
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen catalytic process for γ-valerolactone production from levulinic acid and formic acid†
Evelyn
Vega Sánchez
a,
J. Francisco Javier
Tzompantzi-Morales
a,
Luis
Ortiz-Frade
b,
Marcos
Esparza-Schulz
a,
Reyna
Ojeda-López
c,
Raúl
Pérez-Hernández
 d,
Atilano
Gutiérrez-Carrillo
a,
Lázaro
Huerta
e,
Victor H.
Lara
a,
Leticia
Lomas-Romero
*a and
Lucero
González-Sebastián
*a
d,
Atilano
Gutiérrez-Carrillo
a,
Lázaro
Huerta
e,
Victor H.
Lara
a,
Leticia
Lomas-Romero
*a and
Lucero
González-Sebastián
*a
aDepartamento de Química, Universidad Autónoma Metropolitana-Iztapalapa, Av. San Rafael Atlixco No. 186, Ciudad de México, C.P. 09340, Mexico. E-mail: lucero.gs@xanum.uam.mx; Fax: +52 58044666; Tel: +525520932342
bDepartamento de Electroquímica, Centro de Investigación y Desarrollo Tecnológico en Electroquímica S.C. Parque Tecnológico Querétaro, Sanfandila, Pedro de Escobedo, C.P. 76703, Querétaro, Mexico
cInstituto Tecnológico del Valle de Etla (ITVE), Tecnológico Nacional de México (TecNM), 68230, Oaxaca, Mexico
dInstituto Nacional de Investigaciones Nucleares, Gerencia de Ciencias Ambientales, Carretera México-Toluca S/N, La Marquesa, Ocoyoacac, Estado de México C.P. 52750, Mexico
eInstituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Ciudad de México, C.P.70360, Mexico
First published on 20th January 2025
Abstract
A highly efficient and environmentally friendly process for the hydrogenation of biomass-derived levulinic acid (LA) using formic acid (FA) as a hydrogen donor to produce γ-valerolactone (GVL) has been developed. This method achieves a remarkable 99% yield of GVL in an aqueous medium under mild, additive-free conditions (150 °C, 1.5 hours, 0.5 mol% [Ru]). These conditions represent the best reported so far for producing GVL from LA and FA using a ruthenium bifunctional catalyst (MO-Ru: Ru–Mg/Al, MO: mixed oxide). A significant synergy between Ru and Mg/Al was observed, enhancing the selective activation of formic acid and the subsequent hydrogenation of levulinic acid. This effect is attributed to the combined catalytic action of Ru species and the medium-strength acidic and basic sites found on the MO-Ru surface, which together promote selective reaction steps in the FA activation and LA hydrogenation processes. The production of GVL from levulinic acid and formic acid, both derived from cellulose hydrolysis, is a key reaction in the valorization of biomass into renewable fuels and chemicals. The application of this methodology not only enhances the economic viability of the process but also eliminates the need for energy-intensive separation of levulinic acid from the aqueous mixture of levulinic acid and formic acid. Additionally, a possible reaction mechanism for the hydrogenation of levulinic acid was proposed.
1. Introduction
Nowadays, petroleum is the primary non-renewable carbon resource used to produce the vast majority of fuels, chemicals, and fundamental materials for essential processes, all of which are crucial for improving people's lives. However, society's heavy dependence on this non-renewable fossil fuel gives rise to concerns about its rapid depletion. In addition to the environmental deterioration caused by its extensive burning for energy supply has compelled us to look for renewable alternatives to fossil fuels to ensure a sustainable supply of fuels and chemicals.1–5In this context, biomass, as a natural resource, offers a promising opportunity to serve as a source of organic carbon to produce biofuels and value-added chemicals, or as a direct renewable energy source.6–8 Approximately 180 gigatons of biomass are generated annually from plants, food crops, and waste from human food and animal feed. In general, biomass is constituted by cellulose (40–50%), lignin (15–30%), hemicellulose (20–30%), and other minor components.9,10 The conversion of biomass-derived platform molecules into high-value-added chemicals is a crucial approach for achieving sustainability. For instance, cellulose, the most abundant biomass component in nature, serves as the starting point for producing a range of high-value-added chemicals, including levulinic acid (LA), 1,4-pentanediol (1,4-PDO), ethyl levulinate (EL), and other biomass-derived carbohydrates.11–13 Among these, levulinic acid stands out as one of the most important platform molecules, and its hydrogenation process to produce γ-valerolactone (GVL) is considered a key step in the lignocellulose biorefinery industry. GVL is widely used as a nontoxic and green solvent, serving as an important monomer in the polyester and bioplastic industries, as well as a fuel additive for gasoline and diesel. Additionally, it can be processed to yield high-grade fuels and valuable chemical intermediates such as 1,4-pentanediol, 2-methyltetrahydrofuran, alkenes, etc.14–17 Considering its great potential, the development of efficient hydrogenation methodologies to convert LA to GVL has received considerable attention in recent years.
Generally, the LA catalytic hydrogenation to give GVL is achieved by using H2, secondary alcohols or formic acid as a H2 donors in the presence of a metal catalyst.14,15,17
In particular, formic acid is a promising compound that represents a potential source of H2 energy (4.4 wt% H2) and it is produced in equimolar stoichiometry with levulinic acid from biomass derivatives.18 In order to create a more cost-effective and streamlined biomass hydrogenation process, a series of catalytic methodologies has been designed to replace the external use of molecular hydrogen (H2) with in situ hydrogen generated through the activation of formic acid improving atom economy, and avoiding the energy-costly separation of LA from the mixture of LA and FA.19–21
In general, formic acid can be activated by two competitive routes: dehydrogenation (a) and dehydration (b). Both processes are thermodynamically favored.22 However, to use formic acid as a hydrogen donor source, it is necessary to employ a catalyst capable of favoring the activation of FA via pathway (a) to generate molecular hydrogen and CO2. In addition, the latter can serve to pressurize the system mitigating greenhouse gas emissions,19 while also avoiding CO production, as it could lead to catalyst poisoning.
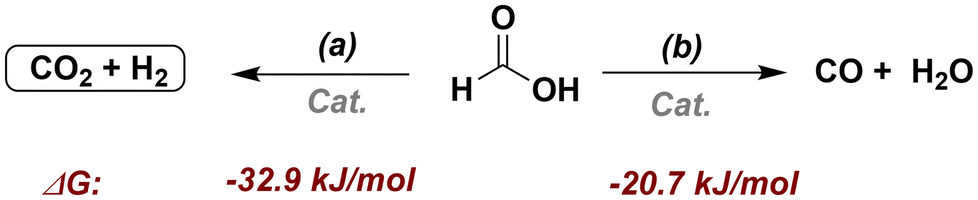
Until now, the catalytic hydrogenation of LA using formic acid as a hydrogen source has been developed with various catalysts, including noble metals such as Pd, Ru, and Au, as well as more cost-effective options like Ni, Cu, Co, Mg, Al, Mn, Zr, and others. In general, catalytic systems employing precious metals tend to perform effectively under relatively milder conditions compared to those based on non-precious metals.5,16,23
Interestingly, catalysts based on ruthenium have shown remarkable catalytic activity in the activation of formic acid (FA) and its subsequent use as a direct hydrogen source in the conversion of LA into GVL. For instance, Guo et al. reported the successful hydrogenation of LA with formic acid into GVL using the homogeneous catalyst RuCl3/PPh3/pyridine at 150 °C for 12 hours.19 One year later, the same group reported the effective conversion of LA and formic acid into GVL catalyzed by the heterogeneous system RuCl3 immobilized on functionalized silica.20 In 2013, the Garcia group achieved the production of GVL through the hydrogenation of LA using formic acid in an H2O/Et3N mixture at 130 °C for 24 hours, catalyzed by Ru nanoparticles generated in situ from [Ru3(CO)12].24 More recently, in 2018, a highly efficient and reusable heterogeneous Ru/ZrO2 catalyst was reported for selectively catalyzing the hydrogenation of LA with FA, yielding a 73% yield of GVL at 150 °C, 12 h and 1 atm of He, with the presence of potassium formate.25 During the same year, Feng et al. introduced a novel approach to produce GVL from LA using solely FA as the exclusive hydrogen source, resulting in significant LA conversion and an 80.75% yield in GVL using Et3N at 160 °C and 3 h under nitrogen atmosphere.21 Although all these catalytic systems can activate FA to produce hydrogen and subsequently carry out the hydrogenation of LA to yield GVL in high amounts, they require large quantities of a base, high pressure, and the strict absence of air to enhance the reaction and prevent catalyst deactivation. Therefore, from both economic and engineering points of view, the development of an efficient catalytic system to produce GVL from levulinic acid and formic acid (which is formed in equimolar amounts with LA by the acidic hydrolysis of biomass) as an in situ source of hydrogen, under greener conditions, remains a matter of concern.
Layered double hydroxides or hydrotalcites (HT) and their mixed oxides (MO) catalytic systems are fascinating due to their potential in green synthetic applications, which have been deeply studied in different biomass upgrading reactions.26,27 Hydrotalcytes are a class of brucite-type octahedral layered inorganic materials with the general formula [M2+1−xM3+x(OH)2]x+(An−)x/n·yH2O. The excess positive charge, resulting from the substitution of M2+ with M3+, is balanced by carbonate anions in the interlayer space. Similar to brucite [Mg(OH)2], the layers are constructed through the condensation of octahedral MO6 units (M2+ or M3+), forming anionic clays where the OH groups point towards the interlayer region and are shared by three octahedral cations.28,29 Mixed oxides (MO) are derived from hydrotalcites (HT) through calcination at 450–600 °C. During this process, the HT structure undergoes dehydration, dehydroxylation, and decarbonization of its interlayer components, transforming into a compact structure where oxide ions create a coordination sphere around the metal ions.30–34
These types of heterogeneous catalysts have attracted particular attention because of their structure and Lewis and Brønsted acid–base tunability, high adsorption capacity, and special microenvironments.29,35 In addition, several transition metals can be grafted onto hydrotalcite in its brucite-like layers due to the isomorphic characteristic substitution of Mg2+ or Al3+ ions at the octahedral sites. These sites are considered active for various organic transformations, thus increasing the versatility of the catalysts.
