
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnalysis of the charge generation and recombination processes in the PM6:Y6 organic solar cell†
Saied Md
Pratik
 ,
Grit
Kupgan
,
Jean-Luc
Brédas
* and
Veaceslav
Coropceanu
*
,
Grit
Kupgan
,
Jean-Luc
Brédas
* and
Veaceslav
Coropceanu
*
Department of Chemistry and Biochemistry, The University of Arizona, Tucson, Arizona 85721-0041, USA. E-mail: coropceanu@arizona.edu; jlbredas@arizona.edu
First published on 4th December 2024
Abstract
Closing the efficiency gap between organic solar cells and their inorganic and perovskite counterparts requires a detailed understanding of the exciton dissociation and charge separation processes, energy loss mechanisms, and influence of disorder effects. In addition, the roles played by excitations delocalized over two or more (macro)molecules and by localized triplet states remain to be well-defined. To address these issues, we have combined molecular dynamics simulations with density functional theory calculations to provide a comprehensive analysis of charge generation and charge recombination in the representative PM6:Y6 blend, describe loss mechanisms, and assess the influence of disorder on the electronic processes. The results allowed the identification of Y6 excimer-like states that can efficiently dissociate into states with hole–electron separation distances larger than those in conventional donor:acceptor interfacial charge-transfer states. They also point to the appearance of low-energy defect states upon formation of Y6 twisted conformations, which can negatively impact the Y6 chemical stability and device performance. Importantly, it is found that the local triplet states formed via non-geminate recombination can efficiently transfer back to triplet CT states, opening the way to eventual dissociation into free charges. Overall, our work provides valuable insight into the charge dynamics within PM6:Y6 active layers.
Broader contextSignificant progress has recently been achieved in the field of organic photovoltaics thanks to the emergence of non-fullerene molecular acceptors. Power conversion efficiencies of organic solar cells (OSCs) now reach the 20% mark, which is nearly double the top efficiency of fullerene-based devices but remains significantly below the performance of inorganic and perovskite devices, which exceeds 25% efficiency. Closing this efficiency gap demands to develop a complete understanding of the fundamental electronic processes taking place in the OSC active layers and therefore to address simultaneously the key aspects related to exciton dissociation, charge separation, energy loss mechanisms, formation of triplet electronic states, delocalization of singlet excitons, and role of disorder and defect states. Here, by considering the widely studied PM6:Y6 blend as a model system and combining molecular dynamics simulations with density functional theory calculations, our theoretical work brings forth the required comprehensive description of all these key aspects. Thus, our study offers unprecedented insight and guidance for ongoing efforts to enhance both the efficiency and stability of OSCs. |
1. Introduction
Donor:acceptor (D:A) blends comprising the PM6 donor polymer and the Y6 non-fullerene acceptor (NFA) (see Fig. 1a) have garnered considerable attention in the field of organic photovoltaics.1–8 Single-junction PM6:Y6 organic solar cells (OSCs) have represented an important stepping stone in the quest for high power conversion efficiencies (PCEs), as they reached PCEs up to ∼15.7% with notable metrics including a high short-circuit current density (Jsc) of 25.3 mA cm−2, an open-circuit voltage (Voc) of 0.83 V, and a fill factor (FF) of 74.8%.1,6 This remarkable performance has been attributed namely to a negligible barrier for charge separation combined with a low density of traps within the material.9,10 Subsequent efforts focused on chemical modifications of Y6 to further enhance the PCE. A number of modified versions of Y6, such as N3,11 Y11,12 Y18,13 BTP-eC9,14 and L8-BO,15 among others, have been developed; devices based on PM6 and these modified versions exhibit efficiencies reaching and now even surpassing 20% when incorporated into more complex active-layers, such as ternary blends.14–18 These advancements underscore the potential for continued progress and optimization in the organic photovoltaics arena.19,20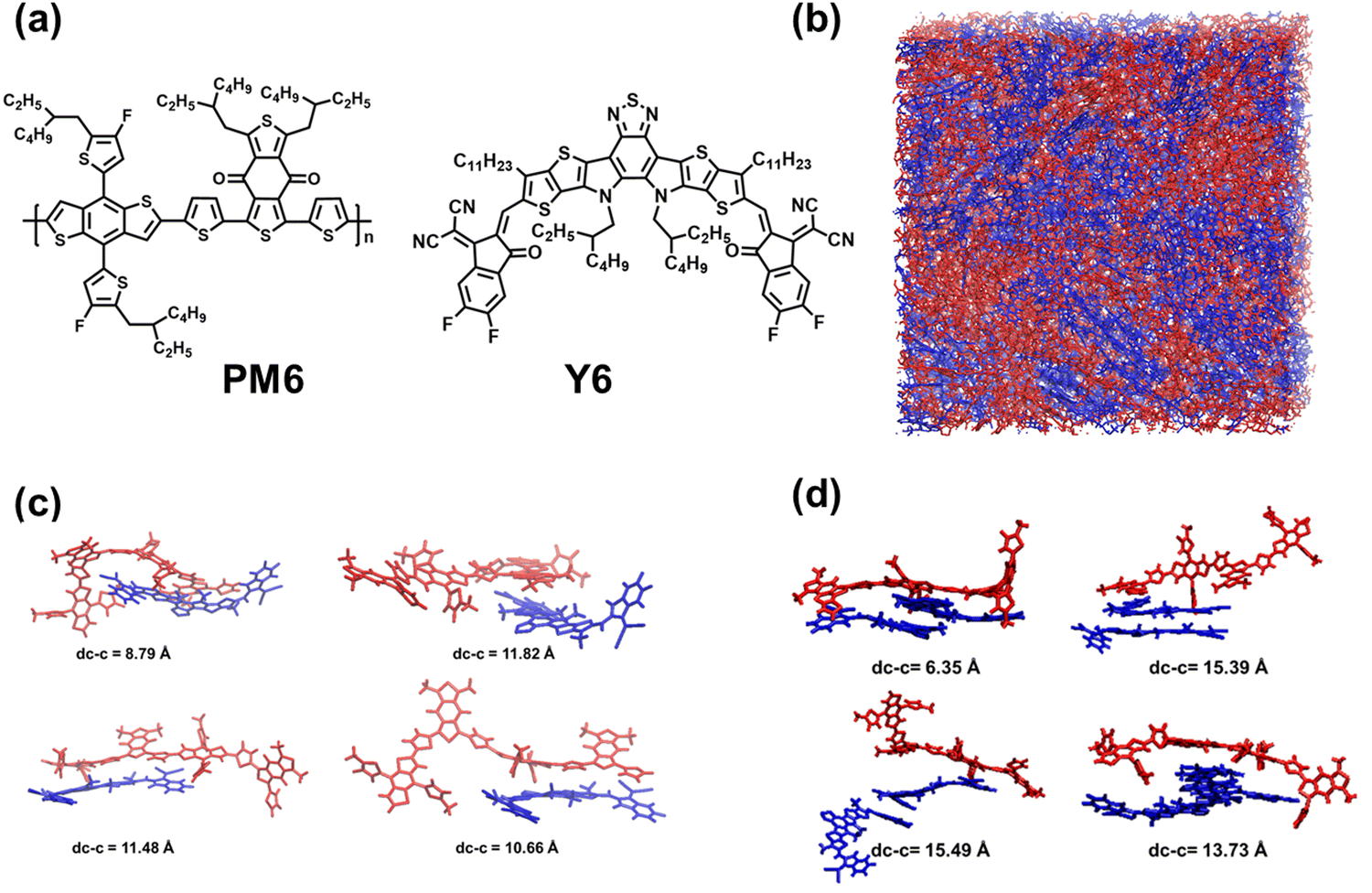 | ||
| Fig. 1 (a) Chemical structures of the PM6 polymer donor and Y6 acceptor. (b) Snapshot of a molecular dynamics (MD) simulation of the PM6:Y6 blend, consisting of 10 twenty-mer PM6 chains and 200 Y6 molecules. PM6 chains are shown in red and Y6 molecules, in blue. Representative donor–acceptor pairs extracted from the MD simulations, along with the corresponding center-of-mass to center-of-mass distances (dc–c) between PM6 and Y6, shown for (c) PM6:Y6 and (d) PM6:2Y6 configurations. | ||
The introduction of NFAs helped significantly reduce the non-radiative voltage losses (ΔVnr), from 0.4–0.6 eV in fullerene-based OSCs21,22 to, for instance, 0.25 (ref. 15)–0.27 eV (ref. 6) in PM6:Y6, 0.24 V in PM6:L8-BO,15 0.17 V in PM6:Y11,23 or 0.15 V in PBDB-T:SM16.24 These values, however, are still larger than those observed in GaAs (ΔVnr = 0.027 V)25 and perovskite (ΔVnr = 0.034 V)26 solar cells whose PCEs are greater than 25%.26,27 It is worth noting that Voc in GaAs devices is 1.12 V, while it does not exceed 0.9 V in the best performing OSCs.16,28 These lower Voc values are not only related to non-radiative voltage losses but also to the respective values of the active-layer donor ionization potential and acceptor electron affinity. In addition, the Jsc and FF values in GaAs solar cells are 29.8 mA cm−2 and 87.7%, respectively.28,29 While OSCs with Jsc over 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mA cm−2 and FF of about 82% have also been reported, they have not been obtained simultaneously in the same device.30,31 Therefore, to make OSCs more competitive with respect to inorganic and perovskite solar cells, it goes without saying that a further increase in Voc should be achieved in concert with an increase in the device Jsc and FF parameters.32 To realize this goal, it is essential to gain a comprehensive understanding of the charge generation and recombination processes in OSCs. In this context, being able to answer the following questions is of special interest: (i) what are the main factors controlling charge generation and charge recombination? (ii) Are there other electronic states that impact exciton dissociation in addition to the commonly invoked local (i.e., monomolecular) excitations (LEs) and interfacial charge-transfer (CT) states? (iii) What is the impact of the presence of low-energy LE triplet states?
mA cm−2 and FF of about 82% have also been reported, they have not been obtained simultaneously in the same device.30,31 Therefore, to make OSCs more competitive with respect to inorganic and perovskite solar cells, it goes without saying that a further increase in Voc should be achieved in concert with an increase in the device Jsc and FF parameters.32 To realize this goal, it is essential to gain a comprehensive understanding of the charge generation and recombination processes in OSCs. In this context, being able to answer the following questions is of special interest: (i) what are the main factors controlling charge generation and charge recombination? (ii) Are there other electronic states that impact exciton dissociation in addition to the commonly invoked local (i.e., monomolecular) excitations (LEs) and interfacial charge-transfer (CT) states? (iii) What is the impact of the presence of low-energy LE triplet states?
