
Collagen-decorated electrospun scaffolds of unsaturated copolyesters for bone tissue regeneration†
Heloísa Bremm
Madalosso
a,
Camila
Guindani
 b,
Bianca Chieregato
Maniglia
c,
Pedro Henrique
Hermes de Araújo
a and
Claudia
Sayer
*a
b,
Bianca Chieregato
Maniglia
c,
Pedro Henrique
Hermes de Araújo
a and
Claudia
Sayer
*a
aDepartment of Chemical Engineering and Food Engineering, Federal University of Santa Catarina, Campus Trindade, 88040-900, Florianópolis, Brazil. E-mail: claudia.sayer@ufsc.br
bChemical Engineering Program/COPPE, Federal University of Rio de Janeiro, Cidade Universitária, CP: 68502, Rio de Janeiro, 21941-972 RJ, Brazil
cSão Carlos Institute of Chemistry, University of São Paulo – USP, Campus São Carlos, 13566-590, São Carlos, SP, Brazil
First published on 20th February 2024
Abstract
Many efforts have been devoted to bone tissue to regenerate damaged tissues, and the development of new biocompatible materials that match the biological, mechanical, and chemical features required for this application is crucial. Herein, a collagen-decorated scaffold was prepared via electrospinning using a synthesized unsaturated copolyester (poly(globalide-co-pentadecalactone)), followed by two coupling reactions: thiol–ene functionalization with cysteine and further conjugation via EDC/NHS chemistry with collagen, aiming to design a bone tissue regeneration device with improved hydrophilicity and cell viability. Comonomer ratios were varied, affecting the copolymer's thermal and chemical properties and highlighting the tunable features of this copolyester. Functionalization with cysteine created new carboxyl and amine groups needed for bioconjugation with collagen, which is responsible for providing biological and structural integrity to the extra-cellular matrix. Bioconjugation with collagen turned the scaffold highly hydrophilic, decreasing its contact angle from 107 ± 2° to 0°, decreasing the copolymer crystallinity by 71%, and improving cell viability by 85% compared with the raw scaffold, thus promoting cell growth and proliferation. The highly efficient and biosafe strategy to conjugate polymers and proteins created a promising device for bone repair in tissue engineering.
1. Introduction
Scaffolds for tissue engineering consist of 3D-assembled materials acting as an artificial extra-cellular matrix (ECM) capable of supporting the attachment of cells and guiding their regeneration, thereby ensuring cell viability and growth.1 For this reason, in addition to biologically desired properties, these scaffolds require an ideal morphological structure, with their architecture governing the physiology and delivery of nutrients to cells and promoting vascularization and diffusion of nutrients and oxygen.2 To this end, choosing a proper technique to assemble these 3D structures allows for the achievement of desired mechanical characteristics, high cell adhesion, and high porosity as required for tissue engineering devices.3,4 In this context, electrospinning has emerged as a widely applied process to design scaffolds with topographical features, and microscale to nanoscale fibers with facile processability, allowing a high control of scaffold properties such as fiber diameter and scaffold porosity, resembling the natural ECM.5–8 This technique also enables the interconnection between the porous fiber structures, facilitating cell migration and nutrient transportation during healing processes, matching the requirements for tissue engineering applications.2 Regarding the materials applied to compose these 3D-assembled structures, aliphatic polyesters derived from macrolactones have gained attention owing to their suitable and tunable properties, such as biocompatibility and biodegradability.9,10 They can be synthesized using approaches with a high control of molecular weight and dispersity, such as ring-opening polymerization (ROP),11 which can be catalyzed by enzymes (e-ROP), such as Novozym 435 (an immobilized enzyme, Candida antarctica lipase fraction B). For biomedical applications, the use of enzymes is particularly important because it results in polymers without any remaining toxicity, avoiding several purification steps, which are required when chemical catalysts are applied.12 Lipases are capable of acting as a catalyst for e-ROP of several lactones and macrolactones, such as ε-caprolactone,12,13 ω-pentadecalactone,14 and globalide,15 resulting in faster reactions under lower temperatures.11However, applying homopolymers derived from lactones in biomedical devices still presents some drawbacks, which are mainly related to their high hydrophobicity, low hydrolysis rate, and poor resorbability. To overcome these problems, copolymerization with unsaturated macrolactones followed by conjugation with biomolecules appears to be an effective approach to obtaining materials with tailored features, mainly related to mechanical behavior, crystallinity, hydrolysis, biodegradation rates, and thermal properties.16 Besides, polymer functionalization can also lead to desirable polymer properties, allowing specific interactions within biological systems. The direct functionalization during e-ROP is highly challenging since the reaction does not tolerate the presence of functional groups such as hydroxyl or amines, which act as initiators of this reaction.17 A feasible and fast alternative for post-polymerization modification is the click reactions, such as thiol–ene. In this reaction, combinations of alkenes and thiols occur with high reaction rates, forming harmless products.17,18 Thus, choosing specific functional groups to add to the copolymer structure and tuning the ratio between the monomers allows the design of polymers with desired physicochemical properties or affinity for different human tissues.19 These possibilities turn functionalized polyesters into great candidates for developing biofunctional materials.
The incorporation of amino acids (AA), proteins, and peptides in a polymer chain represents an excellent alternative to tune the biomaterial's properties, allying features of conventional artificially degradable polymers and available peptides with enhanced hydrophilicity and biocompatibility.20 Cysteine, a hallmark of anti-oxidative stress, consists of a hydrophilic and reactive thiol-containing amino acid with the ability to react with oxygen and nitrogen species, being easily bound on polymer chains via thiol–ene chemistry.21,22 The previous functionalization with amino acids such as cysteine on a polyester chain introduces hydrophilic functional groups (i.e., amines and carboxylic acids), which enabled the conjugation between the polymer and biomacromolecules, potentially improving the material's performance for biomedical applications. In this sense, collagen, a right-handed bundle of three polypeptide chains, appears as a great candidate to be covalently bound to these hydrophilic functional groups. Since it is one of the major proteins in the ECM, distributed in connective tissues, it plays a key role in creating biological and structural integrity in the designed scaffold.8,23 Besides, its amorphous structure can reduce the polymer crystallinity, and its hydrophilic behavior can reduce the overall polymer hydrophobicity, providing improved cell adhesion and proliferation.24,25
Collagen has been used as a potential material to build porous lattice sponges to support the in vitro growth of many types of tissues.26 Collagen-based scaffold materials can be designed either using collagen as the main component of the structure,27,28 or as a functional biomolecule employed to functionalize a specific surface. In the first case, a cross-linking agent, such as glutaraldehyde-based agents, is usually employed to achieve the required mechanical properties and stability. However, concerns related to cytotoxicity and bio incompatibility have been raised regarding these materials.26 In the latter case, collagen could be combined with several types of materials and can create biocompatible structures with high porosity and adhesion. Materials such as polymers derived from natural monomers,29–32 natural ceramics such as hydroxyapatite and tricalcium phosphate,32,33 or composite materials have been widely employed as main components of scaffolds to further collagen-based surface functionalization. These collagen combinations can be developed via polymeric blends, coatings involving physical adsorption in porous structures,31,34 or collagen covalently binding through bioconjugation.30 Bioconjugation appears to be an advantageous strategy to bind collagen to scaffolds due to the high stability of the formed covalent bonds and the more efficient collagen attachment when compared to other functionalization methods.29
In this context, the present work proposes the design of a novel 3D-assembled scaffold for bone tissue engineering. Herein, we accomplished the synthesis of the copolyester poly(globalide-co-ω-pentadecalactone) (PGlPDL), followed by its 3D-assembling into scaffolds by the electrospinning technique. The 3D scaffolds were then functionalized with cysteine via photo-initiated thiol–ene reactions, aiming to provide NH2 and COOH functional groups for further bioconjugation with collagen via carbodiimide chemistry, aiming to improve the hydrophilicity and cell viability. PGlPDL was synthesized via enzymatic ring-opening polymerization (e-ROP) using Novozym 435 as a catalyst. The effect of the comonomer ratio on the physicochemical properties of the copolymer was extensively studied. The resultant materials were analysed regarding their chemical structures, crystallinity, water contact angle, and melting temperatures. The interactions of the designed scaffolds with pre-osteoblast cells were evaluated in vitro in order to determine the material biocompatibility and mineralization performance. To the best of our knowledge, this work reports the bioconjugation of PGlPDL electrospun scaffolds with collagen for the first time. This study can be a starting point for designing bone-regenerator materials with proper characteristics, contributing to developing more effective medical devices.
2. Experimental
2.1 Material preparation
Dichloromethane P.A. 99.8% (DCM), propanone P.A. and ethanol P.A. were purchased from Vetec Química Fina Ltda. (Rio de Janeiro – Brazil) and used without previous purification. Toluene P. A. was purchased from Dinâmica Química Contemporânea Ltda. (São Paulo – Brazil) and used as received. The enzyme Novozym 435 was donated by Novozymes A/S (Barigui – Brazil), which corresponds to the commercial lipase B from Candida Antarctica immobilized on cross-linked polyacrylate beads. Esterification activity assays were performed according to a methodology proposed in the literature35 and resulted in 28.5 U g−1. Enzymes were dried under vacuum before polymerization for 24 h at 65 °C and stored in a desiccator over silica and 4 Å molecular sieves. The monomer ω-pentadecalactone was purchased from Sigma Aldrich (São Paulo – Brazil), and globalide was kindly donated by Symrise (São Paulo – Brazil). Both monomers were dried before polymerization for 24 h at 80 °C under vacuum conditions. After drying, they are also stored in a desiccator over silica and 4 Å molecular sieves.Cysteine hydrochloride was purchased from Vetec Química Fina Ltda. (Rio de Janeiro – Brazil), and the hydrolyzed collagen type I and Verisol® collagen, a pure bioactive peptide derived from collagen optimized for skin health, were purchased from Iberoquímica do Brasil (São Paulo – Brazil). Verisol® collagen has a shorter chain when compared with the hydrolyzed collagen, resulting from further enzymatic hydrolysis during its processing, which is expected to increase its bioavailability. They were employed without any pre-treatment. The photoinitiator Omnirad 2959 (1-[4-(2-hydroxyethoxyl)-phenyl]-2-hydroxy-2-methylpropanone) was kindly donated by IGM resins do Brasil Ltda. (São Paulo – Brazil). N-Hydroxysuccinimide (NHS, 98%) and N-(3-dimethylaminopropyl)-N′-ethylcarbodimide hydrochloride (EDC) were purchased from Sigma Aldrich (São Paulo – Brazil). Sodium chloride (NaCl), potassium chloride (KCl), disodium hydrogen phosphate (Na2HPO4), and potassium phosphate monobasic (KH2PO4) were purchased from Sigma Aldrich (São Paulo – Brazil) and employed to prepare the PBS and phosphate buffered solutions (at 0.15 M and 0.1 M, respectively).
2.2 Synthesis of poly (globalide-co-ω-pentadecalactone) via enzymatic ring-opening polymerization
The copolymer synthesis involving the comonomers globalide and ω-pentadecalactone was carried out in a 25 mL vial in a proportion of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of the total monomers to the solvent (toluene). To evaluate the influence of each comonomer on the final copolymers’ properties, different monomer feed mass ratios (Gl:PDL) were employed in the copolymer formulations: 100:0; 75:25, 50:50, 25:75, and 0:100. Monomers, solvent, and enzyme were weighed in a precision balance (ATX224 Shimadzu, Japan) and the reaction system flask was placed inside a sand bath to control the reaction temperature. The concentration of the enzyme Novozym 435 was fixed at 5 wt%, relative to the total amount of the monomers. After reaching the reaction temperature of 65 °C, the reaction proceeded for 2 h under magnetic stirring. The reaction conditions were chosen based on previous studies,13–15 where similar macrolactones were employed in the synthesis of copolyesters. Novozyme 435 and its amount were chosen based on the results of previous works36 that elucidated the effect of enzyme amount and enzyme nature on the kinetics of e-ROP.
