
New VVO2, VVO, VVOVVO and electrogenerated VIVOVVO systems of valproic acid hydrazones: a study of catalytic activity†
Roumi Patraa,
Debopam Sinhaab,
Sandip Mondalc and
Kajal Krishna Rajak *a
*a
aInorganic Chemistry Section, Department of Chemistry, Jadavpur University, Kolkata, 700032, India. E-mail: kajalrajak@gmail.com; patra.roumi141@gmail.com; debopamsinha@hotmail.com
bDepartment of Chemistry, Vijaygarh Jyotish Ray College, Kolkata, 700032, India.. E-mail: debopamsinha@vjrc.ac.in
cDepartment of Chemistry, Darjeeling Govt. College, Darjeeling, 734101, India. E-mail: sandipmondal30@gmail.com
First published on 15th January 2025
Abstract
The reaction between [VO(acac)2] and two valproic acid hydrazide ligands, H2L1 and H2L2, resulted in the formation of two mononuclear dioxido complexes [VV(O2)HL1–2] (1, 2) in acetonitrile, two oxidomethoxido complexes [VVO(L1–2)(OMe)(OHMe)] (3, 4) in methanol and the corresponding dinuclear μ-oxidodivanadium complexes [{VVOL1–2}2 μ-O] (5, 6) in dichloromethane. Here, H2L1 is the valproic acid hydrazone of salicylaldehyde and H2L2 is that of 2-hydroxy naphthaldehyde. X-ray crystallographic studies revealed the dual binding mode of the ligands, e.g. the neutral amido form in the dioxido complex 1 and the dianionic iminolato form in the oxidomethoxido complex 3. The redox behaviour of all the complexes was investigated using a combination of experimental and theoretical approaches. The partial reduction of 5 and 6 by constant-potential electrolysis (CPE) resulted in Robin–Day type II mixed-valence species 5a and 6a with a general formula of (L)(O)VIV–O–VV(O)(L). The complexes catalytically oxidized pyrogallol to purpurogallin and a catechol-like system to quinone under ambient conditions. The catecholase-like activity was found to be facilitated by a very rare semiquinone radical intermediate in the vanadium model systems.
Introduction
Valproic acid, a fatty acid,1 serves as a crucial foundational component for numerous pharmaceutically important molecules, particularly those used in the treatment of epilepsy, bipolar disorder, migraines, neurological diseases, cancer, and various other conditions. A growing body of evidence also supports its pharmacological action in treating neurodevelopmental disorders, including ADHD (attention deficit hyperactivity disorder) and cardioprotection, by inhibiting the activity of histone deacetylases (HDACs).2 The interest in hydrazone derivatives can be attributed to their promising bio-potency,3 which arises from their simple synthesis,4 intrinsic conformational flexibility5 and medicinal uses.6On the other hand, the coordination chemistry of vanadium with hydrazones7 is one of the most fascinating research topics owing to their various applications, including anticancer therapeutic potential.8 Vanadium compounds have garnered significant attention in both biomimetic and catalytic applications,9 such as insulin mimicry,10 haloperoxidase activity,11 and epoxidations,12 owing to their unique chemical properties and versatile behaviour. Moreover, a promising area of research in vanadium chemistry relies upon the synthesis of μ-oxidodivanadium(IV,V) complexes13 from their binuclear vanadium(V/V) analogues of the type (VVOL)2O. These complexes exhibit unique properties due to the presence of two different oxidation states of vanadium, which can lead to interesting redox behaviour and catalytic activity. Furthermore, the exploration of valence isomers in these vanadium complexes opens up new avenues for applications in molecular electronics and molecular computing. The diversification of the ligand's electronic environment around the metal core allows for the tuning of electronic properties, which is essential for the development of efficient electronic components and memory devices at the molecular level.14 Traditionally, the Robin–Day mixed valency scheme for complexes refers to type I (valence-trapped), where the odd electron is not delocalised,15 and type III (delocalised), where the odd electron is shared equally over both redox sites.16,13a Between these two extremes, type II (hopping)17 complexes are also found, where some delocalization takes place over both redox centres. Herein, we have explored the extent of electronic communication of the odd electron within type II mixed-valence oxidovanadium(IV/V) species and their predecessors. Additionally, a relevant area of vanadium chemistry has been identified for catecholase-like models.18 Despite the numerous reports on model complexes of catechol oxidase, there are only a few reports on the involvement of semiquinone radicals in the catecholase-like activity of metal complexes.19 However, to the best of our knowledge, no systematic study has been conducted on the catecholase-like activity of vanadium model systems via the semiquinone radical pathway. Furthermore, the catalytic oxidation of pyrogallol, a functional analogue of catechol, by vanadium complexes is rarely explored.20
In this work, we report six vanadium complexes [VV(O2)HL1–2] (1, 2), [VVO(L1–2)(OMe)(OHMe)] (3, 4), and [VV2O3(L1–2)2] (5, 6), as depicted in Chart 1. Schiff base ligands H2L1 and H2L2 were synthesised by the condensation of valproic acid hydrazide and salicylaldehyde or 2-hydroxy naphthaldehyde. In order to characterise the complexes, various physio-chemical techniques were used. The solid-state structures of ligands H2L1–2 and complexes 1 and 3 were confirmed using single-crystal X-ray diffraction. A combination of theoretical and experimental methods was used to examine the redox behaviour of the complexes. We also report type II mixed-oxidation state species 5a and 6a electrogenerated from their respective divanadium(V/V) precursors 5 and 6. The complexes were found to catalyse the oxidation of pyrogallol to purpurogallin and a catechol-like system to quinone under ambient conditions. The analyses revealed that the quinone formation reaction proceeds via the semiquinone radical route, which opens up a new horizon in the study of catecholase activity in vanadium systems. The mechanism has been proposed based on mass spectrometry, UV-Vis, EPR spectroscopy and 51V NMR experiments.
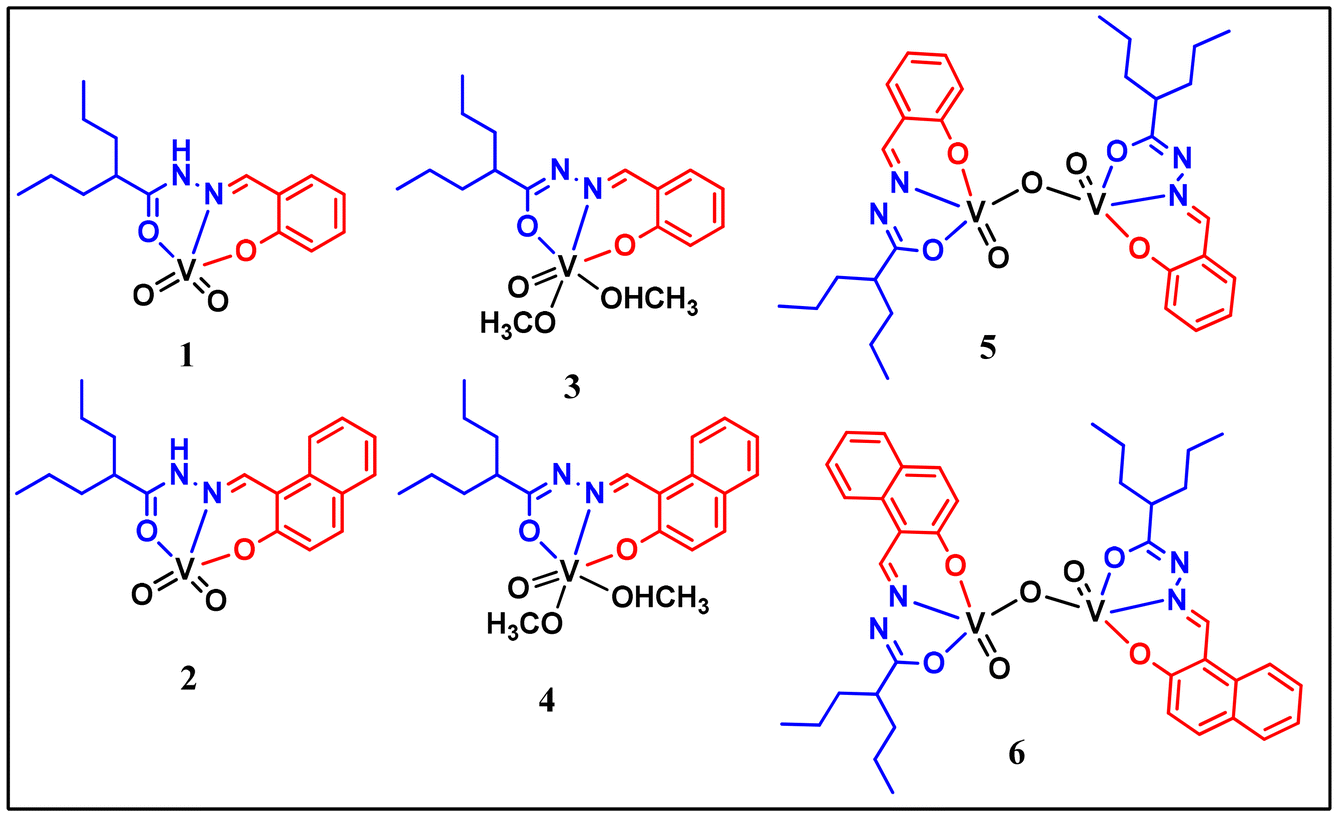 | ||
| Chart 1 Isolated vanadium complexes. | ||
Results and discussion
Syntheses
The ligands H2L1 and H2L2 were afforded by the condensation of valproic acid hydrazide with salicylaldehyde or 2-hydroxy naphthaldehyde. The reactions of [VO(acac)2] with an equimolar amount of the ligands in acetonitrile, in which the ligands existed in the neutral amido form, produced monomeric dioxido complexes (1, 2). A similar reaction performed in methanol yielded monomeric oxidomethoxido complexes (3, 4). Further, the reaction in dichloromethane led to the formation of dinuclear oxidovanadium complexes (5, 6). In both methanol and dichloromethane, the ligands bonded via their iminolate forms. Scheme 1 provides an overview of the synthesis of these complexes. The effect of solvent on the structural evolution of vanadium complexes is well-documented in the literature.21,22 Similarly, due to the tautomeric interconversion of the hydrazone moiety in different solvents, we got different types of crystals. Notably, the single crystals of complex 3 and the crystalline product of complex 4 were formed by the slow evaporation of complexes 5 and 6 in methanol solvent. However, the reverse did not happen when the methoxido complexes 3 and 4 were dissolved in dichloromethane. For the dioxido complexes 1 and 2, no interconversion was observed in different solvents. Constant-potential electrolysis (CPE) of 5 and 6 generated the type II mixed oxidation state species 5a and 6a respectively.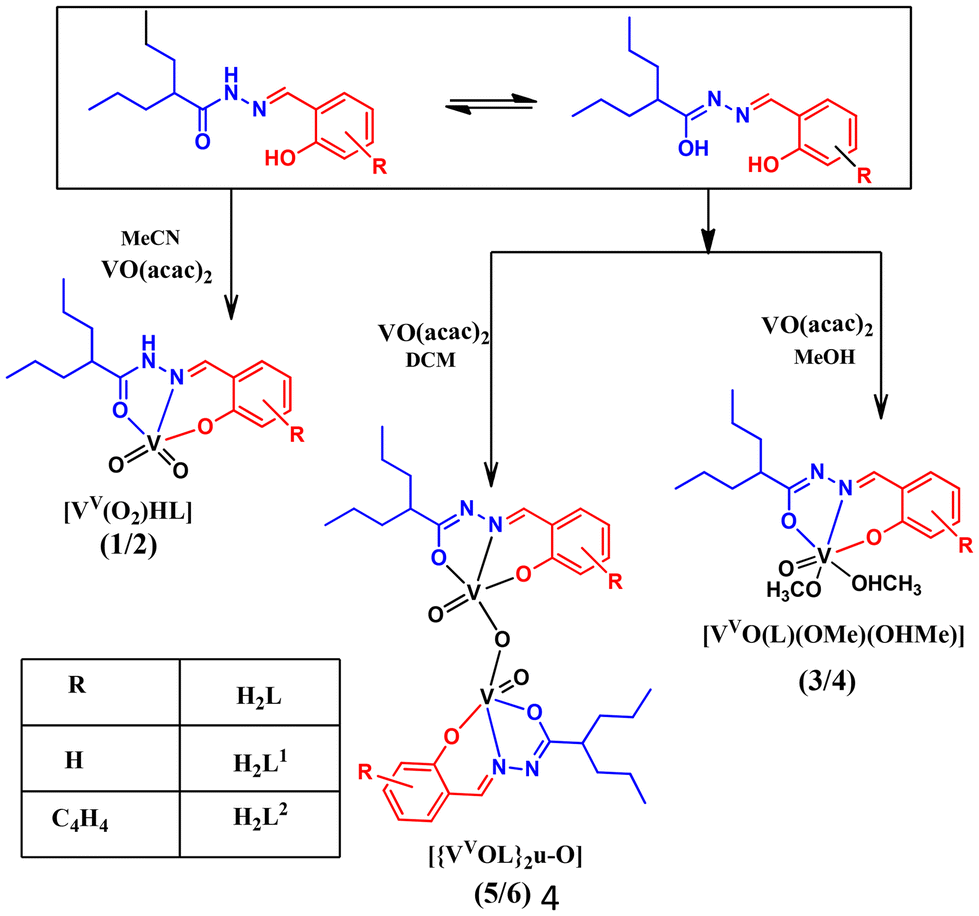 | ||
| Scheme 1 Schematic of the synthesis of the complexes. | ||
IR spectroscopy
The ligands showed a band in the range 3190–2919 cm−1 ascribed to the –OH and –NH moieties.4a The band near 3190 cm−1 was absent in the metal complexes due to coordination. For complexes 1 and 2, the band found near 2970 cm−1 was assigned to –NH stretching. This frequency was absent in the other four complexes. All the complexes showed prominent V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching peaks in the 990–920 cm−1 range, indicating the presence of the VO moiety. The sharp band around 750 cm−1 was assigned to V–O stretching.13 The relevant spectral data and description of the techniques are given in the Experimental section.
