
Exceptional thermal stability of lanthanide-phosphonate frameworks†
Ana D. G.
Firmino
 ab,
Ricardo F.
Mendes
*a,
Flávio
Figueira
a,
João P. C.
Tomé
c and
Filipe A.
Almeida Paz
*a
ab,
Ricardo F.
Mendes
*a,
Flávio
Figueira
a,
João P. C.
Tomé
c and
Filipe A.
Almeida Paz
*a
aDepartment of Chemistry, CICECO–Aveiro Institute of Materials, University of Aveiro, 3810-193 Aveiro, Portugal. E-mail: filipe.paz@ua.pt; Tel: +351 234 401418
bLAQV-REQUIMTE, Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal
cCQE & IMS, Departamento de Engenharia Química, Instituto Superior Técnico, Universidade de Lisboa, Lisboa, Portugal
First published on 3rd September 2024
Abstract
A single-crystal-to-single-crystal transformation (SC–SC) of [Ln(H5btp)]·2H2O [where Ln3+ = Gd3+ (1Gd), Tb3+ (1Tb), Dy3+ (1Dy), Ho3+ (1Ho), Er3+ (1Er), and Tm3+ (1Tm)] led to the formation of [Ln(L)(HL)] (where L = [−(PO3)(C6H3)(PO2)] and HL = [−(PO2H)(C6H3)(PO2)]n) based on a polymeric phosphonate-based organic linker (i.e., a polyMOF). The resulting material has high thermal stability maintaining its crystallinity and structural features up to ca. 800 °C, thus being to date the most thermally-robust and stable MOF. This remarkable feature is attributed to the close compact 3D network maintained by the strong pyrophosphonate bridges formed by the dehydration of the material at high temperatures.
Metal–organic frameworks (MOFs) are highly versatile materials with significant applications in gas storage and separation, catalysis, drug delivery, sensing, and energy storage, owing to their high surface areas, tuneable pore sizes, and diverse chemical functionalities.1–7 Their thermal stability is crucial for these applications, particularly in high-temperature processes, as it ensures structural integrity and long-term functionality.8–10 Factors influencing thermal stability include the choice of metal nodes, the nature of organic ligands, framework topology, and post-synthetic modifications.11,12 Enhancing the thermal stability of MOFs is essential to expand their practical use and improve their performance in demanding environments.7,13 For instance, the petrochemical industry's cracking process requires catalysts with thermal stabilities up to 500 °C, a process that currently relies solely on zeolite materials.14–17 The efficiency of the overall refining process heavily depends on the activity and durability of the catalyst under high temperature and pressure conditions.15,18
Common MOFs are often constructed using carboxylic acids as chelating groups. The use of phosphonic acids has gained traction in the last decade because of their tetrahedral oxygen atoms, which mimic the building units of zeolites, and are able to chelate to multiple metal centres.19–22 This structural feature is key to their exceptional chemical stability and coordination chemistry, leading to greater structural complexity and a wider range of applications when compared to families of MOFs assembled solely with carboxylated ligands.20,23 Consequently, there is increasing interest in examining the physical attributes of phosphonate-based MOFs, such as elastic moduli, responsiveness to stimuli, mesoscale structure, defect content, and thermal stability.24 Despite its importance, the thermal behaviour of these materials has often been overlooked in many reports. Banerjee and collaborators established a benchmark for the thermal stability of MOFs, reporting that a lithium-based MOF remained structurally unchanged when heated up to 610 °C.25 It is important, however, to emphasize that this temperature was achieved through thermogravimetric studies conducted in an inert atmosphere which typically delays the onset degradation of the materials.
Building upon our interest in phosphonate-based MOFs, we investigated the thermal stability of the [Ln(H5btp)]·2H2O system (where H8btp stands for 1,1′-biphenyl-3,3′,5,5′-tetrayltetrakis(phosphonic acid)), and report herein a significant breakthrough: after heating overnight at 400 °C, the MOF undergoes a single-crystal-to-single-crystal (SC–SC) transformation, producing a material with remarkable thermal stability that reaches up to ∼800 °C. This new network exhibits a substantial enhancement in the thermal stability of MOFs, particularly considering that thermogravimetric studies were conducted in the presence of air (a highly oxidative environment that usually promotes onset degradation at low temperatures). In this paper, we not only showcase this exceptional material but also provide several enhancements related to the preparation of the ligand and precursor of [Ln(H5btp)]·2H2O.
