. The porous characteristics of the polymeric framework were studied at 77 K by the N2 physisorption measurements, and the pore size distribution was derived from the nonlinear density functional theory (NLDFT) with a majority of pore diameter ranging from 0.3 nm to 2.3 nm [Fig. S5(b)†]. The BET surface area measurements were further validated using the linear BET plot [Fig. S5(c)†].
Additionally, to determine the robustness of the photocatalyst, the thermogravimetric analysis (TGA) was carried out under an inert atmosphere, and stability tests in different solvents were carried out. Porp-Tz is stable up to 430 °C, signifying its thermal stability [Fig. S6†]. The chemical stability of the photocatalyst was checked in common organic solvents, such as acetonitrile, dimethylformamide, dimethyl sulfoxide, dichloromethane, tetrahydrofuran, ethyl acetate, hexane, boiling water, and 6 M HCl and 6 M NaOH solution [Fig. S7†]. The as-synthesized polymer was immersed under different solvents for 3 days; then, the FTIR spectra of the collected powder were recorded [Fig. S8†]. As shown in Fig. S8,† after the rigorous treatment, FTIR spectra showed no change in the peaks of the collected powders, indicating the chemical stability of the photocatalyst across an extensive array of organic solvents, acids, and bases.24
After structural characterization, the optical properties of the polymeric framework were investigated. UV-vis diffuse-reflectance spectroscopy (DR UV-vis) showed a typical light absorbance behaviour of a porphyrin unit in conjugation with the thiazole unit.19 The Q-band region is observed in the range of 450–700 nm with absorbance at 480, 540, 590, and 670 nm, while the typical Soret band was observed around 360 nm with extended visible light absorption of up to 800 nm, which is typical of porphyrin materials.19 The optical band gap was calculated using a Tauc plot of 1.70 eV [Fig. 2(a) inset]. Photoluminescence (PL) emission spectroscopy was performed at an excitation of 420 nm to assess the transfer efficiency of the photoinduced charge carriers. Fig. 2(b) depicts the PL emission spectra of the Porp-Tz and the corresponding porphyrin amine monomer, where the maximum emission was observed at 720 nm. Predictably, the emission intensity of the Porp-Tz polymer diminishes, indicating that Porp-Tz exhibits a rapid and effective separation of the photogenerated charge carriers.
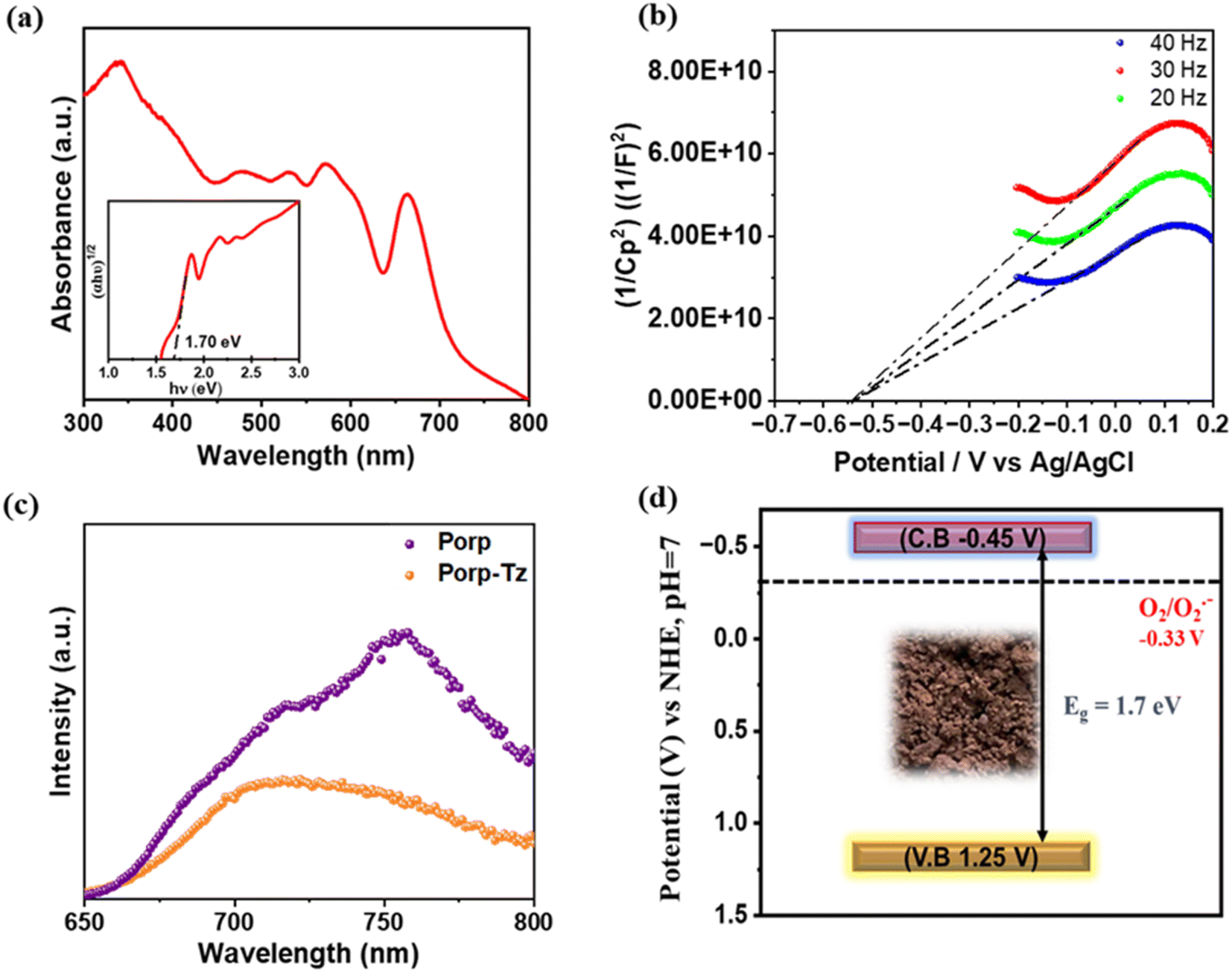 | ||
| Fig. 2 (a) DR UV-vis spectra and Tauc plot (inset); (b) PL spectra; (c) Mott–Schottky plot; and (d) band energy diagram of Porp-Tz. | ||
For a better understanding of the electronic and band structure properties, Mott–Schottky (MS) experiments were performed. A positive slope of the MS curve was observed, indicating that Porp-Tz exhibits a typical n-type semiconductor characteristic [Fig. 2(c)]. The flat band potential was found to be −0.49 V, which is considered to be positioned at 0.1 V below the conduction band (CB) minimum for the n-type semiconductor.
Thus, from the flat band potential, the potential of the conduction band (ECB) of the Porp-Tz was calculated to be +1.25 V (vs. Normal Hydrogen Electrode (NHE), ECB) using the equation Nernst equation (ENHE = EAg/AgCl + 0.197, at pH = 7). The valence band potential (EVB) was calculated from the ECB and Tauc plot, which is estimated to be −0.45 V vs. NHE at pH = 7. Thus, the obtained band energy diagram indicates that CBM and VBM are well-suited for the reduction of oxygen and the oxidation of the benzylamines and THIQs, respectively [Fig. 2(d)]. The optical properties of the polymer Porp-Tz were compared with the monomer Porp [Fig. S9†]. From the UV-vis spectra in Fig. S9(a),† it was evident that the formation of the polymer resulted in better light absorption characteristics, as shown by Porp-Tz. Further from Fig. S9(b),† it was observed that the photocurrent generation significantly increased after the formation of the polymer Porp-Tz, indicating an enhanced charge carrier mobility, which was further validated by a shorter arc radius and lower charge resistance of Porp-Tz in the Nyquist plot, suggesting that the polymer is more favorable for the charge transportation process in photocatalysis [Fig. S9(c)†]. Moreover, in the static PL measurements, the monomer Porp gave an intense emission peak, and it was observed that the emission intensity of Porp-Tz dropped rapidly, inhibiting the charge recombination process in Porp-Tz [Fig. S9(d)†].
Subsequently, the charge transfer dynamics signify a rapid photoinduced charge separation and transfer after polymerization, which is assuredly attributed to the porphyrin and thiazole monomeric units serving as excellent electron-donor and electron-acceptor, respectively.
