
Advanced thermal/environmental barrier coatings of high-entropy rare earth disilicates tuned by strong anharmonicity of Eu2Si2O7†
Shiqiang
Hao
 ab,
Richard P.
Oleksak
a,
Ömer N.
Doğan
a and
Michael C.
Gao
*a
ab,
Richard P.
Oleksak
a,
Ömer N.
Doğan
a and
Michael C.
Gao
*a
aNational Energy Technology Laboratory, 1450 Queen Avenue SW, Albany, OR 97321, USA. E-mail: Shiqiang.Hao@netl.doe.gov; Michael.Gao@netl.doe.gov
bNETL Support Contractor, 1450 Queen Avenue SW, Albany, OR 97321, USA
First published on 15th October 2024
Abstract
Advancing thermal/environmental barrier coating (TEBC) materials with integrated thermal-mechanical functions is paramount for safeguarding SiC-based ceramic matrix composites (CMCs) in high-efficiency gas turbines. Herein, we employ a synergistic approach, combining density functional theory (DFT) methods and combinatorial chemistry techniques, to design high-performance and low-cost RE2Si2O7 (RE = rare earth elements) TEBC materials tailored for enhanced compatibility with SiC-based CMCs. Expanding on phase stability of alloying pure RE2Si2O7, the investigation extends to the mechanical and thermal properties of solid solution systems, including Er1/2Y3/4Yb3/4Si2O7, Gd1/4Er1/4Y3/4Yb3/4Si2O7, and Eu1/4Er1/4Y3/4Yb3/4Si2O7. The solid solution systems exhibit a major reduction in lattice thermal conductivity relative to their pure counterparts, achieving ultralow values of 0.25 to 0.39 W m−1 K−1 at 1500 K. Furthermore, the coefficients of thermal expansion (CTE) of these solid solutions are precisely tuned within the desired range for SiC (4.4 to 5.5 × 10−6 K−1), while maintaining good mechanical properties. In particular, the addition of Eu2Si2O7 demonstrates to be an important variable to the tuning of CTE and lattice thermal conductivity by leveraging its strong anharmonicity, presenting a pioneering avenue for fine-tuning material properties. In summary, this research not only identifies promising TEBC materials with superior thermal properties, but also introduces a valuable computational material design methodology for the rapid discovery of complex materials for harsh environments.
Introduction
In the endless pursuit of increased gas turbine efficiencies, silicon carbide based ceramic matrix composites (CMCs) represent the most promising materials available to replace Ni-based superalloys and push the operational temperature of the turbine. Environmental degradation represents a major challenge to using these CMCs in the hottest portions of gas turbines. As such, environmental barrier coatings (EBCs) have been developed in parallel with silicon carbide-based CMCs. The EBC is applied to the CMC surface and protects them from corrosive reaction with the harsh combustion environment.The role of EBCs as a key enabler for CMCs in gas turbines has led to their continuous development and improvement over the past 35 years.1 Through these efforts, rare earth disilicates RE2Si2O7 (RE = rare earth element) have emerged as the most promising class of EBC materials due to a combination of good CTE match with silicon carbide, good phase stability, and resistance to environmental degradation by steam and molten deposits such as calcia–magnesia–alumina–silica (CMAS). As a result, current state-of-the-art EBCs utilize a rare earth disilicate layer, most notably Yb2Si2O7, as part of the overall coating system. To allow for even further temperature capability of the CMC, and considering the relatively high thermal conductivity of Yb2Si2O7, two additional thermal barrier coating (TBC) and transition layers are usually applied above the EBC layer. Collectively, this multilayer TEBC system protects the CMC from the harsh combustion environment.
Recently it was recognized that mixing rare earths in the RE2Si2O7 EBC system can significantly reduce the thermal conductivity and potentially improve additional coating properties.2 This has spurred significant interest in multicomponent (high entropy) rare earth disilicates for being used as a single TEBC layer by eliminating the need for a thermal barrier and transition layers.3 The first requirement that must be fulfilled for a successful multicomponent rare earth disilicate TEBC is phase stability. Early reported effort in the literature is to dope Yb2Si2O7 with Lu and Sc, which have similar ionic radii to Yb, and β-Lu2Si2O7/β-Sc2Si2O7 are stable over a wide temperature range.1 However, Lu and Sc are extremely expensive, and vulnerable to possible supply chain disruptions. Utilizing affordable earth-abundant rare earths can not only significantly reduce the material cost, but also offer other potential benefits. For example, incorporation of rare earths with relatively lower molecular weight has been shown to improve resistance to CMAS corrosion.4,5 In short, there are countless multicomponent rare earth disilicates remaining to be discovered, and among them is likely to be a material that will help define the next generation TEBC system.
Due to the vast composition space accessible in the rare earth disilicate system, computational approaches to guide material discovery are essential. Density functional theory (DFT) based methods can be used to identify compositions likely to form a single monoclinic phase, followed by calculation of important material properties such as CTE, lattice thermal conductivity, and elastic moduli.6 Furthermore, such approaches can enable precise tuning of critical properties by leveraging unique electronic structures of the various constituents. Herein, we demonstrate such an approach to identify promising candidate TEBC materials in the rare earth disilicate system. Strong anharmonicity of Eu2Si2O7 is identified and utilized to fine-tune CTE and additional properties through alloying with other relatively inexpensive rare earths. The end results are new low-cost high-performance multicomponent RE2Si2O7 materials for high temperature coatings and other applications.
Methods
Density functional theory (DFT) calculations
The Vienna ab initio simulation package7 was utilized for conducting DFT calculations on RE2Si2O7 (RE = Y, Eu, Gd, Er, and Yb) and solid solutions involving rare earth element alloying. Total energies and optimized structural configurations were determined using the projector augmented wave method8 to model electron–ion interactions. The Perdew–Burke–Ernzerhof generalized gradient approximation9 was employed for the exchange–correlation functional. The calculations achieved numerical convergence of total energies to approximately 1 meV per atom, with a basis set energy cutoff of 550 eV. Dense k-meshes, corresponding to 4000 reciprocal atom k-points in the Brillouin zone, were utilized. Atomic positions and cell volumes were relaxed using the conjugate gradient method. For magnetic rare earth elements like Gd, a ferromagnetic spin configuration was applied, with an initial magnetic moment of 5 μB.Cluster expansion
The cluster expansion approach was used to construct an effective Hamiltonian for energy evaluation on the rare earth disilicate structure. We used the ATAT toolkit10 to obtain the optimal effective cluster interactions from fully relaxed total energies of ordered input structures. In a typical simple ternary cluster expansion, the energy is expressed as with a spin variable σi with a value of ±1, and 0 that denotes the ternary type of atom sitting on each site i, where mα and Jα are multiplicities and effective interactions of cluster type α. By fitting 156 ordered input structures, the final cluster expansion contains 21 interaction coefficients, including sixteen two-body interactions, resulting in a leave-one out cross-validation score as 3.5 meV per cation, which is only approximately 7% of the most favorable formation energy of −46 meV. The formation energy ΔH of a A1−x−yBxCy with respect to the energies of constituents A, B, and C is defined as ΔH(σ) = Etot(σ) − (1 − x − y)EAtot − xEBtot − yECtot, where Etot(σ), EAtot, EBtot, and ECtot are total energies of phases A1−x−yBxCy, A, B, and C, respectively. For example, the formation energies of Er1−x−yYxYbySi2O7 (A1−xBxCy) are calculated with respect to the references of the β phase of Er2Si2O7(A), Y2Si2O7(B), and Yb2Si2O7(C).
with a spin variable σi with a value of ±1, and 0 that denotes the ternary type of atom sitting on each site i, where mα and Jα are multiplicities and effective interactions of cluster type α. By fitting 156 ordered input structures, the final cluster expansion contains 21 interaction coefficients, including sixteen two-body interactions, resulting in a leave-one out cross-validation score as 3.5 meV per cation, which is only approximately 7% of the most favorable formation energy of −46 meV. The formation energy ΔH of a A1−x−yBxCy with respect to the energies of constituents A, B, and C is defined as ΔH(σ) = Etot(σ) − (1 − x − y)EAtot − xEBtot − yECtot, where Etot(σ), EAtot, EBtot, and ECtot are total energies of phases A1−x−yBxCy, A, B, and C, respectively. For example, the formation energies of Er1−x−yYxYbySi2O7 (A1−xBxCy) are calculated with respect to the references of the β phase of Er2Si2O7(A), Y2Si2O7(B), and Yb2Si2O7(C).
Utilizing converged effective cluster interactions, we employed canonical Monte Carlo simulations to compute the order–disorder transition temperatures.11 In the canonical ensemble, the alloy's energy and concentration (with a fixed total number of atoms) experience fluctuations, while external impositions maintain temperature and chemical potentials. Scanning across temperature and chemical potentials allowed us to identify two-phase regions in the phase diagram, discerning them through composition discontinuities as a function of chemical potential. Our simulation employed a 30 × 30 × 30 unit cell, covering temperatures from 25 to 800 K with a 25 K interval. Equilibration involved 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Monte Carlo steps, followed by averaging over 100000 steps. Monitoring composition changes with chemical potential at a given temperature, we determined the thermodynamic state as a function of temperature and the chemical potential differences of the constituents.
