
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioremediation of uranium contaminated sites through the formation of U(VI) phosphate (bio)minerals†
Callum
Robinson
 a,
Sam
Shaw
a,
Jonathan R.
Lloyd
a,
James
Graham
b and
Katherine
Morris
*a
a,
Sam
Shaw
a,
Jonathan R.
Lloyd
a,
James
Graham
b and
Katherine
Morris
*a
aRAdioactive waste Disposal and Environmental Remediation (RADER) Facility and Williamson Research Facility, Department of Earth and Environmental Sciences, The University of Manchester, Manchester, M13 9PL, UK. E-mail: katherine.morris@manchester.ac.uk
bNational Nuclear Laboratory, Central Laboratory, Sellafield, Seascale, Cumbria, CA20 1PG, UK
First published on 3rd February 2025
Abstract
Operations at uranium (U)-mining and nuclear facilities have left a global legacy of significant radionuclide contamination in groundwaters which must be managed to minimize environmental harm. Uranium groundwater contamination is present at several sites globally, including Oak Ridge National Laboratory and Hanford, USA and Sellafield nuclear site, UK. In situ phosphate biomineralisation offers a promising method for radionuclide (including 90Sr and U) remediation at these sites. Typically, phosphate-generating amendments are injected into the subsurface to sequester select radionuclides in groundwaters by precipitation of poorly soluble Ca-phosphate phases and subsequent adsorption and/or incorporation of radionuclides to these poorly soluble phases, a remediation route being explored for both U and 90Sr. In this study, we investigate the mechanisms of U-phosphate precipitation in two phosphate-generating amendments (Ca-citrate/Na-phosphate and glycerol phosphate) under conditions relevant to Sellafield, UK. Using aerobic batch sediment experiments, we show both Ca-citrate/Na-phosphate and glycerol phosphate amendments are effective at enhancing removal of U(VI) from representative groundwaters (from 94% to >97%). Aqueous geochemical data coupled to speciation modelling highlighted that precipitation of U(VI) phosphate phases was the likely mechanism of U(VI) removal from groundwaters. Further X-ray absorption spectroscopy (XAS) analysis of solids confirmed U was present as a highly insoluble uranyl orthophosphate-like phase after treatment with both Ca-citrate/Na-phosphate and glycerol phosphate amendments. These data provide underpinning information on U-phosphate remediation in Sellafield relevant conditions thus expanding the range of treatment options for radionuclide contaminated groundwaters and defining the transport and fate of U during phosphate biomineralisation.
Water impactGlobally, U-contaminated groundwater poses a significant hazard at several nuclear sites and developing credible remediation technologies is vital for decommissioning operations. In this study, we broaden the treatment envelope for in situ biomineralisation technologies; Ca-citrate/Na-phosphate and glycerol phosphate, to remediate U(VI) from oxic groundwaters, through the formation of low solubility U(VI) phosphate biominerals. |
1. Introduction
Globally, production of nuclear weapons and nuclear power over the last 70+ years has led to a legacy of radioactively contaminated land at a number of nuclear facilities including Sellafield, UK and Hanford, USA which are contaminated from nuclear fuel cycle operations such as reprocessing and waste management. At these sites, subsurface sediments and groundwaters are contaminated with several different radionuclides.1–3 Subsurface migration of radionuclides, typically from a point source of contamination into a dilute groundwater, may pose a significant environmental hazard and needs to be proactively managed, often with non-invasive or in situ approaches as the subsurface is often inaccessible. Here, U is an important contaminant as it is typically the most significant radionuclide by mass at nuclear facilities and U-mining sites.3 U mobility in the environment is largely controlled by redox and pH conditions. Under oxic conditions U(VI) dominates, typically as the relatively mobile UO22+ uranyl cation under acidic to circumneutral pH.4 At circumneutral to alkaline pH, carbonate present in groundwaters can form aqueous U(VI) carbonate complexes (e.g. [UO2(CO3)3]4−(aq)) potentially enhancing U solubility.5 Additionally, complexation by ligands such as citrate6 and humic acids7 as well as cation (e.g. Ca2+) competition for adsorbed sites on subsurface sediments may also enhance the solubility and mobility of U.8 For radionuclide transport, sorption of U(VI) in oxic subsurface environments typically dominates U removal from solution via the formation of sorption complexes to geomedia (e.g. Fe oxides, clays and organics).4,9–11 These sorption complexes may be susceptible to facile U-remobilization if conditions change and typically, incorporated or precipitated phases are considered more recalcitrant to longer term transport. By contrast, under anoxic conditions poorly soluble U(IV) phases such as UO2 (uraninite) and nanoparticulate U(IV) phases dominate.4,5 Indeed, anoxic conditions have been extensively explored for treatment of U(VI)-groundwater contamination, and can be stimulated through in situ remediation approaches.4 For example, bioreduction technologies have extensively explored at field scale in the remediation of U-contaminated land at the Rifle field site. Here, several campaigns were undertaken where the subsurface was amended with acetate injections to simulate the reduction of U(VI) to U(IV) by metal-reducing bacteria.12–14 Typically, when the active injections stopped, a gradual increase in U concentrations in groundwaters was observed due to oxidative remobilization of U(IV) to U(VI) during groundwater recharge.14 Overall, the poor resistance of bioreduced U(IV) phases to oxidative remobilization over the longer term15,16 makes exploration of other end-states for U in oxic contaminated land desirable. Past studies have focused on improving the stability of in situ bioreduced U(IV) phases to re-oxidation through incorporation and precipitation of U(IV) mineral phases including phosphates (e.g. U(IV) bearing ningyoite-like phases16) and Fe(II) bearing phases (e.g. magnetite).17 Whilst incorporation of U(IV) provides additional buffering to oxidation, U(IV) is still susceptible to eventual oxidative remobilization.15–17 Exploring alternative in situ remediation strategies for U(VI) and other co-contaminants under oxic conditions adds to the toolkit of approaches that can be used to tackle these challenges.18Previous studies have focused on deployment of phosphate-mineral generating solution amendments (e.g. Ca- or U-phosphates). Typically, the reagents are injected and subsequently slowly release phosphate into the subsurface leading to the formation of poorly soluble uranyl- and Ca-phosphate phases such as autunite (Ca(UO2)2(PO4)2·10–12H2O)-, uranyl phosphate ((UO2)3(PO4)2(H2O)4)- and hydroxyapatite (Ca10(PO4)6(OH)2)- like precipitates.19–25 These uranyl phosphate phases have been shown to be recalcitrant to re-dissolution under environmental conditions providing a stable end-point which is robust to reoxidation reactions.26 Additionally, Ca-phosphate phases have been shown to enhance U and 90Sr removal from groundwaters across a range of environmental conditions by sorption and/or incorporation of the radionuclide into Ca-phosphates.27–29 For example, polyphosphate amendments have been deployed at Hanford to remediate U contaminated sediments.30 Soluble polyphosphates injected into the subsurface, underwent slow abiotic hydrolysis, releasing free phosphate as PO43− into solution.24,31 The PO43− then reacted with aqueous Ca2+ and mobile U(VI) leading to the formation of recalcitrant uranyl phosphate phases.23,31 U removal in this scenario can be further enhanced by formation of Ca-phosphates which provide additional sorption sites for U(VI) in sediments.25,32–34
In addition to abiotic phosphate treatments, phosphate (bio)remediation approaches are also highly relevant. Microbially mediated degradation of glycerol phosphate has been explored to treat 90Sr, 99Tc and U contamination under both reducing and oxic conditions.16,21,35,36 Indigenous microbes in sediments can release phosphate from glycerol phosphate under oxic conditions using the phosphatase enzyme.4,19,21,37 Aqueous phosphate is then available to react with U(VI) and Ca2+ in solution, forming recalcitrant U(VI)- and Ca-phosphate (bio)minerals.19–21,37 Additionally, Ca-citrate/Na-phosphate amendments have also been explored for U remediation at laboratory scale.22 Here, the Ca-citrate complex is slowly degraded under oxic conditions by citrate-utilizing bacteria, releasing aqueous Ca2+ into solution which can then react with co-injected Na-phosphate to precipitate Ca-phosphates.22,38 Aqueous U(VI) also present within the groundwater, is then removed from solution through both uranyl phosphate precipitation and sorption to Ca-phosphates.22 River water, both an accessible on-site water source, and a potential additional biomass source was shown to enhance the rate of phosphate (bio)mineralization compared to synthetic groundwater only experiments presumably due to enrichment with indigenous microbes.39
Although past work has successfully demonstrated bioremediation of U(VI) contaminated groundwaters through phosphate (bio)mineralization,21,22 the fundamental precipitation pathways and applicability of these technologies across different site conditions is worthy of further research. In this study we explore the potential for Ca-citrate/Na-phosphate and glycerol phosphate bioremediation amendments to sequester U(VI) from oxic batch experiments using both representative Sellafield synthetic groundwater and local river (Calder River) water. U speciation at experimental endpoints was investigated in solids using X-ray absorption spectroscopy. Here, both amendments led to enhanced removal of U(VI) from solution over the 31-day oxic incubation when compared to sediment only controls and U(VI) was sequestered as a highly insoluble uranyl phosphate phase. Our results, combined with past work demonstrating Sr-removal using similar phosphate mineralisation approaches29,35,38,39 offer a positive prospect for co-treatment of U and 90Sr contaminated groundwaters using these in situ approaches.