:1 of the total monomers to the solvent (toluene). To evaluate the influence of each comonomer on the final copolymers’ properties, different monomer feed mass ratios (Gl:PDL) were employed in the copolymer formulations: 100:0; 75:25, 50:50, 25:75, and 0:100. Monomers, solvent, and enzyme were weighed in a precision balance (ATX224 Shimadzu, Japan) and the reaction system flask was placed inside a sand bath to control the reaction temperature. The concentration of the enzyme Novozym 435 was fixed at 5 wt%, relative to the total amount of the monomers. After reaching the reaction temperature of 65 °C, the reaction proceeded for 2 h under magnetic stirring. The reaction conditions were chosen based on previous studies,13–15 where similar macrolactones were employed in the synthesis of copolyesters. Novozyme 435 and its amount were chosen based on the results of previous works36 that elucidated the effect of enzyme amount and enzyme nature on the kinetics of e-ROP.
After the polymerization reaction, the copolymers were purified, being first solubilized in dichloromethane (DCM), followed by the enzyme removal by filtration and precipitation of the polymer chains in cold ethanol:acetone (3:1) solution to separate the copolymers from residual monomers or oligomers. For the precipitation, the cold mixture of ethanol and acetone was added to the polymer solution in a proportion of 1:6 (v/v). After the precipitation, copolymers were dried in an oven at 60 °C overnight. The purification followed the same procedure proposed by Guindani and coworkers13 and was confirmed by NMR analysis since no monomer peaks remained in the spectrum after the purification step. Typical yields of the copolymerization reactions were 70%.
2.3 Scaffold preparation via electrospinning
Scaffold conformation was carried out in an electrospinning apparatus and a fixed collector using operational conditions of 10 kV of tension, 15 cm of distance tip-to-collector, and a flow rate of 1 mL h−1. All copolymers were solubilized in chloroform to design 5 different doping solutions. In the first electrospinning tests, the solution concentration of all copolymer ratios was set at 25 wt%, which ranges into the typical polymer concentration employed in the electrospinning process.37 However, samples containing higher fractions of ω-pentadecalactone in the copolymer composition resulted in damaged fibers due to the high solution viscosity. For this reason, copolymers containing high amounts of globalide (GL:PDL of 100:0, 75:25, and 50:50) were employed in a concentration of 25 wt%, while the copolymers using the Gl:PDL mass ratios of 25:75 and 0:100 were solubilized in a concentration of 15 wt%. The copolymer concentrations were set based on the adequate viscosity range of the solutions that allowed electrospinning without the formation of beads. The electrospinning assays were carried out for 60 min.
2.4 Thiol–ene functionalization of poly (globalide-co-ω-pentadecalactone) scaffolds with cysteine
Scaffold functionalization assays involved polymers with various globalide contents (Gl:PDL ratios of 100:0, 75:25, 50:50, and 25:75). The thiol–ene reactions were photoinitiated by the hydrosoluble photoinitiator 1-[4-(2-hydroxyethoxy)phenyl]-2-hydroxy-2-methyl-1-propan-1-one (Omnirad 2959), in an aqueous media of 10 mL, using an excess of cysteine (5 times the stoichiometric amount required to functionalize all double bonds). The copolymer scaffolds were immersed in a solution containing cysteine and the photoinitiator, and the thiol–ene reaction was carried out for 5 h in a UV camera. After the reaction, the cysteine-functionalized PGlPDL scaffolds (PGlPDL-cys) were washed several times with distilled water to remove the remaining initiator and non-reacted cysteine.
2.5 Scaffold bioconjugation with collagen via EDC/NHS chemistry
For bioconjugation, the two types of collagens were separately solubilized in deionized water at a concentration of 1 mg mL−1. A PBS buffer solution was prepared at 0.15 M (pH 7.4), and a phosphate-buffered saline was prepared at 0.1 M (pH 6). The scaffolds functionalized with cysteine (PGlPDL-cys) containing 25% of PDL in the copolymer composition were immersed in 10 mL of a buffer phosphate-buffered saline (0.1 M, pH 6) for 10 min in a soaking step to enable the buffer to penetrate the fibers before activation. The activation of functional carboxyl groups consisted of immersing the fibers in a phosphate-buffered saline (0.1 M, pH 6) containing EDC + NHS, in a proportion of 1:3:1 (COOH:EDC:NHS) for 30 min. The stoichiometry was calculated considering 20% of double bonds functionalized with cysteine. Scaffolds were removed from the solution containing EDC + NHS and immersed in a PBS solution (45 mL) + collagen (5 mL, at 1 mg mL−1), which were retained for 24 h. All reaction steps were carried out at 25 °C. After the reaction, the collagen-decorated scaffolds (PGlPDL-hydrolyzed and PGlPDL-Verisol®) were washed several times using distilled water in order to remove the remaining non-reacted cross-linkers and collagen.
2.6 Copolymer characterizations
Poly(globalide-co-ω-pentadecalactone) 1H NMR (CDCl3 200 MHz): δ (ppm) 5.6–5.22 (m, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 4.14–3.97 (m, CH2O(CO)), 2.45–2.19 (m, CH2(CO)O), 2.18–1.82(m, CH2(CHCH)), 1.78–1.47, 1.45–1.08 (m, CH2).
CH), 4.14–3.97 (m, CH2O(CO)), 2.45–2.19 (m, CH2(CO)O), 2.18–1.82(m, CH2(CHCH)), 1.78–1.47, 1.45–1.08 (m, CH2).
000000 g mol−1. Chloroform was employed as an eluent with a volumetric flow rate of 1 mL min−1 at 40 °C. For the analysis, samples weighing 15 mg were dissolved in 4 mL of chloroform.
| γL = (1 + cosθ) = 2(γdL × γdS)1/2 + 2(γpL × γdS)1/2 | (1) |
It is important to denote the separation of γs into two components, one dispersive (γd) and the other polar (γp), through the relation γs = γd + γp. Subscripts S and L represent the solid and the liquid surfaces, respectively. In this way, inserting the values of θ obtained for the three solvents used, which have known values of γs, γd, and γp, the equation is solved in the SCA-20 software (USA). The contact angle assays were performed on the copolymer fibers before and after functionalization with cysteine and bioconjugation with collagen.
2.7 Cell viability and mineralization assays
Cell viability and mineralization tests were performed before and after the thiol–ene functionalization to determine cysteine's influence on biological scaffold properties. Similarly, these tests were performed after the bioconjugation of PGlPDL-cys scaffolds with collagen. To prepare the cell culture, pre-osteoblast cells of murine lineage MC3T3-E1 (American Type Culture Collection-ATCC) were cultured in minimal essential medium (α-MEM, Gibco), together with 10% fetal bovine serum and 1% (v/v) streptomycin/penicillin. After reaching confluence, the cells were trypsinized and resuspended in α-MEM, then seeded in 24-well plates with 5 × 104 cells per well density containing the scaffolds. One aluminum foil (without scaffold presence) was used as a control. The scaffolds containing the cells were maintained in an osteogenic medium (α-MEM supplemented with 10% fetal calf serum (FCS), 50 μg ml−1 of ascorbic acid, and 6mM of β-glycerophosphate). Cell viability was evaluated using the classic MTT assay following the methodology described by Faria and coworkers.38 Briefly, the samples incubated in the cell culture at 7 days (37 °C and 5% CO2 atmosphere) were added to 1.0 mg mL−1 of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5–diphenyltetrazolium) and then incubated at the same conditions for 4 h more. During this process, a tetrazolium salt, a highly coloured compound (formazan), upon reduction with NADH is produced, which reflects the cellular dehydrogenase activity. After 4 h of incubation, the formazan crystals were dissolved in 2-propanol and stirred until complete dissolution. Absorbance was read at 560 and 690 nm on a spectrophotometer (SpectraMaxM3. USA) to determine the concentration of mitochondrial dehydrogenase. Cell viability was expressed as the percentage of the average of 3 experiments, compared to the control (aluminum foil) for the period analyzed, 7 days (100%).
The formation of the mineralized matrix after 7 days of the samples in the cell culture was evaluated with the alizarin Red (AR) staining methodology described by Gregory, C. A.; Gunn, W. G.; Peister, A.; Prockop.39 To this end, the samples containing the cells were rinsed with PBS to remove possible nonspecific precipitated phosphate particles. The formation of mineralized nodules was quantified by dissolving the content of the wells after staining with AR in acetic acid and neutralizing with ammonium hydroxide. The absorbance at 405 nm was read with a spectrophotometer. Aluminum foil was used as a control.
3. Results and discussion
3.1 Synthesis and fiber conformation of poly(globalide-co-ω-pentadecalactone) (PGlPDL)
:PDL mass ratio was set at 100:0, 75:25, 50:50, 25:75, and 0:100. The mass ratio of the comonomers was varied to evaluate the influence of each monomer on the resultant copolymer properties. Other reaction parameters, such as temperature and time, were maintained constant in the experiments. The reaction yields after purification ranged from 65 to 90%. The general diagram of the polymerization reaction is illustrated in Fig. 1(a).