O stretching peaks in the 990–920 cm−1 range, indicating the presence of the VO moiety. The sharp band around 750 cm−1 was assigned to V–O stretching.13 The relevant spectral data and description of the techniques are given in the Experimental section.
Crystal structure
O2 bond lengths around 1.22 Å confirmed the existence of the amido form. The inter-molecular hydrogen bonding interaction (Fig. S2†) between the acyl oxygen and imine hydrogen (O2⋯H–N2) further verified the stability4a of the anti-isomer over the syn isomer.
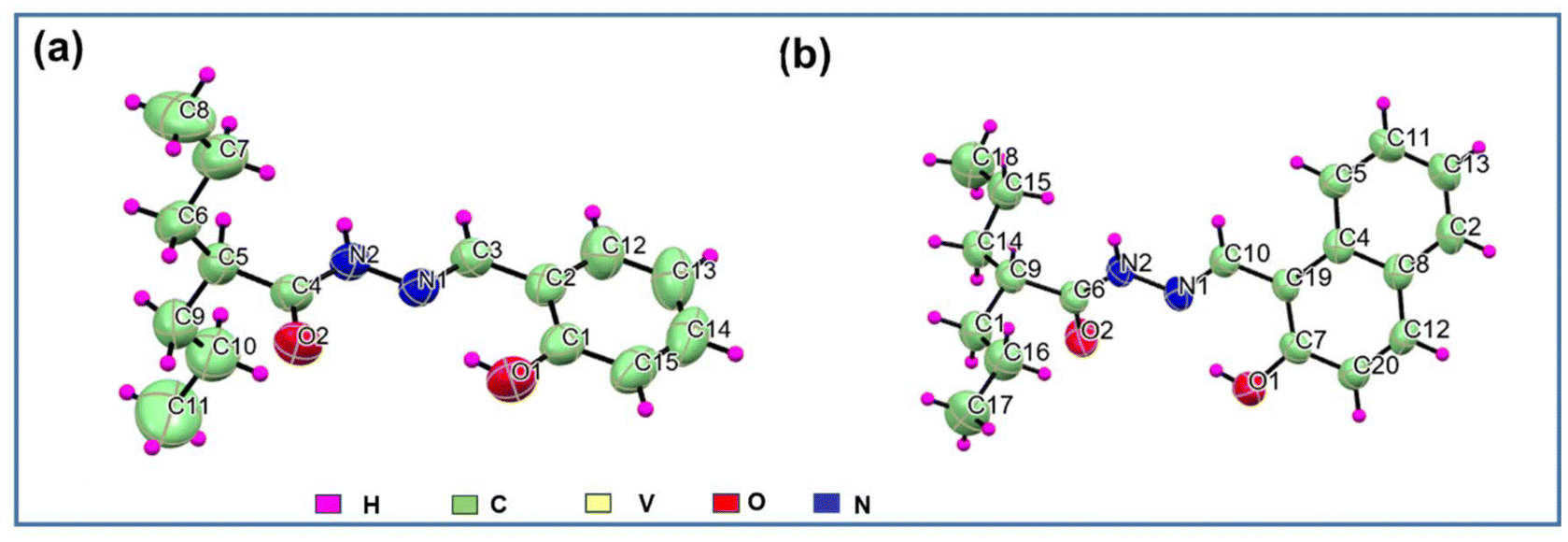 | ||
| Fig. 1 ORTEP diagram (50% ellipsoid probability) and atom-numbering scheme for ligands (a) H2L1 (the disordered part has been omitted for clarity) and (b) H2L2. | ||
Complex 1: Complex 1 was crystallised in the P21/n space group. In the complex, the V atom assumed a five-coordinated distorted square pyramidal geometry.7b The basal plane was defined by the phenolate oxygen (O1), azomethine nitrogen (N1) and acyl oxygen (O2) atoms along with one of the oxo (O4) atoms (Fig. 2). The other terminal oxo (O3) atom occupied the apical position. Here, the HN2–C4 bond length was around 1.322(3) Å, consistent with binding through the neutral amido form of the corresponding hydrazone. The shortest V1O3 and V1O4 distances were 1.6318(15) Å and 1.6069(17) Å, respectively, which are typical of VO double bonds. The τ value of 0.37 suggested a distorted square pyramidal geometry. The angles in the basal planes ranged from 73°–154° and that of the apical oxo O atom and the basal donor atoms was in the range of 96°–119°. The relevant crystallographic data and bond parameters are presented in Table S1.† In complex 1, the N–H⋯O and C–H⋯O hydrogen bonds form the 1D molecular chain (Fig. S2c†).
 | ||
| Fig. 2 ORTEP diagram (50% ellipsoid probability) and atom-numbering scheme for complex 1. | ||
Complex 3: The coordination geometry of complex 3 could be described as a distorted octahedral structure, with the doubly bonded oxo group and methanol in the axial position. The N2O donor set from the ligand consisting of phenolate oxygen (O1), azomethine nitrogen (N1) and acyl oxygen (O2) atoms along with the methoxide oxygen atom (O4) defined the equatorial plane (Fig. 3). The molecule was crystallised in the P21/c space group. The overall charge balance and respective C4O2 and N2–C4 bond lengths suggested coordination through the iminolate form. The O3V1–O5(methanol) axis was almost linear. The trans disposition of the oxo group causes the elongation of the V1–O5(methanol) bond length, weakening the methanol coordination.13b Here, various weak interactions lead to the one-dimensional molecular chain (Fig. S2d†).
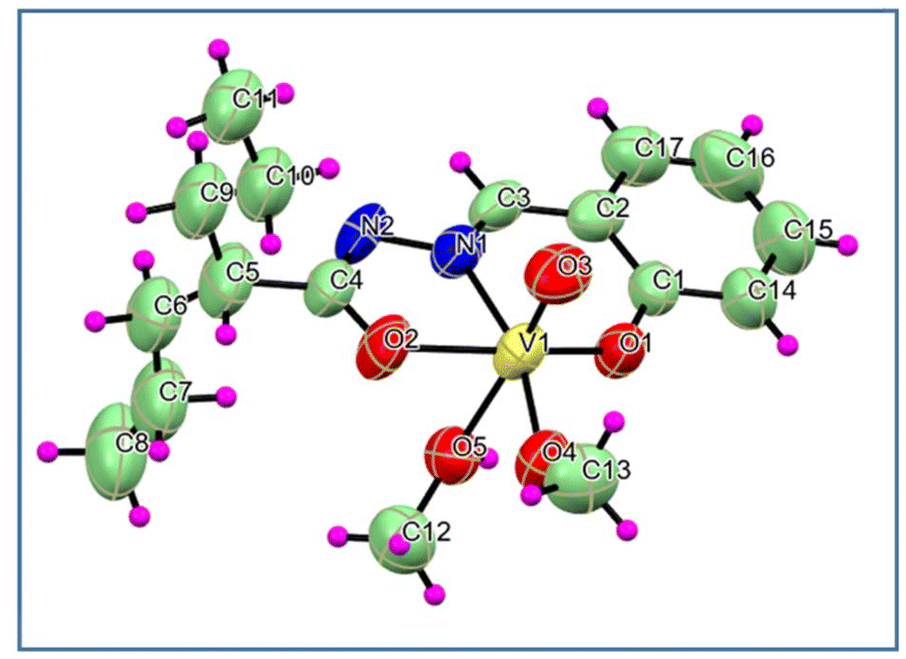 | ||
| Fig. 3 ORTEP diagram (50% ellipsoid probability) and atom-numbering scheme for complex 3. The disordered part has been omitted for clarity. | ||
Complex 2 has a geometry similar to Complex 1. As the data were not crystallographically publishable, the structure is given in the supporting information to illustrate bond connectivity. In Complex 6, the ligand binds in its iminolate form. The structure is provided in the supporting information to depict the bond connectivity only (Fig. S1†).
Electronic absorption spectra
The UV-Vis absorption spectra of the ligands and complexes (1–6) were recorded in CH2Cl2 at 298 K at a concentration of ∼20 μM. The electronic spectrum of H2L1 exhibited three absorption bands. The band at 322 nm corresponded with the intra-ligand n → π* transition. The twin shoulders at 290 nm and 280 nm could be assigned to the π → π* transition of the aromatic rings and azomethine moiety. Similarly, for the ligand H2L2, two bands at around 355 nm and 321 nm with a weak shoulder at 368 nm and 308 nm, respectively, were observed.4a The extended π conjugation in H2L2 resulted in the red shifting of the λmax value (Fig. S3†).The electronic spectra of complexes 1–6 are depicted in Fig. S4.† The set of complexes of each ligand possessed a particular pattern with respect to their structural motif. The spectra of all complexes of H2L1 i.e., 1, 3 and 5 were nearly identical in appearance. The bands of this ligand near 280 nm and 322 nm were also observed in the metal complexes with some variation. For vanadium(V) complexes, no d-d bands were observed. A broad band in the lower-energy region was observed around 380–388 nm due to LMCT transitions.13b,21a Similar was the case of H2L2 (complexes 2, 4 and 6). The bands at around 293 nm and 336 nm could be assigned to the ligand-centred transition. The absorption band of these complexes at around 412 nm was due to the LMCT transition. In the near-IR range between 1182 and 1314 nm, complexes 5− (5a) and 6− (6a) exhibited a characteristic absorption band of the [V2O3]3+ core (Fig. 4) corresponding to the intra-valence charge transfer transition (IVCT)16 between the mixed-valence vanadium nuclei.