2. Materials and methods
2.1 U Biomineralisation microcosms
Batch microcosm experiments were set up using well characterised, representative Sellafield sediments sampled from Peel Place quarry; (54°23′49.2′′N 3°25′59.9′′W) characterised as clay poor quaternary outwash sand29,40 and Calder River water (54°26′25.1′′N 3°28′42.2′′W),41 both collected in November 2021. Previous XRD characterization of Peel Place quarry sediments used in the current work showed they were dominated by quartz (SiO2), with some feldspars (albite (NaAlSi3O8)) and mica (muscovite (KAl2(AlSi3O10)(OH)2)), and XRF showed a major elemental composition of SiO2 (90.4%), Al2O3 (4.3%), K2O (1.9%), Fe2O3 (1.4%) MgO (0.7%) and Na2O (0.6%).29 Experiments were set up with sediment and either synthetic groundwater or Calder River water, using a sterile 500 ml conical flask capped with a porous bung and with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 sediment to groundwater ratio (∼400 ml of headspace). The synthetic groundwater comprised in mg l−1; MgSO4·7H2O, 49.5; CaSO4, 9.53; KCl, 5.22; NaCl, 11.7; CaCl2·2H2O, 91.2; NaNO3, 27.2; NaHCO3, 82.3.29 Synthetic groundwater was sterilized by filtration (0.22 μm) and adjusted to pH 6.5 using HCl. U(VI) was added to synthetic groundwater/Calder River water as a uranyl chloride spike in 0.001 M HCl to give a final concentration of 50 μM/12 ppm, which is representative of the upper bracket of reported U concentrations in Sellafield groundwaters.1,36,42,43
:10 sediment to groundwater ratio (∼400 ml of headspace). The synthetic groundwater comprised in mg l−1; MgSO4·7H2O, 49.5; CaSO4, 9.53; KCl, 5.22; NaCl, 11.7; CaCl2·2H2O, 91.2; NaNO3, 27.2; NaHCO3, 82.3.29 Synthetic groundwater was sterilized by filtration (0.22 μm) and adjusted to pH 6.5 using HCl. U(VI) was added to synthetic groundwater/Calder River water as a uranyl chloride spike in 0.001 M HCl to give a final concentration of 50 μM/12 ppm, which is representative of the upper bracket of reported U concentrations in Sellafield groundwaters.1,36,42,43
To investigate U(VI) phosphate bioremediation under oxic conditions, two different treatment options were explored; Ca-citrate/Na-phosphate and glycerol phosphate. For the Ca-citrate/Na-phosphate experiment, concentrated Ca-citrate/Na-phosphate amendment solutions (50 mM CaCl2·2H2O, 125 mM Na3citrate·2H2O and 100 mM Na2HPO4·H2O) were diluted in to experiments with either synthetic groundwater or Calder River water, to give a final amendment concentration of 1 mM Ca2+, 2.5 mM citrate and 10 mM phosphate informed by past work in this area.27,29,44 For glycerol phosphate, 10 mM of 0.22 μm filter sterilized glycerol phosphate was spiked into microcosms with either synthetic groundwater or Calder River water. Microcosms were maintained in the dark at room temperature (approximately 18–22 °C) and periodic sampling at 0, 1, 3, 7, 14, 21 and 31 days. Experiments were run in triplicate and sediment only controls were also prepared containing 50 μM (12 ppm) U in synthetic groundwater and Calder River water.
2.2 Geochemical analysis
Aseptic technique was employed during microcosm sampling and after sampling of the sediment slurry, solids were separated by centrifugation (8000 rpm, 10 minutes). The supernatant was analysed for pH (Jenway 3520, Fisherbrand FB68801 electrode) and aliquots were taken for ion chromatography, inductively coupled plasma mass spectrometry (ICP MS) analysis of U (Agilent 8900) and atomic emission spectroscopy (ICPAES) analysis of Ca (Agilent 5800) with ICP-MS and -AES samples prepared by dilution into 2% HNO3. Experiments were run in triplicate and ICP-MS certified calibration standards were submitted blind alongside blanks containing 2% HNO3 only to ensure analytical precision and error were assessed throughout. Citrate and glycerol phosphate were analyzed using ion chromatography, with samples diluted in deionized water (Dionex ICS 5000). Solution inorganic phosphate concentrations were determined by spectrophotometry.45 Thermodynamic modelling of microcosms was conducted using PHREEQC version 3 (ref. 46) using the ThermoChimie (V10a) database47 using aqueous concentration and pH data from the experimental systems. Solubility constants for U(VI) phosphates and complexation constants for U(VI)-glycerol phosphate were added to PHREEQC using relevant data from the literature.48,492.3 16S rRNA microbial community analysis
DNA was extracted from 0.2 g of sediment slurry using a DNeasy PowerSoil Pro Kit (Qiagen, Manchester, U.K). Sequencing of PCR amplicons of 16S rRNA was conducted with the Illumina MiSeq platform (Illumina, San Diego, CA, USA) targeting the V4 hyper variable region (forward primer, 515F, 5′-GTGYCAGCMGCCGCGGTAA-3′; reverse primer, 806R, 5′-GGACTACHVGGGTWTCTAAT-3′) for 2 × 250-bp paired-end sequencing (Illumina).50,51 PCR amplification was performed using the Roche FastStart High Fidelity PCR System (Roche Diagnostics Ltd, Burgess Hill, UK) in 50 μL reactions under the following conditions: initial denaturation at 95 °C for 2 min, followed by 36 cycles of 95 °C for 30 s, 55 °C for 30 s, 72 °C for 1 min, and a final extension step of 5 min at 72 °C. The PCR products were purified and normalised to ∼20 ng each using the SequalPrep Normalization Kit (Fisher Scientific, Loughborough, UK). The PCR amplicons from all samples were pooled in equimolar ratios. The run was performed using a 4.5 pM sample library spiked with 4.5 pM PhiX to a final concentration of 12% following the method of Schloss and Kozich.52 For QIIME2 analysis, sequences were imported into QIIME2 q2cli v2021.04.53 The sequences were trimmed with cutadapt, visually inspected with demux, and denoised with DADA2 (ref. 54) to remove PhiX contamination, trim reads, correct errors, merge read pairs and remove PCR chimeras. Representative ASV sequences and their abundances were extracted by feature-table.55 QIIME2 plugins were executed with DADA2 quality settings “- -p-trunc-len-f” of 230 and “- -p-trunc-len-r” of 220. Taxonomy was assigned using the Silva 138 (ref. 56) (99% identity clusters) database using the feature-classifier classify-sklearn function.2.4 X-ray absorption spectroscopy (XAS)
To investigate the speciation of U over time, select microcosms were set up at elevated U-concentrations to allow XAS analysis. Microcosms were set up as described but with sediment:synthetic groundwater ratio of 1:20 and 113 μM (27 ppm) U in solution to yield elevated concentrations of U (several hundred ppm) in the solids at microcosm end-points. XAS analysis was conducted on experimental end points (31 days) for sediment only control microcosms, Ca-citrate/Na-phosphate and glycerol phosphate amendments by mounting the sediment pellet into a cryovial and storing at −80 °C prior to analysis. Additional XAS analysis of a solution sample from the glycerol phosphate 50 μM (12 ppm) experiment was taken after 14 days of incubation when the solution U concentration was approximately 7 ppm to explore U solution phase speciation. A uranyl orthophosphate standard ((UO2)3(PO4)2(H2O)4) was synthesised following from the method of Yagoubi et al.57 and confirmed by XRD prior to XAS analysis (ESI† Fig. S7).
