
Pd@BTL–Cd core–shell nanoparticles as plasmonic photocatalysts for the reductive amination of furfural in water†
Jyoti
Rohilla
 a,
Sahil
Thakur
a,
Sahil
Sharma
b,
Raghubir
Singh
*b and
Varinder
Kaur
*a
a,
Sahil
Thakur
a,
Sahil
Sharma
b,
Raghubir
Singh
*b and
Varinder
Kaur
*a
aDepartment of Chemistry, Panjab University, Sector 14, Chandigarh-160014, India. E-mail: var_ka04@yahoo.co.in; var_ka04@pu.ac.in
bDepartment of Chemistry, DAV College, Sector 10, Chandigarh-160011, India. E-mail: raghu_chem2006@yahoo.com; raghubirsingh@davchd.ac.in
First published on 20th January 2025
Abstract
This work reports the step-wise fabrication of a core–shell plasmonic nanocomposite Pd@BTL–Cd consisting of a BTL–Cd shell and a palladium nanoparticle core. BTL–Cd is the [Cd(BTL)·CdCl2] complex where the heptadentate framework of the bis-compartmental ligand encapsulated two Cd(II) centres in separate pockets. Pd@BTL–Cd has been found to be highly efficient for the photocatalytic conversion of furfural (a biomass-derived aldehyde) to furfuryl amine via reductive amination in aqueous ammonia at room temperature. The improved photocatalytic performance of the nanocomposite and its functioning in visible regions in contrast to parental species are attributed to the synergistic functioning of the core and the shell. The inclusion of the Cd–BTL nanoshell lowers the overall band gap of the material while the Pd nanocore generates in situ hydrogen species during photocatalysis. The optimization of catalytic conditions revealed that 10 mg of the fabricated photocatalyst can offer 99% conversion and a high turnover number in 4 h. The efficacy of the catalyst can be retained for up to 5 cycles with high selectivity for the formation of furfuryl amine (98%) in the presence of visible light (λ = 445 nm). Pd@BTL–Cd is also catalytically effective for the reductive amination of other aldehydes.
1. Introduction
Plasmonic nanoparticles have been proven as excellent materials for generating electromagnetic ‘hot spots’ and catalyzing numerous organic reactions in the presence of visible light.1 However, plasmonics is limited to noble metals having sufficient nanoscale roughness and therefore its utility is narrowed to confined application horizons.2 Palladium nanoparticles have received considerable interest as plasmonic photocatalysts as they hinder the recombination of electron–hole pairs by serving as charge acceptors.3 Some important examples include the use of plasmone-based hybrid materials such as Pd@B–BO3 and TiO2@Au–Pd for the photocatalytic Suzuki coupling reaction4 and hydrogen production: g-C3N4/Cu2O–Pd heterojunctions for photocatalytic conversion of CO2 and H2O to CO and H2O2, respectively,5 and Au and Pd nanoparticle co-deposited Bi2NbO5F for photocatalytic conversion of CH4 to C2H4.6 The amalgamation of Pd nanoparticles in the Cd-MOF works as an antenna to amplify light absorption across the visible region and catalyze photo-oxidation of benzylamine.7 However, the irregularly shaped and inappropriate-sized cavities of the MOFs usually prevent the entry of nanoparticles inside the channels and lower their catalytic efficiency. Therefore, step-by-step construction of a layer around the nanoparticles to harvest more and more light for improved photocatalysis is a better solution to these problems.In the case of Pd nanoparticles, the construction of a core–shell nano-architecture improves the photocatalytic activity by accelerating the movement of charge and suppressing the charge combination. This type of phenomenon is seen in Pd@CdS where Pd nanoparticles generate a Schottky barrier and enhance charge separation.8 Moreover, cadmium-based photocatalysts also possess suitable band gaps for visible light absorption.9 The main limitations of CdS-based catalysts include their reduced photocatalytic activity and insufficient photostability due to significant photo-corrosion and rapid recombination of charge carriers.10 These are also inappropriate for usage at large scales in industrial applications due to acute toxicity and pollution caused due to their solubility in the aqueous medium. The encapsulation of Cd2+ ions in an organic framework is expected to generate a hydrophobic protective layer to make it water-insoluble. Therefore, cadmium complexes are the best replacements for CdS shells to avoid their persistence in water bodies.11
Herein, a bis-compartmental tripodal ligand (BTL) with two separate encapsulating pockets (inner and outer) has been used to form cadmium azametallatrane (BTL–Cd) through the inner pocket with an additional protective covering formed by the outer pocket. Herein, both the pockets were occupied by Cd2+ with CdCl2 in the outer pocket which acted like claws for binding Pd nanoparticles. The interaction through Cl constructed a uniform shell around the Pd core producing Pd@BTL–Cd core–shell nanoarchitectures. This heterostructure exhibited enhanced photocatalytic performance in the visible region due to the localized surface plasmon resonance (LSPR) and simultaneously generated in situ hydrogen species on the palladium core. The inclusion of the Cd complex nano-shell lowered the overall band gap of the material, thus enhancing its photocatalytic capabilities for the conversion of biomass to valuable products.
Biomass conversion is gaining momentum for the production of bio-based chemicals like furfural (FAL), 2-methylfuran,12 alkyl levulinates,13 and furan dicarboxylic acid (FDCA) for prioritizing shifting from petrochemicals to bio-refineries.14 In the current work, Pd@BTL–Cd catalyzes the reductive amination of furfural to furfuryl amine in the presence of visible light. Furfuryl amine plays a vital role in pharmaceuticals, agrochemicals, resins, and pesticides.15 Earlier, Pd-based materials like Pd/SiO216 and Pd/HZSM-53 have been reported for catalyzing the formation of furfuryl amine but they rely on the external use of hydrogen gas and a high reaction temperature. In contrast, in situ generated hydrogen species are considered more safe and reliable than the externally used H2 gas.17 The use of formic acid,18 ethanol,19 and water17 is already reported for the in situ generation of hydrogen species. However, the furfural amination reaction utilizing photocatalysis in the absence of hydrogen gas remains largely unexplored. The current work reports the formation of furfuryl amine from furfural in the presence of the core–shell nanostructured photocatalyst Pd@BTL–Cd using water for the in situ hydrogen generation. Moreover, an atomistic understanding of the mechanism is also demonstrated to understand the internal activity of the as-synthesized photocatalyst.
2. Experimental
2.1. Materials
All experiments were carried out using standard apparatus and commercially available chemicals. Schlenk technique under dry nitrogen was used for the synthesis of the BTL ligand. All the solvents used were dried and stored under nitrogen. Tris-2-aminoethylamine, 2-hydroxybenzaldehyde, sodium methoxide and sodium borohydride (SRL), palladium(II) chloride (Merck), cadmium(II) chloride (CDH), dichloromethane (Advent), hexane (Advent), magnesium(II) sulphate (Qualigens), furfural (Sigma Aldrich), aqueous ammonia (SRL), and furfuryl amine (SRL) were commercially purchased and used without any further purification.2.2. Physical measurements
Powder X-ray diffraction (PXRD) measurements were done using a PANalytical X'PertPRO X-ray diffractometer with Ni-filtered Cu Kα radiation (λ = 1.54059 Å) in the 2θ range from 5° to 80° with a slow scan rate at room temperature. FT-IR spectra were recorded on a Thermo Scientific Nicolet IS50 FT-IR spectrometer in solid state. Thermogravimetric (TGA) analyses were done using an SDT Q600 (V20.9 Build 20) instrument (Artisan Technology Group, Champaign, IL), wherein the samples were heated from 25 °C to 800 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere with a flow rate of 20 mL min−1. The morphology of the samples was analysed using a scanning electron microscope JEOL (JAPAN) JSM 6100 and a transmission electron microscope (TEM) JEOL JEM 2100 Plus. Energy dispersive X-ray spectra (EDAX) and elemental mapping data were also acquired using the same TEM instrument. For electron microscopy measurements, 1 mg of the sample was taken in a vial along with 1 mL of ethanol. After sonication for 1 h, the suspension was drop cast on a piece of Si wafer (for SEM measurements) and a carbon-coated Cu grid (for TEM measurements). These were allowed to dry for 10 h at room temperature before mounting onto the respective sample holders. Subsequently, the SEM and TEM measurements were performed under vacuum conditions. Mass spectra were recorded with a Maldi-TOF Synapt XS HD Mass Spectrometer. Solution NMR spectra were recorded at 25 °C on a Bruker Avance II FT NMR (AL 500 MHz) (1H, 13C). Chemical shifts are reported in parts per million relative to tetramethyl silane (TMS) for 1H and 13C NMR. A Rigaku XTA Lab SuperNova, single source (Mo Kα, λ = 0.71073) at offset/far, HyPix3000 diffractometer was used to collect crystallographic data. The crystal was kept at 293(2) K during data collection. The structure was solved using Olex2 with the ShelXT structure solution program using intrinsic phasing and refined with the ShelXL refinement package using least squares minimization. X-ray photoelectron spectroscopy (XPS) measurements were performed on a Thermo Scientific NEXSA photoemission spectrometer using Al-Kα (1486.6 eV) X-ray radiation. XPS data were acquired with a spot size of 400 μm having a standard lens mode. The obtained data from the instrument were plotted and deconvoluted using Avantage software. The binding energies were calibrated using the C 1s level (284.6 eV) as the internal standard reference. The Brunauer–Emmett–Teller (BET) specific surface area and nitrogen adsorption–desorption isotherms of the samples were obtained on a Quantachrome ASIQwin gas adsorption analyzer (version 5.21) system at 77 K temperature. Electronic spectral measurements were carried out using an EI 2375 double-beam spectrophotometer in the range of 200–800 nm. Electrical measurements were done by using Metrohm Autolab s.v. cyclic voltammetry potentiostat. A FLASH-2000 organic elemental analyzer was used for C, H, and N elemental microanalyses. Density-Functional Theory (DFT) computations were carried out with Gaussian 03, using the B3LYP hybrid method. The fluorescence measurements were carried out on a PerkinElmer LS-55 fluorescence spectrophotometer with a slit width of 6 nm, whose set range of emission intensity was 0 to 1000 (a.u.). The catalytic product (Furfuryl Amine) was analysed using a GC-MS system (Agilent 7890 GC 7000 MS) equipped with a non-polar capillary column (Agilent DB-5), temperature-programmed desorption (TPD) measurements were conducted using the ChemBET TPR/TPD instrument manufactured by Quantachrome, USA, equipped with a TCD detector measurements were conducted using the ICP-MS Agilent 7850 instrument, HORIBA Jobin Youn iHR 550 spectrographs equipped with a grating of 1800 lines per mm and a Peltier cooled CCD detector.2.3. Synthesis of the bis-compartmental tripodal ligand (BTL)
The BTL was synthesized by a slight modification of a previously reported method.20 In brief, tris-2-aminoethylamine (2.00 ml, 13.30 mmol) and 2-hydroxybenzaldeyde (4.16 ml, 39.9 mmol) were heated to reflux in dry methanol (30 mL) for 24 h to afford a clear yellow solution. Then, sodium borohydride (1.11 g, 39.9 mmol) was added slowly in about 1 h under nitrogen with continuous stirring. The contents were further heated to reflux for 6 h. After the completion of the reaction, the solvent was reduced using a rotary evaporator. 100 ml distilled water was added to the contents and the product was extracted in dichloromethane (3 × 100 ml) using a separating funnel. The extract was dried over anhydrous magnesium sulphate. The solvent from the extract was evaporated which afforded an oily product. The obtained oil was washed with hexane to remove unreacted aldehyde and amine followed by vacuum drying. The thick oil was dissolved in methanol and left to stand at room temperature in a flat-bottomed flask for crystallization. All the attempts to grow the crystals of BTL by evaporating its solution failed. However, the crystals of its hydrochloride salt were obtained from one of the reaction mixture flasks kept for the crystallization of its metal complex. The obtained crystals were suitable for single-crystal X-ray diffraction.