Photocatalytic activity
Instigated by the optical properties of Porp-Tz and inspired by the light absorption efficiency, the photocatalytic activity was evaluated by facilitating the organic transformation reactions in a selective manner with the concurrent production of H2O2 in oxygen-saturated acetonitrile solution.25–27 23 W table lamp (White LED) was chosen as a light source to regulate this photo-redox transformation.Photocatalytic homocoupling of benzylamine
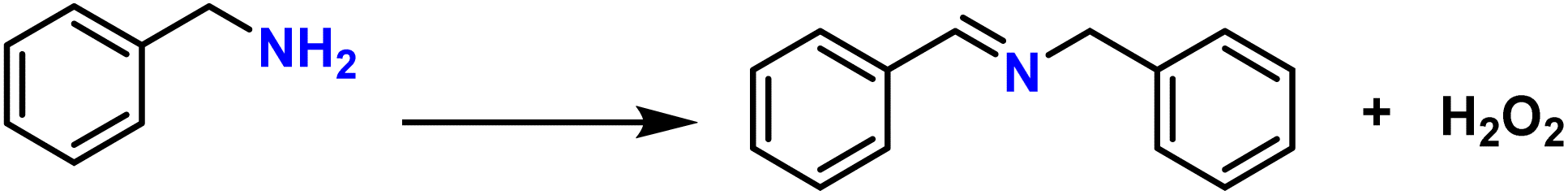
To probe the catalytic activity of Porp-Tz, 1 mmol of benzylamine was chosen as the substrate model compound. Benzylamine undergoes aerobic oxidative homocoupling through the synergetic role of photogenerated holes and electrons to yield corresponding dibenzylimines (DBI) along with the two-electron oxygen reduction in the concurrent production of H2O2. Complete conversion of benzylamine was achieved in 1 h with >99% selectivity along with the simultaneous production of 25.13 mmol g−1 h−1 of H2O2 [Fig. 3(a)]. Porp-Tz exhibited enhanced photocatalytic activity in the visible region among the other metal-free photocatalysts for the co-production of H2O2 alongside the synthesis of benzylamine [Table S2†]. Further, the photocatalytic aerobic homocoupling of benzylamine was carried out by employing the monomer Porp and observed that Porp exhibited insignificant photocatalytic activity when evaluated against the polymeric framework [Fig. S10†]. The photocatalytic performance of Porp was relevant to its optical and electrochemical results when compared with the Porp-Tz. Furthermore, it was observed that the photocatalytic performance under atmospheric conditions is comparable with the oxygen environment [Fig. 3(b)]. To ascertain the natural tendency of the catalyst, the reaction was performed under natural sunlight. Surprisingly, the complete conversion was observed in 0.5 h along with the production of 19.8 mmol g−1 h−1 of H2O2. Based on the above experimental results, it can be inferred that the Porp-Tz exhibits excellent photocatalytic efficiency and applicability under an ambient atmosphere. In the aforementioned photocatalytic reaction, no coupled product (DBI) or H2O2 was observed in the absence of a catalyst or without light irradiation [Fig. 3(c)]. Out of sheer interest, we conducted the heterocoupling of benzylamine with benzyl alcohol, achieving complete conversion in 1 h and H2O2 production rate of 18.7 mmol g−1 h−1. However, surprisingly, the selectivity of the benzylimine was reduced [Fig. S11†].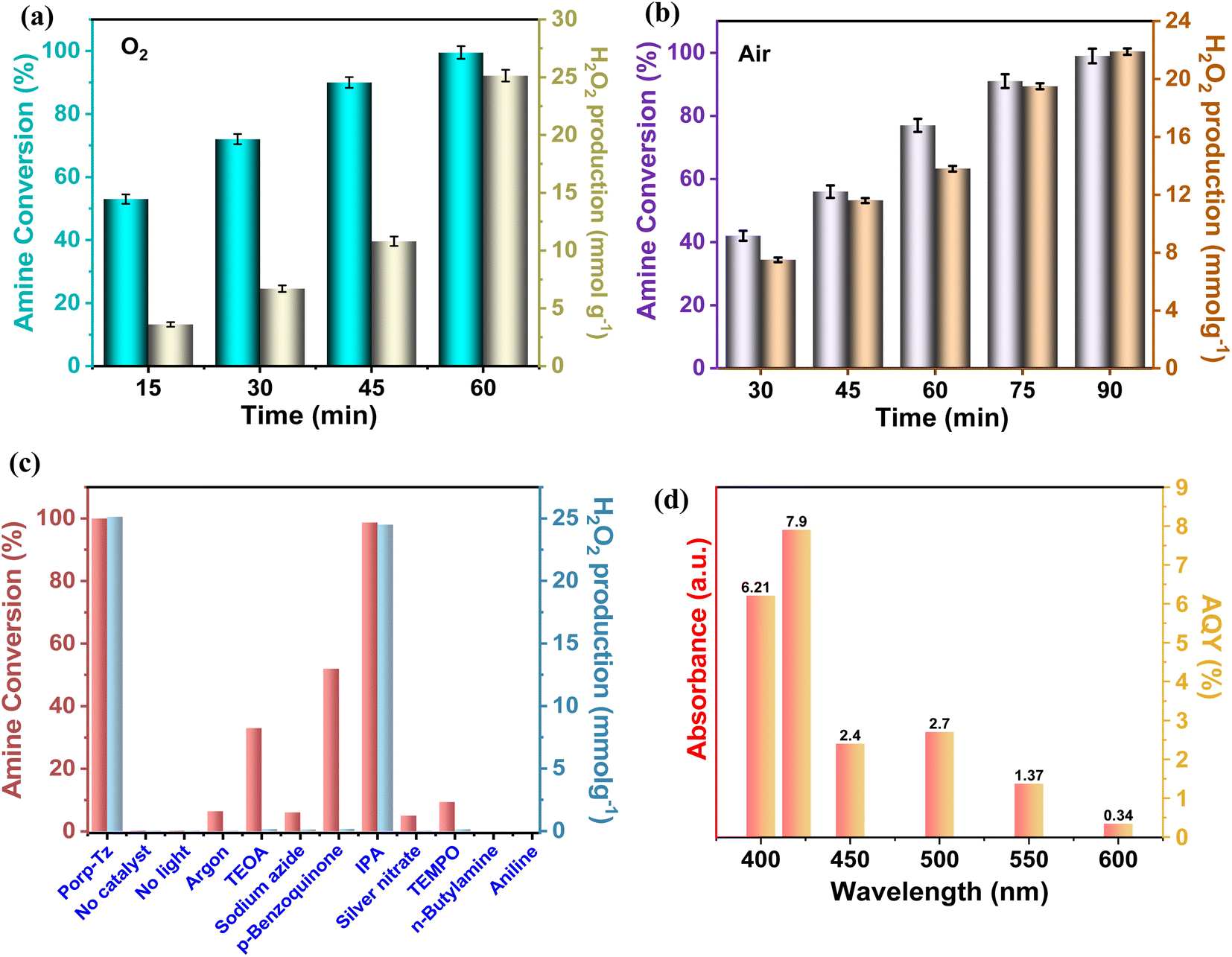 | ||
| Fig. 3 (a) Time-dependent amine conversion and H2O2 in O2; (b) time-dependent amine conversion and H2O2 production in air; (c) controlled experiments (TEOA – hole scavenger, sodium azide-singlet oxygen quencher, p-benzoquinone-superoxide quencher, IPA-hydroxyl radical scavenger, and silver nitrate-electron scavenger); and (d) apparent quantum yield of Porp-Tz at different wavelengths. | ||
Moreover, for the analysis of the apparent quantum yield (AQY%) of H2O2, homocoupling of benzylamine reactions was carried out across various monochromatic light emissions of different wavelengths, ranging from 400 nm to 600 nm with the highest value (AQY%) found to be 7.9% at 420 nm wavelength [Table S4†]. This resonates with the region of maximum absorbance in the DR UV-vis spectra [Fig. 3(d)].
