
Artificial light harvesting gel based on saponification-triggered gelation of aggregation-induced emissive BODIHYs†
Durgendra
Yadav
a,
Vishwa Deepak
Singh
b,
Ashish Kumar
Kushwaha
a,
Anjani
Kumar
a and
Roop Shikha
Singh
 *a
*a
aDepartment of Chemistry, Institute of Science, Banaras Hindu University, Varanasi-221 005, UP, India. E-mail: roopshikha22singh@gmail.com
bDepartment of Chemistry, D.A.V. P.G. College, Gorakhpur-273001, UP, India
First published on 3rd September 2024
Abstract
The present work provides a detailed study on saponification-triggered gelation of ester-based BODIHYs (B1 and B2) derived from ethyl 4-(2-(benzo[d]thiazol-2-yl(cyano)methylene)-hydrazinyl)-benzoate (L1) and diethyl 5-(2-(benzo[d]thiazol-2-yl(cyano)methylene)hydrazinyl)-isophthalate (L2). The ligands and BODIHYs display good emission in the solution and solid states. This study describes the gelation of BODIHYs for the first time, wherein stable gels GL2 and GB2 were prepared via saponification-triggered gelation of L2 and B2, respectively. The gelation and optical properties of the ligands and BODIHYs were compared through single-crystal X-ray diffraction studies. This work further explores the prospect of artificial light harvesting (ALH) via fabrication of ALHSs in the solution {B1/rhodamine B (RhB) and B2/RhB} and gel states (GB2/RhB). It was observed that in the presence of RhB, the emission intensities of BODIHYs and the gel decreased but those of RhB increased. The significant overlapping between the absorption spectrum of RhB and emission spectra of aggregates/gel suggests the possibility of energy transfer via noncovalent interactions. In these systems, B1, B2 and GB2 served as donors, whereas RhB served as an acceptor.
Introduction
The increasing demand for energy from natural resources emerging from the rising global population has caused a worldwide energy shortage.1–3 The current energy crisis has necessitated a surge in sustainable alternative energy resources that could eventually reduce the reliance on fossil fuels.4–6 Solar energy, the driving force behind photosynthesis in natural green plants and some photosynthetic bacteria, is the most sought-after renewable energy resource.7,8 Harnessing solar energy to power chemical reactions through luminescent materials,9 photocatalysis,10,11 fluorescent sensors12,13 and bioimaging14 has drastically changed the renewable energy landscape. Inspired by nature, several artificial light harvesting systems (ALHSs) using porphyrin assemblage molecular polymers,15–17 dendritic macromolecules,18,19 organic gels,20–22 nanocrystals,23–25 coordination compounds,26–28etc. have been developed, which have opened a new avenue for mitigating global energy sustainability. The simulation of a natural photosynthetic apparatus is based on the incorporation and precise orientation of multiple donors and one acceptor, which would lead to the efficient utilization of solar energy through energy transfer between an excited state donor (D) and a proximal ground state acceptor (A) via a non-radiative Förster resonance energy transfer (FRET) process.29,30 This implies that donor–acceptor spatial organization in supramolecular assemblies facilitated by non-covalent interaction can be categorically utilized for exploring energy transfer in ALHSs. In this context, supramolecular gelatinous materials show more potential than conventional solution based ALHSs owing to their stability as well as tighter and ordered arrangements of donor molecules.31–33 The three-dimensional network of a gel can effectively avoid self-quenching of the excited state by preventing the proximity between acceptor molecules by providing a suitable distance. In recent years, the major focus of the researchers has been on the molecular self-assembly of small molecules to form low molecular weight gelators (LMWGs) considering their implementation in optoelectronics,34 adhesive materials35 and drug delivery.36–38 The properties of LMWGs are usually fine-tuned by their design and molecular architecture by alternating noncovalent or weak interactions, including hydrogen bonding,39 π–π stacking,40 van der Waals,41 dipole–dipole,42 and electrostatic forces.43 Despite the apparent advantages, ALHSs based on supramolecular LMWGs have attracted very little attention. Further, solid-state emission has become indispensable for the practical application of functionalized luminescent materials, which has led to a great surge in the exploration of aggregation-induced emission (AIE) materials.44,45 The abovementioned facts have led to an understanding that despite the challenges, the exploration of AIE supramolecular LMWGs is more conducive to the development of efficient ALHSs.46To achieve the exigent demand of AIE in LMWGs, we focused our attention on boron difluoride (–BF2) coordinated luminescent materials due to their potential applications in diverse areas, such as material chemistry,47 biological chemistry48 and optoelectronics.49 Among these, boron dipyrromethene (BODIPY) emerges as an obvious choice; however, their applications are greatly limited by aggregation-caused quenching (ACQ) and small Stokes shift, which results in self-absorption of its fluorescence.50 In this direction, Aprahamian and Gilroy et al. reported a new class of boron-difluoride (BF2)–hydrazone complexes (BODIHYs) exhibiting solid-state luminescence and aggregation-induced emission (AIE).51–55 The AIEgens display valuable photophysical properties with an increasing concentration or upon aggregation, as AIE alleviates close packing in the molecules via various pathways: conformational planarization, J-aggregate formation, E/Z isomerization, twisted intramolecular charge transfer (TICT), excited-state intramolecular proton transfer (ESIPT), and restriction of intramolecular motion (RIM).56
Therefore, to develop AIE-based LMWGs with efficient light harvesting ability, we design and synthesize two BODIHYs using a benzothiazole–hydrazone chelating platform. As it is well known that the peripherally substituted ester(s) are suitable for gelation57 and the phenyl ring may favour the process by increasing the planar π-interactions, the gelation of the BODIHYs has been achieved by incorporating several ester substituents (–COOC2H5) on the phenyl ring. Herein, we describe the synthesis thorough characterizations and photophysical properties of BODIHYs (B1 and B2) and their corresponding ligands ethyl 4-(2-(benzo[d]thiazol-2-yl(cyano)methylene)-hydrazinyl)-benzoate (L1) and diethyl 5-(2-(benzo[d]thiazol-2-yl(cyano)methylene)hydrazinyl)-isophthalate (L2). B1, B2 and L1 are AIE active. Gelation was triggered in B2 and L2 by saponification through NaOH. ALHSs in an aqueous and organic medium are constructed utilizing these assemblies as donors, while rhodamine dye (RhB) is used as an acceptor. To the best of our knowledge, this is the first report dealing with gelation in BODIHYs produced by saponification and light harvesting applications.
Experimental section
Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 (ethyl acetate/hexane) as an eluent to produce L1 as a yellow powder (758 mg). 1H NMR (500 MHz, CDCl3): δ = 14.41 (s, 1H, N-H), 8.09 (d, J = 6 Hz, 3H, H3 and H5), 7.99 (d, J = 8 Hz, 1H, H8), 7.60 (t, J = 6.5 Hz, 1H, H7), 7.52 (t, J = 8 Hz, 1H, H6), 7.46 (d, J = 8 Hz, 2H, H4), 4.40–4.35 (q, J = 6.5 Hz, 2H, H2), 1.40 ppm (t, J = 6.5 Hz, 3H, H1). 13C NMR (125 MHz, CDCl3): δ = 166.0, 159.6, 152.6, 145.1, 133.4, 131.4, 127.5, 127.0, 126.8, 123.2, 122.1, 116.2, 115.1, 108.9, 61.1, 14.4 ppm. HRMS (m/z calcd for C18H14N4O2S [M]): 350.0837; found [M + H]+: 351.1029.
:1 hexane/ethyl acetate as an eluent to give B1 as a yellow crystalline powder (548 mg). 1H NMR (500 MHz, CDCl3): δ = 8.32 (d, J = 8 Hz, 1H, H5), 8.11 (d, J = 9.5 Hz, 2H, H3), 7.90 (d, J = 8 Hz, 1H, H8), 7.89 (d, J = 8 Hz, 2H, H4), 7.74 (t, J = 8 Hz, 1H, H7), 7.66 (t, J = 7 Hz, 1H, H6) 4.41–4.36 (q, J = 6.5 Hz, 2H, H2), 1.40 ppm (t, J = 6.5 Hz, 3H, H1). 13C NMR (125 MHz, CDCl3): δ = 166.0, 157.7, 147.7, 142.7, 130.7, 129.6, 129.1, 128.4, 122.8, 120.6, 120.3, 114.3, 61.2, 14.4 ppm. 11B NMR (160.4 MHz, CDCl3): δ = 0.48, 0.29, 0.09 ppm (t, 1B). 19F NMR (470.6 MHz, CDCl3): δ = −129.3, −129.2, −129.2, −129.1 ppm (q, 2F). HRMS (m/z calcd for C18H13BF2N4O2S [M]): 398.0820; found [M + H]+: 399.1034.