Particularly, ruthenium-grafted hydrotalcite materials have been reported as highly active catalysts for oxidation, cyclocondensation, alkylation, methanation, hydrogen transfer of carbonyl compounds, and hydrogenation of CO2 reactions, among others.36–40
Herein, we present an efficient and environmentally friendly process for hydrogenating levulinic acid with formic acid to selectively yield GVL over activated ruthenium-grafted calcinated hydrotalcite (MO-Ru). This process eliminates the need for additional base or inert gas (Ar, He, N2) to pressurize the system or prevent catalyst deactivation, which is typically required in such reactions. In this catalytic hydrogenation, the MO-Ru plays a dual role as both catalyst and base. GVL is produced through the sequential activation of formic acid to produce H2 and CO2, followed by the consecutive hydrogenation of levulinic acid. The activity and reusability (5 cycles) of the Ru–Mg/Al mixed oxide were appreciable, yielding γ-valerolactone in high yields of up to 99% in water at 150 °C for 1.5 hours.
2. Experimental section
2.1 General information
All reagents and materials were used as received. RuCl3·3H2O, Mg(NO3)2·6H2O, Al(NO3)3·9H3O, Na2CO3, NaOH, HCO2H (88%), CDCl3, Et3N and levulinic acid were purchased from Sigma-Aldrich, USA, and used without further purification. Double-distilled Milli-Q deionized water was used for the synthesis of the catalysts and in catalytic screenings. The deuterated solvent used was CDCl3; chemical shifts (δ) are reported in ppm and coupling constants in Hz. The following abbreviations were used to indicate the multiplicity of signals in 1H NMR spectra: (s) singlet, (d) doublet, (t) triplet, (at) apparent triplet, (m) multiplet, (dd) double doublet, and (bs) broad signal.2.2 Synthesis of hydrotalcite Ru–Mg/Al (HT-Ru) and mixed oxide Ru–Mg/Al (MO-Ru)
Ru–Mg/Al hydrotalcite was synthesized via a coprecipitation method using Mg(NO3)2·6H3O, Al(NO3)3·9H2O, RuCl3·3H2O, Na2CO3, and NaOH as raw materials. The synthesis involved the preparation of two solutions:Solution 1. In a 250 mL Erlenmeyer flask, 11.53 g (45 mmol) of Mg(NO3)2·6H2O, 5.439 g (14.5 mmol) of Al(NO3)3·9H2O, and 0.1037 g (0.5 mmol) of RuCl3·3H2O were dissolved in 50 mL of distilled water.
Solution 2. In a separate 250 mL Erlenmeyer flask, 5.1996 g (74.7 mmol) of Na2CO3 and 7.9173 g (130 mmol) of NaOH were dissolved in 50 mL of distilled water.
Subsequently, solution 1 was added dropwise to solution 2 over approximately 10 minutes with continuous stirring. The resulting mixture was then heated to 60 °C and stirred for 24 her. After this period, the material was filtered and washed with distilled water until the pH of the filtrate was neutral. Finally, the product was dried in a furnace at 80 °C for 24 hours yielding 4.34 g of Ru–Mg/Al hydrotalcite as a light grey powder. The mixed oxide Ru–Mg/Al (MO-Ru) was then obtained by calcining the Ru–Mg/Al hydrotalcite at 450 °C for 4 h in a muffle furnace.
Synthesis of Mg–Al hydrotalcite sample (HT) with Mg/Al molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was done as per the above-mentioned procedure without use of RuCl3·3H2O solution.
:1 was done as per the above-mentioned procedure without use of RuCl3·3H2O solution.
2.3 Characterization of MO-Ru catalyst
XRD analysis of MO-Ru and its precursor HT-Ru was performed using powder X-ray diffraction (XRD) with a Philips X'Pert instrument equipped with Cu-Kα radiation (45 kV, 40 mA). SEM-EDS images and emission spectra for both materials were obtained using a Zeiss SUPRA 55 VP microscope with secondary electron and Oxford detectors. FTIR spectra of the samples were recorded using a Perkin–Elmer 600 spectrometer using the attenuated total reflectance (ATR) method. The absorbance peaks are reported in reciprocal centimeters (cm−1). X-ray photoelectron spectroscopy analyses of HT-Ru and MO-Ru were carried out in an ultra-high vacuum (UHV) system scanning XPS microprobe PHI 5000 VersaProbe II, with an Al Kα X-ray source (hν = 1486.6 eV) monochromatic with 200 μm beam diameter, and a multi-channel detector (MCD) analyzer. The XPS spectra were obtained at 45° to the normal surface, and constant analyzing energy (CAE) E0 = 117.40 and 11.75 eV survey surface and high-resolution narrow scan. The peak positions were referenced to the background Ag 3d5/2 photopeak at 368.20 eV, with an FWHM of 0.56 eV, and C 1s hydrocarbon groups at 285.00 eV, Au 4f7/2 at 84.00 eV central peak core level position. The XPS spectrum was fitted with the program MultiPak PHI software41 and SpectrData Processor, SDP v 4.1.42 Temperature-programmed desorption of CO2 (TPD-CO2) and NH3 (TPD-NH3) was conducted using a BELCAT-B apparatus. The samples were first activated under an Ar flow at 300 °C for 30 minutes to eliminate adsorbed environmental contaminants and then cooled to 100 °C. Subsequently, either CO2 (5%)/Ar at 40 cc min−1 or NH3 (5%)/Ar was injected for 30 minutes. After this, the gas flow was switched to Ar (UHP) at 40 cc min−1 for 30 minutes to remove physisorbed CO2 or NH3. The desorption process was carried out by heating the samples from 100 °C to 800 °C at a rate of 10 °C min−1 under an Ar flow (40 cc min−1). The desorbed CO2 and NH3 were monitored via mass spectrometry using BELLMass equipment, tracking mass 44 (m/z = 44) for CO2 and mass 17 (m/z = 17) for NH3. The textural measurements were performed with a Micro 200 (3P Instruments Micromeritics, 85235 Odelzhausen I Germany) at liquid nitrogen temperature (−196 °C). Before measurement, the samples were degassed at 150 °C for 12 h. The specific surface area was calculated by the multiple point Brunauer–Emmett–Teller (BET) method. For mesoporous materials, the relative pressure interval used was 0.05 to 0.25. Pore size distribution curves were computed by using the nonlocal density functional theory (NLDFT) method.2.4 Catalytic experiments
The catalytic hydrogenation of levulinic acid (LA) using formic acid (FA) was carried out in a 75 mL stainless steel Parr autoclave. In a typical experiment, the reactor was charged with 1 mL of LA (9.8 mmol), 1 mL of FA (19.6 mmol), 100 mg of catalyst MO-Ru (9.8 μmol of [Ru]), and 4 mL of distilled water. The reaction was conducted at various temperatures and reaction times. After completion, the reaction mixture was extracted with ethyl acetate or dichloromethane (3 × 5 mL), and the solvent was removed under vacuum. Experiments involving triethylamine (Et3N) were performed similarly, replacing water with 3 mL of Et3N. After the workup, the resulting GVL batches were centrifuged, and no deposition of metal nanoparticles was observed. Catalysis products were quantified with a GC-MS Agilent 6890N chromatograph equipped with a 30 m DB-1MS Agilent capillary column, coupled to an Agilent Technologies 5973 mass spectrometer equipped with an inert mass selective detector and 1H NMR. NMR experiments were recorded at 300 K on Bruker Avance DMX-500 (500 MHz) spectrometer using TMS or residual proton solvents as internal standard. Gaseous samples were collected using a gas bag and analyzed with a mass spectrometer (Pfeiffer Vacuum Omnistar). This procedure was employed solely to confirm the presence of molecular hydrogen resulting from the decomposition of formic acid and is not considered reliable for quantitative analysis.2.5 Catalyst recovery
