
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-coordinate bis-phosphine and monophosphine Ni(0) complexes: synthesis and reactivity in C–S cross-coupling†
M. Trinidad
Martín
a,
Carlos J.
Carrasco
a,
Nazaret
Santamaría
a,
Celia
Maya
b,
Auxiliadora
Prieto
*a,
Agustín
Galindo
 *a and
M. Carmen
Nicasio
*a
*a and
M. Carmen
Nicasio
*a
aDepartamento de Química Inorgánica, Universidad de Sevilla, 41012 Sevilla, Spain. E-mail: mnicasio@us.es
bInstituto de Investigaciones Químicas (IIQ), Departamento de Química Inorgánica and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Consejo Superior de Investigaciones Científicas (CSIC) and Universidad de Sevilla, Avenida Américo Vespucio 49, 41092 Sevilla, Spain
First published on 3rd January 2025
Abstract
Preformed Ni(0) complexes are rarely used as precatalysts in cross-coupling reactions, although they can incorporate catalytically active nickel directly into the reaction. In this work, we focus on the preparation and the catalytic application of low-coordinate Ni(0) complexes supported by bulky monophosphine ligands in C–S cross-coupling reactions. We have prepared two families of Ni(0) complexes, bis-phosphine aducts of the type [Ni(PR2Ar′)2] (Ar′ = m-terphenyl group) and monophosphine derivatives of the type [Ni(PR2Ar′)(DVDS)] (DVDS = divinyltetramethyldisiloxane). DFT calculations were used to account for the atypical bent structures displayed by the bis-phosphine Ni(0) complexes. Monophosphine-Ni(0) complexes display catalytic activity superior to bis-phosphine Ni(0) adducts, which suggests that the former facilitate the generation of highly reactive monoligated PNi(0) species. Furthermore, the reactivity of monophosphine-Ni(0) precatalysts outperform that observed with Ni(II) precatalysts with the same phosphine ligands, supporting a more facile activation step to the same catalytic species. This enhanced reactivity allows for the use of lower catalyst loadings (1–5 mol%) and for carrying out the challenging coupling between aryl chlorides and alkylthiols.
Introduction
The quest for more sustainable solutions to precious metal catalysis has increased interest in processes catalyzed by first row transition metals, such as nickel.1 In organic reactions catalyzed by nickel, low-coordinate Ni(0) species are often postulated as key intermediates.1c,2 These reactive species are usually generated in situ, either by reduction of a Ni(II) precursor with an external reductant in the presence of the ligand,3,4 or by reaction of Ni(COD)2 (COD = 1,5-cyclooctadiene), the most often used Ni(0) source,5 with the ligand. Although this method is operationally convenient, inefficient reduction and/or formation of species with a different metal–ligand ratio can negatively affect the reaction outcome.1c,6 An alternative approach is to use a preformed Ni(0) complex, in which the ligand is coordinated to the metal, as a precatalyst. The group of Montgomery has successfully applied this strategy in Ni-catalyzed cycloaddition and C–H functionalization reactions.6b,7 However, despite the advantage of not needing activation to enter the catalytic cycle, the use of single-component Ni(0) precatalysts in cross-coupling chemistry is uncommon.8 Among Ni-catalyzed cross-coupling reactions, those involving the formation of C–S bonds have received less attention.9 Most described catalyst systems rely on the use of supporting chelating ligands. These ligands are less susceptible to being displaced by nucleophilic thiolate anions, preventing catalyst deactivation.10 With the exception of NHC-ligated Ni complexes, which demonstrated their catalytic competence in C–S cross-coupling,11 we noted the absence of protocols based on Ni with non-chelating ancillary ligands such as monophosphines.12 Building on our previous experience on Ni-catalyzed C–S bond formation,11b we developed13 a family of Ni(II) precatalysts of the type [Ni(allyl)Cl(PMe2Ar′)]. These complexes effectively catalyzed the coupling of aromatic and aliphatic thiols with different electrophiles, including aryl tosylates for the first time (Scheme 1A). However, the C–S couplings with aryl chlorides required high catalyst loadings (10 mol%), and proceeded mainly with aromatic thiols. We envisaged that the use of preformed Ni(0) precatalysts could facilitate these challenging transformations. In this regard, Doyle and co-workers14 have demonstrated that monoligated PNi(0) accelerates oxidative addition and transmetallation steps, while bis-ligated P2Ni(0) species prevent catalyst deactivation.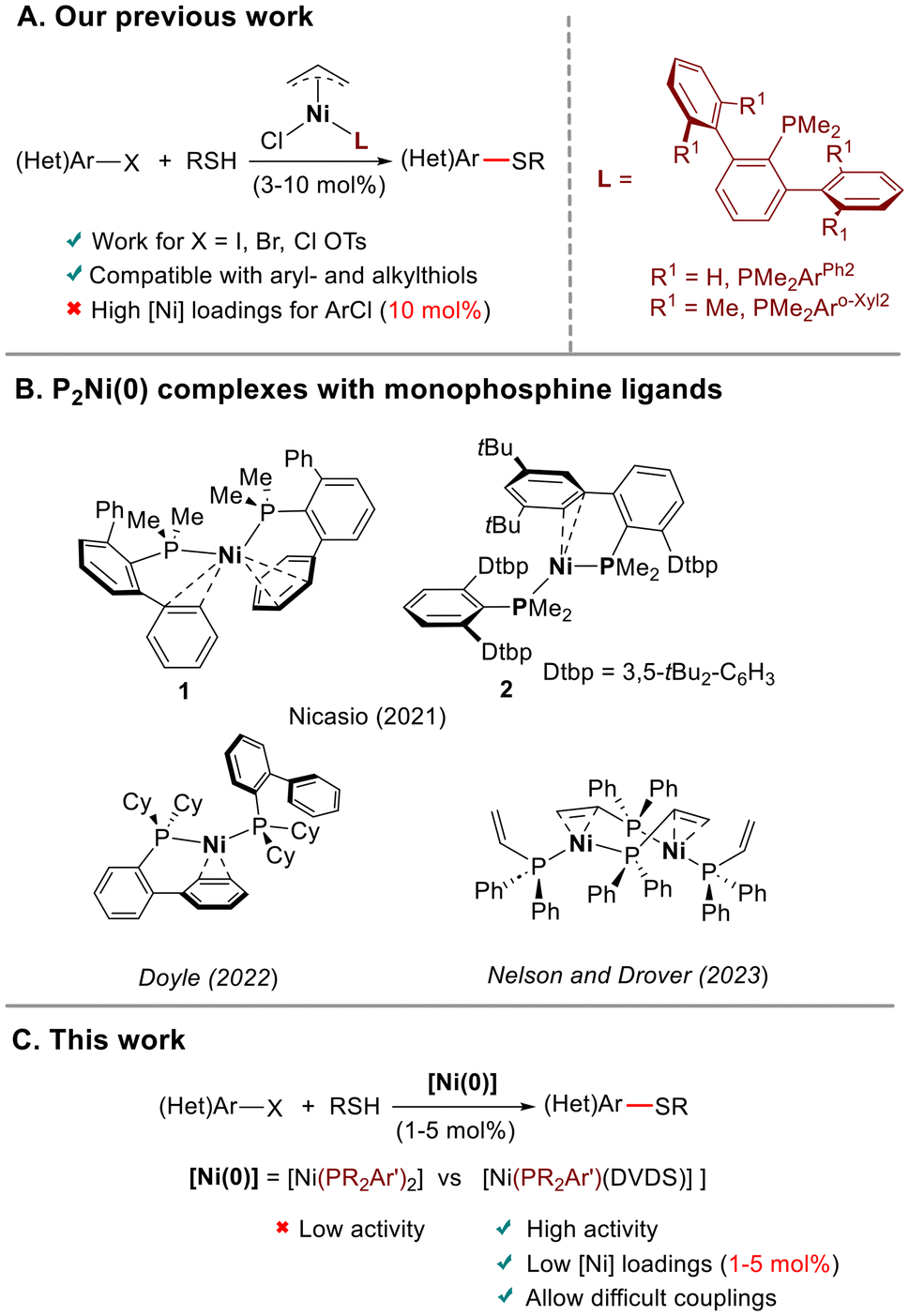 | ||