An improved, and more efficient, procedure to obtain H8btp is described in the ESI† (see Scheme S1). The protocol utilizes a (i) less expensive catalyst (CuCl2·2H2O), (ii) reduces the reaction time to just about 5 hours, and (iii) increases the yield from 70% to 93%. This new approach saves on reaction costs and opens up the possibility of easy preparation of the ligand at the gram scale. All the structural characterisation details can be found in the ESI† (Fig. S1–S11).
The [Ln(H5btp)]·2H2O family of MOF materials was prepared using an adaptation of a previously reported synthetic method,26 using instead lanthanide oxides (Ln2O3) as the metal sources: Gd3+ (1Gd), Tb3+ (1Tb), Dy3+ (1Dy), Ho3+ (1Ho), Er3+ (1Er), and Tm3+ (1Tm). We have further explored the use of smaller lanthanide ions showing that the system remains isotypical throughout the entire lanthanide series (Fig. S12 in the ESI†), without observing a modification on the overall crystallinity and morphology (Fig. S14–S20 in the ESI†).
The average particle size of the obtained materials followed an interesting trend. Lanthanides with higher ionic radius tend to form larger crystals (needles ranging from 30 to 100 μm), with a progressive decrease in the apparent particle size observed for the smaller metal cations: the average crystallite size follows the trend Gd(1Gd) > Tb(1Tb) > Dy(1Dy) > Ho(1Ho) > Er(1Er) > Tm(1Tm) (Fig. 1 and Fig. S14–S20 in the ESI†). This type of size distribution behaviour depending on the metal centre is not usual for LnOF materials and, to the best of our knowledge, was only reported by our group for the [Ln2(H3bmt)2]·H2O series, in which the metallic centre exhibits an uncommon low coordination number octahedral coordination environment, which might induce a progressive reduction in crystal size with the decrease of the metallic ionic radii.27
 | ||
| Fig. 1 Powder X-ray diffraction, scanning electron microscopy images, and particle size distribution of [Ln(H5btp)]·2H2O [Ln3+ = Gd3+ (1Gd), Tb3+ (1Tb), Dy3+ (1Dy), Ho3+ (1Ho), Er3+ (1Er), and Tm3+ (1Tm)]. The average particle size was calculated using a Gaussian function and is given in μm. Histogram analysis was performed by measuring the size of more than 100 particles for each sample. | ||
[Ln(H5btp)]·2H2O (1)26 crystallises in the monoclinic space group C2/c, with the asymmetric unit being composed of half of a metal centre (having a slightly distorted octahedral coordination environment) and half of a H5btp3− residue, with only one disordered crystallisation water molecule. The 3D network is constructed by Ln3+ phosphonate chains distributed in a zigzag fashion along the c-axis, being connected by the organic linker. This allowed the assembly of a microporous material with a pore arrangement resembling a lozenge shaped tubular structure. The planar rearrangement of the organic linker in the structure, with no torsion angle between the two aromatic rings, promoted the presence of weak π–π interactions [distance between centroids of 3.856(6) Å], alongside with hydrogen bonding interactions between the crystallisation water molecules and the phosphonate groups.
Considering the structural similarities between the materials reported in this work and previous yttrium-based compounds, we decided to test their resistance with increasing temperatures. The thermal stability of [Tb(H5btp)]·2H2O (1Tb) was investigated between ambient temperature and ca. 1200 °C (Fig. 2–bottom). The structural features were further investigated using in situ variable temperature powder X-ray diffraction studies (VTPXRD; Fig. 2–top) performed in the temperature range of 25–1200 °C. Between ambient temperature and ca. 250 °C the crystallisation water molecules are liberated from the framework, with a concomitant decrease in mass (ca. 4.83%) that correlates well with the release of the two molecules (calculated as ca. 5.41%) (Fig. 2 and Table S4, ESI†). This simple dehydration is accompanied by a shift in the second reflection to a higher value of 2 theta, i.e., the (110) plane, indicating as expected a reduction in the unit cell dimensions, particularly along the [100] and [010] directions. Based on the structural features of 1Tb, this transformation promoted proximity between the adjacent terbium-phosphonate chains of the framework. The second weight loss, corresponding to 6.05%, occurred between ca. 250 and 650 °C. Based on our previous data, in this temperature range, the phosphonate groups of the organic ligand in the framework may undergo rearrangement, leading to the formation of pyrophosphonates28 (Fig. S23 in the ESI†) with the consequent release of water molecules (in this case, the observed weight loss is equivalent to approximately two water molecules). This behaviour was accompanied by another shift in the second reflection to a higher value of 2θ, leading to the formation of a new crystalline phase that remains stable up to ca. 800 °C. By analysing the DSC data (Fig. S24 in the ESI†) we are able to observe the presence of an endothermic peak at ca. 335 °C that corresponds to the temperature at which this transformation is observed. Such behaviour was observed for the entire lanthanide series reported in this paper (Fig. S26–S31 in the ESI†). We note that MOFs, especially those prepared with phosphonic acid organic linkers, tend to become amorphous at high temperatures (usually around 400 °C). At these temperatures, the solvent molecules, many times coordinated to the metal centres, are removed and the structure tends to collapse. In the present case the increase in temperature leads, however, to a single-crystal-to-single-crystal (SC–SC) transformation of [Ln(H5btp)]·2H2O (1), through the formation several pyrophosphonate bridges between adjacent phosphonate groups (Fig. 3).
