
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral single-atom-bridged diphosphorus ligands: synthesis, complexation and catalysis†
Javier Eusamio
 ab and
Arnald Grabulosa
*ab
ab and
Arnald Grabulosa
*ab
aDepartament de Química Inorgànica i Orgànica, Secció de Química Inorgànica, Universitat de Barcelona, Martí i Franquès, 1-11, E-08028, Barcelona, Spain. E-mail: arnald.grabulosa@ub.edu
bInstitut de Nanociència i Nanotecnologia (IN2UB), Universitat de Barcelona, E-08028, Barcelona, Spain
First published on 6th February 2025
Abstract
The synthesis, complexation and main catalytic applications in enantioselective homogeneous catalysis of enantiopure single-atom-bridged diphosphorus ligands ((R1R2)P–X–P(R3R4); X = CR2, NR, O) is reviewed, covering the literature up to the beginning of 2025. The information is organised by ligand type, with unsubstituted methylene-bridged (–CH2–) and substituted amino-bridged (–NR–) diphosphorus ligands being by far the most common type of ligands. The perspective review is completed by the analysis of all reported crystal structures of bidentate monometallic complexes with the ligands. The bite angles, metal–phosphorus distances and buried volumes (Vbur) are given in the ESI.
 Javier Eusamio | Javier Eusamio obtained his BSc in Chemistry at the University of Barcelona (UB) in 2020 and a MSc in Applied Materials Science in 2021. He is currently finishing his PhD in the Inorganic and Organic Chemistry Department of UB, under the supervision of Dr Arnald Grabulosa. His research interests focus on the synthesis of diphosphorus ligands and their coordination to late transition metals for homogeneous catalysis. In parallel, he pursues a MSc in Computational and Mathematical Engineering. |
 Arnald Grabulosa | Arnald Grabulosa obtained his PhD in Inorganic Chemistry from the University of Barcelona (UB) in 2005 with Prof. Muller. After postdoctoral periods with Profs Gros (Nancy, France), Kamer and Clarke (St Andrews, UK), in 2017 he earned an Assistant Professorship at the Inorganic and Organic Chemistry Department of UB. His research focuses on coordination and organometallic chemistry with novel phosphorus ligands for catalysis and other applications. |
Introduction
The development of new chiral ligands for enantioselective homogeneous catalysis1 has been tremendous during the last four decades and shows no signs of diminution. Despite the plethora of coordination motifs currently in use, diphosphorus ligands2 constitute the most important class of ligands for many reactions. Hundreds, if not thousands of chiral diphosphorus ligands have been described in the literature, many of them with complicated, yet captivating structures. However, this beauty comes with a price as they are obtained by lengthy, multistep syntheses.One of the key parameters of diphosphorus ligands is the bite angle,3 whose structural and catalytic effects have been studied and exploited to a great effect. Another parameter is the bulkiness of the ligands, and recently the buried volume has emerged as a way to parametrize4,5 the steric hindrance of a ligand (Fig. 1A).6–8
 | ||
| Fig. 1 (A) Representation of the bite angle and buried volume, and the value ranges of complexes with chiral PXP ligands coordinated in bidentate fashion. (B) Typical coordination motifs in complexes with PXP ligands. (C) 3D scatter plot of the buried volumes (%Vbur), bite angles, and average P–M distances of the 47 reported crystal structures of monometallic complexes coordinated to chiral single-atom-bridged diphosphanes in a bidentate way. N-bridged diphosphanes (PNP) are shown in red, while C-bridged diphosphanes (PCP) are represented in blue. (D) Slice of plot C showing only the average P–M distance vs. the bite angle. | ||
The simplest bridge is a single atom, and this is incarnated by the ligand 1,2-bis(diphenylphosphino)methane (dppm), a ligand synthesised as early as 1959 by Issleib and Müller9 and used to prepare countless coordination and organometallic compounds. This simplicity can turn into advantage, because it is instructive to recall that some of the best ligands in enantioselective hydrogenation10 are methylene-bridged chiral diphosphanes, some of them with a single stereogenic centre. In addition, the coordination of single-atom-bridged diphosphorus ligands is far from simple because other coordination modes apart from the expected bidentate coordination forming four-membered chelates are known (Fig. 1B).11,12
In 2017 S. Mansell published a fascinating perspective on the catalytic applications of diphosphorus ligands with single-atom linkers.11 Inspired by this publication and our own work in the field, we herewith review the enantiopure single-atom-bridged diphosphorus ligands described so far, with a general formula ((R1R2)P–X–P(R3R4); X = CR2, NR, O), abbreviated as PXP. Although they constitute a relatively small subset of ligands, some of them excel in enantioselective catalysis, particularly in hydrogenation.
There are many ways to classify diphosphorus ligands. In the case of single-atom-bridge ligands (PXP), a convenient way is depicted in Scheme 1.
 | ||
| Scheme 1 Classification of the PXP ligands used in this perspective article. | ||
The ligands are firstly classified by the bridging atom between the phosphorus atoms. In the case of the chiral ligands treated in this review, only carbon, nitrogen and oxygen examples have been described so far. Then the carbon- and nitrogen-based ligands are subdivided according to the substitution or not of the atom in the bridge. The unsubstituted, methylene-bridged ligands can be then subdivided into P-stereogenic (denoted as P*, type 1) or not (type 2). There are only a few PCP ligands with a substituted methylene bridge, which form group 3. The same is true for PNP ligands with an unsubstituted nitrogen bridge (NH, group 4) but there are many ligands with a substituted nitrogen bridge, which constitute group 5. Finally, there is the small subset of ligands with an oxygen bridge (POP), which constitutes group 6.
This classification according to the bridging atom has been conceived as a way to systematise the different ligands present in this review, but it can be observed in Fig. 1(C and D) that the bridging atom does indeed greatly affect the geometry of the ligand and, consequently, its coordination and its behaviour in catalysis.
Fig. 1C is a 3D scatter plot of all the crystal structures of chiral PXP diphosphorus ligands. Only monometallic bidentate complexes, with a bidentate coordination of the ligand have been considered. Interestingly, the difference between PNP and PCP ligands, represented as red and blue points respectively, can be readily observed, as the two types of ligands appear in two differentiated clusters. This can be observed even better in Fig. 1D, when a 2D slice of the previous plot is represented. As expected, the smaller the P–M distance the bigger the bite angle, although this trend is clearer with N-bridged ligands. It can also be observed that there is a noticeable correlation between buried volume and bite angle, with the bulkier ligands generally having wider bite angles (ESI, Fig. S3†). Interestingly, L73 (Fig. 3) presents the smallest bite angle and second smallest buried volume at a sphere radius of 3.5 Å (Table S1†), even though the diphosphane contains two apparently bulky BINAP moieties (Fig. 3). On the other hand, the bulkier diphosphane corresponds to MaxPHOS (L54, Fig. 3), of which multiple crystal structures have been analyzed. As expected, when coordinated to nickel(II) center, L54 is significantly bulkier and has a smaller P–M distance due to the smaller size of the nickel cation. In general, it appears that complexes with PNP ligands appear across a wider range of values than those with PCP ligands (ESI, Fig. S1–S4†). However, this could be because there are more structures for the former type of ligands, and that the metal centers of the complexes are also more varied.
All the ligands described so far are collected in Fig. 2 (PCP ligands) and Fig. 3 (PNP ligands), with the number used to refer to them in this perspective.
 | ||
| Fig. 2 C-bridged diphosphorus ligands (PCP). | ||
 | ||
| Fig. 3 N- and O-bridged chiral diphosphorus ligands (PNP and POP, respectively). | ||
A quick glance at Fig. 2 and 3 shows that most of the ligands are either methylene-bridged (types 1 and 2) or with a substituted nitrogen bridge (type 5). It can be observed that many of the ligands contain a P-stereogenic group, especially in the more recently reported ligands.13,14 Apparently, it is an excellent motif in the four-membered chelated structures formed in the coordination of single-atom bridged ligands.
The reader may wonder why there are so few examples of oxygen-bridged ligands or whether other bridges, for example with sulphur, are possible. The answer is that oxygen (POP) and sulphur (PSP) ligands undergo a phosphorotropic equilibrium between the bis(phosphorus(III)) tautomer (PXP) and the phosphorus(II)–(III) tautomer (PPX, Scheme 2).15
 | ||
| Scheme 2 Phosphorotropic tautomerism between PXP (left) and PPX (right). | ||
This equilibrium is reminiscent to that present between secondary phosphane oxides (SPOs) and phosphinous acids16 and depends on the bridging atom and the substituents present on the phosphorus atom. In the case of nitrogen, the equilibrium almost always lies towards the PNP side, and this has been exploited to prepare a plethora of ligands. In contrast, for oxygen and sulphur, it usually lies towards the PPX side, explaining their rarity. Like in the case of SPOs, however, it is possible to stabilise the PXP tautomer by coordination and so they should not be discarded for catalysis,16–19 although to the best of our knowledge this has not been used in enantioselective catalysis.
The review summarises the chemistry involved in the preparation and complexation of the PXP ligands and their main applications in enantioselective catalysis. In addition, the crystal structures of all the coordination and organometallic complexes described so far with the ligands have been analysed to extract the bite angles, phosphorus-metal distances and buried volumes (Vbur),7 given in the ESI.† The review covers the primary literature up to the beginning of 2025.
C-bridged diphosphorus ligands (PCP)
There are, to our knowledge, 52 examples of enantiopure C-bridged diphosphorus ligands described in the literature (L1–L52, Fig. 2). Interestingly, most of them contain an unsubstituted methylene bridge (L1–L41), with only 11 examples in which the bridge is substituted (L42–L52, Fig. 2).20–22 In all cases, one of the phosphorus is part of a cycle, and there are no examples in the literature with acyclic substituted or disubstituted carbon bridges.We have firstly divided this section into methylene bridged ligands and ligands with a substituted bridge, with the former subsection being the one with the most ligands. For this reason and taking into account that most of the PCP ligands are P-stereogenic, the methylene-bridged diphosphanes have been further divided into alkyl-substituted P-stereogenic, aryl-substituted P-stereogenic, and non-P-stereogenic. This layout appears to be the most natural, since most of the alkyl-substituted P-stereogenic ligands show common synthetic strategies, and the same applies with their aryl-substituted counterparts. Furthermore, this classification provides a good overview of some of the most relevant synthetic methodologies for obtaining P-stereogenic ligands, which could be of interest since the control of the configuration of a stereogenic phosphorus is a challenging topic.13,14
Methylene-bridged P-stereogenic diphosphorus ligands
Immediately after, Imamoto and coworkers applied the previously reported methodology for the synthesis of fully-alkyl methylene-bridged diphosphanes and developed the well-known MiniPHOS ligands (L1–L7), which are nowadays still regarded among the best ligands for enantioselective hydrogenation of functionalized olefins.25
Their synthesis (Scheme 3) started with the preparation of alkyldimethylphosphane–boranes from PCl3 in two simple steps, followed by the stereoselective deprotonation of one of the methyl groups through the Evans method (enantioselective deprotonation), followed by reaction of the carbanion with one equivalent alkyldichlorophosphane followed by addition of methylmagnesium bromide and boronation to protect the newly formed phosphane moiety. Since the addition of the second phosphane is not stereoselective (a P-stereogenic atom is formed), this reaction yielded two diastereomers, the optically active one, and the meso form. Luckily, in most cases they could be separated through recrystallization to obtain the pure protected ligands (Scheme 3), which could then be deboronated with triflic acid for their complexation. The obtained free diphosphanes were air-sensitive but configurationally stable at room temperature.
 | ||
| Scheme 3 Synthesis of MiniPHOS ligands (L1–L7). | ||
Among the four original MiniPHOS (L1–L4) ligands,25 it appeared that t-Bu-MiniPHOS (L1) was the one that afforded the best results in the initial enantioselective catalytic reactions studied (Michael additions and hydrogenation, Fig. 5). Shortly after,26 they synthesized two new MiniPHOS (L5 and L6) ligands, this time bearing 1-adamantyl substituents. Interestingly, ligand L5 is the first (and one of the very few) reported C1-symmetric, methylene-bridged P-stereogenic ligands. A few years later,27 a new ligand L7, bearing t-octyl substituents, was also developed by a different procedure, using an optically pure secondary methylphosphane–borane as starting material.27
Surprisingly, when the coordination of L1 to rhodium(I)-diene moieties was explored, it was observed that, due to their small bite angle, a bischelated complex bis-C1 (Fig. 4) tends to form instead of the expected monochelated complex (C1), regardless of stoichiometry. This is a behaviour that has also been observed with other chiral PXP ligands.28–30
 | ||
| Fig. 4 Monochelated complex (C1) bischelated complex (bis-C1) (top). Equilibrium between the observed dihydrido species and the proposed hemilabile intermediate (bottom). Counter anions (PF6− or BF4−) have been omitted for clarity. | ||
Interestingly, the formation of this bischelated complex could be easily avoided by using [Rh(cod)2]SbF6 as a metal precursor instead of [Rh(nbd)2]SbF6.31
In the case of L1, the mechanism of the catalytic activity of the bischelated complex in enantioselective hydrogenation was thoroughly studied by Imamoto and Gridnev.27,32–34 A hemilabile behaviour was proposed to justify the coordination of the substrate to the octahedral dehydrogenated complex,27 although no key intermediate species could be observed (Fig. 4).
Shortly after the publication of the MiniPHOS ligands (L1–L4), a new P-stereogenic ligand was reported by Hoge and coworkers in 2004.35 This diphosphane presented a C1-symmetry, with only one P-stereogenic phosphorus, which bear a t-butyl and a methyl substituent—like MiniPHOS (L1)—, while the other phosphorus has two t-butyl substituents. Given that three out of the four substituents of the ligand were t-butyl, the ligand was named TriChickenFootPHOS (TCFP, L8). This design is of great interest because it follows a three-hindered quadrant strategy, where only one of the quadrants (the one with the non-bulky methyl substituent) of the catalyst is not sterically hindered. This fact allows for the preferential coordination of the catalytic substrates to form only one species, which could explain the great success of TCFP (L8) as a ligand for enantioselective hydrogenation (Fig. 5).36,37 Interestingly, despite the resemblance with the MiniPHOS (L1) ligands, the synthesis of TCFP (L8) is completely different, since it was not prepared as an optically pure compound, but in racemic form and then the enantiomers of the diphosphane-borane were separated through preparative HPLC on a chiral stationary phase.
 | ||
Fig. 5 Selected substrates used in Rh-catalysed enantioselective hydrogenation with PCP ligands and their corresponding enantioselectivities. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (Z)-isomer. (Z)-isomer. | ||
In terms of synthetic viability, the MiniPHOS (L1–L7) ligands have experienced an unforeseen problem that greatly affected their availability: the sudden and global shortage of sparteine as a cheap and easily available chiral auxiliary.38,39 For this reason, Imamoto and coworkers developed in 2010 a new procedure that allowed for the gram-scale synthesis of optically pure t-butylmethylphosphane–borane using (−)-bornyl chloroformate as the chiral auxiliary, and allowing the obtention of both enantiomers through the inversion of the (S) enantiomer of the secondary phosphane-borane (Scheme 4).27 Through this improved methodology, they were able to synthesize many ligands without relying on sparteine, like MiniPHOS (L1–L7), BisP*, or TCFP (L8), among many others.27,31
 | ||
| Scheme 4 Synthesis of TCFP (L8) and BulkyP* (L9) ligands using (−)-bornyl chloroformate as a chiral auxiliary. | ||
A few years after, Imamoto and coworkers reported a new methylene-bridged ligand, BulkyP* (L9, Scheme 4),40 which resembled TCFP but exchanging the substituents of the non-P-stereogenic phosphorus from t-butyl to 1-adamantyl, furnishing a ligand with a much more pronounced steric effect, as its name implies. Thanks to this fact, the ligand turned out to be an air-stable, crystalline solid, making its handling more convenient. This ligand, like t-Bu-MiniPHOS (L1) and TCFP (L8), showed an excellent performance in the hydrogenation of functionalized alkenes (Fig. 5).
As it can be seen in Fig. 5, many short-bridged diphosphanes with a carbon backbone excel in the enantioselective hydrogenation reaction, proving their usefulness with a wide variety of conditions and substrates. The typical benchmark substrates (Z)-MAC, DMI and MAA have been hydrogenated with almost perfect enantioselectivity with many PCP ligands. The differences arise when more challenging substrates are explored, but t-Bu-MiniPhos (L1) and TCFP (L8) are especially outstanding among the broad range of ligands covered in this review. It seems clear that the rigidity of the four-membered chelate ring, and the electron-richness of the ligands are key factors that produce very high enantioselectivities. It is notable that most of the best ligands contain the t-butyl substituent as a bulky, electron-releasing substituent for the phosphorus atom.
 | ||
| Scheme 5 The original Jugé-Stephan methodology applied to the synthesis of (S,S)-DIPAMP (a–g). The opposite diastereomer could be obtained by switching the order of addition of the organolithium reagents (first MeLi and second (o-An)Li). Synthesis of P-stereogenic chlorophosphanes (h), secondary phosphanes (i), and iodomethylphosphanes (j). | ||
The Jugé-Stephan method relies on the use of (1R,2S)-(−)-ephedrine or its enantiomer as the chiral auxiliary to confer the chirality on the phosphorus atom. Starting from PhP(NEt2)2, the addition of the ephedrine and borane generates a chiral intermediate oxazaphospholidine–borane (b), a heterocycle that is then selectively opened with an organolithium reagent (c), by P–O scission. After that, the P–N bond is cleaved through an acidic methanolysis (d), giving a P-stereogenic phosphinite-borane that can then be converted to a phosphane–borane with another organolithium reagent, in our case methyllithium (e). This product can be further reacted to synthesize a plethora of phosphorus compounds, or deprotected and coordinated as a monophosphane. In Scheme 5, the synthesis of DIPAMP42—an ethylene-bridged ligand—is depicted (f and g), which was the first diphosphane to be obtained through the Jugé-Stephan method.
This method can also be applied to the synthesis of methylene-bridged diphosphanes, since the methyl substituent of the starting phosphane–borane can be easily deprotonated and further reacted. In fact, 15 out of the 41 reported methylene-bridged chiral diphosphorus compounds have been synthesized following the Jugé-Stephan method (L10–L24, Fig. 6), showcasing the versatility of the procedure.
 | ||
| Fig. 6 Methylene-bridged PCP ligands prepared by the Jugé-Stephan method. | ||
Out of the 15 described ligands obtained through the Jugé-Stephan method, the first one to be synthesized was L10 by Mezzetti and coworkers in 2002.43 In this contribution, the authors were exploring the synthesis of bulky P-stereogenic monophosphanes to obtain new highly symmetric, sterically hindered ligands. Initially, the synthesis of mesitylmethylphenylphosphane-borane was attempted, but it could only be obtained if the mesityl substituent was introduced first into the oxazaphospholidine–borane ring (Scheme 5b), and the phenyl substituent was introduced in the second place (c). Regardless of this, the yields were very low, making the procedure synthetically unsuitable (Scheme 6A). If the reaction was performed inverting the order of these reagents, no product was formed, probably due to the steric hindrance of the mesityllithium reagent (Scheme 6B).