000 Monte Carlo steps, followed by averaging over 100000 steps. Monitoring composition changes with chemical potential at a given temperature, we determined the thermodynamic state as a function of temperature and the chemical potential differences of the constituents.
Elastic constants and mechanical properties
We employed strain-energy techniques, akin to our prior investigation,12,13 for evaluating the elastic constants. The overall energy of the crystal system subjected to strain, denoted as δ, can be expressed as a Taylor series in terms of the strain tensor relative to that of the unstrained crystal.14 | (1) |
 | (2) |
For the monoclinic systems, the thirteen independent elastic constants can be derived by imposing thirteen distinct distortions.15 These parameters encompass six diagonal components Cii (i = 1, 2, 3, 4, 5, 6) and seven off-diagonal components, namely C12, C13, C15, C23, C25, C35, and C46, as enumerated in eqn (3):
 | (3) |
To determine the elastic constants, we subjected the DFT-relaxed equilibrium structures to six different levels of strains: ±0.7%, ±1.4%, ±2.1%. Subsequently, we derived the elastic constants from the resulting strain-induced changes by employing a fourth-degree polynomial fit on the DFT energies associated with each strain. It is important to highlight that we deliberately applied relatively small magnitudes of strain to the lattice. This decision was made to mitigate the influence of higher-order phonon–phonon interactions on the energy landscape, thereby avoiding potential phonon mode instabilities not accounted for by the quasi-harmonic approximation.
It is crucial to emphasize that ensuring the mechanical stability of a structure requires the calculated elastic constants to meet the mechanical stability criterion.16 The criteria for mechanical stability in a monoclinic system are delineated in eqn (4). By employing these elastic constants, one can compute the bulk and shear moduli as detailed in eqn (5).
 | (4) |
With the above elastic constants, the average polycrystalline elastic properties, such as bulk modulus (B) and shear modulus (G), can be calculated using the Voigt–Reuss–Hill methods (see eqn (5)). It is known that Voigt's scheme gives the upper bound with the elastic constant matrix input, while Reuss’ method provides the lower bound by evaluating the elastic compliance matrix (inverse to the elastic-constant matrix), and Hill made an average on the Voigt and Reuss results.
 | (5) |
On the basis of elastic constants Cij and elastic compliance constants Sij (inverse matrix of the elastic constant), the elastic anisotropy can be evaluated through directional dependence of Young's and shear moduli. The directional Young's and shear moduli are evaluated by:17,18
 | (6) |
Phonon dynamics
To explore the impact of temperature on the elastic constants, we investigated the system's Helmholtz free energy as a function of temperature, considering the contribution of vibrational entropy. To ensure a meaningful incorporation of phonon energy into the Helmholtz free energy, it is crucial to compute vibrational frequencies throughout the entire Brillouin zone. The representation of phonon vibrational frequencies was achieved by plotting the phonon dispersion along high-symmetry lines within the Brillouin zone. The calculation of phonon modes involved the utilization of finite atomic displacement methods,19 wherein the force constant matrix is generated by analyzing the total energies of various configurations where individual atoms in the cell undergo displacement from their equilibrium positions. The dynamical matrix at any given![[q with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0071_20d1.gif) vector point can be determined by considering the masses of different species based on the force constant matrix:
vector point can be determined by considering the masses of different species based on the force constant matrix: | (7) |
To assess the Helmholtz free energy F(T,V) at different temperatures using the phonon dispersion, we represent the summation over phonon modes as an integration across a densely sampled grid of crystal momenta q-vector in reciprocal space. This integration involves the phonon density of states, denoted as g(ω),14
 | (8) |
Coefficient of thermal expansion and quasi-harmonic approximation
Similar to the mechanical strain, thermal energy can also induce strains within the crystal lattice. The source of thermal expansion in solid-state materials arises from the anharmonicity of the crystal lattice. The predominant factor contributing to this anharmonicity in thermal expansion is the interaction between phonons and strain, leading to changes in the dimensions of the unit cell with temperature variations.20 To calculate the coefficient of thermal expansion, we applied the quasi-harmonic approximation (QHA) in phonon calculations, to account for the temperature-dependent behavior of crystal lattices. The quasi-harmonic approximation extends the traditional harmonic approximation, which assumes that lattice vibrations are purely harmonic and not influenced by temperature. Consequently, the energy associated with a single phonon mode remains unaffected by the occupation of other phonon modes. This implies that phonon frequencies can vary with volume, indicating that vibrational properties should be evaluated not only at the equilibrium volume determined by DFT but also at volumes that have been isotropically expanded or contracted.In materials where each crystal axis experiences uniform expansion or contraction, the coefficient αij simplifies to a diagonal matrix αV. This coefficient, known as the volumetric thermal expansion coefficient (αV), describes volumetric strain in response to temperature changes, as expressed by the equation ΔV = αVΔT. It is essentially the trace of the thermal expansion tensor, measured in units of K−1. To facilitate comparison with experimentally reported CTE values, we divide the calculated volumetric CTE values by 3 to obtain the apparent bulk coefficient of thermal expansion, also known as linear CTE. This simplification streamlines the anisotropic CTE tensor for easier comparison. In this study, we present the linear thermal expansion coefficient as a function of temperature without explicitly specifying the complete tensor of thermal expansion coefficients.
Debye–Callaway model for lattice thermal conductivity
Various approaches have been utilized for calculating lattice thermal conductivity, including second-order, third-order force constants,21 and molecular dynamics. Molecular dynamics and third-order force constant methods are often computationally demanding, whereas the Debye–Callaway model, as demonstrated through numerous comparisons of accurate lattice thermal conductivity with experimental data,22,23 is both simple and sufficiently accurate. Therefore, we opt for the Debye–Callaway model,24 incorporating lattice vibrational properties as outlined in the phonon dynamics section. In the Debye–Callaway formalism, we consider crucial three-phonon processes, encompassing normal phonon scattering and Umklapp scattering.25 The three-phonon processes encompass various interactions, including the creation of a third phonon through the scattering of two phonons and the annihilation of two phonons into a single one. These processes contribute to the complex dynamics of lattice vibrations and play a crucial role in determining the thermal transport properties of materials. While four-phonon processes are significant in certain thermoelectric materials, the likelihood of phonon creation and annihilation in four-phonon processes is considerably lower than in three-phonon processes. Therefore, we continue to regard three-phonon processes as the primary contributors to thermal conductivity, consistent with the traditional perspective. The overall lattice thermal conductivity by Debye–Callaway model can be expressed as the sum of contributions from one longitudinal κLA and two transverse κTA and κTA′ acoustic phonon branches: κLatt = κLA + κTA + κTA′. The partial conductivities κi (i corresponds to TA, TA′ and LA modes) are given by: | (9) |
In this expression, Θi is the longitudinal or transverse Debye temperature, 1/τiN is the scattering rate for normal phonon processes, 1/τiR is the sum of all resistive scattering processes, and 1/τic = 1/τiN + 1/τiR, x = ℏω/kBT, and Ci = kB4/2π2ℏ3vi, here, ħ is the Planck constant, kB is the Boltzmann constant, ω is the phonon frequency, and vi is the longitudinal or transverse acoustic phonon velocity. The resistive scattering rate is the sum of scattering rates due to Umklapp phonon–phonon scattering (1/τiU), mass-difference impurity scattering (1/τim), boundary scattering (1/τiB), and electron–phonon scattering (1/τie–ph). For pure single crystalline compounds, we only consider the intrinsic thermal conductivity and ignore the effect of impurity scattering. Due to relatively large band gaps, we thus neglect electron–phonon scattering contributions especially for temperatures above approximately 100 K. The resistive scattering rate is mainly determined by the Umklapp phonon–phonon processes (1/τiR ≅ 1/τiU). The normal phonon scattering and Umklapp can be written as,


where γ, V, and M are the Grüneisen parameter, the volume per atom, and the average mass of an atom in the crystal, respectively. The Grüneisen parameter can be defined as,

 , characterizing the relationship between phonon frequency and volume change. To obtain the Grüneisen parameters, we utilize the phonon dispersions calculated within the above mentioned quasi-harmonic approximation.
, characterizing the relationship between phonon frequency and volume change. To obtain the Grüneisen parameters, we utilize the phonon dispersions calculated within the above mentioned quasi-harmonic approximation.