U LIII XAS analyses were conducted on beamlines B18 and I20 at the Diamond Light Source, Harwell, UK. Uranyl orthophosphate standard spectra were collected in transmission mode with remaining spectra collected in fluorescence mode. Spectra were calibrated (yttrium foil), background subtracted and normalized using ATHENA.58 Analysis of the EXAFS was conducted using ARTEMIS58 and statistical evaluation of shell by shell fitting was conducted.59
3. Results and discussion
3.1 Microcosm aqueous biogeochemistry
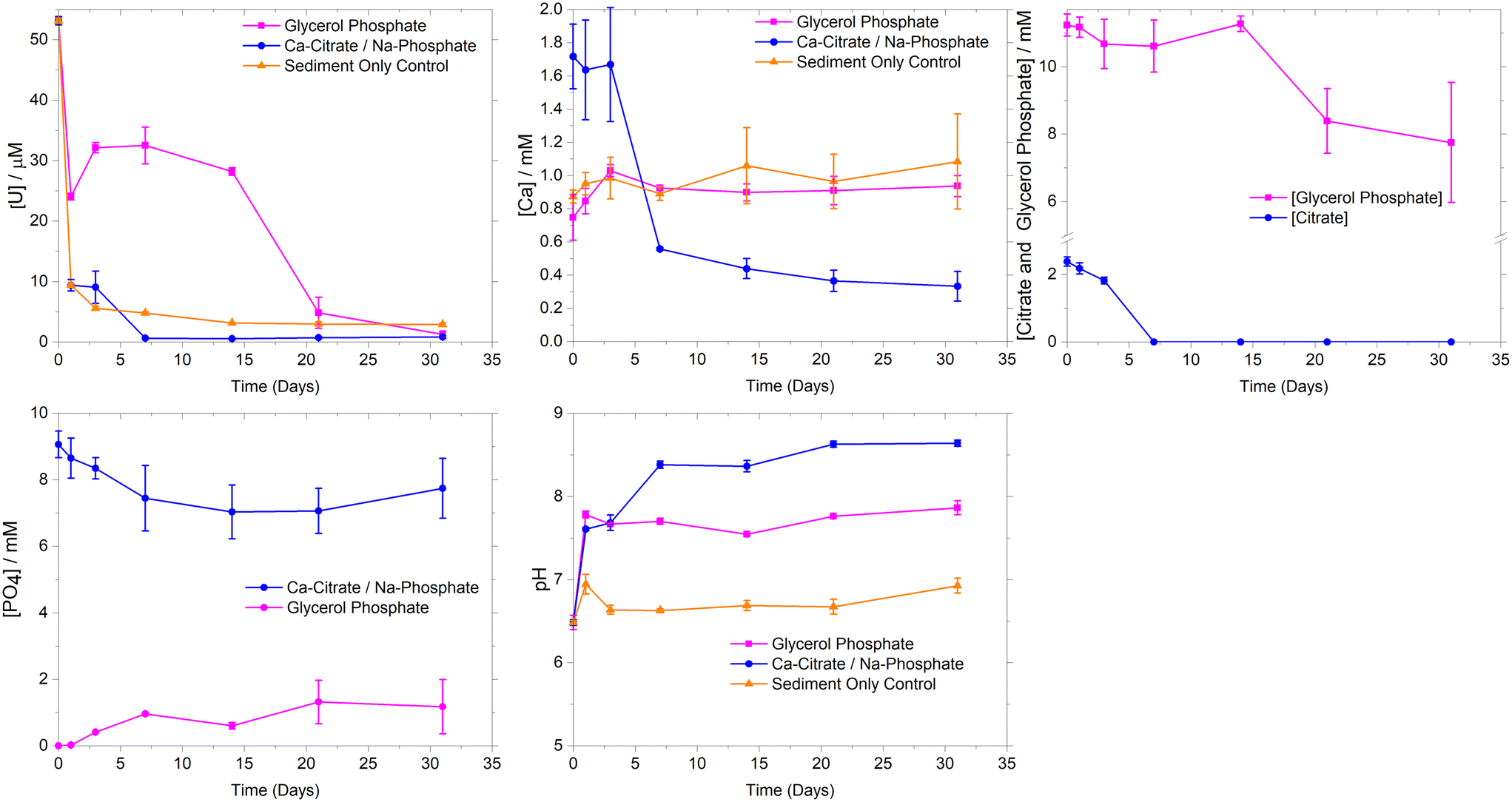 | ||
| Fig. 1 Aqueous geochemical data from synthetic groundwater microcosms amended with 1 mM Ca2+, 2.5 mM citrate with 10 mM phosphate, 10 mM glycerol phosphate and the synthetic groundwater sediment only sorption control. Microcosms were run in triplicate with error bars representing ±1σ. | ||
Experiments using Calder River water showed similar U sorption to the synthetic groundwater systems (ESI† Fig. S2). Here, there was rapid removal (>98%) of U over the first 7 days. This occurred concurrently with Ca2+ and phosphate removal during 1–7 days, consistent with removal trends in synthetic groundwater experiments and suggesting that the addition of river water did not significantly change U removal or Ca phosphate precipitation rates.
To further explore U behavior, PHREEQC geochemical modelling was conducted using aqueous data from the synthetic groundwater experimental systems (ESI† Table S1). Initially, modelling suggested the synthetic groundwater system was oversaturated with respect to several U(VI) phosphate phases (autunite (Ca(UO2)2(PO4)2·10–12H2O), uranyl orthophosphate ((UO2)3(PO4)2·4H2O) and chernikovite ((H3O)2(UO2)2(PO4)2·6H2O)) for the first 7 days of the experiment, and then became undersaturated. This is consistent with precipitation of uranyl phosphate phases controlling U(VI)-solubility in these systems.25,34,61 As well as oversaturation of U(VI)-phases, U(VI) sequestration is also possible through adsorption to newly precipitated Ca-phosphate phases and mineral surfaces within the sediment. Whilst crystalline hydroxyapatite was predicted to be oversaturated for the duration of the experiment, experimental studies typically show poorly ordered Ca-phosphates initially form in microbially mediated precipitation experiments with recrystallization and mineral dissolution potentially occurring to precipitate crystalline hydroxyapatite like phases over the medium term.27,62 In the current study, modelling predicted initial under-saturation of poorly ordered brushite (CaHPO4·2H2O), before it became oversaturated as Ca-citrate degradation released free Ca2+ leading to brushite oversaturation. Indeed, the modelled aqueous Ca-speciation showed the Ca(cit)− complex dominated (∼75–70%) over days 1–3 where Ca-removal was limited, whilst at day 7 when citrate degradation was essentially complete, the aqueous speciation was predicted to be Ca2+ (36%), Ca(PO4)− (25%) and Ca(HPO4) (38%). Overall modeling data suggest U sequestration maybe occurring through precipitation of both U-phosphates and sorption to Ca-phosphates, with U-phosphate formation likely to be the dominant U(VI) sequestration mechanism.
Geochemical modelling of the synthetic groundwater system was undertaken using the aqueous experimental data (ESI† Table S1) to further explore the precipitation processes. The ThermoChimie (V10a) database47 was augmented with uranyl and glycerol phosphate complexation constants.49 Modelling suggested that at day 0, U aqueous speciation was dominated (98%) by the UO2(GlyPO4)22− species, however after 1 day, U(VI) carbonate species dominated (87%) likely driven by the increase in solution pH from day 0 (pH 6.5) to day 1 (pH 7.8). This solution speciation presumably led to the transient (day 0–14) increase in U solubility when compared to the sediment only control, where U-carbonate complexes were less significant as the pH was below 7.0.21 Additionally the formation of aqueous U(VI) complexes with both glycerol phosphate and carbonate were predicted by geochemical modeling. Here, our modeling showed an increasing trend in aqueous complexation of UO2(GlyPO4)22−, reaching ∼20% at day 14 with the remaining U as U(VI)-carbonates. To further investigate aqueous U(VI) speciation, solution phase EXAFS analysis at day 14 was conducted on a sample with only 7 ppm U using an ultra-dilute spectroscopy beamline. Here, a uranyl tri-carbonate species model provided a good fit, but the addition of a P shell of 1 P at 3.26 Å was both statistically significant and improved fitting parameters. This suggested an XAS detectable contribution from U(VI)-phosphate aqueous species, presumably UO2(GlyPO4)22−, which was also observed at significant levels (20%) in the geochemical modelling (ESI† Fig. S1 and S5).