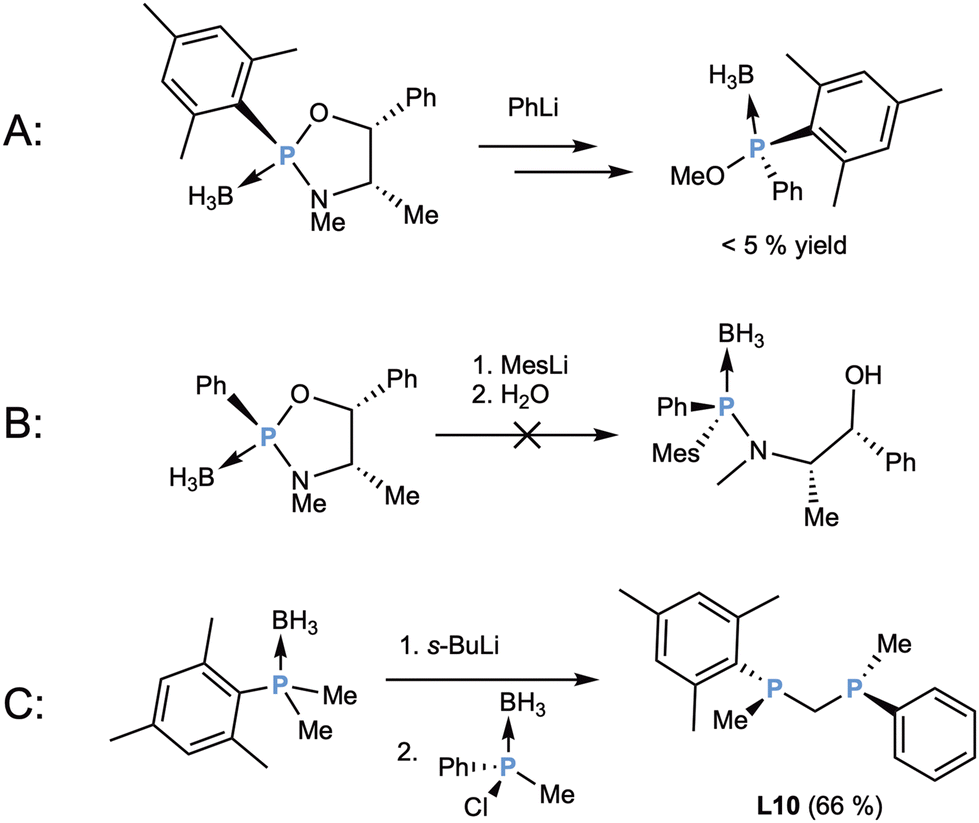 | ||
| Scheme 6 (A and B) Attempted synthesis of mesitylmethylphenylphosphane-borane. (C) Synthesis of L10. | ||
For this reason, the focus was shifted towards the non-chiral mesityldimethylphosphane-borane, which was obtained in good yield, and it was deprotonated with s-BuLi and reacted with the chloromethylphenylphosphane-borane (Scheme 5h),44 forming the new P–C bond. The optically active diastereomer was formed with a good diastereomeric ratio in relation to the meso compound (83:17) and could be isolated in good yield and acceptable enantiopurity (86% ee) (Scheme 6C). Interestingly, when the ligand complexation with [Rh(cod)2]BF4 or [Rh(nbd)2]BF4 was attempted, no product was formed, but reacting the diphosphane with [RhCl(cod)]2 in the presence of NH4PF6 afforded the expected [Rh(cod)(L10)]PF6 (Fig. 7, Complex I). When testing the complex in the enantioselective hydrogenation reaction, the yields were low and the obtained product was racemic, presumably due to the instability of the complex, which degraded giving metallic rhodium.
 | ||
| Fig. 7 General representation of the described complex types with methylene-bridged diphosphanes prepared by the Jugé-Stephan method. | ||
A few years later, Jugé and coworkers synthesized the dppm* ligands L11 and L12, which were conceived as the P-stereogenic, optically pure variants of the classical ligand dppm.45,46 Interestingly, these two are the only short-bridged ligands synthesized by the Jugé-Stephan methodology that are C2-symmetric. This is because, as discussed in the synthesis of L10, the Jugé-Stephan methodology has limitations in terms of the substituents that can be introduced in the oxazaphospholidine–borane. On the other hand, the addition of another P-stereogenic moiety requires the use of a P-stereogenic chlorophosphane, which are synthetically challenging to obtain and handle in optically pure form due to configurational instability (Scheme 3h).47,48
Despite these limitations, Jugé and coworkers developed L11, where two of the phenyl groups of dppm are replaced by 3,5-dimethylphenyl (xylyl) groups, and L12, where o-anisyl groups were used instead. Besides, thanks to the versatility of the Jugé-Stephan methodology, both enantiomers of each ligand could be obtained since, in contrast to the Evans methodology, by altering the order of the substituents the opposite enantiomer could be obtained.
Interestingly, L12 can be viewed as the methylene-bridged version of the well-known ligand DIPAMP, which is famous for its excellent catalytic results in the early days of enantioselective hydrogenation.42 For this reason, it is surprising that the use of L11 and L12 as ligands for catalytic applications has not been reported. Instead, Jugé and coworkers studied their coordination properties to form C3-symmetric trinuclear Pd clusters45,46 (Fig. 7, Complex F) and 1D coordination polymers with Ag and Cu.49 The monomers for these coordination polymers adopted what is known as an “A-frame” coordination (Fig. 7, Complexes C and D), which is not uncommon in coordination chemistry, and short-bridged ligands being no exception.11
In 2021, Jugé and coworkers published a contribution where they combined a phospholyl moiety with a P-stereogenic fragment, connected through a methylene bridge (L13–L15).50 These three compounds, which were later coordinated to rhodium(I), are not strictly diphosphanes, since the P-stereogenic atom remains boronated, and coordinates to the rhodium through the hydrogen atoms of the borane group, forming a 6-membered ring instead of the much more strained 4-membered ring from single-atom diphosphanes. Nonetheless, they have been included because they are relevant to the topic of chiral single-atom bridged diphosphorus ligands, both from their synthetic and coordination points of view.
The incorporation of the phospholyl moiety to the P-stereogenic fragment was performed starting either by reaction of deprotonated methylphosphane (Scheme 5e) with a cyanophosphole, furnishing ligands L13–L15 or by the reaction of a iodomethylphosphane derived from the secondary phosphine-borane (Scheme 5i and j) with lithium phospholide, to give L15.
These ligands were coordinated to rhodium to yield complexes [Rh(L)(cod)]BF4 (L = L13–L15), with the BH3 coordinating to the Rh through two of its hydrogen atoms (η2-BH3) (Fig. 7, Complex J). When the complexes were tested in enantioselective hydrogenation with benchmark substrate methyl 2-acetamidoacrylate (MAA), their performance was not very good, with poor enantioselectivities (<20% ee) and low conversions in some cases. Although this represented the first case of a chiral κ2-BH3 coordinated complex used in enantioselective catalysis, the application of P-stereogenic phospholyl ligands in their more canonical, deboronated version has not been explored.
More recently, Jugé and coworkers have also developed a new methodology to obtain P-stereogenic phosphinites through an N → O phosphinyl migration.51 This migration proceeded after the deboronation of the previously opened oxazaphospholidine–borane ring, via a phosphorane intermediate. Among the wide range of studied phosphanes, one example featuring the synthesis of a new methylene-bridged diphosphane L16 was provided (Scheme 7), although its optical purity was not determined, and the ligand was not complexed or further studied.
 | ||
| Scheme 7 Synthesis of ligand L16. ee n.d. | ||
Around the same time, our group also made a contribution to the field of P-stereogenic methylene-bridged diphosphanes.52 In this initial article, we explored the synthesis of diphosphanes starting from optically pure P-stereogenic methylphosphane-boranes with 1-naphthyl, 2-biphenylyl, or 9-phenanthryl substituents, which were accessed through the Jugé-Stephan methodology (Scheme 5a–e) and had been used in hydrovinylation of olefins.53 The boronated monophosphanes could be easily deprotonated using an organolithium reagent and quenched with a chlorophosphane, in the same manner as with ligands L10–L12, but using achiral chlorophosphanes. This afforded a family of modular ligands comprised of aryl–aryl diphosphanes (L17, L20, and L23) and aryl–alkyl ones (L18, L21, and L24). This last group was later expanded with in a more recent contribution, where the t-butyl substituents were introduced (L19 and L22).29 In this case, it was observed that the bulkiness of the substituents allowed only for one of the phosphorus to be boronated after work-up. Interestingly, this phosphorus turned out to be that with the t-butyl substituents, after a spontaneous deboronation of the initially protected P-stereogenic phosphorus.
After deprotecting the ligands, their coordination with palladium(II) moieties was studied. Upon reacting them with [Pd(cod)Cl2], the expected neutral complexes were obtained for diphosphanes L17–L18, L20–L21, and L23–L24 (Fig. 7, Complex G).
After that, we wanted to explore their behaviour in cationic complexes, and the same ligands were thus coordinated to the metal precursor [Pd(μ-Cl)(η3-allyl)]2 in the presence of NH4PF6 acting as a halogen scavenger (Fig. 7, Complex H). The expected complexes could also be obtained, although it was observed that in the case of the aryl–aryl ligands, they were not the thermodynamically stable species. In solution, [Pd(η3-allyl)(L17)]PF6 quickly evolved into a dimeric, more complex A-frame species, where the diphosphane acted as a bridge between two different palladium atoms (Fig. 7, Complex E). This behaviour is probably favoured by the narrow bite angle and the four-membered chelate ring inherent to mononuclear complexes with short-bridged diphosphanes. In some cases, the geometry of the compound is so strained that the ligand adopts a hemilabile behaviour, decoordinating from the metal centre and favouring its coordination with a different palladium atom. Interestingly, aryl–alkyl diphosphanes did not form these A-frame complexes, suggesting that the coordination of the alkyl-substituted phosphorus with the palladium is stronger.
It was later observed that these A-frame complexes further decomposed into mononuclear bischelated and neutral [Pd(L)Cl2] complexes. The bischelated complexes (Fig. 7, Complex B) are also common with short-bridged ligands. A crystal structure was obtained for the complex with L19, which was the first one obtained for a bischelated complex with a C1-symmetric diphosphane, and it was interesting to see that the ligands presented a cis coordination, with the more sterically hindered substituents on the same side. This unexpected behaviour (from a steric point of view) could also be observed with the analogous rhodium(I) complex bearing ligand L20.29
After palladium, the coordination of these ligands to ruthenium and rhodium was also explored. L20 and L21 were reacted with [RuCl(μ-Cl)(η6-p-cymene)]2 in the presence of a halogen scavenger. Interestingly, the substituents on the non-P-stereogenic phosphorus greatly influenced the coordination behaviour of the ligands, as had been observed with palladium. In the case of ruthenium, L21 showed a much more pronounced trend to form chelated complexes, forming the cationic complex even when no halogen scavenger was added, with one of the chlorines acting as the counteranion (Fig. 7, Complex L). When NH4PF6 was used, the expected complex was obtained (Fig. 7, Complex M). On the other hand, applying these same conditions to L20 produced the neutral complex with the ligand acting in a hemilabile fashion, with only the non-P-stereogenic phosphorus coordinated to the ruthenium (Fig. 7, Complex K). Only when the stronger halogen scavenger TlPF6 was used the chelated cationic complex was observed. However, it was also determined that the solvent plays a key role in the coordination of these ligands, since it was noticed that the hemilabile complex turned into the corresponding chelate when dissolved in methanol.54
In the case of rhodium, a more predictable behaviour can be observed for ligands L17–L22, since depending on the equivalents of ligands employed with respect to rhodium, both the bischelated (Fig. 7, Complex A) or monochelated complexes (Fig. 7, Complex I) could be obtained starting from metal precursor [Rh(cod)2]BF4. Contrasting with what had been reported by Imamoto and coworkers for L1,31 the diene of the precursor did not influence the final coordination of the rhodium, and neither did the counteranion, as had been reported by Mezzetti and coworkers for ligand L10.43 Instead, the monochelated/bischelated behaviour could be regulated by carefully controlling the stoichiometry, order and addition rate of the reagents. When a solution of the rhodium precursor was added dropwise on a solution of the diphosphane solution, the bischelate was preferentially formed. On the other hand, if the solution of the ligand was added dropwise to a solution of the rhodium complex, the pure monochelated complexes could be obtained.
The rhodium monochelated complexes of ligands L17–L22, as well as the bischelate of L21, were tested as catalysts in the enantioselective hydrogenation reaction. Almost all complexes showed quantitative conversions to the desired products, but enantioselectivities were moderate at best (<50% ee).
Around the same time, Dong, Zhang, and coworkers developed the new methylene bridged diphosphane L25 with a P-stereogenic phosphorus substituted by a chiral ferrocenyl moiety.55 The rationale behind this design was to follow the three-hindered quadrant strategy which had produced successful results with some ligands, like the aforementioned TCFP (L8).35 Additionally, this new ligand L25, nicknamed t-Bu-Wudaphos, would have the advantage of being easier to synthesize and to handle, since it makes use of Ugi's amine as a more convenient way of introducing a chiral motif into the molecule.
The synthesis of the ligand proceeded by deprotonation of the ortho position of the substituted cyclopentadienyl of ferrocene, which was then reacted with PCl3 to introduce the first phosphorus atom, which will become the P-stereogenic centre. After that, the boronated di-t-butylmethylphosphane was lithiated and added to the mixture, reacting with one of the two remaining chlorines of the initial PCl3 and forming the methylene bridge through a phosphination reaction. Finally, the last chlorine was reacted with methylmagnesium chloride to give the partially boronated ligand as a single diastereomer, thanks to the use of the optically pure Ugi's amine as a chiral auxiliary. After deprotection, the final ligand L25 is obtained as a convenient highly air-stable solid (Scheme 8).
 | ||
| Scheme 8 Synthesis of L25, t-Bu-Wudaphos. | ||
Unfortunately, no coordination studies for this interesting ligand have been described, but it was used in situ with [Rh(nbd)2]BF4 to assess its catalytic performance in enantioselective hydrogenation. The chosen substrates were α-methylene-γ-keto-carboxylic acids due to their acidic nature. It was postulated that the amine of the Ugi's base could interact with acidic substrates through ion pair noncovalent interactions, directing even more the selective coordination of the substrate to the rhodium complex, in addition to the previously mentioned three hindered quadrant strategy. When a varied scope of carboxylic acids was tested, it was observed that the hydrogenations proceeded with excellent enantioselectivities and conversions with a wide variety of substituents (Fig. 5). On the other hand, when the proposed ion pair interaction was disrupted by adding either an external base, or by removing the acidic group from the substrate, a substantial drop in either conversion or enantioselectivity was observed, highlighting the critical role that this interaction plays in the reaction.
Additionally, α,α-disubstituted terminal olefins were also tested as substrates, which resulted in a more challenging substrate due to the similarity of the olefin substituents.56 However, thanks to the ion pair effect, the interaction between the carboxylic acid and the amine could be used to direct the coordination of the olefin, resulting in excellent yields and enantioselectivities (Fig. 5).
Methylene-bridged non-P-stereogenic diphosphanes
The same year that the MiniPHOS ligands (L1–L4) were disclosed by Imamoto,25 Werner and coworkers also published a very interesting contribution which explored the synthesis of C2- and C1-symmetric methylene bridged diphosphanes.57 Although the focus of their work was not set on obtaining chiral compounds, four out of the eleven ligands that they developed contained the chiral substituent (R)-menthyl, which conferred optical purity to the compounds (L26–L29). The synthesis of these diphosphanes proceeded using stannylated iodomethanes ICH2SnR3 (R = Ph, Me) as starting materials. By reacting these compounds with n-BuLi and dimenthylchlorophosphane, Men2PCH2SnR could be obtained (Men = menthyl). Then, addition of an organolithium reagent results in a transmetallation, producing SnR4 and the lithiated phosphane, which was then reacted with the appropriate chlorophosphane R′2PCl to produce ligands L26 (R′ = Men), L27 (R′ = i-Pr), L28 (R′ = Cy), and L29 (R′ = Ph). Out of the four synthesized ligands, only L26 presents C2 symmetry, with all the substituents on both phosphorus being menthyl groups. It is interesting to note that, in the same contribution, the synthesis arsinophosphanes was also explored, and the first crystal structure of a chiral compound of this kind was obtained for the ligand Men2PCH2AsCy2.57After that, the coordination to rhodium(I) of various diphosphanes and arsinophosphanes was explored, although unfortunately none of the chiral ligands L26–L29 were studied. Nonetheless, the typical coordinative behaviour of achiral, methylene-bridged diphosphanes was observed, obtaining bischelated complexes, chloro-bridged dimers, and a η6 coordination to benzene and toluene to Rh(I), similar to the catalytic systems that have been used by Willis, Weller, and coworkers for the hydroacylation reaction with short-bridged diphosphanes.58,59 A few years later, in 2004, the Schrock-Osborn complexes [Rh(L27)(cod)]PF6 and [Rh(L29)(cod)]PF6 were synthesized by the same group and used as precatalysts in the enantioselective hydrogenation of α-acetamidocinnamic acid methylester, a functionalized trisubstituted olefin.60 Although the reaction yielded the hydrogenated product quantitively, the enantioselectivities were moderate (29–69%) in all cases, regardless of the ligand and the reaction conditions.