Yield: 85% (oil). M.Pt. 218 °C (crystals). Elemental analysis: calculated for C27H36N4O3: C, 69.80, H, 7.81, N, 12.06, found: C, 69.71, H, 7.65, N, 12.01. FTIR (cm−1): 3292 (broad, O–H), 2824 (N–H), 1589 (C–H), 1440 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1243 (C–N). 1H NMR (500 MHz, solvent CDCl3), δ (ppm) 2.46 (t, 6H1, 3J(1H–1H) = 6 Hz), 2.59 (t, 6H2, 3J(1H–1H) = 6 Hz), 3.86 (s, 6H3), 6.66 (m, 6H5,6), 6.85 (d, 3H7, 3J(1H–1H) = 8 Hz), 7.04 (t, 3H8, 3J(1H–1H) = 8 Hz). 13C NMR (125 MHz, solvent CDCl3), δ (ppm) 46.1 (C2), 52.5 (C3), 54.3 (C1), 116.3 (C8), 119.0 (C6), 122.4 (C4), 128.4 (C7), 128.7 (C5), 158.7 (C9). MS: m/z 465.3 [L + H]+, 527.3 [L + 2[CH3OH] + H]+, 359 [L − C7H7O + 2H]+.
C), 1243 (C–N). 1H NMR (500 MHz, solvent CDCl3), δ (ppm) 2.46 (t, 6H1, 3J(1H–1H) = 6 Hz), 2.59 (t, 6H2, 3J(1H–1H) = 6 Hz), 3.86 (s, 6H3), 6.66 (m, 6H5,6), 6.85 (d, 3H7, 3J(1H–1H) = 8 Hz), 7.04 (t, 3H8, 3J(1H–1H) = 8 Hz). 13C NMR (125 MHz, solvent CDCl3), δ (ppm) 46.1 (C2), 52.5 (C3), 54.3 (C1), 116.3 (C8), 119.0 (C6), 122.4 (C4), 128.4 (C7), 128.7 (C5), 158.7 (C9). MS: m/z 465.3 [L + H]+, 527.3 [L + 2[CH3OH] + H]+, 359 [L − C7H7O + 2H]+.
2.4. Synthesis of the cadmium complex (BTL–Cd)
To a 20 mL ethanolic solution of BTL (0.464 g, 1.00 mmol), sodium methoxide (0.162 g, 3.00 mmol) was added. The resulting solution was heated at reflux for approx. 15 min with continuous stirring to dissolve the contents. To this solution, cadmium chloride (0.366 g, 2.00 mmol) was added and the mixture was heated to reflux for 12 h. Then, the solvent was reduced under vacuum and 100 ml distilled water was added. The product was extracted with dichloromethane (3 × 100 ml) and the extract was dried over anhydrous magnesium sulphate. The magnesium sulphate was filtered and the filtrate was evaporated to afford a solid product. It was washed with hexane and vacuum-dried. The crystals were grown by gas phase diffusion of ether in the DMF solution of the product.
Yield: 81%. M.Pt. 248 °C. elemental analysis: calculated for C27H34Cd2Cl2N4O3: C, 42.77, H, 4.52, N, 7.39, found: C, 42.61, H, 4.45, N, 7.31, FTIR (cm−1): 2844 (N–H), 1255 (C–N), 1594 (C–H), 1478 (CC). Raman (cm−1): 104 (Cd–Cl), 338 (Cd–N), 1024, 1426 (Cd–O). 1H NMR (500 MHz, solvent DMSO), δ (ppm) 2.52 (broad singlet, 6H1,10,19), 2.79 (broad singlet, 3H2,11,20), 2.95 (broad singlet, 3H2,11,20), 3.99 (s, 6H3,12,21), 6.62 (broad singlet, 3H5,14,23), 6.96 (s, 3H8,17,26), 7.10 (s, 6H6,7,15,16,24,25) 8.01 (s, 1 OH). 13C NMR (125 MHz, solvent CDCl3), δ (ppm) 30.7 (C2,11), 35.7 (C20), 51.9 (C3,12,21), 64.8 (C1,10,19), 120.4 (C22), 124.7 (C4,13), 124.7 (C7,16,25), 162.8 (C27), 128.8 (C6,8,15,17,24,26), 130.7 (C9,18), 130.7 (C5,14,23). MS: m/z 577.17 [M − CdCl2 + H]+, 465.29 [L + H]+, 723.02 [M − Cl]+, 725.02 [M − Cl]+, 687.05 [M − 2Cl − H]+, 689.05 [M − 2Cl − H]+.
2.5. Synthesis of core–shell nanoparticles (Pd@BTL–Cd)
The nanoparticles of cadmium complex (BTL–Cd NPs) were synthesized by the bottom-up approach of the reprecipitation method.21 A stock solution of BTL–Cd (2.7 mM) was prepared in DMSO and 0.1 mL of the stock solution was injected into 100 mL of de-ionized water. The injection of the stock solution was accompanied by continuous sonication for 30 minutes to ensure the formation of a stable dispersion of nanoparticles in the aqueous medium. The so-fabricated BTL–Cd NPs were redispersed in 50 mL of de-ionized water and palladium chloride was added followed by the addition of sodium borohydride with vigorous stirring for the reduction of Pd2+ to Pd0.22,23 After the completion of the reduction, the product was centrifuged and washed with ethanol. It was dried in an oven at 60 °C for 3 h to obtain bimetallic core–shell nanoparticles Pd@BTL–Cd. The synthesized material was characterized by using different techniques like FTIR spectroscopy, FESEM, HRTEM, elemental mapping, and XPS. Yield: 78%. FTIR (cm−1): 2826 (N–H), 1255 (C–N stretching), 1593 (C–H aromatic compound), 1455 (CC phenyl ring). Raman (cm−1): 112 (Cd–Cl), 321 (Cd–N), 395 (Pd–Cl), 668 (Pd–O), 1028 and 1430 (Cd–O).
2.6. Photocatalytic reaction
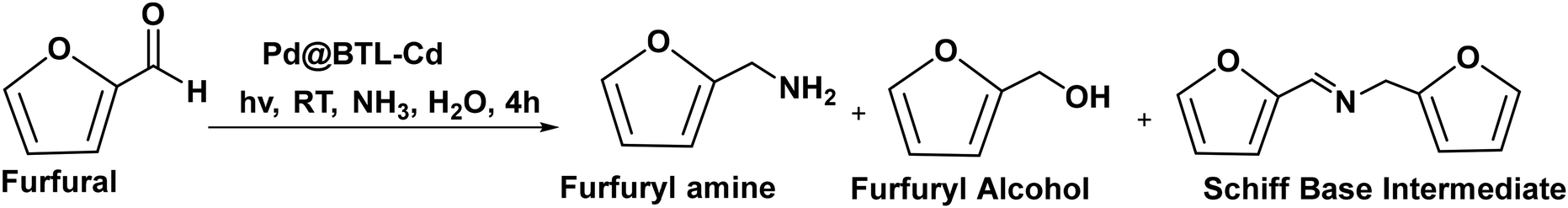
The photocatalytic reaction was carried out as follows: in a 25 mL round bottom flask, furfural (0.48 g, 5.0 mmol), ammonia solution (0.28 g, 8.0 mmol), and Pd@BTL–Cd (10 mg) were mixed with 15 mL distilled water. The reaction mixture was stirred for 4 h under the irradiation of visible light. The progress of the reaction was monitored by thin-layer chromatography. After completion of the reaction, the contents were centrifuged and the product was separated from the catalyst by filtration. The catalyst was washed twice with ethanol and distilled water and dried for the next reaction cycle. The obtained product was analyzed for the formation of furfuryl amine using a GC-MS system by following the procedure as follows. Initially, the oven temperature of the system was kept at 50 °C for 5 minutes. Then, the temperature was ramped at a rate of 10 °C min−1 until it reached 200 °C. The system was held at 200 °C for 5 minutes followed by an increase in temperature to 300 °C at 10 °C min−1 and kept running at this temperature for 20 minutes. The peaks of furfuryl amine, Schiff base intermediate, and furfuryl alcohol were observed in the chromatogram at 15.148, 22.89, and 15.473 min of retention time. The following equations were employed to calculate the selectivity (%) of furfuryl amine and the conversion (%) of furfural.19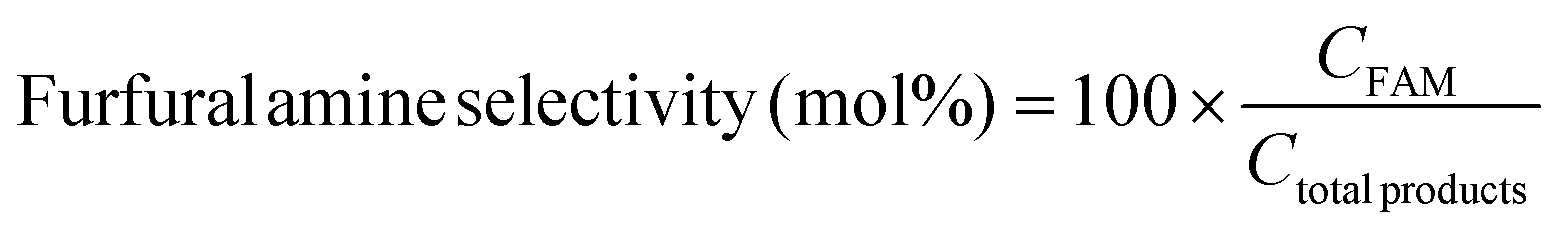
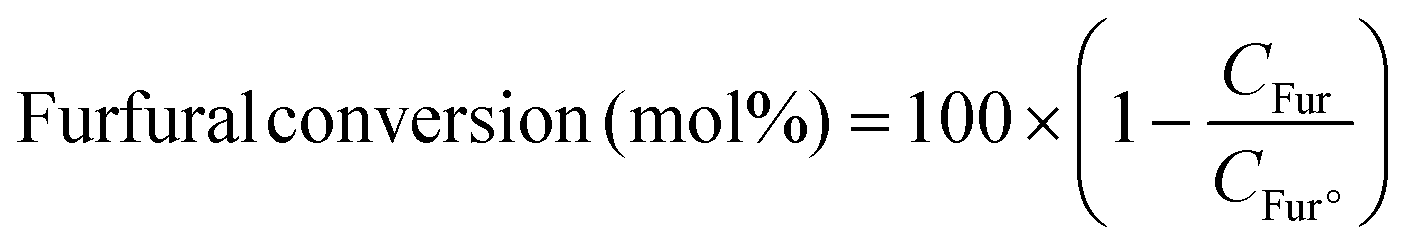
In these equations, CFAM represents the concentrations of furfuryl amine at a specific reaction time (t) and Ctotal products represents the concentration of total products. CFur° denotes the initial concentration of furfural (before the reaction) and CFur denotes the concentration of furfural (after the reaction).