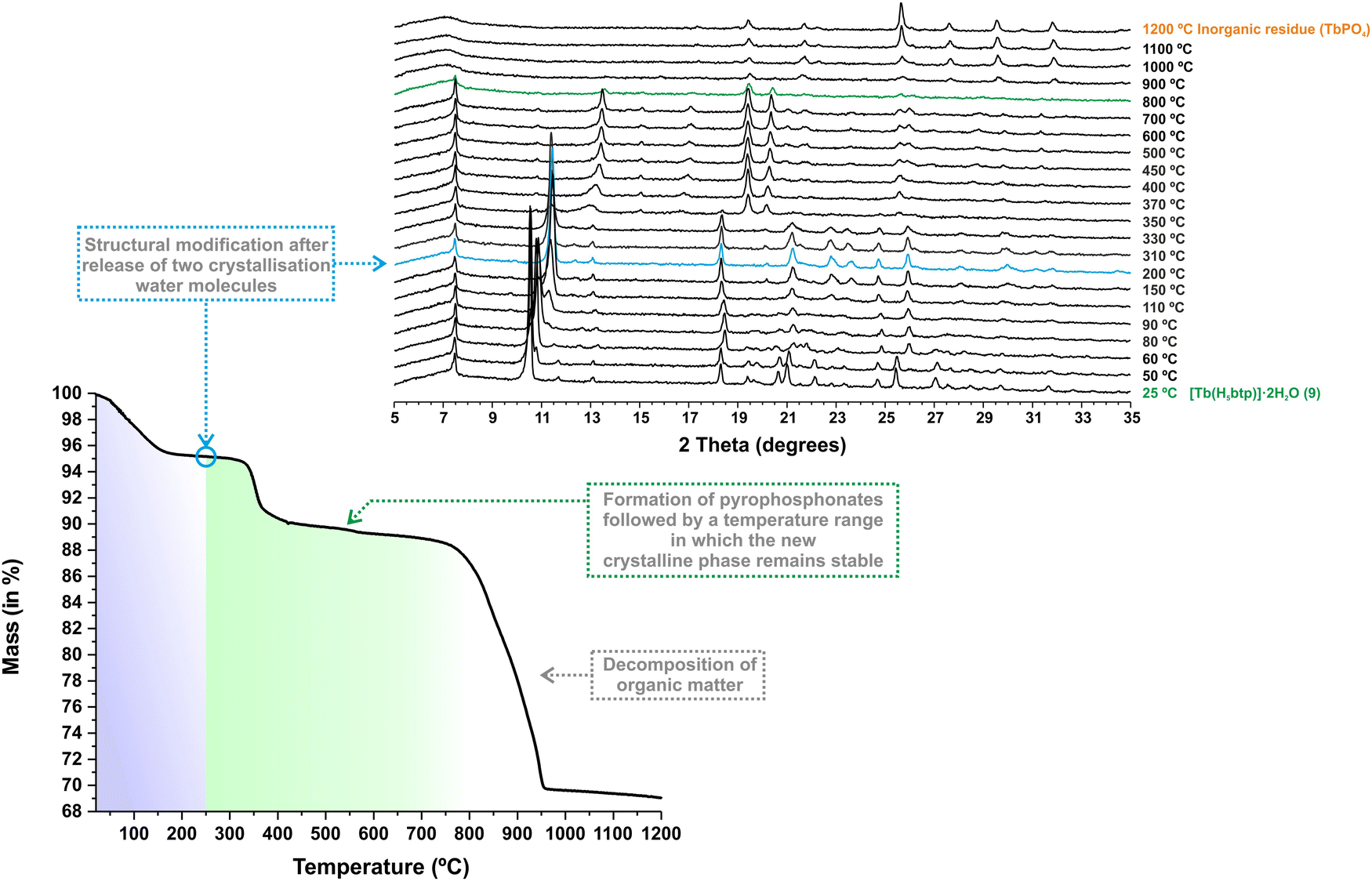 | ||
| Fig. 2 (Bottom) Thermogram and (top) variable-temperature powder X-ray diffraction studies of [Tb(H5btp)]·2H2O (1Tb) collected between ambient temperature and ca. 1200 °C. | ||
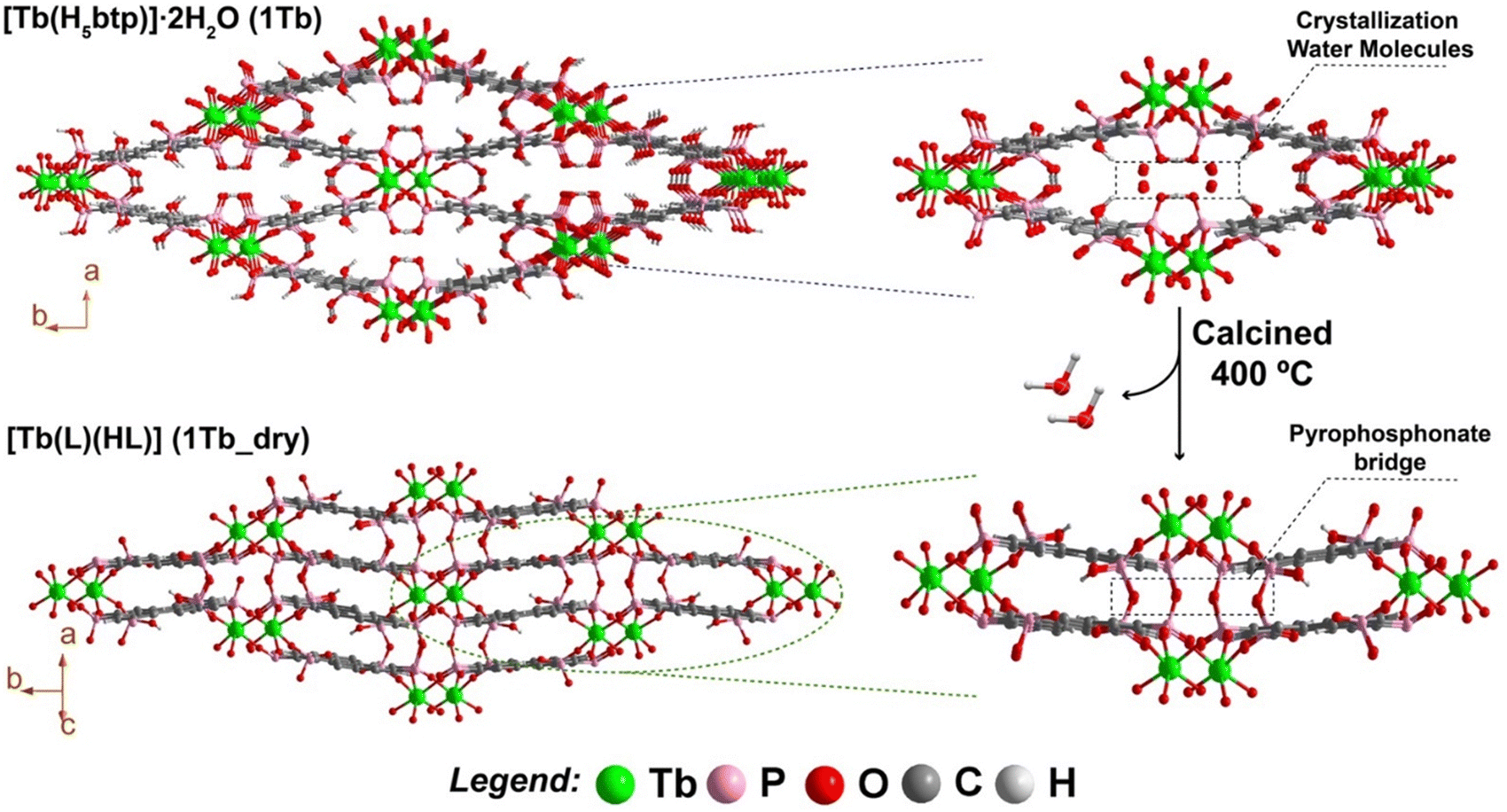 | ||
| Fig. 3 Schematic representation of the single-crystal-to-single-crystal transformation of [Tb(H5btp)]·2H2O (1Tb) into [Tb(L)(HL)] (1Tb_dry) at 400 °C, emphasising the formation of pyrophosphonate bridges. | ||
SC–SC transformations based on dehydration are common, and we have observed this behaviour in a related family of MOF materials: [Y(H5btp)]·5.5H2O is transformed into [Y(H5btp)]·2.5H2O by simple removal of the solid from the mother solution (filtration); the latter frameworks can be further dehydrated and consequently transformed into (H3O)[Y2(H5btp)(H4btp)]·H2O by heating overnight at 110 °C.29 These transformations were possible by the sole removal of crystallisation water molecules from the framework, with no major changes to the coordination backbone of the MOFs. The structural features of the new high-temperature crystalline phase, 1Tb_dry, were unveiled by single-crystal X-ray diffraction studies on calcined crystals at ca. 400 °C (hence the various problems related with the structural model), with the compound being formulated as [Tb(L)(HL)] (where L = [−(PO3)(C6H3)(PO2)] and HL = [−(PO2H)(C6H3)(PO2)]) crystallising in the centrosymmetric space group P21/n (Fig. 3). The SC–SC transformation of 1Tb into 1Tb_dry can therefore be described in two steps: (i) in the first step the crystallisation water molecules are removed from the network, with the process being accompanied with a decrease of the Ln⋯Ln distances of the terbium-phosphonate chains, ultimately bringing the phosphonic acid groups closer to each other; (ii) the second step consists the formation of the pyrophosphonate bridges by condensation of neighbouring POH groups (with the release of two water molecules