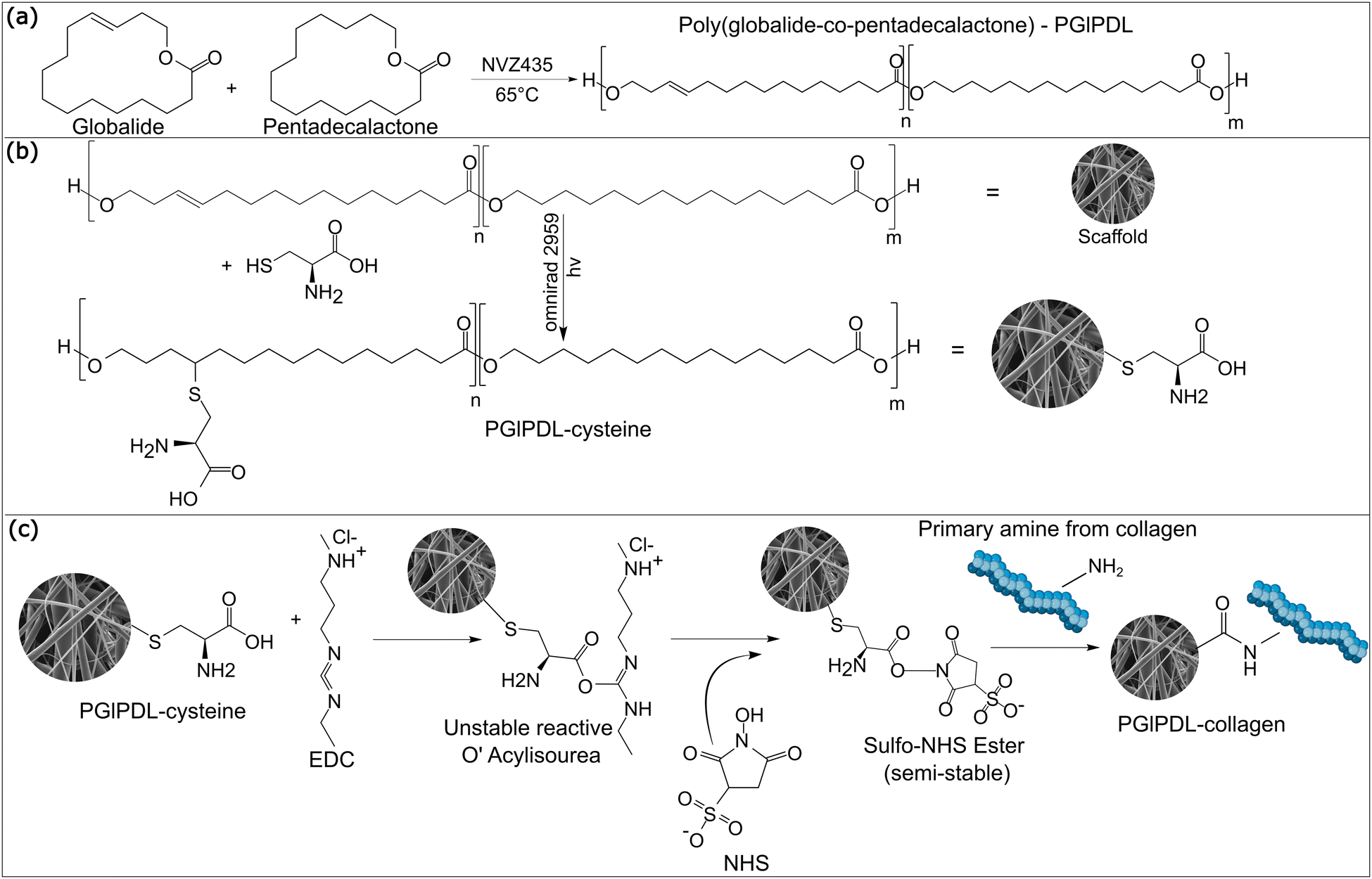 | ||
| Fig. 1 Representation of (a) enzymatic ring-opening copolymerization between Gl and ω-PDL, (b) thiol–ene functionalization of PGlPDL scaffolds with cysteine, and (c) bioconjugation of PGlPDL-cys scaffolds via EDC/NHS chemistry. | ||
Fig. 2 shows the 1H NMR spectrum of the copolymer with a 75:25 (Gl:PDL) monomer ratio. The monomer conversions were calculated based on the methylene peak (4.08–4.18 ppm). This peak is presented in the monomers and the final copolymers. In the latter, the peak was shifted to the right (3.98–4.10 ppm). The evaluation of monomer conversion was performed with the copolymers before purification by comparing the methylene peaks between the monomer and the final copolymer (Fig. S1, ESI†). The copolymer spectra did not present any peaks corresponding to the monomer, indicating the absence of any unreacted monomer in the analyzed samples. For this reason, independent of the initial comonomer ratio, all e-ROP reactions reached 100% conversion. Table 1 summarizes the 1H NMR spectroscopy and GPC results of the copolymers with different Gl:PDL ratios. The molecular weight distributions are presented in Fig. S2 (ESI†).
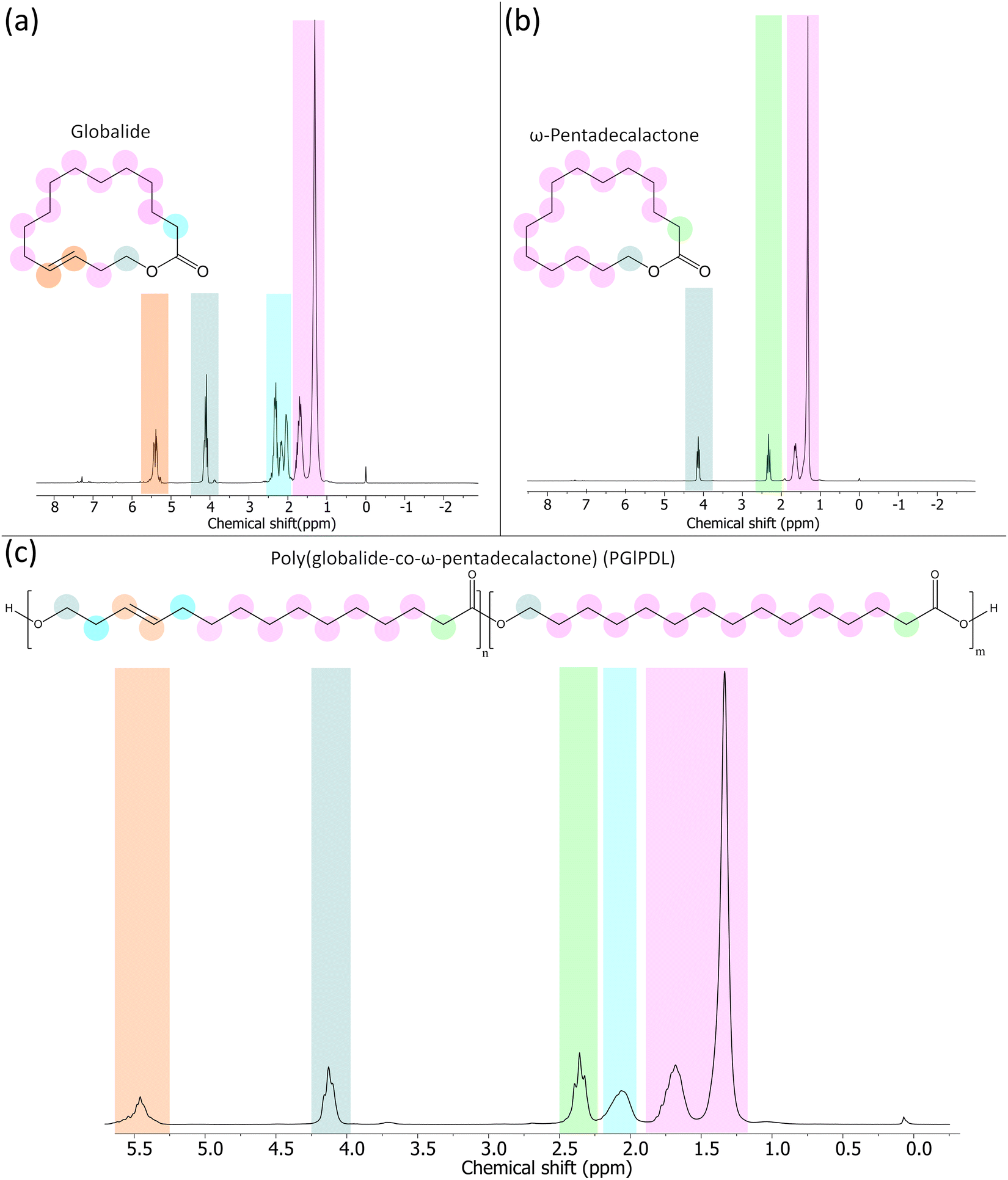 | ||
| Fig. 2
1H NMR of (a) globalide, (b) ω-pentadecalactone, and (c) PGlPDL at 75:25 (Gl:PDL) monomer ratio. | ||
:PDL ratios based on 1H NMR and GPC data
| Initial Gl:PDL (mol ratio) |
NMR Gl:PDL copolymer composition (mol ratio) |
M n | N (polymerization degree) | Đ | Double bonds per chain |
|---|---|---|---|---|---|
| 100:0 |
100:0 |
8440 | 35 | 3.96 | 35.0 |
| 75:25 |
70.4:29.6 |
9220 | 39 | 4.63 | 27.5 |
| 50:50 |
44.8:55.2 |
13860 |
58 | 4.31 | 26.0 |
| 25:75 |
22.1:77.9 |
13600 |
57 | 5.87 | 12.5 |
| 0:100 |
0:100 |
9930 | 41 | 7.24 | 0 |
By analyzing Table 1, it is possible to infer that both monomers were successfully copolymerized, as indicated by the excellent match of the experimental copolymer compositions with the theoretical ones. The resulting Mn values of all copolymer compositions ranged from 8.000–14.000 g mol−1, and no clear tendency with copolymer composition could be evidenced. Similar results were obtained by Tinajero-Diaz, Ilarduya and Muñoz-Guerra40 in the copolymerization of Gl and ω-PDL, with Mn in the range of 9.000–12.000 g mol−1 for different copolymer compositions, without apparent correlation between Mn and the measured composition. In the copolymerization of Gl and ε-CL via e-ROP, Guindani, Dozoretz, Veneral, Silva, Araújo, Ferreira, and Oliveira13 also reported full incorporation of both monomers in the copolymer composition, where none of the monomers reacted preferentially, despite the different sizes of macrolactones employed in their work. In that case, an increase in Mn was observed with increasing globalide content, and it was explained by the larger size of Gl compared to ε-CL.
Intermediate copolymer compositions led to a broad molecular weight peak with a tendency to bimodality (Fig. S2, ESI†). The increased ω-PDL content in copolymer compositions led to higher weight average molecular weights and dispersities and also higher viscosities, easily observed at room temperature. The high viscosity reduces chain mobility, enclosing them for longer periods near the enzyme's active site. As a result, these chains react for longer times, growing more and reaching higher molecular weights than the chains located far away from the enzyme's active site. The higher the molecular weight, the more limited the mobility of the chains, and the more pronounced this effect becomes. Then, significant differences in the chain size are obtained, resulting in high dispersities and bimodal distributions. Polymerization via e-ROP of macrolactones generally results in non-monomodal molecular weight distributions, as reported in the literature in e-ROP of poly(ε-caprolactone),41 poly(globalide-co-ε-caprolactone)13 and PPDL.14
The polymerization degree and the number of double bonds per mol, also displayed in Table 1, were calculated using the information on monomer conversion and copolymer composition obtained from the 1H NMR results of each copolymer. In this case, the weight of each copolymer was divided by the molecular weight of each comonomer (in the proportion determined by NMR). Then, the number of repeat units of each copolymer was determined, also leading to the estimation of the number of double bonds per mol of the copolymer.
Copolymer thermal properties, such as melting temperature (Tm), melting enthalpy (ΔHm), and degree of crystallinity, are presented in Table 2. The degree of crystallinity was calculated based on a 100% crystalline sample of PPDL.42 DSC thermograms are shown in Fig. S3 (ESI†). The melting temperatures of the pristine homopolymers (before electrospinning), 44 °C for PGL and 96 °C, agree with those from the literature, 46 °C15 and 97 °C, respectively.14 The lower Tm of PGl is due to unsaturation, which is responsible for a more irregular and less packed structure. An increase in melting temperature can be observed with the increase in PDL content in the copolymer composition. For instance, Tm increased by almost 20 °C by replacing 25 wt% of Gl by PDL in the copolymer synthesis, as shown in Table 2. These results evidence a great advantage of copolymerization in tuning the polymer thermal properties by employing different monomer ratios. In addition, no double melting behavior was observed in any copolymer compositions (Fig. S3, ESI†), indicating that there is no competition between the melting and recrystallization during the second heating run of DSC. The presence of a single intermediate melting temperature in the copolymers indicates isomorphic crystallization, suggesting that they are indeed random copolymers. Several studies, including those on enzyme-catalyzed copolymerization of lactones and macrolactones, consistently yielded copolymers with random microstructures.13,43–45 This observed microstructure is attributed to the rapid transesterification induced by Novozym 435 during enzymatic copolymerization. Notably, the enzymatic mechanism does not promote the formation of well-organized copolymers, such as alternate or diblock structures. The study of thermal properties of the copolyesters aimed at selecting the comonomer composition resulted in a proper Tm for the proposed application, with a high number of double bonds for subsequent functionalization.