Around the same time, Faraone and coworkers carried out a study on the effects of the bridging atom on short-bridged chiral diphosphanes, where they explored methylene, nitrogen and oxygen bridges.61 To accomplish it, they synthesized C2-symmetrical ligands with the phosphorus containing a binaphthyl moiety, which conferred the chirality to the system. In the case of the methylene-bridged ligand L30, the diphosphane was obtained by reacting bis(dichlorophosphino)methane with (R)-binaphthol (Fig. 8A). When coordinated to the Pd(η3-1,3-diphenyallyl) precursor, it was observed that both monomeric and dimeric complexes were formed in solution (Fig. 8B). When the ligand was tested in the enantioselective allylic alkylation reaction, the resulting enantioselectivity was very low (<10% ee), probably due to the mixture of species formed in solution. It is interesting to note that, when these results are compared with the similar, ethylene-bridged counterpart of L30 for the same reaction the performance was much better, achieving 90% conversion and 73% ee.62 This poses the question of whether the more rigid 4-member chelate hindered the performance of L30 in comparison with its more flexible, 5-member chelate analogue.
 | ||
| Fig. 8 (A) Synthesis of ligand L30. (B) Pd species observed in solution. PF6− counteranions have been omitted for clarity. | ||
A few years later, Jackson and a coworker presented the ligand Ph-BPM L31,63 which was conceived as a short-bridged version of the well-performant diphosphane Ph-BPE, with an ethylene bridge.64 The diphospholane ligand was synthesized starting from the secondary phospholane–borane adduct, which was reacted with its mesylate-substituted derivative in the presence of n-BuLi to form the diboronated ligand, which could then be deprotected with DABCO yielding the final L31 (Scheme 9A).
 | ||
| Scheme 9 (A) Synthesis of ligand L31. (B) One-pot synthesis and complexation of ligand L41. The same procedure is applied for the synthesis of ligands L32–L41. | ||
The [Rh(cod)(L31)]BF4 was then obtained and used as a precatalyst in the enantioselective hydrogenation reaction, providing excellent conversions and enantioselectivities of benchmark substrates with catalysts loadings of as low as 0.01 mol% (Fig. 5). With these positive results, the ligand was also tested in ruthenium catalyzed imine hydrogenation, with overall good yields and selectivities, although not as good as in the previous reaction.
More recently, Pringle and coworkers developed a set of highly modular methylene-bridged diphosphanes with a chiral motif, that could be either a BINAP (L32–L35), a substituted BINAP (L36–L37), or an ortho substituted phospholane (L38–L41).65 To synthesize them, the chiral chlorophosphane was reacted with TMS(CH2)PR2,66 where R could be a phenyl, i-propyl, cyclohexyl, or a t-butyl substituent. With this flexible methodology, they were able to synthesize 10 new ligands using only a few different building blocks (Scheme 9B).
Interestingly, when coordination of these ligands to Rh was attempted with [Rh(nbd)2]BF4 or [Rh(cod)2]BF4 as metal precursors, only ligands L38–L41, which bore a phospholane moiety, were able to yield crystals suitable for X-ray diffraction. Ligands L32–L37, with a BINAP or BINAP-derived moiety, could only produce a crystal for when coordinated to Pt, obtaining the neutral complex [Pt(L35)Cl2]. In both cases, the obtained structure corresponded to the ligand with t-butyl substituents, which seems to be a good substituent for crystallisation purposes.29 Pringle and coworkers demonstrated the facile obtention of Rh complexes by performing, in a one-pot procedure, the synthesis of ligand L41 and its complexation (Scheme 9B).
When the ligands were tested in the enantioselective hydrogenation of the three benchmark substrates MAC, MAA, and DMI, it was observed that the diphosphanes that gave the best enantioselectivities also had t-butyl substituents. Out of the 10 ligands used, L41 turned out to be the best performing one with the studied conditions (Fig. 5).
Diphosphanes with a substituted methylene bridge
In 1995, Pietrusiewicz, Majoral, and coworkers developed the first chiral short-bridged diphosphane with a substituted carbon bridge.67 This ligand L42 was achieved through a very unconventional reaction involving a P–Zr adduct. Starting from the optically pure (R)-1-phenyl-2,5-dihydrophosphole, the addition of [Cp2ZrHCl] resulted in the formation of a chiral α-zirconated phospholane complex. After that, addition of PPh2Cl cleaved the Zr–C bond, resulting in the final diphosphane through an inversion of configuration at the carbon centre.67,68 This ligand was later coordinated with the typical Ru precursor [RuCl(μ-Cl)(η6-p-cymene)]2 to form a hemilabile complex (Scheme 10). The complexed ligand was then further derivatized either by oxidising the uncoordinated phosphorus or by imination through a Staudinger reaction, obtaining a P(III)–P(V) ligand. This derivatized complexes were tested as catalysts in the Diels–Alder reaction, providing good activity but low to moderate diastereo- and enantioselectivity.69 However, the basic L42 ligand has never been studied in catalysis.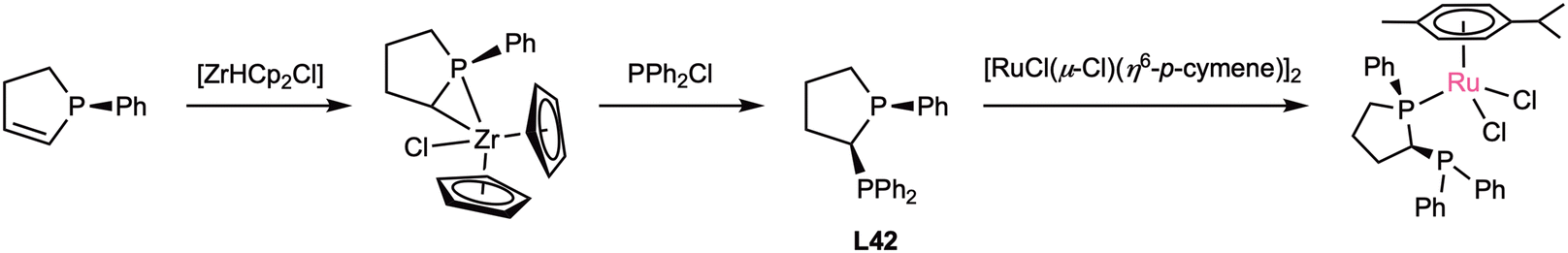 | ||
| Scheme 10 Synthesis and complexation of L42. | ||
That same year, the alkyl-substituted, short-bridged ligand L43 was developed by Marinetti and coworkers.70 This ligand is very interesting because it makes use of a four-membered phosphorus heterocycle (phosphetane)71 to provide the carbon atom for the ligand bridge. This uncommon structure was obtained by deprotonating the phosphetane oxide with n-butyllithium and reacting the carbanion with diphenylchlorophosphane oxide, to give the oxidized diphosphane, which was then reduced with trichlorosilane maintaining the optical purity (Scheme 11B). The initial phosphetane oxide was obtained through the classical McBride method, where a phosphenium cation is formed by the reaction of a dichlorophosphane with AlCl3, which is then reacted with t-butyl ethylene to give the final phosphetane through a rearrangement. In the work of Marinetti and coworkers,70 the initial dichlorophosphane used was the optically pure dichloromenthylphosphane, which produced two different diastereomers that could be separated by fractional recrystallization,72 providing the source of chirality for the final diphosphane (Scheme 11A).
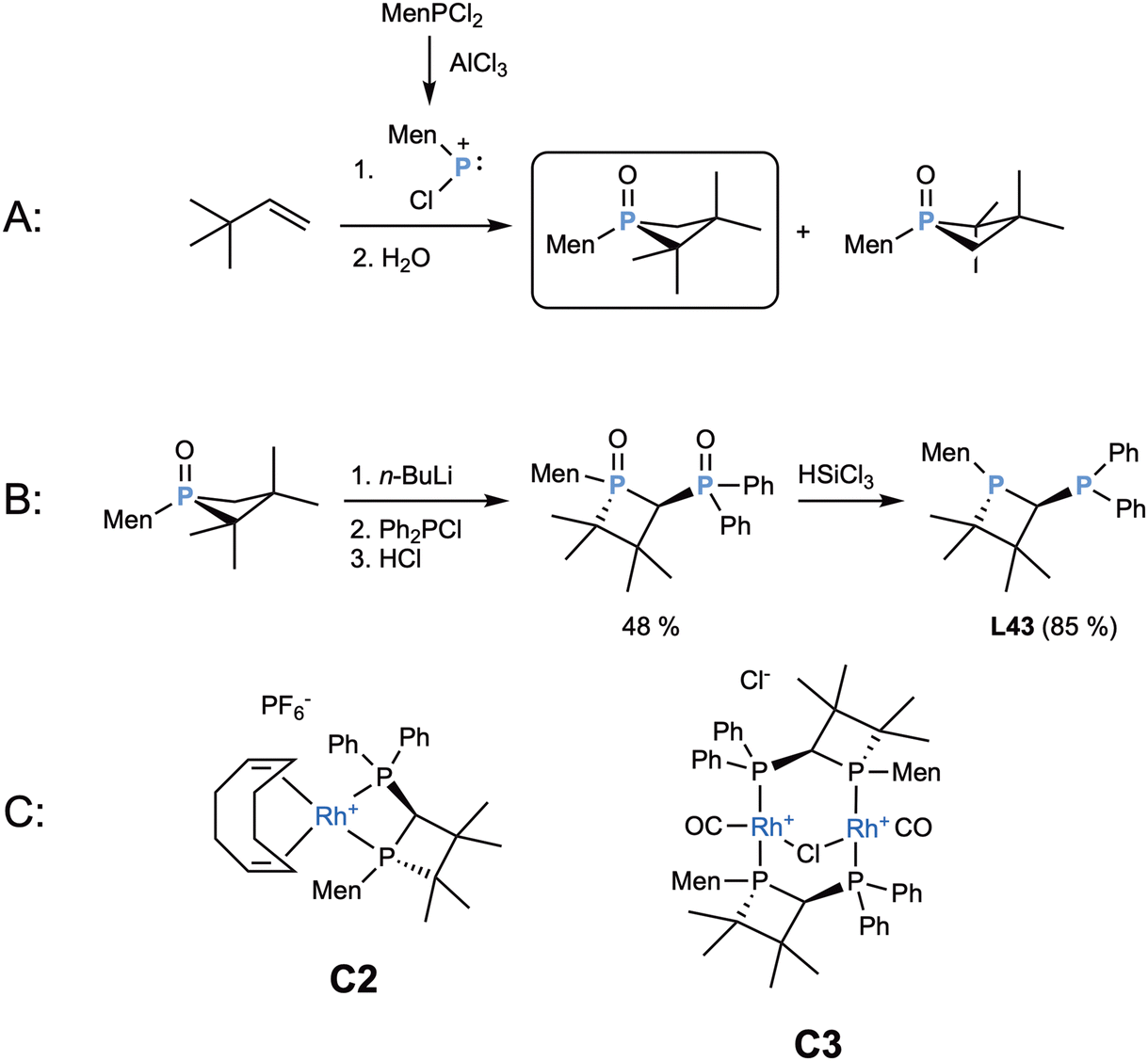 | ||
| Scheme 11 (A) Synthesis of chiral phosphetane oxide. (B) Synthesis of ligand L43. (C) Rhodium complexes with ligand L43. | ||
This ligand L43 was then coordinated to rhodium, obtaining the classical [Rh(cod)(L10)]PF6 (C2) and a less common A-frame dinuclear structure with a bridging chlorine between the two Rh atoms C3 (Scheme 11).
Shortly after, Zhang and coworkers developed a new family of ligands based on the binaphthyl motif.20 It is interesting to note that, for these ligands, L44–L47, the initial alcohol groups of the BINOL were replaced by methyl groups, removing any heteroatoms (apart from phosphorus) from the molecule, which is quite an unusual feature for ligands featuring a BINAP moiety. In fact, Zhang and coworkers had previously developed the first account of a ligand with this fragment, the C2-symmetric binapine, with a two-carbon bridge.73 In the case of L44–L47, the unusual BINAP structure without oxygens allowed for the deprotonation of one of the carbon atoms adjacent to the phosphorus, to which a chlorophosphane could be linked to form the new P–C bond (Fig. 9, top).
 | ||
| Fig. 9 Cyclic monophosphanes (middle), C2-symmetric, ethylene-bridged diphosphanes (left) and C1-symmetric, methylene-bridged diphosphanes (right). | ||
After obtaining the ligands, the typical precatalysts of the type [Rh(cod)(L)]BF4 could be obtained the four ligands L44–L47. When the complexes were tested in the enantioselective hydrogenation reaction, they generally provided good to excellent conversions and enantioselectivities when tested with α- and β-dehydroamino acid derivatives, and with α-arylenamides (Fig. 5). Interestingly, the catalytic reactions showed a strong dependence on the non-P-stereogenic phosphorus substituents, with the phenyl- and cyclohexyl-substituted diphosphanes (L44 and L46, respectively) providing the best results. Unfortunately, no explanation for this behaviour could be provided, and no further studies have been carried out with these diphosphanes to try to elucidate this observation.
Around the same time, Zhang and coworkers also developed another chiral diphosphane with a substituted carbon bridge.22 This new ligand L48 was designed as a TCFP (L8) analogue, in a similar way to the previously mentioned t-Bu-Wudaphos L25 (Scheme 8), which was also reported by Zhang and coworkers a few years later.55 In the case of L48, however, the idea was to replace the P-stereogenic phosphorus with a t-butylphospholane fragment, expanding on the idea of the three hindered quadrant strategy. Similarly to what had been done with ligands L44–L47,20 which were based on their predecessor binapine,73 L48 was based on the previous experience of the group with the diphospholane ZhangPhos.74 By changing only the last synthetic step, the chiral monophospholane synthon could be reacted with t-butylchlorophosphane to obtain L48 (Fig. 9, middle).
After that, the ligand was tested in enantioselective hydrogenation of the typical benchmark substrates and a wide scope of derivatives, giving full conversions and >94% ee in the 25 substrates tested, with 15 of them presenting 99% ee or more (Fig. 5).
The same year, Tang and coworkers published a contribution where they presented a new family of ligands, with a structural similarity to the previously discussed L48, but with very different electronic properties, since the phospholane was changed by an oxaphospholane, and the attached cyclohexyl was instead a phenyl.21 On top of that, they studied how different substituents on the non-P-stereogenic phosphorus and on the ortho position of the phenyl ring could affect the catalytic performance of the ligands, developing four variants L49–L52. The strategy followed was similar to the one that Zhang and coworkers also used for L44–L48, where one cyclic monophosphane could be used to obtain the C2-symmetric ligand (in the case of Tang and coworkers, the well-performing BIBOP),75 or the C1-symmetric short-bridged one (Fig. 9, bottom). In this case, the synthesis proceeded in a similar way as with L44–L49, where the P-adjacent carbon was deprotonated with an organolithium and reacted with a chlorophosphane. The ligands were named POPs, with L49 specifically known as MeO-POP, since it was the one that performed best.
Initially, the ligands were coordinated to [Rh(nbd)2]BF4 to obtain the classical precursors [Rh(L)(nbd)]BF4 without issues. Interestingly, an X-ray crystal structure of the complex with MeO-POP (L49) could be obtained, where a bite angle of 73.7° could be observed. This value is bigger than the typical ∼72° bite angle observed for short-bridged ligands with a carbon bridge, a fact that is probably explained by rigidity and steric hindrance imposed by the bicyclic bridge, on top of the already bulky t-butyl substituents of the phosphorus. When the complexes were tested in the enantioselective hydrogenation of 2-acetamido-3-phenylacrylic acid, it was observed that MeO-POP was clearly the ligand providing the highest enantioselectivity (99% ee), while its counterpart with cyclohexyl substituents, L51, was by far the worst performing one (54% ee). On the other hand, L50 and L52 showed very similar selectivities (87% and 92% ee, respectively), indicating that the methoxy substituent of the phenyl ring attached to the oxaphospholane played a pivotal role in the catalysis. Unfortunately, the mechanistic underpinnings behind this observation have not been explored. Instead, efforts were focused on expanding the substrate scope of the catalysis, showing that the ligands were especially well-suited for hydrogenating β-(acylamino)acrylates, both as (E)- and (Z)-isomers (Fig. 5).
N-bridged diphosphorus ligands (PNP)
Several outstanding ligands with a carbon bridge have been described in the previous section. Most of them contain an unsubstituted methylene bridge (Scheme 1, groups and 2) because the preparation of chiral diphosphanes with a substituted methylene bridge, although leading to interesting diphosphanes (Scheme 1, group 3), is by no means straightforward.Changing the bridging atom from carbon to nitrogen opens a completely different scenario,76 giving PNP ligands that are often called diphosphazanes.77–80 The “parent” compound would be bis(diphenylphosphino)amine (dppa),81 which is analogous to dppm but with a NH bridge and was described for the first time almost 60 years ago.82,83
The NH bridge can be deprotonated to create anionic ligands or, more importantly for the purpose of the present perspective, alkylated with a group, which can be chiral, providing an easy entry to chiral PNP ligands. This has given many ligands (32 in total, L53, L56–L86, Fig. 3), described in the following sections. Interestingly, unsubstituted, chiral NH-bridged ligands were unknown in the literature, until the relatively recent discovery of the important P-stereogenic ligand MaxPHOS in 2010,84 described in the next section.