| Scheme 1 (A) Our previous work on C–S cross-coupling using preformed Ni(II)-allyl precatalysts with terphenyl phosphine ligands. (B) P2Ni(0) complexes with monophosphine ligands. (c) This work. | ||
Bis-ligated P2Ni(0) complexes with monophosphine ligands are rare.15 We recently16 isolated and structurally characterized two P2Ni(0) supported by terphenyl phosphines, which displayed bent structures (Scheme 1B). Shortly thereafter, the group of Doyle17a described the preparation of analogous P2Ni(0) derivatives using Buchwald biaryl phosphines and Nelson and Drover17b reported the preparation of dimeric species supported with allyl- and vinyldiphenylphosphines (Scheme 1B). To further explore the reactivity of these unusual low-coordinate P2Ni(0) species,18 here we describe the synthesis and characterization of two new P2Ni(0) complexes supported by the ligands PMe2Arm-Xyl2 (Arm-Xyl2 = 2,6-bis-(3,5-Me2-C6H3)-C6H3) and P(OPh)2ArPh2 (ArPh2 = 2,6-Ph2-C6H3), which exhibit bent P–Ni–P structures. We have analyzed the reasons that favor the bending in P2Ni(0) complexes using DFT calculations. Furthermore, we have prepared [Ni(PR2Ar′)(DVDS)] complexes by displacement of one of the phosphines in P2Ni(0) complexes with the diolefin ligand divinyltetramethyldisiloxane, DVDS. The latter could serve as synthons of monoligated PNi(0) species by diene extrusion. The behavior of both types of Ni(0) complexes as precatalysts in C–S cross-coupling has been examined. This study highlights the superior reactivity of monophosphine-ligated complexes, which outperform the results obtained with our terphenyl phosphine-supported Ni(II)-allyl precatalysts (Scheme 1C).
Results and discussion
Synthesis and characterization of [Ni(PR2Ar′)2] and [Ni(PR2Ar′)(DVDS)]
As we described earlier,16 we succeeded in preparing the bis-phosphine complex [Ni(PMe2ArPh2)2] 1, in good yields, by the reaction of Ni(COD)2 with 2 equiv. of the less bulky terphenyl phosphine PMe2ArPh2 (ArPh2 = 2,6-Ph2C6H3) in THF at room temperature. However, when increasing the steric bulk of the terphenyl moiety, by placing tBu groups at meta positions of the flanking phenyl rings, no displacement of COD ligand occurred, albeit the complex [Ni(PMe2ArDtbp2)2] 2, (ArDtbp = 2,6-(3,5-tBu2C6H3)2C6H3) could be prepared in moderate yield by heating the reaction under hydrogen pressure.16We further expanded the family P2Ni(0) complexes by reacting Ni(COD)2 with the ligands PMe2Arm-Xyl2 (Arm-Xyl2 = 2,6-(3,5-Me2C6H3)2C6H3) and P(OPh)2ArPh2. Compounds 3 and 4 were obtained as air-sensitive reddish solids in good yields from the direct displacement of COD by the corresponding P-based ligand (Scheme 2A). However, terphenyl phosphines with ortho-substitution on the flanking rings or with P-bound substituents larger than methyl failed to produce any products by this synthetic route. We then focused on synthetic strategies that avoid the use of Ni(COD)2 as the metal precursor. We found that complex 2 could be obtained with high yields by reducing Ni(acac)2 with AlMe3 in the presence of 2 equiv. of PMe2ArDtbp2 (Scheme 2B). Using this strategy, we could observe, by 31P NMR, the formation of the complex [Ni(PMe2Aro-Xyl2)2], bearing the phosphine ligand with ortho substitution on the flanking rings of the terphenyl moiety. However, the instability of this species in solution prevented its isolation and full characterization.
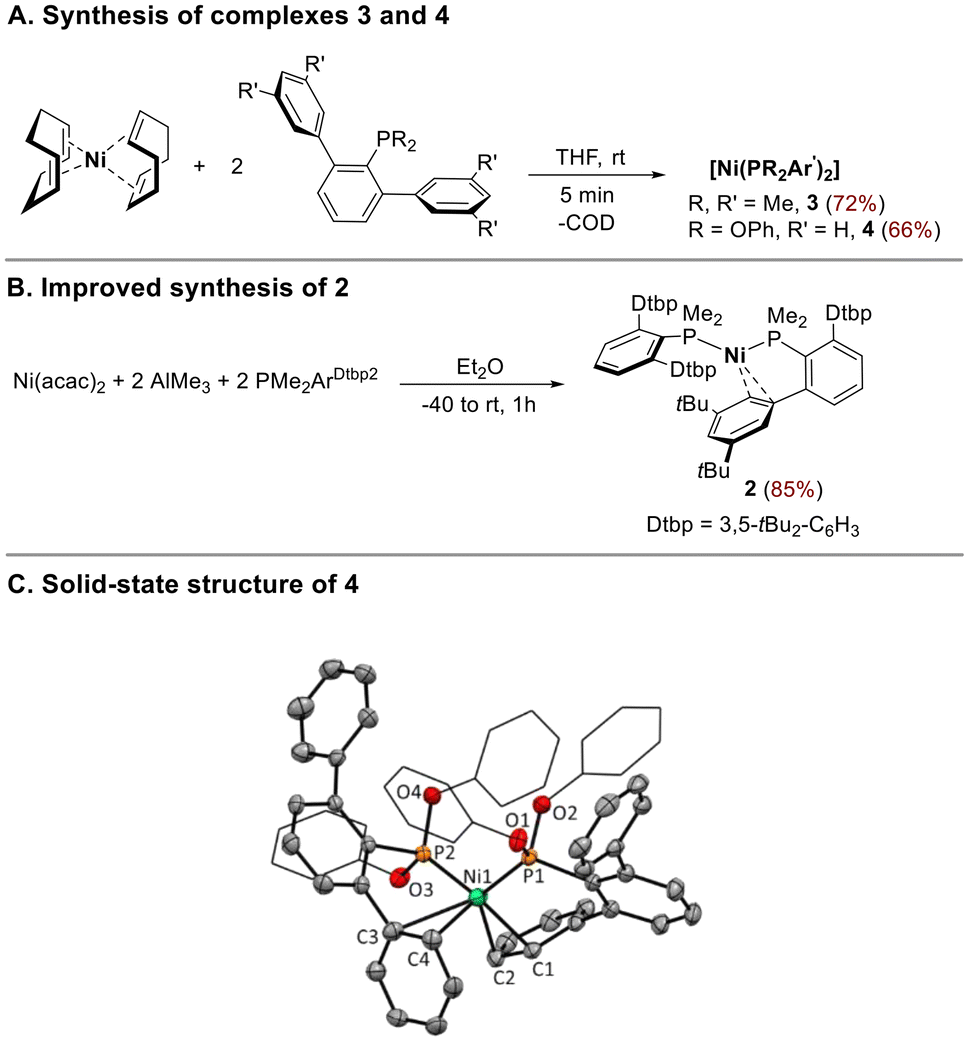 | ||
| Scheme 2 (A) Synthesis of NiP2 complexes 3 and 4. (B) Improved synthetic route for [Ni(PMe2ArDtbp2)2], 2. (C) Molecular structure of 4. Hydrogen atoms are omitted for the sake of clarity. Thermal ellipsoids are shown at a 50% probability. Selected bond lengths [Å] and angles [°]: Ni–P1 2.1253(5), Ni–P2 2.1074(5), Ni–C1 2.086(2), Ni–C2 2.211(2), Ni–C3 2.614(2), Ni–C4, 2.4261(2), P1–Ni–P2 115.37(2). | ||
The new complexes 3 and 4 displayed NMR features similar to those described for adducts 1 and 2. Room temperature 31P{1H} NMR spectra of 3 and 4 in C6D6 show the equivalence of both phosphorus ligands, as indicated by the presence of sharp singlets at −0.5 and 160.4 ppm, respectively. Additionally, the 1H and 13C{1H} NMR spectra of 3 clearly show the presence of a virtual coupled triplet for the methyl groups bonded to the phosphorus nuclei.