Substrate scope
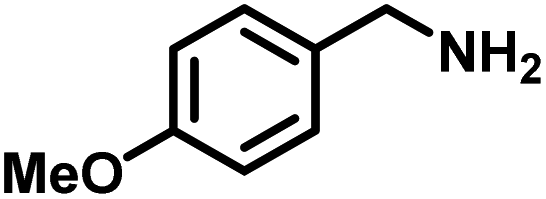
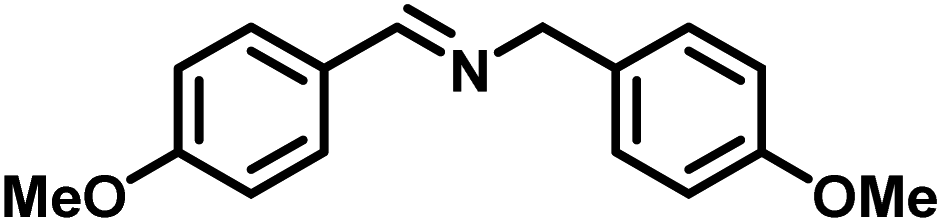
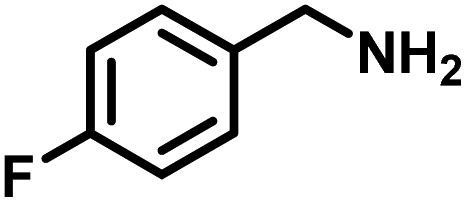
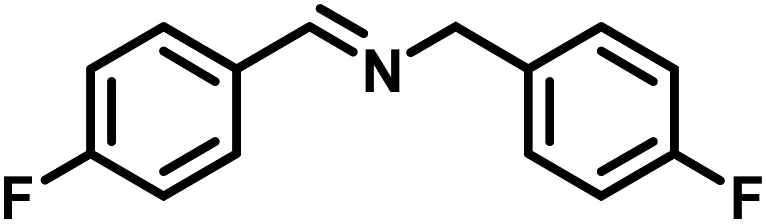
Upon successful optimization of the reaction conditions for carrying out oxidative benzylamine coupling, other derivatives of benzylamine were investigated to demonstrate the universal applicability of the photocatalyst [Table 1]. It is evident from the table that the maximum conversion (>99%) of 4-methoxy benzylamine was observed in 1 h with the maximum yield of 28.5 mmol g−1 h−1 of H2O2, and for 4-chloro benzylamine, only 82% of the conversion was noted along with the lowest production rate of H2O2 [Table 1]. This observation was consistent with the electron-donating and electron-accepting electronic effects of the derivatives.|
|
||||
|---|---|---|---|---|
| S. No. | Substrate | Product | Benzylamine conversion (%) | H2O2 (mmol g−1) |
| a Reaction conditions: Porp-Tz (5 mg); benzylamine substrates (1 mmol); ACN (20 mL); O2; RT; 1 h; and White LED (23 W). | ||||
| 1 |
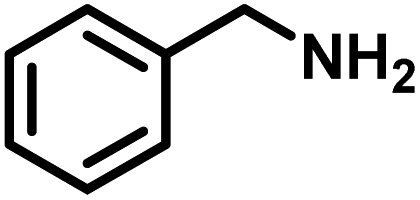
|
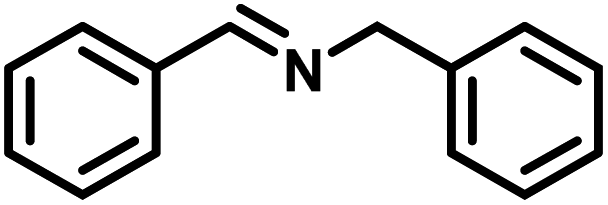
|
>99 | 25.13 |
| 2 |
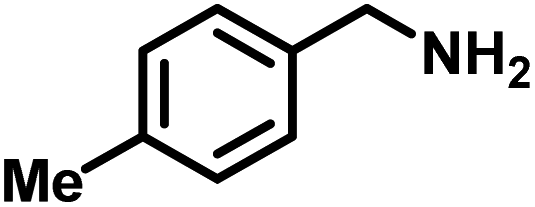
|
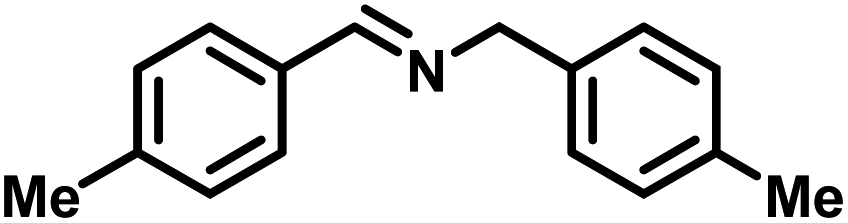
|
>99 | 25.45 |
| 3 |
|
|
>99 | 28.50 |
| 4 |
|
|
92 | 20.13 |
| 5 |
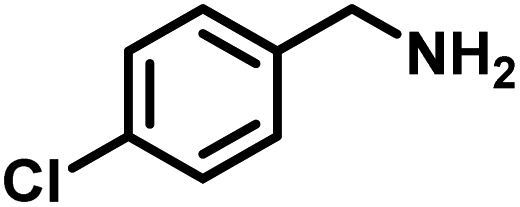
|
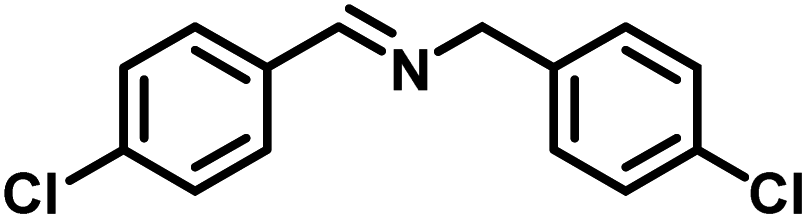
|
82 | 13.60 |
| 6 |
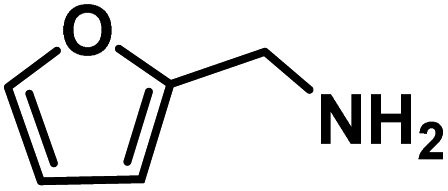
|
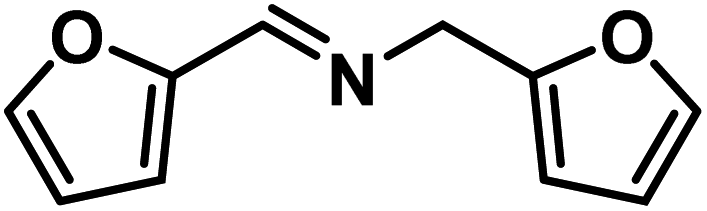
|
64 | 0.80 |
Photooxidative semi-dehydrogenation of 1,2,3,4-tetrahydroisoquinoline
The remarkable photocatalytic performance of the Porp-Tz encouraged us to explore its photocatalytic capabilities further. Herein, semi-dehydrogenative oxidation of 1,2,3,4- tetrahydroisoquinolines (THIQs) and simultaneous 2e− oxygen reduction reactions were achieved to obtain more value-added 3,4-dihydroisoquinolines (DHIQs) with high regioselectivity along with the co-production of H2O2. As shown in Fig. 4, the Porp-Tz has good photocatalytic performance with the maximum conversion of THIQs of 91% in 2 h with 94.1% regioselective formation of the desired product DHIQs along with the 13.34 mmol g−1 h−1 concurrent production of H2O2. This kind of synergistic transformation has been reported for the first time among metal-free POPs.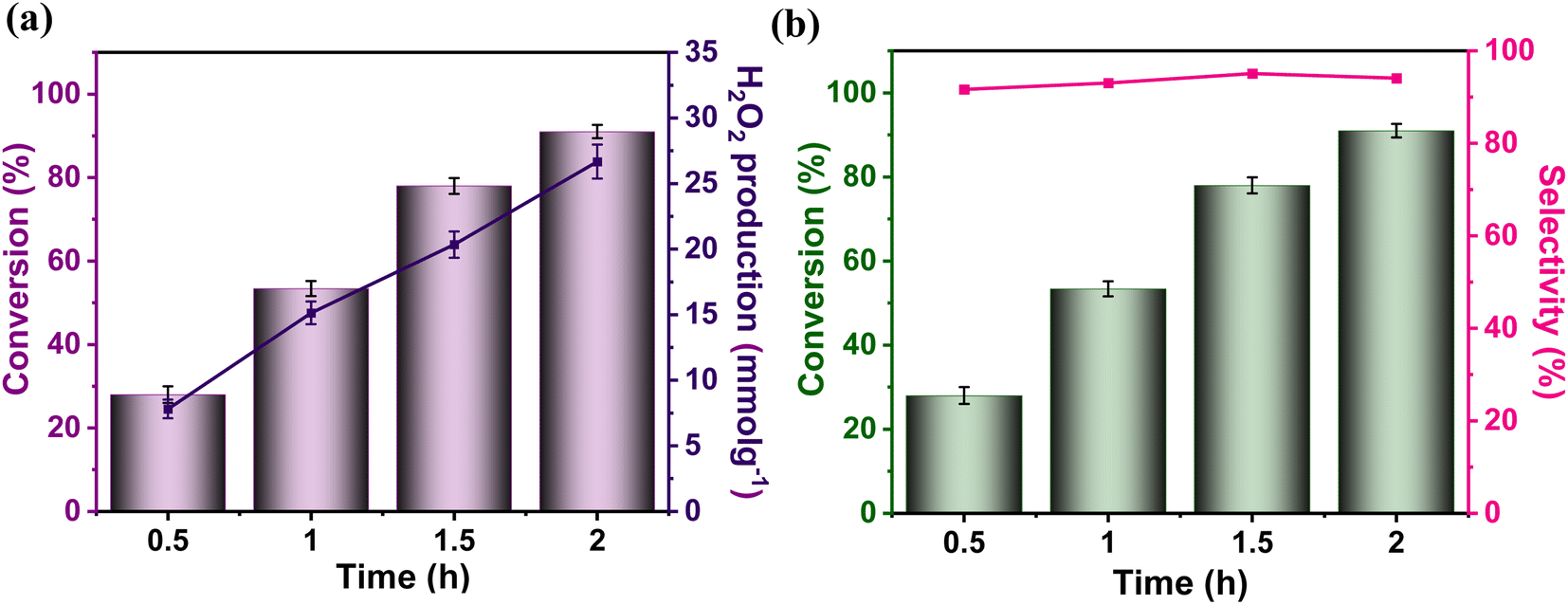 | ||
| Fig. 4 (a) Time-dependent THIQ conversion and H2O2 production, and (b) DHIQ selectivity of Porp-Tz. | ||
Other derivatives of THIQ were also investigated for a future scope using the photocatalyst Porp-Tz. As shown in Table 2, electron-donating substituents, such as –OMe and –Me, increase the dehydrogenation process, and electron-withdrawing substituents, such as –NO2 and –Br, decrease the photocatalytic activity due to the electronic effect.