Unsubstituted nitrogen bridge
The large steric bias between the t-butyl and methyl groups, along with the rigid backbone of a methylene bridge has given PCP ligands producing exceptionally high enantioselectivities in enantioselective catalysis,85 as seen in the previous sections. These ligands are prepared by several methods, but important synthons are t-butylmethylphosphane-borane and t-butyldimethylphosphane-borane, both of nucleophilic character after deprotonation.Inspired by this chemistry, the group of Riera and Verdaguer86 envisaged that the aminophosphino-borane P1 and the phosphinous acid-borane P2 could be convenient electrophilic synthons for the preparation of new ligands (Fig. 10) with the t-butyl/methyl substituents. It should be recalled that their most logical precursors, chlorophosphanes, are difficult to obtain in optically pure form, due to lack of configurational stability.47
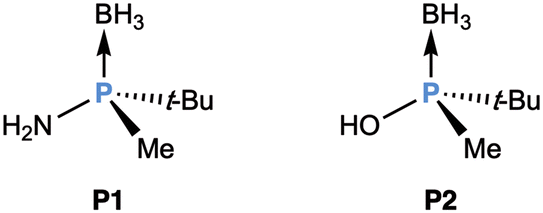 | ||
| Fig. 10 Structures of amino t-butylmethylphosphane-borane (P1) and t-butylmethylphosphinous acid-borane (P2). | ||
Inspired by early work of Kolodiazhnyi87 racemic t-butylchlorophosphanes were reacted with several enantiopure amines, followed by boronation, producing the corresponding aminophosphane-boranes in low diastereomeric ratios, by a partial DKR process (Scheme 12).84
 | ||
| Scheme 12 Preparation of diastereomeric mixtures of aminophosphane-boranes and first reported preparation of P1 and P1-Me. | ||
Fortunately, in the case of phenylglycinamide, the two diastereomers of the t-butylmethylaminophosphane-borane could be separated by column chromatography and recrystallization. A reductive cleavage with lithium in liquid ammonia furnished the aminophosphane-borane P1 as stable and crystalline solid. The N-methylated compound (P1-Me) could be also prepared by methylation of P1. The compounds P1 and P1-Me are ideal building blocks for enantiopure P*NP and P*NP* ligands (P* denotes a P-stereogenic moiety) and this was exploited with the preparation of several N-methyl substituted diphosphazane-boranes (L·BH3) by deprotonation and reaction with a chlorophosphane (Scheme 13).
 | ||
| Scheme 13 Preparation of N-methyl substituted diphosphazane-boranes (L·BH3). | ||
Interestingly, the reaction with racemic t-BuMePCl furnished a mixture of meso- and C2-symmetric L53, which can be considered a nitrogen (methylated nitrogen) analogue of the ligand t-Bu-MiniPHOS (L1),25,32 discussed earlier in this review (Scheme 3). The reaction with the bulkier t-Bu2PCl only took place with the primary amine of P1 and furnished the so-called ligand MaxPHOS (L54), named after their discoverers, as shown in Scheme 14.88
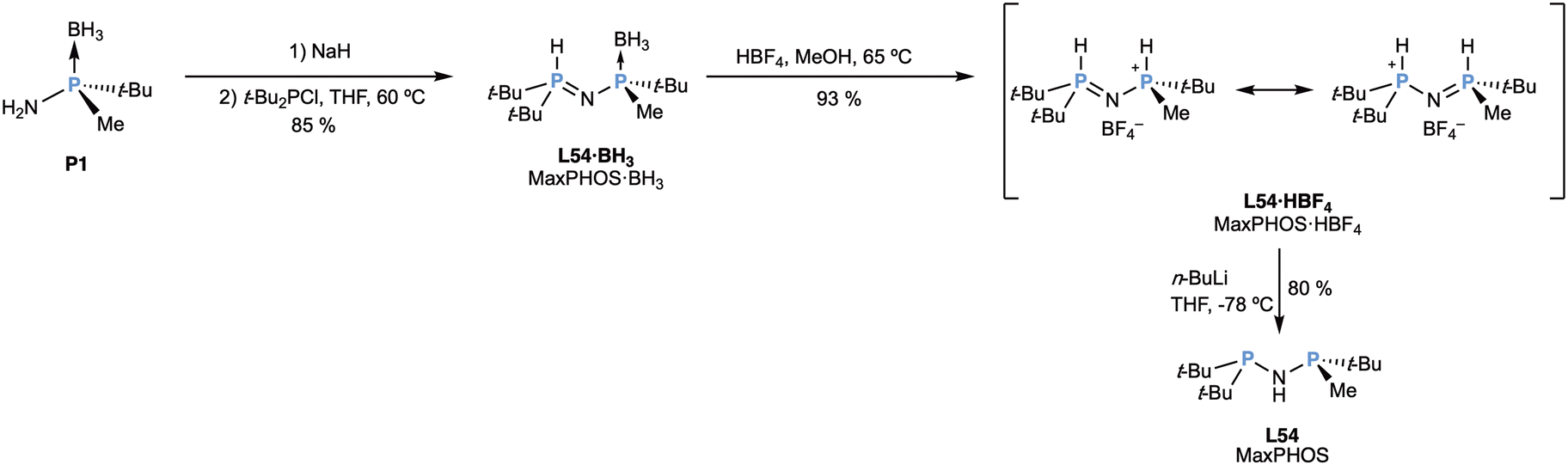 | ||
| Scheme 14 Synthesis of the MaxPHOS ligand (L54). | ||
The reaction of deprotonated P1 with t-Bu2PCl produced L54·BH3, a phosphamide that was exclusively present as the P–H tautomer (formally, a P(V)–P(III) species) as shown in scheme, which prevented the oxidation of the phosphorus atom. The deboronation with tetrafluoroboric acid was possible only under rather harsh conditions, which did not hydrolyse the P–N bonds and rendered the phosphonium salt (L54·HBF4) as a very crystalline, air-stable solid, soluble in medium and high-polarity solvents. The description of the electronic structure of this compound corresponds to a hybrid form between the tautomeric forms of the scheme, since the two P–N distances were found to be almost identical by X-ray crystallography.88 The free MaxPHOS (L54) ligand could be obtained in a later publication88 by deprotonation of MaxPHOS·BH3 (L54·HBF4) at low temperature with n-BuLi, as a very air-sensitive oil. For this reason, in the complexation reactions the salt is usually used, as discussed later. Notably, the free ligand behaves as the NH tautomer, in contrast to the related ligand (t-Bu)2PNHP(t-Bu)2, which is present in a 70:30 mixture of the NH and PH tautomers, respectively, in toluene solution.89 In this regard, switching a single t-butyl group to a methyl is enough to displace completely the tautomeric equilibrium towards the NH form. Interestingly, MaxPHOS (L54) ligand can be considered the analogous of Hoge's TCFP (L8) ligand (Scheme 4),35 replacing the methylene bridge with a NH bridge.
Very recently,90 the same group has used the same synthetic methodology to prepare the so-called MAdPHOS ligand (L55), bearing two 1-adamantyl groups in the non-stereogenic phosphorus atom (Scheme 15).
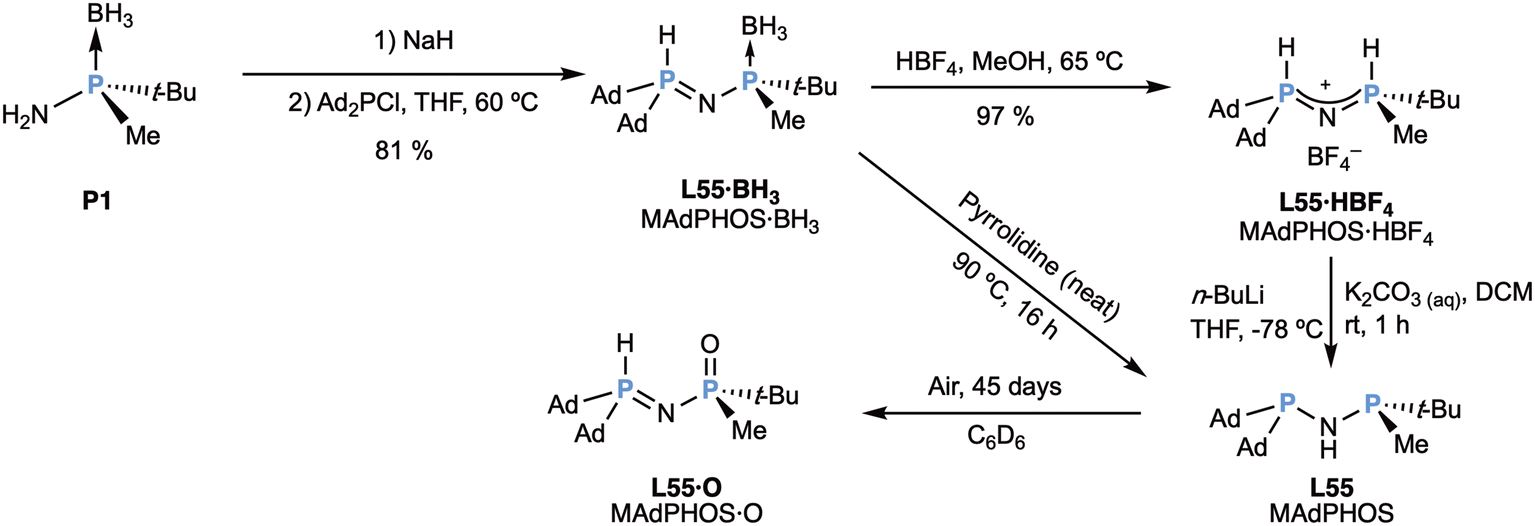 | ||
| Scheme 15 Preparation of the MAdPHOS (L55) ligand. | ||
This ligand was prepared because of the beneficial effects of 1-adamantyl groups as phosphorus substituent,91 demonstrated by Imamoto with his BulkyP*(L9) ligand (Scheme 4),40 which resulted to be solid and air-stable, compared to the very air-sensitive TCFP (L8, Scheme 4), bearing t-butyl groups.
The precursors MAdPHOS·BH3 (L55·BH3) and MAdPHOS·HBF4 (L55·HBF4) were prepared (Scheme 15) in an analogous way to the MaxPHOS (L54) precursors (Scheme 14). The same bonding situation was encountered according to NMR spectroscopy. Free MAdPHOS (L55) could be obtained by deprotonation of MAdPHOS·HBF4 (L55·HBF4) with n-BuLi, as MaxPHOS (L54, Scheme 14), but also with the much milder base potassium carbonate. Interestingly, the ligand could also be obtained by deboronation of MAdPHOS·BH3 (L55·BH3) with pyrrolidine, but in this case the pyrrolidine-borane adduct could not be separated from the ligand. Importantly, free MAdPHOS (L55) is much more stable than MaxPHOS (L54), because it could be stored in the fridge (under nitrogen atmosphere) for 2 months. A sample in an NMR tube in benzene slowly oxidized (45 days) to give exclusively MAdPHOS·O (L55·O), the imino-tautomer with the non-stereogenic phosphorus as a P–H species and the stereogenic phosphorus oxidized.
Both MaxPHOS (L54) and MAdPHOS (L55) were initially developed as chiral ligands for rhodium-catalyzed hydrogenation, inspired by the TCFP (L8) and BulkyP* (L9) ligands, respectively. For this reason, the coordination of the ligands to rhodium(I) moieties has been well studied (Scheme 16).
 | ||
| Scheme 16 Rhodium(I) complexes of MaxPHOS (L54) and MAdPHOS (L55). | ||
Initially, the very stable Schrock-Osborn complex [Rh(cod)(L54)]BF4 was prepared by reaction of the MaxPHOS (L54·HBF4) salt with [Rh(cod)2]BF4 precursor with sodium carbonate to neutralize the equivalent of acid formed (procedure A).84 This method worked well, but a simplified method (procedure B) was developed, which took advantage of the basicity of the departing acetylacetonate ligand92 present in the precursor [Rh(acac)(cod)] to avoid the formation of any salt.88 The same method was employed very recently with the MAdPHOS (L55) ligand with good results.90 An analogous method was used to prepare [Rh(L54)(nbd)]BF4, although this complex was found to be unstable.88 In order to evaluate the electronic features of MaxPHOS (L54) and MAdPHOS (L55), the dicarbonyl complexes were easily obtained as yellow solids by displacement of cod under CO atmosphere (Scheme 16). In addition, the selenides of the two ligands were also prepared by treatment of the free ligands with selenium, to measure the 1J(31P,77Se), which is a measure of the σ-donation of phosphorus ligands.93–95
Some important parameters of MaxPHOS (L54) and MAdPHOS (L55), their related methylene-bridged diphosphanes (TCFP (L8) and BulkyP* (L9), respectively; Scheme 4) and the rhodium(I) complexes are given in Table 1.
| Ligand or complex | Bite angle (°) | νCos (cm−1) | 1JP–Se (Hz) | Ref. |
|---|---|---|---|---|
| TCPF (L8) | — | — | 723, 713 | 88 |
| MaxPHOS (L54) | — | — | 776, 740 | 88 |
| BulkyP* (L9) | — | — | 723, 723 | 96 |
| MAdPHOS (L55) | — | — | 766, 736 | 90 |
| [Rh(L8)(cod)]BF4 | 72.6 | — | — | 35 |
| [Rh(L9)(cod)]SbF6 | 72.5 | — | — | 40 |
| [Rh(L54)(cod)]BF4 | 70.0 | — | — | 84 |
| [Rh(L54)(nbd)]BF4 | 70.1 | — | — | 88 |
| [Rh(L55)(cod)]BF4 | 70.5 | — | — | 90 |
| [Rh(L8)(CO)2]BF4 | — | 2079 | — | 88 |
| [Rh(L54)(CO)2]BF4 | 69.7 | 2088 | — | 88 and 90 |
| [Rh(L55)(CO)2]BF4 | — | 2079 | — | 90 |
The 31P–77Se coupling constant, although it is not free from steric interreference,88 is indicative of the σ-donation of phosphanes, with electron-richer ligands giving smaller coupling constants.95 From the table it can be concluded that the σ-donor capacity follows the order TCFP (L8) > BulkyP* (L9) > MAdPHOS (L55) > MaxPHOS (L54).
From the frequency of the CO stretching vibration (the smaller the value, the higher the σ-donation),97 the order would be TCFP (L8) ≈ MAdPHOS (L55) > MaxPHOS (L54). It seems clear, therefore, that electron withdrawing inductive effect of the NH bridge makes the MaxPHOS (L54) and MAdPHOS (L55) less σ-donating than TCFP (L8) and BulkyP* (L9).
Both MaxPHOS (L54) and MAdPHOS (L55) performed very well in enantioselective hydrogenation of wide range of functionalised olefins under mild conditions (Fig. 11).10,86,88,90
 | ||
| Fig. 11 Asymmetric hydrogenation with MaxPHOS (L54) and MAdPHOS (L55). | ||
The substrates included the benchmark dehydroaminoacids, alkenes containing heteroaryl substituents and many different N-protecting groups and enamides. In some cases, the performance of the two ligands was similar, but in others the bulkier ligand MAdPHOS (L55) outperformed MaxPHOS (L54).10,90
A rhodium complex of MaxPHOS (L54) was also studied by the same group98 as catalytic precursors for the intramolecular Pauson-Khand reaction of 1,6-enynes (Scheme 17).
 | ||
| Scheme 17 Pauson-Khand reaction of enynes catalyzed by Rh-MaxPHOS (L54). | ||
The reaction provided moderate yields of the desired Pauson-Khand (PK) adduct achieving good enantioselectivities, under low CO pressure. A competitive reaction was the [2 + 2 + 2] cycloaddition between the enyne and the triple bond of another enyne. The dicarbonyl complex (Scheme 16) was shown to be catalytically active and was used to demonstrate that the presence of cod was important to attain high selectivity.
The complexation of MaxPHOS (L54) to nickel(II) and palladium(II) moieties has also been explored by Grabulosa, Verdaguer and coworkers.99 The coordination chemistry was explored with simple nickel(II) and palladium(II) precursors (Scheme 18).
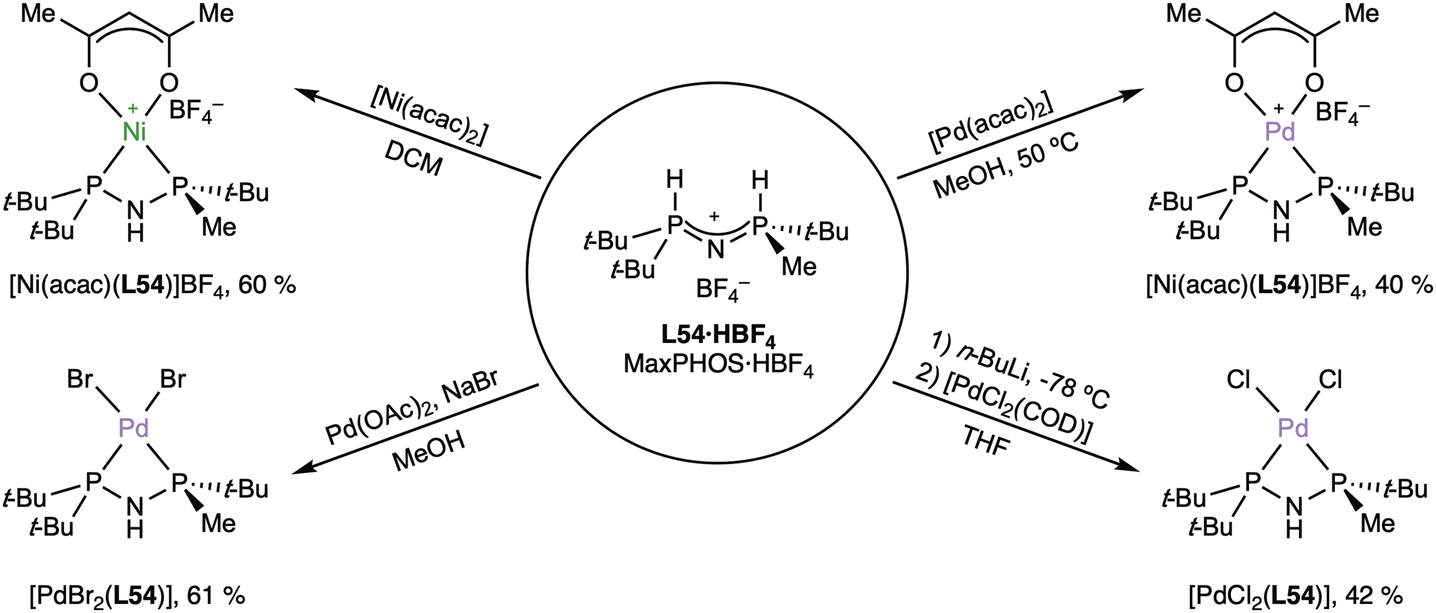 | ||
| Scheme 18 Coordination compounds of Ni(II) and Pd(II) with MaxPHOS (L54). | ||
The reaction of the MaxPHOS salt (L54·HBF4) with [M(acac)2] (M = Ni, Pd), following the “acac method”92 cleanly yielded the pure complexes as air-stable solids, whose X-ray structures were very similar except for the coordination distances. In the same report, the preparation of the neutral dihalido complexes of palladium(II) were reported. While the dibromido complex could be obtained from palladium acetate in the presence of sodium bromide, the method did not work for the preparation of the diclorido complex, which had to be prepared from the free ligand and [PdCl2(cod)].
The reaction of three different η3-allylic palladium(II) dimers with MaxPHOS·HBF4 (L54·HBF4) in the presence of sodium carbonate and ammonium tetrafluoroborate furnished the corresponding allylic complexes (Scheme 19).
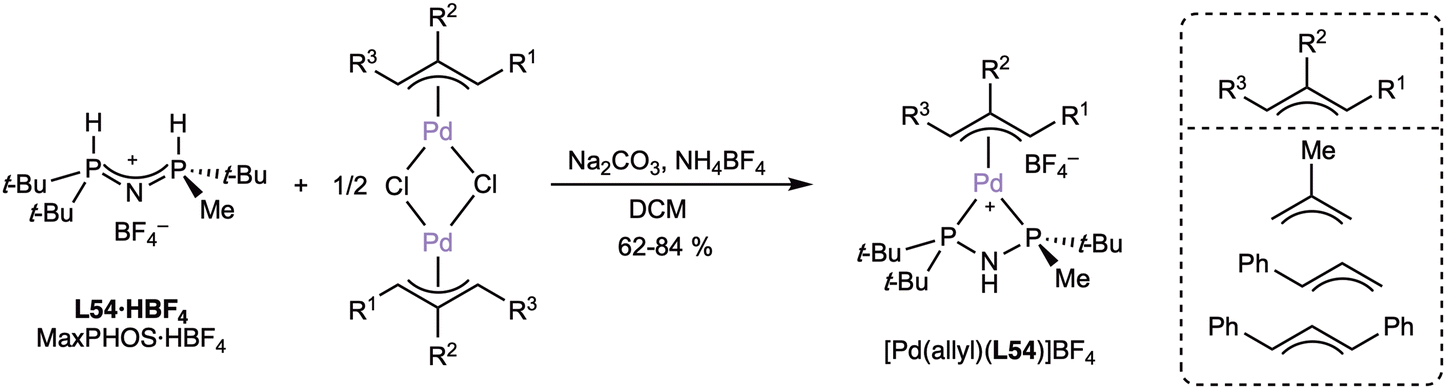 | ||
| Scheme 19 Preparation of palladium(II) allylic complexes with MaxPHOS (L54). | ||
The NMR characterization showed that the three complexes were present as mixtures of isomers, due to the unsymmetrical nature of MaxPHOS (L54),53,99 although the crystal structures contained only one isomer. The allylic complexes were used in enantioselective allylic substitution reactions, but the conversions and enantioselectivities were only moderate.99
The increased stability of MAdPHOS (L55) compared to MaxPHOS (L54) made the same group explore the coordination chemistry towards Ni(0).90 To this end, MAdPHOS (L55) was reacted with [Ni(cod)2] (Scheme 20).