The molecular structure of 4, determined by X-ray diffraction studies (Scheme 2C) is very similar to that of 1, with the two phosphine ligands adopting pseudobidentate modes, k1-P,η2-Carene. Both Ni–Carene interactions were of different magnitude. The shortest separations occurred between Ni–Cortho(C1) (2.086(2) Å) and Ni–Cmeta(C2) (2.211(2) Å) of a flanking phenyl ring of one of the phosphine ligands. The other Ni–Carene interaction was much less pronounced and involved the Cipso(C3) and Cortho(C4) of a side ring of the second phosphine, with distances ranging from 2.461(2) to 2.614(2) Å, much longer to those found in the phosphine adduct 1 (2.322(3) and 2.403(3) Å).16 Overall, the shortest Ni–Carene distances and the P–Ni–P angle (115.37(2) °) were comparable to those found in 1 and in biaryl phosphine analogues.17a Finally, the slightly shorter Ni–P bond distances (2.1074(5) and 2.1253(5) Å) compared to those in 1 (average 2.18 Å) reflected the π-acceptor character of the phosphonite ligand.
One of the phosphorous ligands in P2Ni(0) complexes 1–4 can be easily replaced by the chelating olefin DVDS at room temperature,19 giving the coordinatively unsaturated monophosphine species 5–9 (Scheme 3). Alternatively, these compounds are directly prepared by the reaction of Ni(COD)2 with DVDS (2 equiv.) in the presence of the appropriate terphenyl phosphine at room temperature (Scheme 3). All compounds except 8 were isolated as yellow crystalline solids by addition of pentane to the reaction mixtures. Complex 8 defied all efforts at isolation due to its thermal sensitivity, but its formation could be confirmed by NMR analysis of the reaction mixtures in C6D6 (see ESI† for details).
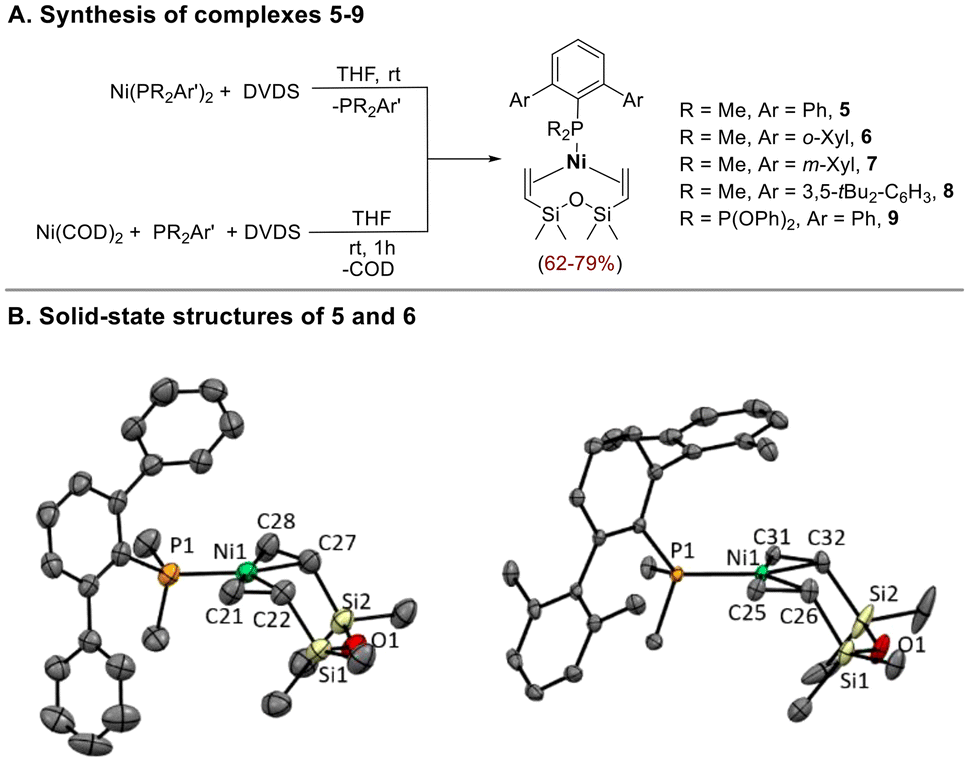 | ||
| Scheme 3 (A) Synthesis of [Ni(PR2Ar′)(DVDS)] complexes 5–9. (B) Molecular structures of 5 (left) and 6 (right). Hydrogen atoms are omitted for the sake of clarity. Thermal ellipsoids are shown at a 50% probability. Selected bond lengths [Å] and angles [°] for 5: Ni1–P1 2.162(1), Ni1–C21 1.986(4), Ni1–C22 2.000(4), Ni1–C27 2.019(3), Ni1–C28 1.995(3), C21–C22 1.398(6), C27–C28 1.293(5), P1–Ni1–C21 92.1(1), P1–Ni1–C22 132.4(1), P1–Ni–C27 133.1(1), P1–Ni1–C28 93.2(1); for 6: Ni1–P1 2.1669(8), Ni1–C25 1.998(2), Ni1–C26 2.008(2), Ni1–C31 2.017(2), Ni1–C32 1.990(2), C25–C26 1.411(4), C31–C32 1.406(3), P1–Ni1–C25 91.67(8), P1–Ni1–C26 132.58(8), P1–Ni–C31 133.74(7), P1–Ni1–C32 92.70(7). | ||
The 1H NMR spectra of 5–9 show two signals for the methyl groups on the silicon atoms of the DVDS ligand and two resonances between 2–3 ppm assigned to the vinyl protons.3,19 Due to unhindered rotation of the phosphine ligand around the Ni–P bond, the PMe2 groups give rise to a doublet and the substituents on the flanking rings of the terphenyl moiety (i.e. Me or tBu) originate a single resonance, both in the 1H and 13C{1H} NMR spectra.