:8 (ethyl acetate/hexane) as an eluent to produce L1 as a yellow powder (758 mg). 1H NMR (500 MHz, CDCl3): δ = 14.41 (s, 1H, N-H), 8.09 (d, J = 6 Hz, 3H, H3 and H5), 7.99 (d, J = 8 Hz, 1H, H8), 7.60 (t, J = 6.5 Hz, 1H, H7), 7.52 (t, J = 8 Hz, 1H, H6), 7.46 (d, J = 8 Hz, 2H, H4), 4.40–4.35 (q, J = 6.5 Hz, 2H, H2), 1.40 ppm (t, J = 6.5 Hz, 3H, H1). 13C NMR (125 MHz, CDCl3): δ = 166.0, 159.6, 152.6, 145.1, 133.4, 131.4, 127.5, 127.0, 126.8, 123.2, 122.1, 116.2, 115.1, 108.9, 61.1, 14.4 ppm. HRMS (m/z calcd for C18H14N4O2S [M]): 350.0837; found [M + H]+: 351.1029.
:1 hexane/ethyl acetate as an eluent to give B1 as a yellow crystalline powder (548 mg). 1H NMR (500 MHz, CDCl3): δ = 8.32 (d, J = 8 Hz, 1H, H5), 8.11 (d, J = 9.5 Hz, 2H, H3), 7.90 (d, J = 8 Hz, 1H, H8), 7.89 (d, J = 8 Hz, 2H, H4), 7.74 (t, J = 8 Hz, 1H, H7), 7.66 (t, J = 7 Hz, 1H, H6) 4.41–4.36 (q, J = 6.5 Hz, 2H, H2), 1.40 ppm (t, J = 6.5 Hz, 3H, H1). 13C NMR (125 MHz, CDCl3): δ = 166.0, 157.7, 147.7, 142.7, 130.7, 129.6, 129.1, 128.4, 122.8, 120.6, 120.3, 114.3, 61.2, 14.4 ppm. 11B NMR (160.4 MHz, CDCl3): δ = 0.48, 0.29, 0.09 ppm (t, 1B). 19F NMR (470.6 MHz, CDCl3): δ = −129.3, −129.2, −129.2, −129.1 ppm (q, 2F). HRMS (m/z calcd for C18H13BF2N4O2S [M]): 398.0820; found [M + H]+: 399.1034.
Results and discussion
Syntheses and characterizations
Syntheses of the hydrazones L1–L2 and their –BF2 complexes, BODIHYs B1–B2, are achieved by following the literature procedures and using BF3·Et2O in the presence of N,N-diisopropylethylamine (DIPEA). A simple scheme showing the synthesis of the ligands (L1–L2) and BODIHYs (B1–B2) is depicted in Scheme 1. The ligands and BODIHYs under investigation are air-stable, non-hygroscopic solids, soluble in common organic solvents, such as diethyl ether, petroleum ether, hexane, acetonitrile, methanol, ethanol, acetone, dichloromethane, chloroform, dimethylformamide, dimethyl-sulfoxide, and insoluble in water. Characterization of these compounds is performed by acquiring NMR (1H, 13C, 11B, 19F), HRMS, electronic absorption and emission spectral data (Fig. 1 and Fig. S1–S8, ESI†). 1H, 13C, 11B, and 19F spectral data are presented in the Experimental section, and spectra are shown through Fig. S1–S6 (ESI†).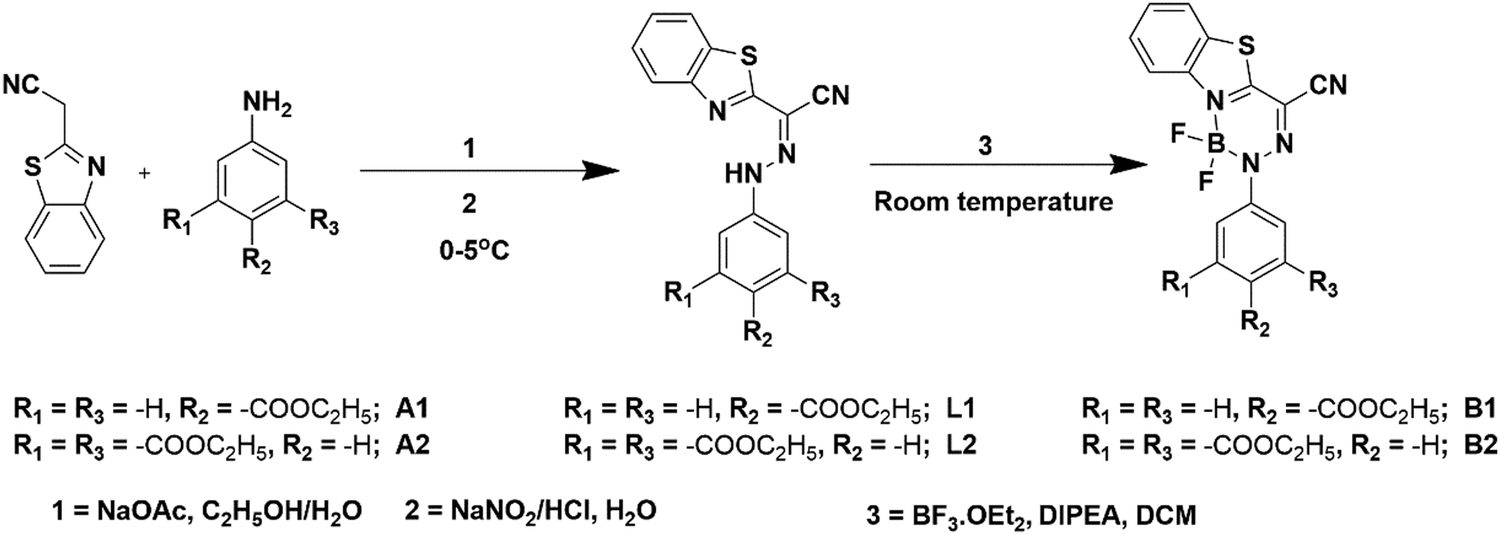 | ||
| Scheme 1 Synthesis of ligands (L1–L2) and BODIHYs (B1–B2). | ||
 | ||
| Fig. 1 Normalized UV-Vis absorption and emission spectra of ligands (L1–L2) (a) and BODIHYs (B1–B2) (b) in THF (c, 5.0 × 10−5 M). | ||
The N–H protons for L1 and L2 resonated as a singlet at δ 14.41 and 14.43 ppm, while aromatic protons appeared as broad multiplets in the range of δ ∼ 8.44–7.24 ppm. In addition, methylene and methyl protons appeared as a quartet and a triplet, respectively, at δ 4.38 (2H), 1.40 ppm (3H) in L1 and 4.44 (4H), 1.45 (6H) in L2. After the formation of BODIHYs, the N–H proton disappeared, and a broad multiplet of aromatic protons resonated in the downfield region in the range of δ ∼ 7.64–8.33 (B1) and 7.65–8.65 ppm (B2). 13C NMR spectroscopic data for the ligand (L1–L2) and BODIHYs (B1–B2) further supported their formation and proposed structures. HRMS strongly supported the formation of the compounds: L1, L2, B1 and B2. As expected in their mass spectra, the compounds under investigation displayed molecular ion peaks at m/z [M + H]+: 351.1029, 423.1266, 399.1034 and 471.1269 (Fig. S7 and S8, ESI†), further confirming their respective formulations.
Photophysical studies
The photophysical behaviour of hydrazone ligands (L1–L2) and BODIHYs (B1–B2) has been examined in dilute solutions through UV/Vis and photoluminescence studies. Ligands under study displayed absorptions (λabs) at 403 (L1) and 392 nm (L2) (THF; c, 5.0 × 10−5 M) due to intramolecular charge transfer (ICT).60 However, the intense ICT absorption bands in B1 and B2 were observable at 428 and 421 nm, respectively. Further, when excited at 403 (L1) and 392 nm (L2), they showed emission maxima at 459 and 435 nm with a Stokes shift (SS) of ∼3026 and ∼2521 cm−1, respectively. The solution fluorescence quantum yield (Φf) of L1 and L2 were found to be 7.6 and 35.3%, respectively. Upon excitation, B1 showed emission maxima at 519 with Stokes shift (SS) and Φf of ∼3955 cm−1 and 4.5%, respectively. However, B2 exhibited emission maxima at 503 nm with Stokes shift and Φf of ∼3832 cm−1 and 8.3%, respectively (Fig. 1). Apparently, blue-shifted absorption and emission as well as higher Φf for L2 and B2 in solution may be attributed to the presence of one additional electron withdrawing ester group (–COOC2H5), which weakens the ICT character in the ground and excited states.61In their solid-state (powder) emission profile, upon excitation at 403 and 392 nm, L1 and L2 both showed a broad band (500–650 nm) with the maxima centred at 548 (with a shoulder at ∼576 nm) and 571 nm, respectively. B1 and B2 showed broad bands (475–700 nm) upon excitation at ∼428 and 421 nm, with maxima centred at 586 and 571 nm, respectively (Fig. S9, ESI†). Similarly, solution B2 displayed a blue-shifted emission maximum relative to solution B1. The red-shifted emission wavelengths of B1 and B2 in the solid state compared to their solution state can be attributed to increased intermolecular interactions in the solid state. Further, the observation of ICT bands in the absorption spectra and broad solubility range of B1–B2 compounds in common organic solvents, such as hexane, benzene, toluene, 1,4-dioxane, CHCl3, tetrahydrofuran (THF), CH2Cl2, DMF, CH3CN, and CH3OH, prompted us to examine the dependence of the excited state on solvent polarity. Going from non-polar (hexane; f = 0.0) to polar (methanol; f = 0.309) solvents, the emission band displayed significant broadening with a decrease in emission intensities. The typical bathochromic shift of the emission maxima in polar solvents (Δλ = 8 nm, B1 and 13 nm, B2) suggested the involvement of the ICT state for BODIHY complexes (B1 and B2) (Fig. S10, ESI†).