per formula unit), resulting in a final distance of metal centres of 7.660(3) Å (Fig. S13 in the ESI†). Remarkably, these pyrophosphonate bridges led to the formation of a polymeric organic linker that interconnect the lanthanide oxide chains running parallel to the c-axis of the unit cell. Notably, the presence of polymeric organic linkers in MOFs (known as polyMOFs) have received great interest in recent years,30,31 increasing the possibility of tailoring the physicochemical properties of existing MOFs, with the more known examples being in the improvement of CO2 adsorption.32,33
The resulting [Tb(L)(HL)] polyMOF remains crystalline at high temperatures and after cooling down to ambient temperature (Fig. 4). In fact, thermogravimetric analysis of 1Tb_dry shows a negligible weight loss up to 500 °C (only about 1.5%, attributed to the removal of adsorbed water at the surface of crystallites-Fig. S25 in the ESI†), proving its thermal stability after the SC–SC transformation. We attribute the resilience of the structural features to the lack of coordinated solvent molecules to the metal centres in the parent 1 materials, which impels that the structural modifications driven by the increasing in temperature are confined solely to the channels of the microporous structure. The formation of pyrophosphonate bridges was already hypothesized by us in a previous study based on powder X-ray diffraction data, in which we have shown that these bridges are formed by the presence of the vibrational band of P–O–P in the FTIR spectra.28 These vibrational modes are once again markedly visible in the FT-IR spectra of the materials isolated at high temperatures (see Fig. S23 in the ESI†). SEM and EDS analysis (Fig. S21 in the ESI†) further evidence the presence of crystals with the same needle-like morphology, with both the Tb and P elements homogeneously distributed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.6 ratio, which agrees well with the crystal structure model presented.