Experimentally, significant U removal only occurred from day 14 despite geochemical modelling suggesting oversaturation of autunite and uranyl orthophosphate between day 3 and day 14. Here, the saturation indices for the glycerol phosphate experiments were significantly lower than at the parallel time points for the Ca-citrate/Na-phosphate system (ESI† Fig. S1). This suggests that U-phosphate precipitation may require a threshold value of oversaturation for precipitation to occur. In the glycerol phosphate system, phosphate levels only began to rise above 1 mM from day 14 as significant glycerol phosphate degradation occurred and with U(VI) removal occurring after this point suggesting a minimum free phosphate concentration greater than approximately 1 mM was required to precipitate uranyl phosphate phases and mirroring past experiments.21 Indeed, in the Ca-citrate/Na-phosphate system, free phosphate is also likely controlling U removal, however, the excess of phosphate (10 mM) with respect to U, from day 0 facilitated rapid precipitation of U-phosphate. Interestingly, in the glycerol phosphate experiment, modeling did not show significant oversaturation of Ca-phosphate phases in these lower Ca2+ experiments and analysis of sediment endpoints with EXAFS (Fig. 2) confirmed that U removal was dominated by precipitation of uranyl phosphate-like phases rather than sorption or incorporation into Ca-phosphates. Overall, glycerol phosphate impacts U-solubility in two ways. Initially, the pH increase from 6.5 to 7.8 on addition of 10 mM glycerol phosphate enhances the solubility of U compared to the sediment only control due to the formation U(VI)-carbonate and U(VI)-phosphate species as predicted by geochemical modelling and confirmed by XAS over the initial 14 days of the experiment. Secondly the slow biodegradation and subsequent release of phosphate from glycerol phosphate into solution causes a delay in U(VI) phosphate precipitation, with U-removal occurring when free phosphate is > approximately 1 mM.
 | ||
| Fig. 2 U LIII EXAFS data collected from uranyl orthophosphate standard and from synthetic groundwater sediments after 31 days of treatment with Ca-citrate / Na-phosphate, glycerol phosphate and sediment only control. Data solid line and best fit dashed line. | ||
3.2 16S rRNA microbial community analysis
Analysis of unaltered sediments showed that a diverse microbial population was present (ESI† Fig. S3 and Table S2) in the starting material (Shannon Diversity Index (H) of 3.7). Here, bacteria affiliated with the genus Perlucidibaca (20%) were most abundant and other common aerobic soil and freshwater bacteria were also detected, for example organisms most closely related to such as Polaromonas eurypsychrophila and Aquirhabdus parva.64–66 By 31 days, the microbial community in the synthetic groundwater sediment only control had modestly reduced diversity compared to the initial sediments, with Shannon Diversity Indeces (H) dropping from 3.7 to 3.3, (Table S2†) with the population then dominated by bacteria from the genus Methylobacterium (43%) (ESI† Fig. S3). Species within this genus have high tolerance for metal contaminants (e.g. Cu, Ni and Zn),67,68 perhaps reflecting the presence of 12 ppm (50 μM) U(VI) in the systems. Initial Calder River water comprised several species (ESI† Fig. S4), however it was not possible to resolve the three most dominant to genus level. Here, 16S rRNA gene sequencing identified species that were members of Acidobacteriota (phylum) (19%), Vicinamibacterales (order) (16%) and Actinobacteriota (phylum) (15%). Members of these phyla/orders have been isolated from a variety of environments including freshwater rivers, soils and sediments. Sequencing also identified representatives of the genera Bosea, Bauldia and Rhizobacter, present at 13%, 12% and 11% abundance respectively. Species from these genera such as Bosea lupini and Rhizobacter profundi are aerobes isolated from environmental samples such as soils, wastewaters and plant biota.69,70 At 31 days Calder River water sediment only control sediments had higher microbially diversity (H 3.3) than initial river water alone (H 2.3), presumably driven by ingrowth of species from sediment. Here, sequencing identified common soil and freshwater bacteria such as organisms most closely related to Bacteriovorax stolpii and Duganella albus.71,72Microbial communities characterised in sediments amended with Ca-citrate/Na-phosphate and glycerol phosphate were less diverse than for the initial unaltered sediments (H Index 3.7, ESI† Table S2) in the synthetic groundwater systems (citrate, 2.3 and glycerol phosphate, 2.8). Synthetic groundwater sediments 14 days after Ca-citrate/Na-phosphate amendment became enriched in bacteria from the genera Cecembia (33%), Labrys (17%) and Phenylobacterium (8%). These include aerobic citrate assimilating species such as Cecembia rubra and Phenylobacterium lituiforme.73,74 Known citrate degraders continued to be enriched in sediments at 31 days. Here, bacteria from the genera Labilithrix (17%) and Sphingomonas (17%) dominated however it was not possible to further resolve the dominant bacteria at genus level. After 14 days treatment with Ca-citrate/Na-phosphate the microbial community within Calder River water sediments became substantially less diverse than for sediment only control sediments with the H index decreasing from initial 3.7 to 1.8. After incubation the dominant bacteria were affiliated with the genus Thauera (38%), which contains aerobic heterotrophic species that are enriched in wastewaters and groundwater aquifers contaminated with inorganic pollutants (e.g. selenate).75 Additionally, the genus contains species capable of utilizing citrate, including Thauera rnechernichensis and Thauera propionica.76,77 Citrate-utilizing species are known within these genera, which are often found in aerobic soils, sediments and freshwater environments. These data suggest that upon addition of citrate, sediment microbial communities became enriched in bacteria capable of citrate utilization in both synthetic groundwater and Calder River water systems, consistent with the removal of citrate (Fig. 1 and ESI† S2).
Following treatment (31 days) with glycerol phosphate, bacteria from the genera Sediminibacterium (37%) and Sphingomonas (35%) dominated in synthetic groundwater and Calder River water sediments respectively. Numerous species within these genera possess phosphatase enzyme activity (e.g. Sphingomonas alpina).78 This suggested that enrichment of the glycerol phosphate degradation community occurred as expected.29 The microbial community changed from day 14 to day 31, coinciding with U removal from solution in both synthetic groundwater and Calder River water systems. For the synthetic groundwater system, organisms affiliated with the genera Rhizobacter (47%) and Arsenicitalea (30%) were most abundant after 14 days. It was not possible to resolve the most abundant bacteria to genus level at 31 days within the Calder River water system, however, species from the genus Sediminibacterium were present at 11% relative abundance. Species within this genus have been shown to proliferate in environments with heavy metal and organic pollutants.79 Here, aqueous U may influence the microbial community and select for bacteria that can persist in environments containing toxic heavy metals such as Sediminibacterium species. Indeed, other genera within the Chitinophagaceae family can tolerate aqueous U.79 Ingrowth of heavy metal tolerant bacteria that have phosphatase enzyme activity is consistent with the addition of glycerol phosphate and aqueous U. The change in microbial community toward heavy metal tolerant species may explain the slower glycerol phosphate degradation observed in the current work compared to similar past work without aqueous U.29
3.3 X-ray absorption spectroscopy
U LIII XAS was conducted on sediment end points from synthetic groundwater experiments to explore U-speciation in the treated sediments (Fig. 2). Analysis of the XANES region showed U was present as uranyl-like U(VI) in all systems, consistent with the oxic experimental conditions (ESI† Fig. S6). EXAFS analysis further explored U speciation after treatment with Ca-citrate/Na-phosphate, glycerol phosphate and in the synthetic groundwater sediment only control. Additionally, XAS was conducted on a solution containing only 7 ppm U from the synthetic groundwater glycerol phosphate amended system at 14 days.The U(VI) sediment only control EXAFS were best fit with 2 O atoms at 1.8 Å consistent with the U-O axial bonding in uranyl, followed by a split shell of 3 O at 2.29 Å and 2.47 Å respectively (ESI† Table S3). Features at higher R in the Fourier transform beyond 3 Å were best fit using 1.7 C atoms at 2.95 Å and 0.5 Fe atoms at 3.45 Å. Overall, this is consistent with uranyl carbonato species sorbed on the surface of Fe-bearing phases (e.g. goethite (FeOOH)).80,81 EXAFS spectra from Ca-citrate/Na-phosphate and glycerol phosphate amended systems were fitted using uranyl orthophosphate, chernikovite or autunite models as these phases were predicted to be oversaturated in modelling of the experiments. U(VI) sorbed/incorporated to Ca-phosphates was also considered in fitting as U has been shown to sorb or incorporate to these phases.25,61,82–84 For the Ca-citrate/Na-phosphate amended system a first shell of 2 O atoms were fit at 1.81 Å consistent with uranyl speciation.84 However, attempts to fit a single shell of equatorial oxygen back scatters expected in autunite and chernikovite (approximately 4 O at 2.26 Å) were unsuccessful. Splitting the equatorial oxygen shell improved fitting parameters with a best fit of 2.7 O atoms at 2.30 Å and 2.3 O at 2.44 Å. Interestingly, the split shell suggested uranyl orthophosphate or surface bond uranyl complexes were forming.82,85 The fit was further improved by the addition of 1 P atom at 3.13 Å, close to the short U-P (2.99–3.05 Å) distance from bidentate coordination of the uranyl equatorial plane with phosphate groups located on the surface of Ca-phosphate mineral phases.61,83,85 At the same time, a short U-P bond is also present in uranyl orthophosphate at ∼3.16 Å (ref. 82, 84) and this phase was predicted in geochemical modelling and is dominant in phosphate containing systems at neutral to mildly alkaline conditions.82,86 Further shells of P in U(VI)-orthophosphate at 3.60 and 3.74 Å present in the synthesized standard were not fully resolved, however the fit was improved by the addition of statistically significant (f-test 100%) 2 P backscatterers at 3.66 Å presumably reflecting an averaged environment for both P shells. The presence of this U–P shell suggests uranyl-orthophosphate rather than a U adsorption complex dominates. Attempts to fit a Ca shell at 3.80–4.01 Å, which occurs in autunite- and hydroxyapatite-like mineral phases were unsuccessful. Instead, the fit was further improved by the addition of 1.3 U backscatterers at 4.00 Å, again consistent with the U–U distance in uranyl orthophosphate.82,84