In this work, to shed more light on these questions, we performed a comprehensive computational analysis of the charge generation and recombination processes in the PM6:Y6 blend. We conducted molecular dynamics (MD) simulations on the blend (see Fig. 1b), followed by time-dependent long-range corrected density functional theory (TD-DFT) calculations on molecular complexes comprising a PM6 oligomer and initially a single Y6 molecule (see Fig. 1c), to explore their excited-state electronic properties. Furthermore, to address the influence of Y6–Y6 interactions on the electronic processes, we expanded our investigations to include complexes, referred to as PM6:2Y6, consisting of a PM6 oligomer and two Y6 molecules (see Fig. 1d). Importantly, we also investigated the impact of the formation of Y6 triplet states on free-charge formation.
2. Results and discussion
(i) Active-layer morphology of the PM6:Y6 blend
We first focus on the active-layer morphology of the PM6:Y6 blend. This exploration is a crucial component as several critical processes, including exciton diffusion and dissociation, charge carrier transport, and charge recombination strongly depend on the morphological characteristics.33–38Fig. 1b gives an illustration of the active-layer morphology as obtained from the MD simulations of a blend consisting of 10 twenty-mer of PM6 and 200 Y6 molecules (see the computational details in the ESI†). To remain unbiased with respect to the initial configuration, we conducted three independent MD simulations and combined the results from all three simulations for our quantum-mechanical calculations. We note here that our simulations primarily focus on the interface between PM6 and Y6, while actual devices often contain crystalline domains of Y6 and aggregated PM6 chains, which can impact the overall electronic processes and thereby contribute to device performance.To analyze the electronic processes occurring at the interfaces between the PM6 and Y6 domains in the blend, we extracted 3000 PM6:Y6 complexes from the MD simulated morphologies (i.e., 1000 extracted from each simulation box) consisting of a dimeric-PM6 unit and a Y6 molecule (see Fig. 1c). Additionally, we extracted 1500 PM6:2Y6 complexes (500 complexes from each simulation box), consisting of a dimeric-PM6 unit and two Y6 molecules (see Fig. 1d). Subsequent excited-state calculations on these pairs were performed at the ωB97X-D/6-31G(d,p) level of theory with an implicit consideration of the medium dielectric (ε = 3.0), see Section SA in the ESI† for computation details. For the sake of comparison, excited-state calculations were also performed on the Y6 monomers and Y6 dimers involved in the investigated PM6:Y6 and PM6:2Y6 complexes (see Sections SB and SD in the ESI†). The variations in geometric configurations of the complexes will lead to distributions of state energies, which we discuss below.
(ii) Singlet and triplet excited states in the PM6:Y6 complexes
To facilitate the discussion of the underlying electronic processes, we first assigned the states in the singlet and triplet manifolds (see Section SC and Fig. S3 in the in the ESI†) as LE, CT, and hybrid LE–CT states. The assignment relied on a fragment-charge difference (FCD) approach that quantifies the extent of charge transfer within the state (referred to as q), as shown in Fig. 2 (see also Fig. S4 and S5 in the ESI†). The q values close to 0 indicate a predominantly LE state, while values approaching 1 point to a CT state where an electron is located on a Y6 molecule and a hole on PM6. We have arbitrarily set a threshold of q ≤ 0.1 to identify LE states and values of q ≥ 0.9 for CT states, while intermediate values of q represent LE–CT hybrid states. We note that the overall classification of the states as LE and CT hardly changes if the threshold value of q is set to q ≤ 0.2 for LE states and q ≥ 0.8 for CT states (see Table S2 in the ESI†). | ||
| Fig. 2 Histogram plots of charges (q) and their distributions as a function of energy for the (a) S1, (b) S2, (c) T1, and (d) T2 states, as obtained over 3000 PM6:Y6 complexes extracted from the MD simulations. The insets in the histogram distributions represent zoomed-in perspectives for the S1 and T1 states. | ||
As shown in Fig. 2, about 83% of the D:A complexes have a q value ≥0.9 in the S1 state, indicating a CT character (1CT1), while the lowest singlet LE states (1LE1) primarily appear in S2 (∼43% of D:A complexes) and S3 (∼20% of D:A complexes), see Fig. S4 and Table S2 in the ESI† for further details. Interestingly, when turning to the triplet manifold, all the T1 states in the PM6:Y6 complexes possess a q value ≤0.1, underlining that they are LE triplet (3LE1) states; these triplet LE states are located on Y6. The second LE triplet (3LE2) states are coming from the T2 states (∼57%) and T3 states (∼22%) of the PM6:Y6 complexes. Conversely, the lowest-lying triplet CT states (3CT1) are predominantly arising from the T3 states (57%) with an additional contribution of about 22% from the T2 states of the complexes.
Here, we are mostly interested in the recombination processes and will only consider the LE states located on Y6. Indeed, the S1 state in PM6 is located about 0.4 eV above that in Y6 and experimental data suggest that the S1 state in PM6 decays mostly via energy transfer to Y6 excitons.4,39,40 The energy distributions of the lowest (singlet and triplet) LE and CT states are shown in Fig. 3; all these distributions (which we recall are related to the variations in geometric configurations of the complexes in the blend) turn out to exhibit Gaussian shapes whose standard deviations are summarized in Table S3 (ESI†). It should be borne in mind that the energy distributions derived from the MD/TDA-DFT calculations account for both static and dynamic disorder contributions;33,41,42 the impact of disorder on the various electronic processes will be discussed in a later section.
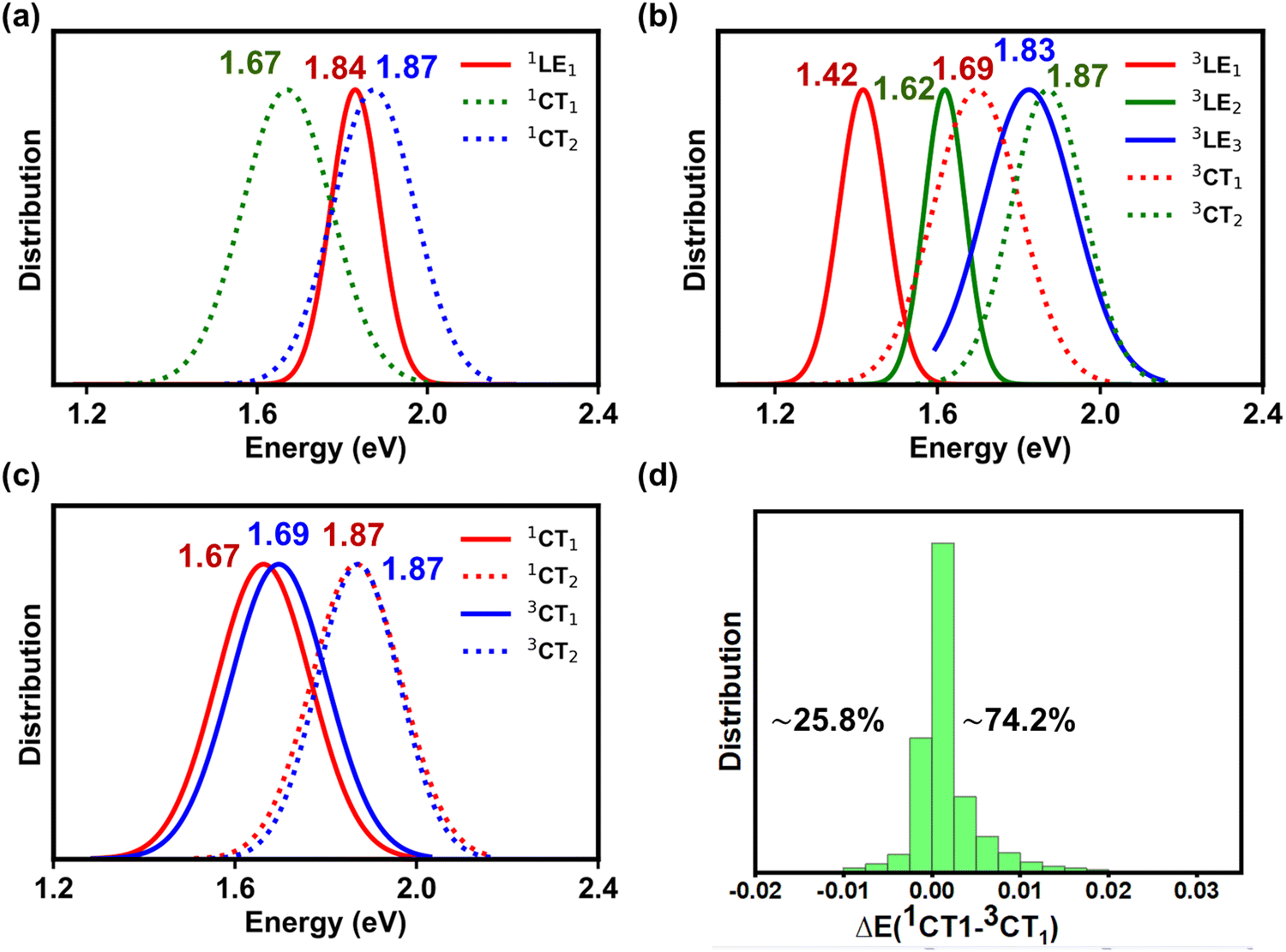 | ||
| Fig. 3 Normalized energy distributions of the (a) singlet CT and LE states, (b) triplet CT and LE states, and (c) singlet and triplet CT states; the numbers on top of the curves indicate the average energy (in eV) of each state. (d) Distribution of the energy offsets (in eV) between the lowest singlet and triplet CT states [ΔE(1CT1–3CT1)] along with the percentages of positive and negative energy offsets. | ||
As shown in Fig. 3a, the lowest singlet 1LE1 states, localized on Y6 (see Fig. S7 and Section SE in the ESI†), have a mean energy value of 1.84 eV. The energy peak for the 1CT1 state distribution is located 170 meV below that of 1LE1 states; based on electro-absorption measurements, Wan et al. have reported a value of 140 meV for this ΔE(1LE1–1CT1) energy difference, which compares well with our theoretical estimate.3 We note that, while the 1LE1 and 1CT1 distributions only slightly overlap, the distribution of the second CT singlet states (1CT2) largely overlaps with that of the 1LE1 states (ΔE(1CT2–1LE1) = 30 meV).
