
Metal–ligand and hydrogen bonding in the active site of Fe(III)-, Mn(III)- and Co(III)-myoglobins†
Marek
Freindorf
 and
Elfi
Kraka
*
and
Elfi
Kraka
*
Chemistry Department, Southern Methodist University, 3215 Daniel Avenue, Dallas, TX 75275, USA. E-mail: ekraka@smu.edu
First published on 27th January 2025
Abstract
We investigated in this work the strength of metal–ligand bonding in complexes formed between Fe(III)-, Mn(III)- and Co(III)-myoglobin and methanol, water, nitrite, and azide, serving as neutral and ionic prototype ligands, for the ε and δ protonation forms of the myoglobin distal histidine. In total, 24 complexes and 12 associated gas phase models were investigated combining a QM/MM protocol with our local vibrational mode analysis at the PBE0/6-31G(d,p)/AMBER level of theory. According to our results, complexes with methanol and water ligands form weaker metal–ligand bonds than those with nitrite and azide ligands. Furthermore, the strength of the metal–ligand bonds depends on the protonation form of the distal histidine. Among the three metals investigated in this study, Fe, the metal found in native myoglobin, turned out to be the most versatile candidate, providing the broadest range of metal–ligand bond strengths. We also analyzed potential hydrogen bonds formed between the ligand and the distal histidine of the heme pocket. The ε tautomer of histidine forms weaker O⋯H type hydrogen bonds whereas the δ tautomer forms stronger N⋯H type hydrogen bonds. Overall, our findings identify the strength of both metal–ligand and hydrogen bonds (fully captured by our local vibrational mode analysis) as a key parameter determining the catalytic activity and function of myoglobins. This is particularly relevant when considering neutral versus ionic ligands and other metals such as Mn or Co as alternatives to Fe. The insights gained through our investigation offer valuable guidance for strategically fine-tuning existing artificial myoglobins and designing new, versatile variants. We hope that our QM/MM – local mode analysis protocol will become a valuable addition to the research community's toolkit.
1 Introduction
Myoglobin (Mb) is a member of the hemoprotein superfamily, which is found in muscles of vertebrates and in almost all mammals. Mb is responsible for storage of oxygen in vertebrates and plays an important role in many physiological functions of the heart and skeletal muscles. It is also one of the most studied proteins disclosing interactions between the protein active site and the surrounding protein environment.1–4 The active site of Mb includes a prosthetic heme group, which involves a protoporphyrin ring and a central Fe atom. The heme group is attached to the protein backbone by a covalent chemical bond with the proximal histidine, while the distal heme pocket can be occupied by small molecules such as O2, NO, H2S and CO, which are responsible for Mb's diverse biochemical activities.Over the past decade a number of artificial Mb, i.e., functionalized, bioengineered, or synthetic Mb proteins have been reported being intended to adapt Mother Nature's unique design to the specific needs across chemistry and beyond with applications in catalysis potentially replacing less sustainable and environmental friendly industrial catalysts; such as in medicine helping maintain oxygen delivery in situations where blood transfusions are limited or unavailable,5 or as biosensors detecting oxygen or other gases.6,7 In addition, they are ideal research models in experiment and theory, providing a controlled model to study heme protein function, protein folding, and oxygen storage mechanisms.
There are a number of reports applying artificial Mbs to various challenging catalytic transformations, bridging the gap between the efficiency of enzymatic reactions and the versatility of transition metal catalysis. Iron porphycene complexes of Mb used for selective CH functionalization reactions offer innovative ways to form CC, CN and CO bonds, such as cyclopropanation, amination, and azide reduction.8–13 The iron porphyrin cofactor makes heme proteins particularly well-suited as catalysts for nitrene transfer reactions, including the reduction of azides to amines.10 An engineered Mb-based catalyst has shown to be capable of catalyzing the cyclopropanation of aryl-substituted olefins with catalytic proficiency and excellent diastereo- and enantioselectivity via transmetalation.14
Replacement of Fe in hemoproteins by different transition metals has been utilized to investigate the role of heme in determining the protein properties for many years,15–17 and the most popular metals used in the modified hemoproteins are Mn and Co.18,19 Some recent examples of the Mn-substituted Mb (MnMb) include the discovery of two redox pathways in MnMb,20 CH bond hydroxylation catalyzed in MnMb,21,22 and oxidation properties of MnMb towards weak CH bonds.23 Similarly, examples of Mn-substituted cytochromes P450 include structural properties of Mn-substituted P450(CYP101),24 theoretical study of the pentacoordinate Mn in cytochrome P450cam,25 catalytic activity of Mn-substituted cytochrome P450(BSb),26 reconstruction of Mn-substituted cytochrome P450(BM3).27–29 The Mn metal was also used in Mn-substitution of an abiological protein in a study of a porphyrin-binding protein with high-valent Mn oxidation states.30
Similarly, recent examples of Co-substituted Mb (CoMb) involve theoretical study of dioxygen bound to CoMb,31 relaxation of ligand binding in CoMb,32 nitrogen generation in CoMb,33 and reconstruction of aqua- and cyano-CoMb.34 Cobalt was also used in metal-substituted cystathionine β-synthase,35 hydrogen generation from Co-substituted microperoxidase-1136 and cytochrome b562,37 reconstruction of CoMb-coupled histidine kinase,38 and Co-substitution in a series of hemoproteins in living cells of E. coli.39 CoMb complexes were used to catalyze electro-catalytic H2 evaluation.40,41 Heme oxygenase cobalt–protoporphyrin complexes were used for CO2 photoreduction.42 The Figg group has also introduced zinc–Mbs as catalyst for photo-induced electron/energy transfer (PET)-reversible addition–fragmentation chain transfer (RAFT) polymerizations.43
In order to systematically explore the large potential of these Fe, Co, Mn–Mbs, fine-tuning them for a specific purpose and/or the development of new design routes for artificial Mbs, the understanding of metal–ligand (ML) and potential ligand hydrogen bonding (HB) in the active Mb pocket at the molecular level is an important prerequisite, which to our best knowledge has been missing so far.
Therefore, inspired by the structural study of Mn- and Co-substituted Mb,18 we explored in this study similarities or differences between native Fe(III)Mb, and the engineered Mn(III)Mb and Co(III)Mb systems with methanol (CH3OH, MET), water (H2O, H2O), nitrite (NO2−, ONO), and azide (N3−, NNN) model ligands in the heme distal pocket, with a focus on assessing and comparing the strength of the ML bonds and the strength of potential HB interactions, formed between the distal histidine and the ligands. In order to account for the influence of the protein environment, we compared the ML bond strengths in the proteins with those of corresponding gas phase models. Sketches of the active sites of Fe(III)Mb, Mn(III)Mb and Co(III)Mb with the four ligands as well the corresponding active site gas phase models, investigated in this work, are presented in Fig. 1. As revealed in Fig. 1, in the six of the investigated protein systems the ligand forms either an O⋯H or an N ⋯H HB with the distal histidine, which we compared with the corresponding HB strengths in the water dimer (H2OHOH) and the water–ammonia pair (HOHNH3). Two tautomeric forms of distal histidine were considered, namely His64ε and His64δ (labeled in the following as “e” and “d”), which led to 24 protein complexes and 12 gas phase models. A special focus of this work was on exploring the relevance of the strength of these ML/HB interactions regarding catalytic activity or function of these Mbs for neutral versus ionic ligands as well as for Co and Mn substituted Fe-heme co-factors.