Results and discussions
Atomic structure of single compounds and solid solutions
The β phase of RE2Si2O7 (RE = Y, Eu, Gd, Er, and Yb) belongs to the monoclinic system with the C2/m space group. The atomic structure of the β phase of Er2Si2O7 is illustrated in Fig. 1, where the Si atoms exhibit tetrahedral coordination with four oxygen atoms forming corner-sharing tetrahedra, and the Er atoms form distorted octahedra surrounded by six oxygen atoms. The calculated lattice parameters of Er2Si2O7a = 6.87 Å, b = 8.97 Å, c = 4.66 Å, α = γ = 90°, and β = 101.61°, which are very close to the documented data in well-known material databases such as the open quantum materials database (OQMD).26 | ||
| Fig. 1 The atomic structure of β phase of monoclinic Er2Si2O7 with the space group of C2/m. The Si atoms are tetrahedrally coordinated with four oxygen-forming corner-sharing tetrahedra. The Er atoms form distorted octahedra with six surrounding oxygen atoms. | ||
To model the crystal structures of Er1/2Y3/4Yb3/4Si2O7, Gd1/4Er1/4Y3/4Yb3/4Si2O7, Eu1/4Er1/4Y3/4Yb3/4Si2O7, we employed the special quasi-random structure (SQS) method to represent the random configuration of rare earth elements in the cation site, with Si and O fixed in the β phase of Er2Si2O7. The SQS provides a systematic way to generate special atomic configurations that mimic the statistical distribution of a random alloy but with a reduced number of atoms compared to a supercell.27 To establish the thermodynamic phase stability of the SQS structures at high temperatures, we initially employed the DFT-based cluster expansion computational methodology to assess the formation energy of Er2−x−yYxYbySi2O7 relative to the pure compounds Er2Si2O7, Y2Si2O7, and Yb2Si2O7. We calculated 156 Er2Si2O7-type ordered structures of Er2−x−yYxYbySi2O7 (0 < x < 2) with fewer than 44 atoms per unit cell, as depicted in Fig. 2. By constructing the convex hull (the lowest-energy set of tie-lines such that no structure lies below the hull), we identified the ground state structures at 0 K as a function of composition. Fig. 2(a) illustrates that the formation energies of the Er2−x−yYxYbySi2O7 structures are mostly negative values, indicating stability relative to the pure compounds Er2Si2O7, Y2Si2O7, and Yb2Si2O7 at zero temperature. Er2Si2O7, Y2Si2O7, and Yb2Si2O7 exhibit a common prototype monoclinic structure characterized by a relatively small lattice mismatch (<0.8%). This limited mismatch leads to a minor strain energy penalty, providing an explanation for the observed negative formation energies.28–30
 | ||
| Fig. 2 (a) The formation energy of mixing of Er2Si2O7, Y2Si2O7 and Yb2Si2O7. The y scale of red dots represents the formation energies evaluated using converged cluster interactions, with each red dot corresponding to a distinct ordered structure. The dots aligned vertically share the same composition. The negative formation energy values suggest the structure is more favorable than the three pure compounds. The unit of formation energy is eV per cation. (b) The ternary phase diagram of alloying of three pure compounds at 0 K. The red circles suggested stable ordered compounds relative to the pure compounds of Er2Si2O7, Y2Si2O7 and Yb2Si2O7. (c) A comparison of DFT calculated energy and cluster expansion energy. (d) Order–disorder transition temperature for Er2/3Y2/3Yb2/3Si2O7, which is identified as the inflection point of the energy curve as a function of temperature. | ||
Utilizing converged cluster interactions as a foundation, we have identified seven stable ordered structures in comparison to the pure single compounds Er2Si2O7, Y2Si2O7 and Yb2Si2O7. These ordered structures include Y1/2Yb3/2Si2O7, YYbSi2O7, Y3/2Yb1/2Si2O7, Er1/2Yb3/2Si2O7, ErYbSi2O7, Er3/2Yb1/2Si2O7, and Er2/3Y2/3Yb2/3Si2O7. Notably, the binary stable ordered structures align with our prior investigation results.6 Beyond assessing the relative stability of these ordered structures, tie lines among them have been illustrated in Fig. 2(b) to create a phase diagram at 0 K. To ensure the prediction accuracy of these ordered structures, a comparison between DFT energy and cluster expansion energy is presented in Fig. 2(c), revealing a well-aligned energy comparison along the diagonal line.
To determine the order–disorder transition temperature of the ternary compound Er2/3Y2/3Yb2/3Si2O7, canonical Monte Carlo simulations were conducted to monitor the enthalpy change as a function of temperature. The results revealed a significant energy increase at 390 K, as illustrated in Fig. 2(d), indicative of a transition to a disordered phase with higher energy in the ternary Er2/3Y2/3Yb2/3Si2O7 system. Given our focus on exploring thermal and mechanical properties in the high-temperature range, it is essential to ensure the stability of a solid solution representative at elevated temperatures, particularly for Er, Y, and Yb occupying the cation site of the Er2Si2O7 prototype. Consequently, we advocate for a composition with a Y and Yb-rich content, specifically Er1/2Y3/4Yb3/4Si2O7, to maintain the solid solution structure in the β phase and prevent phase transformation resulting from additional rare earth element incorporation. Furthermore, the addition of other rare earth species, such as Eu and Gd, is contemplated to fine-tune the thermal properties while predominantly retaining Y and Yb as the cation species. Solid solutions at specific compositions, namely Gd1/4Er1/4Y3/4Yb3/4Si2O7 and Eu1/4Er1/4Y3/4Yb3/4Si2O7, are also under consideration for rare earth incorporation into the cation site of Er2Si2O7. We adopt these ternary systems at these compositions because similar systems have been experimentally proven stable for high-entropy rare earth disilicates. However, the phase diagram from ternary cluster expansion indicates that these ternary alloys can be stable in ordered crystals. The order–disorder transition temperature suggests that Er2/3Y2/3Yb2/3Si2O7 is stable as a solid solution among Er, Y and Yb in the cation sublattice at higher temperatures. Considering vibrational entropy31 and the configurational entropy induced by multiple rare earth species, the order–disorder transition temperature should be even lower. Therefore, we can safely conclude that Er1/2Y3/4Yb3/4Si2O7, Gd1/4Er1/4Y3/4Yb3/4Si2O7, and Eu1/4Er1/4Y3/4Yb3/4Si2O7 will be stable as solid solutions at high temperatures.
Mechanical properties
The elastic constants are defined in relation to an orthonormal coordinate system (x, y, z). The number of independent linear equations corresponds to the number of unknown elastic moduli, establishing a well-defined system of equations that can be easily solved. The solution to these linear equations for Cij provides the values of the elastic constants, as presented in Table 1. It is crucial to emphasize that all elastic constants have undergone verification against the respective stability criteria outlined in the calculation section. The results confirm that all six material systems exhibit mechanical stability concerning all three strains.| Er1/2Y3/4Yb3/4 | Gd1/4Er1/4Y3/4Yb3/4 | Eu1/4Er1/4Y3/4Yb3/4 | Gd2Si2O7 | Er2Si2O7 | Eu2Si2O7 | ||
|---|---|---|---|---|---|---|---|
| C 11 (GPa) | 197.8 | 195.3 | 192.9 | 289.9 | 294.5 | 280.2 | |
| C 22 (GPa) | 220.5 | 234.2 | 235.8 | 196.7 | 201.8 | 185.7 | |
| C 33 (GPa) | 241.6 | 237.6 | 254.5 | 193.9 | 203.5 | 177.7 | |
| C 44 (GPa) | 68.9 | 66.3 | 67.6 | 80.1 | 81.3 | 70.5 | |
| C 55 (GPa) | 84.5 | 83.6 | 84.3 | 85.3 | 84.5 | 79.5 | |
| C 66 (GPa) | 79.1 | 77.2 | 77.1 | 55.9 | 55.3 | 47.9 | |
| C 12 (GPa) | 104.4 | 103.3 | 105.0 | 88.3 | 88.3 | 84.7 | |
| C 13 (GPa) | 108.9 | 107.7 | 109.0 | 108.1 | 106.9 | 99.4 | |
| C 23 (GPa) | 104.7 | 114.1 | 106.8 | 115.7 | 118.1 | 108.8 | |
| C 15 (GPa) | −4.9 | −4.7 | −4.9 | −19.6 | −18.7 | −21.7 | |
| C 25 (GPa) | −0.38 | −0.87 | −0.7 | 37.2 | 36.8 | 37.2 | |
| C 35 (GPa) | 15.8 | 18.1 | −18.1 | 13.1 | 13.5 | 13.1 | |
| C 46 (GPa) | −10.73 | −9.9 | −10.2 | 16.2 | 23.7 | 26.8 | |
| Bulk modulus (GPa) | Voigt | 144.0 | 146.4 | 147.3 | 144.9 | 147.4 | 136.6 |
| Reuss | 139.9 | 140.9 | 143.1 | 139.1 | 141.9 | 130.2 | |
| Hill | 141.9 | 143.7 | 145.2 | 142.1 | 144.7 | 133.4 | |
| Young's modulus (GPa) | Voigt | 179.2 | 177.2 | 181.2 | 178.3 | 181.3 | 163.6 |
| Reuss | 152.4 | 152.3 | 159.8 | 152.9 | 153.3 | 129.1 | |
| Hill | 165.9 | 164.8 | 170.5 | 165.7 | 167.4 | 146.6 | |
| Shear modulus (GPa) | Voigt | 69.3 | 68.2 | 69.9 | 68.8 | 70.0 | 62.9 |
| Reuss | 57.8 | 57.7 | 60.8 | 58.1 | 58.1 | 48.4 | |
| Hill | 63.6 | 62.9 | 65.4 | 63.5 | 64.0 | 55.6 | |
| Vickers hardness (GPa) | Voigt | 7.09 | 6.63 | 6.99 | 6.95 | 6.99 | 6.07 |
| Reuss | 4.60 | 4.51 | 5.10 | 4.72 | 4.53 | 3.05 | |
| Hill | 5.82 | 5.55 | 6.03 | 5.83 | 5.74 | 4.52 | |
| Average wave velocity (m s−1) | 3917.6 | 3919.5 | 4001.7 | 3847.6 | 3710.8 | 3679.9 | |
| Debye temperature (K) | 495.0 | 494.6 | 504.7 | 474.5 | 469.2 | 456.3 | |
The average mechanical properties of the polycrystalline material, encompassing parameters such as bulk modulus, Young's modulus, and shear modulus, are determined using both the Voigt and Reuss methods. These methods rely on the calculated single-crystal elastic constants and can be explicitly defined through energy considerations, drawing on three distinct theoretical contributions: Voigt, Reuss, and Hill. Voigt's approach yields an upper bound estimation when employing the elastic constant matrix as input, while Reuss’ method provides a lower bound estimation by assessing the elastic compliance matrix (the inverse of the elastic constant matrix). Hill's approach takes an average of the results obtained from the Voigt and Reuss methods. The calculated mechanical property results are compiled in the lower section of Table 1.