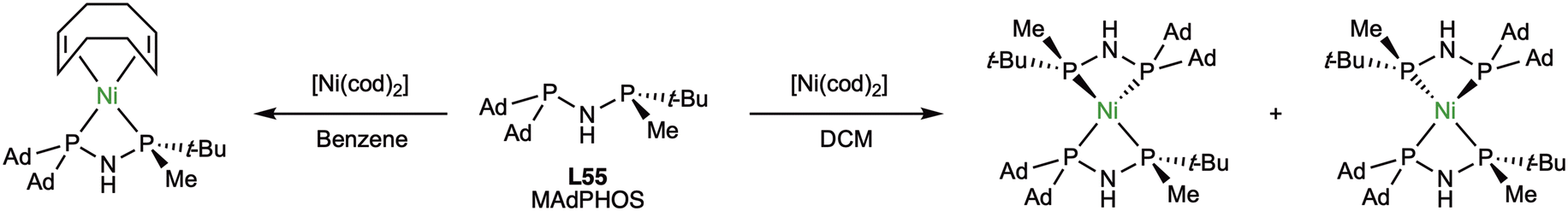 | ||
| Scheme 20 Preparation of Ni(0)-MAdPHOS (L55) complexes. | ||
Using dichloromethane as a solvent, a mixture diastereomers for the bis(chelated) complexes was obtained, while in the case of using benzene, only the monochelated Ni-cod complex was obtained instead, although it was found to be rather unstable.
The coordination of MaxPHOS (L54) to organometallic octahedral moieties has also been explored by Ferrer, Carmona and Grabulosa,100 with the preparation of rhodium(III), iridium(III) and ruthenium(II) complexes. Thus, the reaction of MaxPHOS·HBF4 (L54·HBF4) with [M(acac)ClCp*] (M = Rh, Ir) in refluxing dichloromethane produced the clean substitution of the acac ligand, giving the corresponding complexes in around 90% yields (Scheme 21).
 | ||
| Scheme 21 Reaction of (SP)-MaxPHOS (L54) with [M(acac)ClCp*]. | ||
The characterization of the complexes demonstrated that they were present as a single species. It should be noted that the metal becomes a pseudotetrahedral, stereogenic centre so two different diastereomers, with different metal absolute configuration could be formed. The X-ray crystal structures demonstrated that both complexes had SM configuration, starting from SP-MaxPHOS·HBF4 ((SP)-L54), with a very small bite angle of 68.85° for both complexes.
The complexation to ruthenium(II) through the same method using an acac precursor101 gave only traces of the product,100 but the reaction was successful using the MaxPHOS ligand (L54), the typical ruthenium(II)-p-cymene dimer and ammonium tetrafluoroborate (Scheme 22).
 | ||
| Scheme 22 Preparation of [RuCl(L54)(p-cymene)]. | ||
Again, the compound appeared as a single species and the crystal structure of the hexafluorophosphate salt showed that the absolute configuration of the ruthenium atom was SRu.
An interesting cyclometallation reaction was observed upon halide abstraction on iridium complex (Scheme 23).
 | ||
| Scheme 23 C–H activation of t-butyl group on an iridium-L54 complex. | ||
One of the t-butyl groups of the non-stereogenic phosphorus (P2) in Scheme 23 cyclometallates via intramolecular C(sp3)–H activation. Upon this activation, a new stereogenic centre in P2 is created. NMR studies showed that two species, in 82:18 ratio, were formed and that they are those depicted in the scheme. A plausible mechanism for the formation of the two diastereomers is given in the scheme. Interestingly, this metalation was not observed when the analogous rhodium (Scheme 21) and ruthenium (Scheme 22) complexes were treated with silver tetrafluoroborate.
Although the method of Scheme 12 furnished P1 as an enantiopure compound, it suffered from low yields due to the inefficient DKR and the relatively difficult synthesis of the racemic chlorophosphanes. Inspired by the oxazaphospholidine-based method of Jugé41 the same group devised a strategy using another bifunctional, chiral auxiliary, cis-1-amino-2-indanol, both enantiomers of which are commercially available (Scheme 24).102
 | ||
| Scheme 24 Synthesis of aminophosphane-boranes by cleavage of the chiral auxiliary. | ||
The condensation between (1R,2S)-cis-1-amino-2-indanol and racemic t-butylchlorodiethylaminophosphane, followed by boronation, produced the oxazaphospholidine-borane P3 in good yields and high diastereoselectivity after a single recrystallization. This compound was susceptible to nucleophilic attack by carbanionic reagents and in this case Grignard reagents gave cleaner reaction than organolithiums at 100 °C in toluene, giving good yields of the opened aminophosphane-boranes, P4. Interestingly, X-ray crystallography showed that the opening reaction take place with inversion of configuration at the phosphorus atom, in contrast to the Jugé-Stephan methodology.41 The origin of this difference was studied computationally103 and it could be traced back to the substitution state of the nitrogen atom in oxazaphospholidine-boranes. In the case of 2-phenyloxazaphospholidines with the methylated nitrogen (Jugé-Stephan method41), the reaction takes place with retention of configuration by a two-step frontside SN2@P substitution, with in the case of an unsubstituted nitrogen atom, takes place with inversion of configuration by a single-step backside SN2@P substitution.
The desired primary aminophosphane-boranes with alkyl substituents could be easily obtained by reductive C–N cleavage of the chiral auxiliary. The N-methylated compound P1-Me could be also obtained by permethylation of the precursor and reductive cleavage.
The Li-mediated reduction of the C–N bond could not be applied to substrates with R = aryl, due to the formation of reduced, Birch-type products. Therefore, an acidic cleavage method on the mesylates was developed, furnishing the desired aminophosphane-boranes.
Aminophosphane-boranes, apart from being useful for the preparation of PNP ligands such as MaxPHOS (L54) and MAdPHOS (L55), are important synthons in P-stereogenic chemistry that can give other intermediates in the synthesis of other single-atom-bridged ligands (Scheme 25). The hydrolysis of either P1 or, more conveniently, the aminophosphane-borane with the indanol group, could be carried out under rather forcing acidic conditions,104 furnishing the phosphinous acid-borane P2 as an optically pure compound. This compound was a low-melting point semisolid that presented limited stability, but the same group devised a strategy to stabilize it as a dialkylammonium salt.105
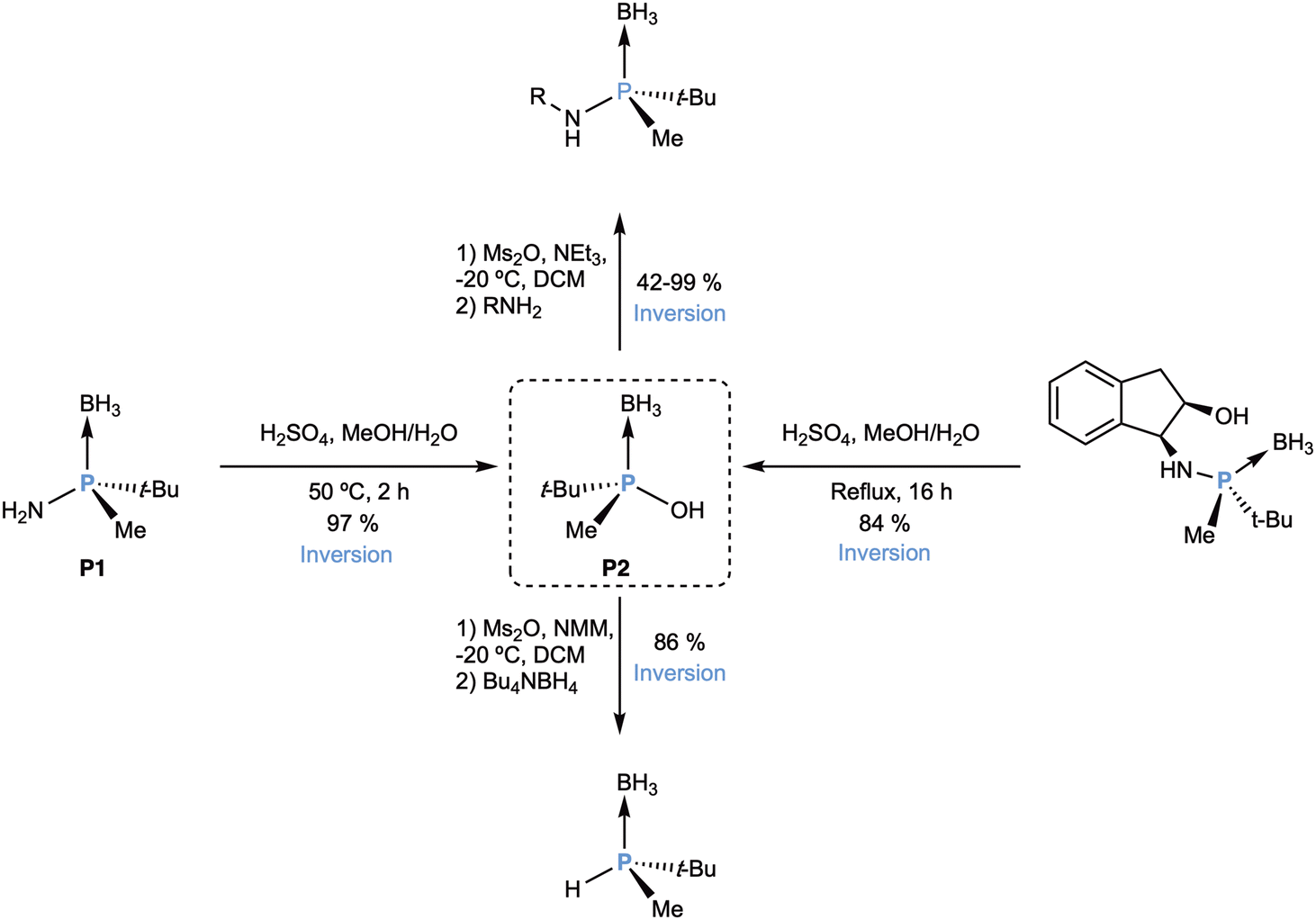 | ||
| Scheme 25 Synthesis and reactivity of phosphinous acid-borane P2. | ||
This compound furnishes a secondary phosphane oxide (SPO) upon deboronation, whose coordination chemistry has been studied.106 In addition, the hydroxyl could be activated by mesylation and the mesylate was a good leaving group in SN2@P reaction by amines, with inversion of configuration. Interestingly, the SN2@P reaction with tetrabutylammonium borohydride was also successful,107,108 giving the important secondary phosphane-borane adduct HP(t-Bu)Me, which has been extensively used by Imamoto14,109 in the synthesis of P-stereogenic ligands.
Substituted nitrogen bridge
As early as 1978, Beck110 reported the synthesis of series of chiral diphosphazanes by reaction of chlorodiphenylphosphane with optically pure amino acids and obtained a surprising variety of ligands which demonstrated their tendency to coordinate in a bidentate fashion (Scheme 26).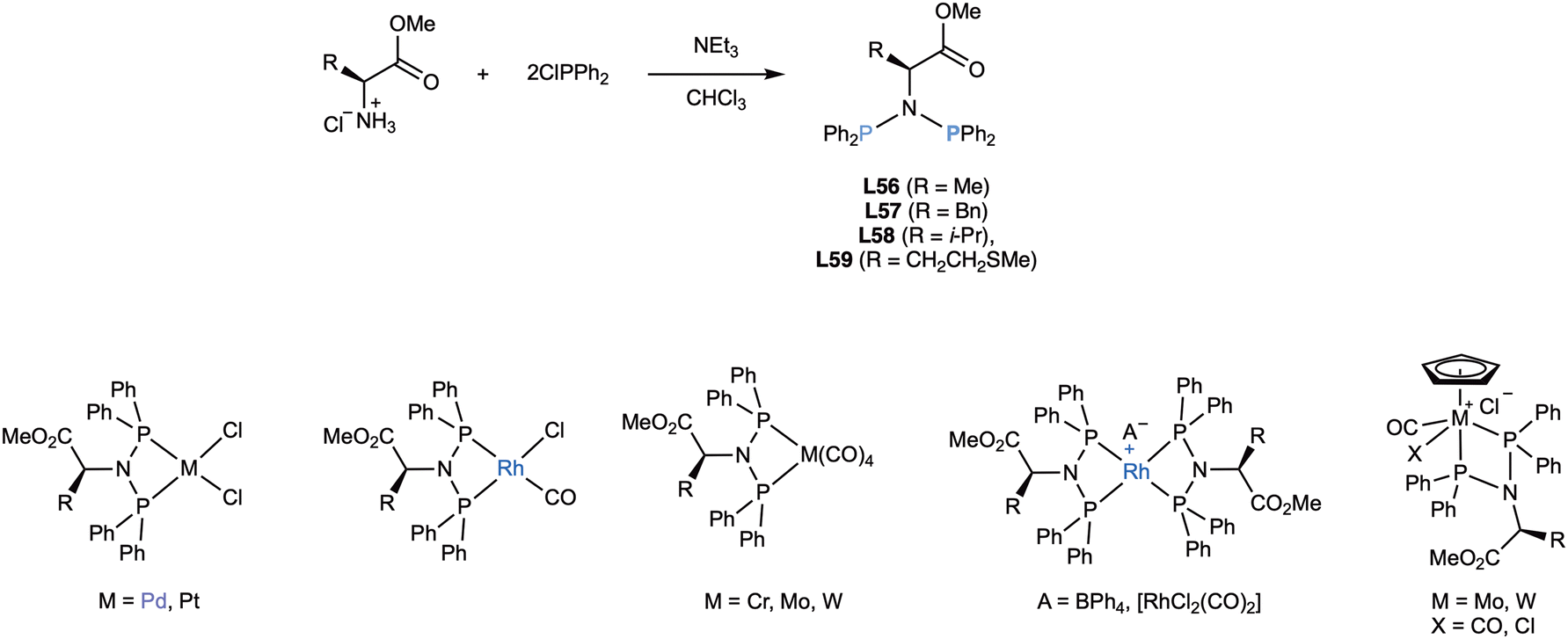 | ||
| Scheme 26 Early synthesis of chiral diphosphazanes (L56–L59) employing natural amino acids and some of their derived complexes. | ||
The ligands were easily obtained by phosphination of natural amino acids (glycine, alanine, phenylalanine, methionine) in 50–70% yields. Their reaction to palladium(II), platinum(II), and rhodium(I) standard precursors produced mononuclear complexes, which were thoroughly characterized.110 Some years later,111 the same ligands were reacted with [MCp(CO)3Cl] (M = Mo, W), producing complexes (X = CO), which led to metal-stereogenic complexes (X = Cl) by thermal or photochemical replacement of CO by chloride. The diastereomers could be separated in the case of the ligand derived from valine (L58). Many years later, Woollins and coworkers112 also revisited the synthesis of L56 (which they named bdppal) and prepared the same [MCl2(L56)] complexes, which were analysed by X-ray diffraction. In addition, they prepared the mono- and dioxidised versions (with oxygen and sulphur) of L56 and studied their coordination to palladium(II) and platinum(II).
In 1981, Payne prepared113 two more diphosphazanes, L60 (peap) and L61 (alap) employing the same method and studied their complexation towards platinum (Fig. 12).113,114
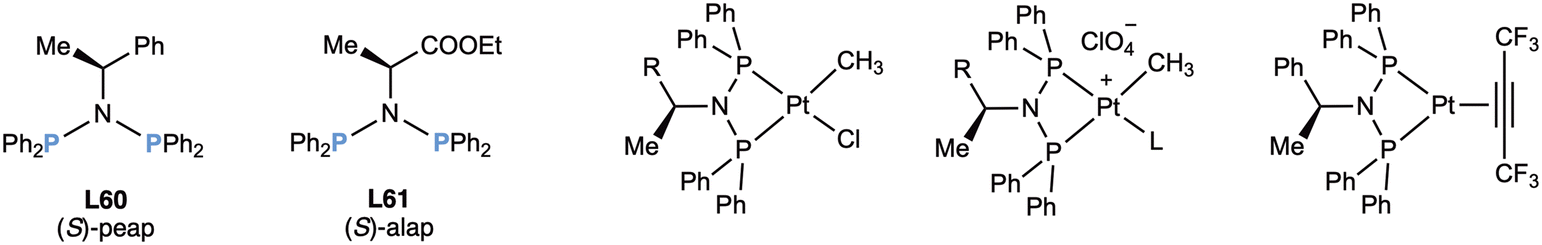 | ||
| Fig. 12 The diphosphazanes peap (L60) and alap (L61) and their first described complexes. | ||
The ligands were prepared in moderate yields from optically pure phenylethylamine and the ethyl ester of alanine and were reacted with [Pt(cod)ClMe] to give the neutral platinum(II) complexes, which were converted into the cationic complexes by halide abstraction in the presence of a monodentate ligand, namely acetone, a p-substituted pyridine or a monophosphane.113 All this array of complexes was characterized in great detail by NMR with the aim of studying the electronic and steric properties of the ligands. In a parallel study,114 the platinum(0) complex (Fig. 12, right) with peap (L60) and acetylene was prepared and its crystal structure determined. A few years later, Basset and coworkers115 reported the use of (S)-peap (L60) to prepare the rhodium cluster [Rh6(CO)10(peap)3], which was found to be inactive in the hydrogenation of dehydroaminoacids.
