
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAspects in cell design for H2O2 electrosynthesis and its integration in tandem systems†
Wenhao Chen
,
Chang Sun
and
Wenchao Sheng
 *
*
State Key Laboratory of Pollution Control and Resource Reuse, College of Environmental Science and Engineering, Tongji University, Shanghai Institute of Pollution Control and Ecological Security, Shanghai 200092, P. R. China. E-mail: wsheng@tongji.edu.cn
First published on 3rd February 2025
Abstract
Hydrogen peroxide (H2O2) is an environment-friendly oxidant with wide applications in daily life and the chemical industry. The electrochemical production of H2O2 through the two-electron oxygen reduction (2e− ORR) process has the advantages of high safety, high energy-efficiency, and environmental sustainability. Prior investigations predominantly concentrated on the intrinsic properties of the catalysts, rather than the performance of the electrodes in real reactors. In this review, the aspects in cell design for H2O2 electrosynthesis will be discussed, including the surface and interface modifications for the carbon electrodes, and the reaction system design for practical H2O2 electrosynthesis, highlighting the critical needs in electrodes and reactors to enhance 2e− ORR performance. Additionally, this review will cover the applications of 2e− ORR integrated tandem systems for chemical synthesis. Finally, current challenges and prospects for future studies in H2O2 electrosynthesis will be presented.
 Wenhao Chen | Wenhao Chen is currently a MSc candidate under the supervision of Prof. Wenchao Sheng in the College of Environmental Science and Engineering at Tongji University. He received his BSc degree from East China University of Science and Technology in 2022, with a major in safety engineering. His current research mainly focuses on the electrode design and device applications of oxygen reduction reactions. |
 Chang Sun | Chang Sun is currently a PhD candidate under the supervision of Prof. Wenchao Sheng in the College of Environmental Science and Engineering at Tongji University. She received her BSc degree from Northeast Forestry University in 2019, majoring in environmental science. Her current research mainly focuses on the fundamental science in oxygen electrocatalysis and applications of renewable energy technologies. |
 Wenchao Sheng | Wenchao Sheng is currently a professor in the College of Environmental Science and Engineering at Tongji University. She obtained her BSc and MSc from the Department of Chemistry at Tongji University, and her PhD from the Department of Chemistry at MIT. After conducting postdoctoral research at the University of Delaware and Columbia University, she joined the faculty of the CESE at Tongji University. Her research interests include fundamental aspects of electrocatalysis in renewable energy storage and conversion technologies, electrochemical synthesis of high-value chemicals, and synthesis and characterization of nanostructured materials for energy applications. |
Broader contextHydrogen peroxide (H2O2) is a versatile green oxidizing agent with growing demand in the global market for its extensive applications in medical disinfection, pulp bleaching, chemical synthesis, wastewater treatment etc. The electrocatalytic two-electron oxygen reduction reaction (2e− ORR) is an environment-friendly and energy-efficient alternative approach compared to the traditional energy-intensive anthraquinone process, owing to its advantages of on-site production, green feedstocks from air, and non-polluting H2O as the only byproduct. With the development of high-performance 2e− ORR catalysts, research focuses have been gradually geared towards practical reactors to facilitate scaled-up H2O2 electrosynthesis. This review presents a comprehensive overview of cell design for H2O2 electrosynthesis and its integration in tandem systems for chemical synthesis, emphasizing key aspects such as electrode surface wettability management, interface microenvironment modification, design of reactor key components, practical challenges, and the integration of the 2e− ORR in tandem systems for synthesizing value-added chemicals. The information and insights delivered in this review could be potentially adopted for other electrocatalytic reactions such as carbon dioxide reduction and nitrogen reduction reactions. |
1 Introduction
Hydrogen peroxide (H2O2) is one of the 100 most important chemical compounds.1 As an environment-friendly oxidant, H2O2 has a growing global market demand for its extensive applications in medical disinfection, pulp bleaching, chemical synthesis, wastewater treatment, etc.2–5 The industrial production of H2O2 primarily relies on the conventional energy-intensive anthraquinone (AQ) process (∼95%), which produces substantial amounts of waste organic pollutants (5,6,7,8-tetrahydroanthrahydroquinone etc.) in addition to carbon emissions, posing a severe risk to water environment safety. Besides, the high-concentration H2O2 (70 wt%) produced through the AQ process presents potential safety risks in transportation.6The electrochemical synthesis of H2O2 based on the two-electron oxygen reduction reaction (2e− ORR), which selectively converts O2 to H2O2, with H2O as the only by-product, offers a green, safe and energy-efficient alternative to the conventional anthraquinone process.7 Recently, numerous catalysts have been developed for the 2e− ORR, including precious metals, non-precious metals and carbon materials.8–10 Precious metal catalysts, such as PtHg4 alloy11 and Pt-based sulfides, selenides and phosphides (PtS1.38,12 Se2-Pt,13 and PtP214), are regarded as the benchmark catalysts for the 2e− ORR due to their outstanding electrocatalytic performance. The high cost and scarcity of precious metal catalysts, however, limit their large-scale applications. In the realm of non-precious metal catalysts, despite their intrinsic instability compared to precious metals, recent innovations of nanostructured materials have resulted in various electrocatalysts with comparable 2e− ORR performance, including Co and Ni based sulfides and selenides (NiSe215 and sc-CoSe216), as well as single-atom and dual-atoms catalysts (Ni-N3S,17 Co1-NBC,18 Co2-DAC,19 and Coln-NC20). In addition, recent studies show that metal-free carbon-based catalysts with adjustable electronic structures, good electrical conductivity and stable chemical properties have great potential in H2O2 electrosynthesis. Various strategies such as heteroatom doping, defect engineering, and structural engineering have been developed and summarized previously.10,21,22
Beyond advancing electrocatalytic materials, the design and optimization of the 2e− ORR system is equally important for highly-efficient H2O2 electrosynthesis. Recent studies have underscored the importance of electrode surface/interface microenvironment modification and reactor configuration innovations to accelerate the reaction kinetics and H2O2 selectivity.23,24 It was revealed that the 2e− ORR performance can be influenced by surface wettability,25 interface proton concentration,26 and interfacial electric fields.27 For instance, electrode wettability management is found to be important for the formation of three-phase interfaces (TPIs), which not only shortens the O2 diffusion path distance from ∼50 μm to ∼50 nm to reach the active sites,28 but also avoids further reduction of H2O2 in the catalytic layer.29 Improper wettability management, such as excessive hydrophobicity, would have negative impacts. In terms of reactor configuration innovations, gas diffusion electrode (GDE) based reactors such as flow-cells, solid–electrolyte cells, and membrane electrode assembly cells have been increasingly reported for different application scenarios. The optimization of the key components of a reactor such as the gas flow channel and electrolyte compartments is critical to enhance the overall reactor performance. For example, the rational design of gas flow channels in a reactor can facilitate O2 transfer by vertical diffusion, increasing the H2O2 production rate.30,31 Meanwhile, GDE-based reactors may encounter additional challenges in real-world applications, such as the Joule heating effect,32 electrode fouling issue,33 and membrane degradation issue, which have not been fully understood yet.
While electro-synthesized H2O2 can be directly used in traditional downstream applications, it's in situ coupling with tandem systems for value-added chemical synthesis has attracted increasing attention lately. The high concentration of H2O2 near the electrode surface can be used to accelerate the synthesis of high-purity chemicals, such as ethylene epoxidation to produce ethylene glycol.34 Besides, the in situ generated reactive oxygen species (ROS) from H2O2 has also been utilized to activate inert reactants such as methane (CH4) and nitrogen (N2).35,36 Hitherto, no comprehensive analysis and review have focused on the optimization of the 2e− ORR system and its integration in tandem systems for high-value chemical synthesis.
In this review, we summarize the latest advances in practical reaction systems for H2O2 electrosynthesis. As illustrated in Fig. 1, the management of surface wettability (①) and interface microenvironment (②) will be discussed in section three, emphasizing the advantages of electrodes with appropriate hydrophobicity, as well as the effects of modifying the interface proton concentration and electric field. Design of reactor key components (③) and reactor configuration (④) will be reviewed in section four, focusing on energy-producing and energy-consuming modes; the challenges and mitigation measures in real-world electrosynthesis will also be discussed. In section five, we will introduce applications of 2e− ORR integrated tandem systems for chemical synthesis (⑤), and explore the H2O2 concentration effect and the ROS effect, highlighting the promising application potential of in situ generated H2O2.