After the reaction was completed, the mixture was cooled to room temperature, and the organic product was extracted with ethyl acetate or dichloromethane (3 × 5 mL). The catalyst was then easily separated by filtration, washed with water and ethanol, dried at 150 °C for 3 hours, and immediately used in subsequent catalytic cycles.3. Results and discussion
3.1 Synthesis and characterization of the catalyst MO-Ru
In general, transition metals can be grafted onto the surface of HT (adsorption ability), within the brucite layer (cation exchange ability), or within the interlayer space (anion exchange ability) to form HT-supported metal catalysts. In this study, ruthenium was specifically targeted for grafting within the brucite layer through cation exchange using the coprecipitation method. RuCl3 was added to a solution containing Mg2+ and Al3+ during the synthesis of HT to produce HT-Ru. Subsequently, HT-Ru was calcined at 450 °C for 6 hours with a heating rate of 2 °C min−1 to produce the MO-Ru catalyst. The structural, textural and morphological characteristics of the as-synthesized MO-Ru and its HT-Ru precursor were further characterized by ICP-AES, FT-IR, XRD, XPS, SEM-EDS, TEM analytical techniques and N2 adsorption/desorption isotherms.The powder XRD patterns of the synthesized precursor HT-Ru and the ruthenium-free hydrotalcite (HT) (synthesized for comparison) exhibited typical diffraction peaks at 2θ values of 11.4°, 23.1°, 34.8°, 39.2°, 46.7°, 639.2°, 46.7°0.9°, 62.2°, and 66.03°, corresponding to the (003), (006), (012), (015), (018), (110), (113), and (116) planes, characteristic of the hydrotalcite structure with interlayer carbonate (see Fig. 1A).43–46 The presence of CO32− anions in the interlayer space of both HT-Ru and HT were confirmed by the characteristic basal spacing of the (003) plane, with d003 values of 7.66 Å and 7.60 Å for HT-Ru and HT, respectively. The slightly broader shape of the (003) and (006) planes in HT-Ru could indicate lower crystallinity compared to the ruthenium-free HT, likely due to the insertion of Ru ions into the HT layer. This decrease in crystallinity is attributed to the larger ionic radius of ruthenium cations compared to aluminum cations in the brucite layers. However, given the similar value of the a parameter calculated for both HT-Ru and ruthenium-free HT, it is highly likely that the ruthenium cation is integrated into the layer network (see Table 1). On the other hand, the XRD profiles of MO-Ru and the ruthenium-free mixed oxide, obtained by calcining their corresponding hydrotalcites at 450 °C, showed diffraction peaks at 2θ values of 43.39° and 62.72° for MO-Ru, and 43.17° and 62.91° for HT without ruthenium. These peaks correspond to the (200) and (220) planes, respectively (Fig. 1B).47
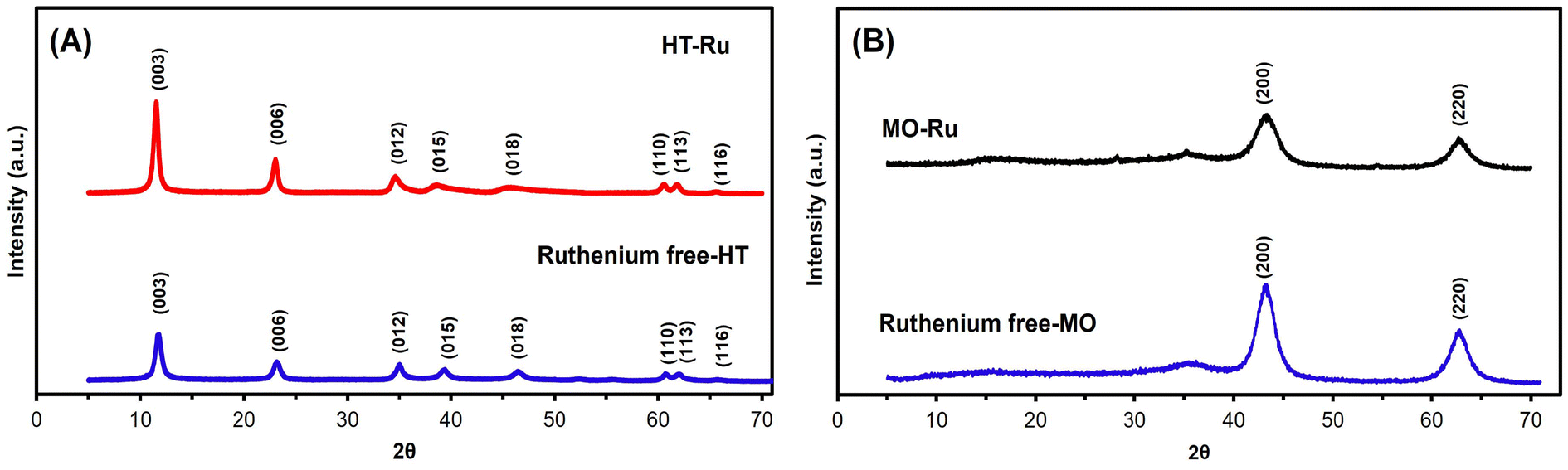 | ||
| Fig. 1 (A) XRD pattern of HT-Ru and ruthenium-free HT (Mg/Al) and (B) XRD pattern of MO-Ru and ruthenium-free MO (Mg/AL). | ||
| Parameter | HT-Ru | Ru-free HT |
|---|---|---|
| a parameter (Å) | 3.053 | 3.036 |
| Lattice parameter c (Å) | 22.998 | 26.108 |
| d interlaminar (Å) | 2.866 | 3.903 |
The FT-IR spectra of MO-Ru and its precursor HT-Ru are represented in Fig. 2. The HT-Ru exhibited multiple absorption bands typical of hydrotalcite observed at 3384, 1606.6, 1548, 1406, 1366.1, 1044, 1020.1, and 555 cm−1.48 The broad vibrational band observed at 3384 cm−1 corresponds to the hydrogen bonding stretching vibration of OH− group in the brucite like layer. The vibrational band observed at 1606.6 cm−1 was assigned to the bending vibration of interlayer water molecules. The peak at 1406 cm−1 and the weak shoulder peak at 1366.1 cm−1 were assigned to the stretching and out-of-plane bending vibration of intercalated CO32− species. The band at 1020.1 cm−1 is also attributed to the CO32− anion since this anion can be coordinated either in a monodentate or bidentate manner. The peak observed at 555 cm−1 belongs to the hydrotalcites frameworks, indicating the translational modes of hydroxyl groups, influenced by the M3+ cations. These observations confirm that the catalysts’ precursor is ordered layered metal hydroxides. Upon calcination of HT-Ru, the CO32− ions and interlayer water molecules are lost, as evidenced by the IR spectra of MO-Ru.
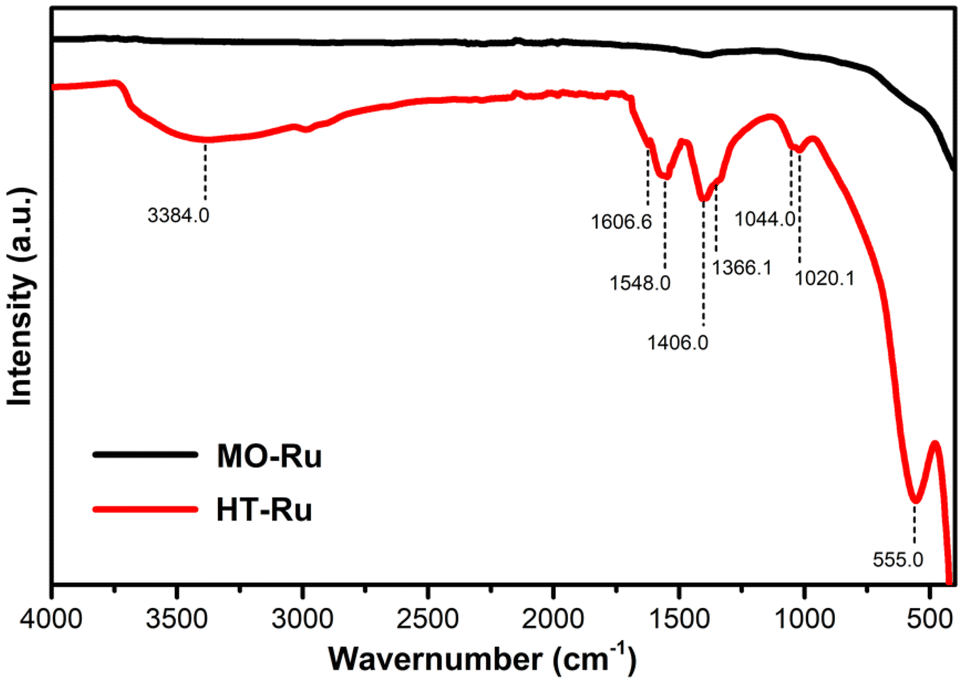 | ||
| Fig. 2 FTIR spectra of MO-Ru and HT-Ru. | ||
The SEM images of MO-Ru and its precursor HT-Ru (ESI, Figure S1 and S2†) showed microparticles resembling flakes and agglomerates. Additionally, energy dispersive X-ray absorption spectroscopy (EDS) revealed the presence of Ru, Mg, Al, C, H, and O atoms on the surface of HT-Ru, while in the MO-Ru sample only Ru, Mg, Al, and O were detected, confirming the loss of CO32− ions and interlayer water molecules in the calcined material. In Fig. 3, the N2 adsorption/desorption isotherms of MO-Ru and HT-Ru are shown. Based on the IUPAC classification, both isotherms are Type IV(a). For the MO-Ru sample, the hysteresis loop could be classified as H2(b) type, suggesting that the calcination process resulted in a solid matrix with a complex porous network composed of cavities of similar sizes connected through necks slightly smaller in size. On the other hand, for the HT-Ru sample, it is challenging to determine the correct classification; however, the resulting matrix comprises large hollow cavities interconnected with each other through necks considerably smaller in size. Consequently, in this latter case, the pore blocking effect is reflected to a much greater extent than in the case represented by the MO-Ru sample. Additionally, the HT-Ru sample exhibits a surface area of 25 m2 g−1, while its calcined form shows an almost threefold increase (79 m2 g−1). This significant enhancement is attributed to the creation of new cavities during the calcination process, which result from the release of H2O and CO32− from the interlaminar region of HT-Ru. Pore size distribution (PSD) and total volume showed different porosity in HT-Ru and MO-Ru. In general, MO-Ru presents a much larger pore size and pore volume than HT-Ru, as can be seen in Table 2.