:3.6 ratio, which agrees well with the crystal structure model presented.
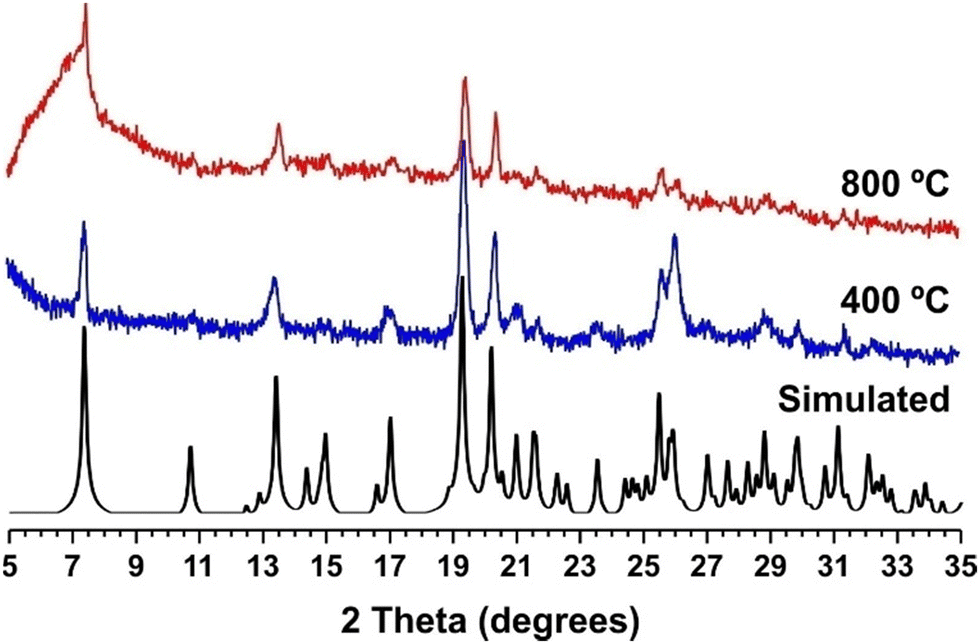 | ||
| Fig. 4 Comparison between the simulated (black line), dehydrated at 400 °C (blue line) and 800 °C (red line) powder X-ray diffraction patterns of [Ln(L)(HL)] (1Tb_dry). | ||
This structural resilience consolidates the [Ln(L)(HL)] family of MOF materials as that with the highest thermal stability reported to date, surpassing in length the known ZIF-8 and UiO-66 compounds (both with thermal stability up to ca. 550 °C),34,35 and also the lithium-based MOF (ca. 610 °C) reported by Banerjee and co-workers.25 This thermal stability is even similar to many zeolite-type materials, with only three minerals being stable to higher temperatures, namely clinoptololite, merlinoite and wairakite.36
In summary, we describe the hydrothermal preparation of a family of 3D LnMOFs, formulated as [Ln(H5btp)]·2H2O [where Ln3+ = Gd3+ (1Gd), Tb3+ (1Tb), Dy3+ (1Dy), Ho3+ (1Ho), Er3+ (1Er), and Tm3+ (1Tm)], from the reaction between lanthanide oxides and the organic ligand [1,1′-biphenyl]-3,3′,5,5′-tetrayltetrakis(phosphonic acid) (H8btp). Both the organic linker and the MOF materials were optimized to obtain better yields and overall cost-effectiveness. This family of compounds has shown outstanding thermal stability (up to ∼800 °C) owing to a peculiar SC–SC transformation that occurs at high temperature. The proximity between the phosphonate groups promoted by the release of water molecules of crystallisation impels a structural rearrangement that forms four pyrophosphonate bridges by local dehydration, with the resulting ligand being polymeric in nature. These bridges, along with the stability of the C–PO3 bond, as well as the remaining intermolecular interactions in this structure, seem to be the reason for the maintenance of the hybrid material that is structurally robust at high temperatures. This communication shows that by tailoring