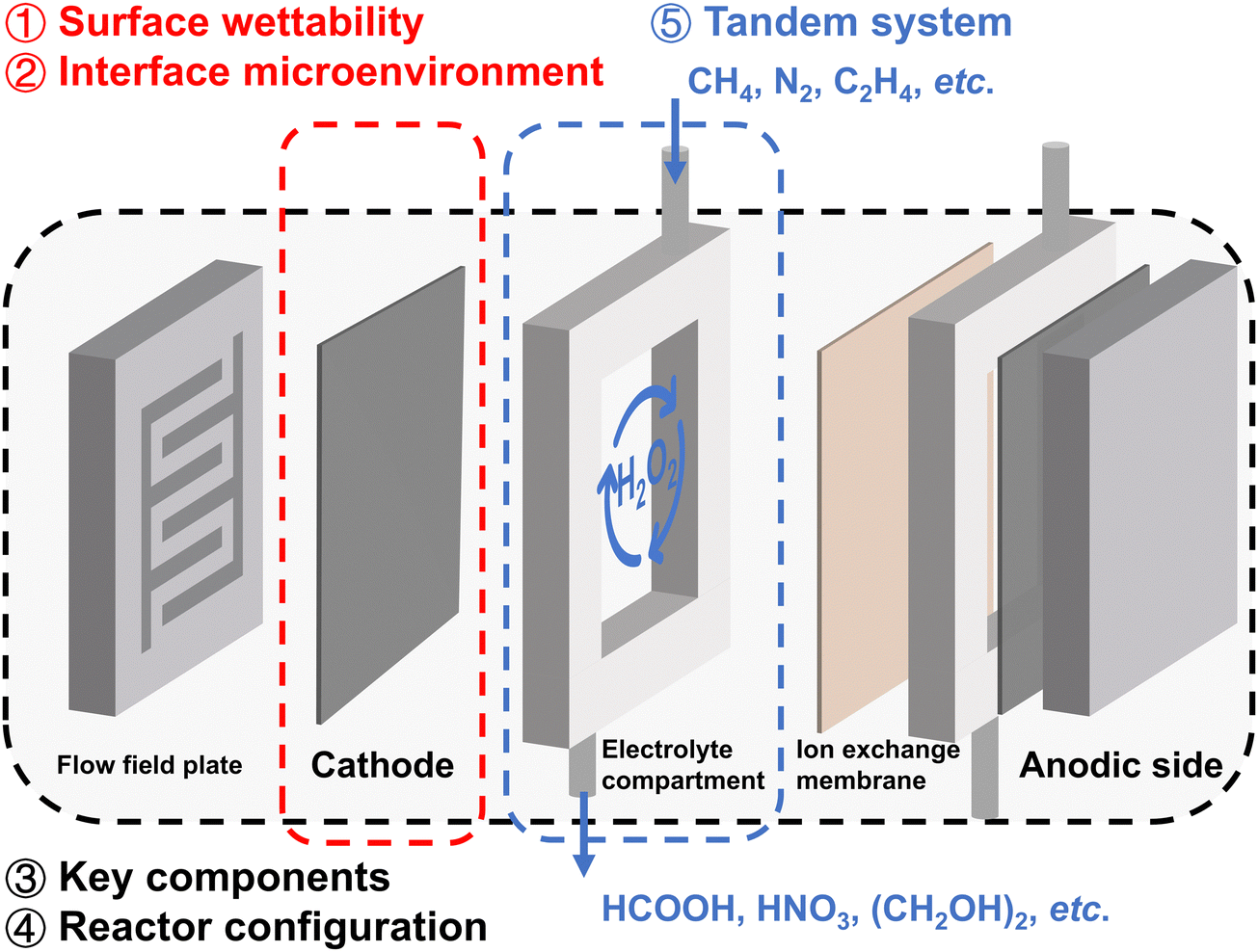 | ||
| Fig. 1 Schematic illustration of the reactor design for H2O2 electrosynthesis and its integration in tandem systems. This review covers the four subtopics (1)–(4) in reactor design and (5) 2e− ORR integrated tandem systems for chemical synthesis. | ||
2 Fundamentals of electrochemical synthesis of H2O2 via the ORR
The oxygen reduction reaction (ORR) is a complex multi-electron transfer process, following either a four-electron pathway to produce H2O (acid in eqn (1) and base in eqn (2)) or a two-electron pathway leading to the production of H2O2 (acid in eqn (3) and base in eqn (4)). The thermodynamic equilibrium potentials are versus the reversible hydrogen electrode (RHE).| O2 + 4H+ + 4e− → 2H2O E0 = 1.23 V | (1) |
| O2 + 2H2O + 4e− → 4OH− E0 = 1.23 V | (2) |
| O2 + 2H+ + 2e− → H2O2 E0 = 0.70 V | (3) |
| O2 + H2O + 2e− → HO2− + OH− E0 = 0.74 V | (4) |
Note that in alkaline electrolyte solutions (pH > 11.6), the product of the two-electron pathway changes from H2O2 to HO2−; the thermodynamic equilibrium potential changes accordingly.
Fig. 2a illustrates the electrocatalytic 2e− ORR process, including the mass transfer of O2 from the bulk electrolyte to the electrode surface, adsorption of O2 at the electrode surface, electrocatalytic reduction of O2 to H2O2, desorption of H2O2, and the mass transfer of H2O2 back to the bulk electrolyte. The adsorption of O2, charge transfer at the interface, and desorption of H2O2 are directly linked to the catalyst performance, while the mass transfer of both O2 and H2O2 is primarily influenced by the reaction reactor configurations.
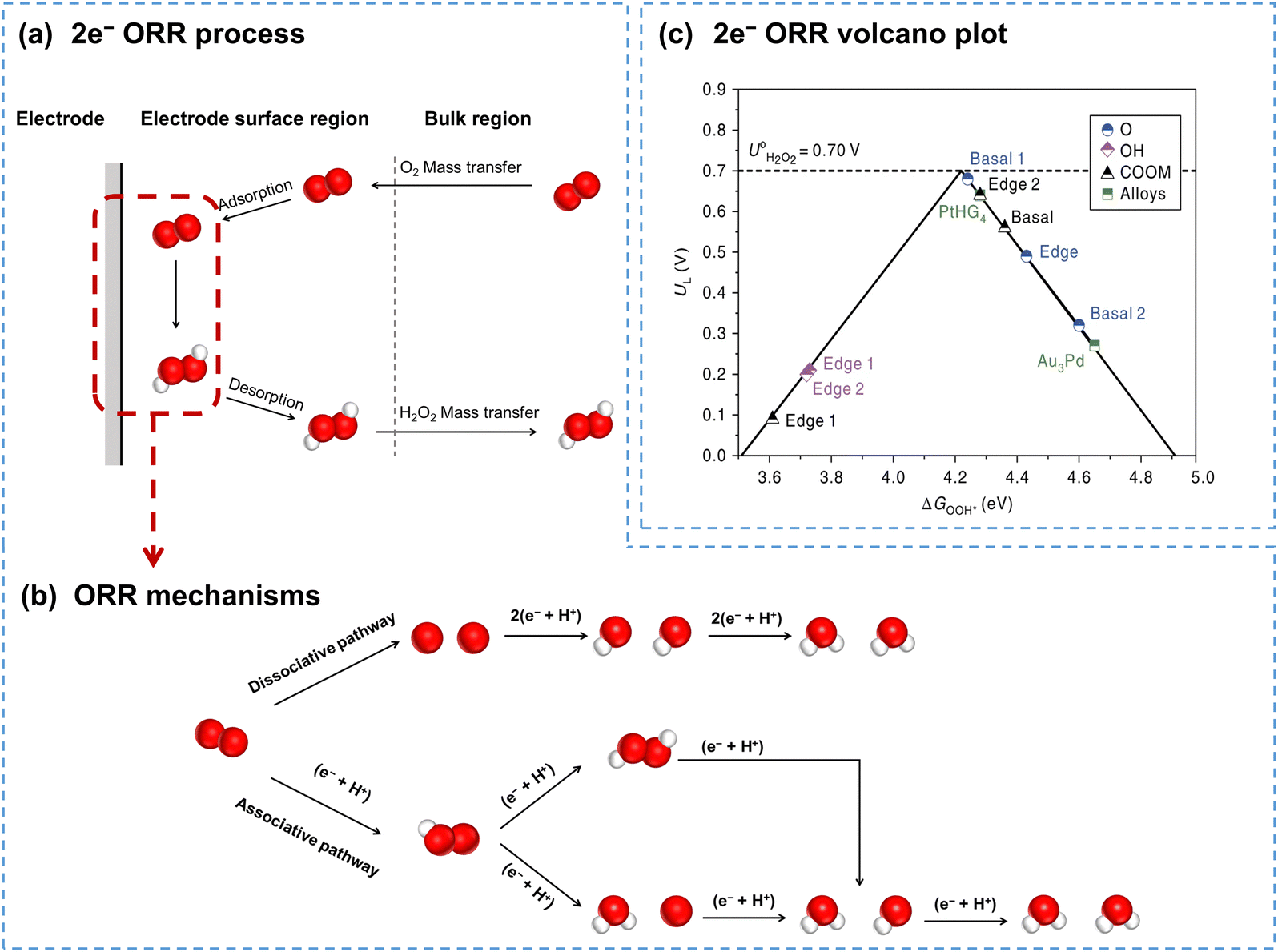 | ||
| Fig. 2 (a) Schematic of the 2e− ORR process; (b) schematic of ORR mechanisms including dissociative and associative pathways; (c) calculated 2e− ORR volcano plot, with the limiting potential plotted as a function of ΔG*OOH, reprinted with permission from ref. 37. Copyright 2018, Springer Nature. | ||
ORR processes involve multiple electron and proton transfers, and several reaction intermediates (Fig. 2b). O2 can be fully reduced to H2O through the dissociative pathway or reduced to H2O2 through the associative pathway. In the associative pathway, O2 is reduced to form the *OOH intermediate first (eqn (5)), which is subsequently reduced to generate H2O2 (eqn (6)). Notably, the *OOH intermediate can also dissociate, and is consequently reduced to H2O (eqn (7)–(9)). Besides, H2O2 can also be further reduced to form H2O (H2O2RR), as shown in eqn (10).
| O2 + * + H+ + e− → *OOH | (5) |
| *OOH + H+ + e− → H2O2 + * | (6) |
| *OOH + H+ + e− → H2O + *O | (7) |
| *O + H+ + e− → *OH | (8) |
| *OH + H+ + e− → H2O + * | (9) |
| H2O2 + 2H+ + 2e− → 2H2O | (10) |
The 2e− ORR mechanism highlights the crucial role of the *OOH intermediate; thus, the binding energy of ΔG*OOH has been generally accepted as the descriptor for the 2e− ORR. Fig. 2c presents the volcano plot of the 2e− ORR on carbon catalysts with various oxygen functional groups. Catalysts on the right branch exhibit weak binding to *OOH, making it difficult for *OOH formation, while the catalysts on the left bind to *OOH too strongly, leading to O–O bond cleavage and ultimately forming H2O. Catalysts with the optimal *OOH binding energy, favoring *OOH formation and further desorption, exhibit an ideal 2e− ORR performance.