Crystals suitable for X-ray diffraction studies of 5 and 6 were obtained by slow recrystallization from petroleum ether/THF mixtures at −20 °C. The structures shown in Scheme 3B are rare examples of crystallographically characterized monophosphine-supported Ni(0) species with DVDS ligand.3,20 In both structures the Ni atom lies in a planar trigonal environment formed by the P atom and the two vinyl groups of the DVDS ligand. The six-membered metallacycle rings adopt a chair conformation. The Ni–P bond lengths (2.162(1) Å in 5 and 2.1669(8) in 6) are slightly shorter than those reported for the related complexes [Ni(PCy3)(DVDS)]20 (2.2060(9) Å) and [Ni(PtBuiPr2)(DVDS)]3 (2.2284(10) Å). The elongated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond distances in the coordinated diene molecule, mean value of 1.40 Å for 5 and 1.41 Å for 6, falls within the range reported for Ni(0)-DVDS complexes bearing σ-donor ligands3,20,21 and proves the metal backdonation.
C bond distances in the coordinated diene molecule, mean value of 1.40 Å for 5 and 1.41 Å for 6, falls within the range reported for Ni(0)-DVDS complexes bearing σ-donor ligands3,20,21 and proves the metal backdonation.
Computational studies
NiP2 complexes with terphenyl phosphines 1–216 and terphenyl phosphonite 4 as well as those supported by biaryl phosphines17a display uncommon bent structures for d10-ML2 complexes.22 Bickelhaupt and coworkers23 have found that the bending of the angle L–M–L in d10-ML2 complexes is a consequence of π-backbonding. When π backbonding is strong, a lower L-M–L angle favors the donor–acceptor orbital interaction with a high-energy donor orbital of the metal fragment, producing an additional stabilization.23b Furthermore, bent ML2 fragments are found when d10-ML2 complexes react with additional π-acid ligands forming triangular planar 16-electron d10-ML3 structures. This is a consequence of the topology of the bent d10-ML2 HOMO, which is made up of a polarized dp orbital specifically suited for a π-backdonation to the extra ligand. Indeed, this is the situation encountered for complexes 1–4 and for P2Ni(0) complexes containing biaryl phosphine ligands.17aHowever, we questioned why bent P–M–P structures occurred for nickel derivatives, whereas P2M(0) complexes of its heavier counterparts, Pd and Pt with terphenyl phosphines, displayed linear geometry.16 To gain information about the reasons behind such different behavior, we carried out computational studies using the complexes [M(C2H4)(PMe2Ph)2] (M = Ni, Pd, Pt) as the model systems (see ESI, Fig. S1†). We calculated the energies of the reaction between [M(PMe2Ph)2] complexes and ethylene (ESI, Table S1†). In this reaction, there are two opposing effects: the stabilization produced by the ethylene coordination and the destabilization caused by the P–M–P bending. Interestingly, the process turned out to be exergonic for the nickel complex (ΔG = −10.6 kcal mol−1), while for the Pd and Pt derivatives the reactions were unfavorable (ΔG = 5.6 and 7.6 kcal mol−1, respectively). These findings suggest that additional coordination of an extra ligand counterbalances the P–M–P bending only if the metal is nickel, in good agreement with the experimental observations and with the results obtained by Bickelhaupt and Radius for related d10-[M(NHC)2] complexes of group 10 metals.23c
Geometry optimizations were performed for [Ni(PMe2ArPh2)2], 1, [Ni(PMe2ArDtbp2)2], 2, [Ni(PMe2Arm-Xyl2)2], 3, and [Ni(P(OPh)2ArPh2)2], 4 (ESI, Fig. S2†). They showed pseudo-tetrahedral geometries that compared well with the experimental ones for 1, 2 and 4. However, we found some discrepancies between the computed and experimental Ni–Carene distances with the flanking phenyl rings (ESI, Table S2†). In these structures, the Ni–Carene separations with the closer phenyl ring were well described by calculations, while for the furthest one the optimized Ni–Carene distances were above 3 Å. Although such inconsistencies can be ascribed to the difference between experimental solid-state and gas-phase calculated structures, they also seem to point to the lability of these Ni–Carene interactions. In fact, NMR spectra of complexes 1–4 show the equivalence of the two phosphorus ligands. Since variable temperature NMR experiments carried out with complex 116 did not provide any information about the dynamic process that occurs in solution (ESI, Fig. S3†), we studied the dynamic behavior of 1 by DFT calculations (see Scheme 4). It is triggered by the temporary cleavage of one of the weak Ni–η2-Carene bonds. Then, the second η2-Ph ring is detached from the metal center with a concomitant loss of the bent disposition towards a lineal arrangement. The ΔG energy difference between the pseudo-tetrahedral structure and the lineal structure in toluene is 9.9 kcal mol−1, in agreement with the observed fluxionality.
 | ||
| Scheme 4 Dynamic process that averages the phosphine environment in solution. | ||
Geometry optimizations were also performed for divinyldisiloxane complexes 5–9 (ESI, Fig. S3†). They showed the typical trigonal-planar structure of the d10-ML3 complexes. A good comparison of the calculated structural parameters with the experimental ones was observed for 5 and 6, except for the Ni–P bond distance, which is slightly overestimated (ESI, Table S3†). The HOMO to HOMO−4 orbitals are the five d orbitals, as expected for a d10-ML3 complex (Fig. S4† for complex 5). In particular, HOMO to HOMO−2 are basically pure d orbitals (HOMO, z2; HOMO−1, yz; and HOMO−2, xz), while backdonations to vinyl groups come from HOMO−3 (xy) and HOMO−4 (x2 − y2). The thermodynamics of the two synthetic pathways for the synthesis of complexes 5–9, namely, starting from P2Ni(0) and from Ni(COD)2 (Scheme 3A), were analyzed. All reactions were exergonic (ESI, Table S4†) in agreement with experimental. For this purpose, the complex [Ni(PMe2Aro-Xyl2)2], not experimentally isolated, was also optimized (ESI, Fig. S2 and Table S2†). This complex is 18.2 kcal mol−1 less stable (electronic energy) than [Ni(PMe2Arm-Xyl2)2], 3, probably due to the steric pressure of ortho methyl substituents that prevents the stabilization of one of the flanking aryl groups (Fig. S2†).