Efficient charge separation is the key factor determining the photocatalytic activity of a semiconducting photocatalyst. Thus, it was essential to determine the HOMO and LUMO energy levels, using the following empirical equations:
| EVB = −[EOxi − E1/2(Ferrocene) + 4.8] eV |
| ECB = EVB + Eg |
3. Results and discussion
3.1. Synthesis and characterization
A bis-compartmental tripodal ligand (BTL) was synthesized by a Schiff base condensation reaction of tris-2-aminoethylamine with 2-hydroxybenzaldehyde followed by in situ hydrogenation by sodium borohydride. Furthermore, the metalation of the synthesized tripodal ligand to obtain BTL–Cd was achieved in ethanol by reacting the tripodal ligand BTL with cadmium chloride dihydrate in the presence of sodium methoxide (Scheme 1). The BTL and BTL–Cd were characterized by FTIR spectroscopy, Raman spectroscopy, 1H-NMR spectroscopy, SCXRD, and ESI-MS.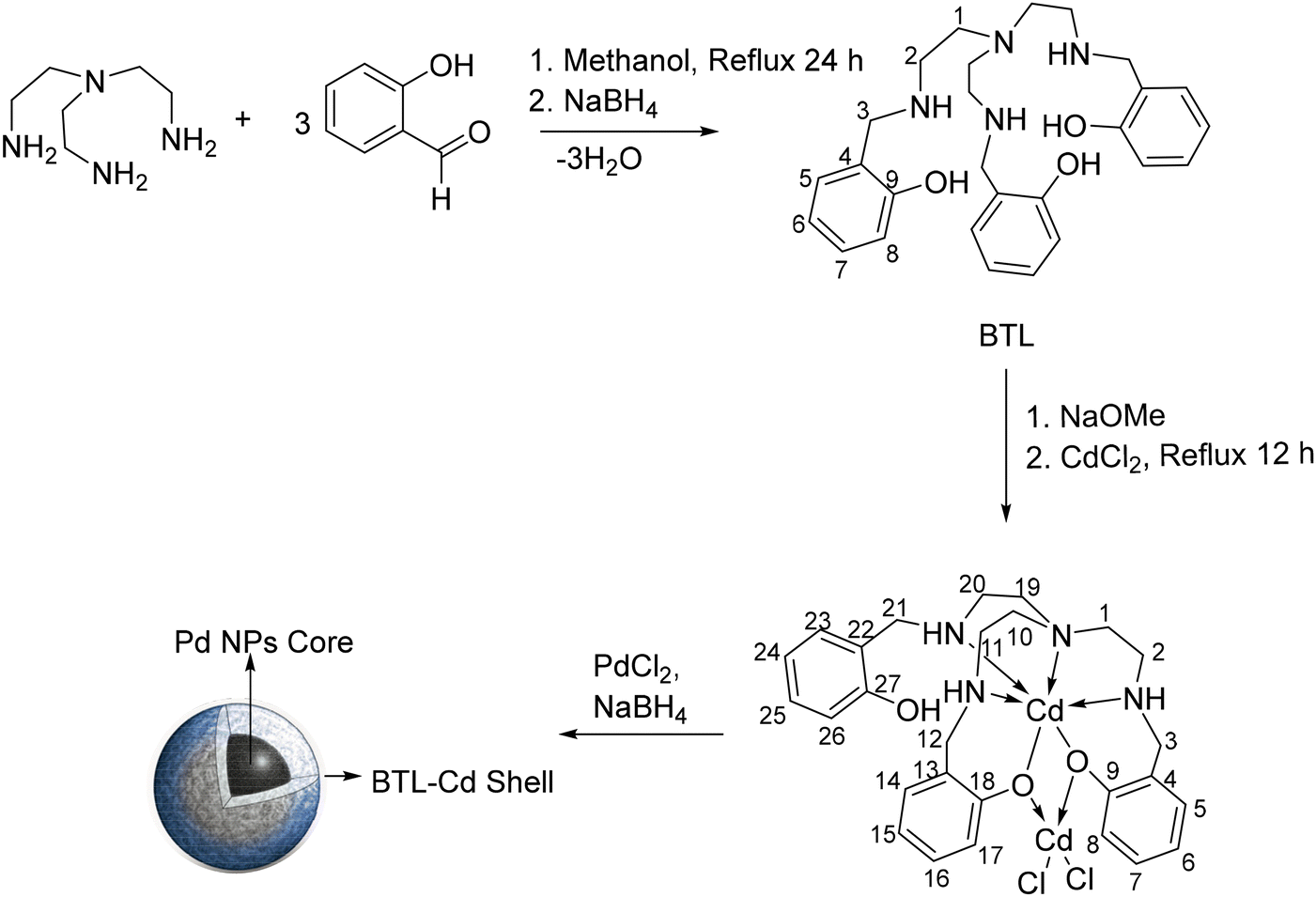 | ||
| Scheme 1 Schematic illustration of the preparation of BTL, BTL–Cd, and Pd@BTL–Cd (the numbering is given to BTL and BTL–Cd structures to assign the NMR signals). | ||
The 1H-NMR spectrum of BTL in CDCl3 exhibited a singlet at 3.85 ppm for three magnetically equivalent protons H3 along with two separate triplets at 2.46 and 2.58 ppm for H1 and H2, respectively (ESI Fig. S1 and S2†). The formation of BTL–Cd was confirmed by the shifting of the H3 signal to 3.99 ppm. The H3, H12, and H21 are expected to show two signals based on the structure of the BTL–Cd complex; however, a single signal was observed for these protons due to magnetic equivalence. Similarly, the signals of aromatic protons of three arms were observed as broad signals revealing their magnetic equivalence even though the coordination of the outer pocket was not uniform. So, it can be inferred that either the Cd sphere encapsulated in the outer pocket dissociated in the solution phase during the NMR study or could not produce much impact on the aromatic signals to split them as separate signals. However, a significant broadening of alkyl and aromatic protons indicated the complexation of BTL with Cd. This broadening is likely a result of restricted mobility and rapid spin relaxation of the skeletal protons within the complex (ESI Fig. S3 and S4†).24,25 The mass spectrum of the BTL and BTL–Cd showed molecular ion peaks at m/z 465.3 and m/z 577.17, respectively (ESI Fig. S5†). The peaks at m/z 723.02 and 725.02 correspond to the fragments formed after the loss of isotopic forms of chloride ions (ESI Fig. S6†).
The molecular structures of the crystalline forms of BTL and BTL–Cd were elucidated by single crystal X-ray diffraction (Fig. 1). Their crystallographic data and structure refining parameters are listed in Table S1.† The selected bond lengths and bond angles for both are summarized in Table S2.† The BTL·3HCl was crystallized in the P21/n space group of the monoclinic crystal system (Fig. 1a). It consisted of a heptadentate donor system distributed uniformly in the tripodal arms to generate two pockets. The inner pocket consisted of the NN3 donor system while the outer pocket was formed by the O3 donor system. BTL–Cd was obtained in its crystalline form in a diffusion tube using DMF–ether as the solvent. The metal complex was crystallized in the P21212 space group of the orthorhombic crystal system. The crystal structure depicted that the inner pocket of the ligand was occupied by Cd1 acquiring hexcoordination with trigonal prismatic geometry. The outer pocket could hold the CdCl2 moiety adopting tetra-coordination around the central metal ion through two phenolic–O donors and two chloro ligands (Fig. 1b). The tetracoordinated Cd2–Cl2, Cd3–O2, and Cd2–O3 bond lengths were found to be 2.421(17), 2.148(3), and 2.184(4) Å, respectively. The angles for the tetrahedral arrangements O2–Cd2–O3 and Cl1–Cd2–Cl2 were determined to be 78.12(13)° and 112.43(6)°, respectively (Fig. 1c).
 | ||
| Fig. 1 ORTEP presentation of (a) BTL ligand, (b) BTL–Cd metal complex with the partial numbering scheme (thermal ellipsoids are drawn at 30% probability, and hydrogen atoms have been omitted for clarity), and (c) magnified view of the trigonal prismatic and tetrahedral geometry of Cd1 and Cd2 around the BTL. | ||
BTL–Cd was converted to the nanoparticle form (BTL–Cd NPs) by the bottom-up approach of the reprecipitation method. The nanoparticles were dispersed in a palladium chloride solution and Pd2+ was reduced to Pd0 through a reduction reaction to prepare core–shell nanoparticles (Scheme 1). The synthesized core–shell nanoparticles (Pd@BTL–Cd) were further characterized by different techniques such as FTIR spectroscopy, FESEM, HR-TEM, XPS, NH3–TPD, etc. In Pd@BTL–Cd, Cd–Cl bonds behaved like claws and could hold the Pd NPs to form a uniform coating on the surface of the particles.