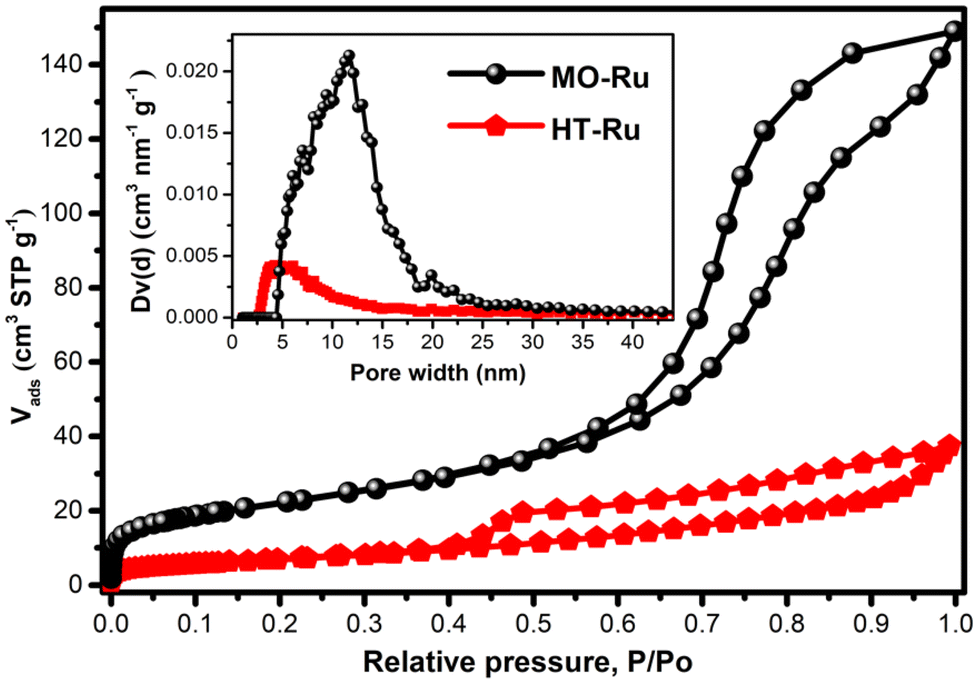 | ||
| Fig. 3 Nitrogen isotherm at −196 °C and PSD for MO-Ru and HT-Ru. | ||
| Catalyst |
A
BETa (m2 g−1) |
V
TOTALb (cm3 g−1) |
D
MESOc (nm) |
|---|---|---|---|
| a Values obtained for the specific surface area using the BET equation. b Values obtained for the total pore volume calculated at 0.95 relative pressure. c Values obtained for the average pore size using NLDFT. | |||
| HT-Ru | 25 | 0.038 | 4.3 |
| MO-Ru | 79 | 0.204 | 11.7 |
ICP-AES analysis revealed that the ruthenium content in MO-Ru was 1 wt%. To obtain information about electronic state of the elements present on the surface of HT-Ru and MO-Ru materials, an XPS study was conducted. Fig. 4 and Table 3 present the results. Gaussian–Lorentzian adjustments and asymmetry software were used for spectra processing, including extra contributions to the initial states corresponding to multiple splitting of the Ru 3d5/2 and Ru 3d3/2 orbitals. In Fig. 5, deconvolutions of Ru 3d and O 1s orbitals for each material are shown, along with the comparison of spectra of Al 2p and Mg 1s core levels between HT-Ru and MO-Ru. In the analysis of the Ru 3d orbital from the MO-Ru sample, ruthenium in two oxidation states with different proportions were observed: Ru4+ (51%), Ru2+ (49%); while in the study of the Ru 3d orbital in the HT-Ru composite, only Ru3+ (100%) was determined.49–51 The binding energy (BE) of Ru 3d5/2 level correspond to Ru4+ at 281.25 eV, Ru3+ at 282.15 eV and Ru2+ at 282.85 eV.36,52
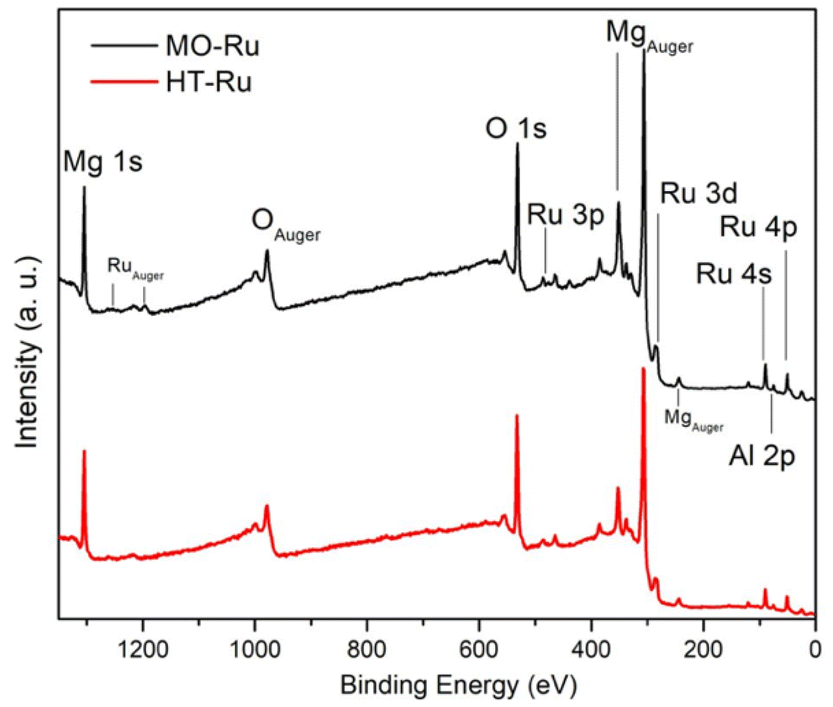 | ||
| Fig. 4 XPS survey spectra of HT-Ru and MO-Ru samples. | ||
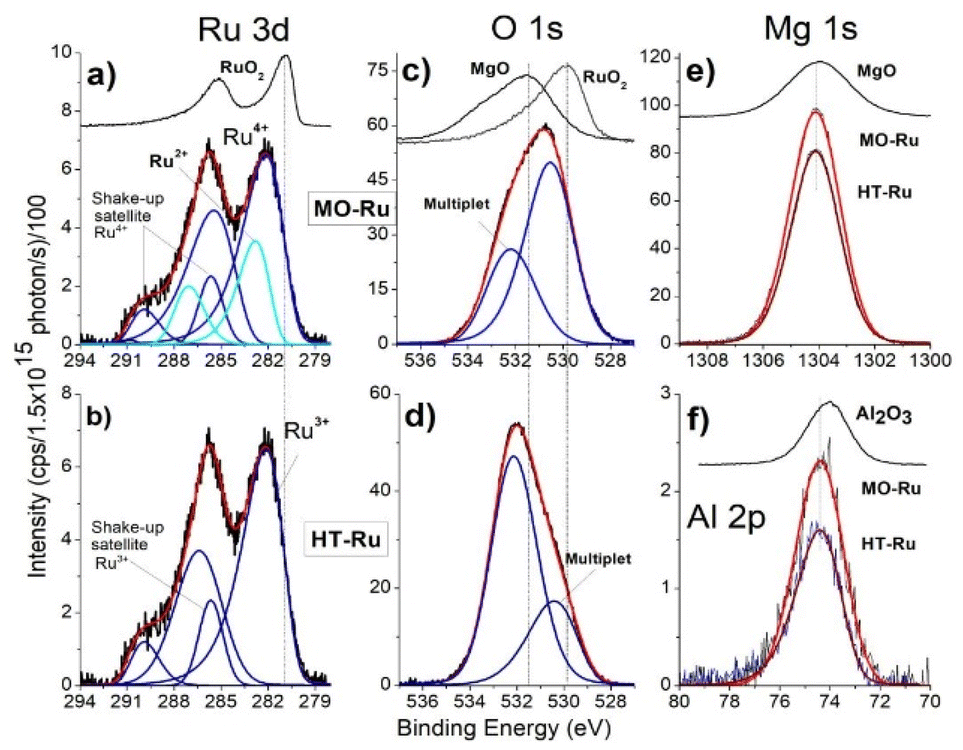 | ||
| Fig. 5 High-resolution XPS spectra of Ru 3d (a and b), O 1s (c and d); Mg 1s (e), and Al 2p (f) orbitals of HT-Ru and MO-Ru samples. Binding energy (BE) values of simple orbitals from reference materials (RuO2, MgO, Al2O3) were added at the top of figures a, c, e, and f for comparison purposes. | ||
| Sample | Ru 3d5/2a | Ru 3d3/2a | Mg 1sa | Al 2pa | O 1sa,b | Valence |
|---|---|---|---|---|---|---|
| Binding Energy (eV) | ||||||
| a Elemental composition used atomic sensitivity factor for Ru 3d (4.529), Mg 1s (1.035), Al 2p (0.256) and O 1s (0.733), respectively. b Main peak of O 1s core level. BE corresponding to the multiplet (minor peak) for HT-Ru at 530.41 eV and for MO-Ru at 532.20 eV. | ||||||
| HT-Ru | 282.15 | 286.40 | 1304.12 | 74.40 | 532.14 | Ru3+ |
| 285.65 | 289.90 | Sat Ru3+ | ||||
| Mg2+ | ||||||
| Al3+ | ||||||
| O2− | ||||||
| MO-Ru | 281.25 | 285.50 | 1304.12 | 74.40 | 530.53 | Ru4+ |
| 285.66 | 290.20 | Sat Ru4+ | ||||
| 282.85 | 287.10 | Ru2+ | ||||
| Mg2+ | ||||||
| Al3+ | ||||||
| O2− | ||||||
| References | ||||||
| RuO2 | 280.90 | 285.22 | 1302.98 | 74.38 | 529.92 | Ru4+ |
| MgO | 530.54 | Mg2+ | ||||
| Al2O3 | 531.61 | Al3+ | ||||
XPS results revealed that ruthenium and oxygen were the species most affected during the calcination process. The oxidation state of Ru3+ shifted to a mixture of Ru2+ and Ru4+, while the nature of oxygen also changed. This change was due to the removal of physiosorbed water on the surface, interlaminar water, and hydroxyl groups chemically bound to the layers, thereby altering the physicochemical properties of the material.
Fig. 5c and d show the spectra corresponding to the O 1s core level, each with two contributions, which can also be associated with the phenomenon of multiplet-splitting of the orbital.53,54 In this context, the smallest multiplet observed in Fig. 5c can be associated with a satellite shake-up, while that observed in Fig. 5d is not, due to the crystal structure not undergoing significant change, along with the presence of only ruthenium in the 3+ oxidation state in HT-Ru. In both materials, the area of contributions is close to ∼70/30 but with inverted binding energies (BE); the BE for MO-Ru are 530.53 eV and 532.20 eV, and for HT-Ru are 532.14 eV and 530.41 eV. As can be seen, the ±BE is located symmetrically with the same chemical shift with respect to the O 1s of MgO at 531.56 eV.