Catalytic studies
Since both P2Ni(0) and PNi(0) could serve as on-cycle species in C–S cross-couplings,10,12 we sought to examine the catalytic performance of bis-phosphine and monophosphine Ni(0) precatalyts and C–S coupling reactions. To investigate the performance of terphenyl phosphines Ni(0) complexes 1–9, we selected the coupling of iodobenzene with thiophenol in the presence of NaOtBu as the model system (Table 1). Initially, we applied the optimized conditions found for the Ni(II)-allyl precatalysts.13 All NiP2 precatalysts 1–4 were active, but none of them provided full conversions even at extending the reaction time (Table 1, entries 1–4). Under these conditions, complex 2, which contains the ligand PMe2ArDtbp2, displayed the higher catalytic activity. This could suggest a more facile dissociation of one of the phosphine ligands in complex 2, leading to reactive monoligated PNi(0) species. In fact, the largest difference between the two Ni–P bond lengths among the NiP2 series was found in the solid state structure of 2 (see Table S2 in ESI† for bond metrics comparisons). We only observed high yields with precatalysts 1 and 2 when using 10 mol% catalyst loadings (entries 5 and 6; see also Table S5 in ESI† for further reaction conditions). Unlike the bis-phosphine complexes, monophosphine NiP(DVDS) derivatives were much more reactive. Therefore, compound 5 provided complete conversions under the best conditions applied for the Ni-allyl precatalysts (Table 1, entry 7). We selected 5 for reoptimization of the reaction parameters (see ESI, Table S5†). We found that by reducing the catalyst loading to 1 mol% and the reaction time to 4 h, the diphenyl sulfide product was produced in quantitative yields (Table 1, entries 8–10). Under the optimized conditions, only precatalyst 6 led to full conversions (entries 11–13). Unfortunately, the thermal instability of 8 prevented us from studying its reactivity in these coupling reactions. Finally, we tested the most active precatalysts 5 and 6 in the coupling of bromobenzene and phenyl tosylate with thiophenol (Table 2 and Table S6 in ESI†). For both electrophiles, quantitative yields were achieved at lower Ni loadings (3 mol%) and shorter reaction times (4 h) than those required for the monophosphine Ni(II)-allyl precatalysts. Overall, these results confirmed that the terphenyl phosphines PMe2ArPh2 and PMe2Aro-Xyl2 were the best performing ligands in these C–S couplings.13 Furthermore, they highlighted the remarkable catalytic activity of complexes 5 and 6, which outperformed that of our Ni(II)-allyl precatalysts. These results point towards a more facile rate of activation of preformed monoligated PNi(0) complexes to catalytically active species.|
|
||||
|---|---|---|---|---|
| Entry | [Ni] (mol%) | Time/h | Conv.b/% | |
| a Reaction conditions: iodobenzene (1.0 mmol), thiophenol (1.1 mmol), NaOtBu (1.2 mmol), DMF (1 mL), T = 100 °C. b Conversion determined by GC using dodecane as internal standard (average of two runs). c Isolated yield of the product. | ||||
| NiP2 | 1 | 1 (3) | 24 | 21 |
| 2 | 2 (3) | 24 | 45 | |
| 3 | 3 (3) | 24 | 11 | |
| 4 | 4 (3) | 24 | 5 | |
| 5 | 1 (10) | 24 | 100 (93)c | |
| 6 | 2 (10) | 24 | (80)c | |
| NiP(DVDS) | 7 | 5 (3) | 6 | 100 |
| 8 | 5 (1) | 6 | 99 | |
| 9 | 5 (0.5) | 6 | 51 | |
| 10 | 5 (1) | 4 | 100 (93)c | |
| 11 | 6 (1) | 4 | 97 (95)c | |
| 12 | 7 (1) | 4 | 77 | |
| 13 | 9 (1) | 4 | 41 | |
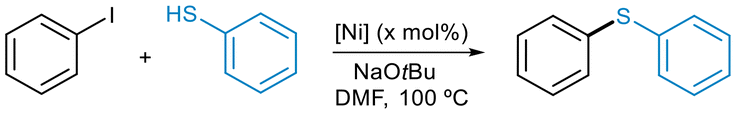
|
|
||||
|---|---|---|---|---|
| Entry | X | [Ni] (mol%) | Time/h | Conv.b/% |
| a Reaction conditions: aryl halide (1.0 mmol), thiophenol (1.1 mmol), base (1.2 mmol), DMF (1 mL) T = 100 °C. b Conversion determined by GC using dodecane as internal standard (average of two runs). c Optimized conditions found for the coupling of bromobenzene with thiophenol catalyzed by Ni(II)-allyl using NaOtBu as the base (see ref. 13). d Isolated yield of the product. e Optimized conditions found for the coupling of bromobenzene with thiophenol catalyzed by Ni(II)-allyl using LiOtBu as the base (see ref. 13). | ||||
| 1c | Br | 5 (5) | 6 | 100 |
| 2 | Br | 5 (3) | 4 | 99 (92)d |
| 3 | Br | 6 (3) | 4 | 94 (91)d |
| 4e | OTs | 5 (5) | 6 | 100 |
| 5 | OTs | 5 (3) | 4 | 92 (79)d |
| 6 | OTs | 6 (3) | 4 | 88 (77)d |
We explored the scope of C–S cross-coupling with a small array of aryl iodides, bromides, and tosylates using the most active precatalysts 5 and 6. All cross-coupled products were obtained in useful synthetic yields under the optimized reaction conditions (Scheme 5). Both catalyst systems were very active in reactions involving sterically hindered substrates, providing the products in high yields (10c and 10d).
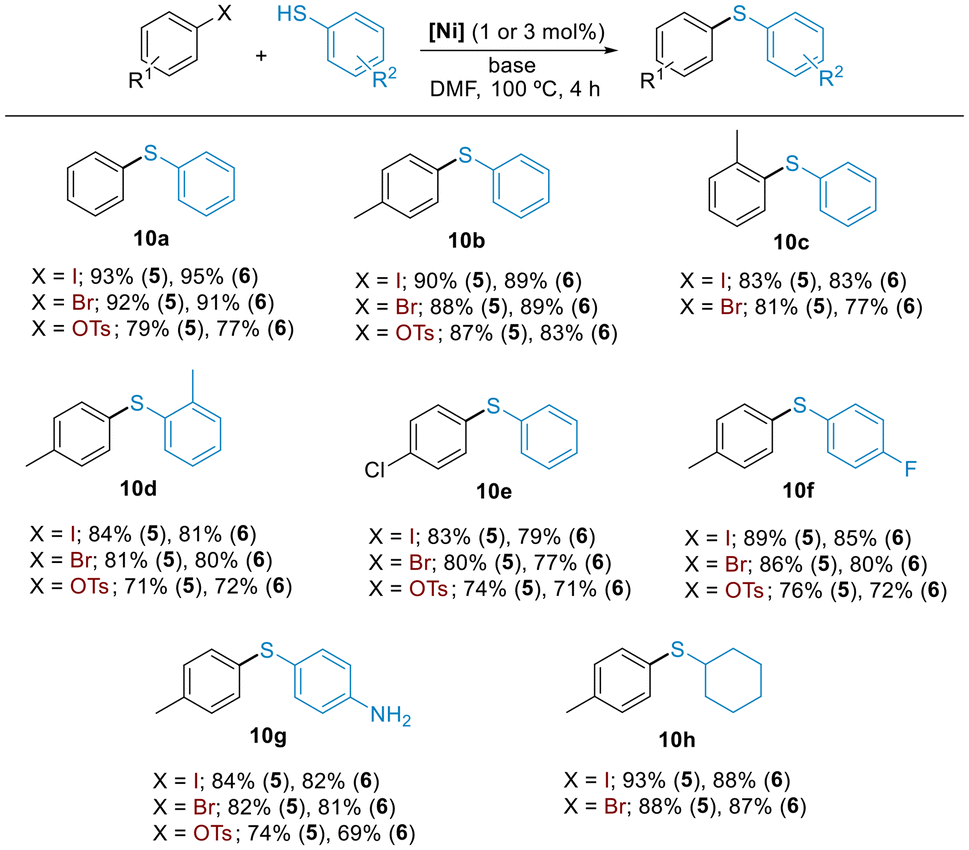 | ||
| Scheme 5 Scope of the arylation of thiols with aryl iodides, bromides and tosylates catalyzed by 5 and 6. Reaction conditions: aryl halide (1.0 mmol), thiol (1.1 mmol), base (NaOtBu for X = I, Br, and LiOtBu for X = OTs; 1.2 mmol), 5 or 6 (1 mol% for reactions with aryl iodides and 3 mol% for reactions with aryl bromides and tosylates), DMF (1 mL), 100 °C, 4 h. Isolated yields of pure products (average of two experiments). | ||
Furthermore, C–I, C–Br or C–OTs bonds were selectively functionalized in the presence of a chloride or a fluoride functionality (10e and 10f). Similarly, free –SH group was efficiently arylated in the presence of a competing protic functionality (10g). Furthermore, aliphatic thiols were also compatible substrates and cyclohexanethiol was successfully arylated using the protocol developed (10h).