The FTIR spectrum of BTL showed strong bands around 1580–1680 cm−1 and 1454 cm−1 assigned to aromatic C–H vibrations. The C–N stretching vibrations were observed in the region 1250–1260 cm−1.26 The O–H vibrational band of the free ligand was observed at 3413 cm−1 while it appeared at 3697 cm−1 for the metal complex (Fig. 2a). The shifting to a high energy region evidenced the binding of the ligand to cadmium metal through –OH.27,28 The broad vibrational band at 2864 cm−1 signified the presence of the N–H in the free ligand.29 It also exhibited a lowering of intensity in the case of the metal complex signifying binding with Cd metal through NH. The formation of Cd–O/Cl bonding in the compound was supported by the Raman spectrum. The peaks at 104 cm−1 could be attributed to Cd–Cl vibrations,30 and the peaks at 1024 cm−1 and 1426 cm−1 could be assigned to Cd–O vibrations.31 A peak at 338 cm−1 was related to the Cd–N vibration32 (ESI Fig. S7†). The FTIR spectrum of Pd@BTL–Cd mimics the spectra of BTL–Cd (Fig. 2a). But the interaction of Pd NPs with BTL–Cd was confirmed through Raman spectroscopy. The peaks at 668 cm−1 were attributed to the vibrations of Pd–O33 and peaks at 395 cm−1 corresponded to the Pd–Cl vibrations.34
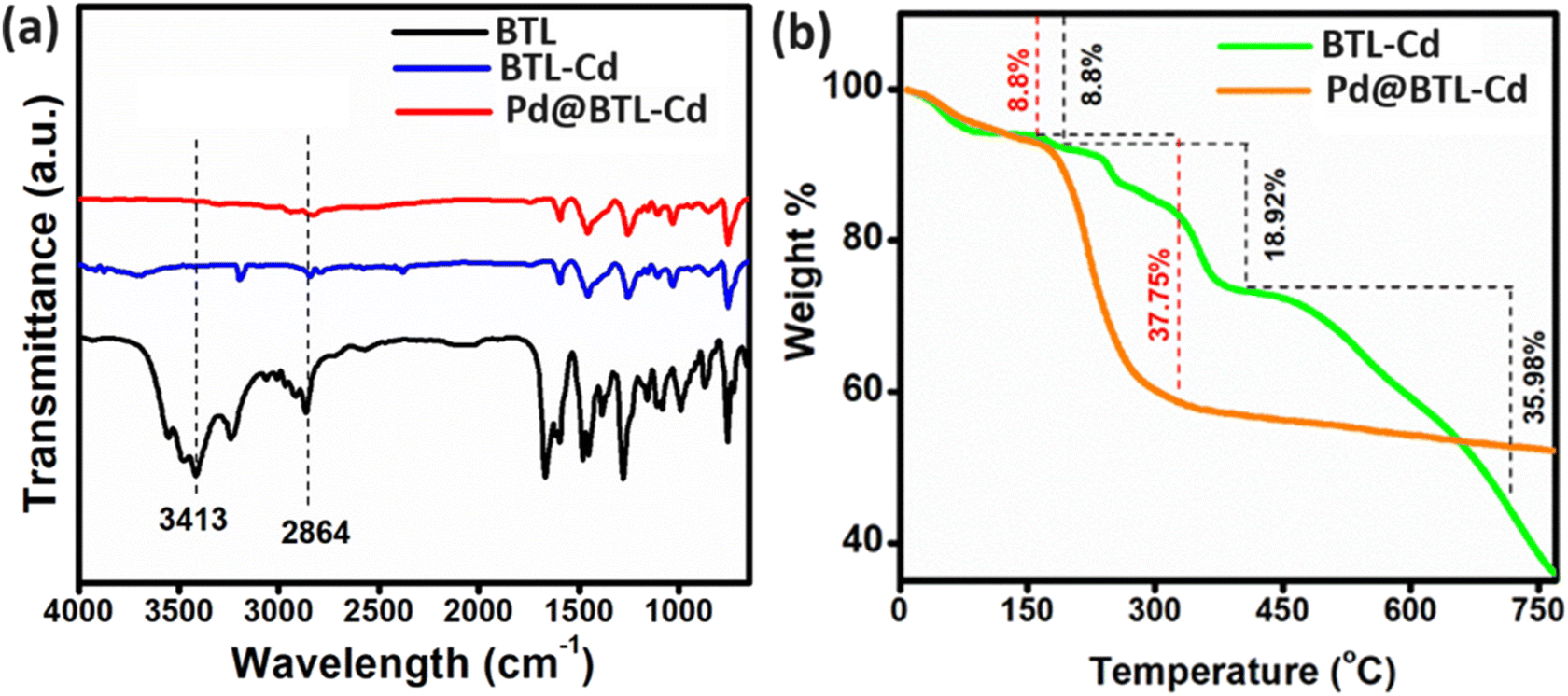 | ||
| Fig. 2 (a) FTIR spectra of BTL, BTL–Cd, and Pd@BTL–Cd and (b) comparison of thermogram of BTL–Cd and Pd@BTL–Cd. | ||
The thermal stability of Pd@BTL–Cd was compared with that of BTL–Cd by using thermogravimetric analysis (Fig. 2b). In both BTL–Cd and Pd@BTL–Cd, weight loss of about 8.8% was observed up to 150 °C due to the presence of absorbed moisture in the materials. The weight loss of 18.92% and 37.75% was observed in the second step when the temperature was raised from 200–450 °C which could be attributed to the decomposition of the organic framework of BTL–Cd and Pd@BTL–Cd, respectively. The results signified that after the introduction of palladium nanoparticles, the organic framework decomposed rapidly but to a lower extent. Subsequently, the structure of BTL–Cd started degrading with complete degradation up to 1000 °C. However, in the case of Pd@BTL–Cd only 47% weight loss was observed up to 350 °C and thereafter, the composite was stable up to 1000 °C.
The porosity of Pd@BTL–Cd was validated through N2 adsorption tests and compared with the BTL–Cd complex through adsorption–desorption isotherm and BET surface area plot (Fig. 3a–d). Employing Brunauer's classification, the adsorption isotherms for Pd@BTL–Cd and BTL–Cd are likely classified as type IV and type II, respectively. The adsorption–desorption isotherms of Pd@BTL–Cd and BTL–Cd displayed H1 and H3 hysteresis loops, respectively, indicating their least porous nature. The surface area and pore volume of the BTL–Cd complex were calculated to be 0.7529 m2 g−1 and 1.22 × 10−3 cm3 g−1, respectively (Fig. 3a and b). The surface area and pore volume of Pd@BTL–Cd were found to be 10.809 m2 g−1 and 0.084 cm3 g−1, respectively (Fig. 3c and d). The increased surface area of Pd@BTL–Cd makes it a highly promising candidate for catalytic applications. Furthermore, the BJH results for BTL–Cd and Pd@BTL–Cd revealed predominant peaks at 6.604 nm and 1.808 nm respectively showing the presence of pores (Fig. 3a and b). BET data clarify that the synthesized material does not have sufficient porosity for gas adsorption and that is why this particular material has been explored for catalysis.
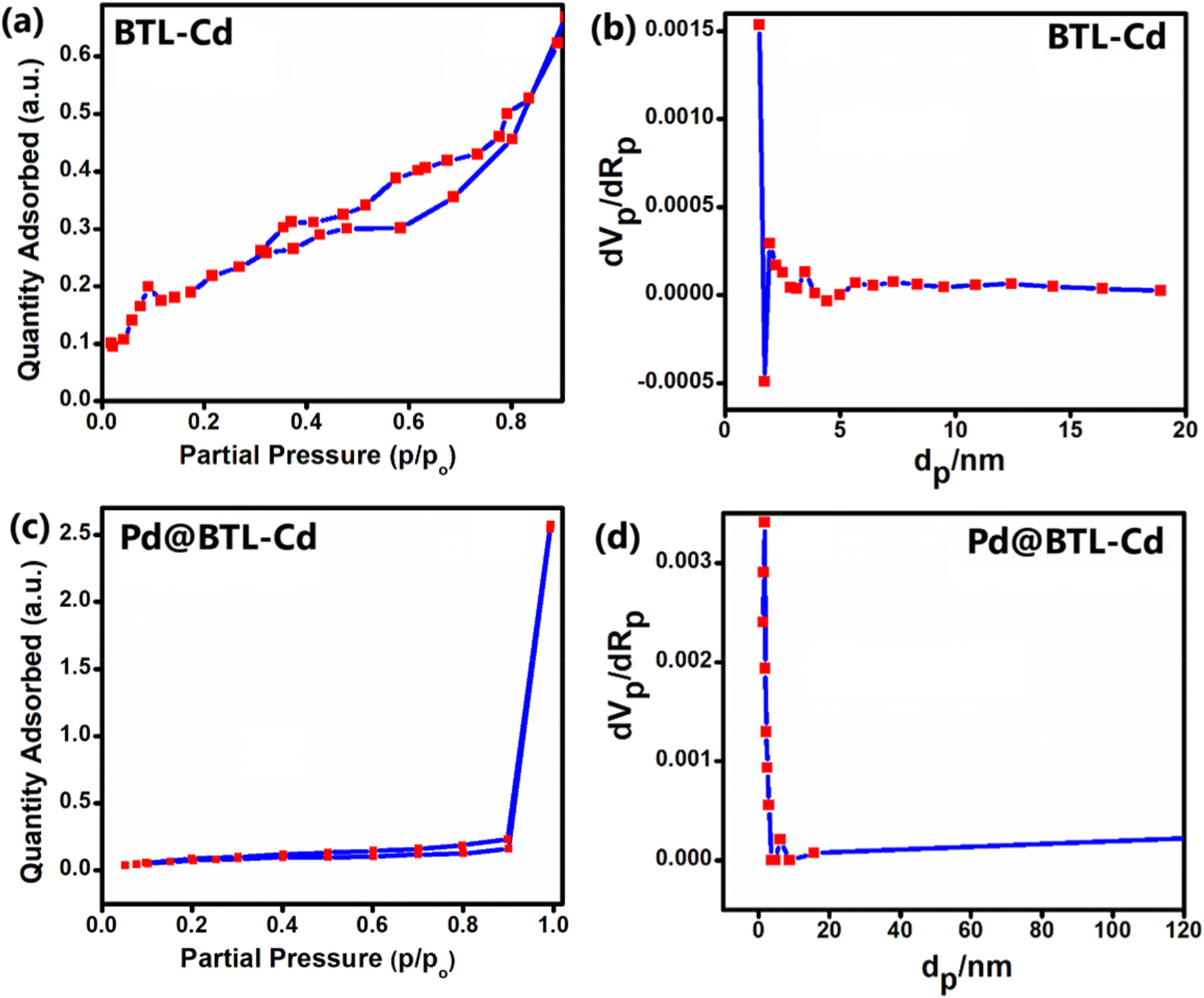 | ||
| Fig. 3 (a) N2 adsorption–desorption isotherms of Pd@BTL–Cd, (b) pore size distribution curves for Pd@BTL–Cd, (c) N2 adsorption–desorption isotherms of BTL–Cd, and (d) pore size distribution curves for BTL–Cd. | ||
The HRTEM images confirmed the formation of a shell over the surface of the palladium nanoparticle core (Fig. 4a and b). At high resolution, a spherical morphology can be seen with a darker palladium nanoparticle core at the centre and a bright BTL–Cd shell outside. The formation of fringes was observed in high-resolution transmission electron microscopy (HRTEM) images with interplanar distances of 0.365 nm, thereby reaffirming the crystalline nature of Pd@BTL–Cd core–shell nanoparticles (Fig. 4c). Energy dispersive X-ray analysis (EDAX) of Pd@BTL–Cd exhibited peaks for carbon, nitrogen, oxygen, palladium, cadmium, and chlorine which confirmed the presence of BTL–Cd and Pd NPs together (Fig. 4d). Elemental mapping of Pd@BTL–Cd confirmed the uniform coating of the BTL–Cd core over Pd NPs (Fig. 4e). The particle size histogram of Pd@BTL–Cd confirmed the successful formation of core–shell nanoparticles with a narrow size distribution, showing a mean particle size of 64.89 ± 0.2 nm (ESI Fig. S8a†). DLS analysis supported the presence of the BTL–Cd shell around the Pd nanoparticle core. The Pd NPs originally ranged in size from 10 to 20 nm, which is consistent with their plasmonic behaviour.35 Following the formation of the BTL–Cd shell, the overall size of Pd@BTL–Cd increased to a range of 60 to 80 nm (ESI Fig. S8b and c†). The FESEM images validate the rough surface of the catalyst with small spherical-shaped nanoparticles under high magnification, revealing a discernible coating over these particles36,37 (ESI Fig. S9†).