| Melting enthalpy (ΔH) (J g−1) | Melting temperature (Tm) (°C) | Degree of crystallinity (XC)b (%) | Contact angle (θ) (°) | Polar component (γps) (mJ m−2) | Dispersive component (γds)(mJ m−2) | Surface free energy (γs) (mJ m−2) | |
|---|---|---|---|---|---|---|---|
| a Thermal properties (in italics) for these conditions were evaluated before electrospinning. Water contact angle and free surface energy were evaluated in the copolymer scaffolds. b Based on a 100% crystalline PPDL sample.42a–c: different letters indicate a statistically significant difference between the samples (Tuckey, p < 0.05)A and B: different capital letters indicate a statistically significant difference between the samples with the same proportion of PDL:PGL but without or with cysteine (Tuckey, p < 0.05). | |||||||
| 100:0a |
65 | 44 | 28 | 107 ± 2a | 0.18 ± 0.03c | 32.65 ± 0.23d | 32.83 ± 0.20d |
| 75:25a |
80 | 63 | 34 | 107 ± 2a | 0.16 ± 0.02c | 32.35 ± 0.20d | 32.41 ± 0.20d |
| 50:50a |
82 | 79 | 35 | 108 ± 1a | 0.24 ± 0.01b | 34.41 ± 0.10c | 34.65 ± 0.10c |
| 25:75a |
97 | 89 | 42 | 109 ± 3a | 0.27 ± 0.05b | 35.31 ± 0.13b | 35.58 ± 0.13b |
| 0:100a |
127 | 96 | 55 | 109 ± 2a | 0.41 ± 0.02a | 36.37 ± 0.09a | 36.90 ± 0.09a |
| 100:0 Gl:PDL-cys |
57 | 43 | 25 | 100 ± 6ª | 0.25 ± 0.05cA | 37.61 ± 0.13bA | 37.86 ± 0.10dA |
| 75:25 Gl:PDL-cys |
90 | 63 | 38 | 102 ± 7a | 0.41 ± 0.02bA | 37.69 ± 0.09aA | 38.10 ± 0.11cA |
| 50:50 Gl:PDL-cys |
98 | 78 | 42 | 112 ± 9a | 0.44 ± 0.05bA | 38.10 ± 0.13bA | 38.54 ± 0.08bA |
| 25:75 Gl:PDL-cys |
127 | 86 | 54 | 110 ± 10a | 0.51 ± 0.02aA | 40.38 ± 0.09aA | 40.89 ± 0.19aA |
| 75:25 Gl:PDL-verisol |
25 | 57 | 10 | 0 | — | — | — |
| 75:25 Gl:PDL-hydrolyzed |
26 | 57 | 11 | 0 | — | — | — |
The crystallinity of the pristine PGl homopolymer is 28% and that of the PPDL homopolymer is 55%, with a fair agreement with literature values of 64%.46 Thus, like the melting temperature, crystallinity also decays with the increasing Gl content, turning the copolymer more amorphous. This characteristic is interesting when dealing with applications requiring bioresorbable materials; when less crystalline, the greater the susceptibility to hydrolysis and enzymatic degradation.47 Thus, the copolymerization strategy can modify the polymer crystallinity besides the melting temperature. All evaluated conditions of different monomer ratios formed semi-crystalline copolymers, with the degree of crystallinity of the homopolymers PGl and PPDL consistent with those from the literature.36,48
:PDL (75:25) raw scaffolds are shown in Fig. 3 (upper left side). SEM images of the raw copolymer scaffolds of all evaluated copolymer compositions are displayed in Fig. S4 (ESI†). All copolymer solutions resulted in homogeneous cylindrical fibers, with diameters ranging from 5.12 to 9.90 μm with no bead formation. The PGl homopolymer resulted in the most uniform fibers with the narrowest fiber diameter distribution, with distributions becoming broader with increasing amounts of PDL in the copolymer. The homopolymer PPDL resulted in fibers with irregular shapes resembling the initial bead conformation. It is worth mentioning that the copolymer concentrations in chloroform were different among the copolymer compositions. Polymers with higher amounts of globalide (100:0, 75:25 and 50:50 Gl:PDL ratios) were dissolved at a concentration of 25 wt%, while the polymers with higher PDL amounts (25:75 and 0:100 Gl:PDL ratios) were dissolved at a concentration of 15 wt%, since higher concentrations of these two polymers did not completely solubilize in chloroform, resulting in unfeasible solutions for electrospinning.
 | ||
| Fig. 3 Morphology, number average fiber diameter (Dn) and water contact angles (θ) of 75:25 Gl:PDL raw scaffolds after functionalization with cysteine (75:25 Gl:PDL-cys) and after bioconjugation with hydrolyzed and with Verisol® collagens, respectively, (75:25 Gl:PDL-hydrolyzed) and (75:25 Gl:PDL-verisol). | ||
The wettability of electrospun fibers (Table 2 and Fig. S4, ESI†) is related to the material's surface energy and roughness. The wettability is generally reduced when the overall surface free energy of the solid surface is lowered; thus, the decrease in surface free energy is recognized as the primary means of developing hydrophobic surfaces.49
The copolymer surface free energy and their components (Table 2) evidenced an increase in the copolymer surface free energy with higher PDL ratios in the copolymer composition. The increase in the surface free energy might be related to the higher molecular weight of the copolymers with higher rates of PDL. The polarity of high molecular weight copolymers is much higher than for low molecular weight copolymers, resulting from higher ratios of Gl because more polar carbonyl groups are constituting the polymer structure and undergoing intramolecular coupling via hydrogen bonds. Thus, surface free energies are expected to be higher with increasing molecular weight of the copolymer.50
Nevertheless, this increase in surface free energy did not lead to a decrease of wettability since no significant differences were observed, and all evaluated fibers resulted in hydrophobic surfaces, with water contact angles ranging from 107 to 109°. This result is attributed to the high roughness provided by the overlapping of electrospun fibers.50 Biomaterials with low water contact angles are required for biomedical applications, especially dealing with biomaterials for tissue engineering. The copolymer hydrophilicity highly affects cell attachment, which increases with the higher surface free energy of the material. For this reason, strategies to increase the hydrophilicity of the designed materials play a key role in their further applications in tissue engineering.
3.2 Functionalization of PGlPDL scaffolds with cysteine
The incorporation of amino acids (AA), such as cysteine, allows for designing biofunctional materials, conceding desired features to conventional degradable polymers and available peptides, such as enhanced hydrophilicity and biocompatibility.51 In the present work, cysteine groups were introduced via thiol–ene click reactions in the unsaturated PGlPDL chains located at the surface of the electrospun fibers (Fig. 1(b)). The main goal of this functionalization was to provide carboxyl groups in the copolymer chain to enable the subsequent bioconjugation with proteins such as collagen through EDC/NHS chemistry. EDC/NHS activates the carboxyl groups of cysteine to further form covalent bonds between carboxyl groups from cysteine and amine groups from collagen. Additionally, cysteine is expected to improve polymer hydrophilicity, reduce its crystallinity, and enhance cell biocompatibility, as observed by Guindani and coworkers.52 Due to the antioxidant potential of cysteine, the functionalized copolymers can also present an enhanced mechanism in healing damaged tissues once the antioxidant character plays a crucial role in combating oxidative stress.19The chemical structures of PGlPDL and PGlPDL-cys scaffolds were evaluated by ATR-FTIR analyses, as illustrated in Fig. 4 for the copolymer with the 75:25 Gl:PDL ratio. ATR-FTIR spectra for all the copolymer compositions before and after functionalizations are presented in Fig. S5 (ESI†).
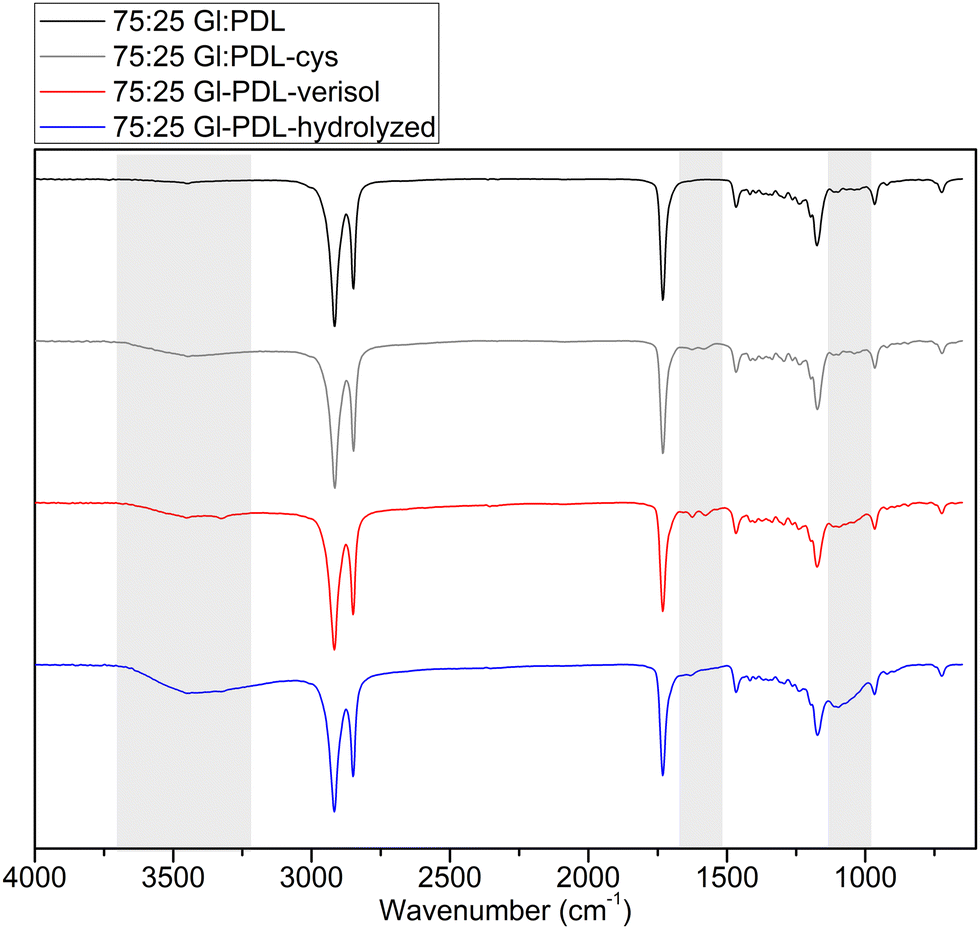 | ||
| Fig. 4 ATR-FTIR spectra of 75:25 Gl:PDL raw scaffolds after functionalization with cysteine (75:25 Gl:PDL-cys) and after bioconjugation with hydrolyzed and with Verisol® collagens (75:25 Gl:PDL-hydrolyzed) and (75:25 Gl:PDL-verisol). | ||
All ATR-FTIR spectra were normalized according to the CO ester peak (1750–1735 cm−1), which remained the same in all samples. The pristine copolymers (before functionalization) presented a band corresponding to the CO stretch at 1750 cm−1 and a second band stretching to C–O bonds at 1300 cm−1, corresponding to ester groups on the polyester chemical composition.5 By analyzing the ATR-FTIR data of functionalized copolymers, two main bands, located in the fingerprint and functional group regions, proved cysteine's presence on the polymer surface. These peaks are related to 1575 cm−1 and 3440 cm−1, corresponding to the presence of primary amines. Besides, both peaks decrease with the decrease in globalide content in the copolymer composition (Fig. S4, ESI†), which agrees with the reduced number of double bonds available for functionalization.
When modifying PGlCL in a bulk approach before electrospinning using N-acetyl-cysteine (NAC), Beltrame and coworkers5 and Guindani and coworkers19 observed a double melting point behavior and a decrease in the melting temperature after functionalization, indicating less stable crystalline domains. Indeed, in these works, the polymer modified with NAC presented a completely amorphous domain because no melting points were observed in the thermograms. This behavior was not observed in the current work because modification with hydrophilic cysteine occurred only at the surface of the fibers, and thus, the number of attached cysteine chains was not enough to decrease the melting temperature of the whole polymer.