EXAFS spectra of sediments amended with glycerol phosphate were very similar to the Ca-citrate/Na-phosphate data suggesting a similar fate for U in these systems. Best fit included 2 O atoms at 1.80 Å, a split equatorial O shell with 2.7 O at 2.29 Å and 2.3 O at 2.49 Å respectively 1.0 P at 3.14 Å and 2 P at 3.70 Å, and 1.3 U backscatterers at 4.00 Å in a uranyl orthophosphate like coordination environment. Overall, these data suggest U removal was dominated by precipitation of a uranyl orthophosphate-like phase. Interestingly, geochemical modelling suggested autunite was the dominant oversaturated phase in both the Ca-citrate/Na-phosphate and glycerol phosphate treatment endpoints but EXAFS data on experimental samples did not support this. Past work has shown that initially, chernikovite forms during U-phosphate biomineralisation at circumneutral pH under lower carbonate conditions.61 This is followed by recrystallization to more stable uranyl orthophosphate and autunite phases.34,61,87 This process may have occurred in these experiments, with initial removal through kinetically favorable chernikovite followed by recrystallisation to uranyl orthophosphate during the 31 day experiment. U removal by hydroxyapatite through both adsorption and incorporation has been observed in past work, however our results do not support this and suggests site specific factors may influence precipitation pathways.25,85 Again, this interpretation is consistent with geochemical data which showed significant U removal prior to substantial Ca2+ removal in both the Ca-citrate/Na-phosphate amendment and glycerol phosphate amended systems which presumably limited the Ca-phosphate available for U adsorption or incorporation. Finally, EXAFS analysis of the very low concentration (7 ppm) solution phase were obtained from the glycerol phosphate amended system at day 14 showed the data were best fit as uranyl carbonate species with 2 O atoms at 1.84 Å, 6 O atoms at 2.45 Å, 3C atoms at 2.93 Å and 2 Ca atoms at 4.05 Å and could include a statistically significant (99%), P shell at 2.3 Å.81,88,89 (ESI Fig. S5; ESI† Table S3). The presence of a U–P shell in the fit suggests a contribution from a U-glycerol phosphate complex (UO2(C3H7O3PO3)22−) as reported in previous studies49 and modelled in our experiments.
3.4 Environmental implications
In oxic U(VI) sediment only and phosphate remediation experiments with citrate/phosphate and glycerol phosphate amendments, removal of U(VI) from solution was dominated by initial rapid sorption to sediments in both synthetic groundwater and Calder River water experiments. Here, sediment only controls showed high (>90%) U(VI) removal and sediment XAS data confirmed a U-carbonate complex inner sphere bound to Fe phases in the sediment. These complexes are typically considered labile and alterations in subsurface biogeochemistry may easily remobilize sorbed U(VI) into groundwaters. Amending experiments with Ca-citrate/Na-phosphate or glycerol phosphate solutions enhanced U removal compared to the sediment only controls. Interestingly, results showed that glycerol phosphate additions may cause a transient increase in U solubility presumably due to increased pH from glycerol phosphate degradation products enhancing soluble U(VI) carbonate complexes with formation of a soluble U-glycerol phosphate complex also identified in both modelling and EXAFS analysis in the solution phase. This further highlights the need to test bioremediation strategies under a broad range of biogeochemical conditions. Despite the transient increase in U solubility in the glycerol phosphate amendment, precipitation of uranyl phosphate-like phases occurred after 14 days of glycerol phosphate treatment as degradation progressed. This may even be advantageous during in situ remediation processes as the slower on set of precipitation may allow the injected solution to disperse further in the contaminant plume and increase overall efficacy. Microbial community analyses showed the ingrowth of close relatives of microbes which utilize citrate or glycerol phosphate in each of the amended treatments. Further analyses showed the ingrowth of close relatives of known heavy metal tolerant bacteria in all experimental end points where U was present at 12 ppm. That coupled to the lower rate of both citrate and glycerol phosphate degradation compared with experiments with no U suggests some toxicity effects may be occurring.XAS analyses from Ca-citrate/Na-phosphate and glycerol phosphate amended systems confirmed U was sequestered into poorly ordered uranyl orthophosphate mineral phases. These have been suggested as favorable end-points for U in contaminated land scenarios due their low solubility under environmental conditions. Indeed, previous laboratory studies have shown the applicability of both Ca-citrate/Na-phosphate and glycerol phosphate amendments to sequester U.20,22,36,90 Our study is consistent with U removal in past work and suggests that both Ca-citrate/Na-phosphate and glycerol phosphate amendments may remediate U(VI) contaminated aquifers under a wide range of biogeochemical conditions.
4. Conclusion
In this study, we explore biomineralisation of U(VI) through precipitation of insoluble phosphate phases. Here, synthetic groundwaters and natural Calder River waters were amended with phosphate generating solutions (Ca-citrate/Na-phosphate and glycerol phosphate) under oxic conditions. Aqueous geochemical data showed high U removal occurred in the synthetic groundwater and Calder River water systems with both the Ca-citrate/Na-phosphate and the glycerol phosphate amendments. Geochemical modeling in combination with aqueous data showed two distinct precipitation pathways were occurring in the different treatments. Ca-citrate/Na-phosphate addition causes rapid oversaturation and precipitation of U(VI)-phosphate phases. However, the glycerol phosphate amendment showed a delayed removal of U, due to the slow ingrowth of aqueous phosphate due to a relatively slow rate of glycerol phosphate biodegradation. Geochemical modelling data highlight the significant difference in U(VI) phosphate saturation between the two amendment systems and the key role aqueous phosphate plays in U(VI) removal. Despite the different treatment pathways, XAS analysis of sediment endpoints showed U was present as highly insoluble uranyl orthophosphate-like phase in both treated systems. Additionally, our data confirmed Calder River water treated experiments mirrored the synthetic groundwater systems and did not significantly enhance removal U when compared to synthetic groundwater. This confirms that Calder River water may be a viable water source onsite amendment injections during field deployment of these techniques as the Calder River runs through the Sellafield site area.Phosphate mineral phases such autunite, uranyl orthophosphate and hydroxyapatite have been suggested as optimal end-points for several priority radionuclides, including U and 90Sr. These phases are recalcitrant to re-dissolution under environmental conditions and remove the need for future subsurface redox control of U. Our study widens the treatment envelope available to Ca-citrate/Na-phosphate and glycerol phosphate treatment techniques, proving fundamental information on the formation of these phases under environmental conditions and demonstrates viable remediation strategies for U contaminated groundwaters at Sellafield.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Callum Robinson: conceptualization, data curation, formal analysis, methodology, software, validation, visualization, roles – writing original draft, writing – review & editing. Sam Shaw: conceptualization, funding acquisition, investigation, project administration, resources, supervision, writing – review & editing. Jonathan R. Lloyd: funding acquisition, investigation, project administration, resources, supervision, writing – review & editing. James Graham: conceptualization, funding acquisition, methodology, supervision, writing – review & editing. Katherine Morris: conceptualization, formal analysis, methodology, validation, writing – review & editing, funding acquisition, investigation, project administration, resources, supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge EPSRC and National Nuclear Laboratory for financial support through EPSRC ICASE PhD studentship (CR; 19000127). We acknowledge EPSRC DTP grant code EP/W524347/1 for finical support. We also acknowledge access to the EPSRC NNUF RADER Facility (EP/T011300/1) and the Diamond Light Source (B18 (RAWDER) SP24074-8 and I20 (P21441-9)) for XAS analysis.References
- Sellafield Ltd, Groundwater monitoring at Sellafield: Annual Data review 2016, Sellaf Ltd., 2016 Search PubMed.