Aggregation-induced emission (AIE)
The excellent solid-state emission of ligands and BODIHYs is anticipated to be accompanied by the AIE attribute. To validate AIE, the absorption and emission behaviour of the ligand L1–L2 and BODIHY B1–B2 are investigated in THF/water binary mixture. In their absorption spectra, the ligands (L1–L2) did not show any major changes up to water fraction (fw) 70%, but as fw approached 80%, a bathochromic shift was observed along with a prominent level off tail, indicating aggregate formation (Fig. S11, ESI†). Variation of water fraction in the range 80–99% caused a red shift (403 to 418, L1; 392 to 401 nm, L2) in the absorption band with quenched absorbance. Further, it was observed that the ligands (L1–L2) displayed analogous emission features toward variation in the water fraction. At fw 0%, L1 displayed emission maxima (λmax) at 459 nm, which shifted to 505 nm at fw 90% with ∼9-fold emission enhancement. Moreover, L2 at fw 0% showed λmax at 435 nm, which red shifted to 537 nm at fw 99% along with significant emission quenching (Fig. S12, ESI†).Further, in their absorption spectra, both BODIHYs (B1–B2) showed insignificant changes of up to fw 70%. However, as fw approached 80%, a bathochromic shift was observable along with the broadening of the absorption band, which in turn indicated the onset of aggregation (Fig. S13, ESI†). Variation of the water fraction in the range of 80–99% led to a red shift (432–445, B1; 421–432 nm, B2) for the absorption band with quenched absorbance. In a large excess of water (fw 99%), they exhibited bathochromically shifted absorption bands relative to THF solution (fw 0%) with a level off tail due to Mie scattering for the aggregate suspension. The emissive behaviour of ensuing aggregates was further investigated by recording the emission profile in an analogous solvent system.
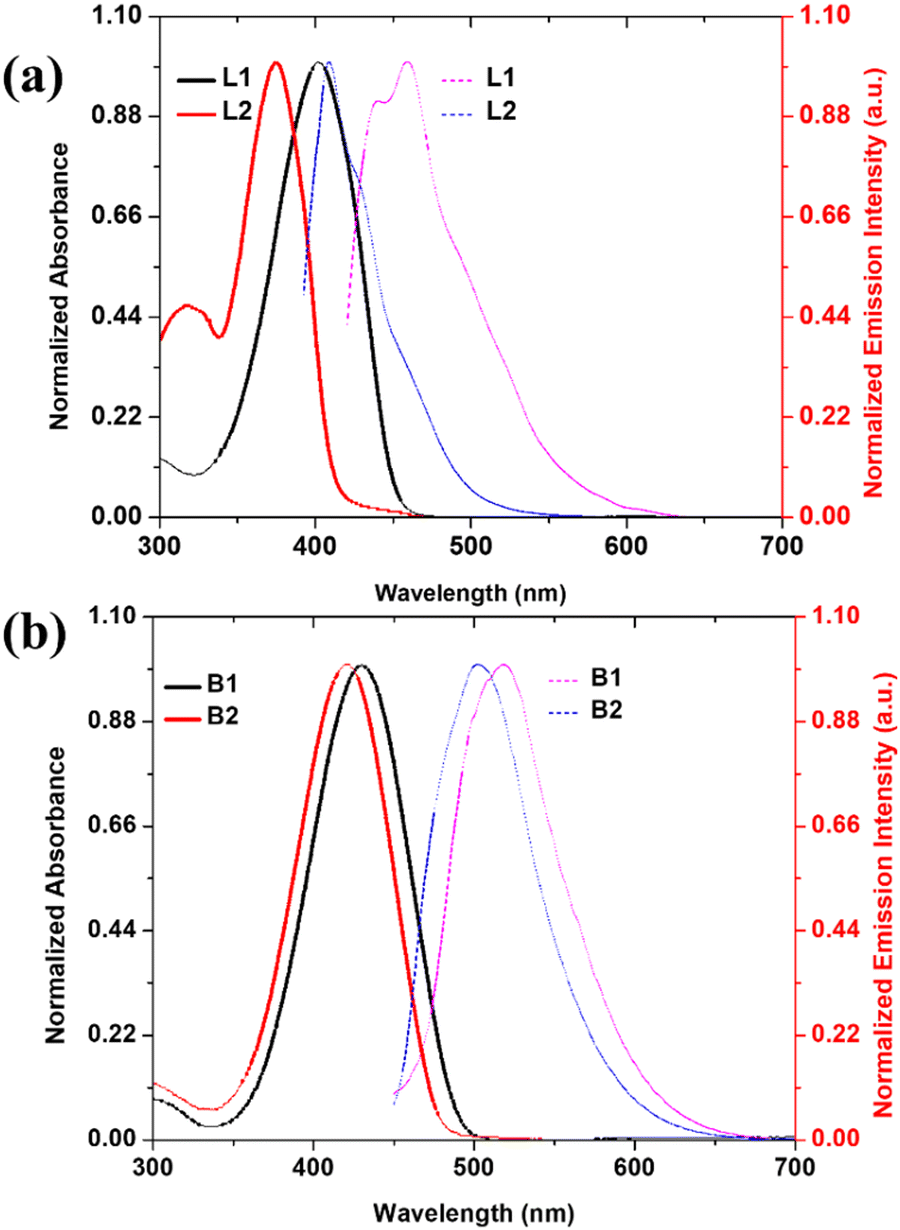
As depicted in Fig. 2, luminophores B1 and B2 showed weak emission in the dilute (THF) solution (fw 0%) at 519 and 503 nm, respectively. The gradual addition of water up to fw 70% caused insignificant changes in their emission behaviour; however, when the fw reached 90%, B1 and B2 exhibited appreciable emission enhancement (∼5- and 8-fold). Interestingly, these luminophores showed emission quenching at fw 99% accompanied by a bathochromic shift to 537 (B1) and 520 nm (B2), which can be attributed to decreased solubility and precipitation. From the above observations, it can be surmised that ligand L2 displays typical ACQ behaviour with an obvious red shift. However, ligand L1 and BODIHYs (B1 and B2) displayed fluorescence enhancement upon aggregation. Thus, these can be classified as AIEgens. These observations have further been supported by DLS studies (Fig. S14a, ESI†). As expected, at fw 90%, the average size of nanoaggregates is found to be ∼200 (L1), 500 (B1) and 150 nm (B2). Further, the Tyndall effect for L1, B1 and B2 in THF/water (fw 0% and 90%) validated the formation of aggregates (Fig. S14b, ESI†).62
 | ||
| Fig. 2 Emission spectra of B1 (a) and B2 (b) in THF/water mixture with varying water fractions (c, 5.0 × 10−5 M); (c) plot between emission intensity and THF/water fractions; and (d) a logarithmic view of time-resolved fluorescence of B1 and B2 (a) in THF (fw = 0%) and THF/water (fw = 90%) (c, 5.0 × 10−5 M). | ||
The morphology of aggregates was analysed by SEM studies on L1, B1 and B2 at fw 90%. With an increasing water gradient, intermolecular interactions in L1, B1 and B2 favour the formation of roughly spherical aggregates (Fig. S15, ESI†). Further, it is well known that restriction of intramolecular rotations (RIR) caused by aggregation is one of the most prominent factors behind the emission enhancement in AIEgens.63 Therefore, the possible mechanism for AIE is investigated by evaluating the viscosity sensitivities of L1, B1 and B2. In this regard, emission spectra of L1, B1 and B2 are obtained in a mixture of CH3OH/glycerol (c, 5.0 × 10−5 M) with varying glycerol fractions.