In Fig. 5, it is possible to observe similarities in the thermal behavior of the functionalized (75:25 Gl:PDL-cys) and non-functionalized (75:25 Gl:PDL) copolymers, with the existence of a single-melting temperature. The same behavior was observed for all evaluated copolymer compositions (Fig. S3 and S6 in the ESI†). The melting temperatures (Table 2) remained almost the same after functionalization with cysteine for all evaluated copolymer compositions. By comparing the crystallinity results in Table 2, it is possible to observe an increase for all evaluated samples after functionalization of the scaffolds with cysteine, except for the pure PGl. Despite the second heating run analyses, this fact can be attributed to the electrospinning process performed before functionalization, increasing polymer crystallinity due to the fiber conformation, which could potentially lead to a rearrangement of the polymer chains. Thermal characterization of the pristine polymers was performed before electrospinning (results in italics in Table 2).
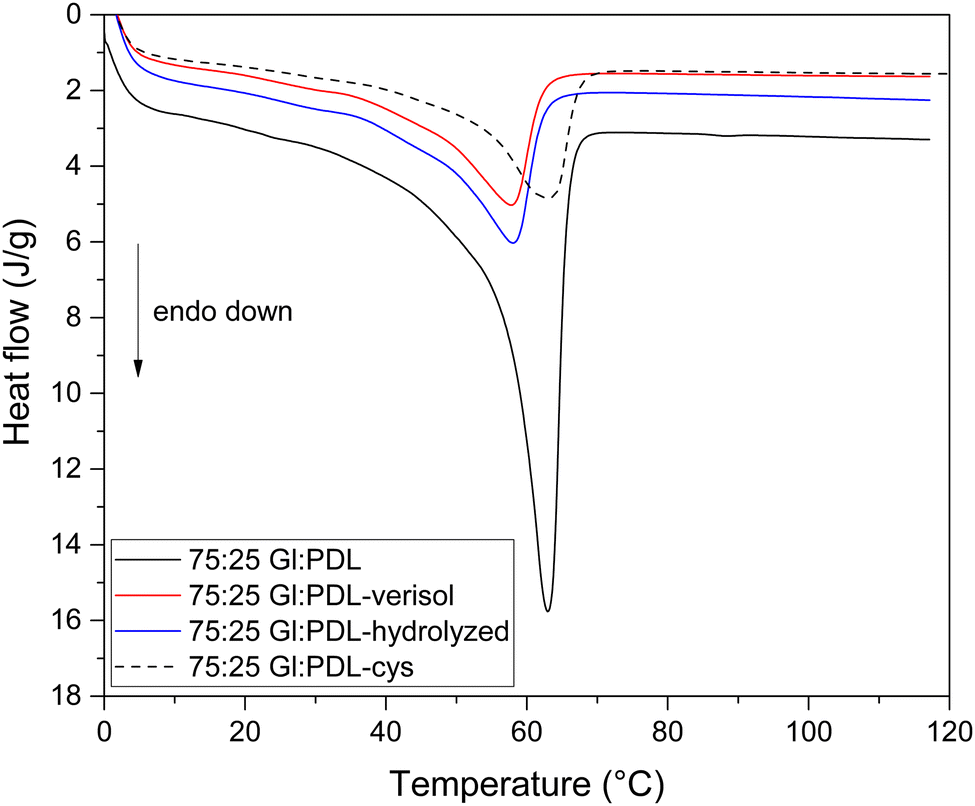 | ||
| Fig. 5 DSC thermograms of 75:25 Gl:PDL raw copolymer and of the scaffolds after functionalization with cysteine (75:25 Gl:PDL-cys) and after bioconjugation with hydrolyzed and with Verisol® collagens (75:25 Gl:PDL-hydrolyzed) and (75:25 Gl:PDL-verisol). | ||
Similar to the thermal behavior, the wettability of the scaffolds was not altered after the functionalization of the surface of their fibers with cysteine, though a small increase could be observed in the estimated surface free energy (Table 2). SEM images in the upper part of Fig. 3 with the comparison between the raw 75:25 Gl:PDL scaffold and the 75:25 Gl:PDL-cys scaffold show that the fiber morphology is preserved after functionalization with cysteine. This same behavior in terms of morphology was observed for all copolymer compositions (Fig. S4 and S7, ESI†).
3.3 Bioconjugation of PGlPDL-cysteine scaffolds with collagen
The proposed bioconjugation of the PGlPDL-cysteine scaffolds with collagen aimed to decrease the surface hydrophobicity, decrease the copolymer crystallinity and improve cell adhesion and cell viability (Fig. 1(c)). Compared to other combination methods between polymers and biomolecules, such as the formation of polymeric blends for further fiber development via electrospinning, bioconjugation creates stable covalent bonds that affect the whole structure of the material (i.e., degree of crystallinity and degradation rate). In polymeric blends, for instance, only surface characteristics could be changed (i.e., hydrophobicity) because the physical and chemical properties of the starting materials remain the same. This way, bioconjugation allows allying the copolymer's mechanical and thermal properties to the bioactive nature of collagen, developing a completely different biomaterial from the starting reactants, with distinguished crystallinity, mechanical properties and degradation rate, prone and easily tuned to meet the requirements for tissue replacement. EDC/NHS chemistry was chosen as a chemical strategy to create amide bonds between the amines from collagen and carboxyl groups from cysteine, enabling a covalent coupling between the copolymer and the collagens. Only one copolymer composition was chosen to proceed with the bioconjugation reaction (Gl:PDL of 75:25). This composition was selected based on the number of double bonds available for cysteine attachment and further bioconjugation and based on the copolymer thermal properties. The ratio of 75:25 satisfactorily increased the melting temperature of the copolymer compared to the homopolymer polyglobalide and also presented a higher number of double bonds available for bioconjugation.
The ATR-FTIR spectra presented in Fig. 4 were normalized according to the C–O ester peak (1750–1735 cm−1). Two bands were highlighted for both collagens to attest the presence of amide bonds: one in the functional group region, at 3460–3420 cm−1, related to the N–H free bond in amides, and another in the fingerprint region (1660 cm−1), related to a CO stretching vibration from amides. The latter is not highly prominent in the spectrum, attesting that the functionalization with collagen was not very high. It is important to emphasize that NMR analyses were also performed in the bioconjugated scaffolds. However, they were not completely soluble in chloroform anymore, indicating that the bioconjugation occurred because the overall solubility of the scaffolds was changed. Then, during the NMR analysis, only the soluble part could be estimated with accuracy. In this sense, NMR was not able to detect and quantify the covalent attachment of collagen on the copolymer chains accurately. For this reason, ATR-FTIR was selected for the qualitative detection of bioconjugation.
The collagen molecule is a cross-linked fibrous protein made of three polypeptides, which in turn, contain around one thousand amino acid residues.53 The chain length is hugely higher than the polymer chain, and it is polar. Even though bioconjugation occurred only with the more superficial chains of the copolymer scaffold, it was enough to change the copolymer crystallinity completely (Table 2). The covalent bond formed between the copolymer and both collagen types hugely reduced the melting enthalpy, corresponding to the endothermic event of Fig. 3 and reduced the melting temperature of the scaffolds by 6 °C. The scaffold with a monomer ratio of 75:25 (Gl:PDL) has its crystallinity changed from 38% to 10% after bioconjugation with collagen. It means that the organization of polymer chains was altered through the attachment of collagen, switching its semi-crystalline structure to an almost amorphous one. In this sense, the bioconjugation proposed in this work was able to reduce the number of crystalline arrangements within the polymeric chains and also reduce the energy needed to overcome the secondary intermolecular forces between the chains in the crystalline phase.54 Despite the changes in crystallinity, the morphological scaffold structures remained the same (Fig. 3).
The overall analysis of thermal properties after bioconjugation of the polymer scaffolds with collagen highlighted one of the most important advantages of covalently binding collagen to polymers over other functionalization techniques, which is the changes in the crystalline structure of the polymer. Despite the satisfactory results related to improvement in cell adhesion and proliferation by changes in the physical surface in other functionalization methods (i.e., physical adsorption31,34), bioconjugation is also able to tune both mechanical and thermal properties of the pre-designed scaffolds, promoting a stable attachment of collagen molecules on the polymer surface. EDC/NHS bioconjugation method, for instance, was employed by Perez-Nava and coworkers30 to bioconjugate collagen to an electrospun poly(vinyl alcohol) scaffold structure, where the resultant scaffolds presented significant improvements in mechanical behavior (i.e., Young's modulus, elongation at break and ultimate tensile strength), which would not be achieved through a simple physical coating, for example. Regarding tissue engineering purposes, the conjugation between collagen and polymers has been proven to be more efficient in terms of mechanical strength, prolonged degradation rate, and cell viability than the simple solvent mixture, as stated by Sadeghi-Avalshahr et al.8
In addition, the carbodiimide chemistry employed in our work has been widely studied for biomaterial fabrication to improve physical and mechanical resistance or bioactive behavior under mild conditions. This bioconjugation method is non-sensitive to light or humidity, involves chemical reactions at room temperature and pressure, occurs in the absence of organic solvents, has high efficiency and the unreacted reagents can be easily removed.30,55 For this reason, it can be carried out in a one-pot strategy, bestowing the present work with several advantages in terms of reaction media conditions aligned with green synthesis concepts.