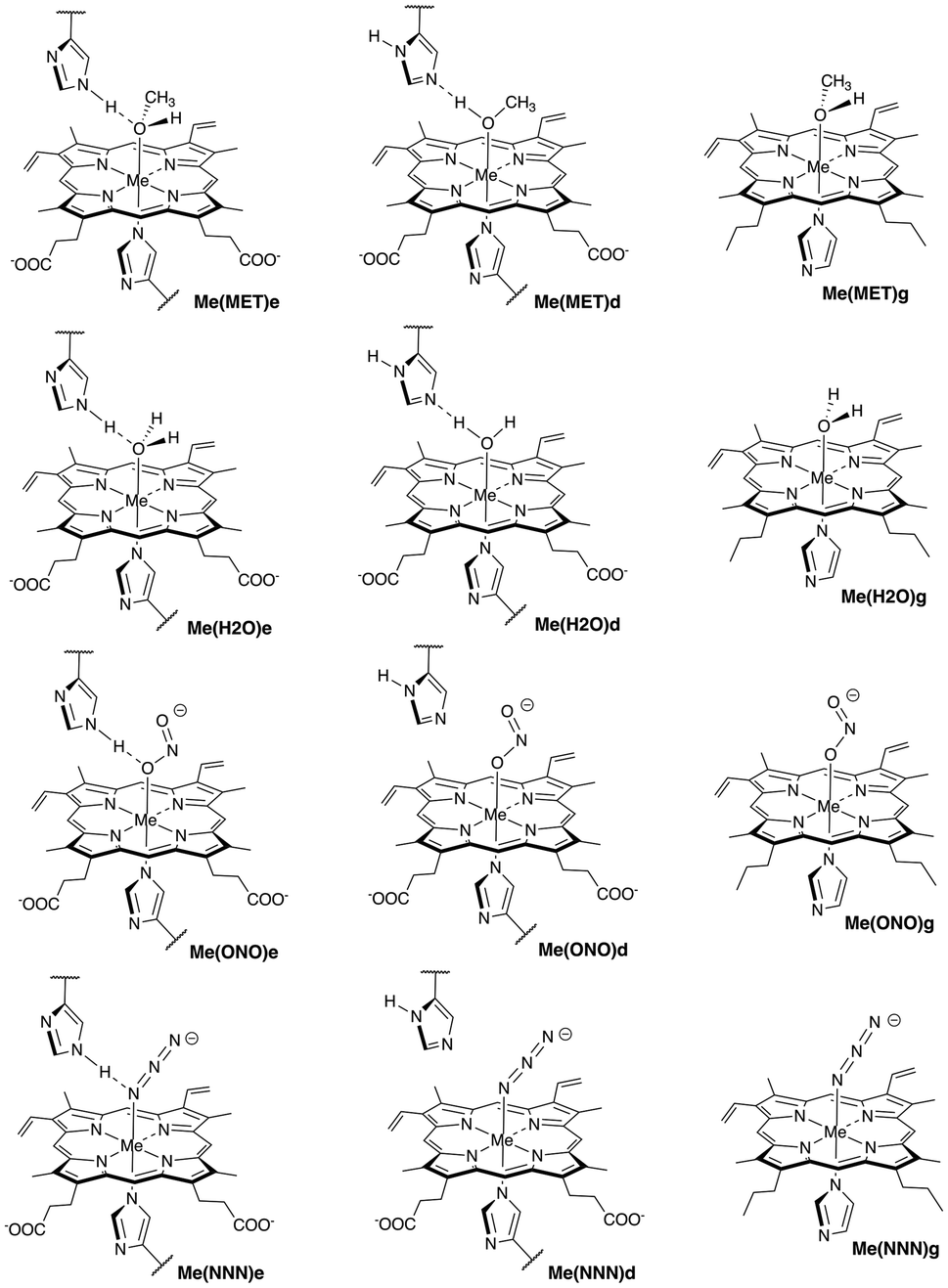 | ||
| Fig. 1 Sketches of active sites in Mb and gas phase models with ligands investigated in this study (Me = Fe, Mn, and Co). Symbols “e” and “d” indicate distal histidine in ε and δ protonation forms, respectively, while symbol “g” indicates gas phase models. For molecular labels, see text. | ||
The following complex notation is used throughout the manuscript (see also Fig. 1): (i) iron complexes: Fe(MET)e, Fe(MET)d, Fe(H2O)e, Fe(H2O)d, Fe(ONO)e, Fe(ONO)d, Fe(NNN)e, and Fe(NNN)d; (ii) manganese complexes: Mn(MET)e, Mn(MET)d, Mn(H2O)e, Mn(H2O)d, Mn(ONO)e, Mn(ONO)d, Mn(NNN)e, and Mn(NNN)d; (iii) cobalt complexes: Co(MET)e, Co(MET)d, Co(H2O)e, Co(H2O)d, Co(ONO)e, Co(ONO)d, Co(NNN)e, and Co(NNN)d; (iv) corresponding heme co-factor ligand gas phase models: Fe(MET)g, Fe(H2O)g, Fe(ONO)g, and Fe(NNN)g; Mn(MET)g, Mn(H2O)g, Mn(ONO)g, and Mn(NNN)g; and Co(MET)g, Co(H2O)g, Co(ONO)g, and Co(NNN)g. We utilized for the protein calculations a hybrid QM/MM (quantum mechanics – molecular mechanics) ansatz,44–48 and for the bond strength assessment we used the local vibrational mode theory (LMA) developed in our group,49,50 complemented with Bader's quantum theory of atoms in molecules (QTAIM) analysis51–53 and natural bond orbital (NBO) analysis.54,55
2 Materials and methods
2.1 Local vibrational mode analysis
In order to assess and compare ML and HB in the Mb complexes investigated in this work a qualified bond strength descriptor is needed. One popular measure to assess bond strength is to use normal mode stretching force constants derived from a normal mode analysis.56 However, as pointed out by Wilson in his pioneering 1941 publication57 normal vibrational modes of polyatomic molecules are generally delocalized, limiting this approach. LMA49,50 originally developed by Konkoli and Cremer58,59 has solved this problem via extracting local vibrational modes and corresponding local mode force constant from the normal vibrational modes.A local vibrational mode an is defined as
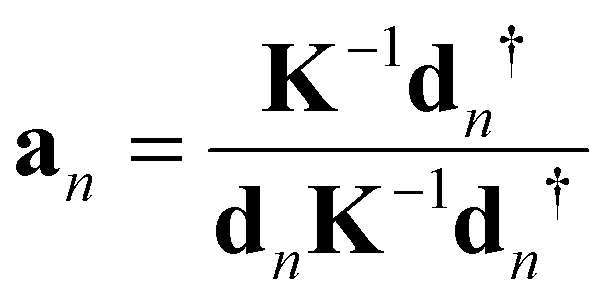 | (1) |
The two ingredients needed for LMA, the diagonal normal mode force constant matrix K in normal mode coordinates Q and the normal mode vectors dn in internal coordinates, can be obtained from a vibrational frequency calculation via the Wilson GF formalism,60,61 which is a routine part of most modern quantum chemistry packages.62 As a consequence, LMA can be applied with minimal computational costs after a routine quantum-chemical calculation of vibrational frequencies, optionally adding measured frequencies as input (a feature opening LMA to the experimental vibrational spectroscopists) to both single molecules in gas phase, solution, or in a protein, but also to periodic systems and crystals.49,50
For each local mode an, one can derive associated local force constants kan describing the local vibration of the atomic fragment under consideration,
 | (2) |
Over the past two decades, we have successfully applied local mode force constants to characterize the strength of covalent bonds and non-covalent interactions across the periodic table as documented in two recent review articles,49,50 including bonding inside the active sites of hemoproteins.64–71 Another important feature of LMA is the characterization of normal mode (CNM) procedure, which decomposes each normal vibrational mode into local mode contributions.49,50,72,73 CNM has advanced the interpretation of vibrational spectra to the next level e.g., identifying which molecular fragments couple in DNA–base pairs or assessing the quality of Stark effect probes with a local probe bond, just to name two examples.67,74–76 A detailed description of the underlying local vibrational mode theory can also be found in the two review articles.49,50
In this work we predominantly focused on local mode stretching force constants ka(AB) reflecting the intrinsic strength the bond/weak interaction between two atoms A and B,77 applied to ML and ligand–histidine HB. For easier comparison we transformed the local mode force constants ka(AB) into relative bond strength orders (BSO) according to the generalized Badger rule derived by Cremer, Kraka, and coworkers78,79via a power relationship in the form of BSO = x(ka)y. Two reference molecules with known BSO and force constants are utilized to obtain the parameters for x and y, with the constraint that a zero value for the force constant ka(AB) equals a zero BSO value. The reference molecules utilized in our study are presented in Table 1. For the Me–ligand bonds we referred to Mayer's bond orders80–82 instead of using BSO = 1 for single and BSO = 2 for double bonds, which in past studies involving transition metal bonding have turned out to be a better choice.49,50,64,68,83
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C
CWe complemented LMA with features of the analysis of the electron density ρ(r) via Bader's QTAIM theory.51–53 In particular the covalent character of the ML and HBs were determined via the Cremer–Kraka criterion,84,85 which is based on the local electron density H(r) = G(r) + V(r), where the kinetic energy density is G(r) (positive, destabilizing) and the potential energy density is V(r) (negative, stabilizing). If at the bond critical point rb of ρ(r) between two bonded atoms AB H(rb) is negative, the character of AB bond is predominantly covalent, whereas a positive H(rb) value indicates a predominantly electrostatic character. In addition we analyzed the atomic NBO charges in ML and HB.54,86
2.2 Calculational details
The starting geometries for the Mn(III)Mb protein systems with methanol (CH3OH), water (H2O), nitrite (NO2−), and azide (N3−) ligands placed in the distal heme pocket (see Fig. 1) were taken from the horse heart X-ray structures of oxidized manganese substituted myomoglobin with methanol (PDB entry: 2O5L), water (PDB entry: 2O58), nitrite (PDB entry: 2O5O), and azide (PDB entry: 2O5 M) ligands in the heme distal pocket.18 The starting geometries for the Fe(III)Mb and Co(III)Mb protein systems were obtained from the corresponding Mn(III)Mb complexes by manually substituting the Mn metal center with Fe and Co, respectively. For all eight complexes hydrogen atoms were added to the experimental protein structures and the proteins were neutralized by counter-ions using AMBER.87 In order to simulate a water environment, the active sites of the proteins were surrounded by a sphere of TIP3P water molecules,88 with a radius of 16 Å. After initial minimization with AMBER, the protein systems were divided into a QM part including the heme co-factor, distal and proximal histidine side chains of Mb, and the ligand (ca. 110 atoms), while the MM part included remaining protein atoms (ca. 3000 atoms) as well as water molecules. Based on previous work on Mb64,68 and supported by the finding of others reporting that the PBE0 functional89,90 shows good performance for the calculation of transition metals complexes,91–94 we used for the QM/MM calculations PBE0 in combination with Pople's 6-31G(d,p) basis set.95 For the MM part we applied the AMBER fore field.87 The QM/MM geometry optimization and frequency calculations were performed with scaled electronic embedding using the ONIOM method.96 All QM/MM geometry optimizations finished as local minima on the potential energy surface, i.e., no imaginary normal mode frequencies were found. The calculations in the gas phase were done in this study using the PBE0/6-31G(d,p) level of