The DFT estimates for the average 1CT and 1LE energies are somewhat higher than their experimental counterparts, which are reported to be ca. 1.29 eV and 1.41 eV, respectively;3,43 this discrepancy could be attributed to the neglect of electron delocalization effects in the calculations discussed in this section (see Section (iv) where we consider the presence of Y6 dimers) and the consideration of too low a dielectric constant (ε = 3) when accounting for the medium effect (some recent experimental data suggest that the dielectric constant in the PM6:Y6 blend could be larger than 5).44
As mentioned above, there are two distinct triplet Y6-based LE states, i.e., 3LE1 and 3LE2, that are located below the 3CT1 state (see Fig. 3b). The mean energy value of the 3CT1 state distribution is about 270 meV higher than that of the 3LE1 states but nearly equal to that of the 3LE2 states. Furthermore, the mean energy value distribution of the 3LE3 states is only 40 meV smaller than that of the 3CT2 states. We note that the 3LE1 and 3LE2 states originate from triplet states located on Y6 while the 3LE3 states originate from PM6 triplet excitations, see Fig. S7 in the ESI.† The implication is that, when 3CT1 states form due to non-geminate recombination processes, there should be a clear path for them to fall into Y6-based triplet LE states, which could then be a major energy-loss mechanism. We will specifically discuss this aspect below.
The singlet and triplet CT energy distributions indicate that the mean value of the triplet 3CT1 energies is larger by some 20 meV than that of the singlet 1CT1 states (see Fig. 3c). However, this does not mean that the 3CT1 states are systematically located above the 1CT1 states. In fact, when the state energies of the PM6:Y6 complexes are examined individually (instead of considering their mean distributions), we find that in most of the complexes (∼74%) the 3CT1 state is in fact located below the 1CT1 state, see Fig. 3d (similarly, in the case of the 1CT2 and 3CT2 states, the 3CT2 state is located below the 1CT2 state in about ∼70% of the complexes, see Fig. S8 in the ESI†). The appearance of 1CT states below their 3CT counter-parts is related to variations in the D:A electronic couplings, as documented earlier by Beljonne and co-workers.4
Due to the amorphous nature of the D:A blends, the energy distributions of the CT and LE states are expected to arise from a combination of dynamic disorder (σD, related to the time-dependent vibrations) and static disorder (σS, related to the time-independent positional disorder).38,41,42,45–48 Here, the σD and σS values were estimated according to the procedure we previously established (see also Section SC in the ESI†).41,42 The results obtained for both LE and CT singlet and triplet states are summarized in Table 1 (see also Fig. S9 in the ESI†). As follows from Table 1, the CT states exhibit similar degrees of static and dynamic disorders, which fall into the range of 70–80 meV; on the other hand, the σS values for both singlet and triplet LE states are smaller than 40 meV. These findings are well supported by recent experimental data: (i) Amassian and co-workers reported, on the basis of scanning tunneling microscopy/spectroscopy measurements, that the magnitudes of the total and static disorders in the 1CT states probed at the top surface of PM6:Y6 films are about 110 ± 9 meV and 55 ± 9 meV, respectively;49 (ii) from temperature-dependent mobility measurement data derived by Shoaee and co-workers one can estimate a σS value of about 80 meV for the CT states.45 We note that the fact that the static and dynamic disorders have comparable magnitudes in the CT states, while in the case of the LE states the static disorder is lower than the dynamic disorder, arises from fundamental differences in the spatial characteristics of the two types of states. The CT states are inter-molecular in nature (spreading across donor and acceptor molecules) and thus influenced by both static and dynamic disorders due to their dependence on inter-molecular interactions; in contrast, the LE states are localized on Y6 molecules and experience disorder primarily driven by dynamic intra-molecular fluctuations.
| 1LE1 | 1CT1 | 1CT2 | 3LE1 | 3LE2 | 3CT1 | 3CT2 | |
|---|---|---|---|---|---|---|---|
| σ T (eV) | 0.08 | 0.11 | 0.10 | 0.07 | 0.07 | 0.11 | 0.09 |
| σ D (eV) | 0.07 | 0.07 | 0.07 | 0.07 | 0.06 | 0.08 | 0.07 |
| σ S (eV) | 0.04 | 0.08 | 0.07 | 0.00 | 0.02 | 0.08 | 0.06 |
(iii) Appearance of defect states
A surprising finding came out of Fig. 2a, which shows that there exist, in about 0.4% of the complexes, Y6-localized S1 states that exhibit q ≤ 0.1 values and have energies as low as 1.17 eV. This energy value is significantly lower than the typical energy of the S1 LE-Y6 states (see Fig. S2 in ESI†). To gain a better understanding of the origin of these low-energy states, we conducted an in-depth investigation of the geometric structure of the Y6 molecules in which they appear. Our analysis revealed that these states emerge when a terminal moiety of the Y6 molecule is significantly twisted with respect to its core, as illustrated in Fig. 4 (see also Fig. S10–S13 in the ESI†). Based on a natural transition orbital (NTO) analysis (which provide a description of the electron and hole wavefunctions in a given