- J. M. Zachara, P. E. Long, J. Bargar, J. A. Davis, P. Fox and J. K. Fredrickson,
et al. Persistence of uranium groundwater plumes: Contrasting mechanisms at two DOE sites in the groundwater-river interaction zone, J. Contam. Hydrol., 2013, 147, 45–72 CrossRef CAS PubMed
.
- K. M. Campbell, R. K. Kukkadapu, N. P. Qafoku, A. D. Peacock, E. Lesher and K. H. Williams,
et al., Geochemical, mineralogical and microbiological characteristics of sediment from a naturally reduced zone in a uranium-contaminated aquifer, Appl. Geochem., 2012, 27(8), 1499–1511 CrossRef CAS
- L. Newsome, K. Morris and J. R. Lloyd, The biogeochemistry and bioremediation of uranium and other priority radionuclides, Chem. Geol., 2014, 363, 164–184 CrossRef CAS
-
G. Choppin, J. O. Liljenzin, J. Rydberg and C. Ekberg, Behavior of Radionuclides in the Environment. Radiochemistry and Nuclear Chemistry, 2013, pp. 753–788 Search PubMed
- F. Y. C. Huang, P. V. Brady, E. R. Lindgren and P. Guerra, Biodegradation of Uranium-Citrate complexes: Implications for extraction of Uranium from soils, Environ. Sci. Technol., 1998, 32(3), 379–382 CrossRef CAS
- J. J. Lenhart, S. E. Cabaniss, P. MacCarthy and B. D. Honeyman, Uranium(VI) complexation with citric, humic and fulvic acids, Radiochim. Acta, 2000, 88(6), 345–353 CrossRef CAS
-
C. R. Bryan and M. D. Siegel, Environmental geochemistry of radioactive contamination. Treatise on Geochemistry, 2003, p. 114 Search PubMed
- N. P. Qafoku and J. P. Icenhower, Interactions of aqueous U(VI) with soil minerals in slightly alkaline natural systems, Rev. Environ. Sci. Biotechnol., 2008, 7(4), 355–380 CrossRef CAS
- P. S. Andersson, D. Porcelli, O. Gustafsson, J. Ingri and G. J. Wasserburg, The importance of colloids for the behavior of uranium isotopes in the low-salinity zone of a stable estuary, Geochim. Cosmochim. Acta, 2001, 65(1), 13–25 CrossRef CAS
- J. A. Davis, D. E. Meece, M. Kohler and G. P. Curtis, Approaches to surface complexation modeling of Uranium(VI) adsorption on aquifer sediments, Geochim. Cosmochim. Acta, 2004, 68(18), 3621–3641 CrossRef CAS
- R. T. Anderson, H. A. Vrionis, I. Ortiz-Bernad, C. T. Resch, P. E. Long and R. Dayvault,
et al. Stimulating the In Situ Activity of Geobacter Species to Remove Uranium from the Groundwater of a Uranium-Contaminated Aquifer, Appl. Environ. Microbiol., 2003, 69(10), 5884–5891 CrossRef CAS PubMed
- J. R. Bargar, K. H. Williams, K. M. Campbell, P. E. Long, J. E. Stubbs and E. I. Suvorova,
et al. Uranium redox transition pathways in acetate-amended sediments, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(12), 4506–4511 CrossRef CAS
- K. H. Williams, P. E. Long, J. A. Davis, M. J. Wilkins, A. L. N'Guessan and C. I. Steefel,
et al. Acetate availability and its influence on sustainable bioremediation of Uranium-contaminated groundwater, Geomicrobiol. J., 2011, 28(5–6), 519–539 CrossRef CAS
- S. M. Hee, J. Komlos and P. R. Jaffé, Uranium reoxidation in previously bioreduced sediment by dissolved oxygen and nitrate, Environ. Sci. Technol., 2007, 41(13), 4587–4592 CrossRef PubMed
- L. Newsome, K. Morris, D. Trivedi, A. Bewsher and J. R. Lloyd, Biostimulation by Glycerol Phosphate to Precipitate Recalcitrant Uranium(IV) Phosphate, Environ. Sci. Technol., 2015, 49(18), 11070–11078 CrossRef CAS PubMed
- G. T. W. Law, A. Geissler, I. T. Burke, F. R. Livens, J. R. Lloyd and J. M. McBeth,
et al. Uranium Redox Cycling in Sediment and Biomineral Systems, Geomicrobiol. J., 2011, 28(5–6), 497–506 CrossRef CAS
- Y. Xie, L. Yu, L. Chen, C. Chen, L. Wang and F. Liu,
et al. Recent progress of radionuclides separation by porous materials, Sci. China:Chem., 2024, 67(11), 3515–3577 CrossRef CAS
- M. J. Beazley, R. J. Martinez, P. A. Sobecky, S. M. Webb and M. Taillefert, Uranium Biomineralization as a Result of Bacterial Phosphatase Activity: Insights from Bacterial Isolates from a Contaminated Subsurface, Environ. Sci. Technol., 2007, 41(16), 5701–5707 CrossRef CAS PubMed
- R. J. Martinez, M. J. Beazley, M. Taillefert, A. K. Arakaki, J. Skolnick and P. A. Sobecky, Aerobic uranium (VI) bioprecipitation by metal-resistant bacteria isolated from radionuclide- and metal-contaminated subsurface soils, Environ. Microbiol., 2007, 9(12), 3122–3133 CrossRef CAS PubMed
- E. S. Shelobolina, H. Konishi, H. Xu and E. E. Roden, U(VI) sequestration in hydroxyapatite produced by microbial glycerol 3-phosphate metabolism, Appl. Environ. Microbiol., 2009, 75(18), 5773–5778 CrossRef CAS PubMed
- J. E. Szecsody, R. Moore, M. Rigal, V. Vermeul and J. Luellen, Use of a Ca-Citrate-Phosphate Solution to Form Hydroxyapatite for Uranium Stabilization of Old Rifle Sediments: Laboratory Proof of Principle Studies, 2016.
- D. M. Wellman, E. M. Pierce, C. C. Bovaird, K. M. Griswold, K. M. Gunderson and S. M. Webb, et al. Laboratory development of polyphosphate remediation technology for in situ treatment of uranium contamination in the vadose zone and capillary fringe, Uranium Compd Isot Appl., 2009, pp. 473–555.
- S. Mehta, Geochemical evaluation of uranium sequestration from field-scale infiltration and injection of polyphosphate solutions in contaminated Hanford sediments, Appl. Geochem., 2017, 84, 133–153 CrossRef CAS
- C. C. Fuller, J. R. Bargar, J. A. Davis and M. J. Piana, Mechanisms of uranium interactions with hydroxyapatite: Implications for groundwater remediation, Environ. Sci. Technol., 2002, 36(2), 158–165 CrossRef CAS PubMed
- P. S. Munasinghe, M. E. Elwood Madden, S. C. Brooks and A. S. Elwood Madden, Dynamic interplay between uranyl phosphate precipitation, sorption, and phase evolution, Appl. Geochem., 2015, 58, 147–160 CrossRef CAS
- J. Szecsody, M. Williams, C. Burns, D. Girvin, R. Moore and J. Mckinley, et al. Hanford 100-N area apatite emplacement: Laboratory results of Ca-Citrate-PO4 solution injection and Sr-90 immobilization in 100-N sediments, Pacific Northwest Natl Lab, 2007, (PNNL-16891).