It is worth noting that β-Er2Si2O7 exhibits noticeably smaller elastic constants Cij including both diagonal and off-diagonal components than β-Er2Si2O7 and β-Gd2Si2O7. For Er1/2Y3/4Yb3/4Si2O7, Gd1/4Er1/4Y3/4Yb3/4Si2O7, and Eu1/4Er1/4Y3/4Yb3/4Si2O7, the elastic constants are almost all lower than the pure cases of Er2Si2O7 and Gd2Si2O7 due to the mixing of rare earth elements. For example, C11 of Er1/2Y3/4Yb3/4Si2O7 is 197.8 GPa compared with the corresponding C11 of 294.5 GPa for Er2Si2O7, and 280.2 GPa for Eu2Si2O7. However, the C22 of the three solid solutions as listed in Table 1 are all much higher than those of C22 201.8 GPa (Er2Si2O7), and 185.7 GPa (Eu2Si2O7).
Table 1 illustrates that Eu2Si2O7 exhibits the smallest moduli compared to all available data. Specifically, the average bulk modulus of Eu2Si2O7 is 133.4 GPa, which is less than the values for Er1/2Y3/4Yb3/4Si2O7 (141.9 GPa), Gd1/4Er1/4Y3/4Yb3/4Si2O7 (143.7 GPa), Eu1/4Er1/4Y3/4Yb3/4Si2O7 (145.2 GPa), and Er2Si2O7 (144.7 GPa). Likewise, the Young's modulus and shear modulus of Eu2Si2O7 are consistently lower than those of comparable compounds. It is established that these moduli reflect a material's resistance to bulk compression, lengthwise tension or compression, and elastic shear deformation for bulk, Young's, and shear modulus, respectively. Smaller moduli values indicate weaker resistance to mechanical changes. The fact that the β phase Eu2Si2O7 shares the same atomic structure as other compounds but possesses the smallest moduli values suggests a deeper physical reason specific to europium compared to other rare earth elements. In the following section, we will demonstrate the pronounced lattice anharmonicity of Eu2Si2O7, revealing the underlying physical cause for its softer elastic modulus compared to other compounds.
Utilizing the strain-energy methods to determine elastic constants at 0 K, the impact of temperature on these constants can be assessed by considering the corresponding free energies for these six oxides. As depicted in Fig. 3, both the diagonal and off-diagonal elements of elastic constants in all six systems demonstrate noticeable softening with increasing temperature. The softening of elastic constants, characterized by varying slopes with temperature, signifies anisotropic responses to external pressure. Typically, the diagonal elements of elastic constants and moduli are anticipated to diminish with rising temperatures, a phenomenon known as softening. This observation can be elucidated by various physical factors. As temperature increases, most materials tend to undergo expansion due to heightened atomic or molecular vibrations. This expansion leads to a larger volume for the material, enhancing its compressibility and consequently diminishing the bulk modulus. Simultaneously, elevated temperatures often result in the attenuation of interatomic or intermolecular bonds within a material. The weakened bond strength renders the material more prone to compression, contributing to a reduction in bulk modulus. Additionally, higher temperatures endow atoms or molecules in a material with greater vibrational energy. This augmented thermal motion disrupts the ordered structure, rendering the material more pliant and less resistant to volume changes. In particular, the diagonal components of the elastic constant in Eu2Si2O7 exhibit more pronounced softening compared to all other systems. Additionally, the moduli of Eu2Si2O7, as illustrated in Fig. 4(a)–(c), not only are smaller than those of other systems but also demonstrate a more rapid decrease with increasing temperature. This distinctive behavior is attributed to the strong anharmonicity of Eu2Si2O7, a topic further addressed in Section 3.5.
 | ||
| Fig. 3 The temperature-dependent elastic constants for the six systems of (a) and (b) Er1/2Y3/4Yb3/4Si2O7 (c) and (d) Gd1/4Er1/4Y3/4Yb3/4Si2O7, (e) and (f) Eu1/4Er1/4Y3/4Yb3/4Si2O7, (g) and (h) Eu2Si2O7, (i) and (j) Er2Si2O7, and (k) and (l) Gd2Si2O7. The left column corresponds to diagonal elements, while the right column pertains to off-diagonal elements. | ||
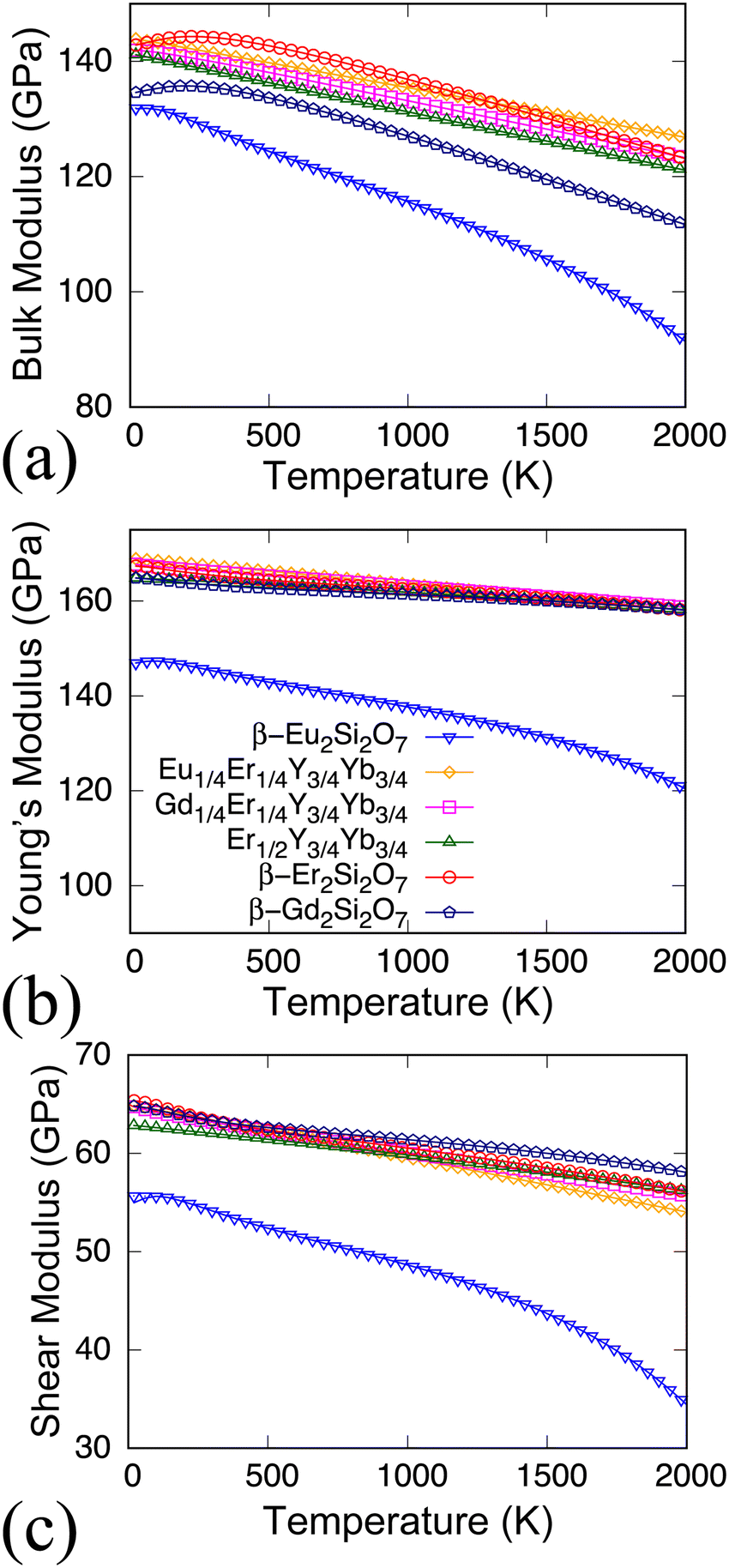 | ||
| Fig. 4 The temperature-dependent (a) bulk modulus, (b) Young's modulus, and (c) shear modulus for the six systems. The identical labels as shown in (b) are employed for the same material across all three figures. | ||
The elastic constants present a captivating opportunity to explore elastic anisotropy, a pivotal factor in the development of microcracks within coatings. Specifically, at a fixed elastic strain, a direction with a high modulus corresponds to high stress and hence high elastic energy, and vice versa. Understanding the anisotropy of elastic properties offers valuable insights into predicting directions with varying levels of fracture energy within the crystal. Fig. 5 visually illustrates the pronounced anisotropic nature of Young's and shear moduli for all systems. In the instances of Er2Si2O7, Gd2Si2O7, and Eu2Si2O7, their Young's and shear moduli display notably similar anisotropic properties. The highest Young's moduli for the pure compounds, illustrated in Fig. 5(a)–(c), are approximately 180 GPa in the (100) plane, 200 GPa in the (010) plane, and 205 GPa in the (001) plane. Likewise, the lowest Young's moduli are approximately 100 GPa in all three systems and across all three planes. Moreover, the highest and lowest Young's moduli of the Er1/2Y3/4Yb3/4Si2O7, Gd1/4Er1/4Y3/4Yb3/4Si2O7, Eu1/4Er1/4Y3/4Yb3/4Si2O7 systems closely align with those of the Er2Si2O7, Gd2Si2O7, and Eu2Si2O7 systems. Regarding shear modulus anisotropy, all six systems, as shown in Fig. 5(d)–(f), exhibit a similar shape in the three planes. Notably, the shear modulus varies significantly along different slip directions in all three planes, indicating a substantial variation in resistance to shear deformation.