excited electronic state), we identified that these Y6 lower-energy states exhibit an intra-molecular charge-transfer character (iY6-CT), see Fig. 4b (as well as Fig. S11 and Section SC in the ESI†). The large dihedral angles related to the appearance of these states could occur in the case of thermal annealing, which facilitates torsional motions and could lead to trapped twisted conformations. Given that torsions of this nature can alter the stability of the chemical bond linking the core and end groups, as reported in a number of NFAs,50,51 it can be expected that these states act as defect states leading, on the one hand, to energy-loss mechanisms and, on the other hand, to reduced chemical stability of the blend, ultimately contributing to a decrease in device efficiency. Along this line, Du et al. reported recently that high-temperature thermal annealing or prolonged annealing durations can indeed negatively impact the performance of PM6:Y6 solar cells.52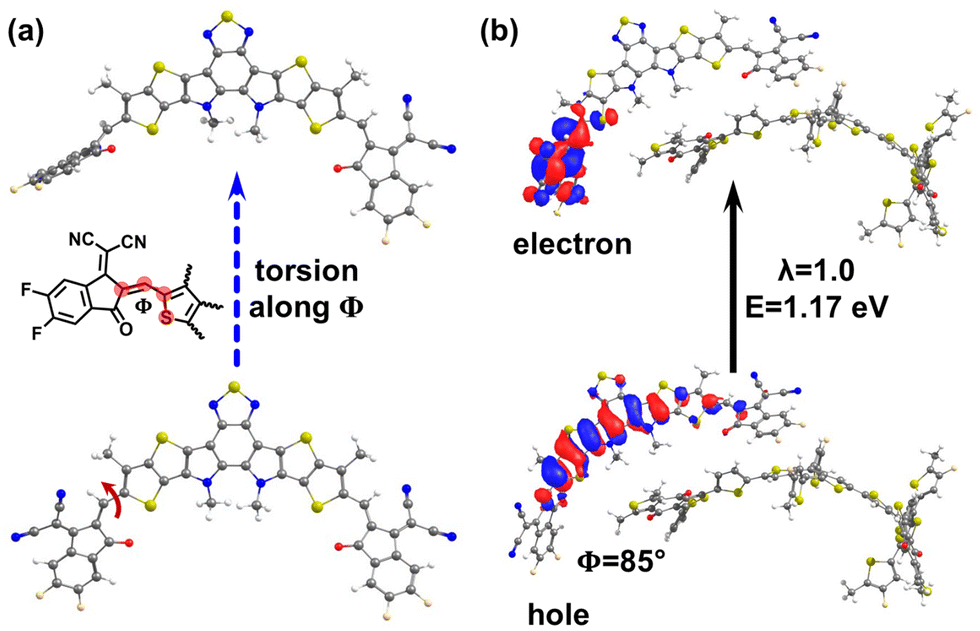 | ||
| Fig. 4 (a) Illustration of the torsional motion leading from a nearly coplanar conformation (bottom) to a twisted conformation (top) of the Y6 molecule. (b) Natural transition orbital (NTO) for the S1 state in a representative PM6:Y6 pair (extracted from the MD simulations) where the Y6 has a large torsion (Φ = 85°) between its core moiety and one of the end moieties; the NTO highlights the intra-Y6 CT nature of the state (λ denotes the weight of the particular NTO in the description of the excited state). | ||
(iv) Consideration of Y6-dimer excited states
It has been suggested that Y6 inter-molecular excimer-like states (denoted hereafter as EX) might act as intermediate states for exciton dissociation.53 Recent studies have shown that EX states have a significant CT character and, as a result, might facilitate exciton dissociation within Y6 domains prior to the excitons reaching the D:A interface.53–56 Therefore, we expanded our analysis to account for the influence of Y6–Y6 interactions on the nature of the LE and interfacial CT states. From our MD simulations, we observe that about 29% of the PM6:Y6 complexes have a second Y6 molecule positioned near the initial Y6 molecule. Subsequently, we randomly selected 1500 complexes comprising a PM6 unit and two Y6 molecules (PM6:2Y6, see Fig. 1d) and conducted the same type of excited-state calculations as before (see Section SD in ESI†). Again, for the sake of comparison, excited-state calculations were also performed on the related Y6–Y6 dimers. The results are presented in Fig. 5 (see also Fig. S14 and S18 in the ESI†).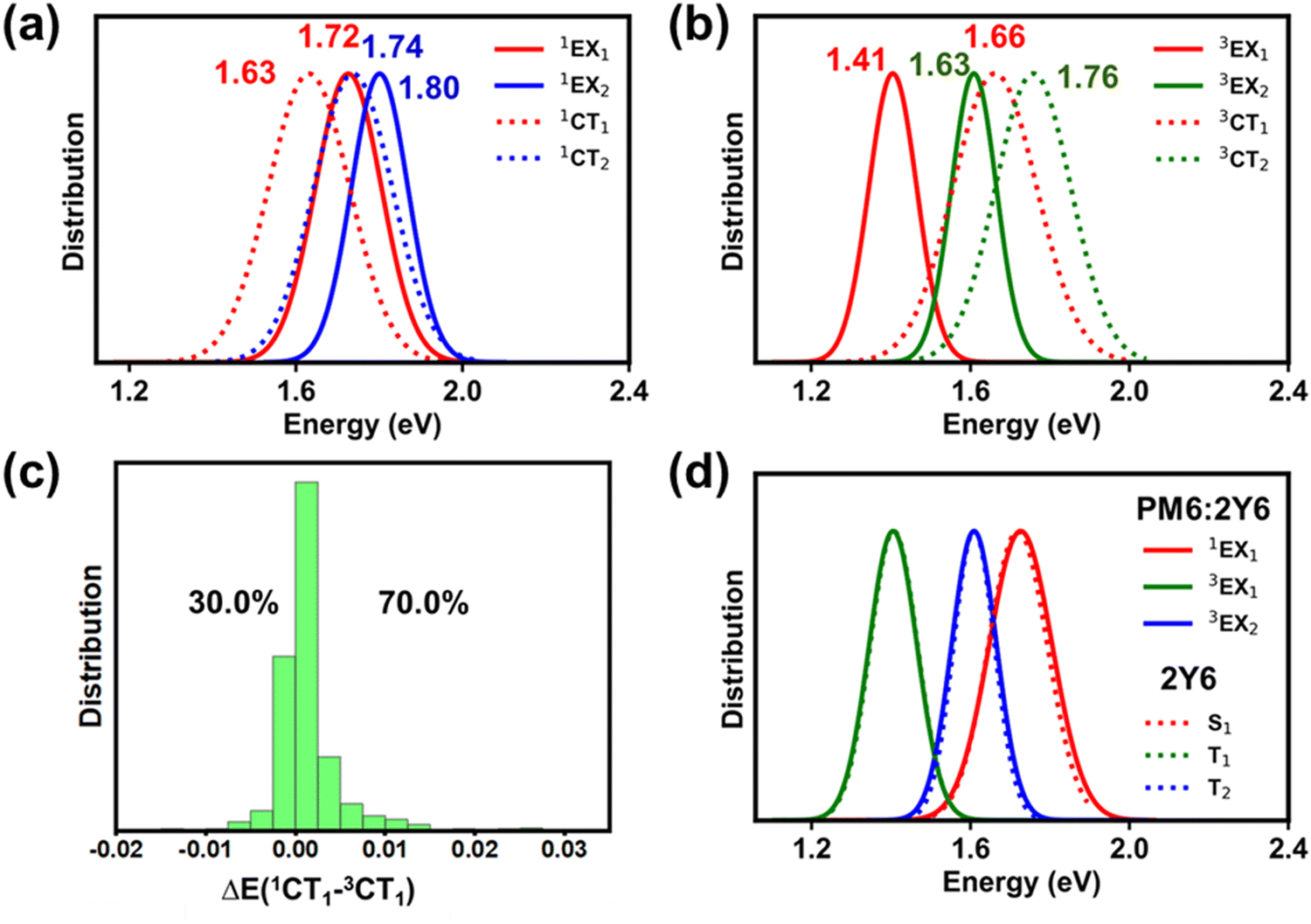 | ||
| Fig. 5 Normalized energy distributions of the (a) singlet and (b) triplet excimeric states and CT states for the PM6:2Y6 complexes. (c) Distribution of energy offsets (in eV) between the singlet and triplet CT states [ΔE(1CT1–3CT1)] across individual PM6:2Y6 complexes, together with the percentage of complexes showing positive and negative energy offsets. (d) Normalized energy distribution plots for singlet and triplet EX state of PM6:2Y6 complexes superimposed on the energy distribution of the EX states in 2Y6 (also see Fig. S20 in the ESI†). | ||
As in the case of PM6:Y6 complexes, we first divided the ensemble of excited states into those localized on Y6 molecules and those forming donor:acceptor CT states (see Fig. 5a and b). As seen from the comparison of Fig. 3 and 5, the lowest-lying Y6-dimer singlet state (1EX1) exhibits a red shift by 120 meV with respect to the Y6 1LE1 state. We note that this feature can be solely attributed to Y6–Y6 interactions as the same red shift is observed in the calculations performed on the 2Y6 dimers, see Fig. 5d. Also, according to the NTO analysis given in Fig. 6 and our previous study,55 the 1EX1 state actually represents a hybridization of Frenkel intra-molecular excitations and Y6 inter-molecular CT excitations. This CT contribution, which is the dominant contribution in many Y6 1EX1 states, is considered to be the key factor facilitating the separation of charge carriers.53–55
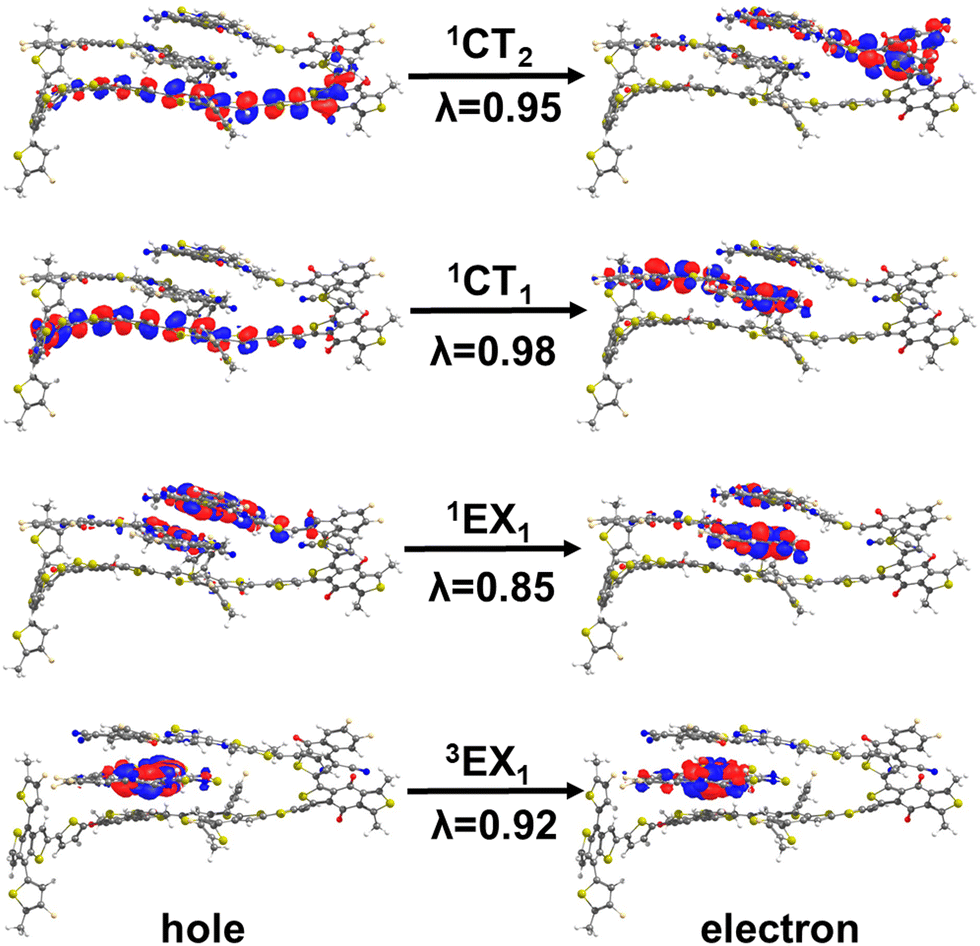 | ||
| Fig. 6 NTO hole and electron wavefunctions for the singlet and triplet excimeric states and CT states for a representative PM6:2Y6 complex (λ denotes the weight of the particular NTO in the description of the excited state). | ||
According to our DFT calculations, the formation of 1EX1 states has only a small effect on the lowest singlet and triplet interfacial CT states (see Fig. 5a, b and Table S3, ESI†). The energies of these states are red-shifted by about 30–40 meV when comparing the results obtained for the PM6:Y6 and PM6:2Y6 complexes. On the other hand, the energy splitting between 1CT and 3CT1 states (ΔE(1CT1–3CT1)) is hardly affected by the Y6–Y6 electronic interactions. As in the case of PM6:Y6 complexes, the 3CT1 states are located below their corresponding 1CT1 states in about 70% of PM6:2Y6 complexes (see Fig. 5c). As seen from Fig. 6, the NTO analyses show that, in PM6:2Y6 complexes, the 1CT1 state is related to electron transfer from PM6 to the closest Y6 molecule while the second interfacial (1CT2) state is related to electron transfer to the second Y6 molecule in the complex. The 1CT2 states overall are higher in energy by ∼0.11 eV with respect to the 1CT1 states.