|
|
|||||
|---|---|---|---|---|---|
| S. No. | Substrate | Product | THIQ conversion (%) | DHIQ selectivity (%) | H2O2 (mmol g−1) |
| a Reaction conditions – Porp-Tz (5 mg); THIQ (1 mmol); ACN (20 mL); O2; RT; 2 h; and White LED (23 W). | |||||
| 1 |
|
|
91.0 | 94.1 | 26.67 |
| 2 |
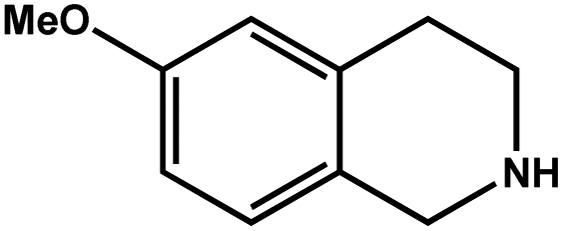
|
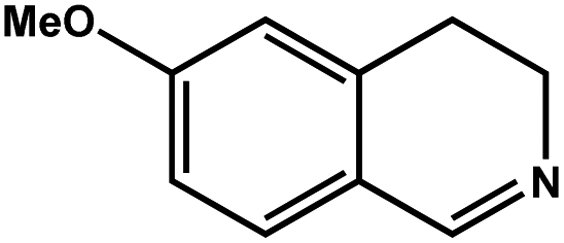
|
93.4 | 93.4 | 27.07 |
| 3 |
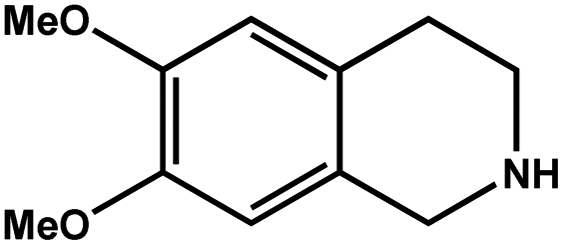
|
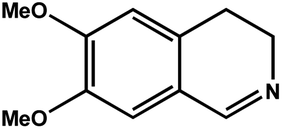
|
92.8 | 92.7 | 29.74 |
| 4 |
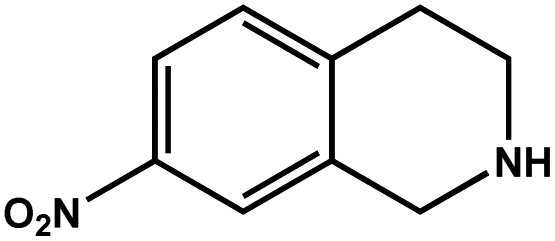
|
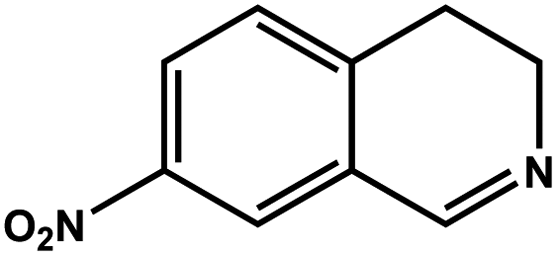
|
57.6 | 93.4 | 18.54 |
| 5 |

|
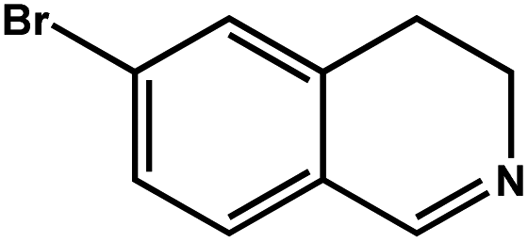
|
72.0 | 94.0 | 20.40 |
| 6 |
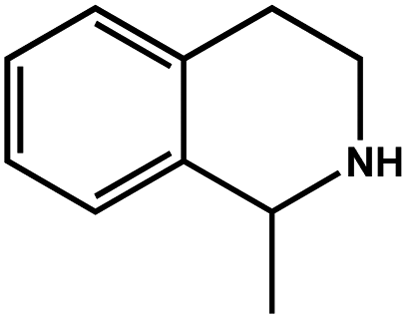
|
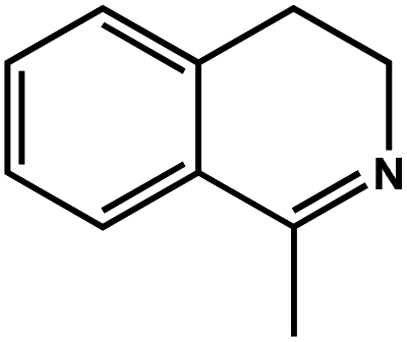
|
94.6 | 97.4 | 26.27 |
| 7 |
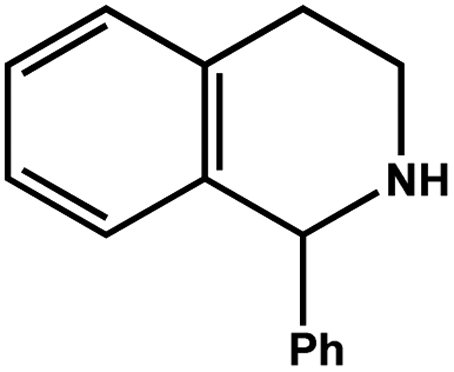
|
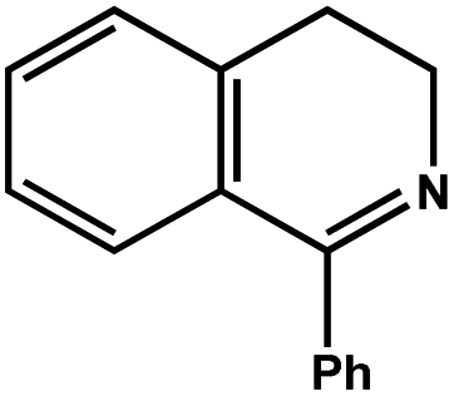
|
78.0 | 98.2 | 24.90 |
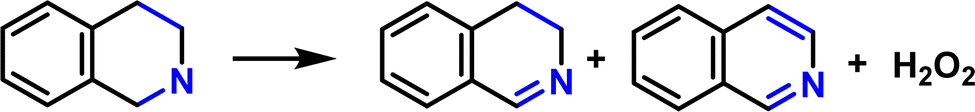
Furthermore, out of curiosity, the dehydrogenation for the substrate having a derivative within the ring consisting of a heteroatom was examined. As expected, for THIQs consisting of a phenyl ring at the ortho position to the heteroatom, the conversion decreases. In this case, both electronic and steric effects play a role. Therefore, Porp-Tz is successfully employed as an excellent photocatalyst for the regioselective semi-dehydrogenation of the partially saturated N-heterocycles along with the simultaneous production of H2O2.