As shown in Fig. S16 (ESI†) emission intensity for L1 (∼6 times), B1 (∼16 times) and B2 (∼15 times) enhanced with increasing glycerol fraction accompanied by a small bathochromic shift in the position of emission maxima (465–490 nm; L1, 522 to 529 nm; B1 and 502–507 nm; B2). Restriction of active intramolecular rotations in viscous solvent resulted in the rigidification of luminophores, thereby impeding the creation of a dark state by blocking the non-radiative decay channels, which further led to enhanced emission.
Additionally, AIE was verified through fluorescence lifetime measurements of THF/water solutions of L1, B1 and B2 at varying water fractions. It was observed that the lifetime increased from 0.20 (fw = 50%), 0.08 and 0.02 (fw = 0%) to 0.77, 0.87 and 0.22 ns (fw = 90%) for L1, B1 and B2, respectively, supporting the emission enhancement upon aggregation (Fig. 2d and Fig. S17, ESI†). The longer lifetime in the aggregated state can be attributed to the lowering of the non-radiative decay through restricted molecular rotations. From the above results, it can be concluded that RIR is responsible for AIE in these systems.
Single-crystal X-ray
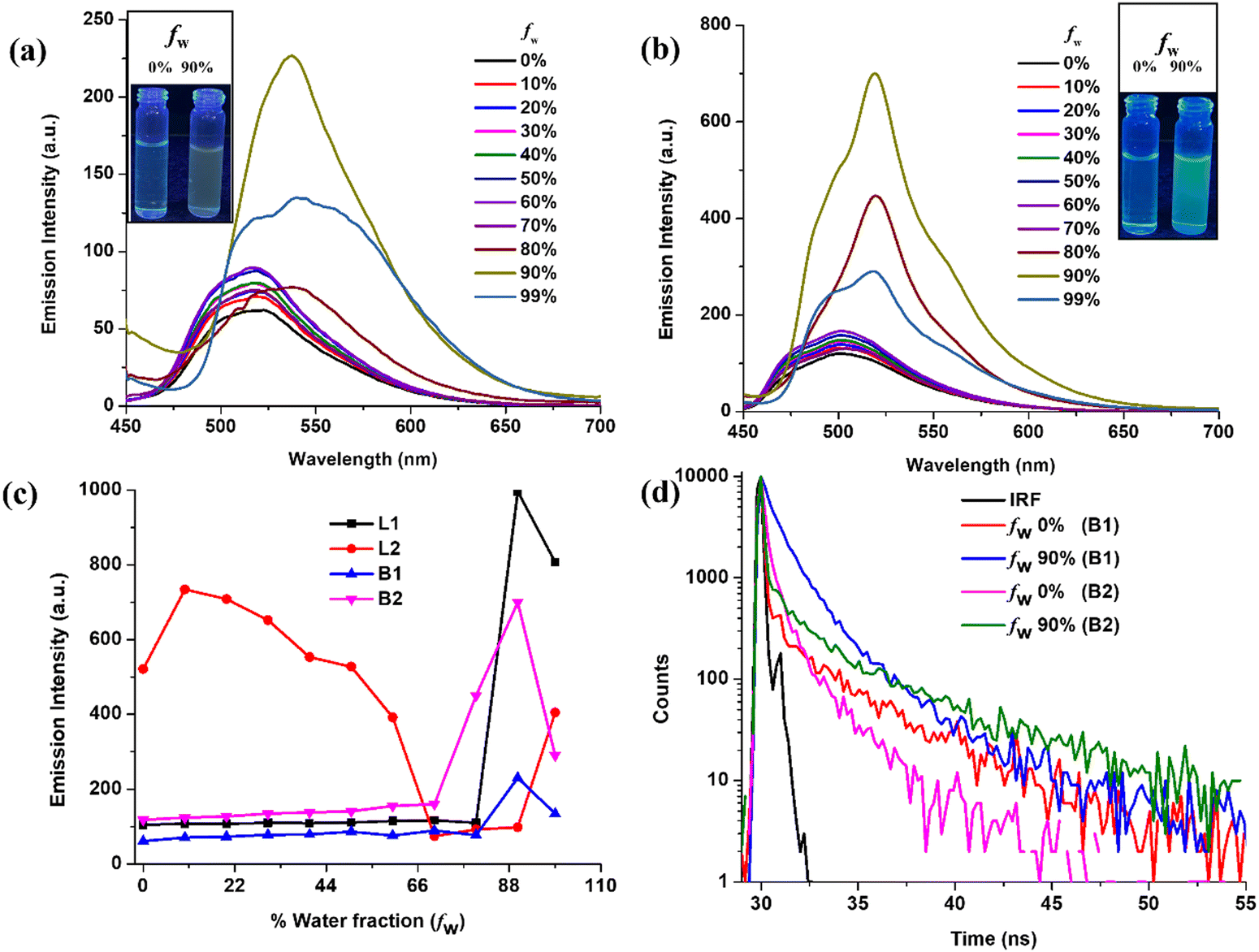
It is believed that structural and packing analyses of the luminophores through single-crystal X-ray crystallography may provide detailed information about the intermolecular interactions, which are the driving forces toward the creation of hierarchical supramolecular structures. We further presumed that the gelation process and corresponding photophysical behaviour may be a cumulative outcome of the self-assembly process in crystallization. The single-crystal structures of the ligands (L1 and L2) BODIHYs (B1 and B2) are unambiguously authenticated by X-ray single-crystal studies, and ORTEP views are shown in Fig. 3. Details about data collection, solution and refinement are presented in Table S1 (ESI†). | ||
| Fig. 3 ORTEP views of L1 (a), L2 (b), B1 (c) and B2 (d) at 50% ellipsoidal probability (H atoms are omitted for clarity). | ||
From the crystal structure of L1 and L2, it is clear that the former has no π–π interactions while the latter has a single π–π interaction (3.337 Å) (Fig. S18, ESI†). The absence of π–π interaction in L1 can prevent the formation of closely packed structures and lead to AIE, unlike ACQ in L2, which shows π–π interactions. Interestingly, L1 forms a ‘V’-shape crystal packing pattern due to short interaction between O and H (2.719 Å). Similarly, the short interaction between N and H (2.640 Å) forms a dimeric structure in L2 (Fig. S18, ESI†). B1 forms two antiparallel chains with head-to-tail and head-to-head arrangement via π–π interactions. Two π–π interactions are illustrated in Fig. 4: one between benzene rings of adjacent benzothiazole moieties (3.359 Å) and the other between carbons of the methylene group and ester moiety (3.398 Å). However, B2 was arranged in a “classical” slipped stacked manner with π–π interaction (3.395 Å) favouring J-aggregation (Fig. 4).
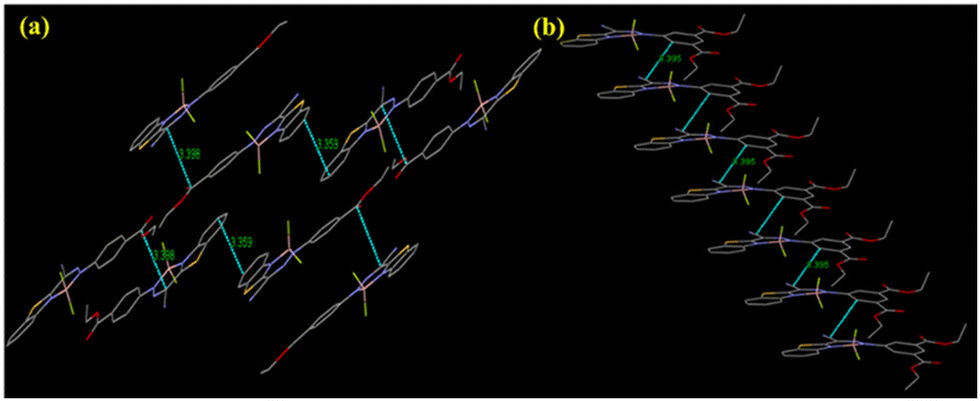 | ||
| Fig. 4 Crystal packing in B1 (a) and B2 (b) via π–π interactions. | ||
Interestingly, B1 has a nearly planar structure, while in B2, the BODIHY core is at 27.52° with the diethyl isophthalate core. This loss of planarity might prevent the very close packing of molecules in nanoaggregates and could be responsible for the greater emission enhancement of B2 in an aggregated state. Further, the selected crystallographic parameters (bond angles and bond lengths) and important short interactions for B1 and B2 are presented in Tables S2–S4 (ESI†).