theory. Using this model chemistry, the calculated FeN bond length of 1.965 Å in the ferric state turned out to be close to the X-ray bond length in bis(1-methylimidazole) (meso-tetramesitylporphinato) Fe(III) (1.970 Å).97Our previous calculations on Fe(III)Mb systems68 in line with experimental data on an Fe(III)Mb–water ligand complex98,99 suggest that the heme co-factor is a high-spin species. Therefore, we calculated Fe(III)Mb complexes with the neutral ligands (i.e., water and methanol) in their quartet electronic state (S = 3/2). In contrast, experimental data on Fe(III)Mb complexes with ionic ligands (i.e., nitrite and azide) suggest a low-spin heme co-factor.100–102 Following this suggestion, we calculated Fe(III)Mb complexes with the nitrite and azide ligands in their doublet electronic state (S = 1/2). Similarly, following suggestions based on experimental data, the Mn(III)Mb protein systems were calculated in this study as high-spin species (S = 2),23,103 and Co(III)Mb protein systems as low-spin species (S = 0).32 Geometry optimizations and vibrational frequency calculations were performed with Gaussian16,104 the LMA analysis was performed using our LModeA program.105 The QTAIM analysis was performed with the AIMALL program106 and NBO charges were calculated utilizing the NBO analysis implemented in Gaussian16. In the following, we present the results of our calculations to three decimal places for most bond properties investigated in this study. To justify this level of accuracy, we performed calculations for Fe(MET)e and Fe(MET)d using the B3LYP/6-311G(d,p)/AMBER level of theory, and a comparison with the PBE0/6-31G(d,p)/AMBER method is provided in Table S1 on page S4 of the ESI.† Additionally, we compared the PBE0/6-31G(d,p)/AMBER level of theory with PBE0/def2-TZVP/SDD(Fe)/AMBER, and the results are presented in Table S7 on page S5 of the ESI.† Table S2 on page S4 of the ESI† shows a comparison of theoretical bond lengths for the heme group in Mn(MET)e and Mn(MET)d with experimental data (PDB entry: 2O5L). Moreover, Tables S3 and S4 on page S5 of the ESI† show the results of calculations for Fe(MET)e and Fe(MET)d using the PBE0/6-31G(d,p)/AMBER level of theory in the doublet, quartet, and sextet electronic states.
3 Results and discussion
In the following ML bonding is discussed for the Fe(III)Mb–ligand complexes and their gas phase analogues, followed by Mn(III)Mb–ligand bonding and Co(III)Mb–ligand bonding. In addition to forming chemical bonds with the metal of the heme cofactor, there is also the opportunity for HB formation with the distal histidine for six protein ligand complexes, as shown in Fig. 1. This will be elucidated in the following sections for the Fe, Co, and Mn protein complexes.3.1 ML bonds in Fe(III)Mb
Fe–ligand bond properties of the investigated Fe(III)Mb–ligand complexes and the corresponding gas phase models are presented in Table 2. Relationships between these properties are shown in Fig. 2 and 3.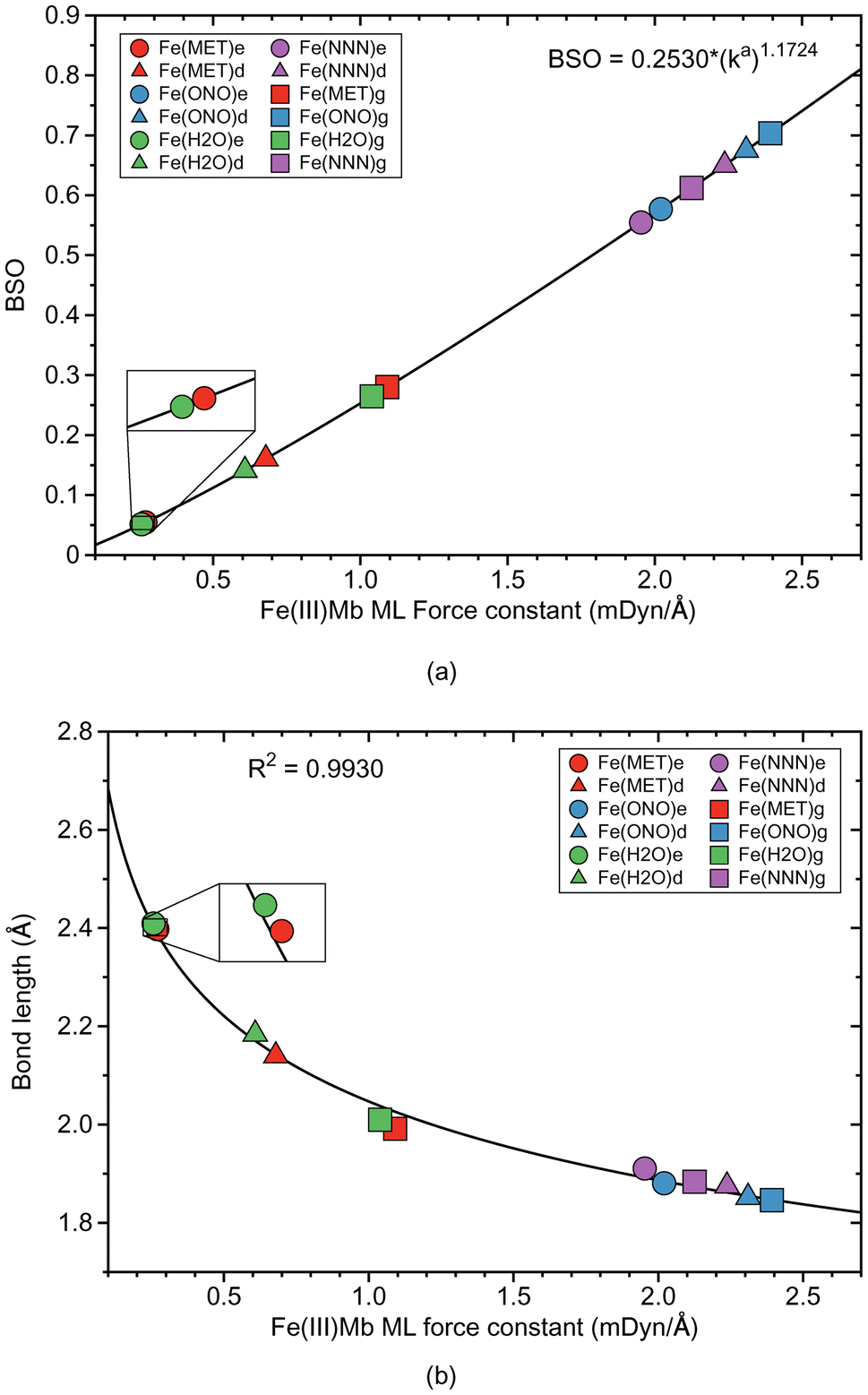 | ||
| Fig. 2 Properties of ML bonds in Fe(III)Mb complexes and corresponding gas phase models. (a) Bond strength order BSO calculated from local mode force constants kavia the generalized Badger rule. (b) Relation between local mode force constant ka and bond length R. For molecular labels, see text. | ||
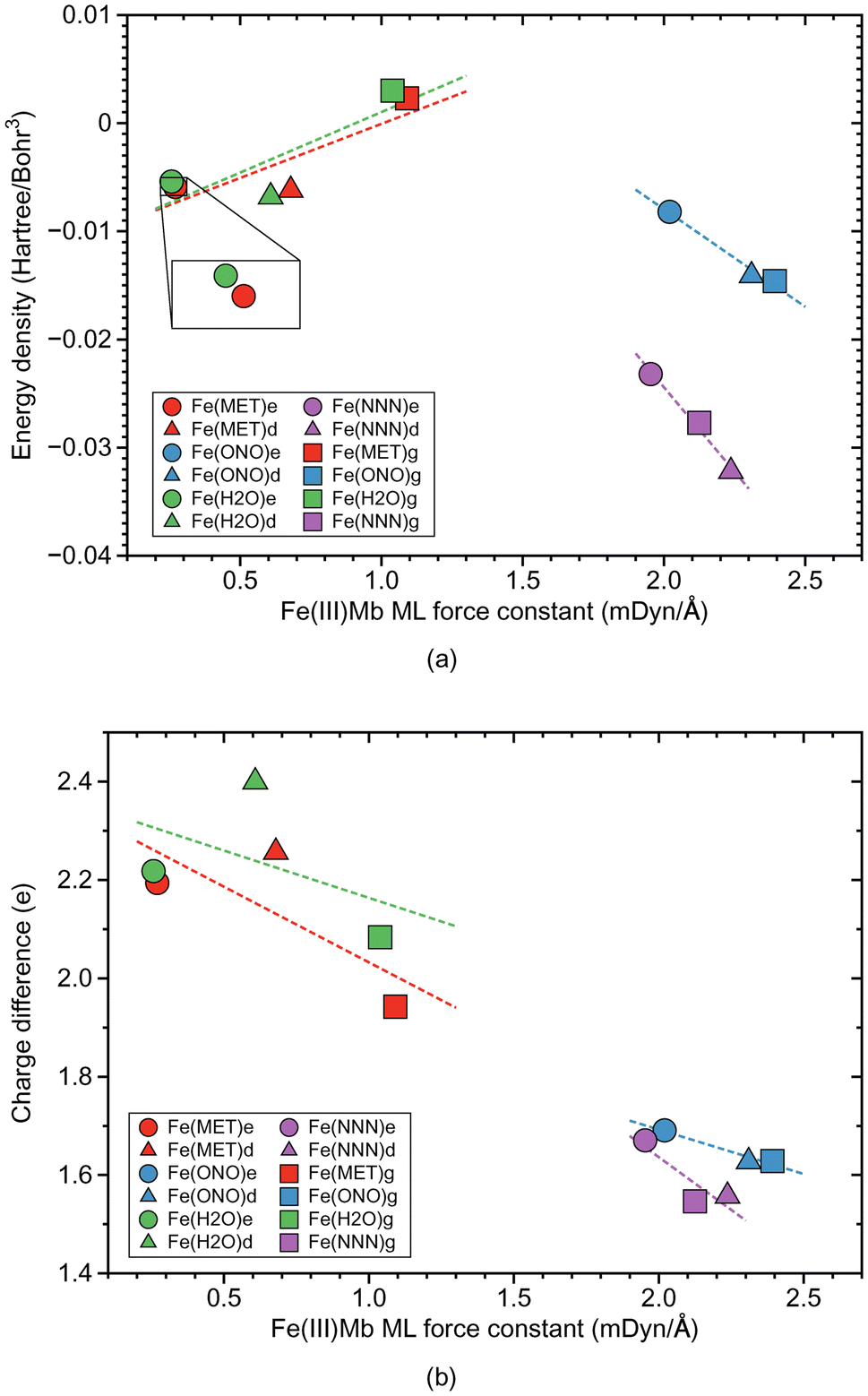 | ||
| Fig. 3 Properties of ML bonds in Fe(III)Mb complexes and corresponding gas phase models. (a) Relation between local mode force constant ka and energy density Hρ. (b) Relation between local mode force constant ka and atomic charge difference. For molecular labels, see text. | ||
| Molecule | Bond | R (Å) | k a (mDyn Å−1) | H ρ (Hr Bohr−3) | Δq (e) | BSO |
|---|---|---|---|---|---|---|
| Fe(MET)e | FeO | 2.398 | 0.270 | −0.0059 | 2.194 | 0.055 |
| Fe(MET)d | FeO | 2.140 | 0.679 | −0.0062 | 2.257 | 0.161 |
| Fe(ONO)e | FeO | 1.881 | 2.020 | −0.0082 | 1.691 | 0.577 |
| Fe(ONO)d | FeO | 1.852 | 2.310 | −0.0141 | 1.627 | 0.675 |
| Fe(H2O)e | FeO | 2.409 | 0.257 | −0.0054 | 2.218 | 0.051 |
| Fe(H2O)d | FeO | 2.184 | 0.608 | −0.0068 | 2.400 | 0.141 |
| Fe(NNN)e | FeN | 1.911 | 1.953 | −0.0232 | 1.671 | 0.555 |
| Fe(NNN)d | FeN | 1.875 | 2.237 | −0.0322 | 1.557 | 0.650 |
| Fe(MET)g | FeO | 1.992 | 1.091 | 0.0023 | 1.942 | 0.280 |
| Fe(ONO)g | FeO | 1.846 | 2.392 | −0.0146 | 1.629 | 0.703 |
| Fe(H2O)g | FeO | 2.010 | 1.039 | 0.0030 | 2.084 | 0.265 |
| Fe(NNN)g | FeN | 1.884 | 2.125 | −0.0277 | 1.546 | 0.612 |
According to Fig. 2a, the Fe–ligand bond strength clusters into two groups; one group for systems with the two neutral ligands, methanol (Fe(MET)e, Fe(MET)d, and Fe(MET)g, with an average ka value of 0.680 mDyn Å−1) and water (Fe(H2O)e, Fe(H2O)d, and Fe(H2O)g, with an average ka value of 0.635 mDyn Å−1). The other group is involving the two ionic ligands, nitrite (Fe(ONO)e, Fe(ONO)d, and Fe(ONO)g, with an average ka value of 2.241 mDyn Å−1) and azide (Fe(NNN)e, Fe(NNN)d, and Fe(NNN)g, with an average ka value of 2.105 mDyn Å−1). Overall, ionic ligands are characterized by considerably stronger Fe–ligand bonding with BSO values between 0.6 and 0.7, whereas as neutral ligands lead to weak Fe–ligand bonds with BSO values in the range of 0.05–0.25. Interesting to note is that for ionic ligands BSO values for proteins and gas phase reference Fe–ligand bonds are comparable whereas for neutral ligands gas phase values are at the stronger end. Moreover, the strength of the Fe–ligand bond in the proteins for the ε protonation form is smaller than for the δ protonation form.
Fig. 2b shows the relationship between the local mode force constant ka and the bond length R for the molecular systems involving the Fe–ligand chemical bond. According to this figure the stronger bond generally correlates well (R2 = 0.9930) with the smaller bond length, which is consistent with the Badger rule.79,107 The covalent character of this bond is expressed in our study by the energy density at the bond critical point Hρ, where the more negative value of the energy density indicates on a more covalent bond character. The relation between those two quantities is presented in Fig. 3a. We find three clusters formed by the individual ligands investigated in this study. The average energy density for the molecular systems with methanol has an Hρ value of −0.0032 Hartree per Bohr3, which indicates on a small covalent character of this bond, similarly as for the molecular systems with water where the average energy density has an Hρ value of −0.0031 Hartree per Bohr3. The Fe–ligand chemical bond has more pronounced covalent character for the molecular systems with the ionic ligands. The average energy density for the molecular systems with nitrite has an Hρ value of −0.0123 Hartree per Bohr3, while for the systems with azide it has an Hρ value of −0.0277 Hartree per Bohr3. Fig. 3b shows the relation between the local mode force constant ka and the charge difference Δq between the Fe atomic charge and the O or N atomic charge of the ligand. Similar as in Fig. 3a, we find three clusters in Fig. 3b formed by the individual ligands. Larger charge differences are observed for molecular systems with water (an average Δq value of 2.234 e) and with methanol (an average Δq value of 2.131 e), and smaller for systems with nitrite (an average Δq value of 1.649 e) and azide (an average Δq value of 1.591 e).
3.2 ML bonds in Mn(III)Mb
ML bond properties of the Mn(III)Mb–ligand complexes and the corresponding gas phase models are presented in Table 3. Relationships between these properties are shown in Fig. 4 and 5.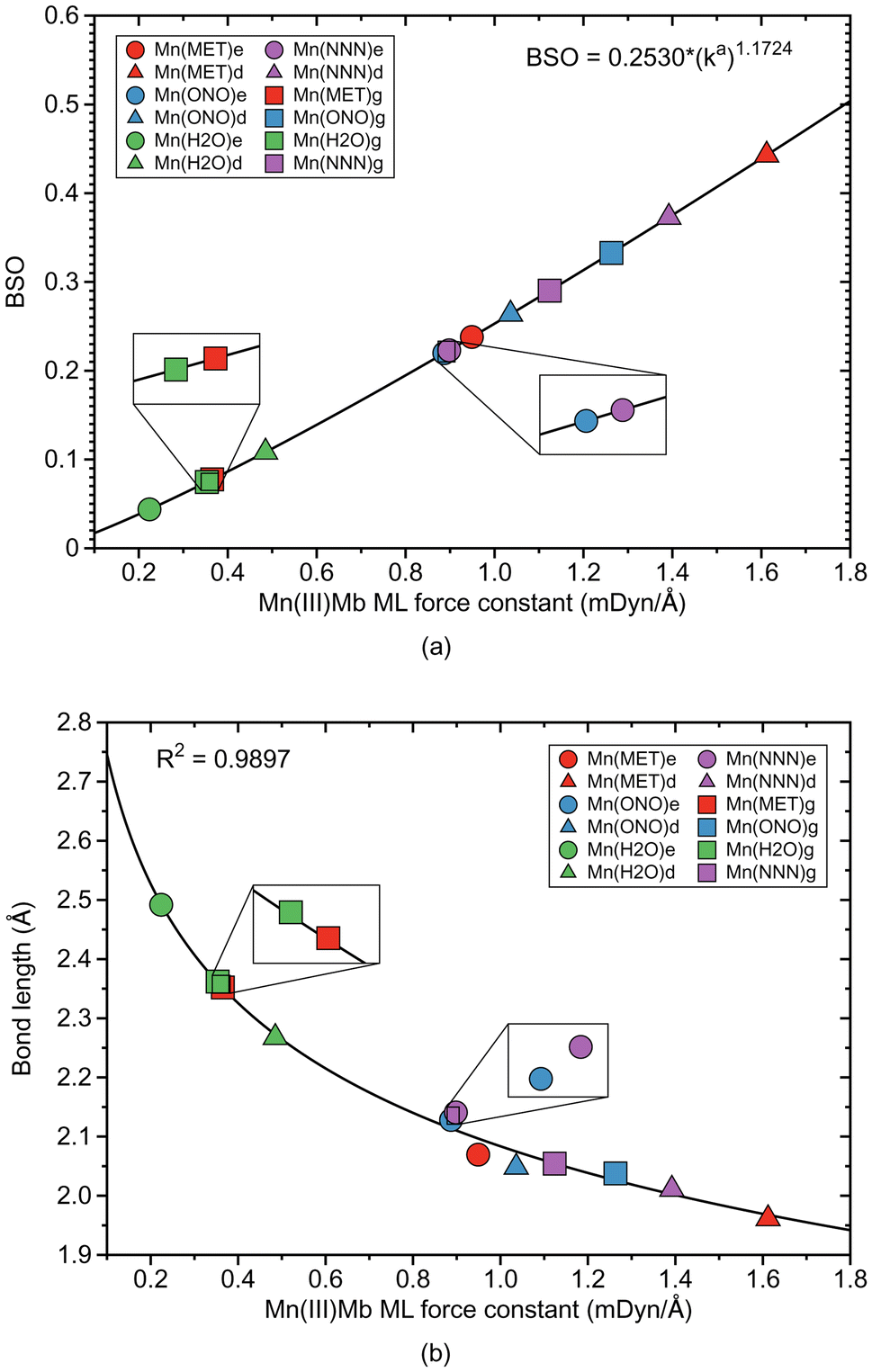 | ||
| Fig. 4 Properties of ML bonds in Mn(III)Mb–ligand complexes and corresponding gas phase models. (a) Bond strength order BSO calculated from local mode force constants kavia the generalized Badger rule. (b) relation between local mode force constant ka and bond length R. For molecular labels, see text. | ||