Dynamics of charge carriers
Photo-electrochemical (PEC), photoluminescence (PL), time-resolved fluorescence decay (TR-PL) experiments, and electron-paramagnetic resonance (EPR) were carried out to investigate the photogenerated charge transfer ability and dynamics of the reaction mechanism. It was observed that under photocurrent response (30 s on/off), Porp-Tz showed a rapid response toward visible light. This indicates that a considerable number of photoexcited holes and electrons undergo separation upon illumination for better utilization of photogenerated charge carriers enhancing the photocatalytic activity [Fig. 5(a)]. Moreover, to obtain a deeper understanding of the charge transfer mechanism, PL and in situ EPR experiments were performed in the presence of the substrate. The PL intensity decreased upon the addition of benzylamine to the photocatalyst [Fig. 5(b)], signifying a reduced recombination rate and an enhanced photogenerated charge transfer process. Furthermore, to gain more insight into the dynamics of the photoinduced charge carriers, the average emission lifetime of the photogenerated charge carriers of Porp-Tz was calculated using the time-resolved photoluminescence (TRPL) spectroscopy technique and was found to be 3.10 ns, which was sufficient to initiate the reaction process vigorously [Fig. 5(c)]. No EPR signal was obtained in the absence of light. However, upon irradiation in an inert atmosphere, a strong intense peak was observed at g = 2.0042 corresponding to the paramagnetic defects in the porphyrin system [Fig. 5(d)], intimating that the photocatalyst promoted the efficient separation of the photoexcited charge carriers. Nevertheless, with the addition of a drop of benzylamine, the EPR signal significantly diminished, facilitating the enhancement in the charge carrier mobility and efficient transfer along the system.19 Based on the PEC, PL, and in situ EPR studies along with the scavenger studies to be discussed in the next section, the mechanistic pathway for the co-production of H2O2 along the oxidation process of benzylamine and THIQs is illustrated.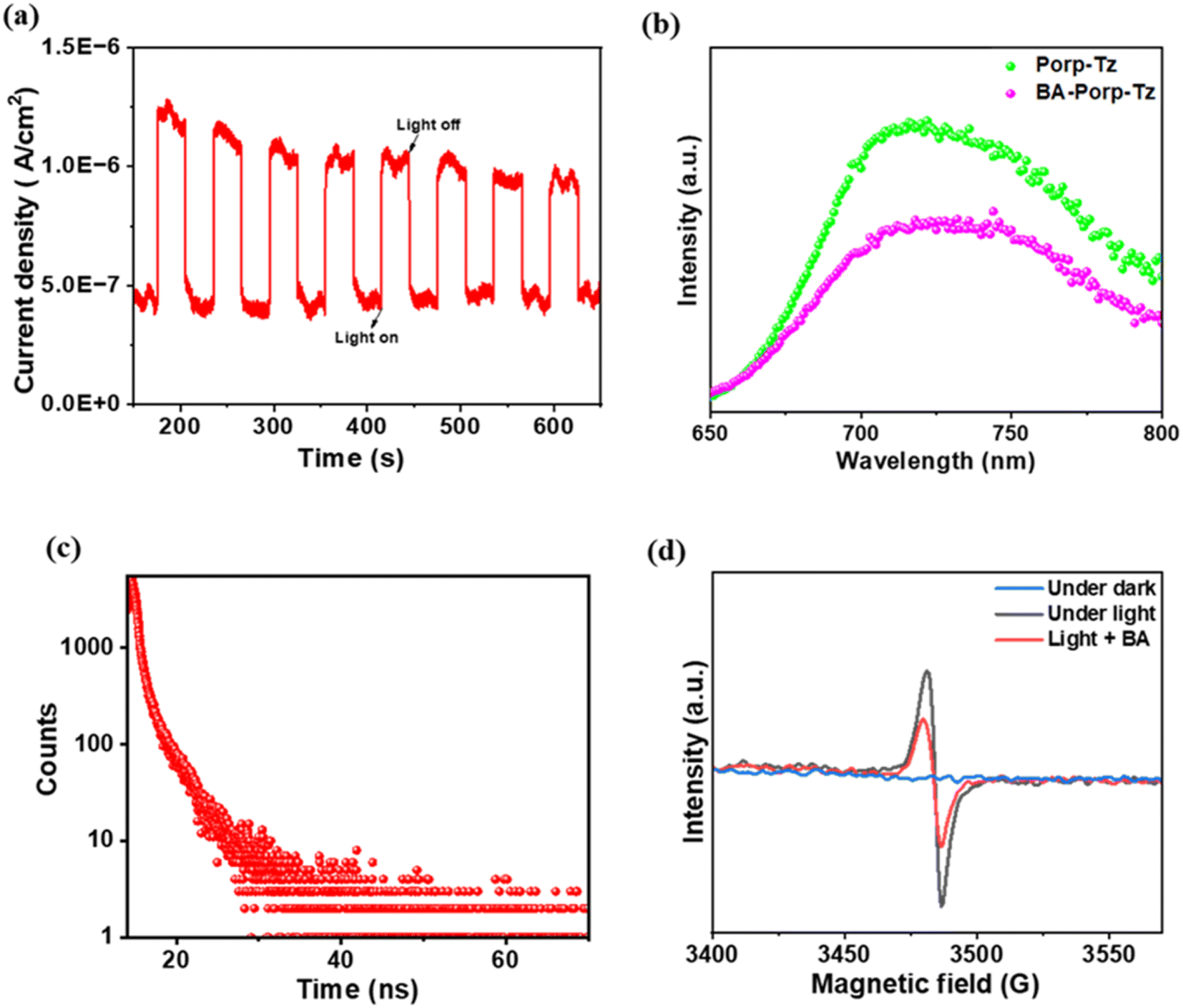 | ||
| Fig. 5 (a) Photocurrent studies of Porp-Tz; (b) PL spectra on the addition of benzylamine; (c) TR-PL spectra of Porp-Tz; and (d) EPR spectra on the addition of benzylamine. BA-benzylamine. | ||
Scavenger studies
Fig. 3(c) depicts that in the argon atmosphere, the benzylamine conversion reduced to 6.4% and H2O2 yield decreased to 0.021 mmol g−1, showing that the oxygen acts as an oxidant to carry out photocatalytic oxidative reactions. However, to gain more insights into the reaction mechanism and understand the co-production of H2O2 and oxidation products, we investigated the radical species formed by carrying out free radical quenching experiments for the homocoupling benzylamines in the presence of different scavengers towards their respective radical species (O2˙−, h+, e−, and ˙OH). As shown in Fig. 3(c), the conversion of benzylamine and the yield of H2O2 exhibit a dramatic decrease upon the addition of NaN3, TEMPO, AgNO3, and TEOA.20,28 No characteristic decrease in the photocatalytic activity was observed after the addition of IPA, indicating that ˙OH was not an active species in this transformation process, which was further validated by employing PL emission spectroscopy [Fig. S12†]. Thus, singlet oxygen, superoxide radicals, electrons, and holes are of substantial importance for oxidative conversions coupled with H2O2 production. Further, to obtain the detailed role of singlet oxygen species, a controlled experiment with 1,5-dihydroxy naphthalene (DHN) was performed.29 In the UV-vis spectra, the adsorption peak at 298 nm corresponding to DHN decreases, and the peak at 419 nm of 5-hydroxy-1,4-naphthalenedione (juglone) progressively increases, suggesting the formation of juglone upon light irradiation24 [Fig. S13†]. Therefore, both superoxide radical and singlet oxygen act as reactive oxygen species (ROS), which was further confirmed by in situ EPR experiments. A quartet signal was specifically derived from the interaction of DMPO with the O2˙− corresponding to the formation of superoxide radical species [Fig. 6(a)].30 2,2,6,6-tetramethylpiperidine (TEMP) was used as a trapping agent for the detection of 1O2, and 5,5-dimethyl-1-pyrroline N-oxide (DMPO) was employed as a trapping agent to detect O2˙−. Upon light irradiation, a specific triplet signal (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1), corresponding to the TEMPO radical, was observed in the presence of the catalyst Porp-Tz, as illustrated in Fig. 6(b), radical was formed due to the reaction of TEMP with the 1O2 revealing the formation of singlet oxygen.30