Given that aryl chlorides were the least reactive substrates in our previous protocol with Ni(II)-allyl precatalysts,13 we sought to determine whether monophosphine Ni(0) precatalysts could facilitate the activation of challenging C–Cl bonds. The catalytic study was carried out with complex 5 as a precatalyst. Gratifyingly, the coupling of chlorotoluene with thiophenol proceeded in quantitative yields applying the reaction conditions found for the Ni(II)-allyl precatalysts, but with half the catalyst loading (Table 3, entry 1). Further reduction of the catalyst loading led to lower conversions (entries 2 and 3).
Encouraged by this result, we targeted more challenging couplings, such as the thioetherification of aliphatic thiols with chloroarenes. Reports on this type of C–S couplings employing Ni catalyst remain rare.24 Under the optimized conditions shown in Table 3, we examined the coupling of (hetero)aryl chlorides with alkylthiols (Scheme 6). Electron-rich and electron-poor aryl chlorides as well as 2-chloropyridine reacted with primary (11a–11c), secondary (11d–11f) and tertiary (11g–11h) alkyl thiols in good isolated yields. The reaction of hindered 2-chloro-toluene occurred to form the product 11i, but with moderate yield. These results highlight the high reactivity of precatalyst 5 in the C–S cross-coupling of both aromatic and aliphatic thiols, significantly improving the results we obtained with Ni(II)-allyl precatalysts.13 Additionally, this study demonstrates the effective steric protection imparted by the terphenyl phosphine ligand to the Ni center, preventing the catalyst deactivation by binding of multiple thiolate ligands.
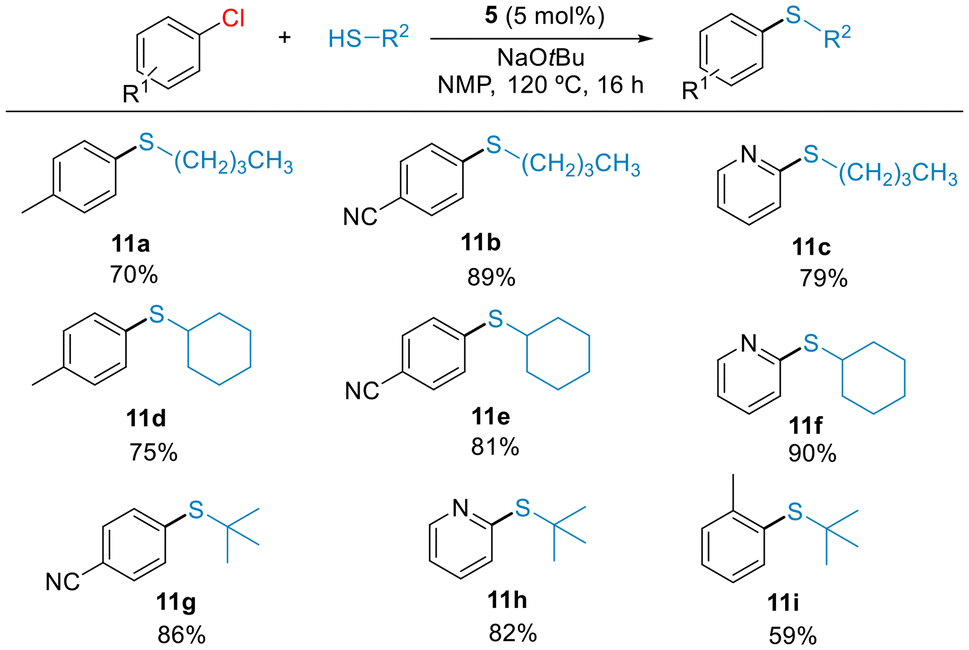 | ||
| Scheme 6 Scope of alkylthiols arylations with aryl chlorides catalyzed by 5. Reaction conditions: aryl chloride (0.55 mmol), thiol (0.5 mmol), NaOtBu (0.6 mmol), 5 (5 mol%), NMP (0.5 mL), 120 °C, 16 h (not optimized). Isolated yields of pure products (average of two experiments). | ||
Conclusions
We have synthesized and structurally characterized two bis-phosphine Ni(0) complexes of the type [Ni(PR2Ar′)2], supported by bulky terphenyl phosphine ligands. The compounds are fluxional in solution, and X-ray diffraction study demonstrates that they are not truly 14-electron species but rather 16-electron species in which one of the phosphorus ligands is bonded in pseudobidentate fashion (k1-P, Ni–Carene). DFT calculations provide an explanation for the preferred bending structures displayed by these d10-ML2 complexes. These studies show that the coordination of the Ni center to one double bond of a side ring of the terphenyl moiety counterbalances the destabilization effect of the P–M–P bending. This Ni–η2-Carene bond is weak and the stepwise dissociation of such an interaction theoretically justifies the dynamic NMR spectra of 1–4.The displacement of one of the phosphorus ligands in P2Ni(0) complexes by diene DVDS is facile, allowing the preparation of a series of monophosphine Ni(0) complexes of the type [Ni(PR2Ar′)(DVDS)]. These complexes can also be synthesized directly from Ni(COD)2. Both synthetic processes are exergonic according to DFT calculations.
Bis-phosphines and monophosphine Ni(0) complexes have been tested as precatalysts in C–S bond formation reactions with the latter showing the best catalytic performance. Among all Ni-DVDS compounds tested, those supported by the parent terphenyl phosphine PMe2ArPh2, 5, or by the phosphine PMe2Aro-Xyl2, 6, are the most active precatalysts. These results suggest that monoligated PNi(0) could be the catalytically reactive intermediates and that monophosphine Ni(0) precatalysts provide a more facile formation of these highly unsaturated species. Moreover, the catalytic performance of 5 and 6 is also superior to that of Ni(II)-allyl precatalysts with the same phosphine ligands, which highlights the advantage of using preformed Ni(0) precatalysts to enhance rate of activation to PNi(0) catalytic species. The increased reactivity of 5 and 6 enables the performance of C–S couplings at lower catalyst loadings (1–5 mol%) and the successful achievement of the challenging thioetherification of aryl chlorides with alkylthiols. Finally, this study also provides some insight into the remarkable ability of terphenyl phosphines to stabilize highly unsaturated monoligated PNi(0) reactive species, preventing their deactivation in the presence of thiolate ligands.