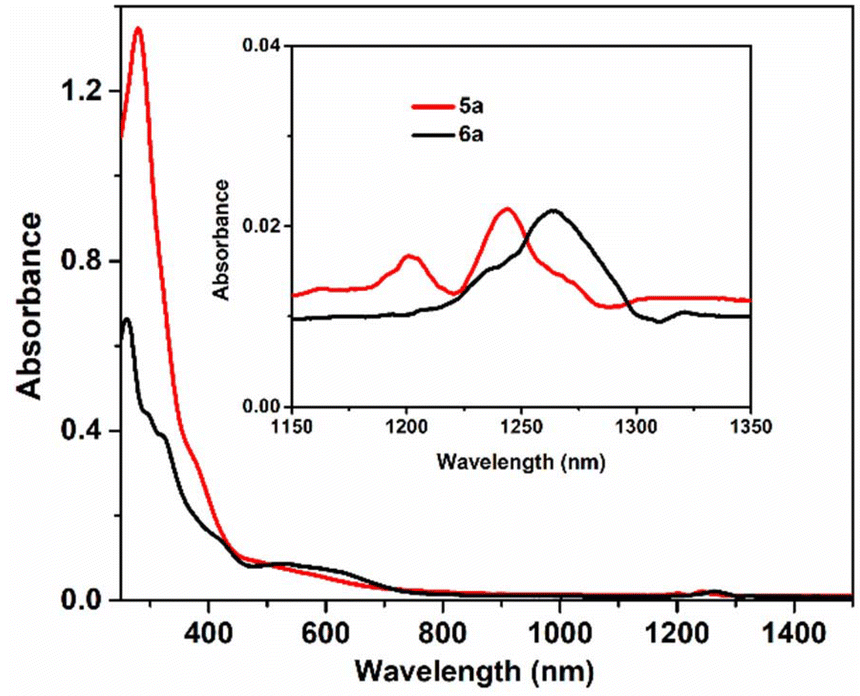 | ||
| Fig. 4 Absorption spectra of 5− (5a) and 6− (6a) in CH2Cl2. | ||
Cyclic voltammetry
The redox activities of 1–6 (referenced to the ferrocenium/ferrocene couple) were investigated by cyclic voltammetry at 295 K in CH2Cl2 containing [N(n-Bu)4]PF6 as the supporting electrolyte. The cyclic voltammograms are illustrated in Fig. 6, and the data are summarized in Table 1. The cyclic voltammograms of the mononuclear complexes (1, 2, 3 and 4) displayed cathodic waves in the range of −0.168 to −0.226 V arising from the VV/VIV reduction couple. The one-electron reversible cathodic process in 1 and 2 was observed at −0.168 and −0.174 V, respectively, depicting VV → VIV reduction. The reversible cathodic processes occurred at considerably lower potentials in the methoxido complexes 3 (−0.244 V) and 4 (−0.212 V). This trend may be attributed to the net decrease in electron density on the metal centre of the dioxido complexes, making the vanadium(V) centre more prone to reduction than those of the methoxy complexes. The redox activities of the dinuclear complexes 5 and 6 were notably different from the rest. The quasi-reversible cathodic waves of complex 5 (Fig. 5) due to the VV^O^VV/VIV^O^VV and VIV^O^VV/VIV^O^VIV redox couples were found at −0.252 and −0.709 V, respectively. Complex 6 also showed similar redox properties to complex 5, as depicted in Fig. 5. One each of reversible and quasi-reversible cathodic waves were observed at −0.204 V and −1.57 V, respectively.16b | ||
| Fig. 5 Cyclic voltammograms of (a) 1, (b) 2, (c) 3, (d) 4, (e) 5 and (f) 6 in CH2Cl2 at 298 K. Conditions: scan rate = 100 mV s−1; 0.20 M [N(n-Bu)4]PF6 supporting electrolyte; platinum working electrode. | ||
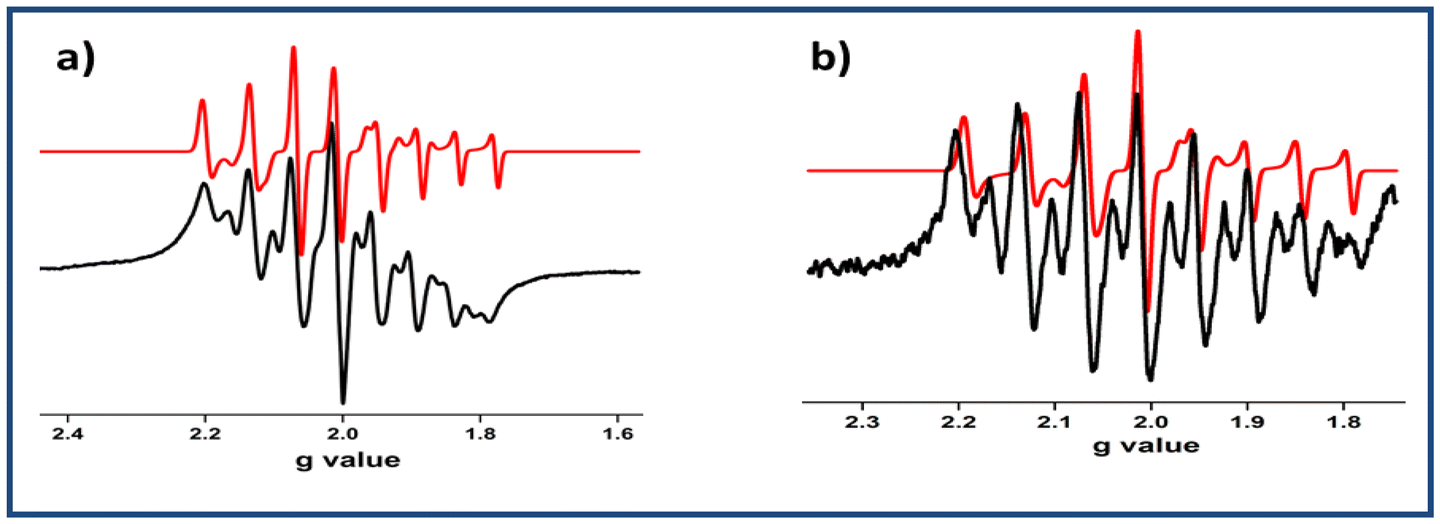 | ||
| Fig. 6 X-band EPR spectra of (a) complex 1− and (b) complex 2− at RT (black: experimental spectrum; red: simulated spectrum). | ||
| Complex | E1/2(V) (ΔE (mV)) |
|---|---|
| ΔE = peak-to-peak separation. | |
| 1 | −0.168 (119) |
| 2 | −0.174 (158) |
| 3 | −0.244 (105) |
| 4 | −0.212 (125) |
| 5 | −0.252 (178), −0.709 (210) |
| 6 | −0.204 (123), −1.57 (192) |
EPR study
All six complexes were diamagnetic in nature and hence EPR-silent. The electrochemically reduced species were found to be paramagnetic. The EPR spectra of all the species were recorded in CH2Cl2 solutions at ambient temperature. The EPR spectra of 1−, 2−, 3− and 4− exhibited the characteristic eight-line pattern due to the presence of extra one unpaired electron spin (S = ½) and the hyperfine coupling of 51V (I = 7/2). Surprisingly, the spectra of 1− and 2− deviated slightly from the ideal eight-line pattern of the V(IV) complexes.7c (Fig. 6 and Table S9†), suggesting the reduction of 1 → 1−or 2 → 2− were due to VV → VIV conversion with some delocalisation of the electron density to the ligand moiety. This diamagnetic [VVO]2+ to paramagnetic [VIVO]2+ shifting was also supported by the spin density data obtained from DFT calculations.Furthermore, to explain the deviated eight-line EPR spectra, we performed cyclic voltammetry experiments with complexes 1 and 2 at different scan rates (Fig. S5†). This study clearly suggested that electrochemical VV → VIV reduction was prominent in this case rather than electron transfer from the metal to the ligand.
At room temperature, the EPR spectra of the solid solutions of the electrochemically synthesized mixed-valence [{VVVIVO2(L)2}-μ-O] species (5a and 6a) consisted of an eight-line pattern (Fig. S6 and Table S8†). In contrast, the frozen glass solutions of the same showed 13 lines at 77 K (Fig. 7 and Table 2). Precisely, in the case of divanadium(IV,V) complexes, a 15-line hyperfine pattern on the EPR time scale suggests a type II or III complex, according to Robin and Day categorization. In a type III complex, the unpaired electron is perfectly delocalized over both the vanadium centres, and in type II, the electron is partially delocalized, i.e. weaker interaction; whereas, an 8-line pattern suggests a valence-trapped state of the odd electron (type I). The occurrence of valence isomerism vs. a valence-trapped state is temperature dependent. In this case, the absence of 15-line patterns indicates that 5a and 6a were not really mixed-valence complexes16 of type III. The unpaired electron spin was not predominantly localized on one of the vanadium(IV) centres but somewhat mixed with the neighbouring vanadium nuclei, which can be represented as [{VVO(L)}{VIVO(L)}-μ-O] ↔ [{VIVO(L)}{VVO(L)}-μ-O]. This anisotropic distribution17 of the unpaired electron spin validates the identity of 5a and 6a as type II mixed-valence complexes.
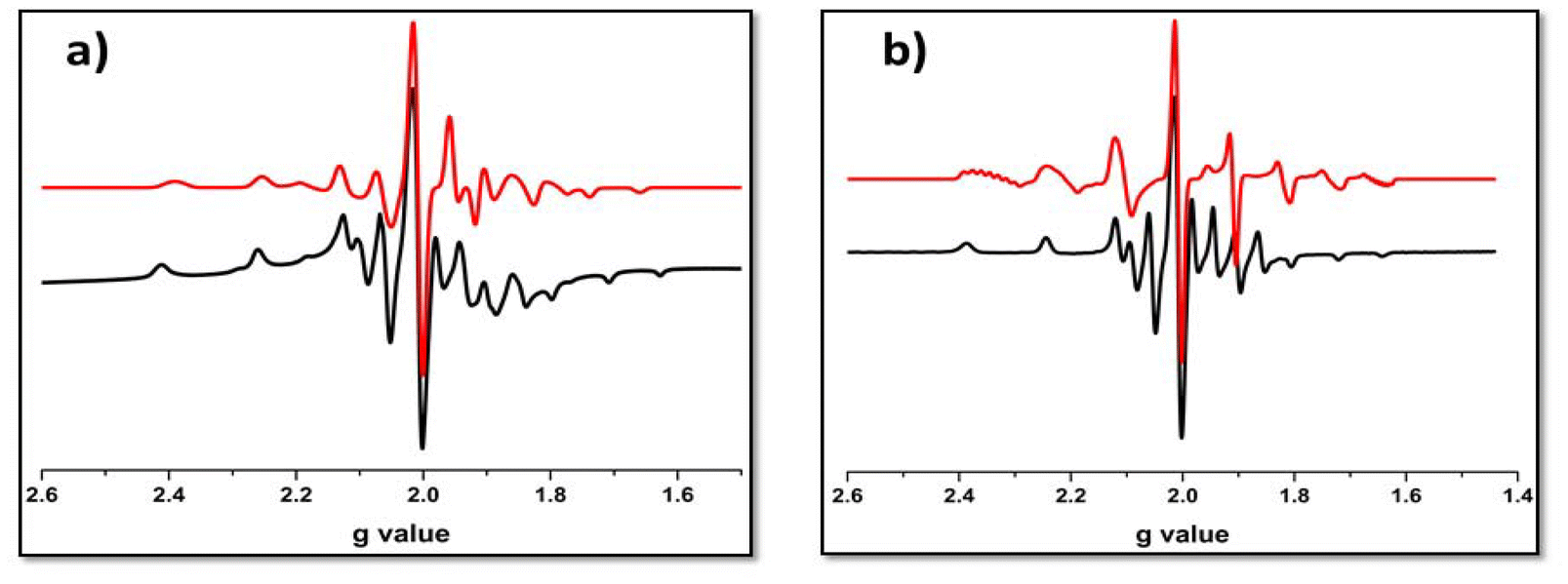 | ||
| Fig. 7 X-band EPR spectra of [V2O3L1–22]3+ complexes (a) 5a and (b) 6a in frozen solutions at 77 K (black-experimental spectrum; red-simulated spectrum). | ||
| Complex | Matrix | g∥ | g⊥ | gav | A∥ (G) | A⊥(G) | Aav (G) | lw (mT) |
|---|---|---|---|---|---|---|---|---|
| 5a | CH2Cl2, 77 K | 1.9576 | 1.9587 | 1.9583 | 468.25 | 230.57 | 309.79 | 2.0 |
| 6a | CH2Cl2, 77 K | 1.9526 | 1.9336 | 1.9399 | 450.05 | 269.97 | 329.99 | 1.25 |
Simulation calculations