Selectivity, activity, and stability are the key parameters for evaluating 2e− ORR catalysts. For fundamental studies, the rotating ring-disk electrode (RRDE) is commonly used, wherein O2 is reduced at the disk electrode, and the produced H2O2 will be detected at the Pt-ring electrode. The H2O2 molar selectivity can be calculated using eqn (11).
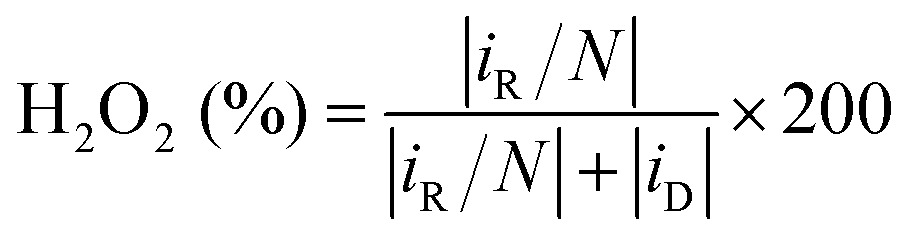 | (11) |
Faraday efficiency (FE), is also an important parameter to evaluate H2O2 selectivity, if the energy consumption is the major concern (eqn (12)).38
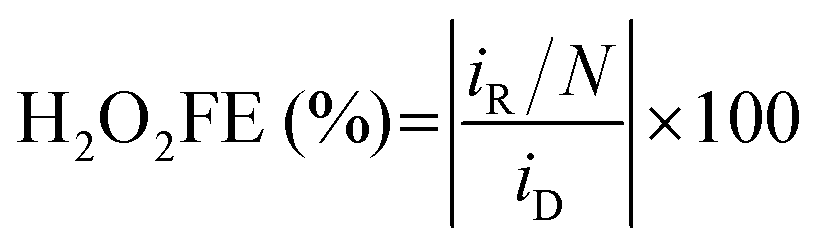 | (12) |
The H2O2 selectivity data obtained using RRDE cannot accurately reflect the selectivity in actual reactors, such as H-cells and flow-cells. The discrepancy arises because H2O2 can be rapidly transferred and detected on RRDE, while in actual reactors, the H2O2 generated on the electrode surface may proceed with further reduction before detection, resulting in a lower detected selectivity.6 Typically, the FE of H2O2 in actual reactors (λFE) is calculated using titration and/or UV-vis spectrophotometry, following eqn (13).
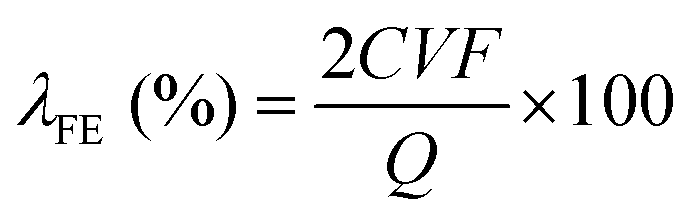 | (13) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1) and total electric charge consumed (C) respectively.
485 C mol−1) and total electric charge consumed (C) respectively.
The 2e− ORR activity of catalysts can be compared using onset potential and overpotential. Typically, the onset potential is defined as the potential at which the ring current density reaches 0.1 mA cm−2. Catalyst stability can be evaluated by comparing the 2e− ORR performance changes before and after the accelerated durability test, or the current density decline during the chronoamperometry test.39 The Pt-ring electrode should be cleaned in a timely fashion, since its surface could be oxidized during the stability test, making the quantification of H2O2 inaccurate.40
3 Carbon electrode surface wettability and interface microenvironment
3.1 Carbon electrode surface wettability management
The management of electrode surface wettability has garnered increasing attention with the continuous development of electrocatalytic reaction systems, especially for reactions involving gaseous reactants or products, for which the optimization of surface wettability is essential. The interaction between gases, electrolytes and catalysts significantly influences the performance of those gas-involving reactions. | ||
| Fig. 3 (a) Electrode surface wetting states: Wenzel state, Wenzel–Cassie coexistent state, and Cassie state, adapted from ref. 41; (b) schematic of species transfer at the electrode scale, and (c) H2O2 production performance of CB/Nafion-thin and CB/PTFE-thin electrodes at different current densities; (b) and (c) are reprinted with permission from ref. 29 copyright 2024, Springer Nature; (d) the evolution of electrode surface wetting states and apparent current efficiency (ACE) during the ORR process; data were taken from ref. 45; (e) the H2O2 current density at 0.60 V measured in both RRDE and GDE cells at different wetting states; data were taken from ref. 46. | ||
Gas-involving electrocatalytic reactions include gas-evolution reactions (GERs) and gas-consuming reactions (GCRs). For GERs, most studies focus on adjusting the electrode surface to the Wenzel state, which facilitates the timely removal of gas bubbles generated during the reaction due to its strong hydrophilicity, and allows full recovery of active sites on the electrode surface.47–49 For GCRs, the surface wettability must be carefully tailored due to the varying diffusion paths of gaseous reactants in reaction systems.
The 2e− ORR is a typical gas-consuming reaction. When O2 diffuses to the electrode surface in the form of dissolved oxygen, hydrophilic electrodes in the Wenzel state are considered advantageous, and are commonly used in fundamental research systems like RRDE and H-cells.25 For instance, a honeycomb carbon nanofiber (HCNF) electrode with a nearly zero contact angle could increase the H2O2 selectivity from 65% to 97% in the RRDE system.50 Recently, by heat-treating the porous carbon materials (HGC) at different temperatures, You et al.51 found that HGC550 treated at 550 °C with the highest hydrophilicity showed superior 2e− ORR performance compared with other electrodes with relatively low hydrophilicity. Further H-cell experiments demonstrated that HGC550 operated stably with 95% H2O2 selectivity for 12 hours, achieving a cumulative H2O2 concentration of up to 4000 ppm, among the top performances using the H-cell configuration.
The O2 mass transfer in the GDE system is more complicated and noteworthy. Initially, gaseous O2 diffuses through the GDE substrate, and further reaches the active sites in the catalyst layer through the gas phase and/or aqueous diffusion (diffusion in gaseous oxygen and/or dissolved oxygen) depending on the wettability of the catalyst layer.46 When the GDE surface is fully hydrophilic (Wenzel state, left in Fig. 3b),29 gas channels are easily flooded by the electrolyte, and O2 can only diffuse in the dissolved form, which results in a sluggish reaction rate because the diffusion coefficient of dissolved oxygen is three orders of magnitude slower than that of gaseous oxygen in GDE.52,53 On the other hand, when the surface is in a fully hydrophobic Cassie state, proton transfer from the electrolyte to the electrode surface will be hindered, which is detrimental to the reaction. Therefore, the GDE surface should be neither excessively hydrophilic nor hydrophobic. When the GDE surface is in a Wenzel–Cassie coexistent state, a gas–solid–liquid three-phase boundary is achieved on the electrode surface, facilitating both O2 and proton transfer, and in turn the efficient production of H2O2 (right in Fig. 3b).29,54,55 In addition, wettability management is also decisive for the efficient mass transfer of the produced H2O2. The reactive electron flux distribution on the hydrophilic electrode surface is in the catalytic layer immersed in the electrolyte, which can easily induce unwanted HER and H2O2RR; when the electrode is in a Wenzel–Cassie coexistent state, the reactive electron flux will be distributed on the three-phase boundary to perform the 2e− ORR. As evidenced in Fig. 3c, the H2O2 yield of the hydrophilic CB/Nafion-thin electrode initially increased and then decreased with increasing current density, while the yield of the CB/PTFE-thin electrode in the Wenzel–Cassie coexistent state increased continuously, which was attributed to the fast mass transfer rate of the produced H2O2.29 The evolution of electrode surface wettability was experimentally observed by Xia et al.45 As shown in Fig. 3d, the electrode surface in the Cassie state exhibited low current during the first 100 seconds. The electrode surface quickly transitioned to a Wenzel–Cassie coexistent state, which exhibited the highest activity and selectivity, lasting for nearly 2000 seconds. As the reaction further progressed, the electrode surface shifted to a Wenzel state, resulting in a sharp drop in current from 400 mA to 100 mA, and a reduction in apparent current efficiency (ACE) from 100% to 55%, which was attributed to the flooding by the electrolyte.