specific structural features in MOF materials, e.g., the close proximity of specific functional groups, it is possible to design new materials with improved properties. In this case, improved thermal stability and robustness opens the possibility of using these compounds in applications that are typically not accessible to MOF materials.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data (including structure factors) for the crystal structure described in the manuscript have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication data No. 2362876.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was developed within the scope of the project CICECO-Aveiro Institute of Materials, (Refs. UIDB/50011/2020, DOI 10.54499/UIDB/50011/2020; UIDP/50011/2020, DOI 10.54499/UIDP/50011/2020; LA/P/0006/2020, DOI 10.54499/LA/P/0006/2020), financed by national funds through the FCT/MCTES (PIDDAC), CQE (UIDB/00100/2020 and UIDP/00100/2020) and IMS (LA/P/0056/2020). FCT is gratefully acknowledged for the PhD grant No. SFRH/BD/84495/2012 (to D. F.) and for the Junior Research Position (https://doi.org/10.54499/CEECIND/00553/2017/CP1459/CT0034) (to R. F. M.). The research contract of F. F. (REF-168-89-ARH/2018) is funded by national funds (OE), through FCT, in the scope of the framework contract foreseen in No. 4, 5 and 6 of article 23 of the Decree-Law 57/2016, of August 29, changed by Law 57/2017, of July 19.Notes and references
- D. Li, A. Yadav, H. Zhou, K. Roy, P. Thanasekaran and C. Lee, Advances and Applications of Metal-Organic Frameworks (MOFs) in Emerging Technologies: A Comprehensive Review, Global Challenges, 2024, 8, 2300244 CrossRef PubMed.
- R. Luque, A. Ahmad, S. Tariq, M. Mubashir, M. S. Javed, S. Rajendran, R. S. Varma, A. Ali and C. Xia, Functionalized interconnected porous materials for heterogeneous catalysis, energy conversion and storage applications: Recent advances and future perspectives, Mater. Today, 2024, 73, 105–129 CrossRef.