In all cases, i.e. complexes 1–6, the highest occupied molecular orbitals (HOMO) were localized mostly on the ligand framework, whereas, the lowest unoccupied molecular orbitals (LUMO) were different from each other as the ligating environment has a significant impact on the energy and stability of the excited state. For the methoxido complexes (3 and 4) and dimeric complexes (5 and 6), the lowest unoccupied molecular orbitals (LUMO) were predominantly localized on the vanadium atom, with minimal contributions from the oxo ligands. However, in the case of the dimeric complexes, there was a small contribution from the bridging oxo ligands towards the LUMO (Fig. 8 for 2, 4, and 6; Fig. S7† for 1, 3, and 5). This is an obvious sign of metal-centred reduction, indicating that the electronic changes primarily occur at the metal centre rather than being significantly influenced by the surrounding ligands. However, in contrast, the LUMOs of 1 and 2 were localized predominantly on the ligand moiety and very little on vanadium. The spin density obtained (Fig. 9) from the Mullikan spin population analysis of 1− and 2− was scattered mainly on the ligand moiety and very little on the vanadium core, suggesting the occurrence of electron density transfer from vanadium to the ligand or electron density reconstruction. Therefore, the cyclic voltammetry (for 1, 2) and EPR studies (for 1− and 2−) suggested a one-electron vanadium centre reduction, which was also observed in the spin population plots of 1 and 2. The spin-density pattern indicated that VV to VIV reduction resulted in further spin delocalization to the ligand framework. The corresponding EPR data is given in Fig. 7. The calculated spin density of 3− and 4− was located mainly on the central vanadium atom, indicating the usual metal-centred reduction. In 5− (5a) and 6− (6a), the spin density was localised mostly on one vanadium atom, with a very small fraction on the second vanadium atom. Thus, the calculations successfully supported the EPR data, which predicted the formation of type II mixed-valence complexes.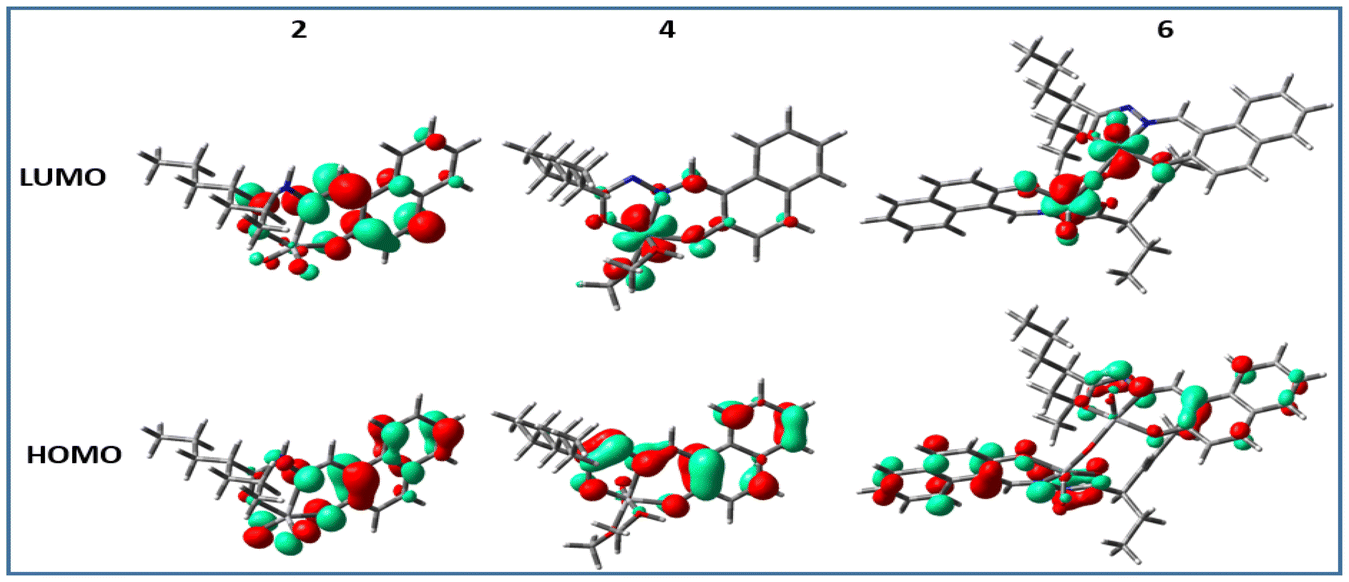 | ||
| Fig. 8 Isodensity plots of the selected frontier orbitals of complexes 2, 4, and 6 (complexes with ligand H2L2). | ||
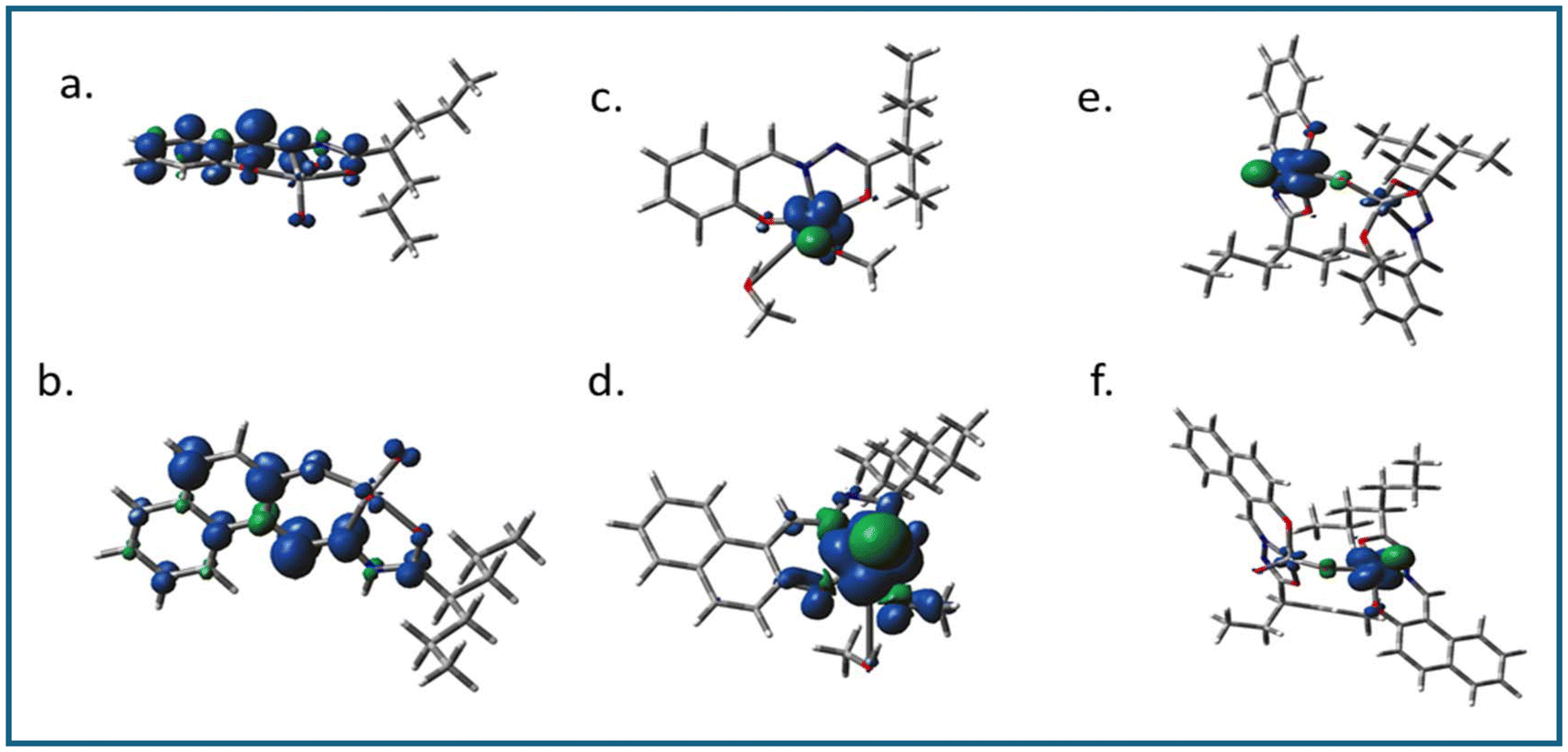 | ||
| Fig. 9 Spin density plots of (a) 1− (b) 2−, (c) 3−, (d) 4−, (e) 5a and (f) 6a. | ||
Catalytic oxidation of pyrogallol
Active oxygen molecules, such as hydrogen peroxide, peroxo compounds and molecular oxygen, are frequently used as primary oxidants23,24 in transition-metal-catalysed oxidation reactions. The complexes catalysed the pyrogallol oxidation reaction in mere aerobic conditions. Complexes 5 and 6 behaved similarly to their respective monomers in methanol i.e. 3, 4. Complexes 1 and 2 showed catalytic activity towards pyrogallol oxidation. A 6 × 10−4 M solution of complex 1 was treated with a 0.006 M solution of pyrogallol, and the course of the reaction was monitored by recording UV-Vis spectra. The spectral changes of complex 1 in methanol are given in Fig. 10. The band near 590 nm gradually decreased, while the band near 388 nm gradually increased, indicating the formation of purpurogallin. The kinetics of pyrogallol oxidation were determined by monitoring the growing absorption band of the product purpurogallin. For this purpose, a 6 × 10−4 M solution of complex 1 was treated with 0.001 M–0.01 M solutions of pyrogallol. Below are the conclusions drawn from the Michaelis–Menten and Lineweaver–Burk equations. Complex 1: k2 = 10.2 min−1, KM = 1.2 × 10−3 and Vmax = 6.1 × 10−3 M min−1 (Fig. 10). Similarly, a 6 × 10−4 M solution of complex 2 was treated with a 0.006 M solution of pyrogallol, and the corresponding spectrum showed that the band near 588 nm gradually decreased, while the band near 402 nm gradually increased. The kinetic parameters of complex 2 are as follows: k2 = 8.5 min−1, KM = 1.12 × 10−3 and Vmax = 5.1 × 10−3 M min−1 (Fig. S8†).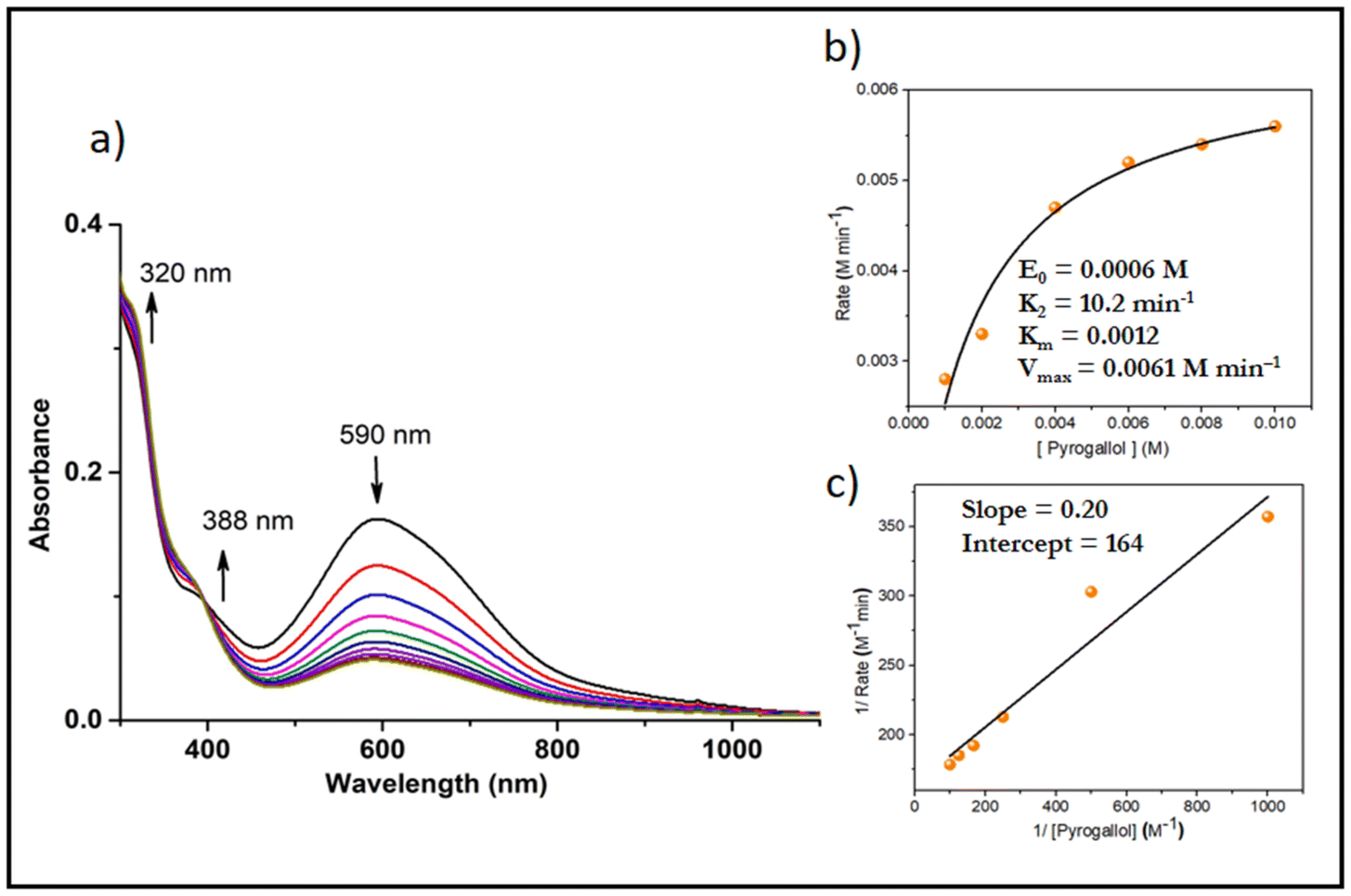 | ||
| Fig. 10 (a) Absorption spectra of a solution containing 6 × 10−4 M complex 1 and 0.006 M pyrogallol recorded at the intervals of 5 min. (b) Plot of reaction rate versus concentration of pyrogallol for the oxidation reaction catalysed by complex 1; (c) Lineweaver–Burk plot. | ||
Likewise, complexes 3 and 4 were also used as active catalysts for the conversion of pyrogallol to purpurogallin. The time-dependent absorbance spectral evolution was the same as those of the corresponding dioxo complexes. The kinetic parameters were evaluated under the same conditions. The results obtained from the conventional Michaelis–Menten and Lineweaver–Burk equations are as follows. Complex 3: k2 = 7.6 min−1, KM = 5 × 10−4 and Vmax = 4.6 × 10−3 M min−1 (Fig. 11); complex 4: k2 = 6.7 min−1, KM = 4 × 10−3 and Vmax = 4 × 10−3 M min−1 (Fig. S9†). Their similar spectral patterns suggest that the reactions proceeded through a common intermediate. Notably, in the literature, kinetic studies of only a few vanadium complexes20 have been reported for pyrogallol oxidation. The catalytic efficacy observed in this study is comparable to the reported data; however, in this case, catalysis proceeds without externally added H2O2.