- S. Handley-Sidhu, J. C. Renshaw, P. Yong, R. Kerley and L. E. Macaskie, Nano-crystalline hydroxyapatite bio-mineral for the treatment of strontium from aqueous solutions, Biotechnol. Lett., 2011, 33(1), 79–87 CrossRef CAS PubMed
- C. Robinson, S. Shaw, J. R. Lloyd, J. Graham and K. Morris, Phosphate (Bio)mineralization Remediation of 90Sr-Contaminated Groundwaters, ACS ES&T Water, 2023, 3(10), 3223–3234 Search PubMed
- V. Vermeul, B. Bjornstad, B. Fritz, J. Fruchter, R. Mackley and D. Mendoza, et al. 300 Area Uranium Stabilization Through Polyphosphate Injection : Final Report. Pacific Northwest Natl Lab, 2009, PNNL–18529.
- D. M. Wellman, J. S. Fruchter and V. Vermuel, Experimental Plan : Uranium Stabilization Through Polyphosphate Injection. Demonstration Project 300 Area Uranium Plume Treatability Demonstration Project. Pacific Northwest Natl Lab, 2006, (PNNL-16101).
- Z. Pan, D. E. Giammar, V. Mehta, L. D. Troyer, J. G. Catalano and Z. Wang, Phosphate-induced immobilization of uranium in hanford sediments, Environ. Sci. Technol., 2016, 50(24), 13486–13494 CrossRef CAS PubMed
- M. R. Baker, F. M. Coutelot and J. C. Seaman, Phosphate amendments for chemical immobilization of uranium in contaminated soil, Environ. Int., 2019, 129, 565–572 CrossRef CAS PubMed
- V. S. Mehta, F. Maillot, Z. Wang, J. G. Catalano and D. E. Giammar, Effect of Reaction Pathway on the Extent and Mechanism of Uranium(VI) Immobilization with Calcium
and Phosphate, Environ. Sci. Technol., 2016, 50(6), 3128–3136 CrossRef CAS PubMed
- A. Cleary, J. R. Lloyd, L. Newsome, S. Shaw, C. Boothman and G. Boshoff,
et al. Bioremediation of strontium and technetium contaminated groundwater using glycerol phosphate, Chem. Geol., 2019, 509, 213–222 CrossRef CAS
- L. Newsome, K. Morris and J. R. Lloyd, Uranium Biominerals Precipitated by an Environmental Isolate of Serratia under Anaerobic Conditions. Janssen PJ, editor, PLoS One, 2015, 10(7), e0132392 CrossRef PubMed
- M. J. Beazley, R. J. Martinez, P. A. Sobecky, S. M. Webb and M. Taillefert, Nonreductive Biomineralization of Uranium(VI) Phosphate Via Microbial Phosphatase Activity in Anaerobic Conditions, Geomicrobiol. J., 2009, 26(7), 431–441 CrossRef CAS
- R. C. Moore, C. Sanchez, K. Holt, P. Zhang, H. Xu and G. R. Choppin, Formation of hydroxyapatite in soils using calcium citrate and sodium phosphate for control of strontium migration, Radiochim. Acta, 2004, 92(9–11), 719–723 CrossRef CAS
- J. E. Szecsody, J. S. Fruchter, M. L. Rockhold, J. P. Mckinley, M. Oostrom and V. R. Vermeul, et al. Sequestration of Sr-90 Subsurface Contamination in the Hanford 100-N Area by Surface Infiltration of a Ca-Citrate-Phosphate Solution, Pacific Northwest Natl Lab, 2009, (PNNL-18303).
- N. T. Smith, J. Shreeve and O. Kuras, Multi-sensor core logging (MSCL) and X-ray computed tomography imaging of borehole core to aid 3D geological modelling of poorly exposed unconsolidated superficial sediments underlying complex industrial sites: An example from Sellafield nuclear site UK, Sb. Geol. Ved, Uzita Geofyz., 2020, 178, 104084 Search PubMed
- Sellafield Ltd. Monitoring our environment: Discharges and Environmental Monitoring. Annual Report 2019/, Sellafield Ltd, 2019.
- H. McKenzie and N. Armstrong-Pope, Groundwater Annual Report 2010, Sellafield Ltd, Sellaf Ltd, 2010.
- L. Newsome, K. Morris, D. Trivedi, N. Atherton and J. R. Lloyd, Microbial reduction of uranium(VI) in sediments of different lithologies collected from Sellafield, Appl. Geochem., 2014, 51, 55–64 CrossRef CAS
- V. R. Vermeul, J. E. Szecsody, B. G. Fritz, M. D. Williams, R. C. Moore and J. S. Fruchter, An Injectable Apatite Permeable Reactive Barrier for In Situ 90Sr Immobilization, Groundwater Monit. Rem., 2014, 34(2), 28–41 CrossRef CAS
- J. K. Heinonen and R. J. Lahti, A new and convenient colorimetric determination of inorganic orthophosphate and its application to the assay of inorganic pyrophosphatase, Anal. Biochem., 1981, 113(2), 313–317 CrossRef CAS PubMed
-
D. L. Parkhurst and C. A. J. Appelo, Description of input and examples for PHREEQC version 3: a computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations, Techniques and Methods. Reston, VA, 2013 Search PubMed
- L. Duro, M. Grivé and E. Giffaut, ThermoChimie, the ANDRA Thermodynamic Database, MRS Online Proc. Libr., 2012, 1475 Search PubMed
- D. Gorman-Lewis, T. Shvareva, K. A. Kubatko, P. C. Burns, D. M. Wellman and B. Mcnamara,
et al. Thermodynamic properties of autunite, uranyl hydrogen phosphate, and uranyl orthophosphate from solubility and calorimetric measurements, Environ. Sci. Technol., 2009, 43(19), 7416–7422 CrossRef CAS PubMed
- A. Koban and G. Bernhard, Complexation of uranium(VI) with glycerol 1-phosphate, Polyhedron, 2004, 23(10), 1793–1797 CrossRef CAS
- J. G. Caporaso, C. L. Lauber, W. A. Walters, D. Berg-Lyons, C. A. Lozupone and P. J. Turnbaugh,
et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(1), 4516–4522 CrossRef CAS PubMed
- J. G. Caporaso, C. L. Lauber, W. A. Walters, D. Berg-Lyons, J. Huntley and N. Fierer,
et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms, ISME J., 2012, 6(8), 1621–1624 CrossRef CAS PubMed
- J. J. Kozich, S. L. Westcott, N. T. Baxter, S. K. Highlander and P. D. Schloss, Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq illumina sequencing platform, Appl. Environ. Microbiol., 2013, 79(17), 5112–5120 CrossRef CAS PubMed
- E. Bolyen, J. R. Rideout, M. R. Dillon, N. A. Bokulich, C. C. Abnet and G. A. Al-Ghalith,
et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2, Nat. Biotechnol., 2019, 37(8), 852–857 CrossRef CAS PubMed
- B. J. Callahan, P. J. McMurdie and S. P. Holmes, Exact sequence variants should replace operational taxonomic units in marker-gene data analysis, ISME J., 2017, 11(12), 2639–2643 CrossRef PubMed
- D. McDonald, J. C. Clemente, J. Kuczynski, J. R. Rideout, J. Stombaugh and D. Wendel,
et al. The Biological Observation Matrix (BIOM) format or: How I learned to stop worrying and love the ome-ome, GigaScience, 2012, 464(1), 1–6 Search PubMed
- C. Quast, E. Pruesse, P. Yilmaz, J. Gerken, T. Schweer and P. Yarza,
et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools, Nucleic Acids Res., 2013, 41, D590–D596 CrossRef CAS PubMed
- S. Yagoubi, C. Renard, F. Abraham and S. Obbade, Molten salt flux synthesis and crystal structure of a new open-framework uranyl phosphate Cs3(UO2)2(PO 4)O2: Spectroscopic characterization and cationic mobility studies, J. Solid State Chem., 2013, 200, 13–21 Search PubMed
- B. Ravel and M. Newville, {ATHENA} and {ARTEMIS} Interactive Graphical Data Analysisusing {IFEFFIT}, Phys. Scr., 2005, 1007 Search PubMed
- L. Downward, C. H. Booth, W. W. Lukens and F. Bridges, A variation of the F-test for determining statistical relevance of particular parameters in EXAFS fits, AIP Conf. Proc., 2007, 882, 129–131 CrossRef CAS