:1:1), corresponding to the TEMPO radical, was observed in the presence of the catalyst Porp-Tz, as illustrated in Fig. 6(b), radical was formed due to the reaction of TEMP with the 1O2 revealing the formation of singlet oxygen.30
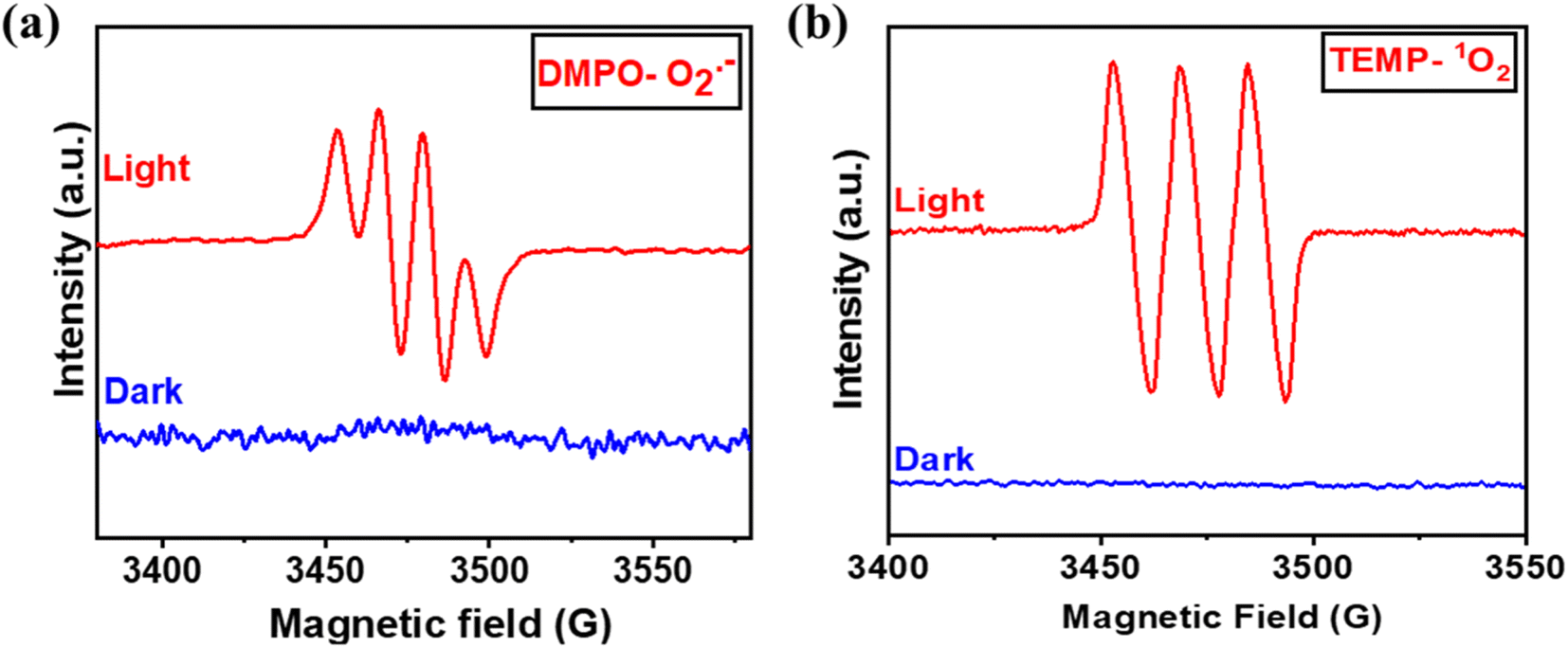 | ||
| Fig. 6 (a) Determination of O2˙− using DMPO as trapping agent; and (b) 1O2 using TEMP as trapping agent by applying EPR spectrometer. | ||
The above interesting results can be summarised only by scrutinizing the mechanistic pathway approach of the photocatalytic reaction.
Reaction mechanism
The scavenger and in situ EPR experiment clearly illustrate the formation of both ROS species, and it is also evident that O2˙− and 1O2 are the prerequisites of reactive oxygen species for oxidative transformations along with the concurrent H2O2 production. The photocatalyst Porp-Tz was excited to Porp-Tz* after irradiation under visible light, but this excited state was quenched through the single electron transfer (SET) or the energy transfer process generating ROS species. Oxygen being a good electron acceptor is activated by the excited catalyst (Porp-Tz*) to generate the superoxide radicals via SET and the singlet oxygen via the energy transfer process, as shown in Scheme 2. Herein, two proton donor systems were employed for the selective production of desired products: one is the oxidative homocoupling of benzylamine and the second is the oxidative semi-dehydrogenation of 1,2,3,4-tetrahydroisoquinoline. Herein, the production of H2O2 entirely depends on the proton donors, as the hydrogen transfer process during the generation of cationic radicals from the substrate compound is the key step in the formation of H2O2.28 A plausible reaction mechanistic pathway is discussed in more detail for both photocatalytic processes.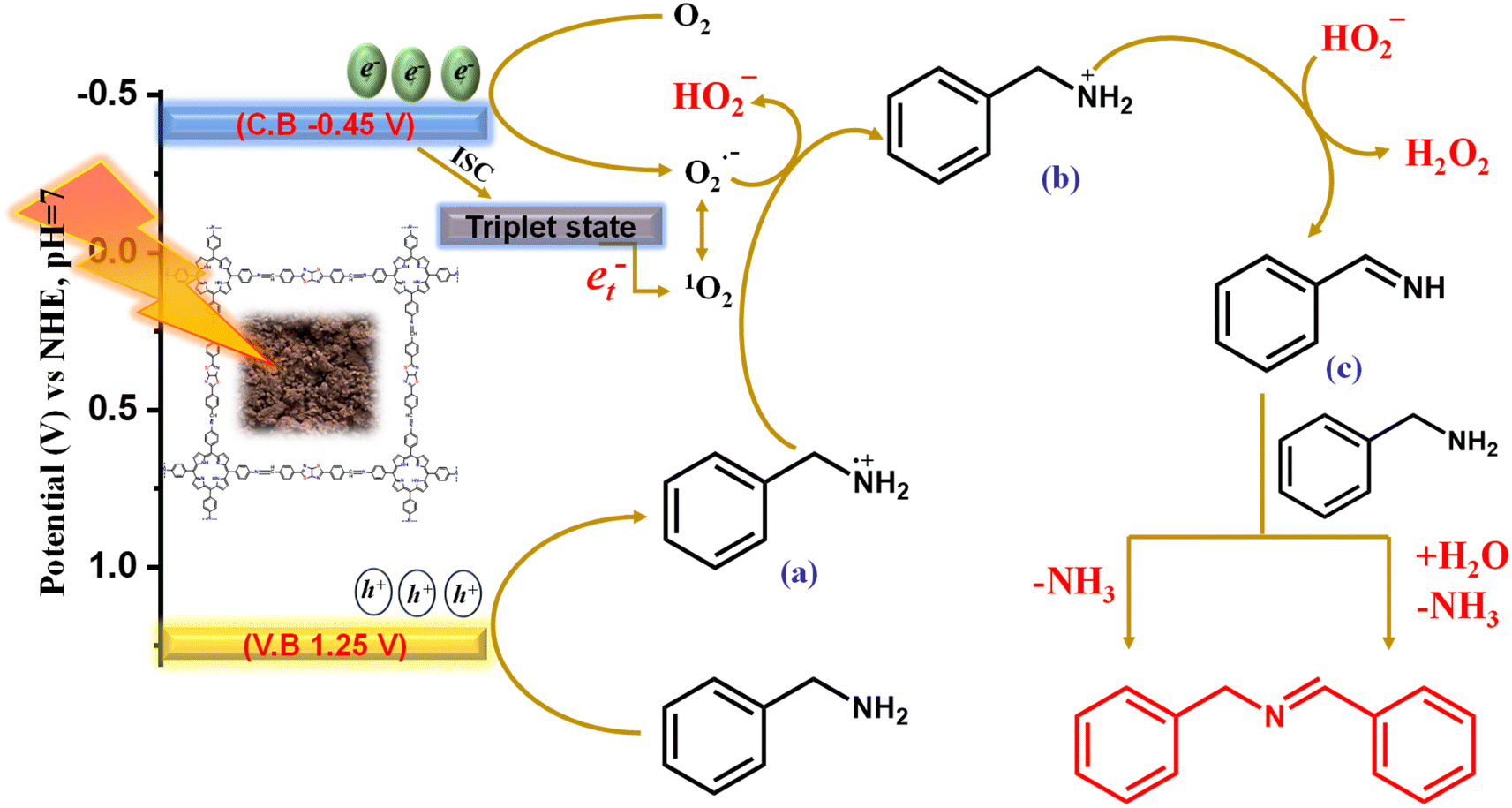 | ||
| Scheme 2 Mechanistic reaction pathway of oxidative coupling of benzylamine catalysed by Porp-Tz. | ||
Photooxidative homocoupling of benzylamine
As illustrated in Scheme 2, the more negative potential of the oxygen surface site makes the electrons available for the reduction of molecular oxygen to the superoxide radical species. Conversely, the molecular oxygen (O2) is also converted to singlet oxygen through the energy transfer process (et−) between photogenerated excitons and the O2via intersystem crossing (ISC).31 As the superoxide radical and singlet oxygen are interconvertible in the presence of holes, both participate in the mechanism process.26 Meanwhile, in the valence band, the adsorbed benzylamine traps photogenerated holes to form a benzylamine radical cation intermediate (a). Usually, the amine radical cation formed exhibits four modes of activity: (1) back electron transfer reaction; (2) hydrogen atom abstraction from (a) when a strong hydrogen atom acceptor is present to produce iminium ion; (3) deprotonation yielding α-amino radical; and (4) cleavage of C–C bond α to the nitrogen atom forming a neutral free radical and iminium ion.32,33 Among these, the second mode of employing imine radical cation as a source of hydrogen atom abstractor has been of great interest for many visible light-mediated organic transformation reactions, and we have employed this mode for the conversion of benzyl cation radical to benzyl iminium ion (b) via proton abstraction from (a) using an efficient proton abstractor O2˙−. The proton abstraction step was verified using aniline and n-butylamine as the substrate, and it was evident that the reaction did not proceed in this case as no benzylic proton (α-H) was available for abstraction in the formation of (b) [Fig. 3(c)]. Further, the iminium ion is deprotonated by hydroperoxyl ion (HOO−) for the production of H2O2 in the process of the generation of benzylimine (c). This is the primary pathway for the production of H2O2 from molecular O2. Eventually, the H2O2 produced was analyzed by applying an iodometric method for