The ligand peap (L60), almost exclusively the S enantiomer, has been employed to prepare many complexes for several applications and the ligand itself has been used for NMR studies.116–118 Most of the complexes reported so far contain the free peap (L60) and are given in Fig. 13, but there are others, not shown, with the ligand oxidized, forming a sulphide or a selenide.119–123
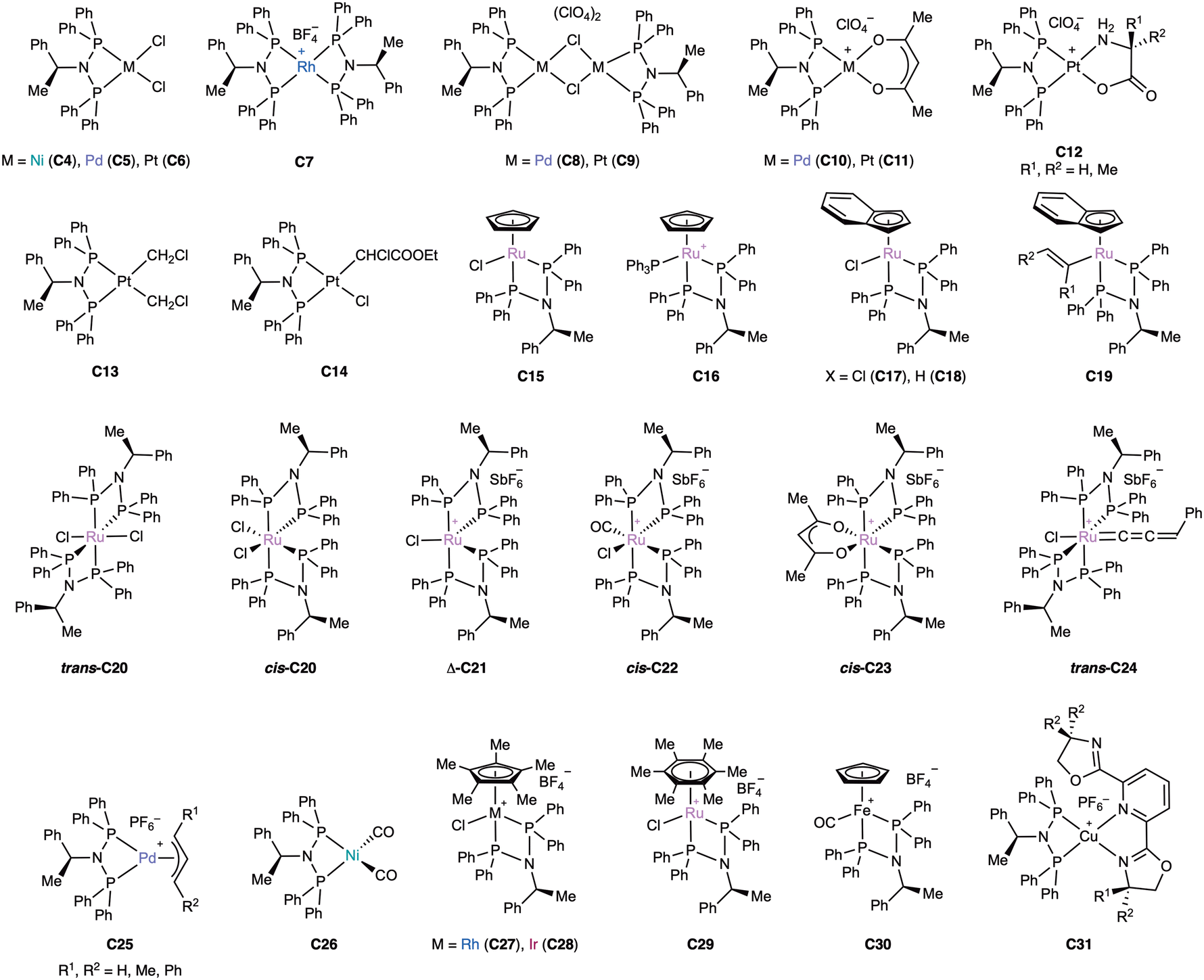 | ||
| Fig. 13 Most of the described complexes with the peap ligand (L60). | ||
Square-planar complexes C5–C7 were obtained by Krishnamurthy28 with standard metallic precursors. The nickel complex C4 was not obtained until much later by Hadjichristidis124 by treating (S)-peap (L60) with [NiCl2(PPh3)2], and has been more recently used to prepare maleonitriledithiolate complexes that act as electrocatalysts for hydrogen evolution.125 A few years later, the corresponding dibromide complex was prepared from [NiBr2(dme)].126 The platinum dichloridocomplex (C6) was hydrolysed in wet acetone in the presence of silver triflate to give the bis(aqua) complex, which was used in hydration of alkynes in micellar media.127 Interestingly, bis(chelated) complex C7 was obtained with [Rh(cod)2]BF4 regardless of the ratio ligand:metal, demonstrating the marked tendency of peap (L60) to produce chelated complexes.28 Slightly later Navarro,128 also described C5 and C6 and observed that one of the P–N bonds of was cleaved when the complexes were treated with alcohols. The same group studied further the chemistry of C5 and C6.129 Treating them with silver perchlorate produced the dimers C8 and C9, which reacted with thallium acetylacetonate to give the acac complexes C10 and C11, respectively. Platinum complex C9 was reacted with α-amino acids L- and D-alanine, giving α-amino acidato complexes C12. When racemic alanine was used, no discrimination was observed in the formation of C11. The reaction of C6 with diazomethane furnished bis(chloromethyl) complex C13 but, in contrast, the reaction with ethyl diazoacetate resulted only in the insertion to only one of the Pt–Cl bonds, giving a mixture of two diastereomers due to the formation of a stereogenic carbon atom in C14.
Krishnamurhthy130 reacted (S)-peap (L60) with [RuCpCl(PPh3)2] in toluene at 100 °C and found that a mixture of neutral complex C15 and cationic complex C16 formed and isolated them in 80 and 5% yields respectively. At the same time, Gamasa131 prepared series or indenylruthenium(II) complexes. The starting complex C17 was prepared from [RuCl(indenyl)(PPh3)2] in toluene at reflux. This complex was converted into the hydride C18 in 85% yield by treatment with sodium methoxide in methanol. This method exploits the by β-hydrogen elimination of the methoxy complex formed in situ. Complex C18 was characterized by X-ray diffraction. This complex was reacted with alkynes providing several alkenyl complexes C19, some of which were converted to ruthenium carbenes (alkenylalkylidenes) by protonation with tetrafluoroboric acid. Their catalytic activity in ring closing metathesis of diethyldiallylmalonate was investigated but they were found to be inactive. Further work of the same group132 focused on five- and six-coordinated ruthenium(II) compounds. Treatment of [RuCl2(dmso)4] with (S)-peap (L60) stereoselectively produced trans-C20, which could be photochemically isomerized to the less stable cis-C20. Halide abstraction of trans-C20 with silver hexafluoroantimonate gave the cationic, five-coordinated, 16-electron complex Δ-C21, which was characterized by X-ray diffraction and was present as a single stereoisomer. This complex was a good intermediate to other complexes and hence carbonyl complex cis-C22 was selectively produced, which slowly isomerized to the trans isomer. The acac complex cis-23 was also prepared. Finally, reaction of Δ-C21 with 1-phenyl-2-propyn-1-ol afforded the allenylidene derivative trans-C24.
Krishnamurhthy122 described the complexation of (S)-peap (L60) to allylpalladium moieties and studied in detail, by NMR, the dynamics of complexes C25 in solution. They found that when the allyl group was symmetrical (R1 = R2 in C25, Fig. 13) the complexes existed as a single species in solution, in the case of unsymmetrical allyl moieties (R1 ≠ R2 in C25, Fig. 13), an equimolar mixture of diastereomers was observed. The palladium-catalysed catalytic allylic alkylation of (E)-1-phenyl-2-propenyl acetate was investigated and while good conversions were found, the regioselectivity was 96:4 favouring the achiral (linear) product.122
Valderrama133 expanded the range of organometallic compounds by the preparation of nickel(0) complex C26 from (S)-peap (L60) and nickel tetracarbonyl and piano-stool complexes C27–C29 by reaction of the ligand with typical organometallic precursors. In addition, the interesting iron(II) complex C30 was prepared from [FeCp(CO)2I] and silver tetrafluoroborate. Several crystal structures were obtained.
There is an interesting example of copper(I) complex (C31) containing a pybox ligand reported by Gamasa.134 It was prepared from a dimeric precursor, and it was found to be stable in air.
These early-prepared ligands (Fig. 12) remained the sole examples of enantiopure diphosphazanes in the literature for quite some time. The outlook of the 1994 excellent review of Krishnamurthy and coworkers78 when referring to the coordination chemistry of (mainly achiral) diphosphinoamines affirmed that “it would also be interesting to incorporate chiral entities into acyclic diphosphazane ligands and use such chiral diphosphinoamines to synthesize transition metal complexes that may function as homogeneous catalysts”. This statement proved to be true in no small part due to the work of his own group in the subsequent years.
In 1995 a first article on the preparation of diphosphazanes derived from (S)-methylbenzylamine appeared (Scheme 27).28
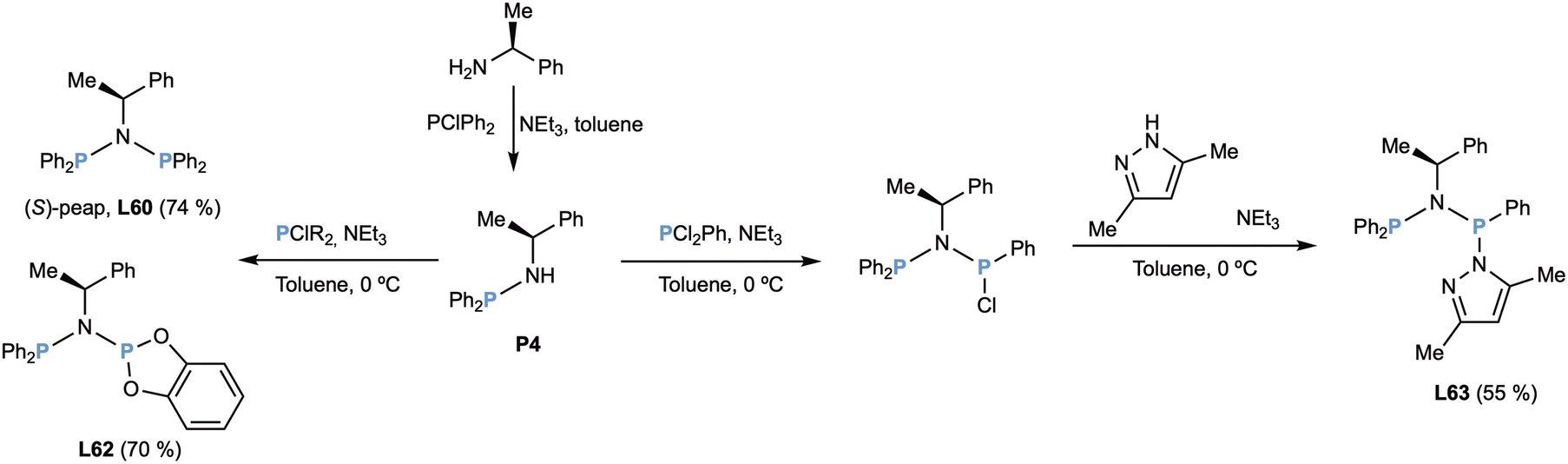 | ||
| Scheme 27 Preparation of chiral diphosphazanes L60 (peap), L62 and L63. | ||
The key optically pure aminophosphane P4 was easily prepared by condensation of methylbenzylamine and chlorodiphenylphosphane. The nitrogen was phosphinated again with a chlorophosphane to give the known ligand peap (L60) and the new ligand L62 in good yields. Interestingly, the reaction with dichlorophenylphosphane gave a chlorinated intermediate with a stereogenic phosphorus atom, with a small chiral induction. The derivatization with dimethylpyrazole furnished ligand L63 in moderate combined yield of the two diastereomers. Pleasingly, the two diastereomers could be separated by fractional crystallization.28
There has been some organometallic chemistry, carried out by the same group, with ligand L63. In the first report,135 it was used to obtain a rather unusual heptacoordinated molybdenum complex [Mo(κ3-N,P,P-L63)(CO)2I2], in which the ligand coordinates through both phosphorus atoms and the pyrazolic nitrogen. Much later, the same ligand was employed to prepare and study the structure and dynamics of allylpalladium complexes [Pd(η3-C3H4R)(L63)]PF6 (R = Me (crotyl), Ph (cinnamyl)).136 Interestingly, the ligands coordinated in N,P fashion instead of the expected P,P fashion. In contrast, with the monosulphide of L63 (in the non stereogenic phosphorus), the P,S coordination was observed. A follow-up study was published, in which the allylic complexes with 1,3-dimethylallyl and 1,3-diphenylallyl were also prepared and studied.137 Interestingly, in the case of the later allyl group, a mixture of N,P and P,P coordinations was found. The ligand was used in the catalytic allylic alkylation of the model substrate rac-1,3-diphenyl-2-propenyl acetate, but despite full conversion being achieved, only 30% ee was observed.137
At the same time,130,138 optically pure (SC,RP)-L63 was coordinated to ruthenium(II) and the neutral complex [RuCpCl(κ2-P,P-L63)] could be isolated in a diastereomerically pure form as the isomer with RRu configuration at the ruthenium atom according to the X-ray crystal structure.130 Many other ruthenium-Cp complexes with achiral ligands were described in the same report.138 There is also a report139 in which the ligand was reacted with [Ru3(CO)12] to furnish a phosphido cluster by cleavage of the P–Npyrazole bond, with the pyrazolate moiety adopting an unusual triply bonding μ3-η1-η1-η1 coordination mode.
The same group,130,140,141 applying methods previously developed to prepare achiral or racemic unsymmetrical diphosphazanes142,143 prepared ligands introducing the typical 2,2-binaphthol moiety (Scheme 28).
 | ||
| Scheme 28 Preparation of diphosphazanes with the racemic binaphthol moiety.130 The same group had published achiral or racemic ligands before.143 | ||
They mainly used racemic binol, although they mentioned that the ligands could also be prepared starting from the optically pure binaphthol and they prepared them in optically pure form in a subsequent study.144 The synthesis of the ligands were based on classic condensations between P–Cl and O–H moieties in the presence of triethylamine and gave ligand L64 with bis(dichlorophosphino)methylamine, whose diastereomers (meso and rac) could not be separated. For the preparation of unsymmetrical diphosphazanes, a multistep synthesis, always based on condensations, was devised and gave ligands L65–L68. Interestingly, with the aim of obtaining an enantiopure ligand, diphosphazane L69 with the optically pure group in the bridge was prepared as a mixture of diastereomers, but they could not be separated by fractional recrystallization and the same happened with the monosulphide. The separation was finally possible from the palladium complex [PdCl2(L69)]. Finally, some ruthenium-cyclopentadienyl complexes with the achiral or racemic ligands were also prepared130 and used to prepare ruthenium carbonyl clusters by reaction with [Ru3(CO)12].144
The same group a few years later described the synthesis of the enantiopure versions of ligand L65 (Scheme 28) and described the synthesis of another enantiopure ligand (L70), which had been described before143 as a racemate (Scheme 29).141
 | ||
| Scheme 29 Preparation of diphosphazane L70 with complexation to allylpalladium moieties. | ||
The allylpalladium complexes were prepared with the racemic ligands and their structures were studied by NMR and X-ray structural analysis. The enantiopure ligands were used in the catalytic allylic alkylation of the model substrate rac-1,3-diphenyl-2-propenyl acetate with dimethyl malonate and N,O-bis(trimethylsilyl)acetamide (BSA) (Scheme 30).
 | ||
| Scheme 30 Palladium-catalysed allylic alkylation of rac-1,3-diphenyl-2-propenyl acetate. | ||
Full conversion was observed after 24 h, but only 20–44% ee values were obtained.141
Slightly later, a comprehensive study of the complexation of many achiral diphosphazane ligands, but also enantiopure (S)-peap (L60) to form half-sandwich (Cp and Cp*) ruthenium(II) complexes was published by the same group.145 The unsymmetrically-substituted diphosphazanes gave complexes with stereogenic ruthenium atoms. In the case of binaphthyl-containing diphosphazanes, it was the binaphthyl moiety that controlled the stereochemistry of the ruthenium atom. The chlorido complexes could be converted into the hydride complexes by treatment with sodium methoxide in methanol and this reaction proceeded stereoselectively if the substituents of the phosphorus were sterically bulky. Finally, they also used several of the enantiopure ligands developed in their group to study the catalytic enantioselective transfer hydrogenation of 2-acetonaphthone (Scheme 31). They found in generally good yields after 16 h, but with low enantioselectivities (<35% ee).
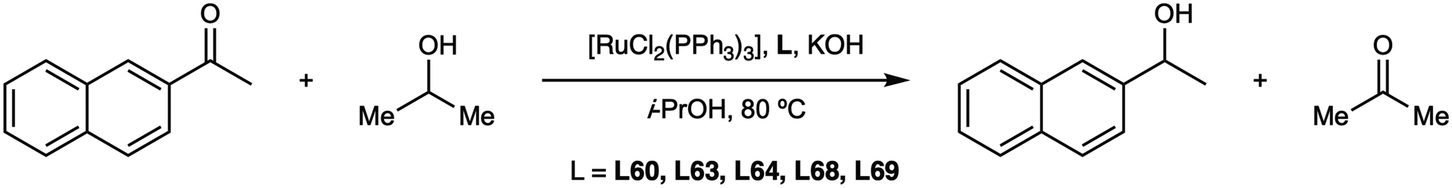 | ||
| Scheme 31 Ruthenium-catalyzed transfer hydrogenation of 2-acetonaphthone. | ||
In 2004, Faraone and coworkers61 reported a few new PNP ligands with a substituted nitrogen bridge with several stereogenic elements to study their combined effect in coordination chemistry and catalysis (Fig. 14).
 | ||
| Fig. 14 PNP ligands prepared by Faraone and coworkers.61 | ||
The bis(diphosphino)alkylamine L71 and the bis(diphosphonito)alkylamines L72 and L73 were obtained in good yields by the condensation of the primary amines and diphenylchlorophosphane or binaphthyl-phosphorochloridite in toluene in the presence of triethylamine. The presence of conformers was detected by NMR due to restricted rotation around the P–N bonds.61 The interesting “mixed” phosphino-phosphonitoamine ligand L74 was obtained in 45% yield taking advantage of the different rate of hydrogen substitution from s-butylamine.28
The ligands were developed for palladium-catalysed allylic alkylation and therefore the synthesis of allylpalladium complexes was studied. It was found that in all cases the standard, mononuclear complexes with typical bidentate coordination of the ligands [Pd(η3-1,3-diphenylallyl)(P–N–P)]X (X = PF6 or OTF) were formed. Detailed NMR experiments to study the number and identity of the different species and their interconversion were carried out. Finally, the application of the ligands in enantioselective allylic alkylation of model substrate 1,3-diphenylallyl acetate with dimethyl malonate and BSA and potassium acetate was studied. Symmetric ligands L71–L73 gave a racemic alkylation product while non-symmetric (phosphino-phosphonito)amine gave the product with only 18% ee.