Finally, in Fig. 5e and f, the spectra for Mg 1s and Al 2p of both samples are shown, which exhibit the same binding energies (BE) of 1304.12 eV and 74.40 eV for Mg 1s and Al 2p, respectively. The binding energies (BEs) of the references used for this analysis are 280.90 eV for Ru 3d5/2 in RuO2, 1304.00 eV for Mg 1s, 531.56 eV for O 1s (with a satellite peak at 533.75 eV) in MgO, and 74.10 eV for Al 2p in Al2O3.
To evaluate the quantity and strength of acidic and basic sites on the surface of the MO-Ru catalyst and compare these with the mixed oxide without ruthenium, NH3-TPD and CO2-TPD measurements were performed. Fig. 6A shows the ammonia desorption profiles, revealing the presence of medium-strength acidic sites in both materials. However, the presence of ruthenium in MO-Ru increases the concentration of acidic sites.
 | ||
| Fig. 6 (A) NH3-TPD profiles for MO-Ru and ruthenium-free MO (Mg/Al) and (B) CO2-TPD profiles for MO-Ru and ruthenium-free MO (Mg/Al). | ||
On the other hand, the CO2-TPD measurements of the Ru-free MO material display a broad CO2 desorption peak from 315 °C to 600 °C and an additional peak at 718 °C, indicating the contribution of basic sites with different natures and strengths. In contrast, the CO2 desorption profile for MO-Ru shows a single peak at 385 °C, corresponding to medium-strength basic sites, Fig. 6B.
In general, the introduction of ruthenium in the mixed oxide increases the total number of acidic sites while unifying the basic site strength, resulting in both acidic and basic sites of medium strength.
3.2 Catalytic studies on the production of γ-valerolactone (GVL) from levulinic acid (LA) and formic acid (FA)
The catalytic studies began by evaluating the activity of MO-Ru under similar reaction conditions as those used by the Feng et al.21 with the Ru (5%)/C catalyst in the hydrogenation of levulinic acid (LA) with formic acid (FA) as the sole hydrogen source and Et3N as base at 160 °C for 3 hours, without inert gas. When comparing the catalytic activities of MO-Ru (86% yield of GVL, Table 4, entry 1) and Ru/C (80.7% yield of GVL, as reported by Feng et al.), MO-Ru achieved higher LA conversion and greater selectivity toward GVL. This outcome may be attributed to the selective activation of formic acid by MO-Ru, which produces H2/CO2 exclusively, as confirmed by gas chromatography analysis that did not detect CO. In contrast, the activation of formic acid by Ru (5%)/C was less selective, generating a mixture of H2/CO2 and CO/H2O, which led to lower selectivity for GVL and the formation of other derivatives, as noted by the authors.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Base | Solvent | LA:FA ratio |
Conv. (%) | Selectivity of GVL (%) |
| a Reaction conditions: MO-Ru (0.098 mmol, based on Ru content), LA (9.8 mmol), formic acid (19.6 mmol), Et3N (3 mL). b MO-Ru (0.098 mmol, based on Ru content), LA (80 mmol), formic acid (80 mmol), H2O (4 mL). c LA (80 mmol), formic acid (80 mmol) and H2O (4 mL). d MO-Mg/Al: mixed oxide without ruthenium. | ||||||
| 1a | MO-Ru | Et3N | — | 1:2 |
89 | 86 |
|
2b |
MO-Ru | — | H 2 O |
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :2 :2
|
100 | 99 |
| 3a | Ru free MOd | Et3N | — | 1:2 |
0 | 0 |
| 4b | Ru free MOd | — | H2O | 1:2 |
0 | 0 |
| 5c | None | — | H2O | 1:2 |
0 | 0 |
Based on these findings, MO-Ru proved to be a highly effective catalyst to produce GVL from LA, utilizing FA exclusively as the H2 source. However, as previously mentioned, MO-Ru material contains not only catalytic metallic sites but also possess acid and basic sites. Therefore, we evaluated the hydrogenation of levulinic acid (LA) with formic acid (FA) without an added base, using water as the solvent. This approach yielded excellent conversion of LA and improved selectivity toward GVL compared to the process conducted in Et3N (Table 4, entries 1 and 2). When the reaction was carried out either in the presence of the mixed oxide without ruthenium (entries 3 and 4) or without any catalyst (entry 5), the hydrogenation of LA with FA did not proceed. These findings indicate that ruthenium is the catalytically active species, which, in combination with the acidic and basic sites present in MO-Ru, may act synergistically to selectively activate FA and efficiently carry out the hydrogenation of LA.
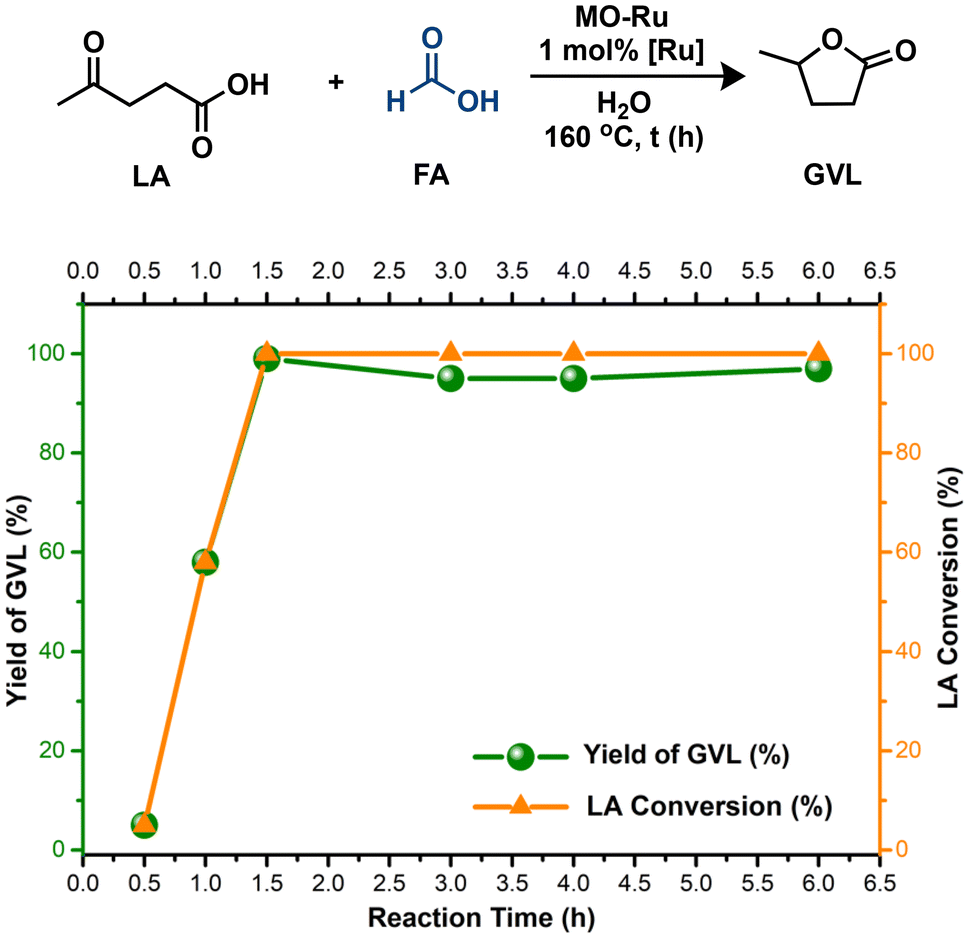 | ||
| Fig. 7 The effect of the reaction time on the conversion of LA into GVL catalyzed by MO-Ru. Reaction conditions: MO-Ru (0.098 mmol, based on Ru content), LA (9.8 mmol), formic acid (19.6 mmol), H2O (4 mL), LA:FA ratio 1:2. | ||
In general, the reaction rate experiences a rapid increase after 30 minutes, which is attributed to the efficient decomposition of FA at this stage, supplying sufficient H2 for the effective hydrogenation of LA. However, although FA decomposition occurs quickly, the generation of GVL requires more time due to the various reactions involved in the entire process. According to these results, 1.5 hours was chosen as the optimal reaction time.