The TD-DFT calculations also show that the triplet excimeric states (3EX) in both PM6:2Y6 and 2Y6 systems remain completely localized on a single Y6 molecule (see Fig. 6), i.e., they are identical to LE-Y6 states. This means that the inter-molecular excitonic couplings between Y6 triplet states are very small, which is in line with the fact in π-conjugated systems, the lowest triplet states are generally more localized than their singlet counterparts.
(v) Exciton dissociation and geminate recombination processes
The key electronic processes that take place in the active layer of organic solar cells are illustrated in Fig. 7. We start with a discussion of the exciton recombination processes. After their photogeneration, the singlet excitons formed on Y6 molecules can (i) either recombine radiatively or non-radiatively to the ground state (GS), (ii) perform an intersystem crossing to LE triplet states, or (iii) upon reaching the PM6:Y6 interface dissociate into CT states. Experimental estimates for the radiative (kr) and nonradiative (knr) rate constants of the 1LE1 → GS transition in Y6 films, have been reported to be 3.6 × 107 s−1 and 6.9 × 108 s−1, respectively.57 | ||
| Fig. 7 Illustration of the major electronic processes taking place in organic solar cells. The rate constants for these processes, as obtained from our calculations or experimental measurements on PM6:Y6, are listed below (for details, see the main text): kr(1LE–GS) ∼ 107 s−1a (ref. 57), knr(1LE–GS) ∼ 108 s−1a (ref. 57), k(1LE–1CT) ∼ 1011–1012 s−1,bk(1CT–1LE) ∼ 109–1010 s−1,bknr(1CT–GS) ∼ 108–1010 s−1c (ref. 3, 9, 58 and 59), kr(1CT–GS) ∼ 106 s−1,bkISC(1CT–3CT)/kISC(3CT–1CT) ∼ 105–106 s−1,bk(3CT–3LE) ∼ 1011–1012 s−1,bk(3LE–3CT) ∼ 108–1010 s−1c (ref. 43), k(3LE–GS) ∼ 105–106 s−1c (ref. 43), a experimental values, b calculated in this work, c calculated in this work based on the experimental state energies. | ||
To compute the rate constants for the exciton dissociation and geminate recombination processes, we had first to evaluate the related electronic couplings and electron-vibrational couplings, see Sections SF and SH in the ESI.† The calculated rate constants are collected in Table 2. Importantly, when deriving the rate constants, we accounted for all relevant initial and final states. For instance, in the case of exciton dissociation, we considered the transition pathways from both LE1 and LE2 states to both CT1 and CT2 states. We also assumed that the LE1 and LE2 states are in thermal equilibrium. In that framework, the exciton dissociation rate constant for the LE → CT transition is estimated to be about 1012 s−1; this value is only slightly smaller for the EX → CT transition (see Table 2). Thus, the calculated rate constants for the formation of both CT1 and CT2 state are very fast. As mentioned above, the electron–hole distance in the CT2 states is larger than that in conventional interfacial CT states, which correspond here to the CT1 states. Since the CT2 states can be regarded as a model representing charge separated (CS) states, our results support the suggestion that the formation of free charge carriers in PM6:Y6 does not necessarily need to proceed via conventional interfacial CT states involving single D and A molecules.53 Clearly, calculations going beyond Y6 dimers are needed to get a better understanding of this issue.
| Rates (s−1) | PM6:Y6 | PM6:2Y6 | PM6:Y6 [+static disorder] |
|---|---|---|---|
| k(1LE–1CT) | 5.05 × 1012 | 7.88 × 1011 | 5.94 × 1012 |
| k(1CT–1LE) | 3.21 × 109 | 2.14 × 1010 | 5.77 × 1010 |
| k nr(1CT–GS) | 3.15 × 108 | 3.32 × 108 | 4.65 × 108 |
| k r(1CT–GS) | 6.31 × 106 | 4.97 × 106 | — |
The calculated energy offset between the LE and CT states, ΔELE–CT, is 0.17 eV and reduces to 0.09 eV in the case of ΔEEX–CT. These relatively small energy offsets lead expectedly to large back electron transfer rate constants for the CT → LE(EX) transitions; indeed, according to our calculations, the rate constant of 1LE-state repopulation from the 1CT states is about 109 s−1; this value further increases to 1010 s−1 in the case of EX states (see Table 2). This result is in fact very important as having a large back electron transfer rate constant is an important factor that acts to reduce the non-radiative voltage losses in organic solar cells.60
The geminate recombination of the 1CT state to GS can occur through both nonradiative and radiative processes. The nonradiative decay rate constant for the 1CT → GS process is calculated to be about 3.0 × 108 s−1, irrespective of whether PM6:Y6 or PM6:2Y6 complexes are considered. We recall that the CT energy in the PM6:Y6 blend is measured experimentally to fall in the range 1.3–1.4 eV.3,9,58,59 Given the exponential dependence of knr(1CT–GS) on the CT energy, we have also estimated the rate by taking 1.3 eV as the energy of the CT state instead of 1.63 eV. In the case of PM6:2Y6 complexes, the decrease in CT energy leads to an increase in knr from 3.3 × 108 s−1 to 2.1 × 1010 s−1. On the other hand, based on calculations performed on both PM6:Y6 and PM6:2Y6 complexes, the radiative rate constant (kr(1CT–GS)) is estimated to be about 5.0 × 106 s−1 (see Table 2). This value decreases by about 50% if the DFT-calculated value for the CT energy is replaced with 1.3 eV, as discussed above for the case of knr.
The non-radiative decay of the 1CT1 state can also take place via intersystem crossing (ISC) transitions to the triplet CT and LE states. ISC can be induced by both spin–orbit and hyperfine interactions. Our DFT calculations indicate that the hyperfine coupling constants in the present systems are about two orders of magnitude smaller than the spin–orbit couplings (SOCs); therefore, only the second mechanism is further discussed. The SOCs computed between the 1CT1 and 3LE states and between the 1CT1 and 3CT states are shown in Fig. 8 in the case of PM6:2Y6 complexes (see also Fig. S22 in the ESI†). The SOC between 1CT1 and 3CT1 is about 0.5 cm−1 (∼0.007 meV) and nearly doubles between the 1CT1 and triplet EX(LE) states. These values are of the same order of magnitude as those found between the lowest singlet and triplet excited states in efficient TADF (thermally activated delayed fluorescence) molecules developed for organic light-emitting diodes.61–63 We stress that the ISC between the 1CT and 3LE states is associated with the same type of geometry reorganizations as in the case of electron transfer between the CT and LE states. Thus, in the calculations of the ISC rate constants between the CT and LE states, the density of the final states (ρf) is approximated by the Franck–Condon-weighted density of states (FCWD) as in electron transfer transitions. In contrast, the ISC from 1CT1 to 3CT1 does not involve any significant geometry reorganization; as a result, defining an FCWD factor in this instance is not feasible. Therefore, in this case, we approximated ρf by ΓT−1 where ΓT is the intrinsic linewidth of the 3CT1 level, which, based on the computed rate constants, is estimated to be 2.0 × 10−4 eV. Following this procedure, we derived rate values of 1.0 × 105 s−1 and 4.0 × 106 s−1 for kISC(1CT1–3CT1) and kISC(1CT1–3LE), respectively. Experimental studies on a series of D:A exciplexes have indicated that kISC values are generally about 106 s−1; also, it was suggested that 1CT → 3LE transitions are faster than the 1CT → 3CT transition.64 Our results are in good agreement with these experimental data. Thus, our calculations indicate that the ISC processes do not contribute significantly to the non-radiative decay of the singlet CT states. As a result, the population of the 1CT1 state is mainly defined by the non-radiative 1CT → 1LE and 1CT → GS transitions as well as by the dissociation of 1CT1 into charge-separated states (1CT → CS); in the PM6:Y6 blend, the latter process was reported to be as fast as 5.5 × 1010 s−1.65 We computed the electron transfer rate from the Y6 molecule in an PM6:Y6 complex to an adjacent Y6 molecule (which corresponds to the initial electron hopping event in the dissociation of the CT state into a CS state). We found rate constants on the order of 1.1 × 1011 s−1 and 1.0 × 1012 s−1 depending on whether electrostatic interactions are or are not taken into account, respectively.