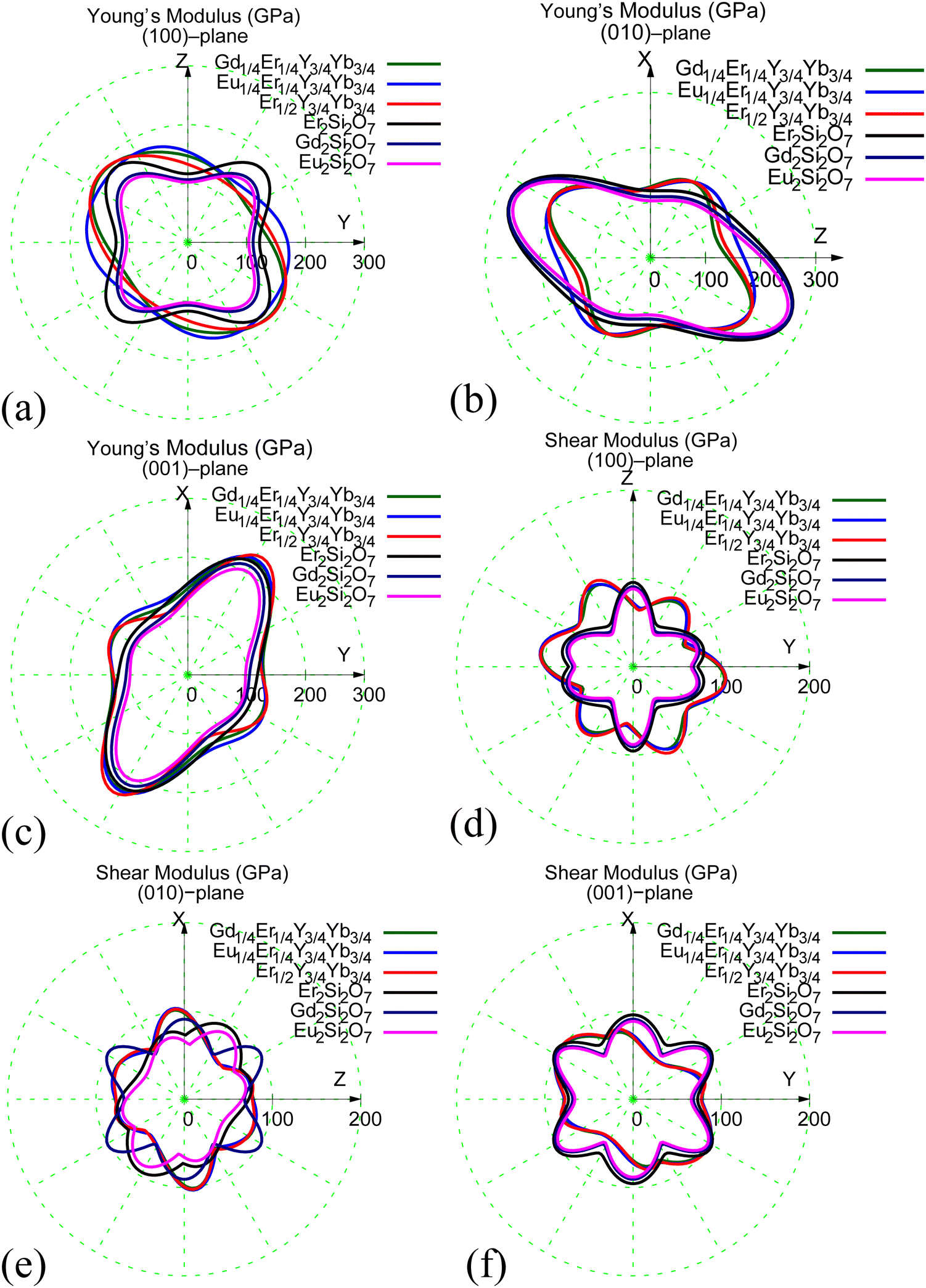 | ||
| Fig. 5 Elastic anisotropy of Young's modulus and shear modulus in different plane of (a) and (d) (100), (b) and (e) (010), and (c) and (f) (001) for all six systems, respectively. | ||
Phonon dispersion
After determining the elastic constants and mechanical properties of the six systems using strain-energy methods, our focus now shifts to thermal properties, including the coefficient of thermal expansion and lattice thermal conductivities. The initial step in investigating thermal properties involves characterizing lattice vibrations through phonon dispersion along the high-symmetry lines in the Brillouin zone. The phonon dispersions, depicted in Fig. 6, reveal that our calculations do not indicate any phonon branch with imaginary frequencies at any points in reciprocal space for the solid solution systems. Consequently, we can confidently assert that all three solid solution systems are dynamically stable at their equilibrium volumes. | ||
| Fig. 6 The phonon dispersion (left panel) and corresponding projected phonon density of states (right panel) of solid solutions of (a) Er1/2Y3/4Yb3/4Si2O7, (b) Gd1/4Er1/4Y3/4Yb3/4Si2O7 and (c) Eu1/4Er1/4Y3/4Yb3/4Si2O7. | ||
To characterize the phonon dispersion behavior of these three solid solution systems, we compare the average values of the Debye temperature, and phonon velocities. By examining the phonon dispersion, the longitudinal Debye temperature can be calculated from Θ = ωD/kB (ωD is the largest acoustic frequency in each direction, kB is the Boltzmann constant). The average longitudinal Debye temperatures are found to be approximately 105 K for three solid solutions Er1/2Y3/4Yb3/4Si2O7, Gd1/4Er1/4Y3/4Yb3/4Si2O7, and Eu1/4Er1/4Y3/4Yb3/4Si2O7. Generally, a lower Debye temperature is associated with a lower lattice thermal conductivity. So, the cation mixing in solid solutions decreases the longitudinal Debye temperatures and consequently lowers lattice thermal conductivity. Similarly, according to the Debye model, with the phonon vibrational energy, the Deby temperature can be derived from  , where integration variable
, where integration variable  , TD Debye temperature, T temperature, n number of phonon energy states, v phonon group velocity, h and kB are respectively Planck and Boltzmann constants. The Debye temperatures were determined to be 495.0 K for Er1/2Y3/4Yb3/4Si2O7, 494.6 K for Gd1/4Er1/4Y3/4Yb3/4Si2O7, and 504.7 K for Eu1/4Er1/4Y3/4Yb3/4Si2O7. The projected phonon density of states depicted in Fig. 6 exhibit a similar trend. It is expected to observe the prominent low frequency peaks in the range of 40–200 cm−1, attributed to the rare earth atoms with the heaviest atomic mass in the cell. Conversely, the local vibration modes of oxygen, the lightest species, are present in all systems, with the peak intensity reaching around 1000 cm−1.