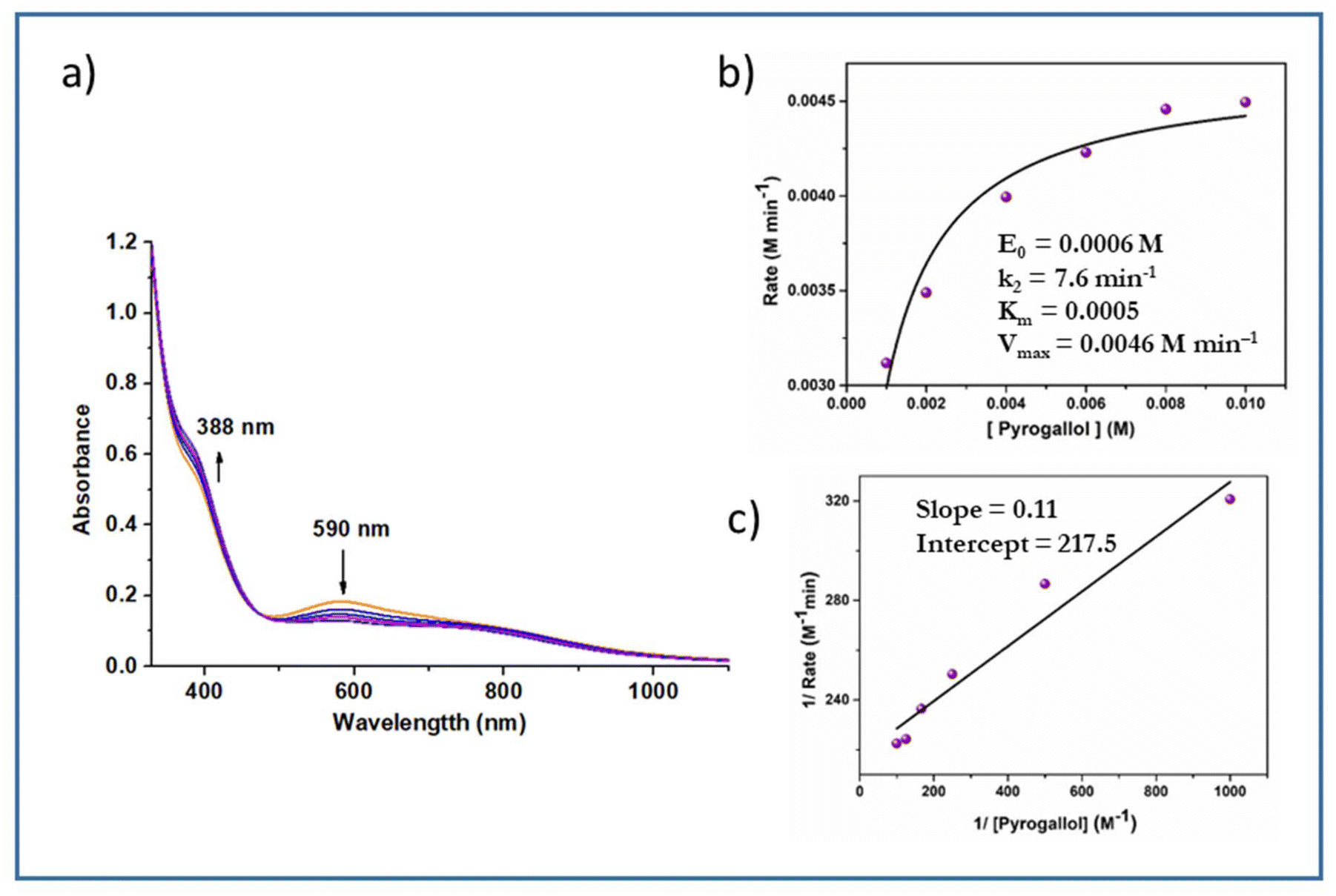 | ||
| Fig. 11 (a) Time-dependent absorption spectra of a solution containing 6 × 10−4 M complex 3 and 0.006 M pyrogallol recorded at time intervals of 5 min. (b) Plot of reaction rate versus concentration of pyrogallol for the oxidation reaction catalysed by complex 3. (c) Lineweaver−Burk plot. | ||
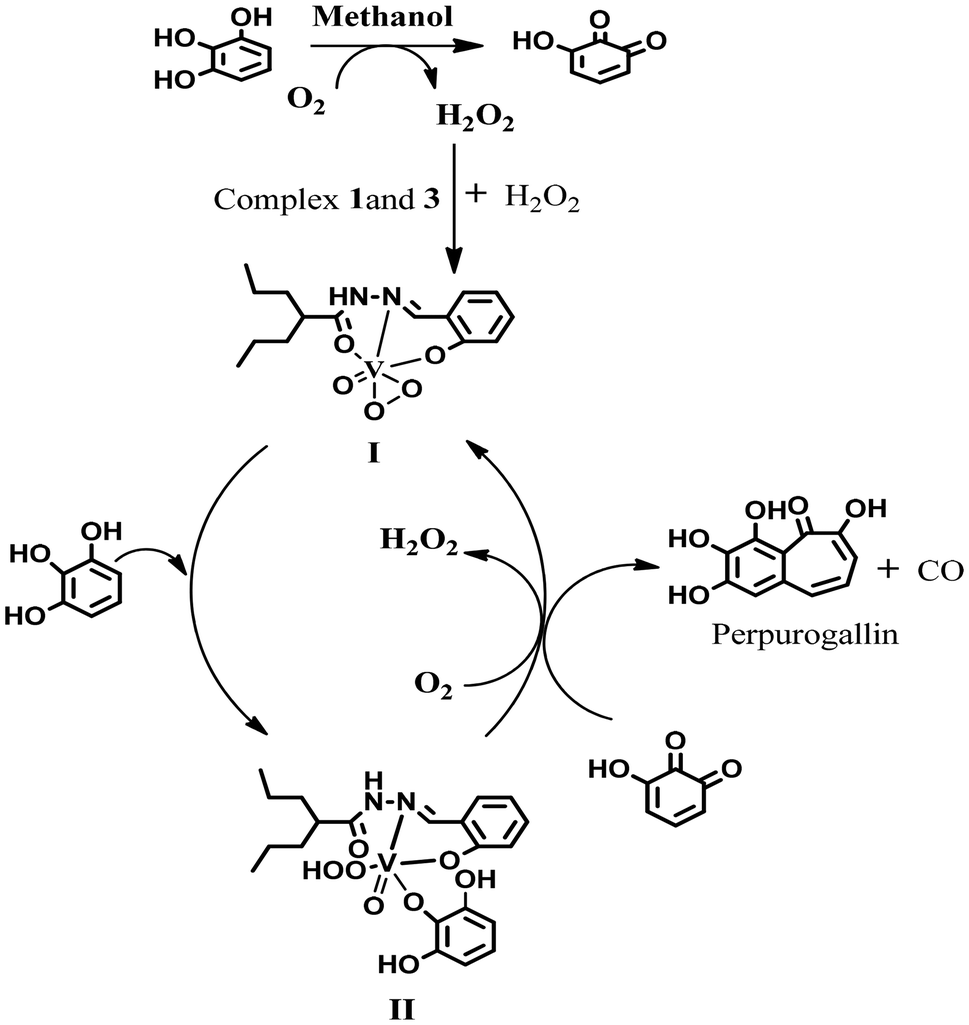 | ||
| Scheme 2 Schematic of pyrogallol oxidation by complexes 1 and 3. | ||
Catechol oxidation
We also examined the catalytic activity of the vanadium complexes towards the oxidation of catechol, a structural analogue of pyrogallol. To study the catecholase activity of all the complexes, we used 3,5-di-tert-butylcatechol (3,5-DTBC) as the model substrate. For catechol oxidation, it was found that the mononuclear oxidomethoxido complexes 3 and 4 were catalytically active, whereas, the dioxo complexes 1 and 2 turned out to be inactive due to the strong ligating environment. Complexes 5 and 6 showed catalytic activity very similar to the methoxido complexes 3 and 4, respectively, because in methanol, 5 and 6 converted to methoxido complexes 3 and 4, respectively. The course of the reaction was monitored by time-dependent UV-Vis spectroscopy. The reaction was initiated by adding 2 × 10−4 M solutions of 3 and 4 to a 0.004 M solution of 3,5-DTBC in methanol in aerobic conditions. The immediate spectral run revealed three peaks at around ∼860 nm, ∼580 nm and ∼410 nm. With the progress of catalytic oxidation, the bands at 860 nm and 580 nm gradually diminished, whereas the bands around 410 nm increased and finally saturated at ∼400 nm due to the formation of quinone over time. The colorimetric shift of the reaction mixture from greenish blue to deep brown and the formation of a single isosbestic point suggested that an intermediate is involved in the conversion of 3,5-DTBC to 3,5-DTBQ. The yield of 3,5-DTBQ was determined after purifying the brown solution obtained after completion of the reaction using column chromatography. The isolated yield was about 56% for complex 3 and 52% for complex 4. To obtain the kinetic parameters, 2 × 10−4 M solutions of the complexes were treated with varying concentrations (0.001–0.01 M solutions) of the substrate. Based on increasing absorption of the quinone band, the rates of the reactions were determined, and the kinetic parameters estimated using the Michaelis–Menten and Lineweaver–Burk equations are as follows. Complex 3: k2 = 25.7 min−1, KM = 2.9 × 10−3 and Vmax = 5.1 × 10−3 M min−1 (Fig. S10†); complex 4: k2 = 22.6 min−1, KM = 2.03 × 10−3 and Vmax = 4.5 × 10−3 M min−1 (Fig. 12).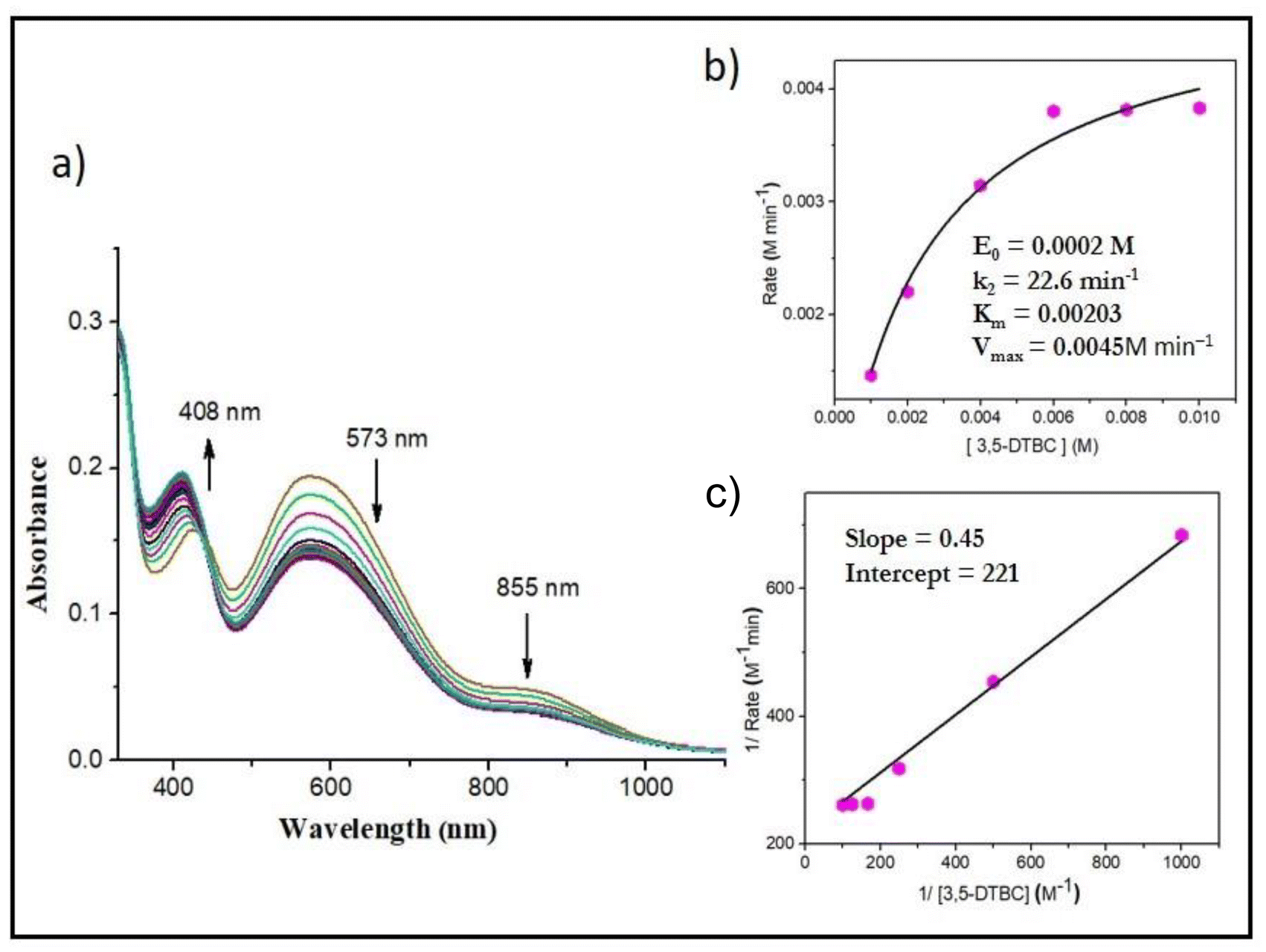 | ||
| Fig. 12 (a) Absorption spectra of a solution containing 2 × 10−4 M complex 4 and 0.004 M 3,5-DTBC recorded at intervals of 5 min. (b) Plot of reaction rate versus concentration of 3,5-DTBC for the oxidation reaction catalysed by complex 4. (c) Lineweaver–Burk plot. | ||
Probable reaction pathway
The active intermediate in vanadium-promoted catechol oxidation is well-reported because catechol binds in the mono-anionic catecholate form.18d,26 However, in our case, 3,5-DTBC binds in the semiquinone form. This was evident from the absorption bands appearing in the near-IR region at ∼860 nm.27 X-band EPR measurements were performed in order to identify paramagnetic species that might have formed in the reaction mixture, as indicated by the absorption spectra. The complexes were observed to be EPR-quiet, as expected for the VV species. Immediately after the addition of 3,5-DTBC to the respective complexes, a strong EPR signal (g ∼ 2.0098) was observed due to the interaction of the [VVO(L1–2)(3,5-DTBSQ˙−)] species with the vanadium nucleus (Fig. 13 and S11†). We also recorded EPR spectra by adding 2,2,6,6-tetramethylpiperidinyl-1-oxy (TEMPO˙) in