Gelation
Because an increase in dipole moment is critical for gelation, we attempted the saponification typically employed for the hydrolysis of fatty esters into –COO−Na+/–K+ salts in the presence of a base (NaOH or KOH) to create polar groups with slight modification. It was found that a 10 mM solution of L1 and B1 in CHCl3 instantly led to yellow colour gelatinous suspensions GL1 and GB1, respectively, in the presence of a methanolic solution of NaOH (Fig. S21 and S22, ESI†). However, ligand L2 and BODIHY B2 formed a stable gel under similar conditions, which was further confirmed by applying the inverted vial method (GL2 and GB2) (based on creating a compact gel in the minimum time, Fig. 5a and e). The weak nature of GL1 and GB1 compared to GL2 and GB2, respectively, is due to the number of peripheral ester groups present on the benzene ring (1 in L1 and B1 while 2 in L2 and B2), which are hydrolysed to carboxylate salts that facilitate gelation through π–π stacking and short intermolecular interactions. | ||
| Fig. 5 (a) L2 and gel GL2 in an inverted vial. (b) AFM, (c) SEM, and (d) TEM for GL2, (e) B2 and GB2 in the inverted vial. (f) AFM, (g) SEM and (h) TEM for GB2. | ||
Owing to the presence of two ester groups on L2 and B2, saponification may create a mixture of Na+-salts. Therefore, it is optimized by varying the ratios of L2 and B2versus NaOH. In this context, we evaluated the photophysical behaviour of L2 and B2 in the presence of NaOH. As shown in Fig. 1, the UV/Vis spectra of L2 and B2 possessed prominent peaks at 393 and 421 nm, respectively. Upon the gradual addition of NaOH, L2 exhibited a significant bathochromic shift accompanied by the emergence of a new band at 453 nm, while B2 showed a gradual decrease in absorption with a small red shift (421–430 nm). Similarly, in fluorescence titration studies, L2 and B2 (c, 1 × 10−2 M) showed emission bands at 437 and 508 nm, respectively (Fig. S23, ESI†). The gradual addition of NaOH with a limit of quantification of 1.0 to 3.2 equiv. (L2) and 1.0 to 2.8 equiv. (B2) led to a decrease in florescence intensity with a red shift (Δλ, 40 and 10 nm). These changes clearly indicate the ratiometric conversion of the esters to carboxylates, leading to the formation of L2 → GL2 and B2 → GB2 through J-aggregation. The gelation was also scrutinized using other common solvents, and the resulting data are presented in Table S5 (ESI†).
Further, the morphological characteristics of gels GL2 and GB2 were investigated by atomic force, scanning electron and transmission electron microscopy (AFM, SEM and TEM). AFM analyses confirmed the interconnected fibrous morphology of GL2 (Fig. 5b) while numerous directional fibres were present in GB2 (Fig. 5f). SEM analyses further attested to the results obtained from AFM studies (Fig. 5c and g). In addition, TEM images showed significant interactions among the fibres in GL2 and GB2, which confirmed that they are involved in the creation of the gel fibres (Fig. 5d and h). The mechanical strength of the gel was characterized by its viscoelastic character and was measured by dynamic rheology. The storage (G′) and loss (G′′) moduli are measured as functions of shear stress (τ), oscillation strain (γ) and angular frequency (ω). The results revealed that the magnitude of G′ is higher than that of G′′ by an order of magnitude up to a certain range of yield stress, showing a true gel nature. The linear viscoelastic (LVE) region is determined by the amplitude sweep of G′ and G′′.
In the present work, the GL2 and GB2 materials show a fair stretch of the LVE region and it is revealed by the frequency sweep measurements at the suggested strains (from amplitude sweep experiments) that G′ > G′′ is maintained throughout along with Δ(G′ ∼ G′′) of 1 order (Fig. S24, ESI†).64 The information regarding structural transformation during the gelation process is collected by 1H-NMR titration of L2 and B2 (0.02 M) in CDCl3 with 2.0 M NaOH in CD3OD (Fig. S25 and S26, ESI†). Upon the gradual addition of NaOH, the loss of the ester group (–CH2CH3: 4H, δ = 4.45; 6H, 1.44 ppm) proton signals were observed. Simultaneously, there is an emergence of two new peaks at δ = 3.70, 1.25 ppm (L2) and 3.38, 1.26 ppm (B2) assignable to the free ethyl group (–CH2CH3). Interestingly, the N–H of L2 at 14.5 ppm also disappeared with the addition of the base.
This conversion is also substantiated by the HRMS of GL2 and GB2 and the presence of suitable molecular ion peaks as well as their isotopic patterns, which matches well with the simulated one (Fig. S27 and S28, ESI†).
Additionally, to correlate the relative gelation efficiencies, centroid–centroid distances (ccds) between the planar rings involved in π–π stacking are investigated. The absence of π–π interactions in L1 relative to that in L2 can be associated with poor gelation efficiency of L1 over L2 (Fig. S18, ESI†). Further, from the crystal structure, it is observed that B1 and B2 are involved in significant intermolecular π-stacking interactions with ccds of 7.998 and 4.712 Å, respectively, between the 6-membered difluoroboron ring (Fig. S29, ESI†). Interestingly, the planar diethyl isophthalate rings involved in π-stacking are arranged in such a manner that they are aligned in one direction in B2 and opposite to each other in B1. From the above observations, it can be surmised that smaller ccd and face-to-face positioning of diethyl isophthalate rings in B2 might lead to a greater extent of π-interactions and consequently higher gelation efficiency in B2 compared to B1 through saponification.
Density function theory (DFT)
For a better understanding of photophysical behaviour and rationalization of the gelation mechanism, theoretical calculations are performed for ligands, BODIHYs and their saponified gels. The frontier molecular orbitals of ligands (L1–L2) and BODIHYs (B1–B2) are shown in Fig. 6 and Fig. S30 (ESI†). Theoretical studies have shown that the HOMO and LUMO of ligands extend mainly over the entire molecule, but the molecular orbital localization differs. This can be observed specifically at the benzene ring (directly attached to ester moiety), where the orbital accumulates around the carbon–carbon double bond at the HOMO state, whereas it is localized over the carbon–carbon single bond at the LUMO state, indicating conjugation within the molecule.65 The band gap between the HOMO (−2.61, L1; −2.49 eV, L2) and LUMO (−6.02, L1; −6.01 eV, L2) for the ligands are 3.41 and 3.52 eV, respectively. However, in both the BODIHYs, HOMO−1, HOMO, LUMO and LUMO+1 are localized over phenyl and benzothiazole units. The band gaps between the HOMO (−6.34, B1; −6.29 eV, B2) and LUMO (−3.05, B1; −2.88 eV, B2) for these molecules are 3.29 and 3.41 eV, respectively. The larger band gap between molecular orbitals is attributed to the presence of two electron withdrawing substituents (–COOC2H5) in L2 and B2, which further supports their blue-shifted absorption band relative to L1 and B1 (Fig. 1).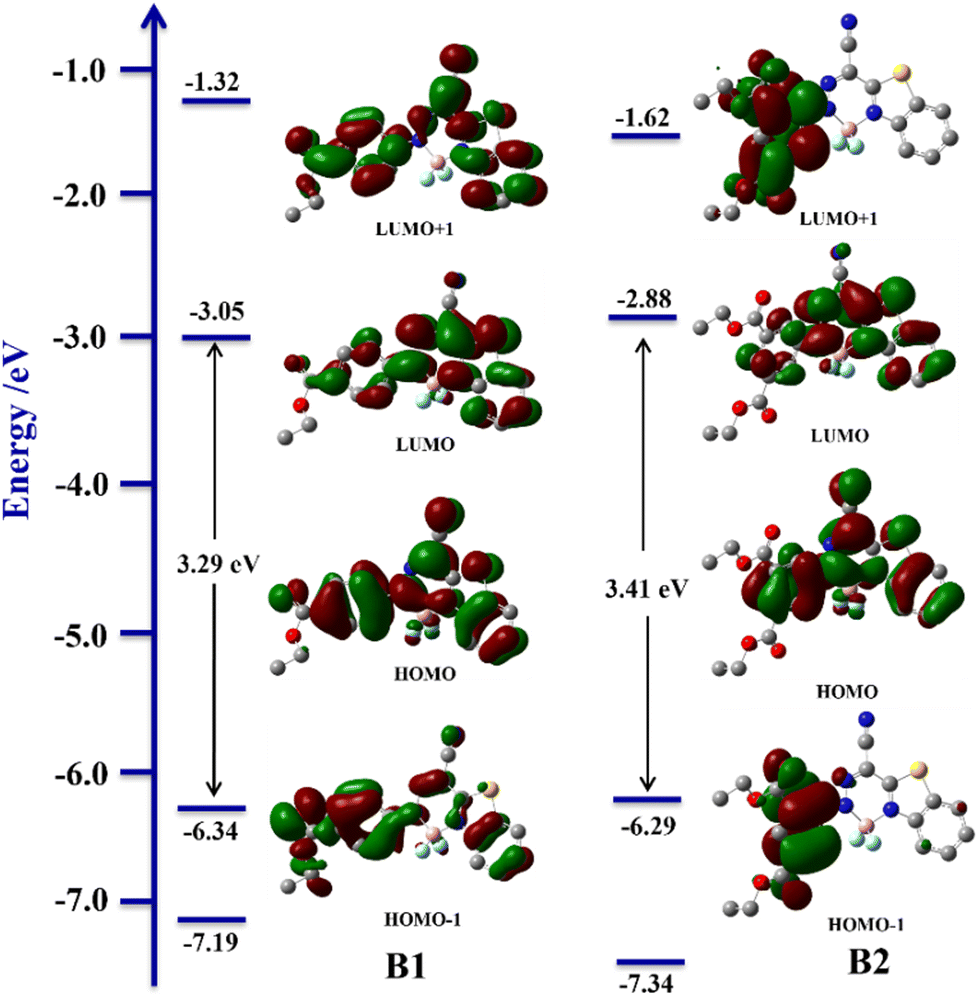 | ||
| Fig. 6 Selected ground state molecular orbitals of B1 and B2 obtained from DFT calculations. | ||
Further, experimental UV/Vis spectra are compared to those from TD-DFT (Fig. S31 and S32, ESI†). For ligands and BODIHYs, the major transition bands originated due to HOMO → LUMO (88%, L1; 85%, L2; 87%, B1; 88%, B2) transition (Table S6, ESI†). Thus, the overall theoretical results validated those obtained from the experimental measurements.