Generally, the carbon electrode wettability can be adjusted by controlling the surface functional groups and/or doping with heteroatoms. Note that the intrinsic activity will also be altered simultaneously, which should be considered when adjusting the electrode wettability. By modifying the hydrophilicity/hydrophobicity of carbon black (CB) electrodes, Xing et al.46 found that the CB electrodes exhibited different trends for their surface hydrophilicity/hydrophobicity in RRDE and GDE configurations (Fig. 3e). In RRDE, the activity of CB increased significantly with increasing hydrophilicity, while increasing its hydrophobicity hardly affected the activity. Nevertheless, the performance of GDEs after both hydrophilic and hydrophobic modifications exhibited a double volcano (M-shaped) correlation with the hydrophilicity/hydrophobicity of CB. Both F-doping and O-doping affected the 2e− ORR performance of carbon electrodes by influencing the number of TPIs. For the hydrophobically modified electrode (F-doped), the number of TPIs initially increased with increasing hydrophobicity, while excessive hydrophobicity would decrease the number of solid–liquid interfaces, thus decreasing the current density. For a hydrophilic modified electrode (O-doped), the activity initially increased due to increased intrinsic activity and increased solid–liquid interfaces, receiving more dissolved oxygen from the electrolyte. However, further hydrophilic treatment leads to electrode flooding, reducing the TPIs. Further stability tests showed that the hydrophobically optimized electrode (C-F-400) maintained a stable current density of 200 mA cm−2 and a FE of 90% for at least 12 hours, whereas the hydrophilic optimized electrode (C-O-6h) exhibited an 80% decrease in current density within 4 hours, due to severe electrode flooding.
The aforementioned studies suggest that appropriate hydrophobic modification is preferred to enhance the overall 2e− ORR performance when using the GDE system. In general, there are two main strategies for hydrophobic modification of electrodes: using hydrophobic polymers and increasing the roughness of the electrode surface.25,56
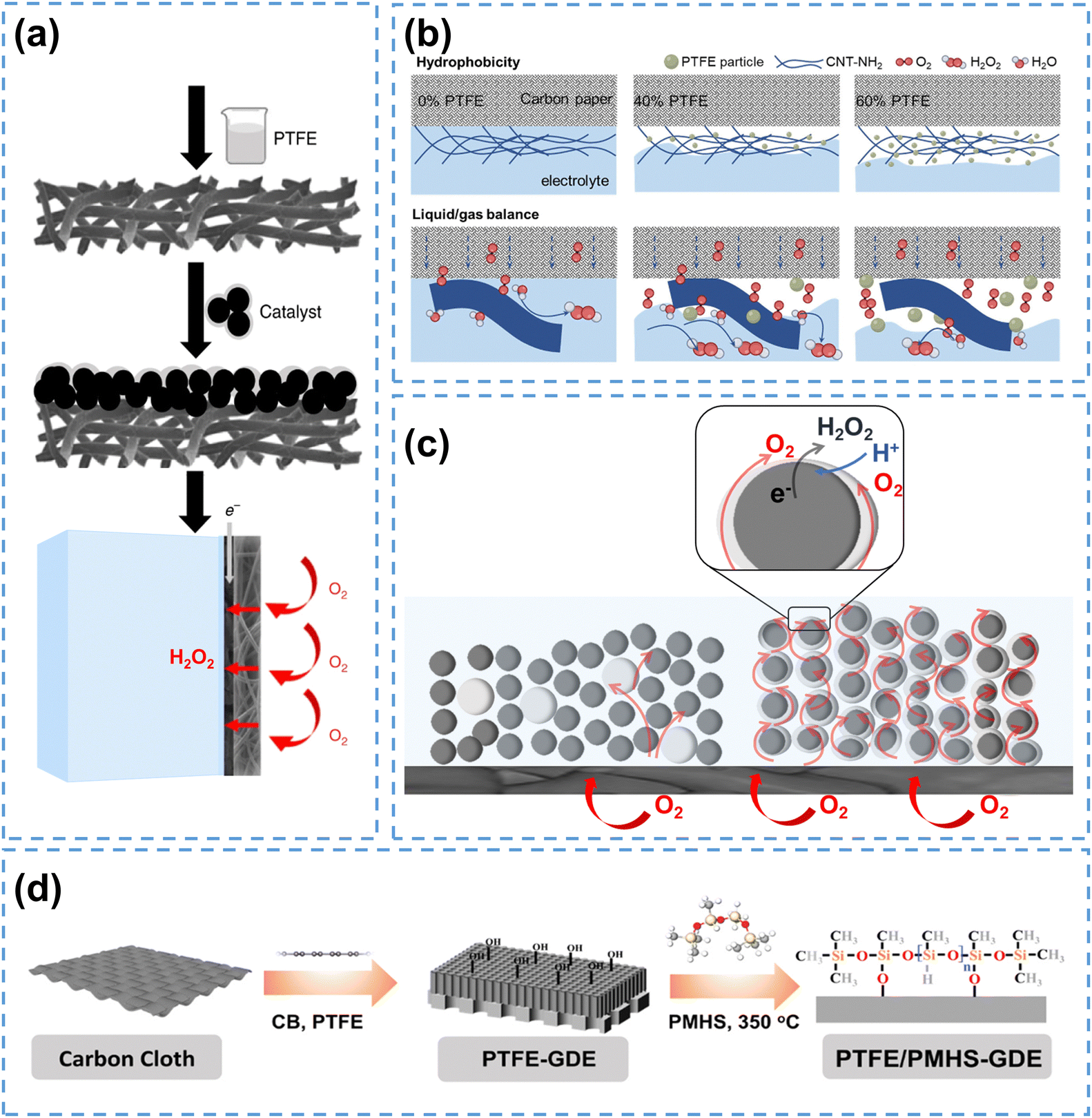 | ||
| Fig. 4 (a) The fabrication process of hydrophobic air diffusion electrodes, reprinted with permission from ref. 57, copyright 2020, Springer Nature; (b) schematic of the CNT-NH2/PTFE electrodes with different PTFE mass loading, reprinted with permission from ref. 58, copyright 2023, Wiley-VCH; (c) schematic of the short-range effect of PTFE NPs and optimization strategy using a thin and intact polymer layer on the catalyst surface, adapted from ref. 59; (d) the PTFE/PMHS-GDE fabrication process, reprinted with permission from ref. 60, copyright 2024, Elsevier. | ||
When subjected to heat treatment above 350 °C, PTFE will melt and bond with the carbon materials, which further enhances the hydrophobicity of carbon electrodes. By calcinating PTFE-containing GDE at 360 °C, Yu et al.61 successfully improved the H2O2 yield to 472.9 mg L−1. Scanning electron microscope images showed that the carbon fibers were tightly bonded with PTFE, and contact angle tests confirmed increased hydrophobicity.
It has to be pointed out that, the calcination process of PTFE will potentially block the active sites since PTFE can encapsulate carbon materials at high temperatures, and its agglomeration can obstruct the pore structure of GDE.62 To solve these issues, PTFE nanoparticles (NPs) are directly used without heat treatment to enhance the hydrophobicity of carbon electrodes and prevent active site blockage. Hu et al.58 physically mixed PTFE NPs with carbon nanotubes (CNT-NH2) to create hydrophobic electrodes. As illustrated in Fig. 4b, the electrode surface without PTFE NPs was in a fully wetted Wenzel state, where O2 could only participate in the form of dissolved oxygen. When the PTFE mass ratio increased to 60%, the electrode surface converted to a completely non-wetted Cassie state, thus hindering the contact between the electrode and electrolyte. When the PTFE mass ratio was 40%, the electrode surface achieved a partially wetted Wenzel–Cassie coexistent state, enabling the formation of TPIs, and thus improving the H2O2 production.
Despite its excellent performance in modifying the surface wettability, the addition of PTFE reduces the ionic conductivity, thus hindering the electrocatalytic reaction. To address this issue, Rawah et al.63 incorporated sodium polystyrene sulfonate (AS-4), an anionic conducting polymer, into the PTFE layer, which established an anionic transport channel and enhanced the ionic conductivity, enabling efficient HO2− transfer. The modified carbon electrode achieved a current density of 250 mA cm−2 at 2.2 V with a H2O2 selectivity above 90%.
Another issue associated with PTFE is that the physical mixing of PTFE NPs with carbon catalysts to construct TPIs may have a short-range effect, which may only modify the hydrophobicity of catalyst particles nearby. This phenomenon has been observed in a recent study using the same strategy to construct TPIs for electrochemical reduction of CO2 (CO2RR),59 and the authors suggested that decorating a thin hydrophobic polymer layer on the catalyst surface could enable the hydrophobic modification for all the catalyst particles, and enhance electrocatalytic performance by forming abundant TPIs. This concept can be potentially applied in H2O2 electrosynthesis, as illustrated in Fig. 4c. Recently, Xia et al.60 employed a novel molecular engineering strategy by binding polymethylhydrosiloxane (PMHS) onto the catalyst surface to achieve hydrophobic modification of the carbon electrode, which exhibited higher H2O2 yield and better stability compared to conventional GDEs (Fig. 4d). Similarly, Wang et al.64 used 1-octadecanethiol (ODT) for surface decoration to prepare a hydrophobic electrode (HMCSs@ODT), which exhibited enhanced 2e− ORR performance. Using hydrophobic polymers offers a general strategy for hydrophobicity management, and further efforts should be focused on developing more stable and environment-friendly hydrophobic polymers for H2O2 production.