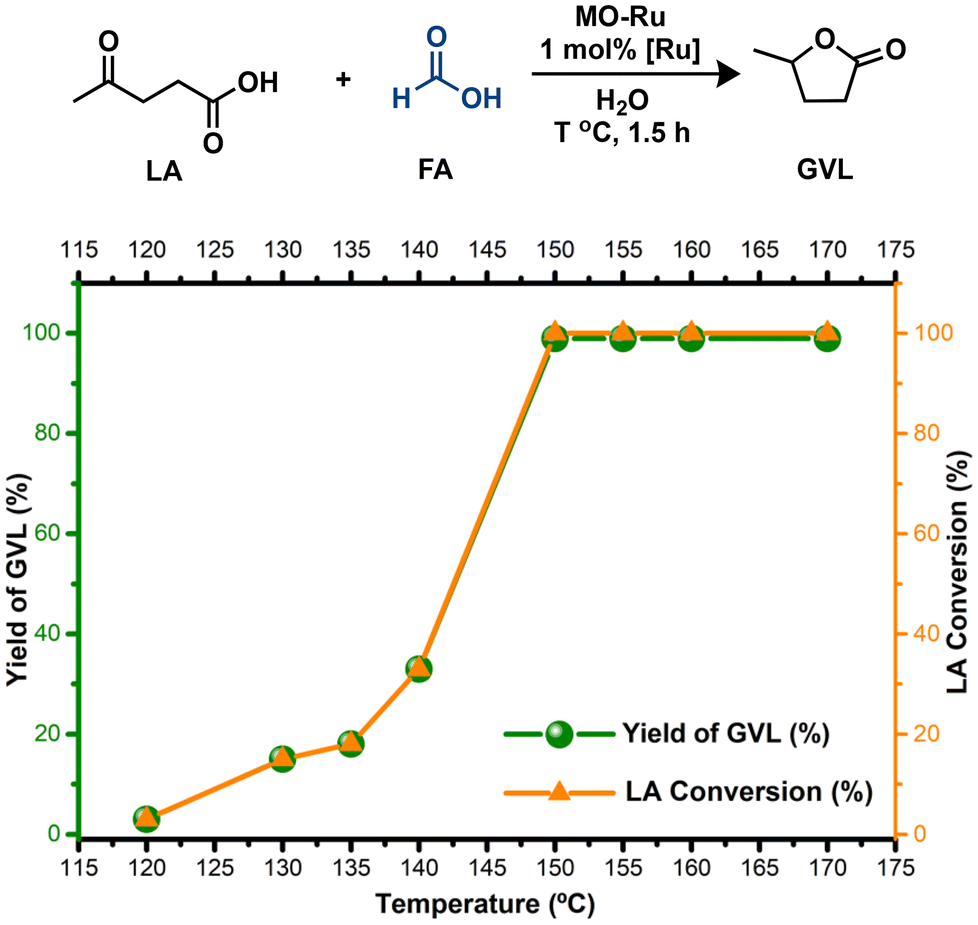 | ||
| Fig. 8 The effect of the reaction temperature on the conversion of LA into GVL catalyzed by MO-Ru. Reaction conditions: MO-Ru (0.098 mmol, based on Ru content), LA (9.8 mmol), formic acid (19.6 mmol), H2O (4 mL), 1.5 h. | ||
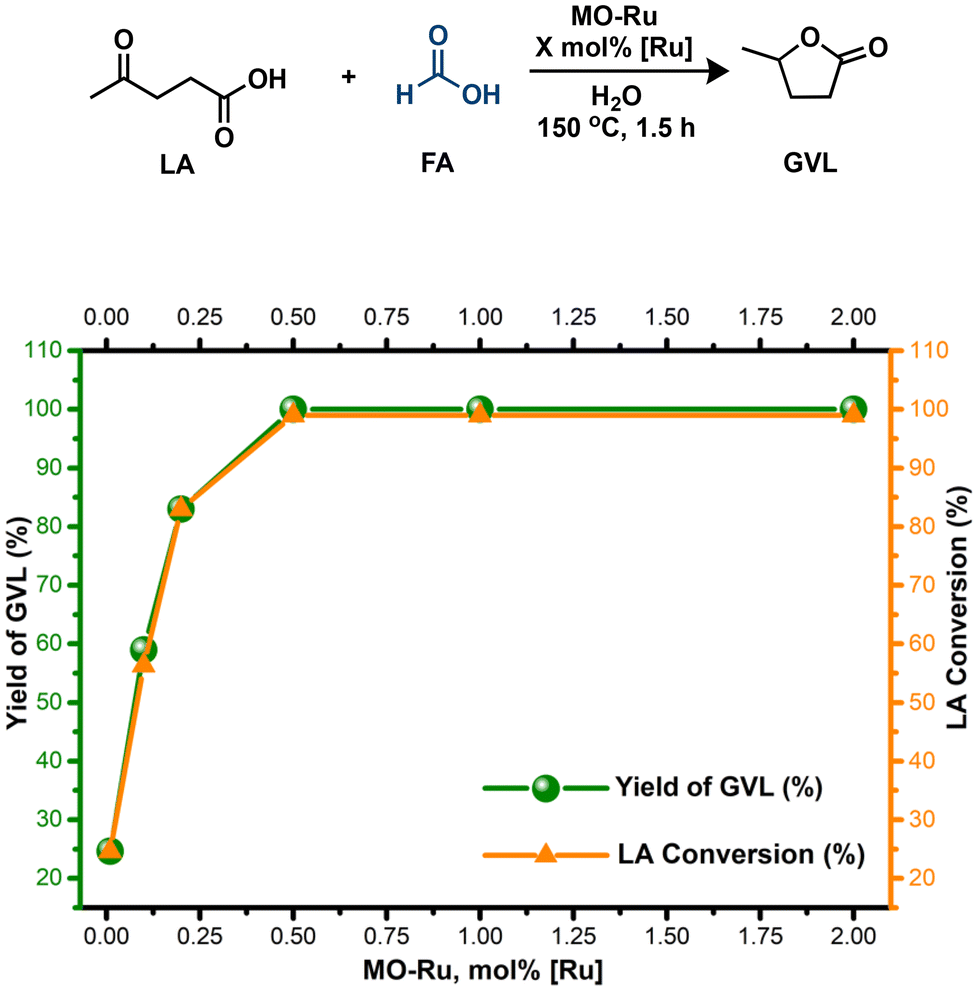 | ||
| Fig. 9 Influence of the catalyst amount (MO-Ru) on the hydrogenation of LA with FA. Reaction conditions: MO-Ru (based on Ru content), LA (9.8 mmol), formic acid (19.6 mmol), H2O (4 mL), 150 °C, 1.5 h. | ||
3.3 Stability and recyclability of MO-Ru catalyst
Since the lifetime of the catalyst is crucial for reducing the cost of a chemical process, its reusability and stability are of great importance for practical applications. To evaluate the reusability of the MO-Ru catalyst, recycling tests for the hydrogenation reaction of LA with FA were conducted over four successive cycles under optimized reaction conditions (0.5% mol [Ru], 150 °C, 1.5 hours). After each catalytic run, the catalyst was separated from the reaction mixture by filtration, washed with ethanol and water, dried at 150 °C for 3 hours, and then used for the next catalytic run. As shown in Fig. 10, the MO-Ru catalyst was reusable at least four times without any loss of catalytic activity and maintained high selectivity toward GVL (99% yield). However, in the fifth run, the catalytic activity decreased to 76%, although the selectivity for GVL remained unchanged. XRD analysis of the MO-Ru recovered after the fifth run, Fig. 11, revealed less defined patterns for the MO-Ru and the emergence of similar peaks to those of HT-Ru (see Fig. 1), which can be attributed to the presence of organic molecules or water within the layers of the material. This behavior, known as the “memory effect”, is a characteristic property of such materials.55 The decrease in activity of the MO-Ru after the fourth run was attributed to the formation of HT-Ru, which exhibits lower catalytic activity compared to MO-Ru.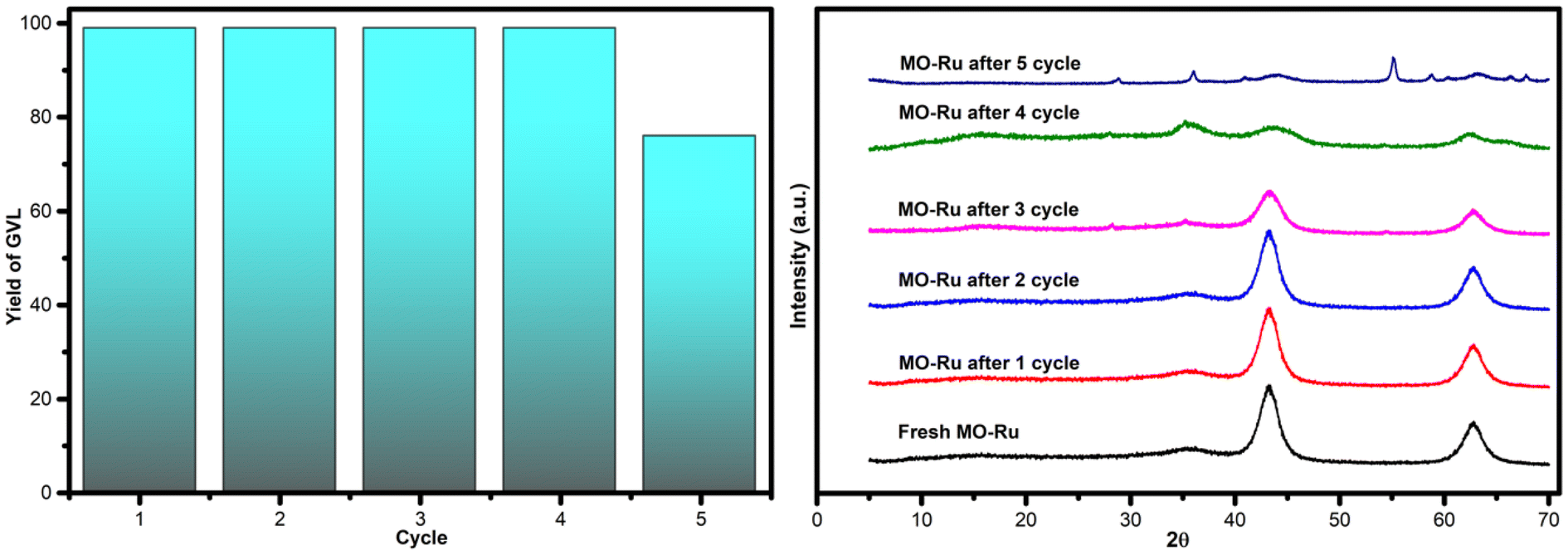 | ||
| Fig. 10 Reuse of the catalyst MO-Ru for hydrogenation reaction of LA with FA and XRD patterns of fresh MO-Ru and MO-Ru after 1, 2, 3, 4 and 5 cycles. | ||
 | ||
| Fig. 11 Test of heterogeneity of the catalyst MO-Ru. | ||
3.4 Catalytic reaction mechanism insights
For the hydrogenation of levulinic acid (LA) with formic acid (FA) to occur, a catalyst capable of efficiently performing both the dehydrogenation of formic acid and the hydrogenation of levulinic acid is required. To investigate the origins of the MO-Ru catalytic performance, several experiments were conducted. First, the hydrogenation of LA using FA as a hydrogen source was tested with a ruthenium-free mixed Al–Mg oxide as the catalyst. As shown in Table 4 (entries 3 and 4), the mixed oxide without ruthenium did not catalyze the hydrogenation of LA. Additionally, the decomposition of FA catalyzed by the ruthenium-free mixed Al–Mg oxide was independently evaluated at 150 °C for 3 hours. Under these reaction conditions, FA activation did not occur. These findings further underscore the critical role of Ru species in the mixed oxide for both FA activation and the hydrogenation of LA. NH3-TPD and CO2-TPD studies revealed that ruthenium species increase the number of acidic and basic sites of medium strength, which may work synergistically to enhance the catalytic activity in both FA activation and LA hydrogenation.Interestingly, when the hydrogenation of LA with FA was catalyzed by MO-Ru, a rapid increase in pressure inside the autoclave was observed, reaching a maximum of approximately 225 psi at 150 °C within the first half-hour of the reaction (see Fig. 12). After approximately 45 minutes, the pressure inside the autoclave began to decrease. In addition, considering the study on the effect of reaction time (Fig. 7), it was observed that GVL was produced after 30 minutes of reaction, achieving the highest conversion at 1.5 hours. It is important to note that the catalytic activation of FA with MO-Ru selectively produces H2 and CO2, as confirmed by gas chromatography analysis, which did not detect any CO. Similarly, the necessity of formic acid as a hydrogen donor in the hydrogenation of LA was demonstrated, as negligible conversion was observed after 3 hours when the reaction was conducted without the addition of formic acid.