Furthermore, the comparison of DFT-optimized structures of saponified products GL1–GL2 and GB1–GB2 is performed (Fig. S30 and S33, ESI†). The HOMO and LUMO of GL1 and GL2 have a distribution similar to that of the ligands, while in the case of GB1 and GB2, similar electron densities are observed in HOMOs. However, in LUMOs, the electron densities are predominantly localized over phenyl and benzothiazole–BF2 units in GB2, unlike in GB1.
As it is well known that an increase in the dipole moment is crucial for saponification-triggered gelation,57 DFT calculations revealed an increase in the dipole moment from 6.85 → 10.39 (L2 → GL2) and 4.58 → 7.13 Debye (B2 → GB2) due to the –COO−Na+ moiety. Further, it is found that GB2 (7.13 Debye) has a higher dipole moment than GB1 (6.63), which in turn suggests the superior gelation efficiency of GB2. Notably, these saponified structures of BODIHYs have two planes in which one plane contains a benzothiazole moiety and the other has an isophthalate ring and Na+ ion. In both cases, Na+ ion is expected to be involved in significant interactions with the first plane, which provides strong evidence of the dominant cation–π interaction in GB2 (1.657 and 4.053 Å) compared to GB1 (1.496 Å) (Fig. S34, ESI†).
Artificial light harvesting systems (ALHSs)
Red fluorescent rhodamine B (RhB) dye has strong absorption in the visible region at 556 nm and emission at 580 nm, making it extensively used in fluorescent studies.66 Both luminophores (B1 and B2) showed strong broad AIE emissions in aqueous solution (fW = 90%) at 537 and 519 nm, respectively. It is apparent that there is a significant overlap between the emission spectra of aggregates of B1 and B2 and the absorption spectrum of RhB, which satisfies one of the most important criteria for FRET between donor and acceptor moieties and construction of ALH assemblies (Fig. 7 and Fig. S35, ESI†). It is worth mentioning that L1 and L2 fail to show FRET when employed as donors against RhB acceptors in ALH assemblies. This is due to poor spectral overlap between the emission of L1 and L2 and the absorption of RhB. Apparently, the incorporation of –BF2 moiety in hydrazone ligands leads to red-shifted emission and increases the possibility of spectral overlap.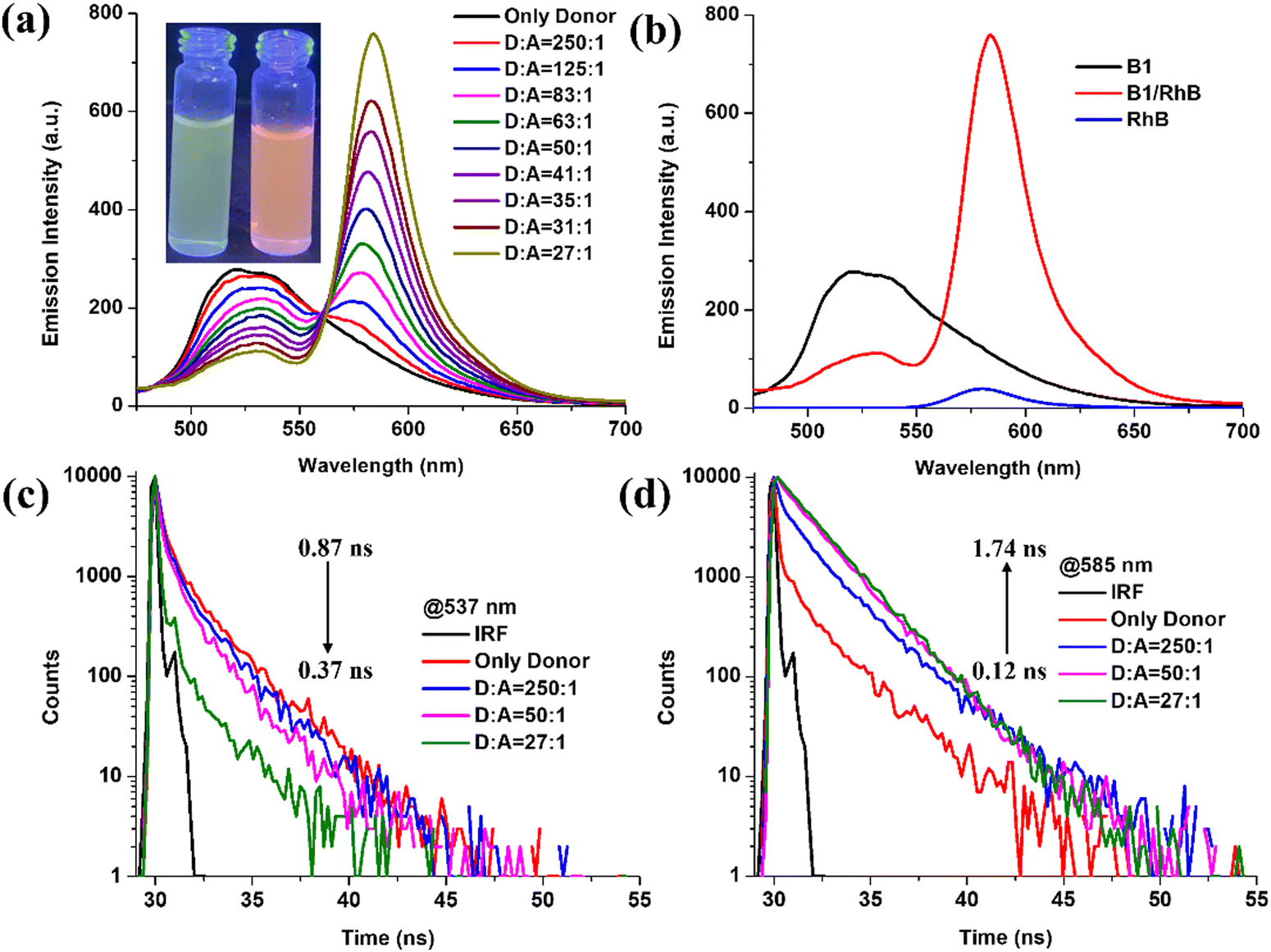 | ||
| Fig. 7 Fluorescence spectra of B1 (c, 5.0 × 10−5 M) with gradual addition of RhB (λex = 421 nm) in THF/water (fw 90%) (c, 0.0, 0.2 × 10−6, 0.4 × 10−6, 0.6 × 10−6, 0.8 × 10−6, 1.0 × 10−6, 1.2 × 10−6, 1.4 × 10−6, 1.6 × 10−6, and 1.8 × 10−6 M); (b) emission spectra of B1 (c, 5.0 × 10−5 M), RhB + B1 (c, 5.0 × 10−5 M, [RhB] c, 1.0 × 10−4 M) and RhB (c, 2.5 × 10−5 M). (c) and (d) Change in the fluorescence decay profiles of B1 in the presence of RhB in THF/water (90%; v/v). | ||
In this direction, aggregates of B1 and B2 were employed for the construction of ALH, wherein B1 and B2 act as donors while RhB acts as an acceptor. Upon gradual addition of RhB into the aggregate of B1 (fw = 90%), the emission intensity of B1 quenched at λem = 537 nm with the synchronous appearance of a new emission at λem = 583 nm upon excitation at λex = 428 nm. Furthermore, on varying the molar ratio of the donor:acceptor (D:A) from 250:1 to 27:1, the ∼5-fold emission enhancement is observed at 583 nm along with ∼3-fold quenching at 537 nm. Similarly, upon gradual addition of RhB to the solution of B2, the intensity of the newly appeared peak at 585 nm increased gradually at different molar ratios, whereas the band at 519 nm experienced emission quenching upon excitation at 421 nm. In contrast, under similar conditions, RhB alone was found to be non-emissive upon excitation at 428 and 421 nm, eliminating the possibility of direct excitation of RhB. Additionally, the gel state of B2 (GB2) is utilised for the construction of ALHSs (Fig. S36, ESI†). With the gradual increase in the molar ratio of RhB from 250:1 to 27:1 (GB2:RhB), the fluorescence intensity of GB2 at 515 nm decreased notably whereas the fluorescence intensity of RhB at 577 nm increased remarkably under excitation at 430 nm.