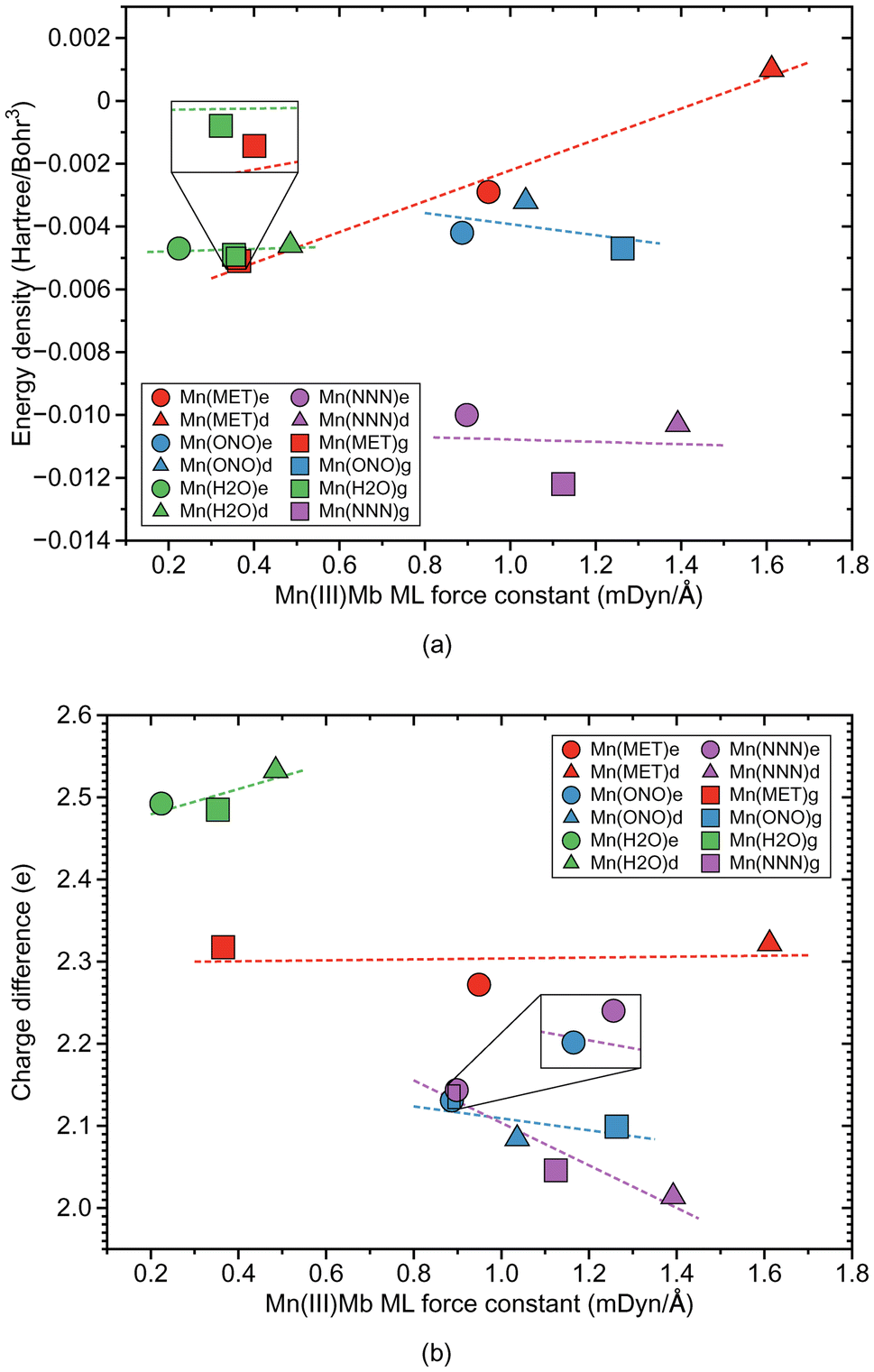 | ||
| Fig. 5 Properties of ML bond in Mn(III)Mb–ligand complexes and corresponding gas phase models. (a) Relation between local mode force constant ka and energy density Hρ. (b) Relation between local mode force constant ka and atomic charge difference. For molecular labels, see text. | ||
| Molecule | Bond | R (Å) | k a (mDyn Å−1) | H ρ (Hr Bohr−3) | Δq (e) | BSO |
|---|---|---|---|---|---|---|
| Mn(MET)e | MnO | 2.070 | 0.949 | −0.0029 | 2.272 | 0.238 |
| Mn(MET)d | MnO | 1.961 | 1.612 | 0.0010 | 2.322 | 0.443 |
| Mn(ONO)e | MnO | 2.128 | 0.887 | −0.0042 | 2.131 | 0.220 |
| Mn(ONO)d | MnO | 2.048 | 1.036 | −0.0032 | 2.084 | 0.264 |
| Mn(H2O)e | MnO | 2.492 | 0.224 | −0.0047 | 2.492 | 0.044 |
| Mn(H2O)d | MnO | 2.268 | 0.485 | −0.0046 | 2.532 | 0.108 |
| Mn(NNN)e | MnN | 2.141 | 0.898 | −0.0100 | 2.144 | 0.223 |
| Mn(NNN)d | MnN | 2.011 | 1.392 | −0.0103 | 2.014 | 0.373 |
| Mn(MET)g | MnO | 2.352 | 0.365 | −0.0051 | 2.317 | 0.078 |
| Mn(ONO)g | MnO | 2.038 | 1.263 | −0.0047 | 2.099 | 0.333 |
| Mn(H2O)g | MnO | 2.362 | 0.353 | −0.0049 | 2.485 | 0.075 |
| Mn(NNN)g | MnN | 2.054 | 1.124 | −0.0122 | 2.046 | 0.290 |
According to Fig. 4a, the Mn–ligand bonds cluster into two groups, one with neutral ligands and one with ionic ligands. The molecular systems with the water ligand have an average force constant ka of 0.354 mDyn Å−1. Similarly as for systems with Fe–ligand bonds, the strength of the Mn–ligand bond in the protein is smaller for the ε protonation form of distal histidine than in the δ form. The strength of the Mn–ligand bond in the systems with methanol is widely distributed keeping a small value for the gas phase model (ka of a value of 0.365 mDyn Å−1), through a medium value for the ε protein system (ka of a value of 0.949 mDyn Å−1), to a relatively big value for the δ protein system (ka of a value of 1.612 mDyn Å−1). Both molecular systems with the ionic ligands have a medium strength, with the average value of the force constant ka of a value of 1.062 mDyn Å−1 for the system with nitrite, and a value of 1.138 mDyn Å−1 for the system with azide. However, the average strength of the Mn–ligand bonds (ka of a value of 0.882 mDyn Å−1), is smaller than the average strength of the Fe–ligand bonds (ka of a value of 1.415 mDyn Å−1). For the neutral ligands forming the bond with Mn, the BSO values range between 0.05 and 0.1, and the BSO values for the ionic ligands are in the range of 0.2–0.45, i.e., they show a larger spread than their Fe–ligand counterpart. According to Fig. 4b, the local mode force constant ka of the Mn–ligand bond relatively good correlates (R2 = 0.9897) with the bond length R, and according to Fig. 5a, the Mn–ligand bonds in all systems have a covalent character, confirmed by negative values of the energy density (an average Hρ value of −0.0047, −0.0040, and −0.0108 Hartree per Bohr3, for the systems with water, nitrite, and azide, respectively), with one exception. The Mn–ligand bond shows electrostatic character for the δ protein system with methanol (Hρ of a value of 0.0010 Hartree per Bohr3), which is also the strongest Mn–ligand bond (ka of a value of 1.612 mDyn Å−1). According to Fig. 5b, the largest atomic charge difference of the Mn–ligand bond is observed in our calculations for molecular systems with water (an average Δq value of 2.503 e), and the smallest values are observed for both systems with the ionic ligands (the average Δq value of 2.105 and 2.068 e, for the system with nitrite and azide, respectively). The charge difference for the system with methanol has a medium value (an average Δq value of 2.304 e).
3.3 ML bonds in Co(III)Mb
Co–ligand bond properties of the Co(III)Mb–ligand complexes and the corresponding gas phase models are presented in Table 4. Relationships between these properties are shown in Fig. 6 and 7. According to Fig. 6a, the Co–ligand bond strengths can be grouped into two clusters, one representing the weaker neutral Co–ligand bonds and one with the stronger Co–ligand ionic bonds, i.e., we find the same trends as for the Fe and Mn systems. The Co–ligand bond strength for the systems with the neutral ligands is substantially smaller (average ka value of 1.262 and 1.273 mDyn Å−1, for water and methanol, respectively) than for the systems with the ionic ligands (average ka value of 2.298 and 2.063 mDyn Å−1, for nitrite and azide, respectively). However, in contrast to the Fe–ligand bonds, we find a larger spread for the neutral system with the BSO range between 0.2–0.45 for the neutral ligands, and 0.5–0.7 for the ionic ligands, respectively. Similarly as in the Fe–ligand bonds, the strength of the Co–ligand bond in the ε protein conformer is smaller than that in the δ conformer. Overall, the average strength of the Co–ligand bonds (average ka of a value of 1.724 mDyn Å−1) is larger than the average strength of the Fe–ligand bonds (average ka of a value of 1.415 mDyn Å−1) and that of the Mn–ligand bonds (average ka of a value of 0.882 mDyn Å−1).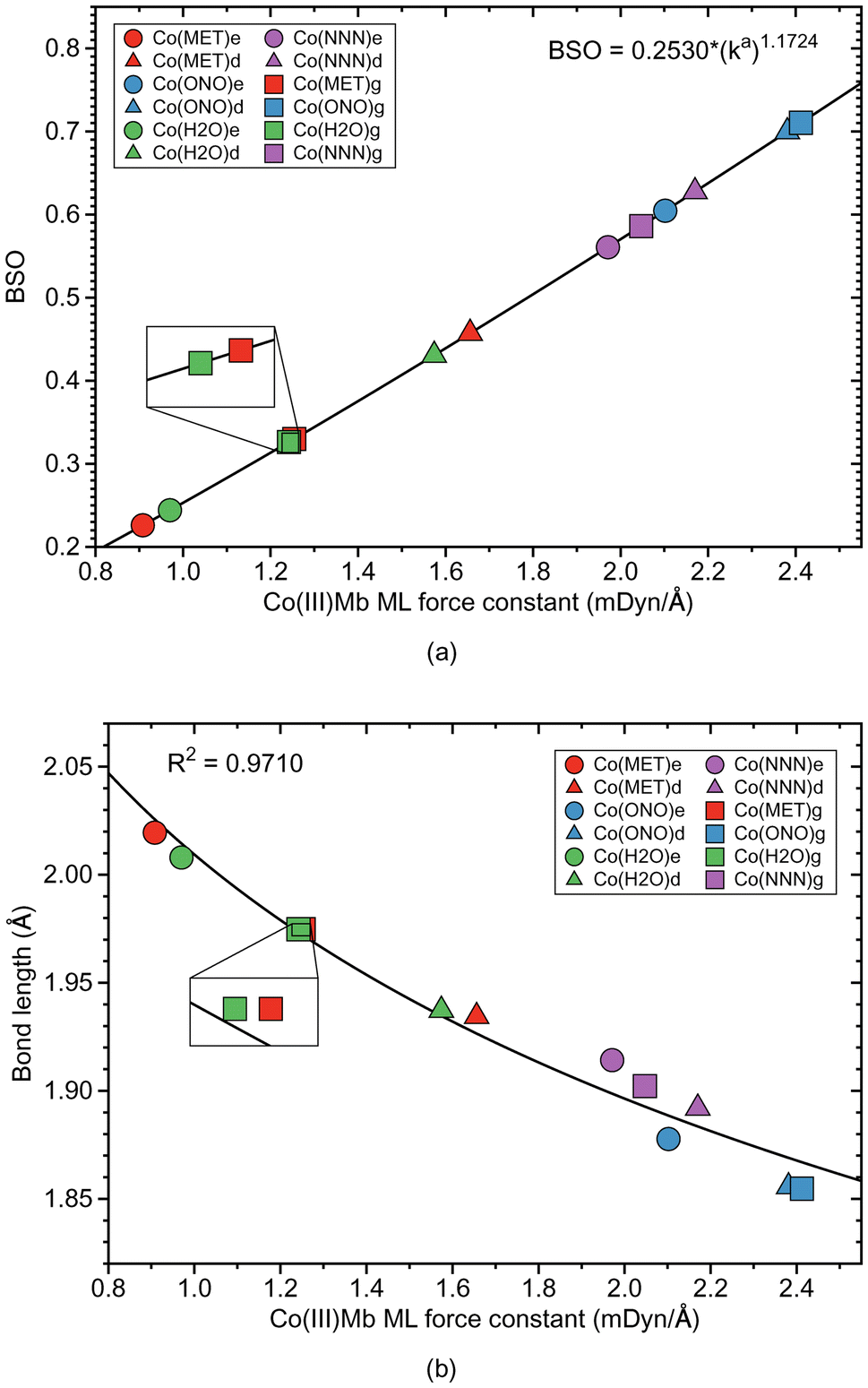 | ||
| Fig. 6 Properties of ML bonds in Co(III)Mb–ligand complexes and corresponding gas phase models. (a) Bond strength order BSO calculated from local mode force constants kavia the generalized Badger rule. (b) Relation between local mode force constant ka and bond length R. For molecular labels, see text. | ||
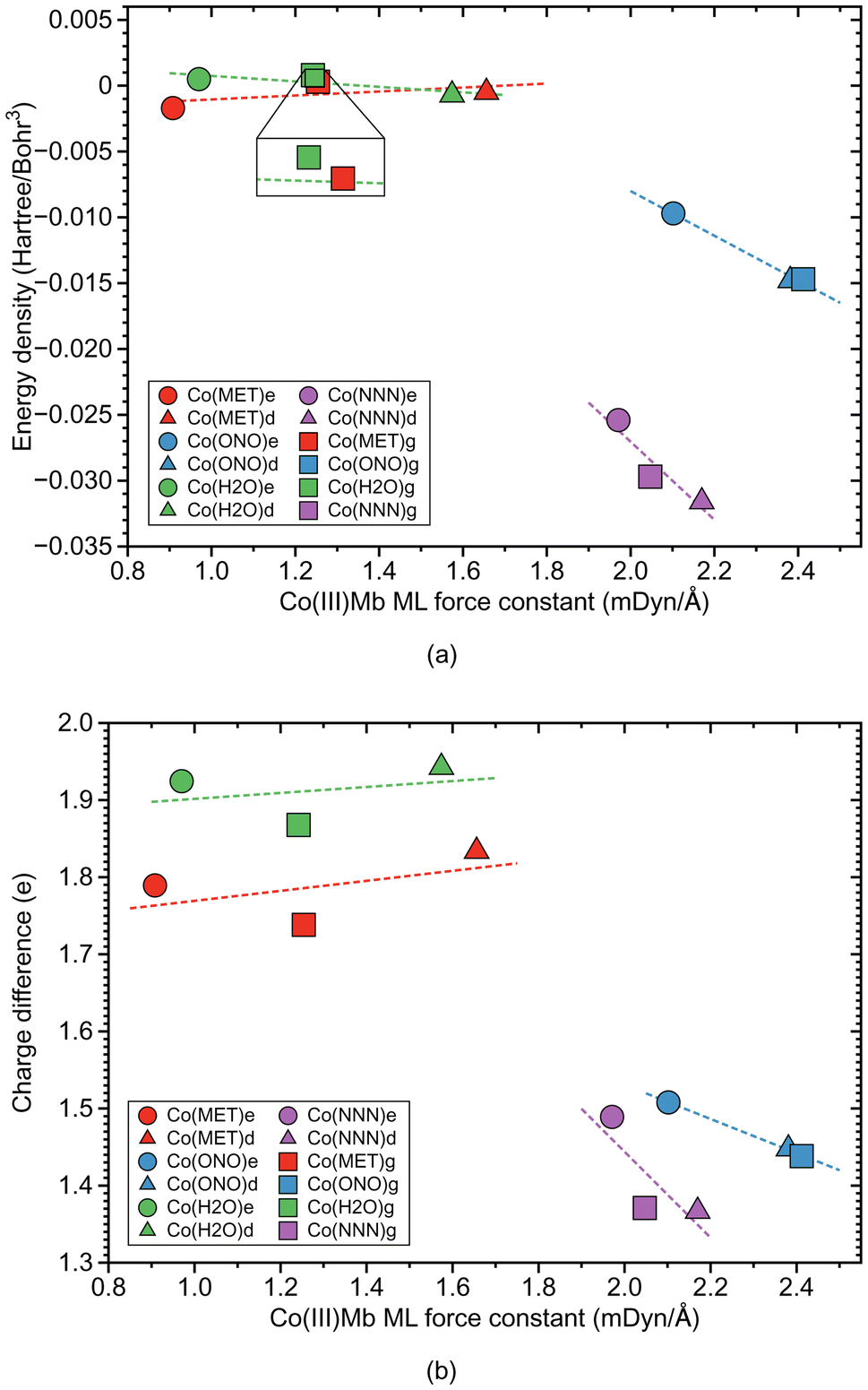 | ||
| Fig. 7 Properties of ML bonds in Co(III)Mb–ligand complexes and corresponding gas phase models. (a) Relation between local mode force constant ka and energy density Hρ. (b) Relation between local mode force constant ka and atomic charge difference. For molecular labels, see text. | ||
| Molecule | Bond | R (Å) | k a (mDyn Å−1) | H ρ (Hr Bohr−3) | Δq (e) | BSO |
|---|---|---|---|---|---|---|
| Co(MET)e | CoO | 2.020 | 0.908 | −0.0017 | 1.789 | 0.226 |
| Co(MET)d | CoO | 1.934 | 1.656 | −0.0005 | 1.834 | 0.457 |
| Co(ONO)e | CoO | 1.878 | 2.102 | −0.0097 | 1.508 | 0.605 |
| Co(ONO)d | CoO | 1.856 | 2.381 | −0.0148 | 1.448 | 0.700 |
| Co(H2O)e | CoO | 2.008 | 0.970 | 0.0005 | 1.924 | 0.244 |
| Co(H2O)d | CoO | 1.937 | 1.574 | −0.0007 | 1.943 | 0.431 |
| Co(NNN)e | CoN | 1.914 | 1.971 | −0.0254 | 1.489 | 0.561 |
| Co(NNN)d | CoN | 1.892 | 2.170 | −0.0316 | 1.367 | 0.628 |
| Co(MET)g | CoO | 1.975 | 1.254 | 0.0003 | 1.738 | 0.330 |
| Co(ONO)g | CoO | 1.855 | 2.412 | −0.0147 | 1.438 | 0.710 |
| Co(H2O)g | CoO | 1.975 | 1.242 | 0.0008 | 1.868 | 0.326 |
| Co(NNN)g | CoN | 1.902 | 2.047 | −0.0297 | 1.371 | 0.586 |
According to Fig. 6b, Co–ligand local mode force constants ka correlate well correlates with the Co–ligand bond lengths R (R2 = 0.9710). Fig. 7a reveals that, similarly as found for the Fe systems, the neutral Co–ligand bonds have an energy density Hρ which is close to zero (an average Hρ value of 0.0002 and −0.0006 Hartree per Bohr3, for water and methanol, respectively), indicating on a predominant electrostatic character of this bond. However, the systems with the ionic Co–ligand bonds display a covalent character (average Hρ value of −0.0131 and −0.0289 Hartree per Bohr3, for nitrite and azide, respectively). As shown in Fig. 7b, we find a similar relationship between the atomic charge difference Δq and the local mode force constant ka for the Co–ligand bonds as for the systems involving the Fe–ligand bonds. The charge difference between the Co and ligand atoms for the neutral ligands is relatively big (average Δq value of 1.912 and 1.787 e, for water and methanol, respectively), when compared to the charge differences for the systems involving the ionic ligands (average Δq value of 1.465 and 1.409 e, for nitrite and azide, respectively).