- J. S. Barbosa, R. F. Mendes, F. Figueira, V. M. Gaspar, J. F. Mano, S. S. Braga, J. Rocha and F. A. Almeida Paz, Bone Tissue Disorders: Healing Through Coordination Chemistry, Chem. – Eur. J., 2020, 26, 15416–15437 CrossRef PubMed.
- F. Figueira, J. P. C. Tomé and F. A. A. Paz, Porphyrin NanoMetal-Organic Frameworks as Cancer Theranostic Agents, Molecules, 2022, 27, 3111 CrossRef PubMed.
- A. K. Bindra, D. Wang and Y. Zhao, Metal–organic frameworks meet polymers: from synthesis strategies to healthcare applications, Adv. Mater., 2023, 35, 2300700 CrossRef PubMed.
- R. F. Mendes, F. Figueira, J. P. Leite, L. Gales and F. A. Almeida Paz, Metal–organic frameworks: a future toolbox for biomedicine?, Chem. Soc. Rev., 2020, 49, 9121–9153 RSC.
- Z. Chang, Recent progress in host–guest metal–organic frameworks: Construction and emergent properties, Coord. Chem. Rev., 2023, 476, 214921 CrossRef.
- C. Healy, K. M. Patil, B. H. Wilson, L. Hermanspahn, N. C. Harvey-Reid, B. I. Howard, C. Kleinjan, J. Kolien, F. Payet and S. G. Telfer, The thermal stability of metal-organic frameworks, Coord. Chem. Rev., 2020, 419, 213388 CrossRef CAS.
- Y. An, X. Lv, W. Jiang, L. Wang, Y. Shi, X. Hang and H. Pang, The stability of MOFs in aqueous solutions—Research progress and prospects, Green Chemical Engineering, 2023 Search PubMed.
- Y.-T. Pan, Z. Zhang and R. Yang, The rise of MOFs and their derivatives for flame retardant polymeric materials: A critical review, Composites, Part B, 2020, 199, 108265 CrossRef.
- S. Afrin, M. W. Khan, E. Haque, B. Ren and J. Z. Ou, Recent advances in the tuning of the organic framework materials–The selections of ligands, reaction conditions, and post-synthesis approaches, J. Colloid Interface Sci., 2022, 623, 378–404 CrossRef.
- S. Mandal, S. Natarajan, P. Mani and A. Pankajakshan, Post-Synthetic Modification of Metal–Organic Frameworks Toward Applications, Adv. Funct. Mater., 2021, 31, 2006291 CrossRef.
- Y.-L. Hou, C. Yang, Z. Yang, H. Zhou, L. Guo, J. Guo and X. Zhang, Building robust metal-organic frameworks with premade ligands, Coord. Chem. Rev., 2024, 505, 215690 CrossRef.
- A. Tanimu, G. Tanimu, H. Alasiri and A. Aitani, Catalytic Cracking of Crude Oil: Mini Review of Catalyst Formulations for Enhanced Selectivity to Light Olefins, Energy Fuels, 2022, 36, 5152–5166 CrossRef.
- M. A. Alabdullah, A. R. Gomez, J. Vittenet, A. Bendjeriou-Sedjerari, W. Xu, I. A. Abba and J. Gascon, A Viewpoint on the Refinery of the Future: Catalyst and Process Challenges, ACS Catal., 2020, 10, 8131–8140 CrossRef CAS.
- Y. Li and J. Yu, Emerging applications of zeolites in catalysis, separation and host–guest assembly, Nat. Rev. Mater., 2021, 6, 1156–1174 CrossRef.
- K. A. D. F. Castro, F. Figueira, F. A. Almeida Paz, J. P. C. Tomé, R. S. da Silva, S. Nakagaki, M. G. P. M. S. Neves, J. A. S. Cavaleiro and M. M. Q. Simões, Copper-phthalocyanine coordination polymer as a reusable catechol oxidase biomimetic catalyst, Dalton Trans., 2019, 48, 8144–8152 RSC.
- Z. Sharifzadeh and A. Morsali, Amine-functionalized metal-organic frameworks: from synthetic design to scrutiny in application, Coord. Chem. Rev., 2022, 459, 214445 CrossRef.
- P. Kumar, K. H. Kim, E. E. Kwon and J. E. Szulejko, Metal-organic frameworks for the control and management of air quality: advances and future direction, J. Mater. Chem. A, 2016, 4, 345–361 RSC.
- P. Tholen, Y. Zorlu, J. Beckmann and G. Yücesan, Probing Isoreticular Expansions in Phosphonate MOFs and their Applications, Eur. J. Inorg. Chem., 2020, 1542–1554 CrossRef.