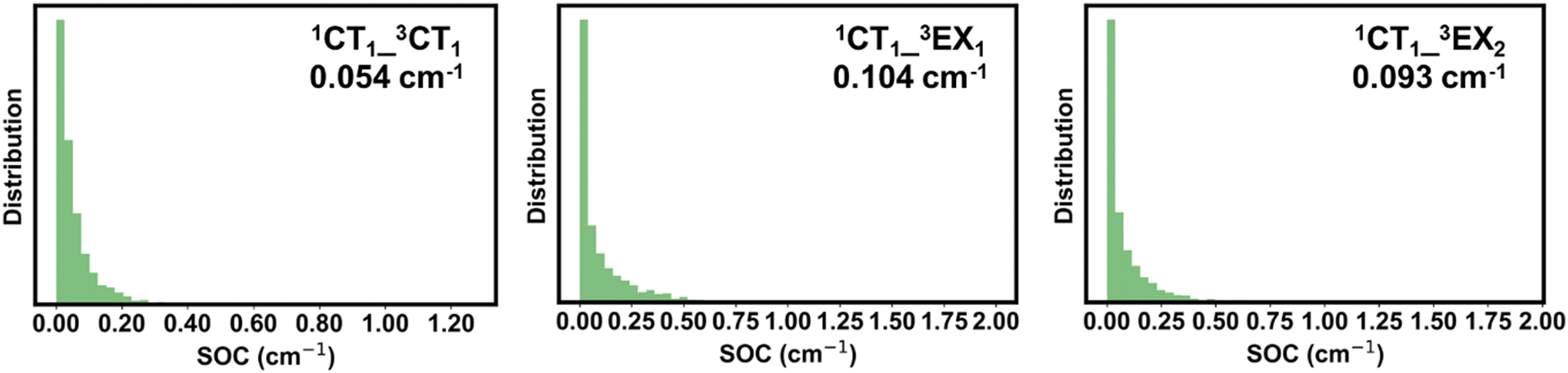 | ||
| Fig. 8 Distributions of spin–orbit couplings (SOC) between the singlet and triplet CT states, and between the singlet CT and triplet EX states in the PM6:2Y6 complexes. The corresponding average values are also given. | ||
We also estimated the impact of static disorder on the rate constants of the non-radiative processes (see Table 2); we note that the radiative transitions are generally only marginally affected by disorder.33 As seen from Table 2, static disorder can alter the non-radiative rates by a factor of up to two times. Thus, the account of static disorder leads to an increase of knr(1CT–GS) by about 50%. If we now take: (i) the value of 3.2 × 106 s−1 for kr(1CT–GS) as computed for PM6:2Y6 complexes but using the experimental value for the CT energy, and (ii) value of 4.8 × 1010 s−1 for knr obtained upon consideration of the disorder effect, we then estimate that the intrinsic photoluminescence quantum efficiency of the 1CT state is about 6.7 × 10−5. This result aligns closely with previously reported values from external electroluminescence quantum efficiency measurements, which range from 2.7 × 10−5 to 4.8 × 10−5.4,6,22
(vi) Non-geminate recombination and the impact of 3LE states
The non-geminate recombination of free charge carriers results initially in the formation of 3CT and 1CT states in the 3:1 ratio expected from spin statistics. As discussed above, the 1CT states can then either transition to 1LE, decay to the GS, undergo ISC transitions, or re-dissociate into free charges. In the case of 3CT states, in addition to re-dissociation and ISC processes, they can also undergo transitions to low-energy triplet LE(EX) states (see Fig. 7). The impact of these local triplet states on the performance of OSCs is a subject of ongoing debates. On the one hand, Friend and co-workers suggested that the decay via local triplet states is responsible for 90% of the non-radiative voltage losses in PM6:Y6 blends.4 On the other hand, recent transient absorption and external quantum efficiency measurements by Tamai and co-workers provide experimental evidence for efficient charge generation from Y6 triplet excitons.66
As described above, our electronic-structure calculations indicate that there are two triplet LE(EX) states located below or near the 3CT1 state. The calculations also show that 3CT1 → 3LE transitions are characterized by very large rate constants in the 1011–1012 s−1 range (5.2 × 1011 s−1 in PM6:2Y6 and 3.3 × 1012 s−1 in PM6:Y6). If instead of the DFT estimated triplet LE and CT energies, we take those reported experimentally, (i.e., E(3CT1) = 1.3 eV and E(3LE1) = 1.14–1.26 eV),43k(3CT–3LE) is only slightly lower, in the range 4.0 × 1011 s−1–2.1 × 1011 s−1.
Once the triplet LE(EX) states are formed, they can either decay to GS, perform a back transition to the 3CT states, or decay via triplet exciton-triplet exciton annihilation (TTA) or triplet exciton-charge annihilation (TCA) processes. Since under one-sun normal illumination conditions the density of triplets is very low, it was reported that TTA is unlikely to affect the performance of OSCs based on PM6:Y6 active layers.67 Our calculations (using the experimental LE state energies) yield for the 3LE1 → GS decay rate constant values that are in the range 3.4 × 105 s−1–6.0 × 104 s−1. Much larger rate constants, 4.3 × 1010 s−1–4.2 × 108 s−1 (see Fig. S24 in the ESI†), are estimated for the 3LE1 → 3CT1 transition, pointing to a very short lifetime for the triplet Y6 excitons in the PM6:Y6 blends. This result is in good agreement with the transient absorption data reported by Friend and co-workers;4 however, the decay of triplet Y6 excitons was attributed to a terminal TCA mechanism that ultimately led the authors to conclude that the decay via local triplet states is responsible for 90% of the non-radiative voltage losses in PM6:Y6. Although we cannot provide at this stage an estimate of TCA decay efficiency, it is reasonable to assume that under one-sun normal illumination conditions this process plays overall a marginal role in the device performance for a reason similar to that discussed above for the TTA process. Therefore, we posit that 3LE1 states in PM6:Y6 are actually not dead-end states since they can undergo fast back transfer to 3CT states and eventually separate back into free charges (CS → 3CT1 → 3LE1 → 3CT1 → CS) or revert to 1CT states (CS → 3CT1 → 3LE1 → 3CT1 → 1CT1). If the latter occurs, the 1CT states will follow the evolutions we described earlier. Therefore, under open-circuit conditions, such cycles could repeat several times until the charges fully separate or recombine (predominantly via the hybrid manifold of singlet LE–CT states). Overall, our results underline that charge separation from Y6 triplet excitons at the PM6:Y6 interface can occur when the energy offset between 3LE1 and 3CT is small, which is in agreement with the recent experimental data from Tamai and co-workers.66
3. Conclusions
Using a combination of classical molecular dynamics simulations and long-range corrected density functional theory calculations, we conducted an in-depth analysis of the charge generation and recombination processes within the PM6:Y6 blend, a representative system for high-performance organic photovoltaics. The main results of our calculations are as follows:• The exciton dissociation rate is extremely fast, on the order of 1012 s−1. This rate is not significantly affected by the presence of excimer-like states. However, the excimer-like states can efficiently form not only conventional interfacial CT states but also states with larger hole–electron separation distances. These results point out that the formation of free charge carriers in PM6:Y6 can take place from delocalized excitonic states without forming first conventional (i.e., single D:single A) interfacial CT states. However, the exact impact of exciton delocalization on charge separation within acceptor (or donor) domains and in donor:acceptor blends needs further investigations.
• The geminate recombination of the 1CT state to the ground state reaches ∼1010 s−1 and can contribute to non-radiative voltage losses. However, the back electron transfer from the 1CT state to the 1LE/1EX state is nearly equally fast (∼109 s−1–1010 s−1) and can act to substantially reduce these losses. Also, the static and dynamic disorders in PM6:Y6 blends were quantified; it is found that static disorder can increase the 1CT–GS non-radiative decay rates by about 50%.
• We identified low-energy defect states associated with the appearance of large dihedral angles between the core and terminal moieties of Y6 molecules, which could arise during thermal annealing processes. These defects could act as low-energy sinks and/or affect the chemical stability of Y6, ultimately reducing the overall device performance. Therefore, the use of more rigid molecules might be critical to improve the stability of the OSC active layers.
• While the non-geminate formation of 3LE states occurs at a high rate (∼1011 s−1 to 1012 s−1), these 3LE states are not necessarily terminal losses. Indeed, we obtain that these states can cycle back into 3CT states, taking advantage of small 3LE–3CT energy offsets, and eventually dissociate into free charges. However, to fully prevent non-radiative losses via local triplet states, blends would need to be designed in which the triplet states are located just above the CT states while maintaining a high CT energy. Conversely, the transition from CT states to local triplet states could also be minimized if the triplet energy is very low so that the electron transfer process takes place in the (unfavorable) inverted Marcus regime; a drawback could be, however, that photostability issues become more prominent in such systems.