the production of yellow-colored I3− whose absorbance was monitored at 350 nm and increased with the reaction time [Fig. S14(a)†]. The benzylimine (c) produced was further converted to the desired product DBI in two reaction pathways: either it reacts with another molecule of benzylamine to form the condensed product DBI with the liberation of NH3 or it reacts with water to form benzaldehyde, which further reacts with benzylamine to give DBI with the liberation of the NH3 molecule. The NH3 liberated was analyzed by carrying out˙− Nessler's reagent test in which a peak at 440 nm was observed in the UV-vis spectra corresponding to the iodomercurate complex formed by the reaction of NH3 with the Nessler's reagent [Fig. S14(b)†]. The product was then analyzed by applying GCMS [Fig. S15†].Photooxidative semi-dehydrogenation of N-heterocycles
After the successful generation of the reactive oxygen species via the photogenerated electrons, the photogenerated holes in the valence band oxidize the THIQs to generate the THIQ cationic radical (a).20 The cationic radical formed again reacts with the reactive oxygen species to form an imine intermediate (b), which further yields DHIQs via the H-transfer pathway along with the co-production of H2O2 [Scheme 3]. H2O2 is produced in the same way as described in Scheme 2. The products were analyzed by applying GCMS [Fig. S16†].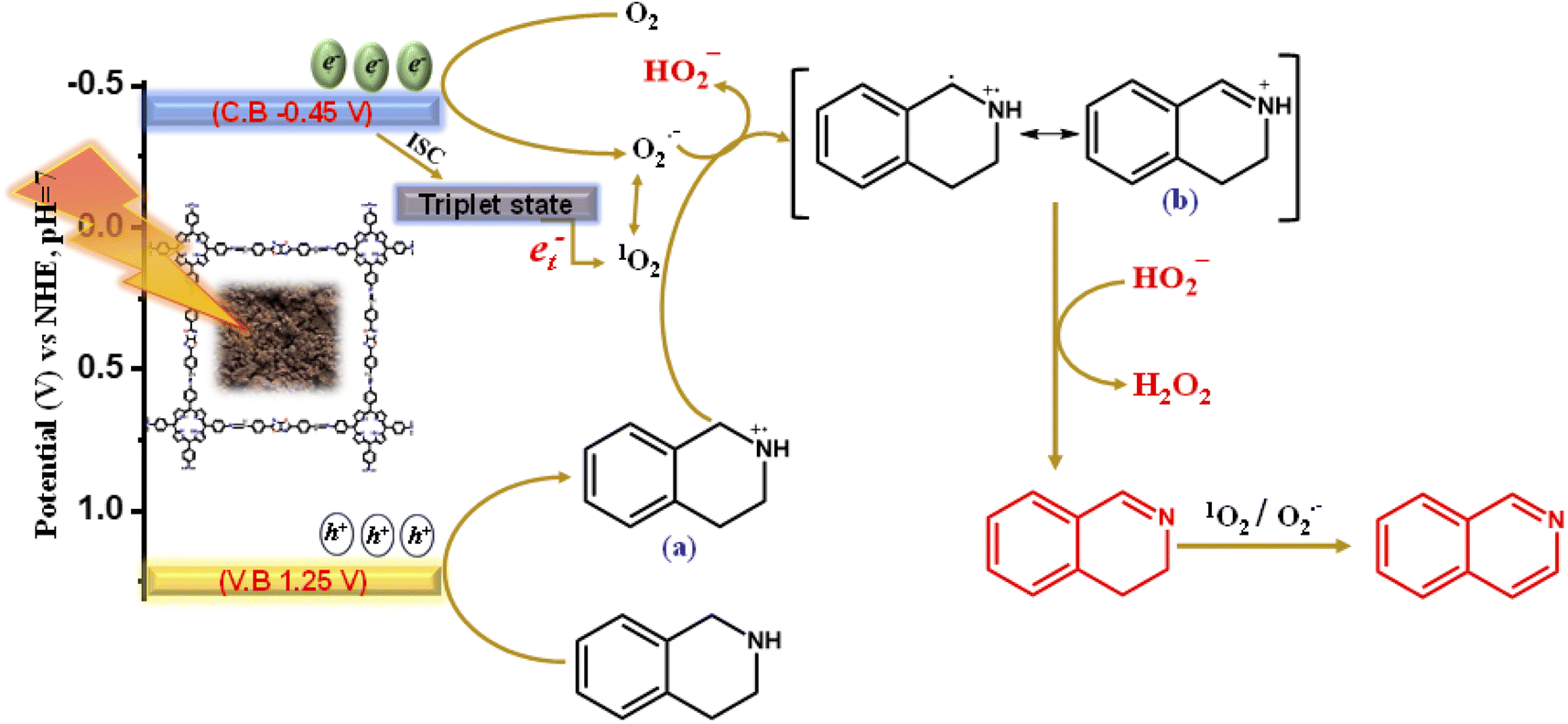 | ||
| Scheme 3 Mechanistic reaction pathway of the complete oxidative dehydrogenation of THIQs catalyzed by Porp-Tz. | ||
Recyclability studies
Recyclability experiments were performed for homocoupling benzylamine to test the reusability of Porp-Tz, and it was investigated that the catalytic ability of the Porp-Tz remained intact even after five cycles without any significant decrease in the conversion and change in the selectivity of the products [Fig. S17†]. FT-IR, N2 physisorption analysis, TGA analysis, DR-UV vis, and PXRD were compared after the recyclability with the pristine one to investigate any change in the properties of the photocatalyst [Fig. S18†]. No observable change was recorded in all the investigations except for a decrease in the BET surface area of 102 m2 g−1. Thus, it was shown that the chemical structure, morphology, and surface compositions of Porp-Tz remained intact after the photocatalytic experiments [Fig. S18†].Conclusion
It can be summarized that the metal-free Porp-Tz developed a multi-catalytic system by employing a porphyrin and dithiazole-linked porous organic polymer to carry out the efficient photo-oxidation of organic substrates to high value-added products with the concurrent production of H2O2 through the proficient separation of the charge carriers. Excellent light absorption, lower band gap, and suitable band positions characterized by Porp-Tz attributed it to outperform the previous reports under visible light for synergistic transformation to high value-added chemicals along with co-production of H2O2. Excitingly, Porp-Tz achieved an AQY of 7.9% at 420 nm for the H2O2 production, which outperformed the previous literature reports to the best of our knowledge. A detailed mechanistic pathway is explained to highlight the role of proton donors and other reactive species in the production of H2O2. This work opens up a new approach towards sustainability from a more economical perspective by the synchronous utilization of both the photogenerated charge carriers maximizing the redox capabilities of the photocatalyst.Data availability
The data supporting this article are included in the ESI.†Author contributions
K. Dhingra: conceptualization, synthesis, investigations, methodology, data collection, writing the draft. N. Saini: data validation and data analysis. A. Kumar: synthesis, data collection and analysis. K. Kailasam: conceptualization, supervision, writing review, and editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
K. Dhingra acknowledges UGC, New Delhi for the fellowship. K. Kailasam thanks INST Mohali, for the funding and instrumental facilities. The authors also thank the Central Research Facility, IIT Delhi (Sonepat campus) for the EPR studies.References
- M. Ikram, A. Raza, S. O. A. Ahmad, A. Ashfaq, M. U. Akbar, M. Imran, S. Dilpazir, M. Khan, Q. Khan and M. Maqbool, Adv. Mater. Interfaces, 2023, 10, 2202172 CrossRef.