, TD Debye temperature, T temperature, n number of phonon energy states, v phonon group velocity, h and kB are respectively Planck and Boltzmann constants. The Debye temperatures were determined to be 495.0 K for Er1/2Y3/4Yb3/4Si2O7, 494.6 K for Gd1/4Er1/4Y3/4Yb3/4Si2O7, and 504.7 K for Eu1/4Er1/4Y3/4Yb3/4Si2O7. The projected phonon density of states depicted in Fig. 6 exhibit a similar trend. It is expected to observe the prominent low frequency peaks in the range of 40–200 cm−1, attributed to the rare earth atoms with the heaviest atomic mass in the cell. Conversely, the local vibration modes of oxygen, the lightest species, are present in all systems, with the peak intensity reaching around 1000 cm−1.
Phonon velocity denotes the speed at which a phonon mode propagates through a crystal. The phonon velocities of acoustic branches, represented by vi, are estimated through the long-wavelength limit of group velocities derived from the phonon dispersion curves. Computed using the formula  , the average phonon velocities for Er1/2Y3/4Yb3/4Si2O7, Gd1/4Er1/4Y3/4Yb3/4Si2O7, and Eu1/4Er1/4Y3/4Yb3/4Si2O7 are 3917.6, 3919.5, and 4001.7 m s−1, respectively. A slower phonon velocity typically indicates sluggish phonon transport, which tends to reduce lattice thermal conductivity. However, since the phonon velocities of the three solid solutions are quite close to each other, a comprehensive evaluation of lattice thermal conductivity should consider all relevant physical quantities.
, the average phonon velocities for Er1/2Y3/4Yb3/4Si2O7, Gd1/4Er1/4Y3/4Yb3/4Si2O7, and Eu1/4Er1/4Y3/4Yb3/4Si2O7 are 3917.6, 3919.5, and 4001.7 m s−1, respectively. A slower phonon velocity typically indicates sluggish phonon transport, which tends to reduce lattice thermal conductivity. However, since the phonon velocities of the three solid solutions are quite close to each other, a comprehensive evaluation of lattice thermal conductivity should consider all relevant physical quantities.
Thermal properties
By examining the phonon dispersions within the quasi-harmonic approximation across five distinct volumes for each system, we can now assess the coefficients of thermal expansion. These coefficients are determined from the volume associated with the minimized free energy as a function of temperature. The free energy is obtained by integrating the phonon energy, as per eqn (8) in Section 2, across lattice vibrational frequencies throughout the entire Brillouin zone, using a densely converged mesh. Fig. 7(a) illustrates the free energies of Er1/2Y3/4Yb3/4Si2O7, with volumes and temperatures as variables, while the analogous shapes of the free energies for other systems are disregarded due to their nearly identical basic forms. The volumes associated with the minimized free energy at each temperature are subsequently represented by the red line in Fig. 7(a). Using this data, the CTE can be determined by taking the first derivative with respect to temperature and depicted in Fig. 7(b). | ||
| Fig. 7 (a) An example of free energy as a function of volume and temperature for Er1/2Y3/4Yb3/4Si2O7 system. The blue dots and blue lines are respectively DFT calculated values and fitted results. The red dots represent the minimum free energy at corresponding volume and temperatures ranging from 0 to 2000 K with a step of 100 K. (b) Coefficients of thermal expansion and (c) lattice thermal conductivity of Eu2Si2O7, Gd2Si2O7, Er2Si2O7, Er1/2Y3/4Yb3/4Si2O7, Gd1/4Er1/4Y3/4Yb3/4Si2O7, and Eu1/4Er1/4Y3/4Yb3/4Si2O7. (d) Comparison of lattice thermal conductivity and linear coefficient of thermal expansion between the calculated systems and 6H-SiC. Blue and red symbols represent properties at 500 and 1500 K, respectively. Solid symbols in (d) denote 6H-SiC, while open symbols represent disilicates, with labels only indicating the rare earth component for brevity. | ||
It is worth noting that the coefficients of thermal expansion at low temperatures (<500 K) are generally modest and experience rapid saturation beyond 500 K across all six systems (Fig. 7(c)). Typically, within the same atomic structure type, rare earth disilicate materials like RE2Si2O7 are expected to exhibit only slight variations in the average CTE. However, the calculated results show that the CTE for Eu2Si2O7 stands out as the highest among the six systems, averaging 8.1 × 10−6 K−1 across a temperature span from 500 to 2000 K. The CTE for Er2Si2O7 and Gd2Si2O7 are nearly the same at 4.2 × 10−6 K−1 over 500–2000 K. It is intuitively expected that the average CTE values for the solid solution systems Eu1/4Er1/4Y3/4Yb3/4Si2O7, Gd1/4Er1/4Y3/4Yb3/4Si2O7, and Er1/2Y3/4Yb3/4Si2O7, fall within the range of the pure cases of Eu2Si2O7, Er2Si2O7, and Gd2Si2O7. These values range from 4.4 to 5.5 × 10−6 K−1, matching CTE of SiC. Notably, our calculated CTE values align closely with experimental measurements for both Y2Si2O7 and Yb2Si2O7.1
Fig. 7(c) illustrates the calculated lattice thermal conductivities for six systems. Generally, for a specific crystal structure, a heavier atomic mass leads to slower phonon group velocities, lower longitudinal Debye temperatures, and consequently lower lattice thermal conductivities. For instance, a comparison between Er2Si2O7 and Gd2Si2O7 reveals that the atomic masses of Er and Gd are 167.3 and 157.3, respectively. As listed in the Table 1, the phonon group velocities for Er2Si2O7 and Gd2Si2O7 are 3710 m s−1 and 3847 m s−1, while the longitudinal Debye temperatures are 469 K for Er2Si2O7 and 500 K for Gd2Si2O7. Therefore, it is understandable that the lattice thermal conductivities of Er2Si2O7 are lower than those of Gd2Si2O7, as depicted in Fig. 7(c). However, it is noteworthy that Eu2Si2O7 exhibits an even lower thermal conductivity than Er2Si2O7, despite Eu (151.9) having an atomic mass much lighter than Er (167.3). We attribute this abnormal behavior to the pronounced lattice anharmonicity in the Eu2Si2O7 structure, an aspect that will be thoroughly addressed in the Section 3.5.
Beyond the pure cases of Eu2Si2O7, Gd2Si2O7, and Er2Si2O7, the lattice thermal conductivities of the remaining three solid solution systems are consistently lower than those of their respective pure counterparts. This reduction of lattice thermal conductivities in the solid solutions is attributed to fluctuations in atomic mass among different atomic species, namely Y (88.9), Eu (151.9), Gd (157.3), Er (167.3), and Yb (173.1). For instance, in the Er1/2Y3/4Yb3/4Si2O7 system, the atomic mass of Y is nearly half that of Er and Yb, leading to pronounced phonon scattering centers due to the substantial mass difference at the cation site. Furthermore, the random distribution of various rare earth species, each with different atomic sizes and electronegativities, induces significant lattice distortion not only at the cation site but throughout the entire lattice. This phenomenon explains the observed trend where increased rare earth additions result in lower thermal conductivity values. From a thermal conductivity perspective, promising candidates for forming thermal barriers with low lattice thermal conductivity include Eu1/4Er1/4Y3/4Yb3/4Si2O7 (0.25 W m−1 K−1 at 1500 K), Gd1/4Er1/4Y3/4Yb3/4Si2O7 (0.33 W m−1 K−1 at 1500 K), and Er1/2Y3/4Yb3/4Si2O7 (0.39 W m−1 K−1 at 1500 K). We also plot the lattice thermal conductivity versus the linear coefficient of thermal expansion for the calculated systems and compare them with 6H-SiC. As depicted in Fig. 7(d), the linear CTE values of Gd1/4Er1/4Y3/4Yb3/4Si2O7 and Eu1/4Er1/4Y3/4Yb3/4Si2O7 show excellent alignment with SiC32 at both low temperatures (e.g. 500 K) and high temperatures (e.g. 1500 K). Remarkably, the lattice thermal conductivities of these two alloyed rare earth disilicate systems are even lower than that of SiC,33 demonstrating their potential suitability for TEBC applications. The detailed thermal conductivity and linear CTE values are tabulated in ESI.†
Lattice anharmonicity
To elucidate the anomalous CTE for Eu2Si2O7 in comparison to other oxides, we scrutinize the phonon dispersion of pure Eu2Si2O7 and juxtapose it with the corresponding dispersions of the two other pure compounds, Er2Si2O7 and Gd2Si2O7. The phonon dispersions and the corresponding density of states are illustrated in Fig. 8. Notably, in the instance of Eu2Si2O7, conspicuous negative frequencies are evident throughout most high symmetry lines in the Brillouin zone. These imaginary modes with negative frequencies, identified as soft phonon modes, serve as a distinctive signature of anharmonicity within the Eu2Si2O7 lattice. Contrastingly, in Er2Si2O7 and Gd2Si2O7, as depicted in Fig. 8(b) and (c), no imaginary modes are observed. This absence of imaginary phonon modes in Eu1/4Er1/4Y3/4Yb3/4Si2O7 (Fig. 6(c)) suggests that the soft modes induced by Eu, rather than Gd or Er, may undergo substantial atomic displacements, thereby leading to more pronounced deviations from harmonic behavior. | ||
| Fig. 8 The phonon dispersion (left panel) and corresponding projected phonon density of states (right panel) of single compounds in the β phase of (a) Eu2Si2O7, (b) Er2Si2O7, and (c) Gd2Si2O7. In (a), three sets of phonon dispersions of Eu2Si2O7 are respectively plotted in red (harmonic approximation), blue (anharmonicity at 300 K) and green (anharmonicity at 1000 K). | ||
To assess the stronger anharmonicity of Eu2Si2O7 compared to Er2Si2O7 and Gd2Si2O7, we analyzed the deviation of the potential energy surface from the harmonic approximation for these three compounds. Specifically, we calculated the system energy deviation relative to the fully relaxed equilibrium states as the lattice underwent a series of C11 and C66 type lattice distortions. Detailed descriptions of these distortions are provided as distortion matrices in the ESI.† The calculated energy deviations were then fitted to a second-order parabolic function using the least squares method, representing the harmonic approximation. To quantify the degree of deviation from this parabolic fit, we utilized the curvature fitting standard errors. A larger standard error indicates a greater deviation from harmonic behavior, signifying stronger lattice anharmonicity. As illustrated in Fig. S1 (ESI†), Eu2Si2O7 exhibits a significantly stronger deviation from harmonic behavior than both Er2Si2O7 and Gd2Si2O7 for both C11 and C66 type distortions, with energy data points not aligning closely with the fitting lines. Specifically, the standard errors for Eu2Si2O7 are 2.602% for C11 and 3.967% for C66 distortions, much larger than those of Er2Si2O7 and Gd2Si2O7, indicating a much stronger lattice anharmonicity in Eu2Si2O7. Note that the energy deviations exhibit similar behavior for other lattice distortions and, therefore, are not plotted.