Experimental
General considerations
All preparations and manipulations were performed under an oxygen-free nitrogen atmosphere using standard Schlenk techniques. The solvents were rigorously dried and degassed before use. Reagents were purchased from commercial suppliers and used without further purification. The dimethylterphenyl phosphines PMe2ArPh2,25 PMe2Arm-Xyl2,13 PMe2Aro-Xyl2,25 PMe2ArDtbp2,26 and Ni(COD)227 were prepared according to procedures described. Solution NMR spectra were recorded on Bruker Avance III 300 MHz and Avance III 500 MHz spectrometers. The 1H and 13C resonances of the solvent were used as the internal standard and the chemical shifts are reported relative to TMS while 31P was referenced to external H3PO4. Elemental analyses were performed by the Servicio de Microanálisis of the Instituto de Investigaciones Químicas (IIQ). X-ray diffraction studies were performed at Centro de Investigación, Tecnología e Innovación de la Universidad de Sevilla (CITIUS) and at the Instituto de Investigaciones Químicas (IIQ).General procedure for the synthesis of [Ni(PR2Ar′)2] complexes 3 and 4
A solution of phosphine (0.2 mmol) in THF (1 mL) was added to a solution of Ni(COD)2 (27.6 mg, 0.1 mmol) in THF (1 mL) under nitrogen. The resulting dark red solution was stirred for 5 minutes and petroleum ether was added to precipitate the corresponding complex. The title compound was obtained as dark red crystals after recrystallization.General procedure for the synthesis of [Ni(PR2Ar′)(DVDS)] complexes 5–9
To a solution of Ni(COD)2 (44.3 mg, 0.16 mmol) in THF (2 mL), 1,3-divinyltetramethyldisiloxane (72 μL, 0.36 mmol) was added. The reaction was stirred for 5 minutes at room temperature, after which a solution of the ligand PR2Ar (0.16 mmol) in THF (2 mL) was added. The mixture was stirred for 1 h and the product was precipitated by adding pentane. The solid was purified by recrystallization in a mixture of THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :petroleum ether (1:2).
:petroleum ether (1:2).
Catalytic procedures for the C–S coupling of aryl iodides/bromides with aromatic/aliphatic thiols
The precatalysts 5 or 6 (0.01 or 0.03 mmol) and the base NaOtBu (1.2 mmol) were dissolved in DMF or (1 mL) in a vial equipped with a J Young tap containing a magnetic bar. The thiol (1.1 mmol) and the aryl iodide/bromide/tosylate (1 mmol) were added in turn under a nitrogen atmosphere. The mixture was stirred at 100 °C for 4 h in an oil bath. The reaction mixture was cooled to room temperature, diluted with ethyl acetate (10 mL) and filtered through a Celite plug. The conversion was determined by GC analysis using dodecane as an internal standard. Pure products were obtained after purification by flash chromatography on silica gel with petroleum ether.Computational details
The electronic structure and geometries of the compounds were investigated by using density functional theory (DFT) at the B3LYP level28,29 using the 6-311G(d) basis set. Molecular geometries were optimized without symmetrical restrictions. Frequency calculations were carried out at the same level of theory to identify all stationary points as minima (zero imaginary frequencies) and to provide the thermal correction to the free energy at 298.15 K and 1 atm. The energies of the reaction between [M(PMe2Ph)2] complexes and ethylene were calculated from the optimized reactants and products using the 6-311++G(d,p) basis set and LANL2DZ30 for the Pd and Pt atoms. The energies of the fluxional behavior of complex 1 were calculated using single-point calculations with the 6-311++G(d,p) basis set. In this case, solution-phase SCF energies were calculated using the CPCM solvation model in toluene.31 The DFT calculations were carried out using the Gaussian 09 program package.32Author contributions
The manuscript was written through contribution of all authors. MTM, CJC and NS designed and conducted experiments and analyzed the data. CM conducted X-ray diffraction analysis. AG carried out the computational studies and contributed to the manuscript writing. MCN and AP conceptualized and supervised the project and drafted the script.Data availability
All data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank financial support from MCIU/AEI/10.13039/501100011033 (Grant PID2020-113797RB-C22) and MCIU/AEI/10.13039/501100011033 and ERDF/EU (Grant PID2023-151417NB-100). The use of computational resources of the Universidad de Granada (cluster Albaicín) is thankfully acknowledged.References
- (a) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed; (b) V. P. Ananikov, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS; (c) C. M. Lavoie and M. Stradiotto, ACS Catal., 2018, 8, 7228–7250 CrossRef CAS; (d) Nickel Catalysis in Organic Synthesis: Methods and Reactions, ed. S. Ogoshi, Wiley-VCH, Weinheim, Germany, 2020 Search PubMed; (e) A. L. Clevenger, R. M. Stolley, J. Aderibigbe and J. Louie, Chem. Rev., 2020, 120, 6124–6196 CrossRef CAS PubMed.
- (a) N. I. Saper, A. Ohgi, D. W. Small, K. Semba, Y. Nakao and J. F. Hartwig, Nat. Chem., 2020, 12, 276–283 CrossRef CAS PubMed; (b) J. B. Diccianni and T. Diao, Trends Chem., 2019, 1, 834–844 Search PubMed; (c) S. Cañellas, J. Montgomery and M. À. Pericàs, J. Am. Chem. Soc., 2018, 140, 17349–17355 CrossRef PubMed; (d) S. Tang, O. Eisenstein, Y. Nakao and S. Sakaki, Organometallics, 2017, 36, 2761–2777 CrossRef CAS; (e) J. A. Garduño, A. Arévalo and J. J. García, Dalton Trans., 2015, 44, 13419–13438 RSC; (f) R. M. Stolley, H. A. Duong and J. Louie, Organometallics, 2013, 32, 4952–4960 CrossRef CAS PubMed.
- (a) E. Richmond and J. Moran, Synthesis, 2018, 499–513 CrossRef CAS; (b) E. Morser, J. Jeanneau, N. Mézailles, H. Olivier-Bourbigou and P.-A. R. Breuil, Dalton Trans., 2019, 48, 4101–4014 RSC.
- An electrochemical method for the synthesis of Ni(0) complexes have been recently reported: C. Z. Rubel, Y. Cao, T. E.-H. Ewing, G. Laudadio, G. L. Beutner, S. R. Wisniewski, X. Wu, P. S. Baran, J. C. Vantourout and K. M. Engle, Angew. Chem., Int. Ed., 2024, 63, e202311557 CrossRef CAS PubMed.
- A variety of bench-stable Ni(0) precursors for in situ ligation protocols have been reported in recent years: (a) V. T. Tran, Z.-Q. Li, O. Apolinar, J. Derosa, M. V. Joannou, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, Angew. Chem., Int. Ed., 2020, 59, 7409–7413 CrossRef CAS PubMed; (b) L. Nattmann, R. Saeb, N. Nöthling and J. A. Cornella, Nat. Catal., 2020, 3, 6–13 CrossRef CAS; (c) L. Nattmann and J. A. Cornella, Organometallics, 2020, 39, 3295–3300 CrossRef CAS; (d) C. Z. Rubel, W.-J. He, S. R. Wisniewski and K. M. Engle, Acc. Chem. Res., 2024, 57, 312–326 CrossRef CAS PubMed.
- (a) E. A. Standely and T. F. Jamison, J. Am. Chem. Soc., 2013, 135, 1585–1592 CrossRef PubMed; (b) A. J. Nett, J. Montgomery and P. M. Zimmerman, ACS Catal., 2017, 7, 7352–7362 CrossRef CAS; (c) M. M. Beromi, G. Benerjee, G. W. Brudvig, D. J. Charboneau, N. Hazari, H. M. C. Lant and B. Q. Mercado, Organometallics, 2018, 37, 3943–3955 CrossRef PubMed.