The wettability of electrodes is directly linked to the wettability of the catalysts, and electrodes composed of hydrophobic catalysts typically exhibit hydrophobic properties. For instance, Cao et al.65 synthesized a highly hydrophobic N-doped carbon catalyst (NPC) with ZIF-8 as the precursor, which enhanced the gaseous oxygen utilization. Consequently, the NPC electrode maintained a FE of 90% under 250 mA cm−2 and operated it stably at 100 mA cm−2 for 200 hours, while the hydrophilic treated NPC electrode experienced severe water flooding in less than 10 hours. Another study illustrated that N-doped carbon catalysts with higher porosity possessed better 2e− ORR performance,66 which probably was also attributed to the higher hydrophobicity.
Considering the high preparation costs of hydrophobic catalysts, it is more practical to engineer microstructures with rough surfaces directly onto the electrode to manage the hydrophobicity. Plasma-enhanced chemical vapor deposition (PECVD) can be used to create carbon structures with precise size and morphology on electrodes,67–69 which has been well-established and may be suitable for large-scale preparation of hydrophobic electrodes for H2O2 electrosynthesis. Wang et al.70 used the PECVD method to grow ordered vertical graphene (VG) arrays on carbon paper, which exhibited enhanced hydrophobicity and 2e− ORR performance compared to those with a disordered graphene electrode, delivering a superior selectivity of 94% and a H2O2 yield of 61.3 mg h−1 cm−2 at 100 mA cm−2. It was reported that subsequent vacuum treatment could lead to the formation of hydrocarbons at the edges of the graphene structure and enhance the hydrophobicity of VG electrodes.71,72 Zhang et al.73 applied similar methods to prepare a hydrophobic self-supporting GDE, which ensured the formation of TPIs during the electrocatalytic process, exhibiting 97% selectivity and improved 2e− ORR activity (Fig. 5a).
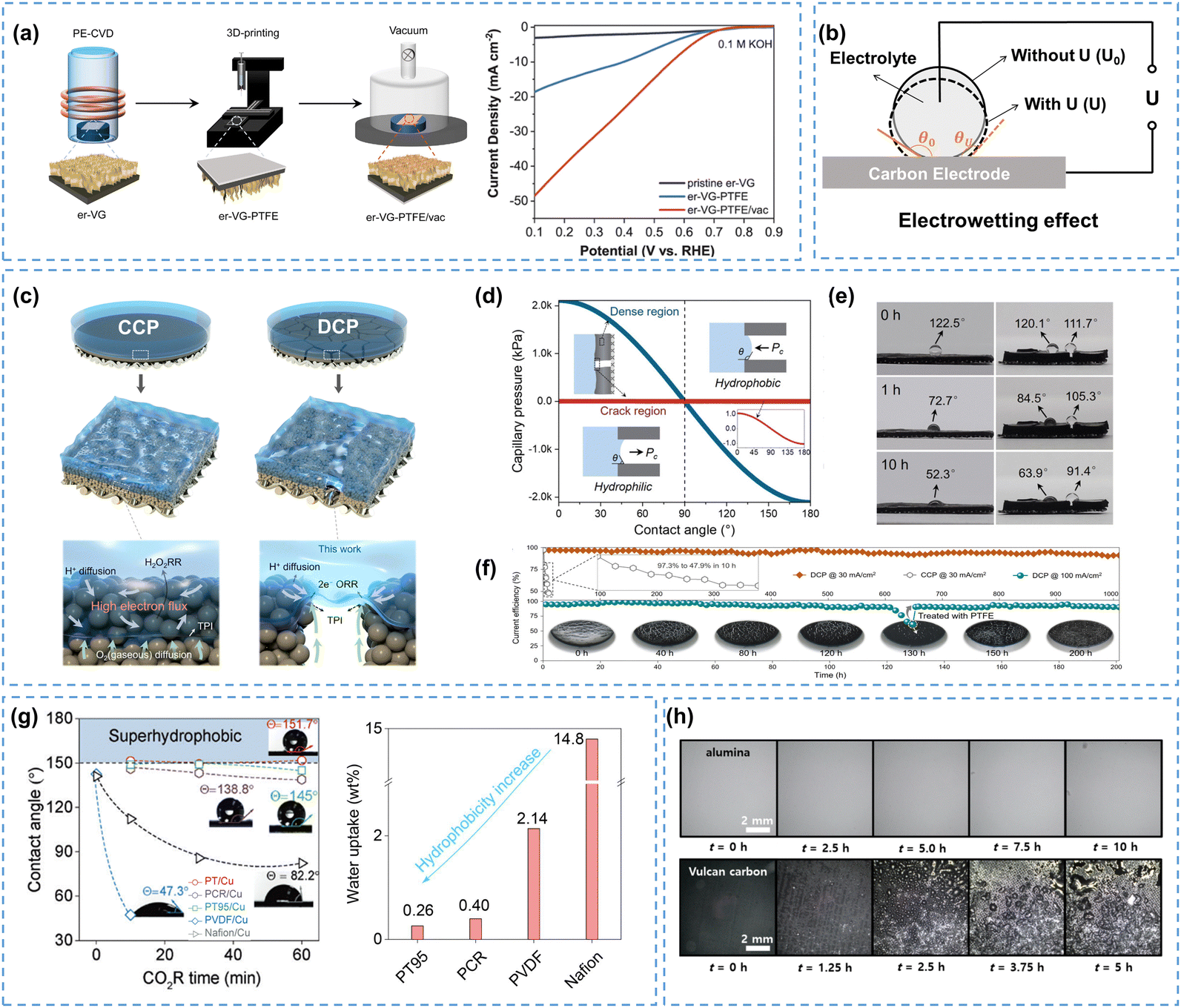 | ||
| Fig. 5 (a) Schematic of the er-VG-PTFE/vac fabrication process, and the LSVs measured in 0.1 M KOH, reprinted with permission from ref. 73, copyright 2023, Elsevier; (b) schematic of the electrowetting effect, adapted from ref. 74; (c) structurally continuous (CCP) and discontinuous (DCP) CB-PTFE film electrodes, (d) calculated capillary pressure for the dense region and crack region of the DCP surface vs. contact angle, (e) contact angles of CCP (left) and DCP (right) electrode surfaces during the reaction, and (f) stability comparison of CCP and DCP electrodes; (c)–(f) are reprinted with permission from ref. 75, copyright 2024, Royal Society of Chemistry; (g) contact angles for five polymer/Cu GDEs at −0.5 A cm−2 (left) and water uptake ability of five polymers (right), reprinted with permission from ref. 76, copyright 2024, Springer Nature; (h) flooding behavior comparison of alumina and carbon-based GDLs during electrolysis, reprinted with permission from ref. 77, copyright 2024, American Chemical Society. | ||
Although many strategies have been developed to enhance electrode hydrophobicity, the electrowetting phenomenon, referring to the transition of an electrode surface from a hydrophobic state to a hydrophilic state due to the electric field-driven reduction of solid–liquid surface tension,74,78 is unavoidable and has often been overlooked. Electrowetting will result in a decreased contact angle on the electrode surface (eqn (14), Fig. 5b),74 which is more pronounced in catalysts and/or electrodes without intentional hydrophobic modifications, posing a risk of electrode flooding.
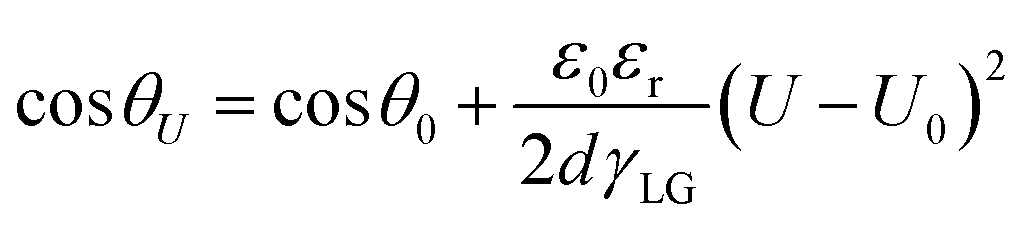 | (14) |
Eqn (14) is the Lippmann–Young equation, where θU and θ0 are the contact angles between liquid and solid at U and U0; ε0 and εr are the absolute dielectric constant in vacuum and the relative dielectric constant of the dielectric; d is the average thickness of the dielectric; γLG is the surface tension coefficient at the interface between liquid and gas.
Suppressing electrowetting is crucial for the industrialization of H2O2 electrosynthesis, and researchers have mainly focused on the electrode microstructure, polymer coatings, and GDL materials to mitigate this inevitable phenomenon. Recently, Cui et al.75 developed discontinuous CB-PTFE films (DCP) with anti-electrowetting properties, which improved the H2O2 production rate compared to traditional continuous CB-PTFE films (CCP), as shown in Fig. 5c. Cracks on the surface of the DCP electrode caused spatial discontinuities that weakened the electric field, thereby reducing the electrowetting effect. The lower capillary pressure in the cracked region (Fig. 5d) prevented the DCP electrode from being excessively hydrophilic or hydrophobic, constructing efficient TPIs. The contact angles of the dense region in both CCP and DCP electrodes were severely dropped from 122.5° to 52.3° and from 120.1° to 63.9°, respectively, exhibiting susceptible electrowetting properties. In contrast, the crack region of the DCP electrode showed stable hydrophobicity after 10 hours of electrolysis (from 111.7° to 91.4°), which further confirmed its excellent anti-electrowetting property (Fig. 5e). No electrode flooding was observed during the 1000-hour stability test for the DCP electrode at 30 mA cm−2, while the CCP electrode was flooded within the initial few hours (Fig. 5f). The overall anti-electrowetting ability of the electrode can also be enhanced by using hydrophobic polymers, as evidenced in previous studies on the CO2RR.76,77 As shown in Fig. 5g, the contact angle of the electrode, modified by the most commonly used Nafion, decreased sharply within only 30 min and ended up at 82.2°. Surprisingly, the PT95-modified electrode exhibited super stable hydrophobicity (151.7°), exhibiting excellent anti-electrowetting properties during the CO2RR. These phenomena may be related to their water uptake abilities (14.8 wt% for Nafion; 0.26 wt% for PT95, shown on the right in Fig. 5g).76 It was also observed that the inherent conductivity of GDL plays a crucial role in electrowetting (eqn (14)), and materials with high conductivity, e.g. carbon, have been proven to be more easily flooded.79 Recently, Haaring et al.77 replaced the generally used carbon powder with non-conductive dielectric alumina to diminish the electrowetting effect, and found that the alumina GDL exhibited excellent resistance to electrowetting (over 10 hours), whereas the carbon GDL experienced severe flooding within only 2.5 hours (Fig. 5h).