The amorphous structure reached through the creation of NH2–COOH bonds in the polymer structure is an important issue when dealing with biomedical applications because it also affects the scaffold biodegradability, which plays an important role for devices where full biodegradation of the biomaterial is expected at the end of the healing process. In general, decreasing the degree of crystallinity by decreasing the glass transition temperature of the polymer tends to increase the biodegradability.47
3.4 Cell viability and mineralization assays
Cell viability in electrospun scaffolds was evaluated in vitro using pre-osteoblast cells, aiming to differentiate in osteoblasts, which are cells responsible for forming bone tissues. These tissues support the vital organs, house and protect the bone marrow, support the skeletal muscles, and are deposits of calcium, phosphate, and other ions. The bone tissue can be classified into compact bone tissue and sponge tissue.56 This work aims to create a bone graft to replace spongy bone tissues. To that end, viability tests were carried out to confirm if the copolymer scaffolds' presence could affect the cells' functioning or cause any disturbance. Quantitative results of metabolic activity were measured by MTT assay (mitochondrial activity). Mitochondria is a fundamental organelle of the organism, and its activity is related to the energy supply for the cell. Any dysfunction occasioned by these organelles can cause damage to cell proliferation and growth, which can potentially cause cell death.57 To a better understanding of the effect of functionalization and bioconjugation, cell viability assays were performed in the raw PGlPDL, PGlPDL-cys, and PGlPDL-collagen scaffolds (Fig. 6).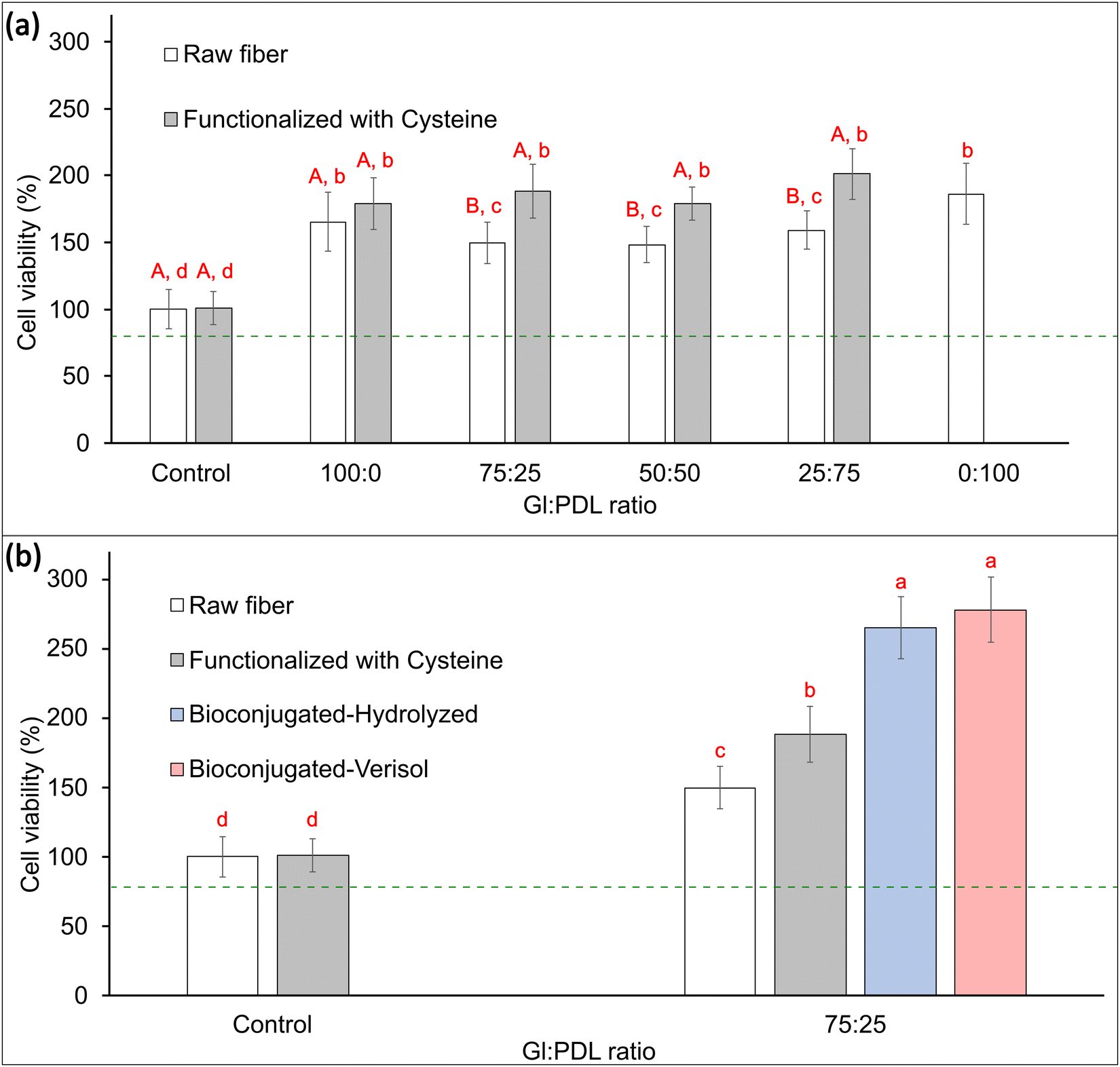 | ||
| Fig. 6 Cell viability of designed scaffolds during 7 days using the MTT assay. (a) Comparison between raw scaffolds and cysteine-modified scaffolds with different copolymer compositions. (b) Comparison between raw, cysteine-modified, and bioconjugated scaffolds. A and B: different capital letters indicate that there is a statistically significant difference between the samples with the same proportion of Gl:PDL, with or without cysteine (Tuckey, p < 0.05). a–d: different letters indicate that there is a statistically significant difference between the samples (Tuckey, p < 0.05). | ||
MTT assays obtained after 7 days of culture in vitro revealed a non-toxicity of all evaluated copolymers. The raw PGlPDL scaffolds did not deleteriously influence the viability compared to the control (aluminum foil) once they presented cell viability higher than 80%. This way, the non-toxicity of the evaluated raw scaffolds toward osteoblast cells can be attested. Besides, all copolymer compositions increased cell growth and differentiation compared with the control. When comparing each copolymer composition (with different monomer ratios), no significant differences among the cell viabilities were observed for samples Gl:PDL (100:0), Gl:PDL (75:25), Gl:PDL (50:50), Gl:PDL (25:75), and Gl:PDL (0:100).
In addition, thiol–ene functionalization with cysteine (Fig. 1(b)) led to a further increase, up to 27%, in cell viability for all evaluated copolymers when compared to the viability of the raw fibers, due to the amino acid character of cysteine. This result highlights the potential of functionalization with AA to improve the performance of scaffolds for tissue engineering.
Furthermore, bioconjugation of 75:25 Gl:PDL-cys scaffolds with collagen almost doubled cell viability (85% increase) when compared to the raw scaffolds. There was no difference in cell viability regarding the type of collagen employed for bioconjugation (Verisol® or Hydrolyzed collagen type I).
Cell adhesion influences diverse cellular processes, including cell–cell cohesion, recognition, signalling, cell viability, and the regulation of cell proliferation. Adhesion molecules, such as proteins, facilitate interactions within the cell microenvironment. In other words, these adhesion molecules are involved in binding within the cells and between the cells and the extracellular matrix (ECM), and their role is to help the cells stick to each other and the surroundings. A high cell adhesion may lead to high cell viability.58 Crystallinity and wettability are keys to modulating cell activity, especially when dealing with bone biomaterials.59 Our collagen-decorated scaffold presented an increase in cell viability of up to 85% when compared to the raw scaffold, and this fact can be attributed to several factors, such as high hydrophilicity, rough surface due to the scaffold conformation technique, the presence of an active biomolecule as collagen, which can act as an adhesion molecule, and the low crystallinity resulting from bioconjugation. The influence of crystallinity on cell adhesion and viability has been widely discussed in the literature,59–62 and it affects cell activity differently depending on the type of cells. Cui and Sinko,61 for instance, synthesized the copolymer poly(caprolactone-co-glycolide) with different crystallinities and observed a significant improvement in efficiency, supporting the growth of osteoblast cells in amorphous copolymers compared to crystalline ones. An opposite behavior was observed for the growth of fibroblasts, which had better support for growth in crystalline structures.
In general, all the scaffolds presented cell viability greater than 100% (control), revealing that a porous surface allied with a material surface with appropriate free energy could induce cell growth and proliferation, ending up in a higher number of cells when compared to the control. Cell viabilities greater than 200% were obtained for both collagen-decorated scaffolds, which may indicate an induction of cell growth and proliferation occasioned by the aspects already mentioned, allied with the presence of a bioactive macromolecule such as collagen, which makes up the final structure of the scaffold. These scaffolds, resembling the extracellular matrix of osteoblastic cells, may create a more compatible structure for cell adhesion, leading to enhanced cell proliferation, as elucidated by Li et al.63
In conclusion, all evaluated scaffolds did not present cytotoxic effects on the cells during the 7 days of investigation. The designed collagen-decorated scaffolds proposed in this work significantly improved osteoblasts’ cell viability and cell growth compared to the control and also promoted cell proliferation, evidencing the potential of the material to act as a scaffold for bone tissue engineering. Previous studies have shown that collagen-based matrices are biologically active because they regulate the activity of osteoblasts and osteoclasts through a variety of signaling pathways and promote the repair of bone defects.63
Studies regarding collagen applied in designing tissue engineering devices ranged from bones,33,64 skin applications,31 silk fibroin scaffolds,65 articular cartilage grafts,66 and organs.67 Each application requires different scaffold properties, such as malleability, rigidity, crystallinity, and hydrophobicity, which are usually achieved by the main-component properties employed to design the scaffold (i.e., ceramic, polymer, or composite) or by cross-linking reactions with collagen. The wide range of applications related to collagen's inherent biocompatibility is also revealed in our study. These biological properties related to collagen, allied with the chemical versatility and non-toxicity of the polymers developed in the present work, highlight our collagen-decorated scaffold as a new biomaterial platform. By tuning the thermal and mechanical properties during the polymer synthesis (i.e., changing the monomer ratio), scaffold thickness during the electrospinning process, and reactant ratio during bioconjugation, our designed scaffold could have its properties easily adapted to other biomaterial requirements. This way, the PGlPDL-collagen scaffold is shown as a promising biomaterial for applications in tissue engineering devices beyond bone repair.
The process of bone repair after damage follows the same pathway as that during normal bone development, which is characterized by several reactions that orchestrate the initiation and ceasing of proliferation, the onset of differentiation, and the beginning of mineralization.68 Mineralization in bone tissues involves stepwise cell–cell and cell-ECM interactions. Alizarin red assessments were performed with the designed scaffolds, and the results are shown in Fig. 7. Microscope images of mineralized scaffolds can be visualized in Fig. 8.