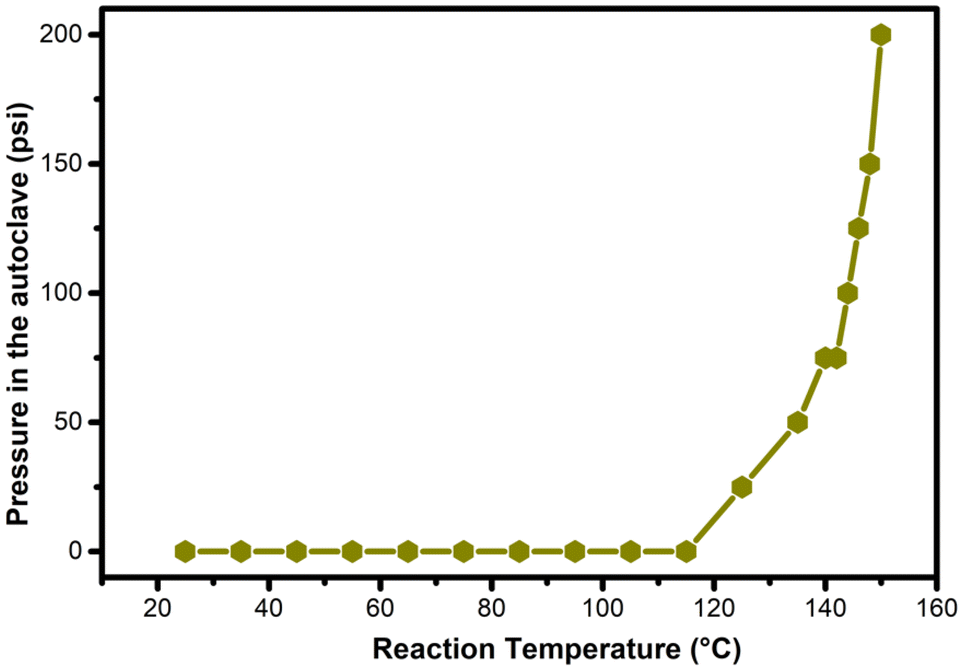 | ||
| Fig. 12 Measurement of the pressure inside the autoclave during the temperature increase in the hydrogenation of LA with FA catalyzed by MO-Ru. | ||
In general, the hydrogenation of LA can proceed through two possible reaction mechanisms: (1) hydrogenation of the ketone group in LA, leading to the formation of the intermediate 4-hydroxypentanoic acid, followed by dehydration and intramolecular esterification, which results in ring closure to yield GVL; or (2) dehydration of LA to α-angelica lactone, followed by its hydrogenation to GVL.
Based on our results and insights from the literature, we propose that the LA hydrogenation process begins with the catalytic activation of formic acid in the first step (I, Fig. 13), followed by the hydrogenation of LA driven by the high pressures of H2 and CO2 generated by MO-Ru (II, Fig. 13). The significant decomposition of formic acid in the initial minutes of the reaction suggests a preferential adsorption of formic acid over LA on the catalytic surface. This preference is likely influenced by the medium-strength basic sites derived from the MgO component of the Mg/Al/Ru mixed oxide catalyst, along with the active ruthenium species. These factors may prevent the dehydration of LA to α-angelica lactone and its subsequent oligomerization, favoring the direct pathway (I) to GVL. In general, the production of α-angelica lactone is favored by the presence of strong acids and high temperatures (>200 °C). Although the hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in α-angelica lactone can lead to GVL, this pathway is not the most accessible under the mild reaction conditions employed in this study. Instead, it is highly likely that the hydrogenation of LA proceeds via the reduction of its ketone group in the first step, generating the intermediate 4-hydroxypentanoic acid. The basic sites on the catalyst may activate the carbonyl group, facilitating this step. Subsequent dehydration and ring closure are influenced by the acidic conditions of the system.5,23,56,57
C bond in α-angelica lactone can lead to GVL, this pathway is not the most accessible under the mild reaction conditions employed in this study. Instead, it is highly likely that the hydrogenation of LA proceeds via the reduction of its ketone group in the first step, generating the intermediate 4-hydroxypentanoic acid. The basic sites on the catalyst may activate the carbonyl group, facilitating this step. Subsequent dehydration and ring closure are influenced by the acidic conditions of the system.5,23,56,57
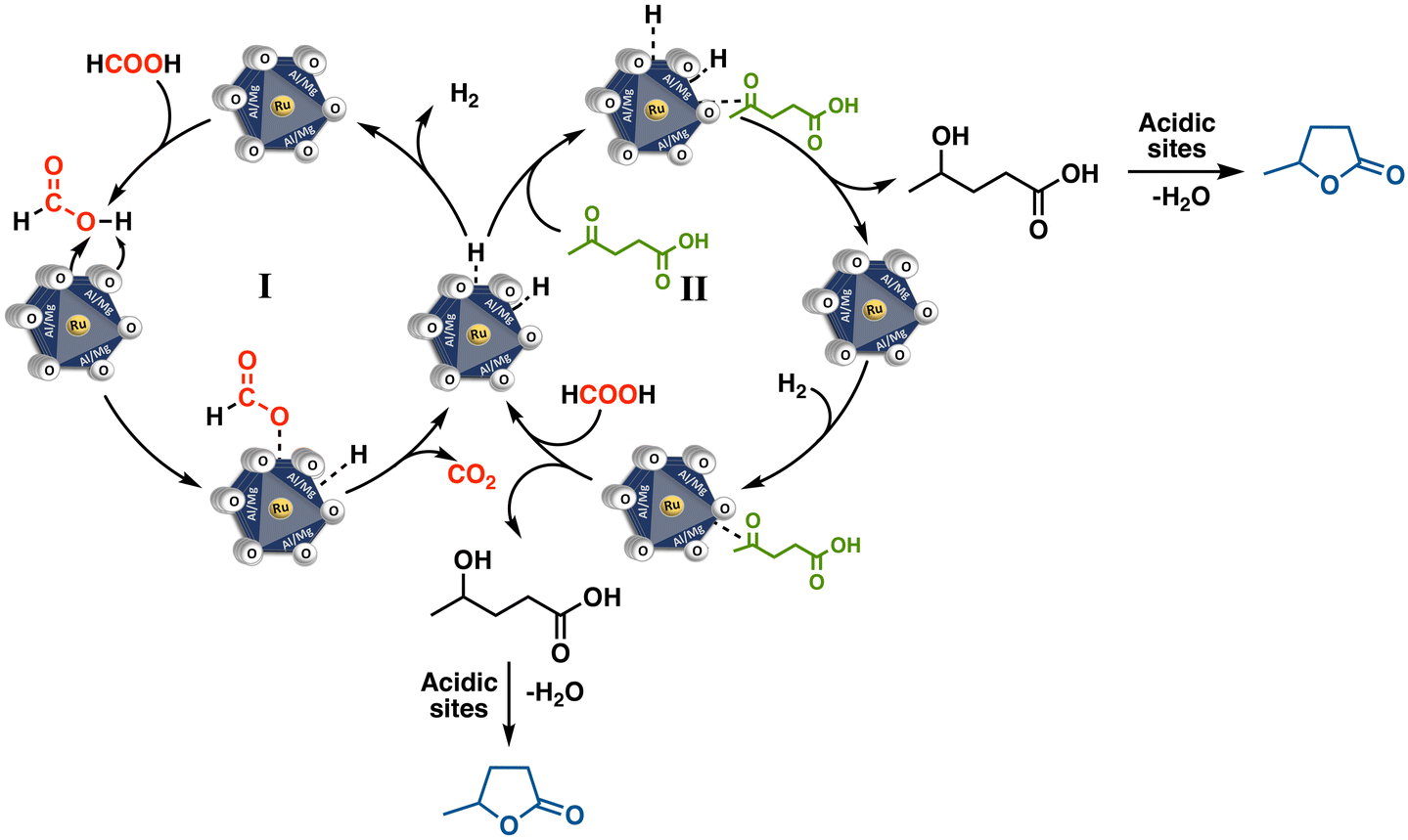 | ||
| Fig. 13 Mechanistic proposal for the hydrogenation of LA with FA catalyzed by MO-Ru. | ||
Notably, no intermediates were detected during the formation of GVL catalyzed by MO-Ru.
4. Conclusion
In summary, we demonstrate that the Ru–MgAl mixed oxide (MO-Ru) is an outstanding bifunctional catalyst. The synergistic interaction between the medium-strength acidic and basic sites on the material's surface and the ruthenium co-catalyst effectively facilitates the hydrogenation of levulinic acid using formic acid as the sole hydrogen source in the aqueous phase. Remarkably, this process does not require the addition of a base or an inert atmosphere, such as N2 or Ar. Under optimal reaction conditions: 150 °C, a formic acid/levulinic acid ratio of 2:1, 0.5 mol% [Ru], and 1.5 hours, complete conversion of levulinic acid was achieved with 99% selectivity toward GVL, significantly outperforming the results reported in the literature under similar conditions.