In a parallel publication, the same group146 studied the coordination of L71, L72 and L74 to other palladium(II) moieties to platinum(II) and rhodium(I) precursors. Starting from typical precursors, they were able to obtain the coordination compounds [M(L76)Cl2] (M = Pd, Pt), [Rh(L71)(cod)]PF6 and [M(L)X2] (M = Pd, Pt; L = L72, L74; X = Cl, I). The crystal structure of palladium and platinum complexes with L71 showed that they were isostructural with very similar geometrical parameters. Ligands L73 and L74 and [Rh(acac)(CO)2] were used in hydroformylation of styrene but no hydroformylation products were observed.
The group of Riera and Verdaguer also reported a few years later a different type of ligands, named ThaxPHOS (L75–L79), easily prepared from oxazaphospholidine P3 (Scheme 24) by deprotonation and phosphination of the NH group (Scheme 32).147
 | ||
| Scheme 32 Synthesis of the ThaxPHOS ligands (L75–L79) and complexation to Co-acetylene moieties. | ||
The ligands were obtained as pure compounds by a simple recrystallization without need to protect the second phosphorus moiety. The deprotection with DABCO and reaction with a cobalt precursor, followed by desilylation with tetrabutylammonium fluoride (TBAF) produced the cobalt-acetylene complexes shown in the scheme as single species, with an “A-frame” structure, as confirmed by X-ray diffraction. These complexes were used in intermolecular enantioselective Pauson-Khand (PK) reactions at 1 bar of carbon monoxide (Scheme 33).147
 | ||
| Scheme 33 Intermolecular PK catalyzed by Co-ThaxPHOS (L75–L77). | ||
The complexes were active in intermolecular PK reactions of several alkynes and norbornadiene, giving the corresponding exo-cyclopentenones. The best alkyne, in terms of enantioselectivity was trimethylsilylacetylene. These results compare very favourably to those obtained much earlier by Gimbert with peap (L60) and other electron-deficient diphosphazane ligands.148
More recently, Kamer and coworkers30,149,150 reported in three consecutive articles, a clever strategy to obtain a group of P-stereogenic diphosphazanes containing other stereogenic elements, collectively abbreviated as JoSoPhos.30
The synthesis started by reproducing earlier work of Kolodyazhnyi,151,152 by preparing phosphanamine P5 in high yield as a 90:10 mixture of diastereomers, using racemic t-butylchlorophenylphosphane and each enantiomer of 1-phenylethylamine (Scheme 34, compare with Scheme 12).30,149
 | ||
| Scheme 34 Preparation of both enantiomers of nucleophilic synthon P5·Li. | ||
The diastereomeric mixtures of P5 were deprotonated with n-BuLi/TMEDA, producing lithium phosphinoamides P5·Li as a solid in the same diastereomeric ratios but upon recrystallization at 80 °C in n-heptane, they could be obtained as a diastereomerically pure solids, (RP,S)-P5·Li or (SP,R)-P5·Li.
This salt is an excellent synthon with a nucleophilic nitrogen, ideally suited to obtain ligands by reaction with chlorophosphanes in moderate yields (Scheme 35).30,149 Therefore, it was reacted at low temperature with 2-chloro-1,3,2-dioxaphospholane (ligand L80), with phosphorochloridites derived from bisphenols (ligands L81–L83) and those of derived optically pure BINOLS (ligands L84 and L85). The stereogenic phosphorus was found to be stable unless it was heated above 80 °C.153
 | ||
| Scheme 35 General procedure to obtain optically pure JoSoPhos (L80–L86) ligands starting from 1·Li and chlorophosphanes (top) and the exact ligands prepared (bottom). For the sake of simplicity, only those ligands prepared from (RP,S)-P5·Li are displayed. | ||
The same group expanded this methodology for the synthesis of P-stereogenic phosphane-diamidophosphite ligand L86, containing five stereogenic centres; two phosphorus and three carbons.150 The required phosphoramidochloridites could be easily obtained by condensation of phosphorus trichloride and the Betti base,154 easily obtainable from cheap reagents.155 The absolute configuration of the phosphorus atoms could be deduced from multinuclear NMR and crystal structure analysis. The ligands were found to be stable under inert atmosphere, but their non-cyclic stereogenic phosphorus atoms were found to be configurationally unstable above 60 °C.
As the JoSoPhos ligands were developed for rhodium-catalyzed enantioselective hydrogenation, their coordination towards rhodium(I) moieties was studied by NMR.30,149,150 Those studies showed that the reaction of (RP,S)-L80 with [Rh(diene)2]BF4 produced the bischelated complex [Rh(L80)2]BF4 in solution as a major product according to 31P NMR spectroscopy, regardless of the metal:ratio employed. On the other hand, bulkier ligand (RP,S,Sa)-L85 produced the expected monochelated complex [Rh(L85)(cod)]BF4 or the bis(chelated) species [Rh(L85)2]BF4 depending on the used rhodium precursor:ligand ratio.156 This kind of behaviour had been observed by Pizzano,157 Vidal-Ferran158 and Grabulosa29 (Fig. 7) for other non-symmetric diphosphorus ligands. A crystal structure determination149 of [Rh(L80)2]BF4 revealed that in the crystal the complex had the expected square-planar geometry around rhodium(I) and that the ligands were in mutual trans arrangement. The differences in coordination behaviour had an impact in hydrogenation, which was studied with a wide variety of substrates (Fig. 15).
 | ||
| Fig. 15 Examples of asymmetric hydrogenation with JoSoPhos ligands L84–L86. | ||
The JoSoPhos ligand library demonstrated high catalytic performance in the enantioselective hydrogenation of several types of di- and trisubstituted enamides and other challenging olefins, with a wide functional group tolerance. In addition, the asymmetric synthesis of an anti-Parkinson drug (rasagiline) was presented, which had an enantioselective hydrogenation as a key step.30
O-bridged diphosphorus ligands (POP)
In the introduction it has been stated that scarcity of oxygen-bridged diphosphorus ligands (POP) is due to their tendency to suffer a phosphorotropic equilibrium between the bis(phosphorus(III)) tautomer (POP) and the phosphorus(II)–(III) tautomer PPO, favouring the latter, especially for electron-rich substituents (Scheme 2).15 There are, however, cases in which the trivalent phosphorus tautomer can be stabilised.Pyrophosphites ((RO)2POP(OR)2) can be considered the analogues of the organic anhydrides of the carboxylic acids and it is known that the alkoxy substituents increase the stability of the POP form.159 After a report on chiral (but racemic) pyrophosphites,160 the first example of an enantiopure POP ligand did not appear until a 2003 in an article of Korostylev and Börner.161 In this contribution, they prepared a small set of enantiopure pyrophosphites by simple two-step procedure (Scheme 36).
 | ||
| Scheme 36 Preparation of enantiopure pyrophosphites based on binaphthol. | ||
After the known preparation of the phosphorochloridites by the standard, solvent-free procedure in neat phosphorus trichloride, its controlled hydrolysis with a half equivalent of water gave the desired ligands L87–L90 as crystalline, relatively stable solids. They used them in situ for enantioselective hydrogenations of model functionalized olefins (MAA and DMI) by treating the ligand with [Rh(cod)2]BF4. Quantitative yields were obtained and the enantioselectivities were only moderate in the best cases, which were achieved with H8-binol derived ligand L90 (48% ee for MAA and 70% ee for DMI). Investigations of the precatalyst solution showed the presence of many unidentified complexes, which were not isolated or further studied.
The following year, Faraone61 reported the same ligand L87, obtained by a different synthetic method (Scheme 37).
 | ||
| Scheme 37 Alternative preparation of pyrophosphite L87. | ||
The synthesis implied the complete hydrolysis of phosphorochloridite derived from binaphthol with water to give phosphonate P6, in tautomeric equilibrium with the corresponding phosphite,16 which was deprotonated and reacted with another equivalent of the phosphorochloridite. The pyrophosphite L87 was obtained in a high yield.
The coordination to palladium-η3-allyl moieties furnished, according to detailed NMR experiments and conductivity, dimeric compounds [Pd(η3-1,3-diphenylallyl)(L87)]2(PF6)2 in contrast to similar, PNP ligands (Fig. 14) which, as discussed before, form monomeric complexes. These dimers contain two palladium(II) centres bridged by two ligands and had been observed and characterised by Grabulosa with methylene-bridged diphosphanes (Fig. 7, complex E).52 In the enantioselective allylic alkylation of 1,3-diphenylallyl acetate with dimethylmalonate, ligand L87 provided the product in a 57% ee, which is much better than with the other ligands of Fig. 14, which is an interesting observation that has not been further explored in the literature.
Conclusions
The considerable corpus of work described in this perspective proves that chiral, single atom-bridged diphosphorus ligands PXP present a rich coordination and organometallic chemistry and have a high potential as ligands in enantioselective homogeneous catalysis. This was proved more than two decades ago by the superb activities and enantioselectivities obtained by the apparently very simple, methylene-bridged electron rich P-stereogenic PCP ligands described in the first part of the review. Clearly, the P-stereogenic moiety is a very powerful motif when forming rigid, four-membered chelate structures. More recently, equally good results have also been obtained with the analogous PNP ligands MaxPHOS (L54) and MAdPHOS (L55) with an unsubstituted nitrogen atom. In contrast, despite the large number of articles published, nitrogen-substituted PNP ligands have given much more modest results in catalysis up to now, with some exceptions, like the recently published JoSoPhos ligands (L84–L86). Despite this, due to their easy synthesis, this is certainly an area where new ligand design ideas could lead to discoveries of new efficient systems. Finally, the area of chiral POP ligands remains essentially unexplored, despite that it has been proven that pyrophosphites can be stabilized in their phosphorus(III) tautomers. Much more work is clearly needed to uncover the potential of PXP ligands in catalysis, but there is little doubt that the future will bring interesting new coordination chemistry and other exceptional ligands.Data availability
No primary research results, software or code have been included. For analysis of crystal structures, crystallographic data from the CCDC has been used.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
We thank MICINN (PID2020-115658GBI100) and AGAUR (2021-SGR-01107) for financial support. The authors thank Prof. Anton Vidal-Ferran for insightful comments about the manuscript that improved its quality.References
- Catalytic Asymmetric Synthesis, ed. T. Akiyama and I. Ojima, Wiley, Hoboken NJ, 2022 Search PubMed.
- Phosphorus Ligands in Asymmetric Catalysis: Synthesis and Applications, ed. A. Börner, Wiley-VCH, Weinheim, 2008 Search PubMed.
- M.-N. Birkholz, Z. Freixa and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099–1118 RSC.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- J. Jover and J. Cirera, Dalton Trans., 2019, 48, 15036–15048 RSC.
- A. C. Hillier, W. J. Sommer, B. S. Yong, J. L. Petersen, L. Cavallo and S. P. Nolan, Organometallics, 2003, 22, 4322–4326 CrossRef CAS.
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC.
- J. Schulz, R. Clauss, A. Kazimir, S. Holzknecht and E. Hey-Hawkins, Angew. Chem., Int. Ed., 2023, 62, e202218648 CrossRef CAS PubMed.
- K. Issleib and D.-W. Müller, Chem. Ber., 1959, 92, 3175–3182 CrossRef CAS.
- A. Vidal-Ferran, A. Grabulosa, X. Verdaguer and A. Riera, in Catalytic Asymmetric Synthesis, ed. T. Akiyama and I. Ojima, Wiley, Hoboken, NJ, 2022, ch. 15, pp. 561–616 Search PubMed.
- S. M. Mansell, Dalton Trans., 2017, 46, 15157–15174 RSC.
- N. T. Coles, A. Sofie Abels, J. Leitl, R. Wolf, H. Grützmacher and C. Müller, Coord. Chem. Rev., 2021, 433, 213729 CrossRef CAS.
- A. Grabulosa, P-Stereogenic Ligands in Enantioselective Catalysis, Royal Society of Chemistry, Cambridge, 2011 Search PubMed.
- T. Imamoto, Chem. Rev., 2024, 124, 8657–8739 CrossRef CAS PubMed.
- F. Flecken and S. Hanf, Dalton Trans., 2024, 53, 17123–17131 RSC.
- A. Gallen, A. Riera, X. Verdaguer and A. Grabulosa, Catal. Sci. Technol., 2019, 9, 5504–5561 RSC.
- T. M. Shaikh, C.-M. Weng and F.-E. Hong, Coord. Chem. Rev., 2012, 256, 771–803 CrossRef CAS.
- P. W. N. M. van Leeuwen, I. Cano and Z. Freixa, ChemCatChem, 2020, 12, 3982–3994 CrossRef CAS.
- D. S. Glueck, Synthesis, 2022, 271–280 CrossRef CAS.
- Q. Dai, W. Li and X. Zhang, Tetrahedron, 2008, 64, 6943–6948 CrossRef CAS.
- W. Tang, A. G. Capacci, A. White, S. Ma, S. Rodriguez, B. Qu, J. Savoie, N. D. Patel, X. Wei, N. Haddad, N. Grinberg, N. K. Yee, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2010, 12, 1104–1107 CrossRef CAS PubMed.
- K. Huang, X. Zhang, T. J. Emge, G. Hou, B. Cao and X. Zhang, Chem. Commun., 2010, 46, 8555–8557 RSC.
- A. R. Muci, K. R. Campos and D. A. Evans, J. Am. Chem. Soc., 1995, 117, 9075–9076 CrossRef CAS.
- T. Imamoto, J. Watanabe, Y. Wada, H. Masuda, H. Yamada, H. Tsuruta, S. Matsukawa and K. Yamaguchi, J. Am. Chem. Soc., 1998, 120, 1635–1636 CrossRef CAS.
- Y. Yamanoi and T. Imamoto, J. Org. Chem., 1999, 64, 2988–2989 CrossRef CAS PubMed.
- A. Ohashi, S. Kikuchi, M. Yasutake and T. Imamoto, Eur. J. Org. Chem., 2002, 2535–2546 CrossRef CAS.
- T. Imamoto, K. Tamura, T. Ogura, Y. Ikematsu, D. Mayama and M. Sugiya, Tetrahedron: Asymmetry, 2010, 21, 1522–1528 CrossRef CAS.
- R. P. Kamalesh Babu, S. S. Krishnamurthy and M. Nethaji, Tetrahedron: Asymmetry, 1995, 6, 427–438 CrossRef.
- J. Eusamio, Y. M. Medina, J. C. Córdoba, A. Vidal-Ferran, D. Sainz, A. Gutiérrez, M. Font-Bardia and A. Grabulosa, Dalton Trans., 2023, 52, 2424–2439 RSC.
- S. Chakrabortty, K. Konieczny, J.-O. Moritz, S. Zheng, S. Tin, B. H. Müller and J. G. de Vries, ACS Catal., 2023, 13, 12030–12040 CrossRef CAS.
- T. Imamoto, Y. Horiuchi, E. Hamanishi, S. Takeshita, K. Tamura, M. Sugiya and K. Yoshida, Tetrahedron, 2015, 71, 6471–6480 CrossRef CAS.
- I. D. Gridnev, Y. Yamamoi, N. Higashi, H. Tsuruta, M. Yasutake and T. Imamoto, Adv. Synth. Catal., 2001, 343, 118–136 CrossRef CAS.
- I. D. Gridnev, M. Yasutake, N. Higashi and T. Imamoto, J. Am. Chem. Soc., 2001, 123, 5268–5276 CrossRef CAS PubMed.
- I. D. Gridnev, M. Yasutake, T. Imamoto and I. P. Beletskaya, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5385–5390 CrossRef CAS PubMed.
- G. Hoge, H. Wu, W. S. Kissel, D. A. Pflum, D. J. Greene and J. Bao, J. Am. Chem. Soc., 2004, 126, 5966–5967 CrossRef CAS PubMed.
- H. Wu and G. Hoge, Org. Lett., 2004, 6, 3645–3647 CrossRef CAS PubMed.
- I. D. Gridnev, T. Imamoto, G. Hoge, M. Kouchi and H. Takahashi, J. Am. Chem. Soc., 2008, 130, 2560–2572 CrossRef CAS PubMed.
- V. M. Foley, R. Cano and G. P. McGlacken, Tetrahedron: Asymmetry, 2016, 27, 1160–1167 CrossRef CAS.
- S. K. Ritter, Chem. Eng. News, 2017, 95, 18–20 Search PubMed.
- Y. Sawatsugawa, K. Tamura, N. Sano and T. Imamoto, Org. Lett., 2019, 21, 8874–8878 CrossRef CAS PubMed.
- S. Jugé, M. Stephan, J. A. Laffitte and J. P. Genêt, Tetrahedron Lett., 1990, 31, 6357–6360 CrossRef.
- W. S. Knowles, M. J. Sabacky, B. D. Vineyard and D. J. Weinkauff, J. Am. Chem. Soc., 1975, 97, 2567–2568 CrossRef CAS.
- F. Maienza, F. Spindler, M. Thommen, B. Pugin, C. Malan and A. Mezzetti, J. Org. Chem., 2002, 67, 5239–5249 CrossRef CAS PubMed.
- D. Moulin, C. Darcel and S. Jugé, Tetrahedron: Asymmetry, 1999, 10, 4729–4743 CrossRef CAS.
- C. Salomon, D. Fortin, C. Darcel, S. Jugé and P. D. Harvey, J. Cluster Sci., 2009, 20, 267–280 CrossRef CAS.
- C. Salomon, S. Dal Molin, D. Fortin, Y. Mugnier, R. T. Boere, S. Jugé and P. D. Harvey, Dalton Trans., 2010, 39, 10068–10075 RSC.
- S. Humbel, C. Bertrand, C. Darcel, C. Bauduin and S. Jugé, Inorg. Chem., 2003, 42, 420–427 CrossRef CAS PubMed.
- C. Bauduin, D. Moulin, E. B. Kaloun, C. Darcel and S. Jugé, J. Org. Chem., 2003, 68, 4293–4301 CrossRef CAS PubMed.
- C. Salomon, D. Fortin, N. Khiri, S. Jugé and P. D. Harvey, Eur. J. Inorg. Chem., 2011, 2597–2609 CrossRef CAS.
- C. Salomon-Bertrand, J. Bayardon, H. Lauréano, S. Jugé, J.-C. Daran and M. Gouygou, J. Organomet. Chem., 2021, 938, 121753 CrossRef CAS.
- A. Jaillet, J. Bayardon, Y. Rousselin and S. Jugé, J. Org. Chem., 2023, 88, 16679–16706 CrossRef CAS PubMed.
- J. C. Córdoba, A. Vidal-Ferran, M. Font-Bardia and A. Grabulosa, Organometallics, 2020, 39, 2511–2525 CrossRef.
- A. Grabulosa, G. Muller, J. I. Ordinas, A. Mezzetti, M. A. Maestro, M. Font-Bardia and X. Solans, Organometallics, 2005, 24, 4961–4973 CrossRef CAS.