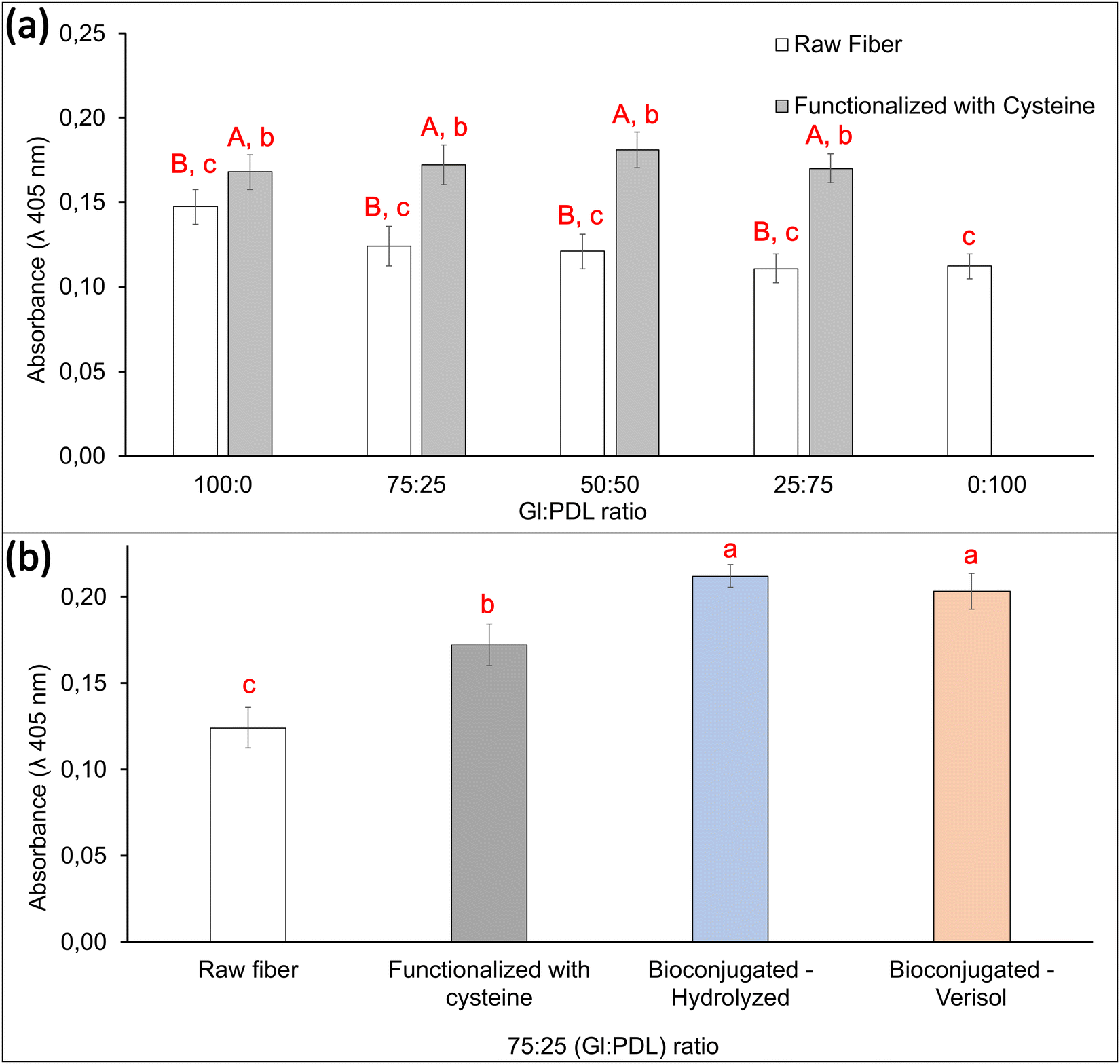 | ||
| Fig. 7 Mineralization assays in the designed scaffolds during 7 days using the alizarin-red method. (a) Comparison between raw scaffolds and cysteine-modified scaffolds with different copolymer compositions. (b) Comparison between raw, cysteine-modified, and bioconjugated scaffolds. A and B: different capital letters indicate that there is a statistically significant difference between the samples with the same proportion of Gl:PDL, with or without cysteine (Tuckey, p < 0.05). a–d: different letters indicate that there is a statistically significant difference between the samples (Tuckey, p < 0.05). | ||
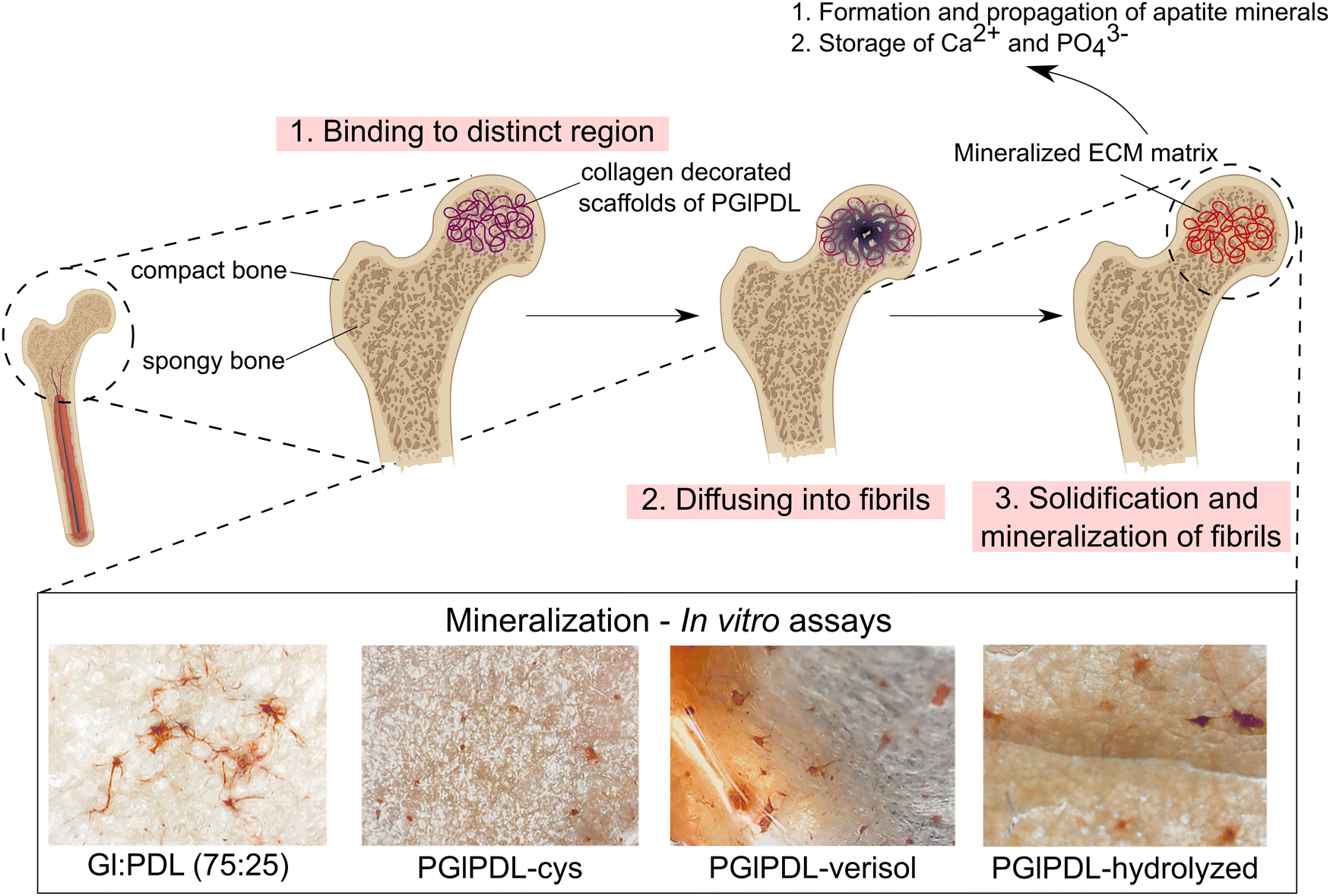 | ||
| Fig. 8 Mineralization process of spongy bone and microscopy images from alizarin-red mineralization assays of the designed scaffolds. | ||
The formation of a mineralized extracellular matrix (ECM) is part of the natural role of osteoblasts. It occurs alongside the release of matrix vesicles, responsible for forming and propagating apatite minerals on collagen fibrils69 (Fig. 8). Thus, mineralization assays can inform about the ability of osteoblasts to store phosphate and calcium ions. Changes in the mineralization mechanism or absence of mineralization when testing the scaffolds could indicate a detrimental influence of these materials on the cells. Alizarin red assays revealed mineralization for all evaluated raw copolymer scaffolds without significant differences among the samples. Following the same trend of cell viability assays, functionalization with cysteine increased the mineralization for all evaluated scaffolds up to 54%. Cysteine can bind calcium, which is released during the growth and mineralization of primary bone cells and promotes the formation of mineral crystals. This protein, which is encoded by the SPARC gene expressed mainly in human bone marrow mesenchymal stem cells, plays essential roles in TGF-β signaling, protein folding, extracellular protein products, and DNA repair.70
Finally, further bioconjugation with both collagen types improved the cell mineralization by 70% when compared to the raw PGlPDL scaffold. Fiber morphology and wettability play important roles in cell viability, proliferation, and mineralization.71 Hydrophilic surfaces created by the incorporation of amide bonds between collagen and the functionalized copolymer favored cell viability, cell growth, and cell mineralization, proving the promising application of the designed material in bone tissue regeneration. Moreover, collagen is an ECM protein that plays a key role in the architecture and tissue regeneration and is responsible for creating the biological and structural integrity of this matrix. By allying its hydrophilic behavior with its fibrous structure and its role in the biological healing and elasticity of all body tissues,8,72 the scaffolds decorated with collagen realistically mimicked the extra-cellular matrix, favoring cell adhesion, proliferation, and mineralization.
4. Conclusions
In this work, we developed homogeneous PGlPDL scaffolds via electrospinning, proposed its functionalization via thiol–ene reaction, and further bioconjugation with collagen via EDC/NHS chemistry, aiming to develop a biomaterial for bone tissue engineering. By varying the monomer ratios in the copolymer synthesis, the polymer properties of the resultant copolymer were easily tuned, reaching 100% monomer conversion and with no preferential reactions with the enzyme active site. The raw materials proved to be non-toxic to osteoblast cells, and their nature and conformation increased the cell viability when compared to the control. The proposed thiol–ene reaction was carried out via a one-pot methodology and led to the formation of carboxylic functional groups on the double bonds of the copolymer chain. The single functionalization did not change the copolymer properties, such as surface hydrophobicity or crystallinity, but increased the cell viability when tested with the pre-osteoblastic cells. Finally, the bioconjugation with collagen, enabled by the previous functionalization with cysteine, changed the organization of the polymer chains, hugely reducing the polymer crystallinity. Also, the proposed bioconjugation with collagen turned the surfaces of the designed scaffolds into hydrophilic. The tests with pre-osteoblast cells revealed a significant increase (up to 85%) in cell viability for the PGlPDL-collagen scaffolds and also a 75% increase in mineralization compared to the raw fibers. This work not only provided a novel polymeric biomaterial platform but also an easy one-pot copolymer functionalization and further efficient bioconjugation with collagen, which improved the features of the designed material to satisfy the required properties for the desired application. Through this bioconjugation, we developed a material that would be suitable for bone repair in tissue engineering devices, inspiring the development of highly efficient and biosafe chemical strategies to conjugate polymers and proteins for biomedical applications. The improvements in cell viability and mineralization due to bioconjugation, allied with the chemical versatility of the designed copolymer, provide the PGlPDL-collagen decorated scaffolds with promising applications in tissue engineering beyond bone repair, such as in developing skin-equivalent devices and organs or cartilages tissues.Author contributions
H. B. M.: investigation, methodology, conceptualization, formal analysis, writing – original draft, visualization. C. G.: investigation, formal analysis, concept development, writing – review and editing. B. C. M.: investigation, formal analysis, writing – original draft., P. H. H. A.: conceptualization, supervision, writing – review and editing. C. S.: formal analysis, conceptualization, project administration, supervision, funding acquisition, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank Symrise (SP, Brazil) for kindly supplying the monomer globalide, the Analysis Center of the Chemical Engineering and Food Engineering Department (EQA-UFSC) for DSC and FTIR analyses, and the Laboratory of Materials from Graduate Program in Materials Engineering for the SEM analyses. C. Guindani thanks FAPERJ (Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro), process number E-26/201.911/2020 and E-26/201.912/2020, for the financial support. B. Maniglia thanks FAPESP (Fundação de Amparo a Pesquisa do Estado de São Paulo), process number 2020/08727-0, for funding this project. The authors also thank CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) for the scholarships and financial support.Notes and references
- F. A. M. M. Gonçalves, A. C. Fonseca, M. Domingos, A. Gloria, A. C. Serra and J. F. J. Coelho, Prog. Polym. Sci., 2017, 15, 1–34 CrossRef.
- I. Jun, H.-S. Han, J. R. Edwards and H. Jeon, Int. J. Mol. Sci., 2017, 19, 1–14 CrossRef PubMed.
- Z. Terzopoulou, A. Zamboulis, I. Koumentakou, G. Michailidou, M. J. Noordam and D. N. Bikiaris, Biomacromolecules, 2022, 23, 1841–1863 CrossRef CAS PubMed.
- I. Jun, H.-S. Han, J. R. Edwards and H. Jeon, Int. J. Mol. Sci., 2017, 19, 1–14 CrossRef PubMed.
- J. M. Beltrame, C. Guindani, M. G. Novy, K. B. Felipe, C. Sayer, R. C. Pedrosa and P. H. Hermes De Araújo, ACS Appl. Bio Mater., 2021, 4, 1552–1562 CrossRef CAS PubMed.
- C. Ulker Turan and Y. Guvenilir, Eur. J. Pharm. Sci., 2022, 170, 106113 CrossRef CAS PubMed.
- S. K. Tiwari and S. S. Venkatraman, Mater. Sci. Eng., C, 2012, 32, 1037–1042 CrossRef CAS.
- A. R. Sadeghi-avalshahr, M. Khorsand-Ghayeni, S. Nokhasteh, A. M. Molavi and H. Naderi-Meshkin, J. Mater. Sci.: Mater. Med., 2017, 28, 1–10 CrossRef CAS PubMed.
- O. Coulembier, P. Degée, J. L. Hedrick and P. Dubois, Prog. Polym. Sci., 2006, 31, 723–747 CrossRef CAS.
- A. E. Polloni, V. Chiaradia, R. J. F. C. Do Amaral, C. Kearney, B. Gorey, D. De Oliveira, J. V. De Oliveira, P. H. H. De Araújo, C. Sayer and A. Heise, Polym. Chem., 2020, 11, 2157–2165 RSC.
- J. Zhang, H. Shi, D. Wu, Z. Xing, A. Zhang, Y. Yang and Q. Li, Process Biochem., 2014, 49, 797–806 CrossRef CAS.
- P. M. Johnson, S. Kundu and K. L. Beers, Biomacromolecules, 2011, 12, 3337–3343 CrossRef CAS PubMed.