the reaction mixture. The EPR spectra showed diminished intensity of the radical peak (Fig. 13). Furthermore, we performed 51V NMR analysis of complex 4 before and after the addition of 3,5-DTBC. We observed that the peak of 51V at −536.26 completely disappeared after the addition of catechol (Fig. S40 and S41†). These results conclusively establish that the reaction predominantly proceeded via a radical intermediate. The presence of distinct peaks at m/z = 597 and 221 (Fig. S13†) in the mass spectra corresponded with intermediate III and step IV, respectively, confirming the progress of the reaction of complex 4, as depicted in Scheme 3.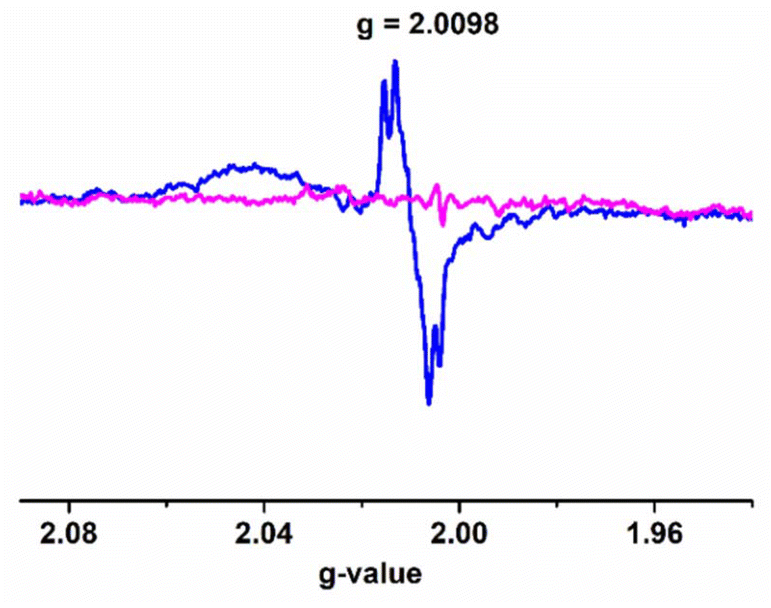 | ||
| Fig. 13 X-band EPR spectra of the intermediate III (4cat) formed during catechol oxidation by complex 4 (blue-intermediate; magenta-4cat with TEMPO). | ||
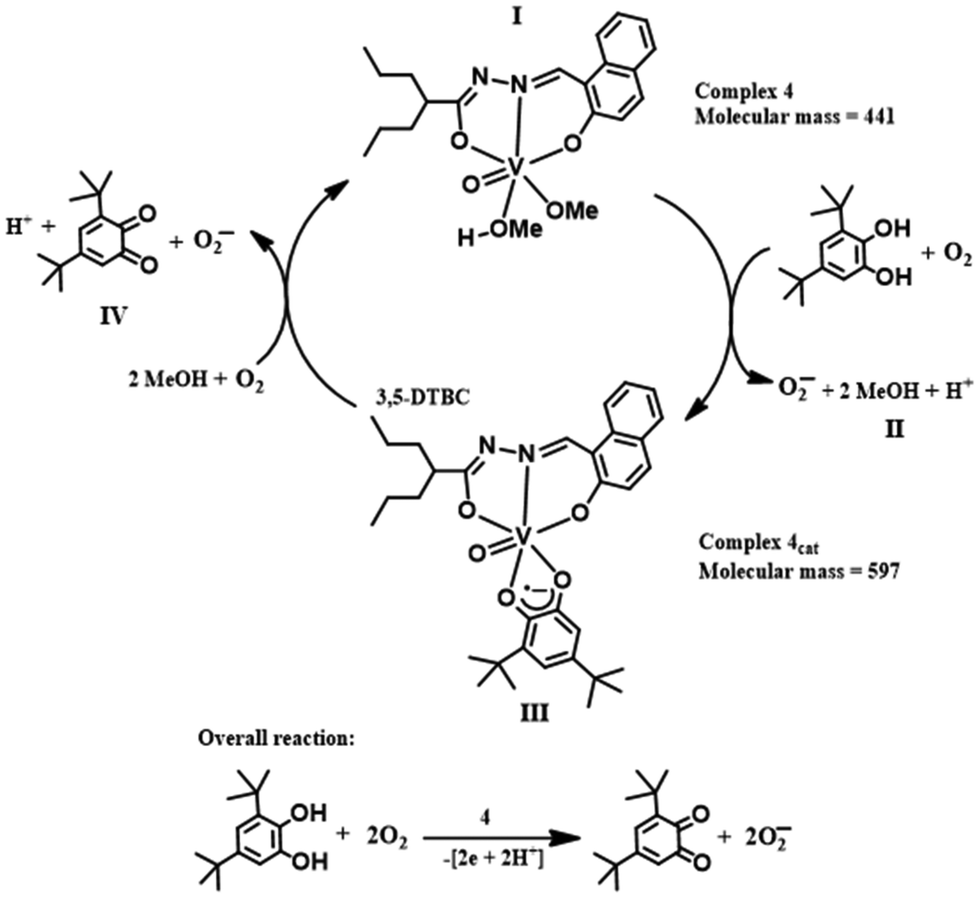 | ||
| Scheme 3 Schematic of catechol oxidation by complex 4. | ||
In general, the catecholase activity of dioxo complexes is completely unknown. Catechol fails to chelate vanadium metal by cleaving one of the VO bonds of the VV(O2) core, in line with Scheme 3. Interestingly, the pyrogallol activity of the dioxo complex prompted us to rethink its catecholase activity. A closer look into the mechanism of pyrogallol activity of complexes 1 and 2 revealed a plausible catecholase mechanism via the generation of a peroxo intermediate, which serves as the active catalyst. Now, in the reaction mixtures containing pyrogallol and the as-prepared complexes, we observed the generation of the peroxo intermediate via the reaction of in situ generated H2O2 by the aerobic auto-oxidation of pyrogallol; but in mixtures containing catechol and complex 1 or 2, the reaction proceeded only after the addition of H2O2. This observation suggests that the generation of H2O2 due to auto-oxidation is more facile in the case of pyrogallol than catechol.28 Herein, we initiated the reaction by adding 2 × 10−4 M solutions of 1 and 2 with a 0.004 M solution of 3,5-DTBC and one drop portion of H2O2 (10−2 M) in methanol.
The time-dependent absorbance spectral evolution is given in Fig. 14. The kinetic parameters were evaluated similarly to those of complexes 3 and 4 except, that H2O2 was added in this case. The results based on the Michaelis–Menten and Lineweaver–Burk equations are as follows. Complex 1: k2 = 37.3 min−1, KM = 1.27 × 10−3 and Vmax = 7.4 × 10−3 M min−1 (Fig. S12†); complex 2: k2 = 34.5 min−1, KM = 1.38 × 10−3 and Vmax = 6.8 × 10−3 M min−1 (Fig. 14).
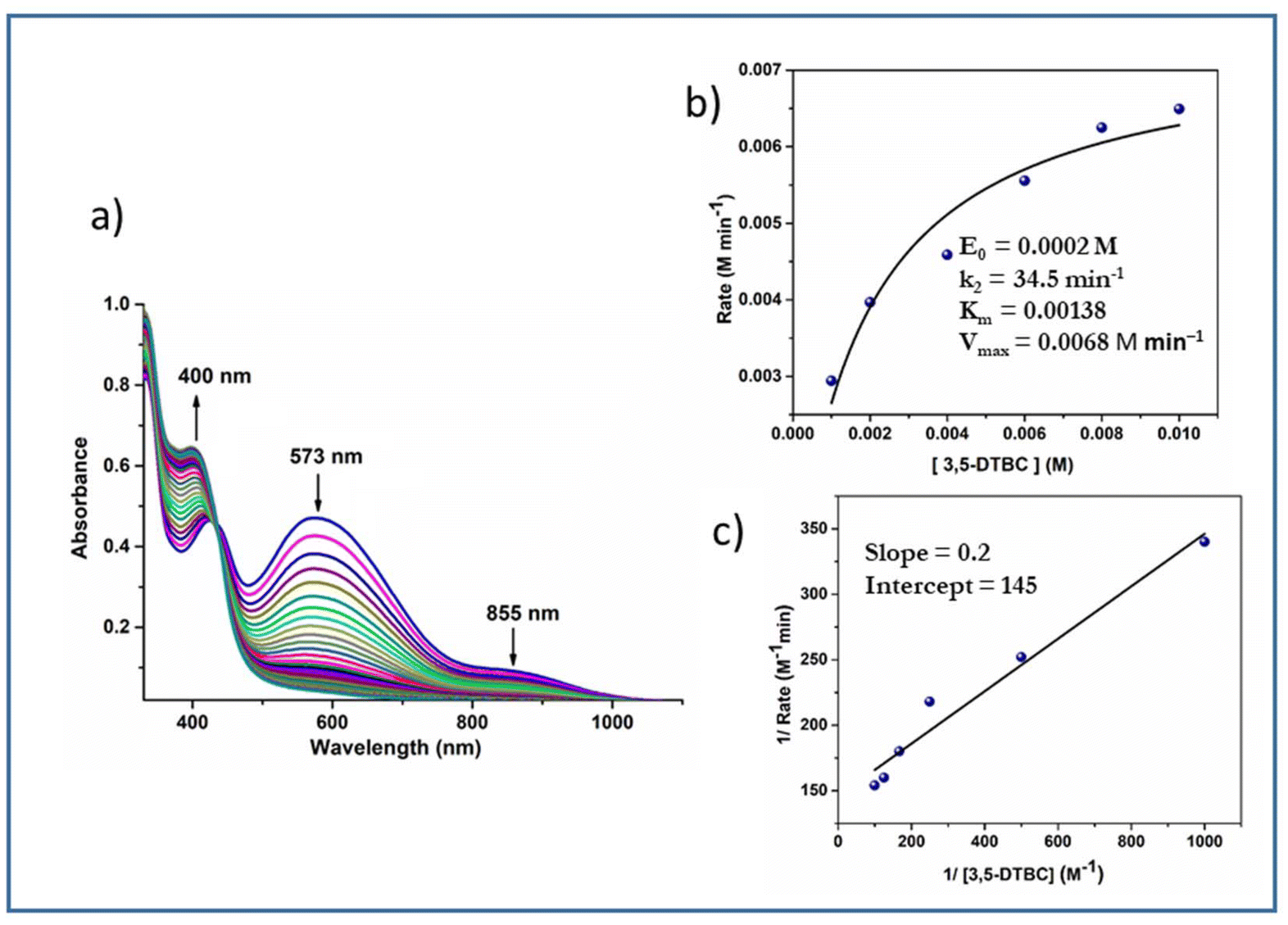 | ||
| Fig. 14 (a) Absorption spectra of a solution containing 2 × 10−4 M complex 2 and 0.004 M 3,5-DTBC in the presence of one drop portion of H2O2 (10−2 M) recorded at intervals of 5 min in methanol. (b) Plot of reaction rate versus concentration of 3,5-DTBC for the oxidation reaction catalysed by complex 2. (c) Lineweaver–Burk plot. | ||
In the case of methoxido complexes 3 and 4, the reaction proceeds due to the chelation of catechol by breaking the two weak V–O bonds originating from the attached methoxy group and the solvent moiety. Therefore, there was no need for external addition of H2O2. On the other hand, the dioxo complexes 1 and 2 require H2O2 to initiate the reaction that generates the peroxo intermediate by replacing the stronger VO bond. The cleavage of the relatively weak peroxo bond is more facile for chelation by catechol. From the mass spectral analysis, we observed the formation of an oxo-peroxo intermediate (m/z = 409). Similarly, a peak at m/z = 597 corresponding to intermediate III (Scheme 3) and a peak at m/z = 221 for 3,5-DTBQ (Fig. S14†) were found. In comparison (Table S10†) with those reported in the literature,18b,c,21b it may be stated that the as-prepared complexes are highly efficient catalysts.