 | ||
| Fig. 4 (a–c) HRTEM images of Pd@BTL–Cd, (d) EDAX of Pd@BTL–Cd showing the presence of carbon, nitrogen, oxygen, cadmium, and palladium, and (e) elemental mapping of Pd@BTL–Cd. | ||
The PXRD analysis of BTL–Cd was performed to demonstrate the bulk purity and crystallinity of the as-synthesized bulk material. The PXRD pattern of BTL–Cd showed similarity with the pattern predicted using Mercury 3.8 software demonstrating the crystalline phase purity of the bulk product (ESI Fig. S10a†). Similarly, Pd@BTL–Cd showed strong diffraction peaks at 40.01°, 46.46°, 67.2° and 81.7°, which corresponded to the lattice planes (111), (200), (220), and (311), respectively.38 These lattice planes are typical Pd planes with a face-centred cubic (fcc) structure (ESI Fig. S10b†). To further uncover the chemical state and composition, XPS was employed to probe the surface property of Pd@BTL–Cd. As shown in the XPS spectrum, the Pd@BTL–Cd nanocomposite is composed of C, O, N, Cl, Pd and Cd elements (Fig. 5a). For the C 1s spectrum, the major peak at 284.60 eV was attributed to surface C–C or C–H bonds, and the second peak at 285.2 eV was ascribed to carbon atoms of C–OH or C–O–C groups present on the surface.35 The major peaks in the N 1s spectrum at 405.16 eV and 400.2 eV showed the presence of C–N–H and N–Cd bonds. The high-resolution deconvoluted spectra of Pd 3d of Pd@BTL–Cd showed peaks at binding energies of 343.1 eV, 341.0 eV, 337.8 eV, and 335.1 eV (Fig. 5b). The characteristic peaks at 337.8 and 335.1 eV are related to the binding energy of metallic Pd, and the binding energy peaks at 343.1 eV and 341.0 eV are ascribed to the presence of Pd2+. It can be inferred from the data that BTL–Cd coordinated with Pd NPs via two chlorine atoms converting Pd NPs into Pd2+ centres. The separate binding energy peaks for Pd0 and Pd2+ reveal the chemical environment of Pd NPs within the composite. Additionally, the peak area ratio of the spin–orbital pair Pd0/Pd2+ is 1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. This ratio reflects the relative interaction between Pd NPs and BTL–Cd, thus generating Pd in dual oxidation states. The peaks present at the deconvoluted spectra of Cd 3d confirmed the presence of Cd2+ at 411.5 eV and 404.7 eV (Fig. 5c). The spectra of C 1s, O 1s, Cl 2p, and N 1s of Pd@BTL–Cd are shown in ESI Fig. S11a–d.†
:1. This ratio reflects the relative interaction between Pd NPs and BTL–Cd, thus generating Pd in dual oxidation states. The peaks present at the deconvoluted spectra of Cd 3d confirmed the presence of Cd2+ at 411.5 eV and 404.7 eV (Fig. 5c). The spectra of C 1s, O 1s, Cl 2p, and N 1s of Pd@BTL–Cd are shown in ESI Fig. S11a–d.†
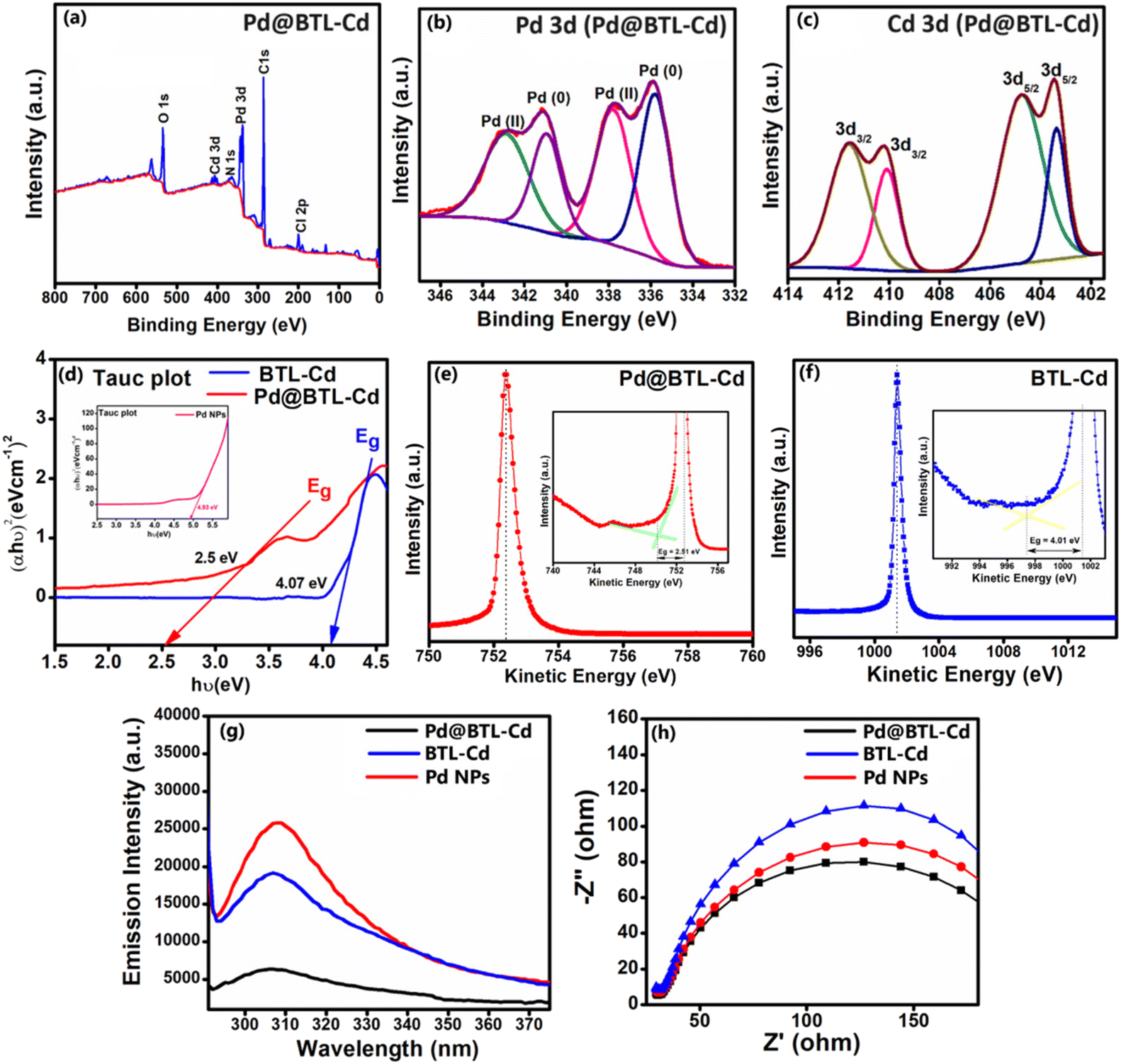 | ||
| Fig. 5 (a) XPS survey spectrum of Pd@BTL–Cd, (b) XPS plots of Pd 3d (Pd@BTL–Cd), (c) XPS plots of Cd 3d (Pd@BTL–Cd), (d) Tauc plot of Pd@BTL–Cd and BTL–Cd showing the band gap value Eg, (e) REELS spectra of Pd@BTL–Cd, (f) REELS spectra of BTL–Cd, (g) photoluminescence response of BTL–Cd and Pd@BTL–Cd, and (h) EIS of BTL–Cd and Pd@BTL–Cd. | ||
To delve into the significance of palladium in catalytic processes, we conducted a thorough analysis of acidic sites in both BTL–Cd and Pd@BTL–Cd using temperature-programmed desorption (TPD) studies (ESI Fig. S12†). The findings revealed that the total acidic content in BTL–Cd stands at 2.151 mmol g−1. Conversely, in the case of Pd@BTL–Cd, this value increases to 2.333 mmol g−1. The detailed calculation of the acidic active sites of Pd@BTL–Cd is provided in ESI Section S1.† The presence of acidic sites in Pd@BTL–Cd led to the initiation of the catalytic reaction and the formation of the desired product, particularly in the amination of furfural. Turnover number (TON) values were also calculated using the amount of acidic and basic sites obtained from NH3–TPD analysis as active reaction sites of the catalyst and its detailed calculations are provided in ESI Section S2.† The existence of Pd and Cd metal ions within Pd@BTL–Cd and BTL–Cd was confirmed through ICP-MS analysis. The practical loadings of Pd and Cd in Pd@BTL–Cd are 10.4% and 4.96%, respectively. In BTL–Cd, the loadings of Cd metal ions are 10.9%.
3.2. Photocatalytic properties of Pd@BTL–Cd
The UV absorption spectra, photoluminescence (PL) spectra, and electrochemical impedance spectra (EIS) of Pd NPs, BTL–Cd, and Pd@BTL–Cd were recorded to compare the photocatalytic properties of the parent compounds and the composite. The absorption bands showed absorption maxima at 209 and 275 nm for BTL–Cd, 224 and 408 nm for Pd0, and 321 and 431 nm for PdCl2. In contrast, Pd@BTL–Cd exhibited four bands centred at 233, 274, 344, and 405 nm. The absence of the band centred at 431 nm confirms the reduction of PdCl2 to Pd0; however, the presence of weak bands at 344 and 405 nm suggests the presence of both Pd0 and Pd2+ in the composite39 (ESI Fig. 13a–d†). Pd@BTL–Cd shows absorbance in the visible region as well as the UV region at optimized concentrations (ESI Fig. S13d†). The plasmonic properties of Pd NPs underscore the potential of Pd@BTL–Cd for light harnessing in visible regions for light-driven photocatalytic activity.To decipher the band gap of the synthesized composite (Pd@BTL–Cd), Tauc plot and reflected electron energy loss spectroscopy (REELS) measurements were employed. The Tauc plot was obtained by using the relationship (αhν)2 = hν − Eg, where α and Eg represent the absorption coefficient and the energy gap of the material, respectively. The resulting linear fit of (αhn)2versus photon energy (hν) provided band gap values of 2.5, 4.07, and 4.93 eV for Pd@BTL–Cd, BTL–Cd, and Pd NPs, respectively (Fig. 5d). The band gap obtained from the Tauc plot was further verified with high surface sensitive REELS measurement (Fig. 5e and f). The band gap values of Pd@BTL–Cd and BTL–Cd were 2.51 eV and 4.01 eV, in good agreement with the results obtained from the Tauc Plot.40 Cd-based materials often exhibit narrow band gaps that absorb visible light indirectly through surface or defect states. The combination of both Pd and Cd centres along with the ligating skeleton lowers the LUMO energy level and significantly raises the HOMO energy level of Pd@BTL–Cd resulting in a decrease in the band gap.