- A. B. Chaplin, C. Fellay, G. Laurenczy and P. J. Dyson, Organometallics, 2006, 26, 586–593 CrossRef.
- C. Chen, S. Wen, M. Geng, S. Jin, Z. Zhang, X.-Q. Dong and X. Zhang, Chem. Commun., 2017, 53, 9785–9788 RSC.
- S. Wen, C. Chen, S. Du, Z. Zhang, Y. Huang, Z. Han, X.-Q. Dong and X. Zhang, Org. Lett., 2017, 19, 6474–6477 CrossRef CAS PubMed.
- J. Wolf, M. Manger, U. Schmidt, G. Fries, D. Barth, B. Weberndorfer, D. A. Vicic, W. D. Jones and H. Werner, J. Chem. Soc., Dalton Trans., 1999, 1867–1876 RSC.
- I. Pernik, J. F. Hooper, A. B. Chaplin, A. S. Weller and M. C. Willis, ACS Catal., 2012, 2, 2779–2786 CrossRef CAS.
- A. B. Chaplin, J. F. Hooper, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 4885–4897 CrossRef CAS PubMed.
- G. Fries, J. Wolf, K. Ilg, B. Walfort, D. Stalke and H. Werner, Dalton Trans., 2004, 1873–1881 RSC.
- G. Calabrò, D. Drommi, G. Bruno and F. Faraone, Dalton Trans., 2004, 81–89 RSC.
- J.-L. Vasse, R. Stranne, R. Zalubovskis, C. Gayet and C. Moberg, J. Org. Chem., 2003, 68, 3258–3270 CrossRef CAS PubMed.
- M. Jackson and I. C. Lennon, Tetrahedron Lett., 2007, 48, 1831–1834 CrossRef CAS.
- M. J. Burk, J. Am. Chem. Soc., 1991, 113, 8518–8519 CrossRef CAS.
- P. G. Pringle, E. L. Hazeland, A. Chapman and H. Sparkes, Chem. Commun., 2015, 51, 10206–10209 RSC.
- J. Cámpora, C. M. Maya, I. Matas, B. Claasen, P. Palma and E. Álvarez, Inorg. Chim. Acta, 2006, 359, 3191–3196 CrossRef.
- M. Zablocka, A. Igau, N. Cenac, B. Donnadieu, F. Dahan, J. P. Majoral and K. M. Pietrusiewicz, J. Am. Chem. Soc., 1995, 117, 8083–8089 CrossRef CAS.
- M. Zablocka, F. Boutonnet, A. Igau, F. Dahan, J. P. Majoral and K. M. Pietrusiewicz, Angew. Chem., Int. Ed. Engl., 1993, 32, 1735–1737 CrossRef.
- A. E. Diaz-Alvarez, P. Crochet, M. Zablocka, V. Cadierno, C. Duhayon, J. Gimeno and J. P. Majoral, New J. Chem., 2006, 30, 1295–1306 RSC.
- A. Marinetti, C. Le Menn and L. Ricard, Organometallics, 1995, 14, 4983–4985 CrossRef CAS.
- J. Xu, Tetrahedron, 2024, 134425 Search PubMed.
- A. Marinetti and L. Ricard, Tetrahedron, 1993, 49, 10291–10304 CrossRef CAS.
- W. Tang, W. Wang, Y. Chi and X. Zhang, Angew. Chem., Int. Ed., 2003, 42, 3509–3511 CrossRef CAS PubMed.
- X. Zhang, K. Huang, G. Hou, B. Cao and X. Zhang, Angew. Chem., Int. Ed., 2010, 49, 6421–6424 CrossRef CAS PubMed.
- W. Tang, B. Qu, A. G. Capacci, S. Rodriguez, X. Wei, N. Haddad, B. Narayanan, S. Ma, N. Grinberg, N. K. Yee, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2010, 12, 176–179 CrossRef CAS PubMed.
- M. Biosca, D. Tarr, O. Pàmies and M. Diéguez, in Adv. Organomet. Chem, Academic Press, 2024, vol. 81, pp. 349–388 Search PubMed.
- M. Witt and H. W. Roesky, Chem. Rev., 1994, 94, 1163–1181 CrossRef CAS.
- M. S. Balakrishna, V. S. Reddy, S. S. Krishnamurthy, J. F. Nixon and J. C. T. R. B. S. Laurent, Coord. Chem. Rev., 1994, 129, 1–90 CrossRef CAS.
- T. Appleby and J. D. Woollins, Coord. Chem. Rev., 2002, 235, 121–140 CrossRef CAS.
- C. Fliedel, A. Ghisolfi and P. Braunstein, Chem. Rev., 2016, 116, 9237–9304 CrossRef CAS PubMed.
- P. Bhattacharyya and J. D. Woollins, Polyhedron, 1995, 14, 3367–3388 CrossRef CAS.
- O. Schmitz-Du Mont, B. Ross and H. Klieber, Angew. Chem., Int. Ed. Engl., 1967, 6, 875–876 CrossRef CAS.
- H. Nöth and L. Meinel, Z. Anorg. Allg. Chem., 1967, 349, 225–240 CrossRef.
- M. Revés, C. Ferrer, T. León, S. Doran, P. Etayo, A. Vidal-Ferran, A. Riera and X. Verdaguer, Angew. Chem., Int. Ed., 2010, 49, 9452–9455 CrossRef PubMed.
- P. Rojo, A. Riera and X. Verdaguer, Coord. Chem. Rev., 2023, 489, 215192 CrossRef CAS.
- A. Cabré, A. Riera and X. Verdaguer, Acc. Chem. Res., 2020, 53, 676–689 CrossRef PubMed.
- O. I. Kolodiazhnyi, E. V. Gryskun, N. V. Andrushko, M. Freytag, P. G. Jones and R. Schmutzler, Tetrahedron: Asymmetry, 2003, 14, 181–183 CrossRef CAS.
- E. Cristóbal-Lecina, P. Etayo, S. Doran, M. Revés, P. Martín-Gago, A. Grabulosa, A. R. Constantino, A. Vidal-Ferran, A. Riera and X. Verdaguer, Adv. Synth. Catal., 2014, 356, 795–804 CrossRef.
- J. S. Ritch, T. Chivers, D. J. Eisler and H. M. Tuononen, Chem. – Eur. J., 2007, 13, 4643–4653 CrossRef CAS PubMed.
- M. Bellido, H. Solé-Àvila, M. Sidro, A. Grabulosa, X. Verdaguer and A. Riera, J. Org. Chem., 2025, 90, 1794–1800 CrossRef CAS PubMed.
- L. Chen, P. Ren and B. P. Carrow, J. Am. Chem. Soc., 2016, 138, 6392–6395 CrossRef CAS PubMed.
- J. Vicente and M. a. T. Chicote, Coord. Chem. Rev., 1999, 193–195, 1143–1161 CrossRef CAS.
- R. P. Pinnell, C. A. Megerle, S. L. Manatt and P. A. Kroon, J. Am. Chem. Soc., 1973, 95, 977–978 CrossRef CAS.
- A. Grabulosa, A. Mannu, G. Muller, T. Calvet and M. Font-Bardia, J. Organomet. Chem., 2011, 696, 2338–2345 CrossRef CAS.
- Z. L. Niemeyer, A. Milo, D. P. Hickey and M. S. Sigman, Nat. Chem., 2016, 8, 610–617 CrossRef CAS PubMed.
- J. Eusamio and A. Grabulosa, unpublished results.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- E. Cristóbal-Lecina, A. R. Costantino, A. Grabulosa, A. Riera and X. Verdaguer, Organometallics, 2015, 34, 4989–4993 CrossRef.
- A. Grabulosa, S. Doran, G. Brandariz, G. Muller, J. Benet-Buchholz, A. Riera and X. Verdaguer, Organometallics, 2014, 33, 692–701 CrossRef CAS.
- J. Tellez, A. Gallen, J. Ferrer, F. J. Lahoz, P. Garcia-Orduna, A. Riera, X. Verdaguer, D. Carmona and A. Grabulosa, Dalton Trans., 2017, 46, 15865–15874 RSC.
- D. Carmona, J. Ferrer, L. A. Oro, M. C. Apreda, C. Foces-Foces, F. H. Cano, J. Elguero and M. L. Jimeno, J. Chem. Soc., Dalton Trans., 1990, 1463–1476 RSC.
- T. León, A. Riera and X. Verdaguer, J. Am. Chem. Soc., 2011, 133, 5740–5743 CrossRef PubMed.
- H. Zijlstra, T. León, A. de Cózar, C. F. Guerra, D. Byrom, A. Riera, X. Verdaguer and F. M. Bickelhaupt, J. Am. Chem. Soc., 2013, 135, 4483–4491 CrossRef CAS PubMed.
- S. Orgué, A. Flores-Gaspar, M. Biosca, O. Pàmies, M. Diéguez, A. Riera and X. Verdaguer, Chem. Commun., 2015, 51, 17548–17551 RSC.
- E. Salomó, A. Prades, A. Riera and X. Verdaguer, J. Org. Chem., 2017, 82, 7065–7069 CrossRef PubMed.
- A. Gallen, S. Orgue, G. Muller, E. C. Escudero, A. Riera, X. Verdaguer and A. Grabulosa, Dalton Trans., 2018, 47, 5366–5379 RSC.
- M. Stankevič and K. M. Pietrusiewicz, J. Org. Chem., 2007, 72, 816–822 CrossRef PubMed.
- E. Salomó, S. Orgué, A. Riera and X. Verdaguer, Synthesis, 2016, 2659–2663 Search PubMed.
- T. Imamoto, Chem. Rec., 2016, 16, 2659–2673 CrossRef PubMed.
- P. W. Lednor, W. Beck, H. G. Fick and H. Zippel, Chem. Ber., 1978, 111, 615–628 Search PubMed.
- H.-G. Fick and W. Beck, J. Organomet. Chem., 1983, 252, 83–93 Search PubMed.
- A. M. Z. Slawin, J. D. Woollins and Q. Zhang, J. Chem. Soc., Dalton Trans., 2001, 621–632 RSC.
- N. C. Payne and D. W. Stephan, J. Organomet. Chem., 1981, 221, 203–221 CrossRef CAS.
- D. H. Farrar and N. C. Payne, J. Organomet. Chem., 1981, 220, 251–270 CrossRef CAS.
- R. Mutin, W. Abboud, J. M. Basset and D. Sinou, J. Mol. Catal., 1985, 33, 47–59 CrossRef CAS.
- D. Sakellariou, S. P. Brown, A. Lesage, S. Hediger, M. Bardet, C. A. Meriles, A. Pines and L. Emsley, J. Am. Chem. Soc., 2003, 125, 4376–4380 CrossRef CAS PubMed.
- S. Cadars, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2005, 127, 4466–4476 CrossRef CAS PubMed.
- S. Cadars, A. Lesage, M. Trierweiler, L. Heux and L. Emsley, Phys. Chem. Chem. Phys., 2007, 9, 92–103 RSC.
- E. Simón-Manso, M. Valderrama, P. Gantzel and C. P. Kubiak, J. Organomet. Chem., 2002, 651, 90–97 CrossRef.
- J. W. Faller, J. Lloret-Fillol and J. Parr, New J. Chem., 2002, 26, 883–888 RSC.
- K. Raghuraman, S. S. Krishnamurthy and M. Nethaji, J. Organomet. Chem., 2003, 669, 79–86 CrossRef CAS.
- S. K. Mandal, G. A. N. Gowda, S. Krishnamurthy, C. Zheng, S. Li and N. S. Hosmane, J. Organomet. Chem., 2003, 676, 22–37 CrossRef CAS.
- T. S. Venkatakrishnan, S. S. Krishnamurthy and M. Nethaji, J. Organomet. Chem., 2005, 690, 4001–4017 CrossRef CAS.
- G. C. Vougioukalakis, I. Stamatopoulos, N. Petzetakis, C. P. Raptopoulou, V. Psycharis, A. Terzis, P. Kyritsis, M. Pitsikalis and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5241–5250 CrossRef CAS.
- W.-Y. Mou, T. Li, B. Xie, D.-L. Zhang, C. Lai, C.-L. Deng, J.-X. Cao, X.-X. Bai and X.-Q. Liu, Inorg. Chim. Acta, 2020, 507, 119587 CrossRef CAS.
- I. Stamatopoulos, M. Plaček, V. Psycharis, A. Terzis, J. Svoboda, P. Kyritsis and J. Vohlídal, Inorg. Chim. Acta, 2012, 387, 390–395 CrossRef CAS.
- F. Trentin, A. M. Chapman, A. Scarso, P. Sgarbossa, R. A. Michelin, G. Strukul and D. F. Wass, Adv. Synth. Catal., 2012, 354, 1095–1104 Search PubMed.
- A. Badía, L. R. Falvello, R. Navarro and E. P. Urriolabeitia, J. Organomet. Chem., 1997, 547, 121–128 CrossRef.
- A. Badía, R. Navarro and E. P. Urriolabeitia, J. Organomet. Chem., 1998, 554, 105–112 CrossRef.
- K. Raghuraman, S. K. Mandal, T. S. Venkatakrishnan, S. S. Krishnamurthy and M. Nethaji, Proc. – Indian Acad. Sci., Chem. Sci., 2002, 114, 233–246 Search PubMed.
- K. Bieger, J. Díez, M. P. Gamasa, J. Gimeno, M. Pavlišta, Y. Rodríguez-Álvarez, S. García-Granda and R. Santiago-García, Eur. J. Inorg. Chem., 2002, 1647–1656 CrossRef CAS.
- J. Díez, M. P. Gamasa, J. Gimeno, Y. Rodríguez and S. García-Granda, Eur. J. Inorg. Chem., 2004, 2078–2085 CrossRef.
- E. Simón-Manso and M. Valderrama, J. Organomet. Chem., 2006, 691, 380–386 CrossRef.
- M. Panera, J. Díez, I. Merino, E. Rubio and M. P. Gamasa, Eur. J. Inorg. Chem., 2011, 393–404 CrossRef CAS.
- R. P. K. Babu, S. S. Krishnamurthy and M. Nethaji, Organometallics, 1995, 14, 2047–2056 CrossRef CAS.
- S. K. Mandal, G. A. N. Gowda, S. Krishnamurthy, C. Zheng, S. Li and N. S. Hosmane, Eur. J. Inorg. Chem., 2002, 2047–2056 CrossRef CAS.
- S. K. Mandal, G. A. Nagana Gowda, S. S. Krishnamurthy and M. Nethaji, Dalton Trans., 2003, 1016–1027 RSC.
- K. Raghuraman, S. S. Krishnamurthy and M. Nethaji, J. Chem. Soc., Dalton Trans., 2002, 4289–4295 RSC.
- T. S. Venkatakrishnan, M. Nethaji and S. S. Krishnamurthy, J. Organomet. Chem., 2006, 691, 224–228 CrossRef CAS.
- F. Robert, Y. Gimbert, M. T. Averbuch, A. Durif and A. Greene, Z. Kristallogr. NCS, 1999, 214, 581–582 CAS.
- S. K. Mandal, G. A. Nagana Gowda, S. S. Krishnamurthy, T. Stey and D. Stalke, J. Organomet. Chem., 2005, 690, 742–750 CrossRef CAS.
- R. P. K. Babu, S. S. Krishnamurthy and M. Nethaji, Heteroat. Chem., 1991, 2, 477–485 CrossRef CAS.
- R. P. Kamalesh Babu, K. Aparna, S. S. Krishnamurthy and M. Nethaji, Phosphorus, Sulfur Silicon Relat. Elem., 1995, 103, 39–53 CrossRef.
- T. S. Venkatakrishnan, M. Nethaji and S. S. Krishnamurthy, Curr. Sci., 2003, 85, 969–974 CAS.
- T. S. Venkatakrishnan, S. K. Mandal, R. Kannan, S. S. Krishnamurthy and M. Nethaji, J. Organomet. Chem., 2007, 692, 1875–1891 CrossRef CAS.
- G. Calabrò, D. Drommi, C. Graiff, F. Faraone and A. Tiripicchio, Eur. J. Inorg. Chem., 2004, 1447–1453 CrossRef.
- S. Orgué, T. León, A. Riera and X. Verdaguer, Org. Lett., 2015, 17, 250–253 CrossRef PubMed.
- D. Konya, F. Robert, Y. Gimbert and A. E. Greene, Tetrahedron Lett., 2004, 45, 6975–6978 CrossRef CAS.
- J.-O. Moritz, S. Chakrabortty, B. H. Müller, A. Spannenberg and P. C. J. Kamer, J. Org. Chem., 2020, 85, 14537–14544 Search PubMed.
- S. Chakrabortty, K. Konieczny, B. H. Mueller, A. Spannenberg, P. Kamer and J. G. de Vries, Catal. Sci. Technol., 2022, 12, 1392–1399 RSC.
- O. I. Kolodyazhnyi, N. V. Andrushko and E. V. Grishkun, Russ. J. Gen. Chem., 2004, 74, 515–522 CrossRef CAS.
- E. V. Gryshkun, N. V. Andrushko and O. I. Kolodiazhnyi, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 1027–1046 CrossRef CAS.
- S. Fernandez, M. Pfeffer, V. Ritleng and C. Sirlin, Organometallics, 1999, 18, 2390–2394 CrossRef CAS.
- C. Cardellicchio, M. A. M. Capozzi and F. Naso, Tetrahedron: Asymmetry, 2010, 21, 507–517 Search PubMed.
- C. Schmitz, W. Leitner and G. Franciò, Eur. J. Org. Chem., 2015, 6205–6230 CrossRef CAS.
- T. Pan, Q. Yuan, D. Xu and W. Zhang, Org. Lett., 2024, 26, 5850–5855 Search PubMed.
- M. Rubio, S. Vargas, A. Suárez, E. Álvarez and A. Pizzano, Chem. – Eur. J., 2007, 13, 1821–1833 Search PubMed.
- H. Fernández-Pérez, S. M. A. Donald, I. J. Munslow, J. Benet-Buchholz, F. Maseras and A. Vidal-Ferran, Chem. – Eur. J., 2010, 16, 6495–6508 CrossRef PubMed.
- A. D. DeBellis, S. D. Pastor, G. Rihs, R. K. Rodebaugh and A. R. Smith, Inorg. Chem., 2001, 40, 2156–2160 Search PubMed.
- M. Mikołajczyk, B. Ziemnicka, J. Karolak-Wojciechowska and M. Wieczorek, J. Chem. Soc., Perkin Trans. 2, 1983, 501–513 Search PubMed.
- A. Korostylev, D. Selent, A. Monsees, C. Borgmann and A. Börner, Tetrahedron: Asymmetry, 2003, 14, 1905–1909 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt03572k |
| This journal is © The Royal Society of Chemistry 2025 |