Furthermore, we explored the catalytic activity of the complexes by extending the substrate scope beyond 3,5-DTBC. We selected other catechol derivatives, including 3-methoxycatechol, 3-methyl catechol and pyrocatechol, and studied the reaction pathways and corresponding kinetic studies. In each case, the reaction proceeded via catechol binding to the metal core in the semiquinone state, as evident from mass spectral and EPR studies. As a representative case, the data for complex 3 is presented here. The catechol-substrate adduct formation was confirmed by ESI-MS analysis (Fig. S21–S23†). The semiquionone nature of the intermediates was evident from the absorption bands in the near IR region at ∼860 nm (Fig. S17–S19†) and the X-band EPR measurements (Fig. S20†). This finding unambiguously proves that the catalytic cycle of the catechol-like systems with the vanadium complexes in this study follows a semiquinone radical pathway. 3-Methoxycatechol: k2 = 20 min−1, KM = 1.2 × 10−3 and Vmax = 4 × 10−3 M min−1 (Fig. S17†); 3-methylcatechol: k2 = 9.5 min−1, KM = 8 × 10−4 and Vmax = 1.9 × 10−3 M min−1 (Fig. S18†); pyrocatechol: k2 = 6.5 min−1, KM = 3.6 × 10−4 and Vmax = 1.3 × 10−3 M min−1 (Fig. S19†). From the experimental results, it can be said that the rate of catechol-quinone oxidation follows the below trend: 3,5-DTBC > 3-methoxycatechol > 3-methylcatechol > pyrocatechol.
Experimental section
Materials and physical measurements
All analytically pure solvents were purchased from commercial sources and used without further purification. VO(acac)2 was prepared according to the literature.17a All the reactions with metal salts were carried out in an open-air atmosphere. Elemental analyses (C, H, and N) were carried out on a Perkin–Elmer 2400 series II analyser. IR spectra (400–4000 cm−1) were obtained using a Perkin–Elmer L-0100 spectrophotometer. Absorption spectra were measured on a PerkinElmer Lambda 25 and a PerkinElmer Lambda 750 spectrophotometer. The 1H NMR spectra were obtained using a Bruker FT 300 MHz spectrometer. High-resolution mass spectra data were collected by employing ESI in the positive ionization mode in a Waters Q-TOF micromass spectrometer. ESI mass spectra were recorded on a Shimadzu LCMS2020 mass spectrometer and a Micromass Q-Tof YA 263 mass spectrometer. On the electroanalytical instrument BASi Epsilon-EC, cyclic voltammetry studies were carried out in CH2Cl2 solutions containing 0.2 M tetrabutylammonium hexafluorophosphate as the supporting electrolyte. A BASi platinum working electrode, a platinum auxiliary electrode, and an Ag/AgCl reference electrode were used in the experiments. The ferrocenium/ferrocene (Fc+/Fc) couple was used as the reference for the redox potential data. The X-band EPR spectra were collected using a Magnettech GmbH MiniScope MS400 spectrometer (fitted with a TC H03 temperature controller), and the microwave frequency was measured with an FC400 frequency counter. The EPR spectra were simulated using the Easy Spin software.Computational details
The non-local correlation functional of Lee–Yang–Parr (B3LYP)29 was used in the DFT30 calculations to optimise the geometrical structures without any symmetry restrictions. The vanadium core electron (1s22s22p6) was described using the calculation approach31 based on the effective core potential (ECP) approximation of Hay and Wadt, while the valence shell was described32 using the corresponding “double-”quality basis set LANL2DZ. We used the 6-31+G basis set for H atoms and the 6-311+G basis set for the non-hydrogen atoms C, N, and O. The Gaussian 09W software package33 and Gauss View 5.1 software were used for all calculations.Crystallographic studies
By slowly evaporating the corresponding solution systems, the complexes were reduced to single crystals suitable for X-ray crystallographic investigation. Using a Bruker AXS SMART APEX CCD diffractometer with a Mo target rotating-anode X-ray source and a graphite monochromator (Mo Kα, = 0.71073) operating at 293 K, the X-ray intensity measurements were recorded. The data were reduced in SAINTPLUS, and the SADABS software was used to apply empirical absorption correction.34 The structure was improved using the full matrix least-square technique on F2. Anisotropic refinement was performed on all non-hydrogen atoms with the exception of some disordered carbon atoms. The Refinements were performed by SHELX 2016 programs using OLEX2 platform.35c The ORTEP and Mercury software were used to create the molecular structure plots.36 Elaborate discussion on the bond parameters of ligand H2L2 and complex 3 is excluded due to the lack of precise data. In both cases, the data were cut at 2 theta = 44 degrees to get optimum results.Constant-potential electrolysis
CPE was carried out at a potential chosen based on the experimental cyclic voltammograms of the reduction reactions of complexes 5 and 6 in order to use the monooxidobridged divanadium VV/VV complexes as precursors to obtain mixed-valence VIV/VV species. The coulometric method was employed in accordance with the redox potential range of the complexes. After that, the experiment was stopped so that the electrolyzed solution could be frozen for later research. To obtain a solid residue, a second sample of the same solution was rapidly evaporated. The frozen solution and solid residue were then used for the EPR experiment.Synthesis of ligands
Schiff base ligands (H2L1–2) were prepared by the condensation of equimolar amounts of valproic acid hydrazide (10 mmol) and salicylaldehyde (H2L1) or 2-hydroxy naphthaldehyde (H2L2) in methanol.H2L1: Anal. calc. for C15H22N2O2: C 68.67%; H 8.45%; N 10.68%; found: C 68.59%; H, 8.38%; N, 10.60%. 1H NMR {300 MHz, CDCl3, δ (ppm), J (Hz)}: 10.86 (NH, s), 9.01 (OH, s), 8.06 (–CHN, s) 7.36–6.94 (4H, ArH), 2.21–2.20 (1H, m, –CH). 1.79–1.33 (8H, m, 4CH2), 0.95–0.92 (6H, m, 2CH3). IR (KBr, νmax/cm−1): 3061(O–H); 2919(N–H); 1659(CO), 1618(CN). 13C NMR (75 MHz, CDCl3) δ 178.53, 157.70, 147.34, 131.77, 131.12, 119.82, 117.46, 116.84, 41.10, 34.42, 20.71, 14.15.
H2L2: Anal. calc. for C19H24N2O2: C 73.05%; H 7.74%; N 8.97%; found: C 72.92%; H, 7.69%; N, 8.91%.1H NMR {300 MHz, CDCl3, δ (ppm), J (Hz)}: 11.52 (NH, s), 9.49 (OH, s), 8.91 (–CHN, s) 8.03–7.23 (6H, ArH), 2.22–2.19 (1H, m, –CH). 1.66–1.28 (8H, m, 4CH2), 0.98–0.86 (6H, m, 2CH3). IR (KBr, νmax/cm−1): 3190 (O–H); 2940 (N–H); 1725 (CO); 1646 (CN).4b 13C NMR (101 MHz, CDCl3) δ 172.21, 158.02, 147.42, 143.37, 133.16, 129.24, 127.74, 123.77, 123.44, 119.80, 118.83, 107.91, 41.35, 34.43, 20.73, 14.16.
Synthesis of complexes
[VVO2(HL1)] (1). Yield: 76%. Anal. calc. for C15H21VN2O4: C 52.33%; H 6.15%; N 8.14%. Found: C 52.28%; H 6.02%; N 8.06%.1H NMR {300 MHz, CDCl3, δ (ppm)}, 11.605 (1H, s, –NH), 8.717 (1H, s, –N
CH), 7.708–6.799 (4H, m, Ar–H), 2.066–2.048 (1H, m, –CH), 1.543–1.249 (8H, m, 4CH2), 0.843 (6H, m, 2CH3). IR (KBr, νmax/cm−1): 2980 (N–H); 1612 (CO); 1542 (CN), 1215 (C–O)enolic; 955 (VO); 748 (V–O).13C NMR (75 MHz, DMSO) δ 153.33, 152.07, 134.47, 133.40, 120.79, 119.00, 118.79, 116.23, 41.96, 35.26, 20.62, 14.37.
[VVO2(HL2)] (2). Yield: 72%. Anal. calc. for C19H23VN2O4: C 57.87%; H 5.88%; N 7.10%; found: C 57.77%; H, 5.72%; N, 7.03%.1H NMR {300 MHz, DMSO-D6, δ (ppm)}, 9.635 (1H, s, –NH), 8.525 (1H, s, –N
CH), 8.278–7.073 (6H, m, Ar–H), 2.309–2.284 (1H, m, –CH), 1.603–1.287 (8H, m, 4CH2), 0.904–0.882 (6H, m, 2CH3). IR (KBr, νmax/cm−1): 2966 (N–H); 1607 (CO); 1548 (CN), 1291 (C–O)enolic; 964 (VO); 753 (V–O).13C NMR (101 MHz, DMSO-D6) δ 158.32, 135.87, 133.21, 129.51, 128.66, 128.21, 124.88, 124.04, 122.45, 121.29, 120.76, 119.14, 42.07, 35.17, 20.68, 14.50.
[VVO(L1)(OMe)(MeOH)] (3). Yield: 68%. Anal. calc. for C17H27VN2O5: C 52.31%; H 6.97%; N 7.18%. Found: C 52.25%; H 6.89%; N 7.09%.1H NMR {300 MHz, CDCl3, δ (ppm)}, 8.900 (1H, s, –N
CH), 7.712–7.128 (4H, m, Ar–H), 2.586–2.238 (1H, m, –CH), 1.606–1.171 (8H, m, 4CH2), 0.907–0.863 (6H, m, 2CH3). IR (KBr, νmax/cm−1): 2402 (broad band of the bound MeOH moiety), 1600 (imine CN), 1304 (C–O)enolic, 950 (VO), 766 (V–O).13C NMR (101 MHz, CDCl3) δ 164.53, 154.20, 135.44, 135.36, 132.29, 122.14, 119.77, 116.94, 41.68, 35.14, 20.91, 14.12.
[VVO(L2)(OMe)(MeOH)] (4). Yield: 70%. Anal. calc. for C21H29VN2O5: C 57.27%; H 6.64%; N 6.36%; found: C 57.19%; H, 6.52%; N, 6.24%.1H NMR {300 MHz, DMSO-D6, δ (ppm)}, 9.78 (1H, s, –N
CH), 8.29–7.44 (6H, m, Ar–H), 2.66–2.52 (1H, m, –CH), 1.68–1.14 (8H, m, 4CH2), 0.93–0.78 (6H, m, 2CH3). IR (KBr, νmax/cm−1): 1596 (imine CN), 1215 (C–O)enolic, 2595 (broad from bound MeOH moiety), 926 (VO), 750 (V–O).13C NMR (101 MHz, CDCl3) δ 166.10, 150.31, 136.39, 136.32, 131.91, 129.39, 129.34, 128.89, 125.20, 121.21, 118.60, 111.96, 41.51, 35.21, 20.75, 13.93.