3.4 Protein–ligand hydrogen bonding
As depicted in Fig. 1, in six of the investigated Mb–ligand complexes, the ligand can form a HB with the distal histidine, namely in Me(MET)e, Me(MET)d, Me(H2O)e, Me(H2O)d, Me(ONO)e, and Me(NNN)e. The ε form of the distal histidine serves as HB donor leading to O⋯H type HBs with the ligand for Me(MET)e, Me(H2O)e, and Me(ONO)e, and for Me(NNN)e to an N⋯H type HB. The δ form of the distal histidine serves as HB acceptor leading to N⋯H type HBs with the ligand for Me(MET)d and Me(H2O)d.Table 5 shows HB properties for the MeMb–ligand complexes (Me = Fe, Mn, Co) along with HB properties of the water dimer (H2OHOH) and the water–ammonia pair (HOHNH3, where water is a hydrogen atom donor) for comparison. Fig. 8, 9, and 10 show HB BSO values calculated from local mode force constants kavia the generalized Badger rule, and the relationship between local mode HB force constant ka and bond length R. The corresponding relationships between HB force constant ka and energy density Hρ, as well as between HB force constant ka and the atomic charge difference between the two atoms engaged in the HB, are presented in Fig. S1–S3 of the ESI.† In the following HBs for the Fe(III)Mb, Mn(III)Mb, and Co(III)Mb–ligand complexes are discussed, followed by the CNM analysis comparing ε and δ hydrogen bonding.
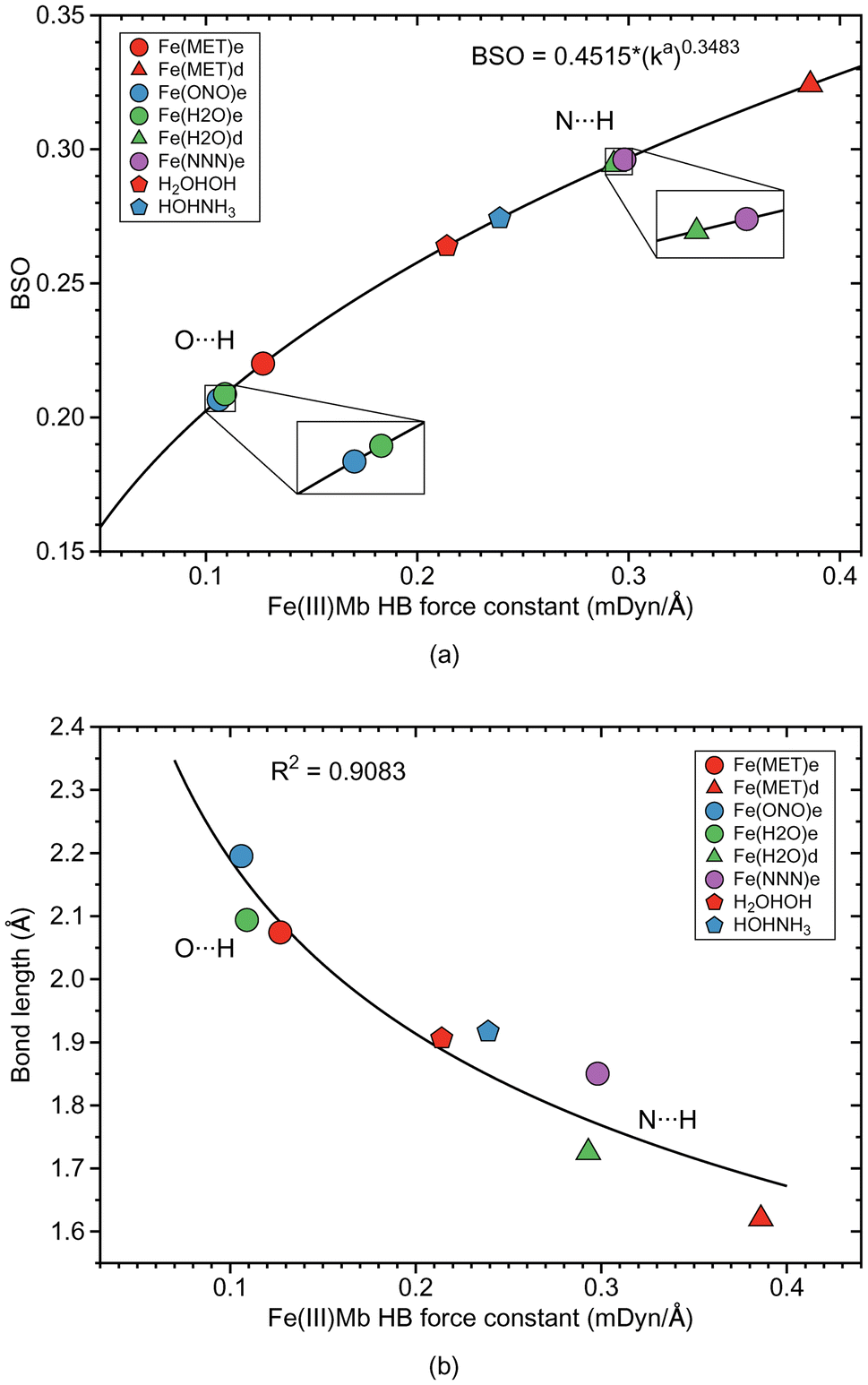 | ||
| Fig. 8 HB properties of Fe(III)Mb–ligand complexes and corresponding properties of the water and water–ammonia molecular complexes. (a) Bond strength order BSO calculated from local mode force constants kavia the generalized Badger rule. (b) Relation between local mode force constant ka and bond length R. For molecular labels, see text. | ||
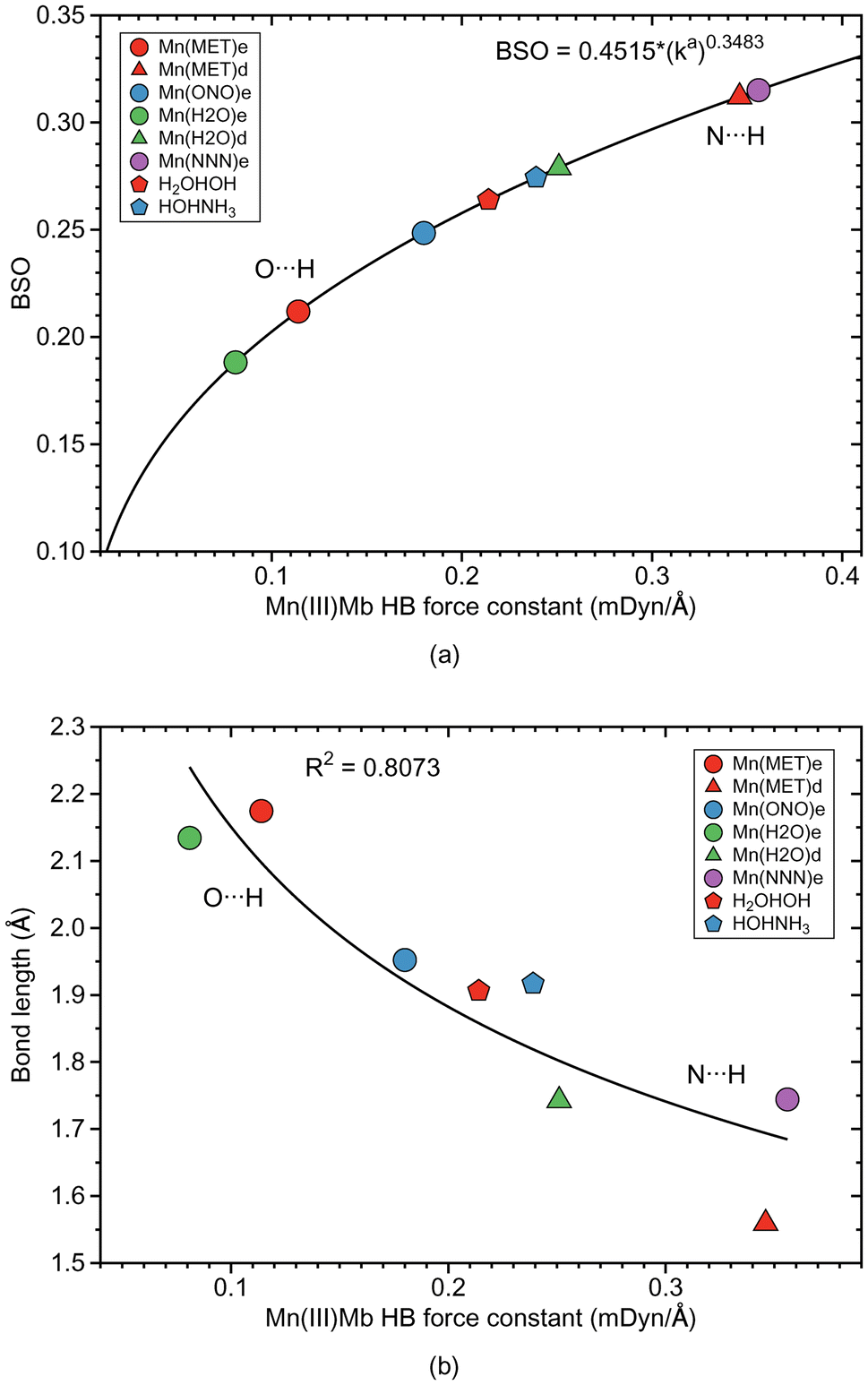 | ||
| Fig. 9 HB properties of Mn(III)Mb–ligand complexes and corresponding properties of the water and water–ammonia molecular complexes. (a) Bond strength order BSO calculated from local mode force constants kavia the generalized Badger rule. (b) Relation between local mode force constant ka and bond length R. For molecular labels, see text. | ||
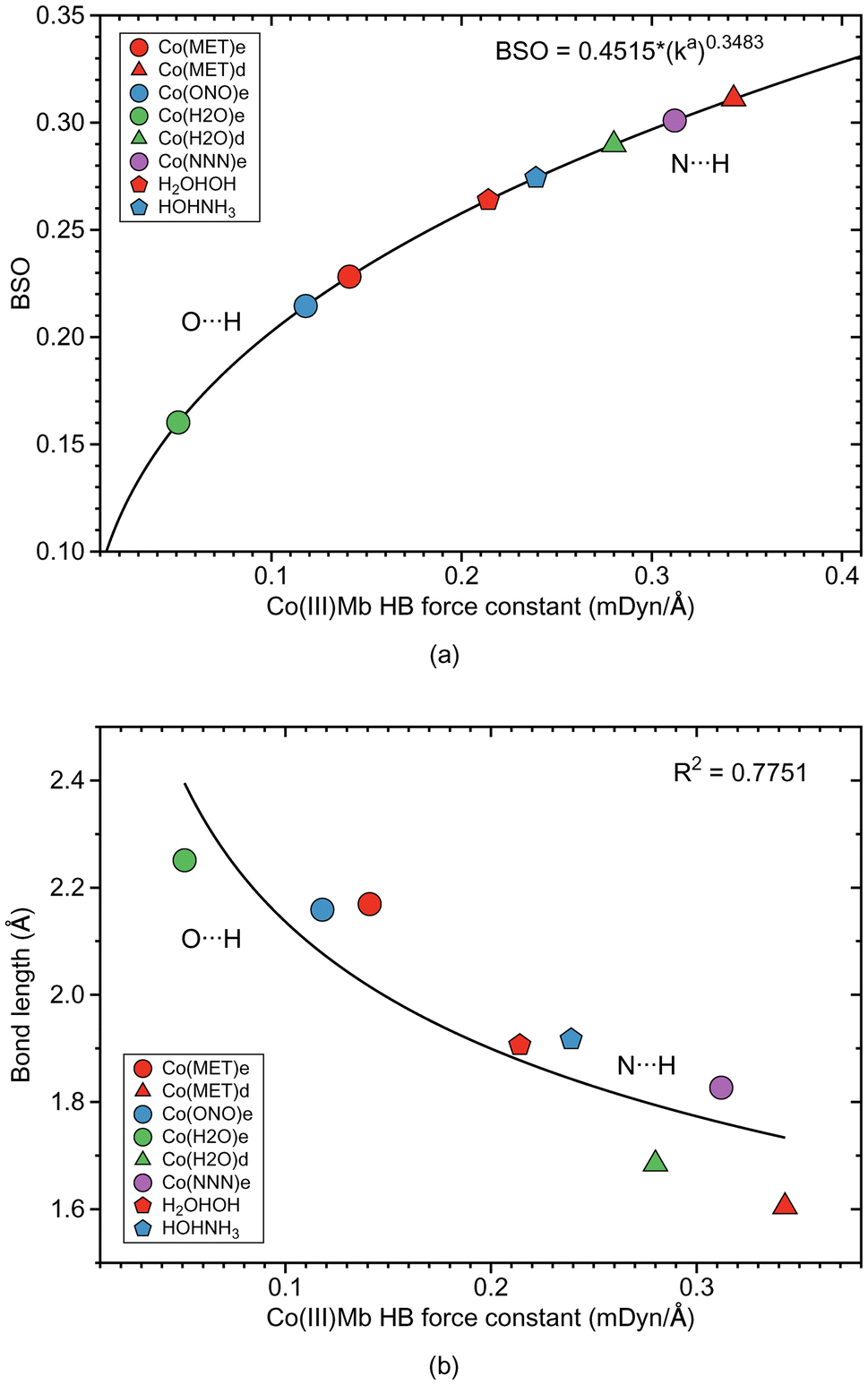 | ||
| Fig. 10 HB properties of CO(III)Mb–ligand complexes and corresponding properties of the water and water–ammonia molecular complexes. (a) Relationship between local mode force constant ka and BSO. (b) Relation between local mode force constant ka and bond length R. For molecular labels, see text. | ||
| Molecule | Bond | R (Å) | k a (mDyn Å−1) | H ρ (Hr Bohr−3) | Δq (e) | BSO |
|---|---|---|---|---|---|---|
| Fe(MET)e | O⋯H | 2.074 | 0.127 | −0.0008 | 1.223 | 0.220 |
| Fe(MET)d | N⋯H | 1.620 | 0.386 | −0.0118 | 1.111 | 0.324 |
| Fe(ONO)e | O⋯H | 2.195 | 0.106 | −0.0006 | 0.994 | 0.207 |
| Fe(H2O)e | O⋯H | 2.094 | 0.109 | −0.0006 | 1.421 | 0.209 |
| Fe(H2O)d | N⋯H | 1.726 | 0.293 | −0.0037 | 1.103 | 0.294 |
| Fe(NNN)e | N⋯H | 1.850 | 0.298 | −0.0011 | 1.018 | 0.296 |
| Mn(MET)e | O⋯H | 2.174 | 0.114 | −0.0004 | 1.235 | 0.212 |
| Mn(MET)d | N⋯H | 1.559 | 0.346 | −0.0210 | 1.117 | 0.312 |
| Mn(ONO)e | O⋯H | 1.952 | 0.180 | −0.0014 | 1.091 | 0.248 |
| Mn(H2O)e | O⋯H | 2.134 | 0.081 | −0.0004 | 1.421 | 0.188 |
| Mn(H2O)d | N⋯H | 1.743 | 0.251 | −0.0035 | 1.112 | 0.279 |
| Mn(NNN)e | N⋯H | 1.744 | 0.356 | −0.0033 | 1.123 | 0.315 |
| Co(MET)e | O⋯H | 2.169 | 0.141 | −0.0004 | 1.170 | 0.228 |
| Co(MET)d | N⋯H | 1.605 | 0.343 | −0.0141 | 1.121 | 0.311 |
| Co(ONO)e | O⋯H | 2.159 | 0.118 | −0.0007 | 0.962 | 0.215 |
| Co(H2O)e | O⋯H | 2.251 | 0.051 | 0.0001 | 1.346 | 0.160 |
| Co(H2O)d | N⋯H | 1.684 | 0.280 | −0.0063 | 1.114 | 0.290 |
| Co(NNN)e | N⋯H | 1.827 | 0.312 | −0.0014 | 0.993 | 0.301 |
| H2 OHOH | O⋯H | 1.906 | 0.214 | −0.0013 | 1.446 | 0.264 |
| HOHNH3 | N⋯H | 1.917 | 0.239 | −0.0016 | 1.657 | 0.274 |
According the Fig. S1a of the ESI,† the weak O⋯H type HBs in Fe(MET)e, Fe(H2O)e, and Fe(ONO)e, exhibit on small covalent character as indicated by their small negative energy density values. On the other hand, the strong N⋯H type HBs of Fe(MET)d and Fe(H2O)d, have more negative energy density values, disclosing more covalent bond character. It is interesting to note that the covalent character of the gas phase reference molecules (H2OHOH and HOHNH3), is relatively small, which indicates on a predominant effect of the protein environment, increasing the covalent character of the N⋯H type HBs and their strength. According to Fig. 8b, the HB bond strength of the Fe(III)Mb proteins shows a weak correlation with the corresponding HB bond length, with the N⋯H bonds being overall shorter than their O⋯H counterparts.