3.2 Carbon electrode interface microenvironment modification
The carbon electrode surface wettability management physically establishes stable TPIs, facilitating efficient O2 mass transfer and ensuring highly efficient and stable H2O2 electrosynthesis. The chemical microenvironment at the electrode–electrolyte interface also affects the activity and selectivity for H2O2 electrosynthesis.The electrocatalytic reaction occurs within the confined nanoscale space between the electrode and electrolyte, known as the electrode–electrolyte interface.80 The electric double layer (EDL) model, which consists of the inner Helmholtz plane (IHP), outer Helmholtz plane (OHP) and diffusion layer, has been used to describe the electrode–electrolyte interface.81
The ions and solvent water molecules adsorbed in the double layer create the EDL microenvironment, which can be optimized to enhance the 2e− ORR performance. The potential of zero charge (PZC) refers to the potential at which an electrode has zero net charge, serving as a key parameter in describing the double-layer structure. When the working potential of the 2e− ORR is higher than the PZC, the electrode becomes positively charged, causing water molecules and anions to aggregate in the IHP due to electrostatic forces, while repelling cations from the electrode interface. Conversely, when the working potential of the 2e− ORR is below the PZC, the electrode carries negative charges, which results in the distribution of water molecules in the IHP with hydrogen atoms in water orienting towards the electrode interface.82 The high degree of hydration and large hydration radius of alkali metal cations (AMCs) make it difficult to reach the IHP at the electrode–electrolyte interface, and it is generally regarded that the hydrated AMCs will accumulate in the OHP under the influence of electrostatic attraction.83
In order to reach industrial-level current density, the working potential has to be set below PZC, which leads to the accumulation of adsorbed AMCs at the electrode surface, causing the cation effects.84,85 Specifically, the cation effects alter the hydrogen bond network connectivity, affect proton transfer, enhance the interface electric field strength, and influence the stability of reaction intermediates, which is directly related to the 2e− ORR performance. Therefore, optimizing the electrode interface microenvironment is essential to enhancing the H2O2 electrosynthesis performance.
Adjusting protons using AMCs. AMCs can be used to modify the proton concentration at an electrode interface.87,88 By adding trace amounts of Na+ into H2SO4 electrolyte, it was found that under industry-relevant current values, negatively charged carbon electrodes could accumulate protons and Na+ at the electrode interface by electrostatic interaction. Molecular dynamics simulations revealed that Na+ was more competitive in occupying the double layer at the electrode interface, effectively repelling protons. This shielding effect of Na+ on protons significantly reduced the proton concentration at the electrode interface, creating an alkaline-like microenvironment that improved H2O2 electrosynthesis, and this effect continued as the Na+ concentration increased. By adding 0.1 M Na+, the overpotential of the carbon electrode decreased by 0.6 V at 400 mA cm−2, and the selectivity increased by nearly 80% (Fig. 6a).86
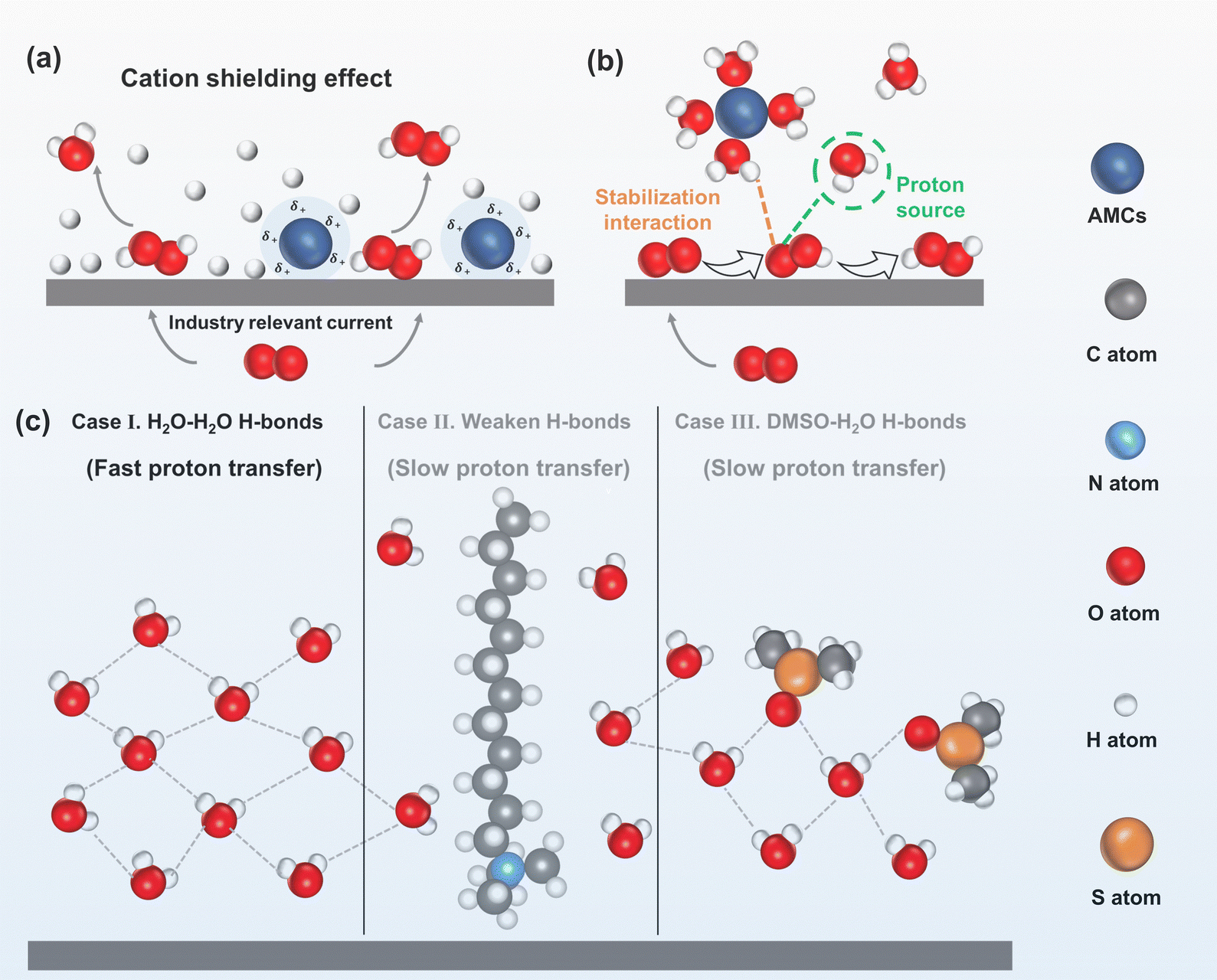 | ||
| Fig. 6 (a) Schematic of the 2e− ORR in acid with/without AMCs under an industry-relevant current, reproduced with permission from ref. 86 copyright 2022, Springer Nature; (b) schematic of the solvated AMCs and adsorbed *OOH, reproduced with permission from ref. 89, copyright 2024, Wiley-VCH; (c) schematic of the interfacial H-bond network: H2O–H2O H-bonds enable fast proton transfer (case I), weakened H-bonds induced by DTAB (case II), and structurally altered H-bonds induced by DMSO (case III); both cases II and III resulted in slow proton transfer; adapted from ref. 90 and 91. | ||
AMCs can also modify the proton transfer rate to the electrode interface. Recently, Cao et al.89 investigated the cation effect on proton transfer rate by adding K+ into the H2SO4 electrolyte. The solvated K+ in the OHP reduced the connectivity of the hydrogen-bonds (H-bonds) network near the electrode surface, and inhibited the transfer of H3O+ to the electrode interface. Molecular dynamics simulations revealed that K+ promoted H2O dissociation at the electrode interface, facilitating the protonation of *O2 or *OOH. Additionally, solvated K+ and nearby H2O could interact with the *OOH intermediate at the electrode interface, and enhance its stability (Fig. 6b).