- C. Guindani, P. Dozoretz, J. G. Veneral, D. M. da Silva, P. H. H. Araújo, S. R. S. Ferreira and D. de Oliveira, J. Supercrit. Fluids, 2017, 128, 404–411 CrossRef CAS.
- A. E. Polloni, J. G. Veneral, E. A. Rebelatto, D. de Oliveira, J. V. Oliveira, P. H. H. Araújo and C. Sayer, J. Supercrit. Fluids, 2017, 119, 221–228 CrossRef CAS.
- C. Guindani, W. A. G. Jaramillo, E. A. Rebelatto, F. W. Tavares, C. Pinto and P. M. Ndiaye, J. Supercrit. Fluids, 2022, 186, 1–11 CrossRef.
- Z. Jiang, H. Azim, R. A. Gross, M. L. Focarete and M. Scandola, Biomacromolecules, 2007, 8, 2262–2269 CrossRef CAS PubMed.
- M. Claudino, I. Van Der Meulen, S. Trey, M. Jonsson, A. Heise and M. Johansson, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 16–24 CrossRef CAS.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- C. Guindani, P. Dozoretz, P. H. H. Araújo, S. R. S. Ferreira and D. de Oliveira, Mater. Sci. Eng., C, 2019, 94, 477–483 CrossRef CAS PubMed.
- S. S. Gupta, V. Mishra, M. Das Mukherjee, P. Saini and K. R. Ranjan, Int. J. Biol. Macromol., 2021, 188, 542–567 CrossRef CAS PubMed.
- I. C. Carvalho, A. A. P. Mansur, S. M. Carvalho, R. M. Florentino and H. S. Mansur, Int. J. Biol. Macromol., 2019, 133, 739–753 CrossRef CAS PubMed.
- M. Suzuki, K. Makimura and S. I. Matsuoka, Biomacromolecules, 2016, 17, 1135–1141 CrossRef CAS PubMed.
- L. Gerrits, R. Hammink and P. H. J. Kouwer, Polym. Chem., 2021, 12, 1362–1392 RSC.
- H. Seyednejad, A. H. Ghassemi, C. F. Van Nostrum, T. Vermonden and W. E. Hennink, J. Controlled Release, 2011, 152, 168–176 CrossRef CAS PubMed.
- A. Irastorza, I. Zarandona, M. Andonegi, P. Guerrero and K. de la Caba, Food Hydrocolloids, 2021, 116, 106633 CrossRef CAS.
- J. Glowacki and S. Mizuno, Biopolymers, 2008, 89, 338–344 CrossRef CAS PubMed.
- Y.-H. Jiang, Y.-Y. Lou, T.-H. Li, B.-Z. Liu, K. Chen, D. Zhang and T. Li, Am. J. Transl. Res., 2022, 14, 1146–1159 CAS.
- W. F. Daamen, H. T. B. Van Moerkerk, T. Hafmans, L. Buttafoco, A. A. Poot, J. H. Veerkamp and T. H. Van Kuppevelt, Biomaterials, 2003, 24, 4001–4009 CrossRef CAS PubMed.
- M. Nieuwoudt, I. Woods, K. F. Eichholz, C. Martins, K. McSweeney, N. Shen and D. A. Hoey, Ann. Biomed. Eng., 2021, 49, 3621–3635 CrossRef PubMed.
- A. Pérez-Nava, A. E. Espino-Saldaña, E. Pereida-Jaramillo, J. Hernández-Vargas, A. Martínez-Torres, M. O. Vázquez-Lepe, J. D. Mota-Morales, B. A. Frontana Uribe and J. Betzabe González-Campos, Process Biochem., 2023, 126, 1–14 CrossRef.
- A. Ravindran Girija, V. Palaninathan, X. Strudwick, S. Balasubramanian, S. Dasappan Nair and A. J. Cowin, RSC Adv., 2020, 10, 26594–26603 RSC.
- A. Gloria, R. De Santis and L. Ambrosio, J. Appl. Biomater. Biomech., 2010, 8, 57–67 CAS.
- S. Teixeira, L. Yang, P. J. Dijikstra, M. P. Ferraz and F. J. Monteiro, J. Mater. Sci.: Mater. Med., 2010, 388, 539–547 Search PubMed.
- Y. Xie, N. Kawazoe, Y. Yangc and G. Chen, Mater. Adv., 2022, 3, 1556–1564 RSC.
- D. Oliveira, A. C. Feihrmann, A. F. Rubira, M. H. Kunita, C. Dariva and J. V. Oliveira, J. Supercrit. Fluids, 2006, 38, 373–382 CrossRef CAS.
- A. E. Polloni, V. Chiaradia, E. M. Figura, J. P. Paoli, D. de Oliveira, J. V. Oliveira, P. H. H. Araujo and C. Sayer, Appl. Biochem. Biotechnol., 2018, 184, 659–672 CrossRef CAS PubMed.
- H. B. Madalosso, R. Machado, D. Hotza and C. Marangoni, Adv. Eng. Mater., 2021, 2001456, 1–26 Search PubMed.
- A. N. de Faria, D. C. Zancanela, A. P. Ramos, M. R. Torqueti and P. Ciancaglini, Cytotechnology, 2016, 68, 1623–1632 CrossRef CAS PubMed.
- C. A. Gregory, W. G. Gunn, A. Peister and D. J. Prockop, Anal. Biochem., 2004, 329, 77–84 CrossRef CAS PubMed.
- E. Tinajero-Díaz, A. Martínez de Ilarduya and S. Muñoz-Guerra, Eur. Polym. J., 2019, 116, 169–179 CrossRef.
- P. M. Johnson, S. Kundu and K. L. Beers, Biomacromolecules, 2011, 12, 3337–3343 CrossRef CAS PubMed.
- B. Lebedev and A. Yevstropov, Macromol. Chem. Phys., 1984, 185, 1235–1253 CrossRef CAS.
- M. Claudino, I. Van Der Meulen, S. Trey, M. Jonsson, A. Heise and M. Johansson, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 16–24 CrossRef CAS.
- I. Van Der Meulen, Y. Li, R. Deumens, E. A. J. Joosten, C. E. Koning and A. Heise, Biomacromolecules, 2011, 12, 837–843 CrossRef CAS PubMed.
- Z. Jiang, H. Azim, R. A. Gross, M. L. Focarete and M. Scandola, Biomacromolecules, 2007, 8, 2262–2269 CrossRef CAS PubMed.
- M. L. Focarete, M. Scandola, A. Kumar and R. A. Gross, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 1721–1729 CrossRef CAS.
- A. Larrañaga and E. Lizundia, Eur. Polym. J., 2019, 109296 CrossRef.
- A. E. Polloni, J. G. Veneral, E. A. Rebelatto, D. de Oliveira, J. V. Oliveira, P. H. H. Araújo and C. Sayer, J. Supercrit. Fluids, 2017, 119, 221–228 CrossRef CAS.
- K. Song, J. Lee, S. O. Choi and J. Kim, Polymers, 2019, 11, 1–15 Search PubMed.
- D. Kołbuk, M. Ciechomska, O. Jeznach and P. Sajkiewicz, RSC Adv., 2022, 12, 4016–4028 RSC.
- P. K. Gupta, R. Gahtori, K. Govarthanan, V. Sharma, S. Pappuru, S. Pandit, A. S. Mathuriya, S. Dholpuria and D. K. Bishi, Mater. Sci. Eng., C, 2021, 127, 112198 CrossRef CAS PubMed.
- C. Guindani, P. Dozoretz, P. H. H. Araújo, S. R. S. Ferreira and D. de Oliveira, Mater. Sci. Eng., C, 2019, 94, 477–483 CrossRef CAS PubMed.
- L. Gerrits, R. Hammink and P. H. J. Kouwer, Polym. Chem., 2021, 12, 1362–1392 RSC.
- S. V. Canevarolo Jr, Ciência dos Polímeros, São Carlos, 2nd edn, 2006, vol. 1 Search PubMed.
- M. Agheb, M. Dinari, M. Rafienia and H. Salehi, Mater. Sci. Eng., C, 2017, 71, 240–251 CrossRef CAS PubMed.
- R. Florencio-Silva, G. Rodrigues, E. Sasso-Cerri, M. J. Simões, P. S. Cerri and B. Cells, BioMed Res. Int., 2015, 421746, 1–17 Search PubMed.
- K. Todkar, H. S. Ilamathi and M. Germain, Front. Cell Dev. Biol., 2017, 5, 1–7 Search PubMed.
- C. Nakamura and H. Kim, in Encyclopedia of Sensors and Biosensors, ed. R. Narayan, Elsevier, 1st edn, 2023, vol. 1, pp. 556–571 Search PubMed.
- A. Rodriguez-Contreras, D. Guadarrama Bello and A. Nanci, Appl. Surf. Sci., 2018, 445, 255–261 CrossRef CAS.
- D. Kołbuk, M. Ciechomska, O. Jeznach and P. Sajkiewicz, RSC Adv., 2022, 12, 4016–4028 RSC.
- H. Cui and P. J. Sinko, Front. Mater. Sci., 2012, 6, 47–59 CrossRef.
- V. Bernardo, G. M. Luz, N. M. Alves and J. F. Mano, Mater. Lett., 2012, 87, 105–108 CrossRef CAS.
- Y. Li, Y. Liu, R. Li, H. Bai, Z. Zhu, L. Zhu, C. Zhu, Z. Che, H. Liu, J. Wang and L. Huang, Mater. Des., 2021, 210, 110049 CrossRef CAS.
- L. Fan, Y. Ren, S. Emmert, I. Vučković, S. Stojanovic, S. Najman, R. Schnettler, M. Barbeck, K. Schenke-Layland and X. Xiong, Int. J. Mol. Sci., 2023, 24, 3744 CrossRef CAS PubMed.
- Y. Shen, S. L. Redmond, J. M. Papadimitriou, B. M. Teh, S. Yan, Y. Wang, M. D. Atlas, R. J. Marano, M. Zheng and R. J. Dilley, Biomed. Mater., 2014, 9, 015015 CrossRef PubMed.
- D. A. Grande, C. Halberstadt, G. Naughton, R. Schwartz and R. Manji, J. Biomed. Mater. Res., 1997, 34, 211–220 CrossRef CAS PubMed.
- Y. Snyder and S. Jana, Biomaterials, 2022, 288, 121675 CrossRef CAS PubMed.
- A. J. Deegan, H. M. Aydin, B. Hu, S. Konduru, J. H. Kuiper and Y. Yang, Biomed. Eng. Online, 2014, 13, 1–17 CrossRef PubMed.
- L. F. B. Nogueira, M. A. E. Cruz, C. B. Tovani, H. B. Lopes, M. M. Beloti, P. Ciancaglini, M. Bottini and A. P. Ramos, Colloids Surf., B, 2022, 217, 112622 CrossRef CAS PubMed.
- U. Ghimire, R. Kandel, S. Shrestha, J. Y. Moon, S. R. Jang, B. K. Shrestha, C. H. Park and S. C. Kim, Colloids Surf., B, 2023, 223, 113152 CrossRef CAS PubMed.
- R. V. Chernozem, M. A. Surmeneva, S. N. Shkarina, K. Loza, M. Epple, M. Ulbricht, A. Cecilia, B. Krause, T. Baumbach, A. A. Abalymov, B. V. Parakhonskiy, A. G. Skirtach and R. A. Surmenev, ACS Appl. Mater. Interfaces, 2019, 11(21), 19522–19533 CrossRef CAS PubMed.
- Y. Xie, N. Kawazoe, Y. Yangc and G. Chen, Mater. Adv., 2022, 3, 1556–1564 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb02665e |
| This journal is © The Royal Society of Chemistry 2024 |