• The present findings point to the CT state as the primary source of non-radiative loss in the PM6:Y6 blend. This calls for further investigations on how to reduce non-radiative recombinations from the CT states. A possibility is to use more rigid D and A components with extended π-conjugated backbones to reduce the electronic coupling with high-frequency vibrations; this coupling is known to be a major factor promoting non-radiative transitions. At this stage, it also remains unclear why adding a second A or D component in ternary blends can in some instances reduce the voltage losses. Further investigations of all these points are warranted to advance the field of organic photovoltaics.
To summarize, this comprehensive theoretical analysis of charge generation and recombination in PM6:Y6 active-layers not only advances our understanding of the electronic processes in organic solar cells but also provides a valuable framework for the design and optimization of next-generation high-performance devices.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Office of Naval Research, Awards No. N00014-20-1-2110 and N00014-24-1-2114, and the College of Science of the University of Arizona for funding this work. We are also grateful to the University of Arizona Institute of Energy Solutions and Office for Research, Innovation, & Impact for support via the Arizona Technology and Research Initiative Fund. The computational work was supported in part by a grant of computer time from the DOD High Performance Computing Modernization Program.References
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- S. Shoaee, H. M. Luong, J. Song, Y. Zou, T.-Q. Nguyen and D. Neher, Adv. Mater., 2024, 36, 2302005 CrossRef CAS.
- P. Wan, X. Chen, Q. Liu, S. Mahadevan, M. Guo, J. Qiu, X. Sun, S.-W. Tsang, M. Zhang, Y. Li and S. Chen, J. Phys. Chem. Lett., 2021, 12, 10595–10602 CrossRef CAS.
- A. J. Gillett, A. Privitera, R. Dilmurat, A. Karki, D. Qian, A. Pershin, G. Londi, W. K. Myers, J. Lee, J. Yuan, S.-J. Ko, M. K. Riede, F. Gao, G. C. Bazan, A. Rao, T.-Q. Nguyen, D. Beljonne and R. H. Friend, Nature, 2021, 597, 666–671 CrossRef CAS.
- D. Neusser, B. Sun, W. L. Tan, L. Thomsen, T. Schultz, L. Perdigón-Toro, N. Koch, S. Shoaee, C. R. McNeill, D. Neher and S. Ludwigs, J. Mater. Chem. C, 2022, 10, 11565–11578 RSC.
- L. Perdigón-Toro, L. Q. Phuong, S. Zeiske, K. Vandewal, A. Armin, S. Shoaee and D. Neher, ACS Energy Lett., 2021, 6, 557–564 CrossRef.
- D. Qian, S. M. Pratik, Q. Liu, Y. Dong, R. Zhang, J. Yu, N. Gasparini, J. Wu, T. Zhang, V. Coropceanu, X. Guo, M. Zhang, J.-L. Bredas, F. Gao and J. R. Durrant, Adv. Energy Mater., 2023, 13, 2301026 CrossRef CAS.
- N. Tokmoldin, C. Deibel, D. Neher and S. Shoaee, Adv. Energy Mater., 2024, 14, 2401130 CrossRef CAS.
- L. Perdigón-Toro, H. Zhang, A. Markina, J. Yuan, S. M. Hosseini, C. M. Wolff, G. Zuo, M. Stolterfoht, Y. Zou, F. Gao, D. Andrienko, S. Shoaee and D. Neher, Adv. Mater., 2020, 32, 1906763 CrossRef.
- Z. Tu, G. Han and Y. Yi, J. Phys. Chem. Lett., 2020, 11, 2585–2591 CrossRef CAS.
- K. Jiang, Q. Wei, J. Y. L. Lai, Z. Peng, H. K. Kim, J. Yuan, L. Ye, H. Ade, Y. Zou and H. Yan, Joule, 2019, 3, 3020–3033 CrossRef CAS.
- Y. Firdaus, V. M. Le Corre, S. Karuthedath, W. Liu, A. Markina, W. Huang, S. Chattopadhyay, M. M. Nahid, M. I. Nugraha, Y. Lin, A. Seitkhan, A. Basu, W. Zhang, I. McCulloch, H. Ade, J. Labram, F. Laquai, D. Andrienko, L. J. A. Koster and T. D. Anthopoulos, Nat. Commun., 2020, 11, 5220 CrossRef CAS PubMed.
- C. Zhu, J. Yuan, F. Cai, L. Meng, H. Zhang, H. Chen, J. Li, B. Qiu, H. Peng, S. Chen, Y. Hu, C. Yang, F. Gao, Y. Zou and Y. Li, Energy Environ. Sci., 2020, 13, 2459–2466 RSC.
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma, C. An, C. He, Z. Wei, F. Gao and J. Hou, Adv. Mater., 2020, 32, 1908205 CrossRef CAS.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- Z. Chen, J. Ge, W. Song, X. Tong, H. Liu, X. Yu, J. Li, J. Shi, L. Xie, C. Han, Q. Liu and Z. Ge, Adv. Mater., 2024, 36, 2406690 CrossRef CAS.
- Y. Jiang, S. Sun, R. Xu, F. Liu, X. Miao, G. Ran, K. Liu, Y. Yi, W. Zhang and X. Zhu, Nat. Energy, 2024, 9, 975–986 CrossRef CAS.
- Z. Sun, H. Ma, S. Yang, Y. Cho, S. Lee, J. Park, T. L. H. Mai, W. Kim, S. Jeong, S. Kim and C. Yang, Adv. Funct. Mater., 2024, 34, 2403093 CrossRef CAS.
- Y. Cho, B. Lee, S. Jung, S. Jeong, J. Park, G. Park, S. Yang and C. Yang, Energy Environ. Sci., 2023, 16, 6035–6045 RSC.
- J. Yao, T. Kirchartz, M. S. Vezie, M. A. Faist, W. Gong, Z. He, H. Wu, J. Troughton, T. Watson, D. Bryant and J. Nelson, Phys. Rev. Appl., 2015, 4, 014020 CrossRef.
- V. Coropceanu, X.-K. Chen, T. Wang, Z. Zheng and J.-L. Brédas, Nat. Rev. Mater., 2019, 4, 689–707 CrossRef.
- S. Liu, J. Yuan, W. Deng, M. Luo, Y. Xie, Q. Liang, Y. Zou, Z. He, H. Wu and Y. Cao, Nat. Photonics, 2020, 14, 300–305 CrossRef CAS.
- H. Lu, W. Liu, H. Jin, H. Huang, Z. Tang and Z. Bo, Adv. Funct. Mater., 2022, 32, 2107756 CrossRef CAS.
- M. A. Green, E. D. Dunlop, J. Hohl-Ebinger, M. Yoshita, N. Kopidakis and A. W. Y. Ho-Baillie, Prog. Photovoltaics Res. Appl., 2020, 28, 3–15 CrossRef.
- J. J. Yoo, S. Wieghold, M. C. Sponseller, M. R. Chua, S. N. Bertram, N. T. P. Hartono, J. S. Tresback, E. C. Hansen, J.-P. Correa-Baena, V. Bulović, T. Buonassisi, S. S. Shin and M. G. Bawendi, Energy Environ. Sci., 2019, 12, 2192–2199 RSC.
- G. Yang, Z. Ren, K. Liu, M. Qin, W. Deng, H. Zhang, H. Wang, J. Liang, F. Ye, Q. Liang, H. Yin, Y. Chen, Y. Zhuang, S. Li, B. Gao, J. Wang, T. Shi, X. Wang, X. Lu, H. Wu, J. Hou, D. Lei, S. K. So, Y. Yang, G. Fang and G. Li, Nat. Photonics, 2021, 15, 681–689 CrossRef CAS.
- B. M. Kayes, H. Nie, R. Twist, S. G. Spruytte, F. Reinhardt, I. C. Kizilyalli and G. S. Higashi, 2011 37th IEEE Photovoltaic Specialists Conference, IEEE, Seattle, WA, USA, 2011.
- Q. Liu and K. Vandewal, Adv. Mater., 2023, 35, 2302452 CrossRef CAS.
- X. Yu, P. Ding, D. Yang, P. Yan, H. Wang, S. Yang, J. Wu, Z. Wang, H. Sun, Z. Chen, L. Xie and Z. Ge, Angew. Chem., Int. Ed., 2024, 63, e202401518 CrossRef CAS.
- G. Wang, J. Wang, Y. Cui, Z. Chen, W. Wang, Y. Yu, T. Zhang, L. Ma, Y. Xiao, J. Qiao, Y. Xu, X.-T. Hao and J. Hou, Angew. Chem., Int. Ed., 2024, 63, e202401066 CrossRef CAS PubMed.
- M. Azzouzi, N. P. Gallop, F. Eisner, J. Yan, X. Zheng, H. Cha, Q. He, Z. Fei, M. Heeney, A. A. Bakulin and J. Nelson, Energy Environ. Sci., 2022, 15, 1256–1270 RSC.
- Z. Zheng, N. R. Tummala, T. Wang, V. Coropceanu and J.-L. Brédas, Adv. Energy Mater., 2019, 9, 1803926 CrossRef.
- Y. Fu, T. H. Lee, Y.-C. Chin, R. A. Pacalaj, C. Labanti, S. Y. Park, Y. Dong, H. W. Cho, J. Y. Kim, D. Minami, J. R. Durrant and J.-S. Kim, Nat. Commun., 2023, 14, 1870 CrossRef CAS PubMed.