Overall we observe the same trends as for the Fe(III)Mb–ligand complexes with the following exceptions. As shown in Fig. 9a, weak O⋯H type HBs are formed between the neutral ligands and the ε form of distal histidine, namely Mn(MET)e and Mn(H2O)e. We find the strongest N⋯H type HB for the ε form of distal histidine with the azide anion (Mn(NNN)e). The strongest HBs of N⋯H type involving the δ form of distal histidine are found for Mn(MET)d and Mn(H2O)d. Whereas similar to Fe(III)Mb, the weaker O⋯H type HB of Mn(H2O)e is paired with a weaker Mn–ligand bond for the ε Mn(III)Mb protein, for Mn(ONO)e and Mn(MET)e the Mn–ligand bond strength lies in the middle range. The ε form of the Mn(III)Mb protein azide complex (Mn(NNN)e) with the strongest N⋯H type HB in this series is paired with a Mn–ligand bond of middle strength.
Generally, for the Co(III)Mb–ligand complexes we observe the same trends as for the Fe(III)Mb–ligand systems with some exceptions. According to Fig. 10a, the weakest HBs of the O⋯H type are found for the ε form of the Co(III)Mb protein (Co(MET)e, Co(ONO)e, and Co(H2O)e). The HB of the N⋯H type for the same ε protein system (Co(NNN)e), is one of the strongest HB in this series. Moreover, both δ Co(III)Mb proteins (Co(MET)d and Co(H2O)d) form relatively strong HBs of the N⋯H type. Similarly as in the Fe(III)Mb and Mn(III)Mb protein systems, the weaker Co–ligand chemical bonds in the Co(MET)e and Co(H2O)e are paired with the weaker HBs of the O⋯H type, however for the azide anion Co(NNN)e, both a strong Co–ligand bond and a strong N⋯H type HB are observed.
3.5 ε versus δ HB visualized by CNM
Additional insights into the difference between ε versus δ ligand–histidine HB determining e.g., the orientation of small molecular ligands in the heme pocket can be gained via our CNM analysis, which decomposes the normal vibrational modes of a molecule into local mode parameters, and as such can identify how the atoms of a specific structural element or functional group move during a specific vibration.49,50 However, it has to be noted that whereas LMA properties can be calculated for a restricted number of local mode parameters of interest, such as the HB force constants ka (HB) and the ML bond constants ka (ML) as in this work, CMN requires the proper choice of a chemically meaningful complete and non-redundant set of Nvib local mode parameters, with Nvib = (3N − 6) for a non-linear and (3N − 5) for a linear molecule being composed of N atoms. This can become unfeasible for systems with a large number of atoms (N > 100), or QM/MM systems, where Nvib is determined from the total number of QM and MM atoms, when performed manually or starting from a large redundant coordinate set and applying some trial and error procedures. Our group has developed two methods in order to help in this situation; (i) the generalized subsystem vibrational analysis (GSVA) developed in our group108,109 which projects out from the full Nvib QM/MM set the important QM vibrations and (ii) LModeAGen67 which offers a convenient way for the generation of local mode parameters based on chemical graph theory.110In the following the CNM analysis for the Fe(MET)e and Fe(MET)d complexes is discussed as an example. Both complexes contain 3049 atoms (103 QM atoms, 2946 MM atoms) leading to a total of Nvib = 9141. After the GSVA procedure extracting the 297 QM normal modes of interest, LModeAGen was applied to generated a set of local mode parameters. In Fig. 11a and b the CNM is shown in the range of 900–3000 cm−1 focusing on the different role of the methanol OH bond. Fig. 11a shows the CNM for Fe(MET)e where one of the distal histidine N–H bonds serves as HB donor and the methanol oxygen as HB acceptor, whereas the methanol OH bond is a spectator bond. Fig. 11b shows the CNM for Fe(MET)d where the distal histidine nitrogen atom serves as HB acceptor and methanol OH bond as HB donor.
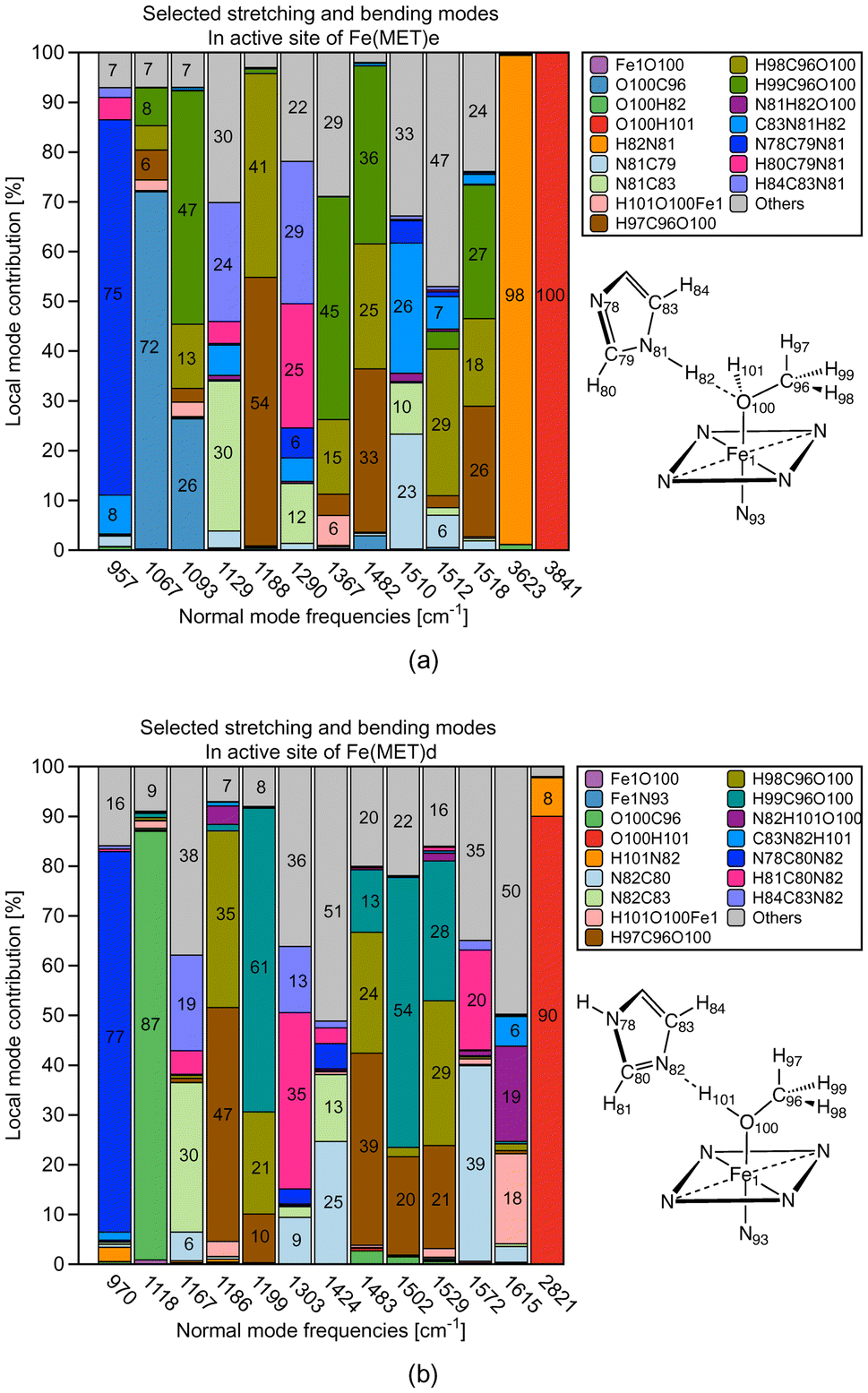 | ||
| Fig. 11 Decomposition of normal vibrational modes into local stretching and bending contributions involved in O⋯H type and N⋯H type HB. (a) Fe(MET)e as representative for O⋯H type HB; (b) Fe(MET)d as representative for N⋯H type HB. For molecular labels, see text. | ||
Fig. 11a clearly confirms the spectator character of the methanol OH bond in Fe(MET)e; the normal mode representing the methanol OH stretching vibration has 100% local OH stretching mode character, in line with the 3841 cm−1 OH stretching frequency, which is in the normal OH stretching range.111 The NH bond of histidine serving as HB donor is with a frequency value of 3623 cm−1 some slightly redshifted,111 but also has 98% local NH mode character. A different picture emerges for Fe(MET)d as revealed by the CNM shown in Fig. 11b. The methanol OH bond of Fe(MET)d serves as HB donor. As a consequence, the OH stretching frequency is considerably redshifted (2821 cm−1). It has 90% local OH stretching character with an 8% admixture of N⋯H local mode stretching; elucidating the different scenarios of methanol OH bonding; qualifying CNM as a helpful tool for artificial Mb designers.