The key to this efficient hydrogenation process lies in the rapid and selective activation of formic acid by MO-Ru, which generates H2, thereby accelerating the hydrogenation step and preventing the formation of α-angelica lactone and other oligomeric by-products resulting from levulinic acid dehydration process. This catalyst demonstrated consistent performance over four cycles without any loss of activity. The additive-free process is straightforward, cost-effective, and environmentally friendly, positioning this material as a promising candidate for efficient GVL production through a green approach.
Author contributions
Evelyn Vega-Sánchez: synthesis, data collection, investigation, methodology; J. Francisco Javier Tzompantzi-Morales: formal analysis, investigation, writing – review; Luis Ortiz-Frade: data curation, investigation; Marcos Esparza-Schulz and Reyna Ojeda-López: data collection and data curation of nitrogen isotherms; Raúl Pérez-Hernández: data collection and data curation of NH3-TPD and CO2-TPD profiles; Atilano Gutiérrez-Carrillo: NMR studies; Lázaro Huerta: data collection and data curation of the XPS analysis; Victor H. Lara: XRD studies; Leticia Lomas-Romero: formal analysis, supervision, investigation, editing; Lucero González-Sebastián: conceptualization, supervision, investigation, writing – review, and editing.Data availability
All data supporting this article are included in the manuscript and/or included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to express our gratitude to M.Sc. Atilano Gutiérrez-Carrillo and Victor H. Lara for their technical assistance. E. V. S. extends sincere thanks to CONACYT (CVU: 1278278) for the M.Sc. scholarship. This work was supported by PEAPDI 2024 (Programa Especial de Apoyo a Proyectos de Docencia e Investigación, CBI, UAM-Iztapalapa).References
- M. Rose and R. Palkovits, Macromol. Rapid. Commun., 2011, 32, 1299–1311 CrossRef CAS PubMed
.
- F. Cherubini, Energy Convers. Manage., 2010, 51, 1412–1421 CrossRef CAS
- D. J. Hayes, Catal. Today, 2009, 145, 138–151 CrossRef CAS
- L. Yan, Q. Yao and Y. Fu, Green Chem., 2017, 19, 5527–5547 RSC
- A. Hijazi, N. Khalaf, W. Kwapinski and J. J. Leahy, RSC Adv., 2022, 12, 13673–13694 RSC
- K. Yan, Y. Yang, J. Chai and Y. Lu, Appl. Catal., B, 2015, 179, 292–304 CrossRef CAS
- F. Valentini, V. Kozell, C. Petrucci, A. Marrocchi, Y. Gu, D. Gelman and L. Vaccaro, Energy Environ. Sci., 2019, 12, 2646–2664 RSC
- T. Flannelly, M. Lopes, L. Kupiainen, S. Dooley and J. J. Leahy, RSC Adv., 2016, 6, 5797–5804 RSC
- D. Zhao, T. Su, Y. Wang, R. S. Varma and C. Len, Mol. Catal., 2020, 495, 111133 CrossRef CAS
- L. Lin, X. Han, B. Han and S. Yang, Chem. Soc. Rev., 2021, 50, 11270–11292 RSC
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Chem. Eng. Res. Des., 2006, 84, 339–349 CrossRef CAS
- H. Heeres, R. Handana, D. Chunai, C. Borromeus Rasrendra, B. Girisuta and H. Jan Heeres, Green Chem., 2009, 11, 1247–1255 RSC
- I. T. Horváth, H. Mehdi, V. Fábos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238–242 RSC
- W. R. H. Wright and R. Palkovits, ChemSusChem, 2012, 5, 1657–1667 CrossRef CAS PubMed
- X.-L. Du, L. He, S. Zhao, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Angew. Chem., Int. Ed., 2011, 50, 7815–7819 CrossRef CAS PubMed
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS PubMed
- J. Eppinger and K.-W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS
- L. Deng, J. Li, D.-M. Lai, Y. Fu and Q.-X. Guo, Angew. Chem., Int. Ed., 2009, 48, 6529–6532 CrossRef CAS PubMed
- L. Deng, Y. Zhao, J. Li, Y. Fu, B. Liao and Q.-X. Guo, ChemSusChem, 2010, 3, 1172–1175 CrossRef CAS PubMed
- J. Feng, X. Gu, Y. Xue, Y. Han and X. Lu, Sci. Total Environ., 2018, 633, 426–432 CrossRef CAS PubMed
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC
- Y. Yang, Y. Sun and X. Luo, Energies, 2022, 15, 8093 CrossRef CAS
- C. Ortiz-Cervantes and J. J. García, Inorg. Chim. Acta, 2013, 397, 124–128 CrossRef CAS
- Y. Gao, H. Zhang, A. Han, J. Wang, H.-R. Tan, E.-S. Tok, S. Jaenicke and G.-K. Chuah, ChemistrySelect, 2018, 3, 1343–1351 CrossRef CAS
- O. Vozniuk, T. Tabanelli, N. Tanchoux, J.-M. M. Millet, S. Albonetti, F. Di Renzo and F. Cavani, Catalysts, 2018, 8, 332 CrossRef
- S. K. Hussain, V. K. Velisoju, N. P. Rajan, B. P. Kumar and K. V. R. Chary, ChemistrySelect, 2018, 3, 6186–6194 CrossRef CAS
- G. Fan, F. Li, D. G. Evans and X. Duan, Chem. Soc. Rev., 2014, 43, 7040–7066 RSC
- S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2013, 15, 2026–2042 RSC
- J. Kameliya, A. Verma, P. Dutta, C. Arora, S. Vyas and R. S. Varma, Inorganics, 2023, 11, 121 CrossRef CAS
- S.-B. Lee, E.-H. Ko, J. Y. Park and J.-M. Oh, Nanomaterials, 2021, 11, 1153 CrossRef CAS PubMed
- S. Kumari, A. Sharma, S. Kumar, A. Thakur, R. Thakur, S. K. Bhatia and A. K. Sharma, Chemosphere, 2022, 306, 135464 CrossRef CAS PubMed
- P. S. Jijoe, S. R. Yashas and H. P. Shivaraju, Environ. Chem. Lett., 2021, 19, 2643–2661 CrossRef CAS
- M. Xu and M. Wei, Adv. Funct. Mater., 2018, 28, 1802943 CrossRef
- Y. Zhao, X. Jia, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung, D. O'Hare and T. Zhang, Adv. Energy Mater., 2016, 6, 1501974 CrossRef
- M. S. Maru, S. Ram, N. H. Khan and R. S. Shukla, Mater. Adv., 2021, 2, 5443–5452 RSC
- K. Motokura, T. Mizugaki, K. Ebitani and K. Kaneda, Tetrahedron Lett., 2004, 45, 6029–6032 CrossRef CAS
- K. Motokura, D. Nishimura, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 5662–5663 CrossRef CAS PubMed
- F. Basile, G. Fornasari, M. Gazzano and A. Vaccari, J. Mater. Chem., 2002, 12, 3296–3303 RSC
- J. A. Martins, A. C. Faria, M. A. Soria, C. V. Miguel, A. E. Rodrigues and L. M. Madeira, Catalysts, 2019, 9, 1008 CrossRef CAS
- Multipack version 9.6.0.15, 2015-02-19, Ulvac-phi, Inc, Physical Electronics USA, 1994–2014.
- B. V. Crist, 17 January 2004 SDP v 4.1 (32 bit) Copyright ©2004, XPS International, LLC, Compiled January 17, 2004, https://www.xpsdata.com.
- A. V. Karim, A. Hassani, P. Eghbali and P. V. Nidheesh, Curr. Opin. Solid State Mater. Sci., 2022, 26, 100965 CrossRef CAS
- S. P. Newman, W. Jones, P. O'Connor and D. N. Stamires, J. Mater. Chem., 2002, 12, 153–155 RSC
- J.-C. Dupin, H. Martinez, C. Guimon, E. Dumitriu and I. Fechete, Appl. Clay Sci., 2004, 27, 95–106 CrossRef CAS
- S. Radha, S. V. Prasanna and P. V. Kamath, Cryst. Growth Des., 2011, 11, 2287–2293 CrossRef CAS
- F. Basile, L. Basini, G. Fornasari, M. Gazzano, F. Trifirò and A. Vaccari, Chem. Commun., 1996, 2435–2436 RSC
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS
- A. Ananth, M. S. Gandhi and Y. S. Mok, J. Phys. D: Appl. Phys., 2013, 46, 155202 CrossRef
- S. Bhaskar, P. S. Dobal, S. B. Majumder and R. S. Katiyar, J. Appl. Phys., 2001, 89, 2987–2992 CrossRef CAS
- L. Jozwiak, J. Balcerzak and J. Tyczkowski, Catalysts, 2020, 10, 278 CrossRef CAS
- P. S. Bagus, E. S. Ilton and C. J. Nelin, Surf. Sci. Rep., 2013, 68, 273–304 CrossRef CAS
- C. Sousa, P. S. Bagus and F. Illas, Chem. Phys. Lett., 2019, 731, 136617 CrossRef CAS
- J. A. van Bokhoven, J. C. A. A. Roelofs, K. P. de Jong and D. C. Koningsberger, Chem. – Eur. J., 2001, 7, 1258–1265 CrossRef CAS
- P. Zhu, M. Gao, J. Zhang, Z. Wu, R. Wang, Y. Wang, E. R. Waclawik and Z. Zheng, Appl. Catal., B, 2021, 283, 119640 CrossRef CAS
- M. N. E. H. Belguendouz, J. Gancedo, P. Rapado, D. Ursueguía, Y. Patiño, L. Faba, A. Bahmani, E. Díaz and S. Ordóñez, Chem. Eng. J., 2021, 414, 128902 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt03345k |
| This journal is © The Royal Society of Chemistry 2025 |