The photoluminescence study revealed a consistent characteristic band for Pd@BTL–Cd but with lower intensity in comparison to those of BTL–Cd and Pd NPs. The lowering of intensity demonstrates the reduced electron–hole pair recombination due to enhanced separation of the pairs with improved photocatalytic activity. The association of Pd NPs and BTL–Cd formed a metal–semiconductor Schottky junction that potentially yielded an excellent photoactive response (Fig. 5g).41,42 The increased photoactive response of Pd@BTL–Cd as compared to BTL–Cd and Pd NPs was also confirmed by electrochemical impedance spectroscopy (EIS). The Nyquist plot illustrated a notably diminished arc for Pd@BTL–Cd in comparison to those of BTL–Cd and Pd NPs suggesting reduced charge-transfer resistance and an accelerated interfacial charge-transfer rate43,44 (Fig. 5h). Therefore, the combination of palladium nanoparticle and BTL–Cd resulted in improved LSPR, enhanced charge transfer, and consequently superior photoactivity.
3.3. Photocatalytic studies
Pd@BTL–Cd has shown highly selective photocatalytic reductive amination of furfural to furfuryl amine (FAM) in the presence of aqueous ammonia (water as a hydrogen source) under visible light (λ = 445 nm). Previous reports suggested that the production of byproducts alongside primary amines is always unavoidable in this reaction.45 The byproducts mainly originate when external H2 is used because the carbonyl group undergoes hydrogenation thus hampering the formation of primary amine. The catalytic efficiency of Pd@BTL–Cd in the synthesis of FAM was demonstrated by using water as a hydrogen source. Furthermore, to avoid the formation of by-products, various reaction parameters such as temperature, substrate/ammonia ratio, reaction time, and catalyst amount were optimized in addition to the controlled release of hydrogen (Table 1 and ESI Fig. S14†). The formation of FAM was analyzed by GC-MS (ESI Fig. S15–S18†). The variation of the catalyst amount (from 0 to 1.25 mol%) showed 71% selectivity for FAM with 80% conversion when 0.25 mol% (5 mg) of the catalyst was used. By increasing the amount of the catalyst from 0.25 to 0.5 mol% (5 to 10 mg), the highest selectivity (98%) and conversion (99%) were observed due to an increase in catalytic sites for the complete conversion of furfural to FAM. Furthermore, an increase in the catalyst amount to 1.25 mol% (25 mg) showed a decrease in the selectivity due to the presence of excess active sites which led to the formation of side products like the Schiff base intermediate, secondary amine, etc. The absence of Pd@BTL–Cd led to only a 15% conversion of furfural, highlighting the effectiveness and efficiency of the catalyst. The variation in the catalyst amount does not enhance the selectivity of furfuryl alcohol to a larger extent; this may be caused by the inactivity of our catalyst towards the hydrogenation of furfural to furfuryl alcohol. However, increasing the catalyst quantity from (0.25 to 1.25 mol%) 10 to 25 mg significantly enhances the selectivity for the Schiff base intermediate, while decreasing the selectivity for FAM. This suggests that 10 mg/0.5 mol% is the threshold value and an increase in the catalyst amount leads to ample active catalytic sites thus dominating the formation of byproducts (Table 1). The impact of reaction temperature was also investigated within the range of 25 °C to 90 °C. The overall conversion remained the same; however, noteworthy changes were observed in the selectivity of the reaction. At a relatively high reaction temperature (of 70 °C), a lower selectivity of 48% was seen leading to the formation of a high number of by-products. Moreover, upon elevating the temperature up to 90 °C, the reaction mixture showed decomposition and resulted in a decrease in the selectivity of furfuryl amine up to 26%.|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Substrate/ammonia ratio | Catalyst amount (mol%) | Solvent | Temp (°C) | Time (h) | Selectivity (%) | Conversion (%) | ||
| Furfuryl amine | Furfuryl alcohol | Schiff base intermediate | |||||||
| 22*: BTL–Cd as the catalyst; 23*: bare Pd NPs as the catalyst; 24*: without the light reaction. | |||||||||
| 1 | 0.6 | 0 | Water | RT | 4 | — | 99 | — | 15 |
| 2 | 0.6 | 0.25 | Water | RT | 4 | 71 | 4 | 25 | 80 |
| 3 | 0.6 | 0.5 | Water | RT | 4 | 98 | <1 | <1 | 99 |
| 4 | 0.6 | 0.75 | Water | RT | 4 | 73 | 9 | 18 | 99 |
| 5 | 0.6 | 1.0 | Water | RT | 4 | 71 | 7 | 22 | 99 |
| 6 | 0.6 | 1.25 | Water | RT | 4 | 70 | 9 | 21 | 99 |
| 7 | 0.6 | 0.5 | Water | 70 | 4 | 48 | 11 | 41 | 99 |
| 8 | 0.6 | 0.5 | Water | 90 | 4 | 26 | 15 | 59 | 99 |
| 9 | 0.6 | 0.5 | Water | RT | 1 | 25 | — | 75 | 69 |
| 10 | 0.6 | 0.5 | Water | RT | 2 | 66 | 10 | 24 | 86 |
| 11 | 0.6 | 0.5 | Water | RT | 5 | 80 | 10 | 10 | 99 |
| 12 | 0.6 | 0.5 | Water | RT | 6 | 58 | 11 | 31 | 99 |
| 13 | 0.6 | 0.5 | Water | RT | 8 | 61 | 13 | 26 | 99 |
| 14 | 0.6 | 0.5 | MeOH | RT | 4 | 75 | — | 25 | 99 |
| 15 | 0.6 | 0.5 | HCOOH | RT | 4 | 81 | 15 | 4 | 99 |
| 16 | 0.6 | 0.5 | H2O/NaBH4 | RT | 4 | 90 | — | 10 | 99 |
| 17 | 0.4 | 0.5 | Water | RT | 4 | 77 | — | 23 | 99 |
| 18 | 1.0 | 0.5 | Water | RT | 4 | 68 | — | 32 | 99 |
| 19 | 1.5 | 0.5 | Water | RT | 4 | 60 | — | 40 | 99 |
| 20 | 2.0 | 0.5 | Water | RT | 4 | 87 | — | 13 | 99 |
| 21 | No ammonia | 0.5 | Water | RT | 4 | — | 99 | — | 26 |
| 22* | 0.6 | 0.5 | Water | RT | 4 | 27 | 10 | 63 | 41 |
| 23* | 0.6 | 0.5 | Water | RT | 4 | 24 | 10 | 66 | 89 |
| 24* | 0.6 | 0.5 | Water | RT | 4 | 21 | — | 79 | 39 |
These outcomes conclude that at room temperature there is an existence of adsorption–desorption equilibrium of the intermediate thus influencing the predominant reaction pathway that leads to the generation of FAM and, in turn, enhances its selectivity.46,47 However, elevated temperatures disturb the equilibrium and catalyze other reaction pathways yielding side products such as hydrogenation of the Schiff base intermediate. Additionally, the hydrogenation of furfural to furfuryl alcohol was observed but to a lesser extent. The reaction mixture at room temperature (25 °C) reveals no/negligible photothermal heating effect. This finding suggests that the increase in temperature resulting from the absorption of light energy and its conversion into heat is minimal. This evidence further reinforces the conclusion that the photocatalytic activity of the material is not significantly influenced by thermal contributions under these conditions.48,49
The reaction was carefully optimized by varying the reaction time to determine the duration that yields the highest selectivity and conversion. Furfural was completely consumed within 2 h. However, the reaction carried out for 60 min showed 25% selectivity for furfuryl amine and a maximum for the Schiff base intermediate (75%). After 4 h of the reaction, the selectivity of FAM increased with a decrease in the selectivity of the Schiff base intermediate. Different solvents were tried as hydrogen sources such as methanol, formic acid, and sodium borohydride. A varied selectivity was found for these solvents like 81% for formic acid, 75% for methanol, and 90% for sodium borohydride. Sodium borohydride, being a potent reducing agent, not only promoted high selectivity but also resulted in the formation of by-products. The substrate/ammonia ratio revealed that at a higher ratio (low concentration of ammonia), a higher amount of Schiff base intermediate was formed thus lowering the selectivity of FAM and vice versa. Hence, under the optimal reaction conditions, the highest selectivity of 98% was obtained using the optimum substrate/ammonia ratio (0.6). In the absence of ammonia, the reaction could not yield FAM, and furfuryl alcohol was formed to a lesser extent. Moreover, the catalytic reaction was also conducted in the dark under optimal reaction conditions, and only 21% selectivity of FAM was detected with 39% conversion (Table 1, entry 24). The plasmonic properties and synergistic functioning of Pd@BTL–Cd make it more selective than the parent compounds BTL–Cd and Pd NPs which showed only 27% and 24% selectivity for the conversion. The photocatalytic reaction was further optimized under UV light (254 nm) to evaluate the performance of Pd@BTL–Cd and no detectable photocatalytic activity was observed. This result underscores the ineffectiveness of the photocatalytic behaviour of the composite under UV light and emphasises the exclusive role played by the combination of BTL–Cd and PdNPs in the amination of furfural.
Various metal catalysts like Pd/CNT,50 Pd/SiO2,16 Pd/HZSM-5,3 Ru/Nb2O5,51 Co@NC-800,52 Ru NPs,53 Ru/TiO2,51 Fe3O4@SiO2–Ni,54 RANEY® Ni,55 Ru/BNC,56 0.5% Ru/P25,17,19 and Ni/CaCO317 have been previously reported for this reaction (ESI Table S3†). Most of the protocols comprise the use of molecular hydrogen as an H source in methanol and require elevated temperatures affording less selectivity for FAM. However, the current photocatalyst Pd@BTL–Cd afforded 98% selectivity of furfuryl amine under visible light with 99% furfural conversion and a high TON (20815) within 4 h using water as the hydrogen source. The magnification experiment was also carried out to assess the catalytic performance of Pd@BTL–Cd at the gram scale. Under optimal conditions, 10 mg of Pd@BTL–Cd exhibited a 98% selectivity for furfuryl amine at a lower concentration of furfural (5 mmol). However, at a higher furfural concentration (15 mmol), the same catalyst amount (10 mg) achieved an 85% selectivity for furfuryl amine which signifies the potential activity of Pd@BTL–Cd for the gram scale reaction also.