4 Conclusions and outlook
Based on comprehensive QM/MM calculations combined with LMA, we investigated the strength of ML bonding with methanol, water, nitrite, and azide as model ligands in the active site of Fe(III)Mb, Mn(III)Mb, and Co(III)Mb. This analysis was conducted for both the ε and δ forms of the distal histidine. In addition to ML bonding, we explored potential HB between the ligand and the distal histidine, a possibility that has not received much attention to date. To account for the effects of the protein environment, we also examined the corresponding gas-phase models of the protein active sites, using the same metals and ligands. HBs in the protein were compared with HBs in the water and water–ammonia molecular complexes.As depicted in Fig. 12a the results of our study show that the two neutral ligands, methanol and water, form relatively weak ML bonds compared to their ionic counterparts, nitrite and azide, forming considerably stronger ML bonds. Moreover, ML bonds are weaker in the protein than in the gas phase models, which indicates that the protein environment influences ML bond formation/cleavage. The strength of the ML protein bonds also depends on the protonation form of the distal histidine, namely for the ε form of this residue the ML bond is generally weaker than that for the δ form (see Fig. 12a).
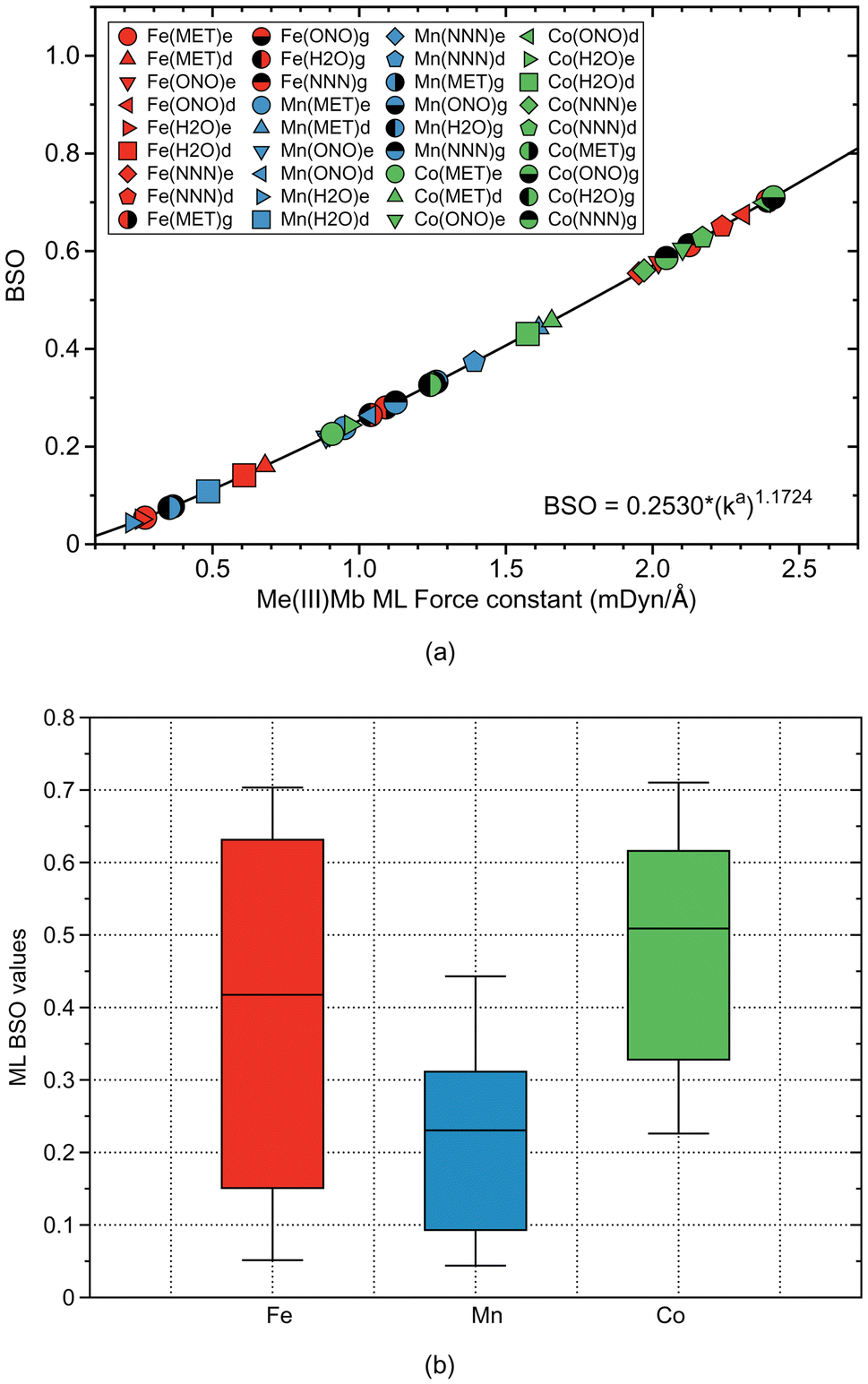 | ||
| Fig. 12 (a) Bond strength order BSO calculated from local mode force constants kavia the generalized Badger rule for all ML bonds of Mb complexes and gas phase models investigated in this study (Me = Fe, Mn, and Co). For molecular labels, see text. (b) Box and whisker plot of BSO values for all ML bonds investigated in this study. | ||
Overall, the average strength of the Co–ligand bonds (average ka of 1.724 mDyn Å−1) is greater than the average strength of Fe–ligand bonds (average ka of 1.415 mDyn Å−1) and that of Mn–ligand bonds (ka of 0.882 mDyn Å−1). These findings provide useful guidelines for fine-tuning of artificial Mbs with specific ML bond strengths. According to Fig. 12b summarizing the strength of the ML bonds for all three metals, the strength of the ML bonds with Fe covers a broader range than the other metals, which indicates that Fe should be selected as a candidate for a catalyst in chemical reactions that require ML bonds of different strengths.
For the ε histidine tautomer (see Fig. 13a) most ligands form O⋯H type HBs, where the distal histidine is the hydrogen atom donor, while for the δ tautomer N⋯H type HBs are formed with the distal histidine as HB acceptor. According to our calculations, the N⋯H type HBs are stronger and shorter in the proteins than in the reference water–ammonia pair, moving the ligands closer to the metal. However, as revealed by Fig. 13b we did not observe a direct correlation between ML and HB bond strengths.
 | ||
| Fig. 13 (a) Bond strength order BSO calculated from local mode force constants kavia the generalized Badger rule for all HB bonds of Mb complexes investigated in this study (Me = Fe, Mn, and Co). (b) Relation between ML and HB bond strengths for all Mb complexes investigated in this study. Zero HB BSO values indicate the lack of HB opportunity. For molecular labels, see text. | ||
The analysis of the HBs formed between the ligands and the distal histidine of Mb adds another layer of useful information, such as on the strength and covalency of HBs formed between ligand and histidine, with O⋯H type HBs overall weaker than their N⋯H type HB counterparts (see Fig. 13a) and how these HBs may influence the ligand orientation in the Mb pocket. We confirmed that stronger HBs are formed with the ionic ligands (nitrite and azide) compared to the neutral ones (methanol and water), which can be useful for the design of ionic reactants and intermediates of catalytic reactions taking place in the active site of Mb and/or its mutations.
A recent experimental investigation shows that replacing the native Mb porphine ring with a porphycene ring increases the protein's catalytic activity for the dehydration of various aldoximes.112 These authors suggest that this enhancement is attributed to the involvement of the distal histidine in the reaction, and that a HB with the distal histidine determines the ligand's correct orientation, significantly reducing the reaction's activation energy. Furthermore, they propose that the hydrogen atom of histidine involved in the HB directly participates in the reaction, underscoring the crucial role of this HB in the overall reaction mechanism. We are currently exploring the mechanistic details with our Unified Reaction Valley Approach (URVA)113,114 combined with LMA, which will provide a comprehensive, holistic picture. The results will be published in a forthcoming article.
In conclusion, our investigation provides in-depth insight into the strength of ML bonds formed between four prototype ligands (methanol, water, nitrite, and azide) and the metal centers in Fe, Mn, and Co myoglobins at the atomic level. As illustrated in the box-and-whisker plot in Fig. 12b, Fe exhibits a broader variation in ML bond strengths compared to Mn and Co. This suggests that Fe, the metal found in native myoglobin, is the most versatile candidate for designing artificial myoglobins for catalytic applications that require variable ML bond strengths. However, Mn, Co or other metals might still be preferable for specific synthetic reasons or in cases, such as PET-RAFT polymerization, where specific excitation of the metal is needed. Additionally, as shown in our study, analyzing potential HB interactions between ligands and the distal Mb histidine provides valuable insights into how small molecular ligands orient within the heme pocket. As revealed by our results this orientation is guided by the strength of the HBs formed with the distal histidine.
Overall, our findings identify the strength of both ML bonds and HBs, fully captured by LMA, as a key parameter determining the catalytic activity and function of Mbs. This is particularly relevant when considering neutral versus ionic ligands and other metals such as Mn or Co as alternatives to Fe. The insights gained through our investigation offer valuable guidance for strategically fine-tuning existing artificial Mbs and designing new, versatile variants. Specifically, bond strength combinations like those illustrated in Fig. 13b provide a practical roadmap for future exploration. We hope that our QM/MM-LMA protocol will become a valuable addition to the research community's toolkit.
Author contributions
Marek Freindorf (https://orcid.org/0000-0001-5285-5455): writing – review and editing, conceptualization, investigation, data curation, methodology; Elfi Kraka (https://orcid.org/0000-0002-9658-5626): writing – review and editing, conceptualization, funding acquisition, supervision.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the SMU O'Donnell Data Science and Research Computing Institute for providing generous computational resources. This work was financially supported by the National Science Foundation, Grant CHE 2102461. We also thank Bangaru Bhaskararao and Juliana Antonio for fruitful discussions.References
- B. M. Hoffman, The Porphyrins, Academic Press, New York, 1979, vol. 7, pp. 403–444 Search PubMed.
- J. S. Olson, Antioxid. Redox Signaling, 2019, 32, 228–246 CrossRef.
- G. A. Ordway and D. J. Garry, J. Exp. Biol., 2004, 207, 3441–3446 CrossRef CAS PubMed.
- I. E. Elkholi, M. E. Elsherbiny and M. Emara, Biochim. Biophys. Acta, 2022, 1877, 188706 CAS.
- E. Zonetff, Y. Wang, C. Jackson, O. Smith, S. Duchi, C. Onofrillo, B. Farrugia, S. Moulton, R. Williams, C. Parish, D. R. Nisbet and L. M. Caballero-Aguilar, Nat. Commun., 2024, 15, 4361 CrossRef PubMed.
- F. Darain, P. Yager, K. L. Gan and S. C. Tjin, Biosens. Bioelectron., 2009, 24, 1744–1750 CrossRef CAS PubMed.
- K. K. Adepu, A. Anishkin, S. H. Adams and S. V. Chintapalli, Physiol. Rev., 2024, 104, 1611–1642 CrossRef PubMed.
- Y. Kagawa, K. Oohora and T. Hayashi, J. Inorg. Biochem., 2024, 252, 112459 CrossRef CAS PubMed.
- K. Oohora, A. Onoda and T. Hayashi, Acc. Chem. Res., 2019, 52, 945–954 CrossRef CAS PubMed.
- M. Tinzl, J. V. Diedrich, P. R. Mittl, M. Clémancey, M. Reiher, J. Proppe, J.-M. Latour and D. Hilvert, J. Am. Chem. Soc., 2024, 146, 1957–1966 CrossRef CAS PubMed.
- K. Yu and T. R. Ward, J. Inorg. Biochem., 2024, 258, 112621.1–112621.18 CrossRef PubMed.
- W.-N. Xu, Y.-D. Gao, P. Su, L. Huang, Z.-L. He and L.-C. Yang, ACS Catal., 2024, 14, 14139–14160 CrossRef CAS.
- Y.-W. Lin, J. Inorg. Biochem., 2024, 257, 112595.1–112595.14 CrossRef PubMed.
- M. Bordeaux, V. Tyagi and R. Fasan, Angew. Chem., 2015, 127, 1764–1768 CrossRef.
- H. M. Key, P. Dydio, D. S. Clark and J. F. Hartwig, Nature, 2016, 534, 534–537 CrossRef CAS PubMed.
- P. Dydio, H. M. Key, A. Nazarenko, J. Y.-E. Rha, V. Seyedkazemi, D. S. Clark and J. F. Hartwig, Science, 2016, 354, 102–106 CrossRef CAS PubMed.
- G. Sreenilayam, E. J. Moore, V. Steck and R. Fasan, Adv. Synth. Catal., 2017, 359, 2076–2089 CrossRef CAS PubMed.
- Z. N. Zahran, L. Chooback, D. M. Copeland, A. H. West and G. B. Richter-Addo, J. Inorg. Biochem., 2008, 102, 216–233 CrossRef CAS PubMed.
- K. L. Stone and S. M. Ahmed, Inorganics, 2016, 4, e12.1–e12.16 CrossRef.
- R. Lin, C. E. Immoos and P. J. Farmer, J. Biol. Inorg. Chem., 2000, 5, 738–747 CrossRef CAS PubMed.
- K. Oohora, Y. Kihira, E. Mizohata, T. Inoue and T. Hayashi, J. Am. Chem. Soc., 2013, 135, 17282–17285 CrossRef CAS PubMed.