- (a) A. J. Nett, W. Zhao, P. Zimmerman and J. Montgomery, J. Am. Chem. Soc., 2015, 137, 7636–7639 CrossRef CAS PubMed; (b) A. J. Nett, S. Cañellas, Y. Higushi, M. T. Robo, J. M. Kochkodan, M. T. Haynes II, J. W. Kampf and J. Montgomery, ACS Catal., 2018, 6, 6606–6611 CrossRef PubMed; (c) M. T. Robo, A. R. Frank, E. Butler, A. J. Nett, S. Cañellas, P. Zimmerman and J. Montgomery, Organometallics, 2022, 41, 3293–3300 CrossRef CAS PubMed.
- N. Hazari, P. R. Melvin and M. M. Beromi, Nat. Rev. Chem., 2017, 1, 0025 CrossRef CAS PubMed.
- (a) For recent reviews see: I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2022, 122, 16110–16293 CrossRef CAS PubMed; (b) V. J. Geiger, R. M. Oechsner, P. H. Gehrtz and I. Fleischer, Synthesis, 2022, 5139–5167 CAS; (c) E. Rufino-Felipe, H. Valdés and D. Morales-Morales, Eur. J. Org. Chem., 2022, e202200654 CrossRef CAS.
- (a) E. Alvaro and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 7858–7868 CrossRef CAS PubMed; (b) G. Yin, I. Kalvet, U. Englert and F. Schoenebeck, J. Am. Chem. Soc., 2015, 137, 4164–4172 CrossRef CAS PubMed.
- (a) Y. Zhang, K. C. Ngeow and J. Y. Ying, Org. Lett., 2007, 9, 3495–3498 CrossRef CAS PubMed; (b) M. J. Iglesias, A. Prieto and M. C. Nicasio, Adv. Synth. Catal., 2010, 352, 1949–1954 CrossRef CAS; (c) A. R. Martin, D. J. Nelson, S. Meiries, A. M. Z. Slawin and S. P. Nolan, Eur. J. Org. Chem., 2014, 3127–3131 CrossRef CAS; (d) M. A. Rodríguez-Cruz, S. Hernández-Ortega, H. Valdés, E. Rufino-Felipe and D. Morales-Morales, J. Catal., 2020, 383, 193–198 CrossRef.
- It has been reported the use of monophosphine ligands in Pd-catalyzed C-S cross-coupling. See: J. Xu, R. L. Liu, C. S. Yeung and S. L. Buchwald, ACS Catal., 2019, 9, 6461–6466 CrossRef CAS PubMed.
- M. T. Martín, M. Marín, C. Maya, A. Prieto and M. C. Nicasio, Chem. – Eur. J., 2021, 27, 12320–12326 CrossRef PubMed.
- J. E. Borowski, S. H. Newmann-Stonebraker and A. G. Doyle, ACS Catal., 2023, 13, 7966–7977 CrossRef CAS PubMed.
- (a) R. J. Rama, M. T. Martín, R. Peloso and M. C. Nicasio, Adv. Organomet. Chem., 2020, 74, 241 CrossRef CAS; (b) V. M. Englert, P. W. Jolly and G. Wilke, Angew. Chem., 1971, 83, 84–85 CrossRef; (c) P. W. Jolly, K. Jonas, C. Krüger and Y.-H. Tsay, J. Organomet. Chem., 1971, 33, 109–122 CrossRef CAS.
- M. T. Martín, M. Marín, R. J. Rama, E. Álvarez, C. Maya, F. Molina and M. C. Nicasio, Chem. Commun., 2021, 57, 3083–3086 RSC.
- (a) S. H. Newman-Stonebraker, J. Y. Wang, P. D. Jeffrey and A. G. Doyle, J. Am. Chem. Soc., 2022, 144, 19635–19648 CrossRef CAS PubMed; (b) M. L. Clapson, D. J. Nelson and M. W. Drover, ACS Org. Inorg. Au, 2023, 3, 217–222 CrossRef CAS PubMed.
- M. T. Martín, C. Maya, A. Galindo and M. C. Nicasio, Adv. Synth. Catal., 2024 DOI:10.1002/adsc.202400765.
- NiP2 complexes reacted with electron-deficient alkenes such as styrene, maleic anhydride, dimethyl fumarate among others, but in all cases mixtures of bis-alkene and mono-alkene complexes were obtained that could not be separated.
- P. B. Hitchcock, M. F. Lappert, C. MacBeath, F. P. E. Scott and N. J. W. Warhurst, J. Organomet. Chem., 1997, 528, 185–190 CrossRef CAS.
- (a) J. Wu, J. W. Faller, N. Hazari and T. J. Schmeier, Organometallics, 2012, 31, 806–809 CrossRef CAS; (b) S. Abe, T. Kosai, T. Limura and T. Iwamoto, Eur. J. Inorg. Chem., 2020, 2651–2657 CrossRef CAS; (c) M. Muhr, J. Hornung, J. Weßing, C. Jandl, C. Gemel and R. A. Fischer, Inorg. Chem., 2020, 59, 5086–5092 CrossRef CAS PubMed.
- A bent P–Pd–P angle was found in [Pd(PCy3)2]: E. Dinjus and W. Leitner, Appl. Organomet. Chem., 1995, 43, 50 Search PubMed.
- (a) W. J. Van Zeist and F. M. Bickelhaupt, Dalton Trans., 2011, 40, 3028–3038 RSC; (b) L. P. Wolters and M. Bickelhaupt, ChemistryOpen, 2013, 2, 106 CrossRef CAS PubMed; (c) F. Hering, J. Nitsch, U. Paul, A. Steffen, F. M. Bickelhaupt and U. Radius, Chem. Sci., 2015, 6, 1426–1432 RSC.
- (a) P. H. Gehrtz, V. Geiger, T. Schmidt, L. Sršan and I. Fleischer, Org. Lett., 2019, 21, 50–55 CrossRef CAS PubMed; (b) R. M. Oechsner, J. P. Wagner and I. Fleischer, ACS Catal., 2022, 12, 2233–2243 CrossRef CAS.
- L. Ortega-Moreno, M. Fernández-Espada, J. J. Moreno, C. Navarro-Gilabert, J. Campos, S. Conejero, J. López-Serrano, C. Maya, R. Peloso and E. Carmona, Polyhedron, 2016, 116, 170–181 CrossRef CAS.
- (a) M. Marín, J. J. Moreno, C. Navarro-Gilabert, E. Álvarez, C. Maya, R. Peloso, M. C. Nicasio and E. Carmona, Chem. – Eur. J., 2019, 25, 260–272 CrossRef PubMed; (b) M. Marín, J. J. Moreno, M. M. Alcaide, C. Maya, E. Álvarez, J. López-Serrano, J. Campos, M. C. Nicasio and E. Carmona, J. Organomet. Chem., 2019, 896, 120–128 CrossRef.
- M. M. Colqhoun, J. Holton, D. J. Thompson and M. V. Twiggs, in New Pathways for Organic Synthesis. Practical Applications of Transition Metals, Plenum Press, New York, 1984 Search PubMed.
- D. J. Becke, Chem. Phys., 1993, 98, 5648–5652 Search PubMed.
- L. W. Yang and R. G. Parr, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, spectroscopic and analytical data, X-ray crystallographic and computational data. CCDC 2405003 for 4, 2405002 for 5, 2405004 for 6. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03375b |
| This journal is © The Royal Society of Chemistry 2025 |