- T. A. Dela Peña, R. Ma, Z. Xing, Q. Wei, J. I. Khan, R. M. Young, Y. Hai, S. A. Garcia, X. Zou, Z. Jin, F. L. Ng, K. L. Yeung, D. F. Swearer, M. R. Wasielewski, J. Wang, H. Cha, H. Yan, K. S. Wong, G. Li, M. Li and J. Wu, Energy Environ. Sci., 2023, 16, 3416–3429 RSC.
- G. Kupgan, X. K. Chen and J. L. Brédas, Mater. Today Adv., 2021, 11, 100154 CrossRef CAS.
- T. Wang, G. Kupgan and J.-L. Brédas, Trends Chem., 2020, 2, 535–554 CrossRef CAS.
- L. Perdigón-Toro, L. Q. Phuong, F. Eller, G. Freychet, E. Saglamkaya, J. I. Khan, Q. Wei, S. Zeiske, D. Kroh, S. Wedler, A. Köhler, A. Armin, F. Laquai, E. M. Herzig, Y. Zou, S. Shoaee and D. Neher, Adv. Energy Mater., 2022, 12, 2103422 CrossRef.
- W. Li, J. Li and M. Sun, Phys. Chem. Chem. Phys., 2023, 25, 9807–9816 RSC.
- S. Karuthedath, J. Gorenflot, Y. Firdaus, N. Chaturvedi, C. S. P. De Castro, G. T. Harrison, J. I. Khan, A. Markina, A. H. Balawi, T. A. D. Peña, W. Liu, R.-Z. Liang, A. Sharma, S. H. K. Paleti, W. Zhang, Y. Lin, E. Alarousu, S. Lopatin, D. H. Anjum, P. M. Beaujuge, S. De Wolf, I. McCulloch, T. D. Anthopoulos, D. Baran, D. Andrienko and F. Laquai, Nat. Mater., 2021, 20, 378–384 CrossRef CAS PubMed.
- N. R. Tummala, Z. Zheng, S. G. Aziz, V. Coropceanu and J.-L. Brédas, J. Phys. Chem. Lett., 2015, 6, 3657–3662 CrossRef CAS.
- N. R. Tummala, S. A. Elroby, S. G. Aziz, C. Risko, V. Coropceanu and J.-L. Brédas, J. Phys. Chem. C, 2016, 120, 17242–17250 CrossRef CAS.
- S.-i Natsuda, T. Saito, R. Shirouchi, Y. Sakamoto, T. Takeyama, Y. Tamai and H. Ohkita, Energy Environ. Sci., 2022, 15, 1545–1555 RSC.
- P. Li, J. Fang, Y. Wang, S. Manzhos, L. Cai, Z. Song, Y. Li, T. Song, X. Wang, X. Guo, M. Zhang, D. Ma and B. Sun, Angew. Chem., Int. Ed., 2021, 60, 15054–15062 CrossRef CAS PubMed.
- S. M. Hosseini, S. Wilken, B. Sun, F. Huang, S. Y. Jeong, H. Y. Woo, V. Coropceanu and S. Shoaee, Adv. Energy Mater., 2023, 13, 2203576 CrossRef CAS.
- S.-U.-Z. Khan, J. Bertrandie, M. Gui, A. Sharma, W. Alsufyani, J. F. Gorenflot, F. Laquai, D. Baran and B. P. Rand, Joule, 2022, 6, 2821–2834 CrossRef CAS.
- K. Tvingstedt, J. Benduhn and K. Vandewal, Mater. Horiz., 2020, 7, 1888–1900 RSC.
- C. Göhler and C. Deibel, ACS Energy Lett., 2022, 7, 2156–2164 CrossRef.
- G. J. Thapa, M. Chauhan, J. P. Mauthe, D. B. Dougherty and A. Amassian, arXiv, 2024, preprint, arXiv:2406.11735 DOI:10.48550/arXiv.2406.11735.
- A. J. Clarke, J. Luke, R. Meitzner, J. Wu, Y. Wang, H. K. H. Lee, E. M. Speller, H. Bristow, H. Cha, M. J. Newman, K. Hooper, A. Evans, F. Gao, H. Hoppe, I. McCulloch, U. S. Schubert, T. M. Watson, J. R. Durrant, W. C. Tsoi, J.-S. Kim and Z. Li, Cell Rep. Phys. Sci., 2021, 2, 100498 CrossRef CAS.
- J. Luke, E. M. Speller, A. Wadsworth, M. F. Wyatt, S. Dimitrov, H. K. H. Lee, Z. Li, W. C. Tsoi, I. McCulloch, D. Bagnis, J. R. Durrant and J.-S. Kim, Adv. Energy Mater., 2019, 9, 1803755 CrossRef.
- X. Du, L. Lüer, T. Heumueller, J. Wagner, C. Berger, T. Osterrieder, J. Wortmann, S. Langner, U. Vongsaysy, M. Bertrand, N. Li, T. Stubhan, J. Hauch and C. J. Brabec, Joule, 2021, 5, 495–506 CrossRef CAS.
- R. Wang, C. Zhang, Q. Li, Z. Zhang, X. Wang and M. Xiao, J. Am. Chem. Soc., 2020, 142, 12751–12759 CrossRef CAS PubMed.
- M. B. Price, P. A. Hume, A. Ilina, I. Wagner, R. R. Tamming, K. E. Thorn, W. Jiao, A. Goldingay, P. J. Conaghan, G. Lakhwani, N. J. L. K. Davis, Y. Wang, P. Xue, H. Lu, K. Chen, X. Zhan and J. M. Hodgkiss, Nat. Commun., 2022, 13, 2827 CrossRef CAS.
- S. Mahadevan, T. Liu, S. M. Pratik, Y. Li, H. Y. Ho, S. Ouyang, X. Lu, H.-L. Yip, P. C. Y. Chow, J.-L. Brédas, V. Coropceanu, S. K. So and S.-W. Tsang, Nat. Commun., 2024, 15, 2393 CrossRef CAS PubMed.
- M. Pranav, A. Shukla, D. Moser, J. Rumeney, W. Liu, R. Wang, B. Sun, S. Smeets, N. Tokmoldin, Y. Cao, G. He, T. Beitz, F. Jaiser, T. Hultzsch, S. Shoaee, W. Maes, L. Lüer, C. Brabec, K. Vandewal, D. Andrienko, S. Ludwigs and D. Neher, Energy Environ. Sci., 2024, 17, 6676–6697 RSC.
- X.-K. Chen, C. Chan, S. Mahadevan, Y. Guo, G. Zhang, H. Yan, K. S. Wong, H.-L. Yip, J.-L. Bredas, S. W. Tsang and P. C. Y. Chow, arXiv, 2024, preprint, arXiv:2304.09408 DOI:10.48550/arXiv.2304.09408.
- A. Karki, J. Vollbrecht, A. L. Dixon, N. Schopp, M. Schrock, G. N. M. Reddy and T.-Q. Nguyen, Adv. Mater., 2019, 31, 1903868 CrossRef CAS.
- Q. Liu, Y. Wang, J. Fang, H. Liu, L. Zhu, X. Guo, M. Gao, Z. Tang, L. Ye, F. Liu, M. Zhang and Y. Li, Nano Energy, 2021, 85, 105963 CrossRef CAS.
- X.-K. Chen, D. Qian, Y. Wang, T. Kirchartz, W. Tress, H. Yao, J. Yuan, M. Hülsbeck, M. Zhang, Y. Zou, Y. Sun, Y. Li, J. Hou, O. Inganäs, V. Coropceanu, J.-L. Bredas and F. Gao, Nat. Energy, 2021, 6, 799–806 CrossRef CAS.
- P. K. Samanta, D. Kim, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS.
- E. Cho, M. Hong, V. Coropceanu and J.-L. Brédas, Adv. Opt. Mater., 2021, 9, 2002135 CrossRef CAS.
- S. M. Pratik, V. Coropceanu and J.-L. Brédas, ACS Mater. Lett., 2022, 440–447, DOI:10.1021/acsmaterialslett.1c00809.
- I. R. Gould, J. A. Boiani, E. B. Gaillard, J. L. Goodman and S. Farid, J. Phys. Chem. A, 2003, 107, 3515–3524 CrossRef CAS.
- C. C. S. Chan, C. Ma, X. Zou, Z. Xing, G. Zhang, H.-L. Yip, R. A. Taylor, Y. He, K. S. Wong and P. C. Y. Chow, Adv. Funct. Mater., 2021, 31, 2107157 CrossRef CAS.
- K. Kohzuki, R. Shirouchi, S.-I. Natsuda, T. Saito, Y. Sakamoto and Y. Tamai, Sol. RRL, 2023, 7, 2300359 CrossRef CAS.
- L. J. F. Hart, J. Grüne, W. Liu, T.-K. Lau, J. Luke, Y.-C. Chin, X. Jiang, H. Zhang, D. J. C. Sowood, D. M. L. Unson, J.-S. Kim, X. Lu, Y. Zou, F. Gao, A. Sperlich, V. Dyakonov, J. Yuan and A. J. Gillett, Adv. Energy Mater., 2023, 13, 2301357 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details; distribution of the singlet and triplet energies of Y6 and 2Y6; distribution of charges for singlet and triplet states in PM6:Y6, PM6:2Y6, and 2Y6, singlet and triplet energies and their LE(EX) or CT nature; disorders; origin of the defect states; distributions of electronic couplings; parameters for rate calculations; and other relevant data. See DOI: https://doi.org/10.1039/d4ee03815k |
| This journal is © The Royal Society of Chemistry 2025 |