Therefore, it can be safely concluded that an efficient FRET process exists between aggregates of B1 and B2 and gel GB2 with RhB; in these systems, AIEgen acts as the energy donor and RhB acts as the acceptor. Further, to obtain better insight into the energy transfer process, the fluorescence lifetime of the donor was measured in the presence of the acceptor. In this direction, fluorescence lifetime decay profiles for B1/RhB and B2/RhB at different ratios are investigated in an aqueous medium (fw = 90%) at 537, 583 nm and 520, 585 nm, respectively. Notably, luminophore B1 (τagg = 0.87 ns) showed a higher fluorescence lifetime relative to B1 in the presence of RhB (D:A = 27:1) (τ = 0.37 ns) (Fig. 7). Further, the fluorescence lifetime of the self-assembly of B2 and RhB (τ = 0.11 ns) were found to be shorter (Fig. S35, ESI†) than the fluorescence lifetime of B2 (τagg = 0.22 ns) at a similar molar ratio (D:A = 27:1). Simultaneously, RhB showed enhanced fluorescence lifetimes of 1.74 (B1) and 1.82 ns (B2) upon excitation at 428 and 421 nm. Additionally, the GB2/RhB dyad showed similar fluorescence lifetime results in the gel state. The decay profile of GB2 monitored at 515 nm steadily decreased from 0.10 to 0.08 ns along with an increase in lifetime from 0.09 to 3.56 ns of RhB at 577 nm (Fig. S36, ESI†). Such a decrease in fluorescence lifetime suggests an energy transfer from donor to acceptor moieties. The energy transfer efficiency and antenna effect for BODIHYs are calculated in the aggregated state (B1 = 58%, 16.4; B2 = 3.4%, 15.6) and gel state (GB2 = 26%, 4.4) at a ratio of 27:1 (ESI†). These results indicate that energy transfer can occur from the AIEgens (B1 and B2) to the RhB in aggregated and gel states. Thus, we conclude that successful FRET may be responsible for the observed sensitization of RhB by B1 and B2. Notably, this antenna effect is similar to the recently reported values for other artificial light-harvesting systems in aqueous and organic environments.
Conclusion
In conclusion, two BODIHYs (B1 and B2) and their corresponding ligands (L1 and L2) are designed, synthesized and thoroughly characterized. With the intent of creating gel via saponification, several ester substituents (–COOC2H5) on the phenyl rings of L1–L2 and B1–B2 are introduced, further leading to the construction of supramolecular gels GL2 and GB2 based on L2 and B2, respectively. The presence of two peripheral ester groups on the benzene rings of L2 and B2, in contrast to those on L1 and B1, enables strong gelation due to increased π–π interaction in the former following hydrolysis under basic conditions. The ligands and BODIHYs display good emission in the solution and solid states. B1 and B2 showed excellent AIE behaviour with ∼5- and 8-fold emission enhancement in the aggregated state, respectively. RIR is the driving force behind the AIE activity, as established through viscosity experiments. A comparative account of optical behaviour in solution and aggregated states is provided through the evaluation of intermolecular interactions visualized through single-crystal XRD data. B1, B2 and GB2 have further been utilized for fabrication of ALHSs: B1/RhB, B2/RhB and GB2/RhB. The energy transfer efficiency and antenna effect for BODIHYs are B1 = 58%, 16.4; and B2 = 3.4%, 15.6 in the aggregated state and GB2 = 26%, 4.4 in the gel state at a ratio of 27:1. Thus, the present work relays the pioneering work in supramolecular gelation of BODIHYs and exploration of those gels for constructing ALHSs. This will enable further exploration of the outstanding optical properties of BODIHYs for the fabrication of novel and more efficient ALHSs.
Data availability
This is to certify that all the data related to the present work have been provided in figures and tables in the manuscript and its ESI.† Crystallographic data have been provided in separate checkCIF files.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors gratefully acknowledge the Science and Engineering Research Board (SERB), New Delhi, India (Scheme ECR/2018/001810) and BHU IOE cell (Seed/Incentive Grant) for providing financial assistance and also to CSIR, New Delhi, India for award of a Senior Research Fellowship to Durgendra Yadav (09/0013(11822)/2021-EMR-I). We are thankful to the Head, Departments of Chemistry, Institute of Science, Banaras Hindu University, Varanasi (UP) India, for extending laboratory facilities. We are also thankful to Dr Mrigendra Dubey, Soft Materials Research Laboratory, Department of Metallurgy Engineering and Materials Science, IIT Indore, India and to Dr Avanish Singh Parmar, Biophysics and Nanotechnology Laboratory, Department of Physics, IIT (BHU) Varanasi, India for providing rheology data.References
- K. K. Jaiswal, C. R. Chowdhury, D. Yadav, R. Verma, S. Dutta, K. S. Jaiswal, B. Sangmesh and K. S. K. Karuppasamy, Energy Nexus, 2022, 7(100118), 2772–4271 Search PubMed.
- N. Gallandat, J. Bérard, F. Abbetab and A. Züttel, Sustainable Energy Fuels, 2017, 1, 1748–1758 RSC.
- D. Gielen, F. Boshell, D. Saygin, M. D. Bazilian, N. Wagner and R. Gorini, Energy Strategy Rev., 2019, 24, 38–50 CrossRef.
- P. J. Megía, A. J. Vizcaíno, J. A. Calles and A. Carrero, Energy Fuels, 2021, 35(20), 16403–16415 CrossRef.
- K. Alper, K. Tekin, S. Karagöz and A. J. Ragauskas, Sustainable Energy Fuels, 2020, 4, 4390–4414 RSC.
- C. J. Quarton, O. Tlili, L. Welder, C. Mansilla, H. Blanco, H. Heinrichs, J. Leaver, N. J. Samsatli, P. Lucchese, M. Robiniusc and S. Samsatli, Sustainable Energy Fuels, 2020, 4, 80–95 RSC.
- J. Lv, J. Xie, A. G. A. Mohamed, X. Zhang, Y. Feng, L. Jiao, E. Zhou, D. Yuan and Y. Wang, Nat. Rev. Chem., 2023, 7, 91–105 CrossRef.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- M. M. Hasan, S. Hossain, M. Mofijur, Z. Kabir, I. A. Badruddin, T. M. Y. Khan and E. Jassim, Energies, 2023, 16(18), 6456 CrossRef CAS.
- R. Vargas, D. Carvajal, L. Madriz and B. R. Scharifker, Energy Rep., 2020, 6(4), 2–12 CrossRef.
- T. Kawawaki, Y. Mori, K. Wakamatsu, S. Ozaki, M. Kawachi, S. Hossain and Y. Negishi, J. Mater. Chem. A, 2020, 8, 16081–16113 RSC.
- Z. Lian, Y. Kobayashi, J. J. M. Vequizo, C. S. K. Ranasinghe, A. Yamakata, T. Nagai, K. Kimoto, K. Kobayashi, K. Tanaka, T. Teranishi and M. Sakamoto, Nat. Sustain., 2022, 5, 1092–1099 CrossRef.
- R. P. Steer, Dalton Trans., 2018, 47, 8517–8525 RSC.
- Y. Wang, N. Han, X. Li, S. Yu and L. Xing, Chem. Phys. Mater., 2022, 1, 281–293 Search PubMed.
- X. Feng, X. Ding, L. Chen, Y. Wu, L. Liu, M. Addicoat, S. Irle, Y. Dong and D. Jiang, Sci. Rep., 2016, 6, 32944 CrossRef CAS.
- Y. X. Hu, W. J. Li, P. P. Jia, X. Q. Wang, L. Xu and H. B. Yang, Adv. Opt. Mater., 2020, 8, 2000265 CrossRef CAS.
- F. Hajjaj, Z. S. Yoon, M. C. Yoon, J. Park, A. Satake, D. Kim and Y. Kobuke, J. Am. Chem. Soc., 2006, 128(14), 4612–4623 CrossRef CAS PubMed.