- A. I. Osman, Y. Zhang, M. Farghali, A. K. Rashwan, A. S. Eltaweil, E. M. Abd El-Monaem, I. M. A. Mohamed, M. M. Badr, I. Ihara, D. W. Rooney and P.-S. Yap, Environ. Chem. Lett., 2024, 22, 841–887 CrossRef.
- T. Zhang, G. Xing, W. Chen and L. Chen, Mater. Chem. Front., 2020, 4, 332–353 RSC.
- X. Guo, L. Wang, L. Wang, Q. Huang, L. Bu and Q. Wang, Front. Chem., 2023, 11 DOI:10.3389/fchem.2023.1116524.
- S. Fajal, S. Dutta and S. K. Ghosh, Mater. Horiz., 2023, 10, 4083–4138 RSC.
- Z. Zhang, J. Jia, Y. Zhi, S. Ma and X. Liu, Chem. Soc. Rev., 2022, 51, 2444–2490 RSC.
- Q.-Y. Liu, J.-F. Li and J.-W. Wang, J. Incl. Phenom. Macrocycl. Chem., 2019, 95, 1–15 CrossRef CAS.
- B. B. Rath, S. Krause and B. V. Lotsch, Adv. Funct. Mater., 2023, 2309060 CrossRef.
- F. Liu, I. Rincón, H. G. Baldoví, A. Dhakshinamoorthy, P. Horcajada, S. Rojas, S. Navalón and A. Fateeva, Inorg. Chem. Front., 2024, 11, 2212–2245 RSC.
- B. Ge, Y. Ye, Y. Yan, H. Luo, Y. Chen, X. Meng, X. Song and Z. Liang, Inorg. Chem., 2023, 62, 19288–19297 CrossRef CAS PubMed.
- K. Wu, X.-Y. Liu, M. Xie, P.-W. Cheng, J. Zheng, W. Lu and D. Li, Appl. Catal., B, 2023, 334, 122847 CrossRef CAS.
- N. Huang, L. Zhai, D. E. Coupry, M. A. Addicoat, K. Okushita, K. Nishimura, T. Heine and D. Jiang, Nat. Commun., 2016, 7, 12325 CrossRef PubMed.
- V. R. Battula, B. Rawat and K. Kailasam, ACS Appl. Polym. Mater., 2023, 5, 1989–1997 CrossRef CAS.
- Y. Wu, X. Deng, R. Cui, M. Song, X. Guo, X. Gong, J. He and P. Chen, J. Colloid Interface Sci., 2024, 656, 528–537 CrossRef CAS PubMed.
- Z. Tian, C. Han, Y. Zhao, W. Dai, X. Lian, Y. Wang, Y. Zheng, Y. Shi, X. Pan, Z. Huang, H. Li and W. Chen, Nat. Commun., 2021, 12, 2039 CrossRef CAS.
- W. Wang, W. Gao, X. Nie, W. Liu, X. Cheng, N. Shang, S. Gao and C. Wang, J. Colloid Interface Sci., 2022, 616, 1–11 CrossRef CAS PubMed.
- W. Gao, Y. Fu, X. Nie, Y. Zhao, C. Sun, N. Shang, X. Cheng, S. Gao, B. Tian and C. Wang, ChemPhotoChem, 2024, e202300294 CrossRef CAS.
- G. Sirvinskaite, J. C. Reisenbauer and B. Morandi, Chem. Sci., 2023, 14, 1709–1714 RSC.
- N. Saini, N. Sharma, D. K. Chauhan, R. Khurana, M. E. Ali and K. Kailasam, J. Mater. Chem. A, 2023, 11, 25743–25755 RSC.
- Y. Chen and J. Jiang, Sustain. Energy Fuels, 2021, 5, 6478–6487 RSC.
- P. Li, Y. Tian, L. Tian and Y. Wang, Org. Biomol. Chem., 2024, 22, 725–730 RSC.
- Z. Xu, X. Cui, Y. Li, Y. Li, Z. Si and Q. Duan, Appl. Surf. Sci., 2023, 613, 155966 CrossRef CAS.
- S. Chen, Q. Wan and A. K. Badu-Tawiah, Angew. Chem. Int. Ed., 2016, 55, 9345–9349 CrossRef CAS PubMed.
- N. Sharma, D. K. Chauhan, N. Saini and K. Kailasam, ACS Appl. Polym. Mater., 2023, 5, 4333–4341 CrossRef CAS.
- F. M. Yap, G. Z. Sheng Ling, B. J. Su, J. Y. Loh and W.-J. Ong, Nano Res., 2024, 3, e9120091 Search PubMed.
- J. Luo, X. Wei, Y. Qiao, C. Wu, L. Li, L. Chen and J. Shi, Adv. Mater., 2023, 35, 2210110 CrossRef CAS.
- L. Li, X. Huo, S. Chen, Q. Luo, W. Wang, Y. Wang and N. Wang, Small, 2023, 19, 2301865 CrossRef PubMed.
- N. Sharma, S. Kumar, V. R. Battula, A. Kumari, A. Giri, A. Patra and K. Kailasam, Chem.–Eur. J., 2021, 27, 10649–10656 CrossRef PubMed.
- R. Liang, J. Luo, S. Lin, Z. Li, Z. Dong, Y. Wu, Y. Wang, X. Cao, C. Meng, F. Yu, Y. Liu and Z. Zhang, Sep. Purif. Technol., 2023, 312, 123291 CrossRef.
- B. Rawat, V. R. Battula, P. K. Nayak, D. Ghosh and K. Kailasam, ACS Appl. Mater. Interfaces, 2023, 15, 53604–53613 CrossRef.
- W.-K. An, X. Xu, S.-J. Zheng, Y.-N. Du, J. Ouyang, L.-X. Xie, Y.-L. Ren, M. He, C.-L. Fan, Z. Pan and Y.-H. Li, ACS Catal., 2023, 13, 9845–9856 CrossRef.
- J. Hu, J. Wang, T. H. Nguyen and N. Zheng, Beilstein J. Org. Chem., 2013, 9, 1977–2001 CrossRef.
- G. Che, W. Yang, C. Wang, M. Li, X. Li and Q. Pan, Inorg. Chem., 2022, 61, 12301–12307 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis procedures and characterization techniques of the catalyst. See DOI: https://doi.org/10.1039/d4ta04445b |
| This journal is © The Royal Society of Chemistry 2024 |