Adjusting protons using organic additives. Cationic surfactants such as dodecyl tricaprylmethylammonium chloride (Aliquat 336), tetrabutylammonium bromide (TBAB), trimethylammonium bromide (DTAB), and cetyltrimethylammonium bromide (CTAB) can also adsorb on the electrode surface by electrostatic interaction as AMCs, and their unique chemical structures can introduce more possibilities. Gyenge et al.92 found that electrostatically adsorbed Aliquat A336 molecules were able to replace protons on the carbon electrode surface, reducing the proton concentration at the electrode interface. The pH value at the carbon electrode interface was estimated to increase from 0.9 to 9.4 in 0.1 M H2SO4 electrolyte, which improved the 2e− ORR performance of the carbon electrode. TBAB was also found to enhance H2O2 production on carbon electrodes through a similar mechanism.93
As water molecules serve as the proton donor for the 2e− ORR in alkaline electrolytes, and the H-bonds network at the electrode interface dominates proton transfer rate to the interface,94 controlling the number or altering the structure of H-bond networks can regulate the proton transfer rate and inhibit the H2O2RR. Fan et al.90 found that DTAB molecules adsorbed on the carbon electrode could repel water molecules away from the electrode interface. Therefore, the intensities of strong H-bonds in water and weak H-bonds in water at the electrode interface decreased from 3.02 to 0.69 and from 4.25 to 0.36 respectively, which weakened the strength of the H-bond network. Consequently, the proton transfer rate in the electrode interface was hindered, which inhibited the H2O2RR and enhanced H2O2 selectivity from 75% to 91% (case II in Fig. 6c).
However, the adsorption of cationic surfactants on the electrode may block the active sites and reduce electrode conductivity. Dimethyl sulfoxide (DMSO) is a strong hydrogen bond acceptor that can form robust H-bonds with water molecules, which was found to alter the original H-bond network structure.95 Recently, Fang et al.96 investigated the effect of DMSO on H2O2 electrosynthesis at carbon electrodes by altering the H-bond network. The result revealed that the DMSO formed stronger H-bonds with H2O (DMSO–H2O H-bonds) compared to H2O–H2O H-bonds. As DMSO could not supply protons, and the stronger H-bonds further inhibited water dissociation, this limited proton transfer to the electrode surface and in turn improved H2O2 selectivity from 60% to 90% (case III in Fig. 6c).
Adjusting protons by engineering carbon electrode microstructures. Although the addition of cationic surfactants and organic solvents to an electrolyte can modify proton at the electrode interface, it inevitably introduces impurities into the H2O2 solution. Designing the microstructure of carbon electrodes enables self-modification of protons at the electrode interface. For instance, Yang et al.97 built an O2-accumulation interface microenvironment by introducing a hydrophobic microporous layer into the GDE substrate, which promoted the proton-consuming rate and inhibited further protonation of H2O2, achieving approximately 100% H2O2 selectivity. Recent studies reported that precisely engineered carbon electrode structures, such as mesoporous hollow nanoreactors (MHNs),98 mesoporous carbon nanosheets (MeCNs),99 and micro/mesoporous N, O co-doped carbon nanosheets (N, O-CNSx),100 showed similar promoting effects by adjusting protons at the electrode interface and improved 2e− ORR performances.
It is worth noting that although an alkaline-like microenvironment at the electrode interface improved 2e− ORR performance, the negatively charged carbon electrode tends to repel OH− from the electrode surface to bulk solution, offsetting the promoting effect of alkaline-like microenvironments.101 Lewis acids were found to accept a pair of electrons from OH−, forming new chemical bonds that could enhance OH− adsorption.102,103 Based on this, Li et al.104 synthesized Lewis acid TiO2 modified CB (CB–xTiO2) catalysts using a thermal decomposition method; TiO2 adsorbed in situ generated OH− and established a stable alkaline-like microenvironment, which improved the H2O2 yield in neutral electrolyte (52.5 mmol L−1 h−1) close to that in alkaline electrolyte (58.4 mmol L−1 h−1).
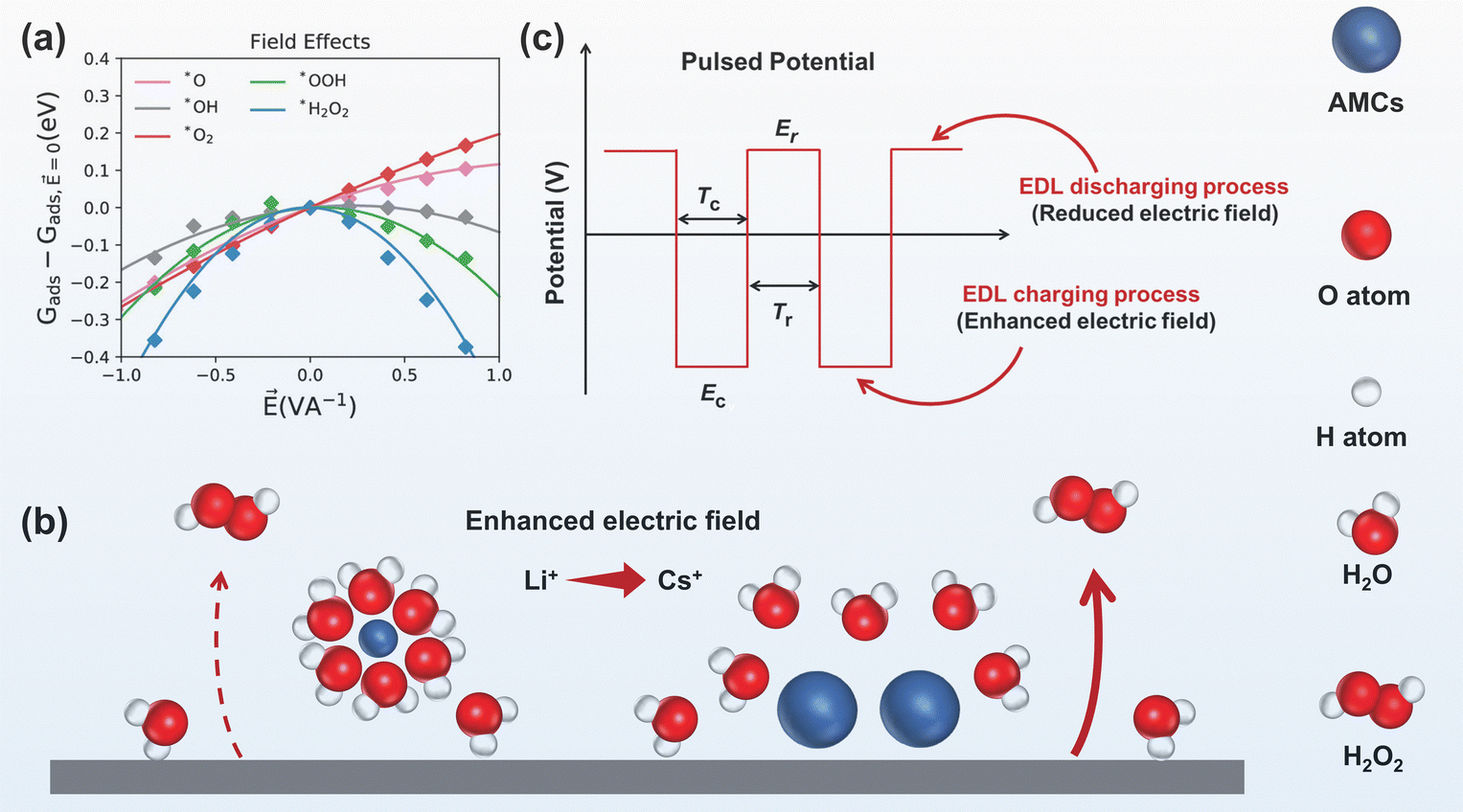 | ||
| Fig. 7 (a) Influence of simulated interfacial electric fields on the adsorption energies of various intermediates during the ORR, reprinted with permission from ref. 105, copyright 2020, American Chemical Society; (b) AMC size effect on the electric field, the large radius AMC (Cs+) enhanced electric field more pronouncedly than the small radius AMC (Li+), adapted from ref. 106; (c) parameters in pulsed potential and their effects on electric field, adapted from ref. 107. | ||
Addition of AMCs not only adjusts the proton at the electrode interface, as discussed in the previous section, but also modifies the electric field strength, which originates from the electrostatic interactions between the negatively charged carbon electrodes and AMCs.108,109 Density functional theory calculations revealed that solvated K+ in the double layer increased the interfacial electric field strength by 0.77–1.44 V Å−1, and the AMC-induced interface electric field effect could shift ΔG*OOH toward the top of the activity volcano plot.85
The AMC-induced interface electric field effect was also found to relate to the size of AMCs. A study showed that the enhancement was in the order of Cs+ > K+ > Li+.106 Li+ with smaller radius and higher charge density, attracted more water molecules and formed a larger hydration layer. Instead, Cs+ formed a smaller hydration layer due to its larger radius, making it easier to approach the electrode surface, which enhanced the interfacial electric field strength (Fig. 7b). Increasing the concentration of AMCs could further enhance the interfacial electric field strength, thus improving H2O2 electrosynthesis performance.
Similarly, adding cationic surfactants to the electrolyte can also modify the interfacial electric field strength through electrostatic effects. Chen et al.110 hypothesized that the adsorption of CTAB could reduce the distance between the OHP and IHP in the double layer, which enhanced the electric field strength at the electrode interface, and in turn optimized the binding energy of the *OOH intermediate. Consequently, the ORR activity increased by three-fold, and the H2O2 selectivity improved from 60% to 93%. The electric field strength at the electrode interface could also be modified by preparing electrodes with specific morphologies.111 For example, by using an Au needle electrode, the electric field strength increased by ten-fold, resulting in significantly improved CO2RR performance.112 This tip-induced electric field strength effect could be applied in the H2O2 electrosynthesis, however, to the best of our knowledge, no relevant research has been conducted in the field of the 2e− ORR.