- X.-W. Lv, C.-C. Weng, Y.-P. Zhu and Z.-Y. Yuan, Nanoporous Metal Phosphonate Hybrid Materials as a Novel Platform for Emerging Applications: A Critical Review, Small, 2021, 17, 2005304 CrossRef PubMed.
- A. D. G. Firmino, R. F. Mendes, D. Ananias, F. Figueira, J. P. C. Tomé, J. Rocha and F. A. Almeida Paz, Pyrene Tetraphosphonate-Based Metal-Organic Framework: Structure and Photoluminescence, Eur. J. Inorg. Chem., 2020, 3565–3572 CrossRef.
- S. Wöhlbrandt, C. Meier, H. Reinsch, E. Svensson Grape, A. K. Inge and N. Stock, A Tetratopic Phosphonic Acid for the Synthesis of Permanently Porous MOFs: Reactor Size-Dependent Product Formation and Crystal Structure Elucidation via Three-Dimensional Electron Diffraction, Inorg. Chem., 2020, 59, 13343–13352 CrossRef PubMed.
- J. Troyano, A. Legrand and S. Furukawa, Mechanoresponsive porosity in metal-organic frameworks, Trends Chem., 2021, 3, 254–265 CrossRef.
- D. Banerjee, S. J. Kim and J. B. Parise, Lithium Based Metal−Organic Framework with Exceptional Stability, Cryst. Growth Des., 2009, 9, 2500–2503 CrossRef.
- D. Ananias, A. D. G. Firmino, R. F. Mendes, F. A. A. Paz, M. Nolasco, L. D. Carlos and J. Rocha, Excimer Formation in a Terbium Metal–Organic Framework Assists Luminescence Thermometry, Chem. Mater., 2017, 29, 9547–9554 CrossRef.
- F. A. A. Paz, S. M. F. Vilela and J. P. C. Tome, Layered Metal-Organic Frameworks Based on Octahedral Lanthanides and a Phosphonate Linker: Control of Crystal Size, Cryst. Growth Des., 2014, 14, 4873–4877 CrossRef CAS.
- P. Silva, F. Vieira, A. C. Gomes, D. Ananias, J. A. Fernandes, S. M. Bruno, R. Soares, A. A. Valente, J. Rocha and F. A. A. Paz, Thermal Transformation of a Layered Multifunctional Network into a Metal-Organic Framework Based on a Polymeric Organic Linker, J. Am. Chem. Soc., 2011, 133, 15120–15138 CrossRef.
- A. D. G. Firmino, R. F. Mendes, M. M. Antunes, P. C. Barbosa, S. M. F. Vilela, A. A. Valente, F. M. L. Figueiredo, J. P. C. Tome and F. A. A. Paz, Robust Multifunctional Yttrium-Based Metal Organic Frameworks with Breathing Effect, Inorg. Chem., 2017, 56, 1193–1208 CrossRef.
- Z. Zhang, H. T. H. Nguyen, S. A. Miller and S. M. Cohen, polyMOFs: A Class of Interconvertible Polymer-Metal-Organic-Framework Hybrid Materials, Angew. Chem., Int. Ed., 2015, 54, 6152–6157 CrossRef.
- K. C. Bentz, K. Gnanasekaran, J. B. Bailey, S. Ayala, F. A. Tezcan, N. C. Gianneschi and S. M. Cohen, Inside polyMOFs: layered structures in polymer-based metal–organic frameworks, Chem. Sci., 2020, 11, 10523–10528 RSC.
- T. H. Lee, B. K. Lee, S. Y. Yoo, H. Lee, W.-N. Wu, Z. P. Smith and H. B. Park, PolyMOF nanoparticles constructed from intrinsically microporous polymer ligand towards scalable composite membranes for CO2 separation, Nat. Commun., 2023, 14, 8330 CrossRef.
- Z. Zhang, H. T. H. Nguyen, S. A. Miller, A. M. Ploskonka, J. B. DeCoste and S. M. Cohen, Polymer–Metal–Organic Frameworks (polyMOFs) as Water Tolerant Materials for Selective Carbon Dioxide Separations, J. Am. Chem. Soc., 2016, 138, 920–925 CrossRef.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, A new zirconium inorganic building brick forming metal organic frameworks with exceptional stability, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef.
- K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. D. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Exceptional chemical and thermal stability of zeolitic imidazolate frameworks, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef.
- G. Cruciani, Zeolites upon heating: Factors governing their thermal stability and structural changes, J. Phys. Chem. Solids, 2006, 67, 1973–1994 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2362876. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc02589j |
| This journal is © The Royal Society of Chemistry 2024 |