In the finite atomic displacement methods, as mentioned in the calculation section, the potential energy of interacting atoms can be represented through a Taylor expansion concerning atomic displacements, expressed as  .34 In the harmonic approximation, U1 and U2 are considered to construct the corresponding force constant and dynamical matrix for phonon dispersion. Given the presence of soft phonon modes in Eu2Si2O7, indicating pronounced anharmonicity, we extend our analysis to eliminate these soft modes and further demonstrate the strong anharmonic nature of Eu in the lattice of the β phase of Eu2Si2O7. To achieve this, we incorporate up to the fourth-order terms, U3 and U4, in the Taylor expansion, which require much more expensive DFT calculations to construct fourth-order force constant matrix. Employing self-consistent methodologies on the basis of the four-order force constant matrix, we compute temperature-dependent phonon frequencies.35 As depicted in Fig. 8(a) by the blue and green lines, the soft modes, initially calculated by the harmonic approximation, have been successfully corrected at both 300 and 1000 K. This correction provides evidence for the robust anharmonicity of Eu2Si2O7 and provide possibilities for further CTE and lattice thermal conductivity calculations.
.34 In the harmonic approximation, U1 and U2 are considered to construct the corresponding force constant and dynamical matrix for phonon dispersion. Given the presence of soft phonon modes in Eu2Si2O7, indicating pronounced anharmonicity, we extend our analysis to eliminate these soft modes and further demonstrate the strong anharmonic nature of Eu in the lattice of the β phase of Eu2Si2O7. To achieve this, we incorporate up to the fourth-order terms, U3 and U4, in the Taylor expansion, which require much more expensive DFT calculations to construct fourth-order force constant matrix. Employing self-consistent methodologies on the basis of the four-order force constant matrix, we compute temperature-dependent phonon frequencies.35 As depicted in Fig. 8(a) by the blue and green lines, the soft modes, initially calculated by the harmonic approximation, have been successfully corrected at both 300 and 1000 K. This correction provides evidence for the robust anharmonicity of Eu2Si2O7 and provide possibilities for further CTE and lattice thermal conductivity calculations.
The relationship between lattice anharmonicity and elastic properties such as bulk or Young's moduli in materials is not straightforward. Strong anharmonicity might lead to a lower bulk modulus. This could happen if the anharmonicity is associated with softer phonon modes, increased lattice disorder that reduce the material's resistance to compression.36 Soft modes can result in reduced stiffness and higher compressibility. In our case, notable soft phonon modes (indicated by negative frequencies) are evident, as illustrated in Fig. 8(a) through red lines. This observation elucidates the relatively smaller values of bulk modulus, Young's modulus, and shear modulus for Eu2Si2O7 compared to those of Er2Si2O7, Gd2Si2O7, and other solid solutions, such as Er1/2Y3/4Yb3/4Si2O7, Gd1/4Er1/4Y3/4Yb3/4Si2O7 and Eu1/4Er1/4Y3/4Yb3/4Si2O7, as outlined in Table 1.
It is known that anharmonicities within the crystal lattice are the fundamental physical reasons of thermal expansion in solid materials. In general, a more pronounced lattice anharmonicity correlates with a heightened interaction between phonons and strain, leading to greater changes in the unit cell volume with temperature. Consequently, this association results in a larger coefficient of thermal expansion, particularly at elevated temperatures. At these higher temperatures, where atoms within a lattice undergo more extensive vibrational amplitudes, the influence of phonon–phonon scatterings becomes more pronounced, inducing a more substantial departure from harmonic behavior. This offers a plausible explanation for the significantly larger CTE values observed in Eu2Si2O7 compared to those in Er2Si2O7 and Gd2Si2O7.
On the other hand, the strong anharmonicity introduces much stronger phonon–phonon scattering mechanisms in a material than harmonic approximation. While anharmonic scattering can impede the propagation of phonons and enhance more efficient phonon–phonon interaction, thus reduce lattice thermal conductivity. To characterize the anharmonicity of a crystal lattice in response to thermal expansion or compression, we compare the average Grüneisen parameters among the three pure compounds with three solid solutions. The Grüneisen parameter, a dimensionless quantity, offers insights into how lattice vibrations (phonons) react to changes in temperature and volume. Generally, larger Grüneisen parameters indicate stronger lattice anharmonicity and lower lattice thermal conductivity.22 By employing finite differential methods based on phonon dispersion at different volumes, we obtain Grüneisen parameters. The calculated average Grüneisen parameter for Eu2Si2O7 (1.2) significantly exceeds those for Er2Si2O7 (0.56) and Gd2Si2O7 (0.53) as plotted in Fig. 9. This discrepancy elegantly explains the lower lattice thermal conductivity observed in Eu2Si2O7 than those of pure Er2Si2O7 and Gd2Si2O7. The physical origin of strong anharmonicity in Eu2Si2O7 compared to Er2Si2O7 and Gd2Si2O7 is out of the scope of this work. One possible reason is the strong electron–phonon coupling in Eu2Si2O7 induced by the unique Eu electronic configuration of [Xe]4f76s2.
 | ||
| Fig. 9 (a) The Grüneisen parameters of all compounds as a function of temperature. (b) Population of bond lengths in all relevant compounds. Open symbols in (b) represent bond lengths between rare earth and oxygen in single compound RE2Si2O7 (RE = Er, Eu, Gd, Y, Yb). Solid symbols denote bond lengths between rare earth and oxygen in solid solution of Er1/2Y3/4Yb3/4Si2O7, Gd1/4Er1/4Y3/4Yb3/4Si2O7, and Eu1/4Er1/4Y3/4Yb3/4Si2O7. | ||
In addition to the Grüneisen parameters, we also analyzed the distribution of bond lengths in all pertinent compounds. Fig. 9(b) illustrates that in individual compounds RE2Si2O7 (RE = Er, Eu, Gd, Y, and Yb), the bond lengths between rare earth elements and oxygen range from 2.26 to 2.28 angstroms for Er, Gd, Y, and Yb, whereas the Eu–O bond length (2.375 angstroms) exceeds these values significantly, indicating pronounced lattice anharmonicity in Eu2Si2O7 system. It's important to note that these rare earth–oxygen bond lengths represent average values among the six RE–O bonds in each material. Similarly, in the solid solution systems of Er1/2Y3/4Yb3/4Si2O7, Gd1/4Er1/4Y3/4Yb3/4Si2O7, and Eu1/4Er1/4Y3/4Yb3/4Si2O7, the bond lengths between rare earth and oxygen exhibit much greater dispersion within each system. Specifically, RE–O bond lengths in Eu1/4Er1/4Y3/4Yb3/4Si2O7 display more pronounced separation compared to those in Gd1/4Er1/4Y3/4Yb3/4Si2O7 and Er1/2Y3/4Yb3/4Si2O7. Particularly, the averaged Eu–O bond length extends to about 2.43 angstroms, significantly longer than the Er-, Gd-, Y-, and Yb–O bonds. This greater RE–O bond length variability implies more pronounced lattice distortions in solid solutions, consequently leading to lower lattice thermal conductivities.