To evaluate the generalization and broaden the applicability of this catalyst (Pd@BTL–Cd), a range of aldehydes, under the optimized reaction conditions, were screened (ESI Table S4†) and the products were verified with 1H and 13C NMR (ESI Fig. S19–S21†). Notably, most of the aldehydes were efficiently converted into their corresponding primary amines in the presence of the synthesized catalyst (Pd@BTL–Cd). Under optimized conditions, 2-thiophene carboxaldehyde and 2-pyridine carboxaldehyde converted into their corresponding amines with 83% and 80% yield. Benzaldehyde was also converted into benzylamine with a 79% yield. These results confirm that Pd@BTL–Cd enables a selective synthesis of amines with their corresponding aldehydes.
3.4. Mechanistic studies
The photocatalytic mechanism and kinetic aspects of this reaction were elucidated based on literature reports, GC-MS analysis, optical studies, and DFT calculations. The central focus of the current work revolves around the controlled hydrogen availability and it utilizes water as a hydrogen source inspired by our prior research.57 Pd@BTL–Cd plays a vital role in the generation of hydrogen ions. The coordination of BTL–Cd with Pd NPs via –Cl bridges makes the surface positively charged which attracts the water molecules for chemisorption. Here, the dual role of Pd NPs can be understood; Pd0 harvests the light and Pd2+ initiates the generation of reactive species. The absorption of light by the Pd plasmons generated active hydrogen ions for the reduction of furfural. This process involves the reaction of furfural with ammonia to form a derivative of amino alcohol, which further undergoes dehydration to form furfuryl imine by Schiff base condensation. In this case, no evidence of short-lived furfuryl imine was observed during the reaction process because of the presence of excessive water and active hydrogen species (ESI Fig. S15†). Initially, the formation of furfuryl amine was less because the stronger nucleophilicity of furfuryl amine reinforced the formation of the N-furfurylidene furfuryl amine intermediate by Schiff base condensation. This intermediate has a comparatively longer life and can be seen in the GC-MS analysis at 22.139 RT (ESI Fig. S15 and S17†). However, with the progress of the reaction, 100% conversion of furfural was observed due to hydrolysis of the intermediate to furfuryl amine (ESI Fig. S18†) showing 98% selectivity for its formation (Fig. 6).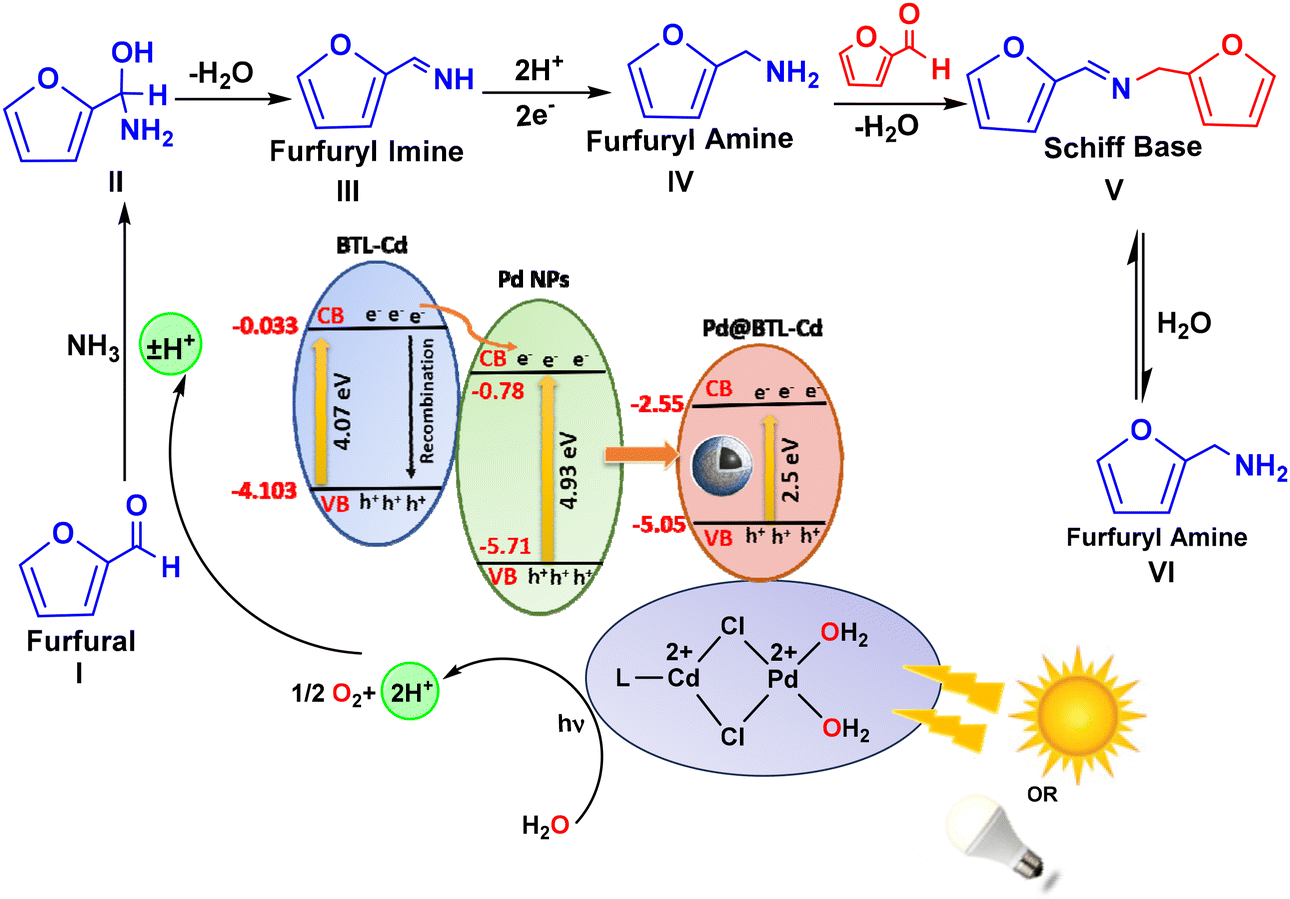 | ||
| Fig. 6 Plausible mechanism for the photocatalytic reductive amination of furfural to furfuryl amine. | ||
The facile generation of hydrogen from water can be understood by careful evaluation of the band gap. A band gap structure of Pd@BTL–Cd can be sketched based on various calculations as shown in Fig. 6. The band gap values determined from the Tauc plot and REELS spectra for Pd NPs, BTL–Cd, and Pd@BTL–Cd were 4.93, 4.07, and 2.5 eV, respectively (Fig. 5d–f). The valence band and conduction band potential for Pd@BTL–Cd were calculated to be −5.05 eV and −2.55 eV, respectively from optical studies. Pd NPs possessed a more negative conduction band potential (−0.78 eV) compared to BTL–Cd (−0.033 eV). Conversely, BTL–Cd had a more positive valence band potential (−4.103 eV) than Pd NPs (−5.71 eV), facilitating the thermodynamically favourable transfer of photo-generated charge carriers. This photosystem exhibited a Type II heterojunction formation between BTL–Cd and Pd NPs as illustrated in Fig. 6. Under visible light irradiation, electrons excite from the valence band of BTL–Cd (VB) to its conduction band (CB), leaving positive holes (h+) behind. The photo-generated electrons subsequently transfer to the CB of Pd NPs. The effective separation of charge carriers in this way increases the effective lifetimes of the generated species and allows prolonged interactions with reactant molecules. As a result, the system exhibits enhanced photoactivity, enabling it to drive photocatalytic reactions more efficiently under visible light irradiation.25
To validate the statement mentioned above, we further examined the optimized geometries and energies of the reactants, intermediates, and products through Gaussian studies (ESI Fig. S22†). Furfural reacted with ammonia to form a derivative of amino alcohol, with a loss of energy of −1.84 kcal mol−1. Dehydration of intermediate II to form imine III involved the gain of 5.53 kcal mol−1 energy. This particular step was an exothermic process which involved the loss of −131.48 kcal mol−1 energy. Furthermore, the conversion of furfuryl amine(III) to Schiff base intermediate(V) was an endothermic process, with a change in energy from −131.48 kcal mol−1 to −114.03 kcal mol−1. The hydrolysis of Schiff base intermediate(V) to furfuryl amine(VI) introduces no change in energy which confirms that this particular step is interconvertible. Furfuryl amine has a ground state energy of −131.48 kcal mol−1 less than the initial substrate used (furfural).
3.5. Heterogeneity, leaching test, and reusability of the catalyst
Heterogeneous catalysts are recognized for their ability to provide the benefit of recyclability and economical method development. Hence, the catalyst was separated from the reaction mixture after each catalytic cycle by centrifugation, washed with de-ionized water and ethanol, and dried in a hot air oven. The catalytic performance of the Pd@BTL–Cd catalyst exhibited very little decrement in selectivity after the first cycle due to the dissolution of unstable species on the surface of the catalyst. The activity of the catalyst was maintained for up to five cycles without much drop in the furfuryl amine selectivity (ESI Fig. S23a†). There is no significant change in the structural and functional characteristics of the recovered catalyst as confirmed by PXRD and FTIR analysis. The PXRD and FTIR spectra of Pd@BTL–Cd confirmed the intact crystallinity and functionality of the catalyst recovered after the 5th cycle but with reduced relative intensity (ESI Fig. S23b and c†). The decline in intensity is attributed to the presence of the adsorbed species (such as amine or ammonia) on the active site of the catalyst. Additionally, a loss in weight of approximately 2 mg was noted after the fifth cycle due to the washing process. To know any potential leaching of Pd0 and Cd2+ ions during the catalytic process, the supernatant solution was recovered through centrifugation after the catalytic reaction and analyzed by using ICP-MS. Remarkably, the ICP-MS analysis revealed only 0.00015% and 0.0055% of Pd0 and Cd2+ ions in the solution, respectively. These findings not only dismissed the possibility of significant leaching of catalytic sites from the framework structure of the Pd@BTL–Cd catalyst but also affirmed that Pd0 and Cd2+ sites remain intact within the framework architecture of Pd@BTL–Cd without undergoing disintegration during the catalytic reaction.4. Conclusion
A straightforward method has been employed to construct a shell of BTL–Cd around a palladium nanoparticle core to fabricate a photosensitized composite. The composite showed an improved photocatalytic activity due to the lowering of the band gap between the HOMO and the LUMO. The Pd NPs helped to generate H-species under mild conditions and without the external use of H2 gas. The synthesized core–shell material turned out to be a promising photocatalyst with high conversion and unparalleled selectivity for the conversion of furfural to furfuryl amine in an aqueous medium. Moreover, this methodology showcases constant reactivity on a gram-scale also. Its notable feature is that it can be recycled and reused up to five times maintaining its catalytic activity and selectivity without any significant deterioration.Data availability
CCDC accession codes 2335270 (BTL) and 2335268 (BTL–Cd) comprise the crystallographic data for this paper.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors are grateful to SERB [No. SPG/2021/000445] and SERB/ANRF [SPM/2023/000057] for providing financial support.References
- A. Gelle, T. Jin, L. de la Garza, G. D. Price, L. V. Besteiro and A. Moores, Chem. Rev., 2019, 120, 986–1041 CrossRef PubMed
.