- W. J. Li, X. Q. Wang, W. Wang, Z. Hu, Y. Ke, H. Jiang, C. He, X. Wang, Y. X. Hu, P. P. Jia, P. Yin, J. Chen, H. Sun, Z. Sun, L. Xu and H. B. Yang, Giant, 2020, 2, 100020 CrossRef.
- W. J. Li, X. Q. Wang, D. Y. Zhang, Y. X. Hu, W. T. Xu, L. Xu, W. Wang and H. B. Yang, Angew. Chem., Int. Ed., 2021, 60, 18761–18768 CrossRef CAS.
- Y. Wang, Y. Lai, T. Ren, J. Tang, Y. Gao, Y. Geng, J. Zhang and X. Ma, Langmuir, 2023, 39, 1103–1110 CrossRef CAS.
- Y. Wang, Y. Gao, J. Tang, T. Ren, J. Zhang, Y. Liang, J. Wei, E. Feng and X. Ma, ACS Appl. Polym. Mater., 2023, 5(5), 3809–3816 CrossRef CAS.
- X. Ma, J. Tang, T. Ren, J. Zhang, Y. Liang, J. Weia and E. Feng, RSC Appl. Interfaces, 2024, 1, 215–221 RSC.
- S. K. Bhaumik, D. Maity, I. Basu, S. Chakrabarty and S. Banerjee, Chem. Sci., 2023, 14, 4363–4374 RSC.
- K. Trofymchuk, A. Reisch, P. Didier, F. Fras, P. Gilliot, Y. Mely and A. S. Klymchenko, Nat. Photonics, 2017, 11, 657–663 CrossRef CAS PubMed.
- Y. X. Li, J. Li, H. B. Zeng, X. J. Zhang, S. Cosnier and D. Shan, Anal. Chem., 2023, 95(6), 3493–3498 CrossRef CAS PubMed.
- V. D. Singh, B. K. Dwivedi, Y. Kumar and D. S. Pandey, New J. Chem., 2021, 45, 1677–1685 RSC.
- B. Shi, Y. Liu, H. Zhu, R. T. Vanderlinden, L. Shangguan, R. Ni, K. Acharyya, J. H. Tang, Z. Zhou, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2019, 141(16), 6494–6498 CrossRef CAS PubMed.
- A. Arrigo, G. L. Ganga, F. Nastasi, S. Serroni, A. Santoro, M. P. Santoni, M. Galletta, S. Campagna and F. Puntoriero, C. R. Chimie, 2017, 20, 209–220 CrossRef CAS.
- H. Q. Peng, L. Y. Niu, Y. Z. Chen, L. Z. Wu, C. H. Tung and Q. Z. Yang, Chem. Rev., 2015, 115, 7502–7542 CrossRef CAS PubMed.
- K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562–4588 CrossRef CAS PubMed.
- X. Ma, B. Qiao, J. Yue, Y. Geng, Y. Lai, J. Zhang, E. Feng, Z. Lia and X. Hana, Mater. Adv., 2022, 3, 4531–4535 RSC.
- X. M. Chen, X. Chen, X. F. Hou, S. Zhang, D. Chen and Q. Li, Nanoscale Adv., 2023, 5, 1830–1852 RSC.
- X. Ma, J. Yue, Y. Wang, Y. Gao, B. Qiao, E. Feng, Z. Li, F. Ye and X. Han, Soft Matter, 2021, 17, 5666–5670 RSC.
- P. Verma, A. Singh and T. K. Maji, Chem. Sci., 2021, 12, 2674–2682 RSC.
- Y. Liang, K. Wang, J. Li, Y. Zhang, J. Liu, K. Zhang, Y. Cui, M. Wanga and C. S. Liu, Mater. Horiz., 2022, 9, 1700–1707 RSC.
- D. K. Smith, Soft Matter, 2024, 20, 10–70 RSC.
- K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellama and M. Marlow, Soft Matter, 2014, 10, 237–256 RSC.
- D. M. Raymond, B. L. Abraham, T. Fujita, M. J. Watrous, E. S. Toriki, T. Takano and B. L. Nilsson, ACS Appl. Bio Mater., 2019, 2(5), 2116–2124 CrossRef CAS PubMed.
- K. Sugiyasu, M. Numata, N. Fujita, S. M. Park, Y. J. Yun, B. H. Kimb and S. Shinkai, Chem. Commun., 2004, 1996–1997 RSC.
- L. Huo, H. Zhang, Y. Wang, Y. Wang, B. Deng and L. Jin, Colloids Surf., A, 2022, 648, 129197 CrossRef CAS.
- J. Raeburna and D. J. Adams, Chem. Commun., 2015, 51, 5170–5180 RSC.
- B. O. Okesolaa and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC.
- E. J. Cho, I. Y. Jeong, S. J. Lee, W. S. Han, J. K. Kang and J. H. Jung, Tetrahedron Lett., 2008, 6, 1076–1079 CrossRef.
- C. Zhu, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, ACS Appl. Bio Mater., 2018, 1(6), 1768–1786 CrossRef CAS.
- Y. Hong, J. W. Y. Lama and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- Z. Li, X. Ji, H. Xie and B. Z. Tang, Adv. Mater., 2021, 33, 2100021 CrossRef CAS.
- A. Tran, M. Leroux, C. Michelin, F. Réveret, D. Boyer and F. Cisnetti, J. Mater. Chem. C, 2023, 11, 14896–14905 RSC.
- S. Leichnitz, K. C. Dissanayake, A. H. Winter and P. H. Seeberge, Chem. Commun., 2022, 58, 10556–10559 RSC.
- D. A. Merkushev, S. D. Usoltsev, Y. S. Marfin, A. P. Pushkarev, D. Volyniuk, J. V. Grazulevicius and E. V. Rumyantsev, Mater. Chem. Phys., 2017, 187, 104–111 CrossRef CAS.
- Z. Liu, Z. Jiang, M. Yan and X. Wang, Front. Chem., 2019, 7, 712–727 CrossRef CAS.
- H. Qian, M. E. Cousins, E. H. Horak, A. Wakefield, M. D. Liptak and I. Aprahamian, Nat. Chem., 2017, 9, 83–87 CrossRef CAS PubMed.
- L. A. Tatum, X. Su and I. Aprahamian, Acc. Chem. Res., 2014, 47(7), 2141–2149 CrossRef CAS PubMed.
- X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981 RSC.
- D. Cappello, A. E. R. Watson and J. B. Gilroy, Macromol. Rapid Commun., 2021, 42, 2000553 CrossRef CAS.
- D. Cappello, R. R. Maar, V. N. Staroverov and J. B. Gilroy, Chem. – Eur. J., 2020, 26, 5522–5529 CrossRef CAS PubMed.
- V. D. Singh, R. S. Singh, R. P. Paitandi, B. K. Dwivedi, B. Maiti and D. S. Pandey, J. Phys. Chem. C, 2018, 122(9), 5178–5187 CrossRef CAS.
- A. Kumar, R. S. Singh, A. Kumar, A. Ali, A. Biswas and D. S. Pandey, Chem. – Eur. J., 2016, 22, 13799–13804 CrossRef CAS PubMed.
- P. Demare and I. Regla, J. Chem. Educ., 2012, 89(1), 147–149 CrossRef CAS.
- A. Kumar, M. Dubey, R. Pandey, R. K. Gupta, A. Kumar, A. C. Kalita and D. S. Pandey, Inorg. Chem., 2014, 53(10), 4944–4955 CrossRef CAS.
- A. Kumar, M. Dubey, A. Kumar and D. S. Pandey, Chem. Commun., 2014, 50, 10086–10089 RSC.
- M. H. S. Marqvorsen, A. Brinkø and H. H. Jensen, Carbohydr. Res., 2020, 487, 107886 CrossRef CAS.
- V. D. Singh, B. K. Dwivedi, Y. Kumar and D. S. Pandey, Dyes Pigm., 2021, 184, 108812 CrossRef CAS.
- R. S. Singh, A. Kumar, S. Mukhopadhyay, G. Sharma, B. Koch and D. S. Pandey, J. Phys. Chem. C, 2016, 120(39), 22605–22614 CrossRef CAS.
- A. Biswas and S. Bera, Langmuir, 2023, 39, 2667–2675 CrossRef CAS PubMed.
- M. F. Zaini, I. A. Razak, M. Z. Anis and S. Arshad, Acta Crystallogr., 2019, 75, 58–63 CrossRef CAS.
- X. Ma, B. Qiao, J. Yue, J. Yu, Y. Geng, Y. Lai, E. Feng, X. Hana and M. Liu, Soft Matter, 2021, 17, 7813–7816 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, 11B, 19F NMR, HRMS, Fluorescence spectra, theoretical studies and tables are provided. CCDC 2337969, 2337970, 2309327 and 2309328. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc01694g |
| This journal is © The Royal Society of Chemistry 2024 |