Overall, we can summarize all calculated properties and provide a comparison between the dominant pure components and the solid solutions. First, regarding thermal properties, the coefficients of thermal expansion of the solid solutions are in good agreement with those of the pure components except Eu2Si2O7. In contrast, the lattice thermal conductivities of the solid solutions are significantly lower than those of the individual pure components. Furthermore, the inclusion of Eu2Si2O7 provides an additional method to fine-tune thermal properties, particularly for lattice thermal conductivity and CTE, due to its strong anharmonicity. Second, the elastic constants and moduli of the pure components fall within a similar range, while the solid solutions align closely with these pure components, showing no significant deterioration attributable to rare earth alloying. Pure components Yb2Si2O7 and Y2Si2O7 are recognized for their EBC application at temperatures up to 1750 K and 2100 K,37 respectively. It is expected that the solid solution samples Er1/2Y3/4Yb3/4Si2O7, Gd1/4Er1/4Y3/4Yb3/4Si2O7, and Eu1/4Er1/4Y3/4Yb3/4Si2O7 exhibit working temperatures around 2000 K.
Conclusion
In summary, our study leverages a combination of DFT methods and combinatorial chemistry techniques to design new low-cost high-performance RE2Si2O7 environmental barrier coatings for protecting SiC-based ceramic matrix composites in gas turbine combustion environments. Through cluster expansion calculations and Monte Carlo simulations, we identify the β-Er2/3Y2/3Yb2/3Si2O7 phase as thermodynamically stable at room temperature, exhibiting an order–disorder transition at 360 K. Expanding on these findings, we investigate the mechanical and thermal properties of solid solution systems, including Er1/2Y3/4Yb3/4Si2O7, Gd1/4Er1/4Y3/4Yb3/4Si2O7 and Eu1/4Er1/4Y3/4Yb3/4Si2O7. Notably, these solid solution EBC systems are predicted to have ultralow thermal conductivity ranging from 0.25 to 0.39 W m−1 K−1 at 1500 K, significantly lower than their pure counterparts. Furthermore, the bulk linear CTE of these solid solutions are predicted to fall within the desired range of 4.4 to 5.5 × 10−6 K−1, matching SiC-based materials.The incorporation of Eu2Si2O7 introduces a distinctive lever to modulate CTE and lattice thermal conductivity, attributed to its robust anharmonicity characterized by its soft modes. Generally, increased lattice anharmonicity boosts phonon–strain interaction, leading to notable volume changes with temperature. Strong anharmonicity also enhances phonon–phonon scattering, hindering phonon propagation and reducing lattice thermal conductivity. This unique feature enables adjustments in CTE while simultaneously reducing lattice thermal conductivity, presenting a novel avenue for fine-tuning material properties.
In conclusion, this computational research identifies new promising RE2Si2O7 EBCs including Er1/2Y3/4Yb3/4Si2O7, Gd1/4Er1/4Y3/4Yb3/4Si2O7 and Eu1/4Er1/4Y3/4Yb3/4Si2O7 superior to the current state of the art such as Yb2Si2O7. Experimental validation will be presented in future work. The combination of DFT methods and combinatorial chemistry techniques represents a valuable computational approach for the rapid discovery of complex materials with balanced properties requirements for harsh environments.
Disclaimer
This project was funded by the U.S. Department of Energy, National Energy Technology Laboratory, in part, through a site support contract. Neither the United States Government nor any agency thereof, nor any of their employees, nor the support contractor, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.Data availability
The code for calculations can be found at VASP – Vienna Ab initio Simulation Package with G. Kresse, J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186. The version of the code employed for this study is version 6.3.1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed in support of the U.S. Department of Energy's (DOE) Fossil Energy and Carbon Management Advanced Energy Materials Research Program. The research was executed through the National Energy Technology Laboratory's (NETL) Research and Innovation Centers Advanced Materials Development Field Work Proposal. This research used resources of the National Energy Research Scientific Computing Center (NERSC), a DOE Office of Science User Facility supported by the Office of Science of the U.S. DOE under Contract No. DE-AC02-05CH11231 using NERSC award ALCC-ERCAP0022624 and ALCC-ERCAP0029917, and computational resources sponsored by the Department of Energy's Office of Energy Efficiency and Renewable Energy and located at the National Renewable Energy Laboratory.References
- D. Tejero-Martin, C. Bennett and T. Hussain, J. Eur. Ceram. Soc., 2021, 41, 1747–1768 CrossRef CAS
.
- L. R. Turcer and N. P. Padture, Scr. Mater., 2018, 154, 111–117 CrossRef CAS
- B. Qian, Y. Wang, J. Zu, K. Xu, Q. Shang and Y. Bai, J. Mater. Res. Technol., 2024, 29, 1231–1243 CrossRef CAS
- G. Costa, B. J. Harder, N. P. Bansal, B. A. Kowalski and J. L. Stokes, J. Am. Ceram. Soc., 2020, 103, 1446–1453 CrossRef CAS
- A. S. Risbud, K. B. Helean, M. C. Wilding, P. Lu and A. Navrotsky, J. Mater. Res., 2001, 16, 2780–2783 CrossRef CAS
- S. Hao, R. P. Oleksak, O. N. Dogan and M. C. Gao, Acta Mater., 2023, 258, 119225 CrossRef CAS
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS
- A. van de Walle, M. Asta and G. Ceder, CALPHAD J., 2002, 26, 539 CrossRef CAS
- A. van de Walle and M. Asta, Modell. Simul. Mater. Sci. Eng., 2002, 10, 521 CrossRef CAS
- S. Hao, R. P. Oleksak, O. N. Dogan and M. C. Gao, Comput. Mater. Sci., 2023, 230, 112541 CrossRef CAS
- S. Hao, Q. J. Hong and M. C. Gao, J. Am. Ceram. Soc., 2023, 106, 7654–7669 CrossRef CAS
-
C. Kittel and P. McEuen, Introduction to Solid State Physics, Wiley, New York, 8th edn, 1996 Search PubMed
- P. Söderlind and J. E. Klepeis, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 104110 CrossRef
- S. F. Pugh, London, Edinburgh Dublin Philos. Mag. J. Sci., 1954, 45, 823–843 CrossRef CAS
- H. Xiang, Z. Feng, Z. Li and Y. Zhou, J. Eur. Ceram. Soc., 2017, 37, 2491–2499 CrossRef CAS
-
J. F. Nye, Physical Properties of Crystals, Their Representation by Tensors and Matrices, Oxford Science Publications, Oxford, 1985 Search PubMed
- L. Chaput, A. Togo, I. Tanaka and G. Hug, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 094302 CrossRef
- E. T. Ritz, S. J. Li and N. A. Benedek, J. Appl. Phys., 2019, 126, 171102 CrossRef
- Z. Tian, K. Esfarjani and G. Chen, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 235304 CrossRef
- L.-D. Zhao, G. Tan, S. Hao, J. He, Y. Pei, H. Chi, H. Wang, S. Gong, H. Xu, V. P. Dravid, C. Uher, G. J. Snyder, C. Wolverton and M. G. Kanatzidis, Science, 2016, 351, 141–144 CrossRef CAS
- G. Tan, S. Hao, J. Zhao, C. Wolverton and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 6467–6473 CrossRef CAS
- D. T. Morelli, J. P. Heremans and G. A. Slack, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 195304–195309 CrossRef
- S. Hao, F. Shi, V. P. Dravid, M. G. Kanatzidis and C. Wolverton, Chem. Mater., 2016, 28, 3218–3226 CrossRef CAS
- https://oqmd.org/materials/entry/1101008 .
- A. Zunger, S.-H. Wei, L. G. Ferreira and J. E. Bernard, Phys. Rev. Lett., 1990, 65, 353–356 CrossRef CAS
- S. Hao, L.-D. Zhao, C.-Q. Chen, V. P. Dravid, M. G. Kanatzidis and C. Wolverton, J. Am. Chem. Soc., 2014, 136, 1628–1635 CrossRef CAS
- S. Hao, Z. Lu and C. Wolverton, Phys. Rev. B, 2016, 94, 014114 CrossRef
- S. Hao, J. He, V. P. Dravid, M. G. Kanatzidis and C. Wolverton, Phys. Rev. Mater., 2019, 3, 106002 CrossRef CAS
- X. Hua, S. Hao and C. Wolverton, Phys. Rev. Mater., 2018, 2, 095402 CrossRef CAS
-
A. Taylor and R. M. Jones, Silicon Carbide – A High Temperature Semiconductor, Pergamon Press, Oxford, London, New York, Paris, 1960 Search PubMed
- https://www.ioffe.ru/SVA/NSM/Semicond/SiC/thermal.html .
- T. Tadano, Y. Gohda and S. Tsuneyuki, J. Phys.: Condens. Matter, 2014, 26, 225402 CrossRef CAS
- T. Tadano and S. Tsuneyuki, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 054301 CrossRef
- Y. Wang, S.-L. Shang, H. Fang, Z.-K. Liu and L.-Q. Chen, npj Comput. Mater., 2016, 2, 16006 CrossRef
- Z. Tian, L. Zheng, Z. Li, J. Li and J. Wang, J. Eur. Ceram. Soc., 2016, 36, 2813–2823 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc03522d |
| This journal is © The Royal Society of Chemistry 2024 |