- A. N. Koya, X. Zhu, N. Ohannesian, A. A. Yanik, A. Alabastri, R. Proietti Zaccaria, R. Krahne, W. C. Shih and D. Garoli, ACS Nano, 2021, 15, 6038–6060 CrossRef CAS PubMed
- C. Dong, H. Wang, H. Du, J. Peng, Y. Cai, S. Guo, J. Zhang, C. Samart and M. Ding, Mol. Catal., 2020, 482, 110755 CrossRef CAS
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. Zhang, Chem. Mater., 2015, 27, 1921–1924 CrossRef CAS
- L. W. Wei, S. H. Liu and H. P. Wang, ACS Appl. Mater. Interfaces, 2023, 15, 25473–25483 CrossRef CAS
- C. Tang, S. Du, H. Huang, S. Tan, J. Zhao, H. Zhang, W. Ni, X. Yue, Z. Ding and Z. Zhang, ACS Catal., 2023, 13, 6683–6689 CrossRef CAS
- R. Su, R. Tiruvalam, A. J. Logsdail, Q. He, C. A. Downing, M. T. Jensen, N. Dimitratos, L. Kesavan, P. P. Wells and R. Bechstein, ACS Nano, 2014, 8, 3490–3497 CrossRef
- P. Kumari, N. Bahadur, L. Kong, L. A. O'Dell, A. Merenda and L. F. Dumée, Mater. Adv., 2022, 3, 2309–2323 RSC
- N. A. Surib, L. C. Sim, K. H. Leong, A. Kuila, P. Saravanan, K. M. Lo, S. Ibrahim, D. Bahnemann and M. Jang, RSC Adv., 2017, 7, 51272–51280 RSC
- J. A. Nasir, Z. ur Rehman, S. N. A. Shah, A. Khan, I. S. Butler and C. R. A. Catlow, J. Mater. Chem. A, 2020, 8, 20752–20780 RSC
- M. Gutiérrez, Y. Zhang and J. C. Tan, Chem. Rev., 2022, 122, 10438–10483 CrossRef PubMed
- S. Thakur, J. Rohilla, K. Kumar, R. Singh, V. Kaur and R. Kamboj, J. Mater. Chem. C, 2024, 12, 1683–1692 RSC
- T. Chhabra, J. Rohilla and V. Krishnan, Mol. Catal., 2022, 519, 112135 CrossRef CAS
- S. Sharma, J. Rohilla, S. Thakur, R. Singh and V. Kaur, Sustainable Energy Fuels, 2024, 8, 3892–3901 RSC
- N. K. Gupta, P. Reif, P. Palenicek and M. Rose, ACS Catal., 2022, 12, 10400–10440 CrossRef
- N. S. Gould, H. Landfield, B. Dinkelacker, C. Brady, X. Yang and B. Xu, ChemCatChem, 2020, 12, 2106–2115 CrossRef
- W. Jia, H. Liu, Y. Feng, J. Zhang, X. Zhao, Y. Sun, Z. Wei, S. Yang, X. Tang and X. Zeng, J. Cleaner Prod., 2022, 345, 131029 CrossRef
- M. Yuan, Y. Long, J. Yang, X. Hu, D. Xu, Y. Zhu and Z. Dong, ChemSusChem, 2018, 11, 4156–4165 CrossRef
- Z. Xue, S. Wu, Y. Fu, L. Luo, M. Li, Z. Li, M. Shao, L. Zheng, M. Xu and H. Duan, J. Energy Chem., 2023, 76, 239–248 CrossRef
- M. G. Leed, N. Wolkow, D. M. Pham, C. L. Daniel, J. L. Dunaief and K. J. Franz, J. Inorg. Biochem., 2011, 105, 1161–1172 CrossRef PubMed
- J. Boucard, T. Briolay, T. Blondy, M. Boujtita, S. Nedellec, P. Hulin, M. Grégoire, C. Blanquart and E. N. Ishow, ACS Appl. Mater. Interfaces, 2019, 11, 32808–32814 CrossRef
- M. Zhao, K. Deng, L. He, Y. Liu, G. Li, H. Zhao and Z. Tang, J. Am. Chem. Soc., 2014, 136, 1738–1741 CrossRef
- F. Li, Y. Yuan, J. Luo, Q. Qin, J. Wu, Z. Li and X. Huang, Appl. Surf. Sci., 2010, 256, 6076–6082 CrossRef
- R. McCaffrey, H. Long, Y. Jin, A. Sanders, W. Park and W. Zhang, J. Am. Chem. Soc., 2014, 136, 1782–1785 CrossRef PubMed
- S. Thakur, J. Rohilla, S. Sharma, R. Singh, R. Kamboj and V. Kaur, Dalton Trans., 2024, 53, 18283–18295 RSC
- H. Y. Nam, G. Lee and S. H. Jhung, Chem. Eng. J., 2024, 153072 CrossRef CAS
- G. S. Kürkçüoğlu, O. Z. Yeşilel, E. Sayın, E. Enönlü and O. Şahin, J. Mol. Struct., 2020, 1219, 128462 CrossRef
- A. Bartyzel, J. Therm. Anal. Calorim., 2017, 127, 2133–2147 CrossRef CAS
- I. Touarssi, S. Oukkass, Z. Habibi, Y. Chaouqi, N. Sefiani, L. Lebrun and M. Hlaibi, Mater. Today: Proc., 2021, 45, 7711–7717 CAS
- L. M. Yang, Y. Liu, L. M. Man, J. R. Zhou, X. P. Liu and C. L. Ni, Vib. Spectrosc., 2017, 93, 23–28 CrossRef CAS
- V. Srihari, V. Sridharan, T. Ravindran, S. Chandra, A. Arora, V. Sastry and C. Sundar, AIP Conf. Proc., 2011, 1349, 845–846 CrossRef
- D. Ishikawa, F. De Souza and C. Téllez, Spectrosc. Lett., 1993, 26, 803–808 CrossRef
- X. Tan, C. Yu, X. Song, L. Ni, H. Xu, Y. Xie, Z. Wang, S. Cui, Y. Ren and W. Li, Nano Energy, 2022, 104, 107957 CrossRef
- M. R. Mian, M. Wakizaka, T. Yoshida, H. Iguchi, S. Takaishi, U. Afrin, T. Miyamoto, H. Okamoto, H. Tanaka and S. I. Kuroda, Dalton Trans., 2021, 50, 1614–1619 RSC
- M. Vadai, D. K. Angell, F. Hayee, K. Sytwu and J. A. Dionne, Nat. Commun., 2018, 9, 4658 CrossRef PubMed
- S. B. Vuggili, K. Kadiya, U. K. Gaur and M. Sharma, Environ. Sci. Pollut. Res., 2021, 28, 46377–46389 CrossRef
- B. Balasubramanian, K. L. Kraemer, N. A. Reding, R. Skomski, S. Ducharme and D. J. Sellmyer, ACS Nano, 2010, 4, 1893–1900 CrossRef
- M. Khan, M. Khan, M. Kuniyil, S. F. Adil, A. Al-Warthan, H. Z. Alkhathlan, W. Tremel, M. N. Tahir and M. R. H. Siddiqui, Dalton Trans., 2014, 43, 9026–9031 RSC
- S. Raja, R. R. Babu, S. C. Mohan, K. Jothivenkatachalam and K. Ramamurthi, Appl. Surf. Sci., 2019, 497, 143737 CrossRef
- A. Kumar, V. Navakoteswara Rao, A. Kumar, A. Mushtaq, L. Sharma, A. Halder, S. K. Pal, M. V. Shankar and V. Krishnan, ACS Appl. Energy Mater., 2020, 3, 12134–12147 CrossRef
- M. L. de Souza, D. P. Dos Santos and P. Corio, RSC Adv., 2018, 8, 28753–28762 RSC
- S. K. Khore, S. R. Kadam, S. D. Naik, B. B. Kale and R. S. Sonawane, New J. Chem., 2018, 42, 10958–10968 RSC
- X. Li, J. Lin, J. Li, H. Zhang, X. Duan, H. Sun, Y. Huang, Y. Fang and S. Wang, ACS Sustainable Chem. Eng., 2021, 9, 7277–7285 CrossRef
- V.-H. Nguyen, M. Mousavi, J. B. Ghasemi, Q. V. Le, S. A. Delbari, A. Sabahi Namini, M. Shahedi Asl, M. Shokouhimehr and M. Mohammadi, J. Phys. Chem. C, 2020, 124, 27519–27528 CrossRef
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2016, 18, 487–496 RSC
- A. W. Heinen, J. A. Peters and H. V. Bekkum, Eur. J. Org. Chem., 2000, 2501–2506 CrossRef
- M. Allegretti, V. Berdini, M. C. Cesta, R. Curti, L. Nicolini and A. Topai, Tetrahedron Lett., 2001, 42, 4257–4259 CrossRef
- F. Wang, C. Li, H. Chen, R. Jiang, L. D. Sun, Q. Li, J. Wang, J. C. Yu and C. H. Yan, J. Am. Chem. Soc., 2013, 135, 5588–5601 CrossRef
- S. Sarina, H. Y. Zhu, Q. Xiao, E. Jaatinen, J. Jia, Y. Huang, Z. Zheng and H. Wu, Angew. Chem., 2014, 126, 2979–2984 CrossRef
- G. Singh, J. Kaishyop, G. Singh, M. J. Gazi, A. Bag, C. Samanta and A. Bordoloi, Mol. Catal., 2023, 535, 112877 CrossRef
- T. Komanoya, T. Kinemura, Y. Kita, K. Kamata and M. Hara, J. Am. Chem. Soc., 2017, 139, 11493–11499 CrossRef PubMed
- F. Unglaube, J. Schlapp, A. Quade, J. Schäfer and E. Mejía, Catal. Sci. Technol., 2022, 12, 3123–3136 RSC
- D. Chandra, Y. Inoue, M. Sasase, M. Kitano, A. Bhaumik, K. Kamata, H. Hosono and M. Hara, Chem. Sci., 2018, 9, 5949–5956 RSC
- M. Manzoli, E. C. Gaudino, G. Cravotto, S. Tabasso, R. N. Baig, E. Colacino and R. S. Varma, ACS Sustainable Chem. Eng., 2019, 7, 5963–5974 CrossRef
- K. Zhou, B. Chen, X. Zhou, S. Kang, Y. Xu and J. Wei, ChemCatChem, 2019, 11, 5562–5569 CrossRef
- H. Zou and J. Chen, Appl. Catal., B, 2022, 309, 121262 CrossRef
- J. Rohilla, S. Thakur, K. Kumar, R. Singh and V. Kaur, ACS Appl. Nano Mater., 2023, 6, 12063–12072 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Additional crystallographic data and structure refinement parameters, FTIR, 1H, 13C, ESI-MS, FESEM, XPS, PXRD, GC-MS, and TON calculations. CCDC 2335268 and 2335270. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03058c |
| This journal is © The Royal Society of Chemistry 2025 |