[VV2O3(L1)2] (5). Yield: 62%. Anal. calc. for C30H40V2N4O7: C 53.74%; H 6.01%; N 8.36%. Found: C 53.63%; H 5.89%; N 8.29%. 1H NMR {300 MHz, CDCl3, δ (ppm)}, 8.810 (1H, s, –N
CH), 7.657–6.973 (4H, m, Ar–H), 2.300–2.266 (1H, m, –CH), 1.484–1.101 (8H, m, 4CH2), 0.995–0.767 (6H, m, 2CH3). IR (KBr, νmax/cm−1): 1595 (imine CN), 1275 (C–O)enolic, 986 (VO), 779 (V–O).13C NMR (75 MHz, CDCl3) δ 161.63, 154.22, 137.02, 135.28, 132.21, 122.11, 119.86, 116.81, 41.51, 35.02, 20.78, 14.02.
[VV2O3(L1)2] (6). Yield: 60%. Anal. calc. for C38H44V2N4O7: C 59.22%; H 5.75%; N 7.27%; found: C 59.08%; H, 5.61%; N, 7.18%.1H NMR {300 MHz, DMSO-D6, δ (ppm)}, 9.70 (1H, s, –N
CH), 8.24–7.31 (6H, m, Ar–H), 2.39–2.31 (1H, m, –CH), 1.58–1.04 (8H, m, 4CH2), 0.99–0.79 (6H, m, 2CH3). IR (KBr, νmax/cm−1): 1590 (imine CN), 1261 (C–O)enolic, 966 (VO), 770 (V–O).13C NMR (101 MHz, CDCl3) δ 161.36, 150.37, 139.73, 136.36, 132.13, 129.39, 129.34, 128.91, 125.21, 121.24, 118.58, 111.96, 41.50, 34.81, 20.60, 14.11.
Conclusion
This study presents an innovative approach to synthesizing distinct vanadium complexes and elucidating their redox behaviour and catalytic activity. By carefully tuning the solvent and employing a flexible ligand pocket (H2L1–2), three unique sets of vanadium(V) complexes were successfully produced. The redox behaviours of all the complexes were explored by combining experimental and theoretical investigations. The identification of type II mixed-valence complexes was particularly notable and was supported by frozen-solution EPR data and electronic spin density calculations, which provided compelling evidence of the nature of the electron delocalization within these complexes. The catalytic activity of these complexes was evaluated based on their ability to facilitate the oxidation of pyrogallol to purpurogallin, as well as the conversion of catechol to quinone. During the transformation of pyrogallol to purpurogallin, in situ generated H2O2 formed the key peroxo intermediate, which was essential for the progress of the reaction. In contrast, the dioxido complexes were found to show catecholase activity only in the presence of external H2O2, which has not been reported so far. Pyrogallol thus appears to be a better H2O2 generator than catechol. Moreover, it is worth highlighting that the oxidation of catechol occurs through the semiquinone radical pathway, which is quite rare among vanadium model systems with catecholase activity.Data availability
The authors hereby confirm that the data of the manuscript or ESI† file will be available upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. P. expresses gratitude to the Council of Scientific and Industrial Research for fellowship grant. D. S. thanks Department of Chemistry at Jadavpur University and Vijaygarh Jyotish Ray College for infrastructural support. S. M. thanks Department of Chemistry, Darjeeling Govt. College, Darjeeling, India for support. K. K. R. acknowledges infrastructure and assistance provided by the Department of Chemistry at Jadavpur University and the CSIR, New Delhi under project No. 01(3066)/21/EMR-II. We sincerely appreciate the assistance provided by Dr Prasanta Ghosh, Department of Chemistry, R. K. Mission Residential College, Narendrapur, Kolkata 103, West Bengal, India.References
- M. K. Mishra, S. Kukal, P. R. Paul, S. Bora, A. Singh, S. Kukreti, L. Saso, K. Muthusamy, Y. Hasija and R. Kukreti, Molecules, 2022, 27, 104 CrossRef CAS PubMed.
- A. Safdar and F. Ismail, Saudi Pharm. J., 2022, 31, 265–278 CrossRef PubMed.
- L. Dong, L. Fang, X. Dai, J. Zhang, J. Wang and P. Xu, Drug Dev. Res., 2022, 83, 131–141 CrossRef CAS PubMed.
- (a) R. Patra and K. K. Rajak, ChemistrySelect, 2020, 5(30), 9477–9485 CrossRef CAS; (b) A. El-Faham, M. Farooq, S. N. Khattab, A. M. Elkayal, M. F. Ibrahim, N. Abutaha, M. A. Wadaan and E. A. Hamed, Chem. Pharm. Bull., 2014, 62, 591–599 CrossRef CAS PubMed.
- X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981 RSC.
- Ł. Popiołek, K. Tuszyńska and A. Biernasiuk, Biomed. Pharmacother., 2022, 153, 113302 Search PubMed.
- (a) J. Szklarzewicz, A. Jurowska, D. Matoga, K. Kruczała, G. Kazek, B. Mordyl, J. Sapa and M. Papież, Polyhedron, 2020, 185, 114589 Search PubMed; (b) P. Noblía, E. J. Baran, L. Otero, P. Draper, H. Cerecetto, M. González, O. E. Piro, E. E. Castellano, T. Inohara, Y. Adachi, H. Sakurai and D. Gambino, Eur. J. Inorg. Chem., 2004, 2004(2), 322–328 CrossRef; (c) M. Shit, S. Bera, S. Maity, T. Weyhermüller and P. Ghosh, New J. Chem., 2017, 41, 4564–4572 RSC.
- A. Levina, A. Pires Vieira, A. Wijetunga, R. Kaur, J. T. Koehn, D. C. Crans and P. A. Lay, Angew. Chem., Int. Ed., 2020, 59, 15834–15838 CrossRef CAS PubMed.
- J. C. Pessoa, S. Etcheverry and D. Gambino, Coord. Chem. Rev., 2015, 301–302, 24–48 CrossRef CAS PubMed.
- A. Butler and J. V. Walker, Chem. Rev., 1993, 93, 1937–1944 CrossRef CAS.
- Y. Hoshino and H. Yamamoto, J. Am. Chem. Soc., 2000, 122, 10452–10453 CrossRef CAS.
- (a) M. Sako, S. Takizawa and H. Sasai, Tetrahedron, 2020, 76, 131645 CrossRef CAS; (b) S. E. Griffin and L. L. Schafer, Inorg. Chem., 2020, 59, 5256–5260 CrossRef CAS PubMed.
- (a) A. Mondal, S. Sarkar, D. Chopra, T. N. Guru Row, K. Pramanik and K. K. Rajak, Inorg. Chem., 2005, 44, 703–708 Search PubMed; (b) S. P. Dash, S. Roy, M. Mohanty, M. F. N. N. Carvalho, M. L. Kuznetsov, J. C. Pessoa and R. Dinda, Inorg. Chem., 2016, 55(17), 8407–8421 CrossRef CAS PubMed; (c) S. Mondal, P. Ghosh and A. Chakravorty, Inorg. Chem., 1997, 36(1), 59–63 CrossRef CAS.
- A. Albino, S. Benci, L. Tesi, M. Atzori, R. Torre, S. Sanvito, R. Sessoli and A. Lunghi, Inorg. Chem., 2019, 58(15), 10260–10268 CrossRef CAS PubMed.
- S. K. Dutta, S. B. Kumar, S. Bhattacharyya, E. R. T. Tiekink and M. Chaudhury, Inorg. Chem., 1997, 36, 4954–4960 Search PubMed.
- (a) S. K. Dutta, S. Samanta, S. B. Kumar, O. H. Han, P. Burckel, A. A. Pinkerton and M. Chaudhury, Inorg. Chem., 1999, 38, 1982–1988 CrossRef CAS PubMed; (b) R. Dinda, P. Sengupta, S. Ghosh and T. C. W. Mak, Inorg. Chem., 2002, 41(6), 1684–1688 Search PubMed.
- (a) R. Patra, S. Mondal, D. Sinha and K. K. Rajak, ACS Omega, 2022, 7, 11710–11721 Search PubMed; (b) M. Mahroof-Tahir, A. D. Keramidas, R. B. Goldfarb, O. P. Anderson, M. M. Miller and D. C. Crans, Inorg. Chem., 1997, 36(8), 1657–1668 CrossRef CAS PubMed.
- (a) C. Balakrishnan, M. Theetharappan, P. Kowsalya, S. Natarajan, M. A. Neelakantan and S. S. Mariappan, Appl. Organomet. Chem., 2017, 31(11), e3776 CrossRef; (b) S. K. Mal, M. Mitra, H. R. Yadav, C. S. Purohit, A. Roy Choudhury and R. Ghosh, Polyhedron, 2016, 111, 118–122 CrossRef CAS; (c) S. Ta, M. Ghosh, K. Ghosh, P. Brandao, V. Felix, S. K. Hira, P. P. Manna and D. Das, ACS Appl. Bio Mater., 2019, 2, 2802–2811 CrossRef CAS PubMed; (d) S. P. Rath, K. K. Rajak and A. Chakravorty, Inorg. Chem., 1999, 38, 4376–4377 Search PubMed.
- (a) A. Guha, T. Chattopadhyay, N. D. Paul, M. Mukherjee, S. Goswami, T. K. Mondal and D. Das, Inorg. Chem., 2012, 51(16), 8750–8759 Search PubMed; (b) L. I. Simándi, T. M. Simándi, Z. May and G. Besenyei, Coord. Chem. Rev., 2003, 245, 85–93 Search PubMed; (c) F. Wendt, C. Näther and F. Tuczek, JBIC, J. Biol. Inorg. Chem., 2016, 21, 777–792 Search PubMed.
- M. R. Maurya, B. Sarkar, F. Avecilla and I. Correia, Eur. J. Inorg. Chem., 2016, 2016, 4028–4044 CrossRef CAS.
- (a) M. R. Maurya, N. Chaudhary, F. Avecilla, P. Adão and J. C. Pessoa, Dalton Trans., 2015, 44(3), 1211–1232 Search PubMed; (b) M. R. Maurya, B. Uprety, F. Avecilla, P. Adão and J. C. Pessoa, Dalton Trans., 2015, 44, 17736–17755 RSC.
- (a) S. A. Patra, M. Mohanty, A. Banerjee, A. S. Kesarwani, F. Henkel, H. Reuter and R. Dinda, J. Inorg. Biochem., 2021, 224, 111582 Search PubMed; (b) S. P. Dash, S. Majumder, A. Banerjee, M. F. N. N. Carvalho, P. Adão, J. C. Pessoa, K. Brzezinski, E. Garribba, H. Reuter and R. Dinda, Inorg. Chem., 2016, 55(3), 1165–1182 Search PubMed.
- J. A. L. da Silva, J. J. R. F. da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2011, 255(19–20), 2232–2248 CrossRef CAS.
- (a) C. R. Waidmann, A. G. DiPasquale and J. M. Mayer, Inorg. Chem., 2010, 49(5), 2383–2391 CrossRef CAS PubMed; (b) R. R. Langeslay, D. M. Kaphan, C. L. Marshall, P. C. Stair, A. P. Sattelberger and M. Delferro, Chem. Rev., 2019, 119, 2128–2191 CrossRef CAS PubMed.
- (a) S. M. Siegel and B. Z. Siegel, Nature, 1958, 181(4616), 1153–1154 Search PubMed; (b) R. Gao, Z. Yuan, Z. Zhao and X. Gao, Bioelectrochem. Bioenerg., 1998, 45(1), 41–45 CrossRef CAS.
- B. Baruah, S. Das and A. Chakravorty, Inorg. Chem., 2002, 41, 4502–4508 CrossRef CAS PubMed.
- C. R. Cornman, G. J. Colpas, J. D. Hoeschele, J. Kampf and V. L. Pecoraro, J. Am. Chem. Soc., 1992, 114, 9925–9933 Search PubMed.
- Y. H. Miura, I. Tomita, T. Watanabe, T. Hirayama and S. Fukui, Biol. Pharm. Bull., 1998, 21(2), 93–96 CrossRef CAS PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98(7), 5648–5652 Search PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 37(2), 785–789 CrossRef CAS PubMed.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52(12), 997–1000 CrossRef CAS.
- (a) M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108(11), 4439–4449 CrossRef CAS; (b) R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109(19), 8218–8224 Search PubMed; (c) R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256(4–5), 454–464 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82(1), 270–283 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng and J. L. Sonnenberg, Gaussian 09, Revision A. 1, Gaussian, 2009 Search PubMed.
- SMART, SAINT, SADABS, XPREP, SHELXTL, Bruker AXS Inc., Madison WI, 1998 Search PubMed.
- (a) G. M. Sheldrick, The SHELX-97 Manual. SHELXL-97, Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71(1), 3–8 Search PubMed; (c) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Cryst., 2009, 42, 339–341 Search PubMed.
- (a) C. K. Johnson, ORTEP Report ORNL-5138, Oak Ridge National Laboratory, Oak Ridge TN, 1976 Search PubMed; (b) C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2410680 (H2L1), 2267633 (H2L2), 2410679 (1) and 2410682 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02030h |
| This journal is © The Royal Society of Chemistry 2025 |