- K. Oohora, H. Meichin, Y. Kihira, H. Sugimoto, Y. Shiro and T. Hayashi, J. Am. Chem. Soc., 2017, 139, 18460–18463 CrossRef CAS PubMed.
- K. L. Stone, J. Hua and H. Choudhry, Inorganics, 2015, 3, 219–229 CrossRef CAS.
- T. M. Makris, K. v. Koenig, I. Schlichting and S. G. Sligar, J. Inorg. Biochem., 2006, 100, 507–518 CrossRef CAS PubMed.
- H. Hirao, K.-B. Cho and S. Shaik, J. Biol. Inorg. Chem., 2008, 13, 521–530 CrossRef CAS PubMed.
- S.-C. Chien, O. Shoji, Y. Morimoto and Y. Watanabe, New J. Chem., 2017, 41, 302–307 RSC.
- N. Kawakami, O. Shoji and Y. Watanabe, ChemBioChem, 2012, 13, 2045–2047 CrossRef CAS PubMed.
- K. Omura, Y. Aiba, H. Onoda, J. K. Stanfield, S. Ariyasu, H. Sugimoto, Y. Shiro, O. Shoji and Y. Watanabe, Chem. Commun., 2018, 54, 7892–7895 RSC.
- K. Omura, Y. Aiba, K. Suzuki, S. Ariyasu, H. Sugimoto and O. Shoji, ACS Catal., 2022, 12, 11108–11117 CrossRef CAS.
- S. I. Mann, A. Nayak, G. T. Gassner, M. J. Therien and W. F. DeGrado, J. Am. Chem. Soc., 2021, 143, 252–259 CrossRef CAS PubMed.
- I. Degtyarenko, R. M. Nieminen and C. Rovira, Biophys. J., 2006, 91, 2024–2034 CrossRef CAS PubMed.
- S. Neya, M. Suzuki, T. Hoshino and A. T. Kawaguchi, Inorg. Chem., 2013, 52, 7387–7393 CrossRef CAS PubMed.
- T. D. Rapson, S. Warneke, M. M. Musameh, H. Dacres, B. C. T. Macdonald and S. C. Trowell, RSC Adv., 2015, 5, 89003–89008 RSC.
- Y. Morita, K. Oohora, E. Mizohata, A. Sawada, T. Kamachi, K. Yoshizawa, T. Inoue and T. Hayashi, Inorg. Chem., 2016, 55, 1287–1295 CrossRef CAS PubMed.
- A. T. Smith, T. Majtan, K. M. Freeman, Y. Su, J. P. Kraus and J. N. Burstyn, Inorg. Chem., 2011, 50, 4417–4427 CrossRef CAS PubMed.
- J. G. Kleingardner, B. Kandemir and K. L. Bren, J. Am. Chem. Soc., 2014, 136, 4–7 CrossRef CAS PubMed.
- D. J. Sommer, M. D. Vaughn, B. C. Clark, J. Tomlin, A. Roy and G. Ghirlanda, Biochim. Biophys. Acta, 2016, 1857, 598–603 CrossRef CAS PubMed.
- K. Kitanishi, M. Shimonaka and M. Unno, ACS Omega, 2021, 6, 34912–34919 CrossRef CAS PubMed.
- L. J. Perkins, B. R. Weaver, A. R. Buller and J. N. Burstyn, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2017625118 CrossRef CAS PubMed.
- M. Meglioli, G. Di Rocco, A. Ranieri, C. A. Bortolotti, M. Sola, G. Battistuzzi and M. Borsari, ChemElectroChem, 2024, 11, e202300821 CrossRef CAS.
- V. Firpo, J. M. Le, V. Pavone, A. Lombardi and K. L. Bren, Chem. Sci., 2018, 9, 8582–8589 RSC.
- R. J. Labidi, B. Faivre, P. Carpentier, J. Perard, P. Gotico, Y. Li, M. Atta and M. Fontecave, J. Am. Chem. Soc., 2024, 146, 28296–28305 CAS.
- I. C. Anderson, D. C. Gomez, M. Zhang, S. J. Koehler and C. A. Figg, Angew. Chem., Int. Ed., 2024, e202414431 Search PubMed.
- C. E. Tzeliou, M. A. Mermigki and D. Tzeli, Molecules, 2022, 27, 2660 CrossRef CAS PubMed.
- M. W. van der Kamp and A. J. Mulholland, Biochemistry, 2013, 52, 2708–2728 CrossRef CAS PubMed.
- V. Guallar and F. H. Wallrapp, Biophys. Chem., 2010, 149, 1–11 CrossRef CAS PubMed.
- A. Warshel and M. Levitt, J. Mol. Biol., 1976, 103, 227–249 CrossRef CAS PubMed.
- A. Warshel and M. Karplus, J. Am. Chem. Soc., 1972, 94, 5612–5625 CrossRef CAS.
- E. Kraka, W. Zou and Y. Tao, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2020, 10, 1480 Search PubMed.
- E. Kraka, M. Quintano, H. W. La Force, J. J. Antonio and M. Freindorf, J. Phys. Chem. A, 2022, 126, 8781–8798 CrossRef CAS PubMed.
- R. F. W. Bader, Monatsh. Chem., 2005, 136, 819–854 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory (International Series of Monographs on Chemistry), Clarendon Press, 1994 Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- F. Weinhold and C. R. Landis, Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective, Cambridge University Press, 2005 Search PubMed.
- J. D. Kelley and J. J. Leventhal, in Problems in Classical and Quantum Mechanics: Normal Modes and Coordinates, Springer, 2017, pp. 95–117 Search PubMed.
- E. Wilson, J. Decius and P. Cross, Molecular Vibrations. The Theory of Infrared and Raman Vibrational Spectra, McGraw-Hill, New York, 1955 Search PubMed.
- Z. Konkoli and D. Cremer, Int. J. Quantum Chem., 1998, 67, 1–9 CrossRef CAS.
- Z. Konkoli, J. A. Larsson and D. Cremer, Int. J. Quantum Chem., 1998, 67, 11–27 CrossRef CAS.
- E. B. Wilson, J. Chem. Phys., 1941, 9, 76–84 CrossRef CAS.
- E. Wilson, J. Decius and P. Cross, Molecular Vibrations. The Theory of Infrared and Raman Vibrational Spectra, McGraw-Hill, New York, 1955 Search PubMed.
- V. Barone, S. Alessandrini, M. Biczysko, J. R. Cheeseman, D. C. Clary, A. B. McCoy, R. J. DiRisio, F. Neese, M. Melosso and C. Puzzarini, Nat. Rev. Methods Primers, 2021, 1, 38 CrossRef CAS.
- W. Zou and D. Cremer, Theor. Chem. Acc., 2014, 133, 1451–1466 Search PubMed.
- M. Freindorf and E. Kraka, J. Mol. Model., 2020, 26, 281-1–281-15 CrossRef PubMed.
- M. Freindorf, A. A. A. Delgado and E. Kraka, J. Comput. Chem., 2022, 43, 1725–1746 CrossRef CAS.
- A. Madushanka, N. Verma, M. Freindorf and E. Kraka, Int. J. Mol. Sci., 2022, 23, 12310–1–12310–25 Search PubMed.
- R. T. Moura, Jr., M. Quintano, J. J. Antonio, M. Freindorf and E. Kraka, J. Phys. Chem. A, 2022, 126, 9313–9331 CrossRef PubMed.
- J. J. Antonio and E. Kraka, Biochemistry, 2023, 62, 2325–2337 CrossRef CAS PubMed.
- M. Freindorf, J. Antonio and E. Kraka, J. Phys. Chem. A, 2023, 127, 8316–8329 CrossRef CAS PubMed.
- M. Freindorf, J. Antonio and E. Kraka, J. Comput. Chem., 2024, 45, 574–588 CrossRef CAS PubMed.
- Y. Dangat, M. Freindorf and E. Kraka, J. Am. Chem. Soc., 2024, 146, 145–158 CrossRef CAS PubMed.
- Z. Konkoli and D. Cremer, Int. J. Quantum Chem., 1998, 67, 29–40 CrossRef CAS.
- Z. Konkoli, J. A. Larsson and D. Cremer, Int. J. Quantum Chem., 1998, 67, 41–55 CrossRef CAS.
- N. Verma, Y. Tao, W. Zou, X. Chen, X. Chen, M. Freindorf and E. Kraka, Sensors, 2020, 20, 2358 CrossRef CAS PubMed.
- M. Quintano, A. A. A. Delgado, R. T. Moura Jr, M. Freindorf and E. Kraka, Electron. Struct., 2022, 4, 044005-1–044005-17 CrossRef.
- M. Quintano, R. T. Moura Jr. and E. Kraka, Chem. Phys. Lett., 2024, 849, 141416 CrossRef CAS.
- W. Zou and D. Cremer, Chem. – Eur. J., 2016, 22, 4087–4099 CrossRef CAS PubMed.
- D. Cremer and E. Kraka, Curr. Org. Chem., 2010, 14, 1524–1560 CrossRef CAS.
- E. Kraka, J. A. Larsson and D. Cremer, Computational Spectroscopy, Wiley, New York, 2010, pp. 105–149 Search PubMed.
- I. Mayer, Chem. Phys. Lett., 1983, 97, 270–274 CrossRef CAS.
- I. Mayer, Int. J. Quantum Chem., 1986, 29, 477–483 CrossRef CAS.
- I. Mayer, J. Comput. Chem., 2007, 28, 204–221 CrossRef CAS PubMed.
- E. Kraka and M. Freindorf, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering - Comprehensive Computational Chemistry, Elsevier, Heidelberg, 2022, pp. 1–27 Search PubMed.
- D. Cremer and E. Kraka, Angew. Chem., Int. Ed. Engl., 1984, 23, 627–628 CrossRef.
- D. Cremer and E. Kraka, Croat. Chem. Acta, 1984, 57, 1259–1281 Search PubMed.
- F. Weinhold, C. R. Landis and E. D. Glendening, Int. Rev. Phys. Chem., 2016, 35, 399–440 Search PubMed.
- D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, D. Ghoreishi, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, S. Izadi, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, R. Salomon-Ferrer, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER, University of California, San Francisco, 2018 Search PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- M. Ernzerhof and J. P. Perdew, J. Chem. Phys., 1998, 109, 3313–3320 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Zhao, Z.-H. Li, W.-N. Wang, Z.-P. Liu, K.-N. Fan, Y. Xie and H. F. Schaefer, J. Chem. Phys., 2006, 124, 184102 CrossRef PubMed.
- C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2009, 11, 10757–10816 RSC.
- S. Li, J. M. Hennigan, D. A. Dixon and K. A. Peterson, J. Phys. Chem. A, 2009, 113, 7861–7877 CrossRef CAS PubMed.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- L. W. Chung, W. M. C. Sameera, R. Ramozzi, A. J. Page, M. Hatanaka, G. P. Petrova, T. V. Harris, X. Li, Z. Ke, F. Liu, H.-B. Li, L. Ding and K. Morokuma, Chem. Rev., 2015, 115, 5678–5796 CrossRef CAS PubMed.
- M. K. Safo, G. P. Gupta, F. A. Walker and W. R. Scheidt, J. Am. Chem. Soc., 1991, 113, 5497–5510 CrossRef CAS.
- G. McLendon and K. Sandberg, J. Biol. Chem., 1978, 253, 3913–3917 CrossRef CAS PubMed.
- S. Della Longa, S. Pin, R. Cortès, A. V. Soldatov and B. Alpert, Biophys. J., 1998, 75, 3154–3162 CrossRef CAS PubMed.
- T. Harami, J. Phys. Chem., 1979, 71, 1309–1318 CrossRef CAS.
- R. Silaghi-Dumitrescu, D. A. Svistunenko, D. Cioloboc, C. Bischin, F. Scurtu and C. E. Cooper, Nitric Oxide, 2014, 42, 32–39 CrossRef CAS PubMed.
- M. Sundararajan and F. Neese, Inorg. Chem., 2015, 54, 7209–7217 CrossRef CAS PubMed.
- R. Langley, P. Hambright, K. Alston and P. Neta, Inorg. Chem., 1986, 25, 114–117 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- W. Zou, R. Moura, Jr., M. Quintano, F. Bodo, Y. Tao, M. Freindorf, M. Z. Makoś, N. Verma, D. Cremer and E. Kraka, LModeA2023, Computational and Theoretical Chemistry Group (CATCO), Southern Methodist University, Dallas, TX, USA, 2023 Search PubMed.
- T. A. Keith, AIMALL, TK Gristmill Software, Overland Park KS, 2017 Search PubMed.
- R. M. Badger, J. Chem. Phys., 1934, 2, 128–131 CrossRef CAS.
- Y. Tao, C. Tian, N. Verma, W. Zou, C. Wang, D. Cremer and E. Kraka, J. Chem. Theory Comput., 2018, 14, 2558–2569 CrossRef CAS PubMed.
- Y. Tao, W. Zou, S. Nanayakkara, M. Freindorf and E. Kraka, Theor. Chem. Acc., 2021, 140, 31-1–31-5 Search PubMed.
- M. Randić, M. Novič and D. Plavšić, Solved and Unsolved Problems of Structural Chemistry, CRC Press, Boca Raton, 2016 Search PubMed.
- M. Freindorf, E. Kraka and D. Cremer, Int. J. Quantum Chem., 2012, 112, 3174–3187 CrossRef CAS.
- S. Kato, M. Abe, H. Gröger and T. Hayashi, ACS Catal., 2024, 14, 13081–13087 CrossRef CAS.
- E. Kraka, J. J. Antonio and M. Freindorf, Chem. Commun., 2023, 59, 7151–7290 RSC.
- E. Kraka, W. Zou, Y. Tao and M. Freindorf, Catalysts, 2020, 10, 691 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Plots of additional properties of HBs in the investigated protein systems, pictures of active sites in the proteins and the gas phase, and optimized coordinates of the active sites. See DOI: https://doi.org/10.1039/d4dt03246b |
| This journal is © The Royal Society of Chemistry 2025 |