- F. Yan, S. Schubert and K. Mengel, Soil pH increase due to biological decarboxylation of organic anions, Soil Biol. Biochem., 1996, 28(4–5), 617–624 Search PubMed
- L. N. Lammers, H. Rasmussen, D. Adilman, J. L. deLemos, P. Zeeb and D. G. Larson,
et al. Groundwater uranium stabilization by a metastable hydroxyapatite, Appl. Geochem., 2017, 84, 105–113 CrossRef CAS
- J. M. Hughes, The many facets of apatite, Am. Mineral., 2015, 100(5–6), 1033–1039 CrossRef
- M. J. Beazley, R. J. Martinez, S. M. Webb, P. A. Sobecky and M. Taillefert, The effect of pH and natural microbial phosphatase activity on the speciation of uranium in subsurface soils, Geochim. Cosmochim. Acta, 2011, 75(19), 5648–5663 CrossRef CAS
- K. Baek, J. H. Han and M. H. Lee, Perlucidibaca aquatica sp. Nov., isolated from fresh water, Int. J. Syst. Evol. Microbiol., 2017, 67(7), 2296–2300 CrossRef CAS PubMed
- T. Xing, T. Yao, Y. Liu, N. Wang, B. Xu and L. Shen,
et al. Polaromonas eurypsychrophila sp. nov., isolated from an ice core, Int. J. Syst. Evol. Microbiol., 2016, 66(7), 2497–2501 CrossRef CAS PubMed
- M. Kim, S. K. Shin and H. Yi, Mucilaginibacter celer sp. Nov. and aquirhabdus parva gen. nov., sp. nov., isolated from freshwater, Int. J. Syst. Evol. Microbiol., 2020, 70(10), 5479–5487 CrossRef CAS PubMed
- M. M. Photolo, L. Sitole, V. Mavumengwana and M. G. Tlou, Genomic and physiological investigation of heavy metal resistance from plant endophytic methylobacterium radiotolerans mamp 4754, isolated from combretum erythrophyllum, Int. J. Environ. Res. Public Health, 2021, 18(3), 1–12 Search PubMed
- J. Kim, G. Chhetri, I. Kim, M. K. Kim and T. Seo, Methylobacterium durans sp. nov., a radiation-resistant bacterium isolated from gamma ray-irradiated soil. Antonie van Leeuwenhoek, Int J Gen, Mol. Microbiol., 2020, 113(2), 211–220 CAS
- S. E. De Meyer and A. Willems, Multilocus sequence analysis of Bosea species and description of Bosea lupini sp. nov., Bosea lathyri sp. nov. and Bosea robiniae sp. nov., isolated from legumes, Int. J. Syst. Evol. Microbiol., 2012, 62(10), 2505–2510 CrossRef CAS PubMed
- L. Jin, S. R. Ko, C. Y. Ahn, H. G. Lee and H. M. Oh, Rhizobacter profundi sp. Nov., isolated from freshwater sediment, Int. J. Syst. Evol. Microbiol., 2016, 66(5), 1926–1931 CrossRef CAS PubMed
- A. R. Snyder, H. N. Williams, M. L. Baer, K. E. Walker and O. C. Stine, 16S rDNA sequence analysis of environmental Bdellovibrio-and-like organisms (BALO) reveals extensive diversity, Int. J. Syst. Evol. Microbiol., 2002, 52(6), 2089–2094 CAS
- H. Lu, T. Deng, F. Liu, Y. Wang, X. Yang and M. Xu, Duganella albus sp. Nov., duganella aquatilis sp. nov., duganella pernnla sp. nov. and duganella levis sp. nov., isolated from subtropical streams in china, Int. J. Syst. Evol. Microbiol., 2020, 70(6), 3801–3808 CrossRef CAS PubMed
- Y. Y. Duan, H. Ming, L. Dong, Y. R. Yin, X. L. Meng and E. M. Zhou,
et al. Cecembia rubra sp. nov., a thermophilic bacterium isolated from a hot spring sediment, Int. J. Syst. Evol. Microbiol., 2015, 65(7), 2118–2123 CrossRef CAS PubMed
- S. Kanso and B. K. C. Patel, Phenylobacterium lituiforme sp. nov., a moderately thermophilic bacterium from a subsurface aquiter, and emended description of the genus Phenylobacterium, Int. J. Syst. Evol. Microbiol., 2004, 54(6), 2141–2146 CrossRef PubMed
-
G. M. Garrity, J. A. Bell and T. Lilburn, BERGEY'S MANUAL OF Systematic Bacteriology. Volume Two The Proteobacteria, 2005, pp. 1145–1194 Search PubMed
- E. Scholten, T. Lukow, I. G. Auling, R. M. Kroppenstedt, F. A. Rainey and H. Diekmann, Denitrif Ier From a Leachate Treatment Plant, Int. J. Syst. Bacteriol., 1997, 1045–1051 Search PubMed
- D. Pal, A. Bhardwaj, S. K. Sudan, N. Kaur, M. Kumari and B. Bisht,
et al. Thauera propionica sp. nov., isolated from downstream sediment sample of the river ganges, Kanpur, India, Int. J. Syst. Evol. Microbiol., 2018, 68(1), 341–346 CrossRef CAS PubMed
- R. Margesin, D. C. Zhang and H. J. Busse, Sphingomonas alpina sp. nov., a psychrophilic bacterium isolated from alpine soil, Int. J. Syst. Evol. Microbiol., 2012, 62(7), 1558–1563 CrossRef CAS PubMed
- R. M. Brzoska, R. E. Edelmann and A. Bollmann, Physiological and Genomic Characterization of Two Novel Bacteroidota Strains Asinibacterium spp. OR43 and OR53, Bacteria, 2022, 1(1), 33–47 CrossRef
- B. C. Bostick, S. Fendorf, M. O. Barnett, P. M. Jardine and S. C. Brooks, Uranyl Surface Complexes Formed on Subsurface Media from DOE Facilities, Soil Sci. Soc. Am. J., 2002, 66(1), 99–108 CrossRef CAS
-
S. D. Kelly, Uranium chemistry in soils and sediments. Vol. 34, Developments in Soil Science. Elsevier Masson SAS, 2010, pp. 411–466 Search PubMed
- J. G. Catalano and G. E. Brown, Analysis of uranyl-bearing phases by EXAFS spectroscopy: Interferences, multiple scattering, accuracy of structural parameters, and spectral differences, Am. Mineral., 2004, 89(7), 1004–1021 CrossRef CAS
-
C. C. Fuller, M. J. Piana, J. R. Bargar, J. A. Davis and M. Kohler, Evaluation of Apatite Materials for Use in Permeable Reactive Barriers for the Remediation of Uranium-Contaminated Groundwater. Handb Groundw Remediat using Permeable React Barriers, 2003, (cv), pp. 255–80 Search PubMed
- A. Krot, I. Vlasova, A. Trigub, A. Averin, V. Yapaskurt and S. Kalmykov, From EXAFS of reference compounds to U(VI) speciation in contaminated environments, J. Synchrotron Radiat., 2022, 29(Vi), 303–314 CrossRef CAS PubMed
- C. C. Fuller, J. R. Bargar and J. A. Davis, Molecular-scale characterization of uranium sorption by bone apatite materials for a permeable reactive barrier demonstration, Environ. Sci. Technol., 2003, 37(20), 4642–4649 CrossRef CAS PubMed
- A. Sanding and J. Bruno, The solubility of (UO2)3(PO4)2 · 4H2O(s) and the formation of U(VI) phosphate complexes: Their influence in uranium speciation
in natural waters, Geochim. Cosmochim. Acta, 1992, 56(12), 4135–4145 CrossRef
- M. F. Fanizza, H. Yoon, C. Zhang, M. Oostrom, T. W. Wietsma and N. J. Hess,
et al. Pore-scale evaluation of uranyl phosphate precipitation in a model groundwater system, Water Resour. Res., 2013, 49(2), 874–890 CrossRef
- S. D. Kelly, K. M. Kemner and S. C. Brooks, X-ray absorption spectroscopy identifies calcium-uranyl-carbonate complexes at environmental concentrations, Geochim. Cosmochim. Acta, 2007, 71(4), 821–834 CrossRef CAS
- S. C. Brooks, J. K. Fredrickson, S. L. Carroll, D. W. Kennedy, J. M. Zachara and A. E. Plymale,
et al. Inhibition of bacterial U(VI) reduction by calcium, Environ. Sci. Technol., 2003, 37(9), 1850–1858 CrossRef CAS PubMed
- R. J. Martinez, C. H. Wu, M. J. Beazley, G. L. Andersen, M. E. Conrad and T. C. Hazen,
et al. Microbial Community Responses to Organophosphate Substrate Additions in Contaminated Subsurface Sediments, PLoS One, 2014, 9(6), 1–11 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ew00846d |
| This journal is © The Royal Society of Chemistry 2025 |