Electrochemical techniques, such as pulse voltammetry, have been used to directly modify the strength of the electric field at the electrode interface, as shown in Fig. 7c. Key parameters include the cathodic pulsed potential (Ec), cathodic pulsed duration (Tc), rest potential (Er) and rest duration (Tr). Different from the constant potential electrocatalysis, pulse electrocatalysis involves repetitive charging and discharging of the electrode and modifies the electric field periodically. Ding et al.107,113 demonstrated that the pulse electrocatalysis method could modify the adsorption of *OOH intermediates by affecting the interface electric field strength. The discharging of the EDL at rest potential resulted in a reduced electric field strength at the electrode interface, decreasing the adsorption strength of the *OOH intermediate, which consequently increased the H2O2 yield by 138%.
4 Reactors for H2O2 electrosynthesis
Managing the surface wettability and modifying the interface microenvironment of carbon electrodes play critical roles in improving the 2e− ORR performance. Moreover, the design of reactor configurations directly influences the practical performance of H2O2 electrosynthesis. In this section, the design of the reactor key components will be further reviewed; the challenges and mitigation measures in real-world electrosynthesis will also be discussed.4.1 Reactor design
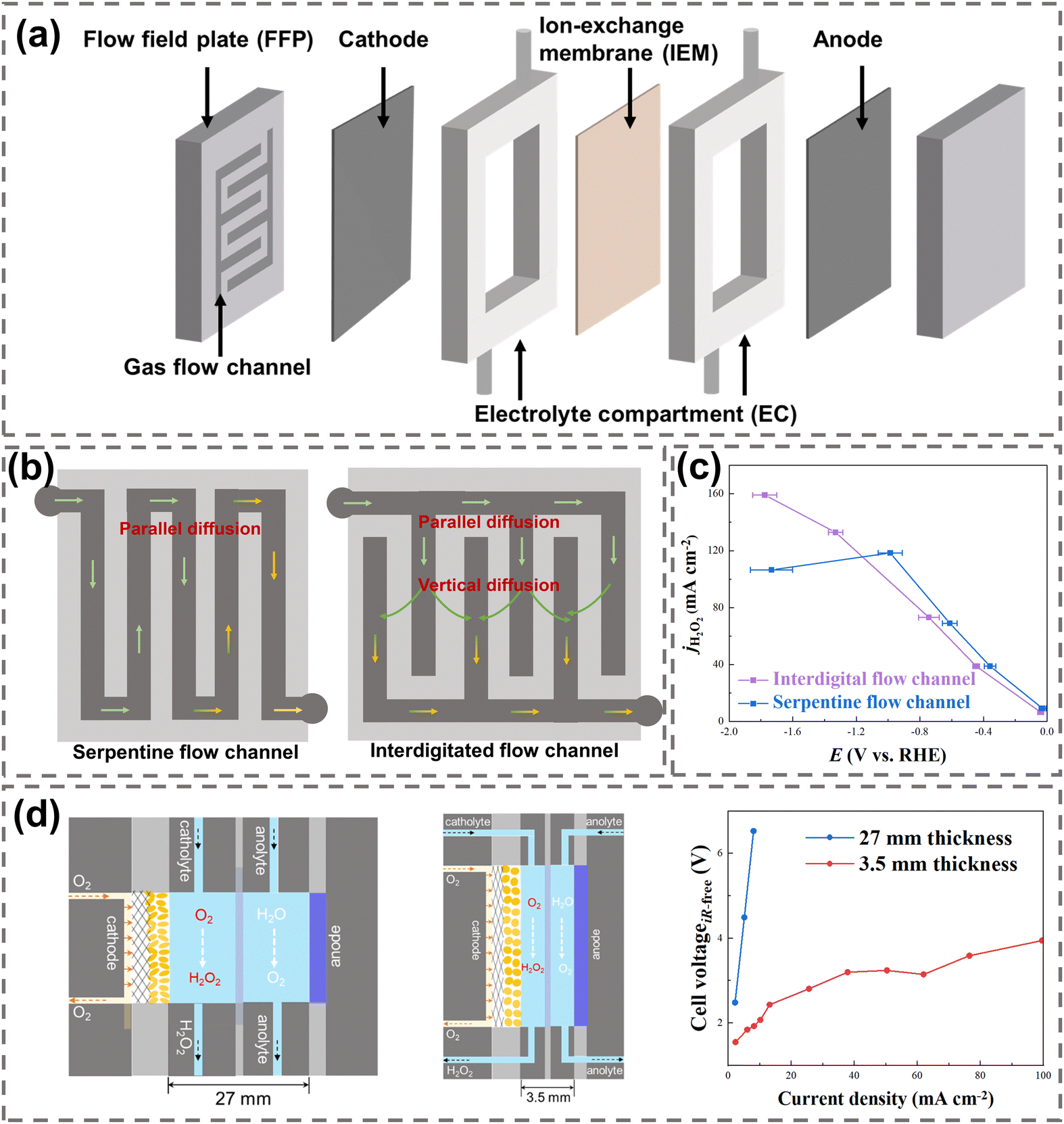 | ||
| Fig. 8 (a) Key components in a typical flow-cell reactor; (b) schematic of the FFPs with a serpentine flow channel (left) and interdigitated flow channel (right), reproduced with permission from ref. 31, copyright 2021, Springer Nature; (c) H2O2 current densities using a serpentine flow channel and interdigitated flow channel; data were taken from ref. 30; (d) schematic of H2O2 electrosynthesis using two reactors with different EC thicknesses (27 mm and 3.5 mm), and their H2O2 electrosynthesis performance, reprinted with permission from ref. 118, copyright 2023, Springer Nature. | ||
The flow field plate (FFP) is a crucial component of the reactor, which supports the reactor structure, conducts electrons, and transports gaseous reactants.24 The geometric design of gas flow channels in the FFP includes a straight parallel channel, serpentine channel, and interdigitated channel.119 A well-designed gas flow channel can effectively regulate the flow rate of O2 to the electrode, ensuring uniform distribution across the electrode surface and reducing pressure loss,120 which subsequently improves the O2 diffusion efficiency and H2O2 production.
Current studies have generally utilized a serpentine flow channel and interdigitated channel for H2O2 electrosynthesis,30,121 as illustrated in Fig. 8b. The serpentine flow channel (left in Fig. 8b) allows O2 to flow from the inlet to the outlet along the path, resulting in the parallel diffusion of O2 to GDE. Nevertheless, a significant loss of gas pressure will be observed in a long serpentine channel, which will reduce the O2 supply near the outlet of the channel and lead to electrode flooding.122 In the interdigitated channel (on the right in Fig. 8b) the O2 at the inlet initially diffuses parallelly to the GDE and is then forced to diffuse vertically into the electrode, which effectively enhances the mass transfer of O2 to the GDE.31 Electrocatalytic performance comparison between the serpentine and interdigitated flow channels showed that the current densities of H2O2 in both channels increased linearly with the rising overpotential at a current density below 100 mA cm−2. Moreover, the current density in the interdigitated channel kept increasing at a current density above 100 mA cm−2, while it decreased in the serpentine channel due to the insufficient O2 diffusion induced electrode flooding (Fig. 8c).30
Materials with high mechanical strength, excellent electrical conductivity, and high durability to corrosion are required for FFP fabrication in H2O2 electrosynthesis. Stainless steel is commonly applied in FFP fabrication due to its high mechanical strength and conductivity, but its durability is unsatisfactory as the generated H2O2 accelerates the corrosion and oxidation of metallic steel, forming metal oxide films that increase the contact resistance between the FFP and GDE.24 Compared to metallic materials, graphite sheets offer high durability for H2O2 electrosynthesis while maintaining high conductivity, but suffer from low mechanical strength and potential gas permeability issues. Developing graphite-based composite materials may address those issues to achieve high strength, conductivity, and durability.
The design of electrolyte compartment (EC) has a significant impact on the internal resistance of the reactor.123 To prevent H2O2 from reacting with EC materials, corrosion-resistant and high-strength plastics materials such as polyether ether ketone (PEEK) and polytetrafluoroethylene (PTFE) are commonly utilized. The thickness of EC directly affects the overall internal resistance of the reactor. By decreasing the EC thickness from 27 mm to 3.5 mm, the internal resistance of the reactor was reduced and the overall electrocatalytic performance was improved, with the cell voltage dropping from 6 V to 2 V at 10 mA cm−2, as shown in Fig. 8d.118 Notably, the addition of solid electrolyte (SE) in the EC could also effectively lower the internal resistance in reactors.124 Styrene-divinylbenzene copolymer microspheres, Dowex 1 × 8 anion conductor resins, and inorganic CsxH3−xPW12O40 proton conductors were often employed as the SE materials due to their high ion conductivity.22,117,125–127
It has to be pointed out that the evaluations of H2O2 electrocatalysts in the laboratories are typically performed with laboratory-scale three-electrode system GDE reactors, focusing on the half-cell reaction rather than the overall reaction performance. The internal resistance of the three-electrode system is directly related to the distance between the reference and working electrodes, rather than the EC thickness. Moreover, laboratory-scale GDE reactors possess a small working electrode area (1 cm−2) with short FFP channels, which could facilitate O2 diffusion. Therefore, in our opinion, the design of the FFP and EC has minimal improvement for H2O2 electrocatalytic in laboratory-scale GDE reactors, but it is very important to optimize those reactor components for industry-level H2O2 production.
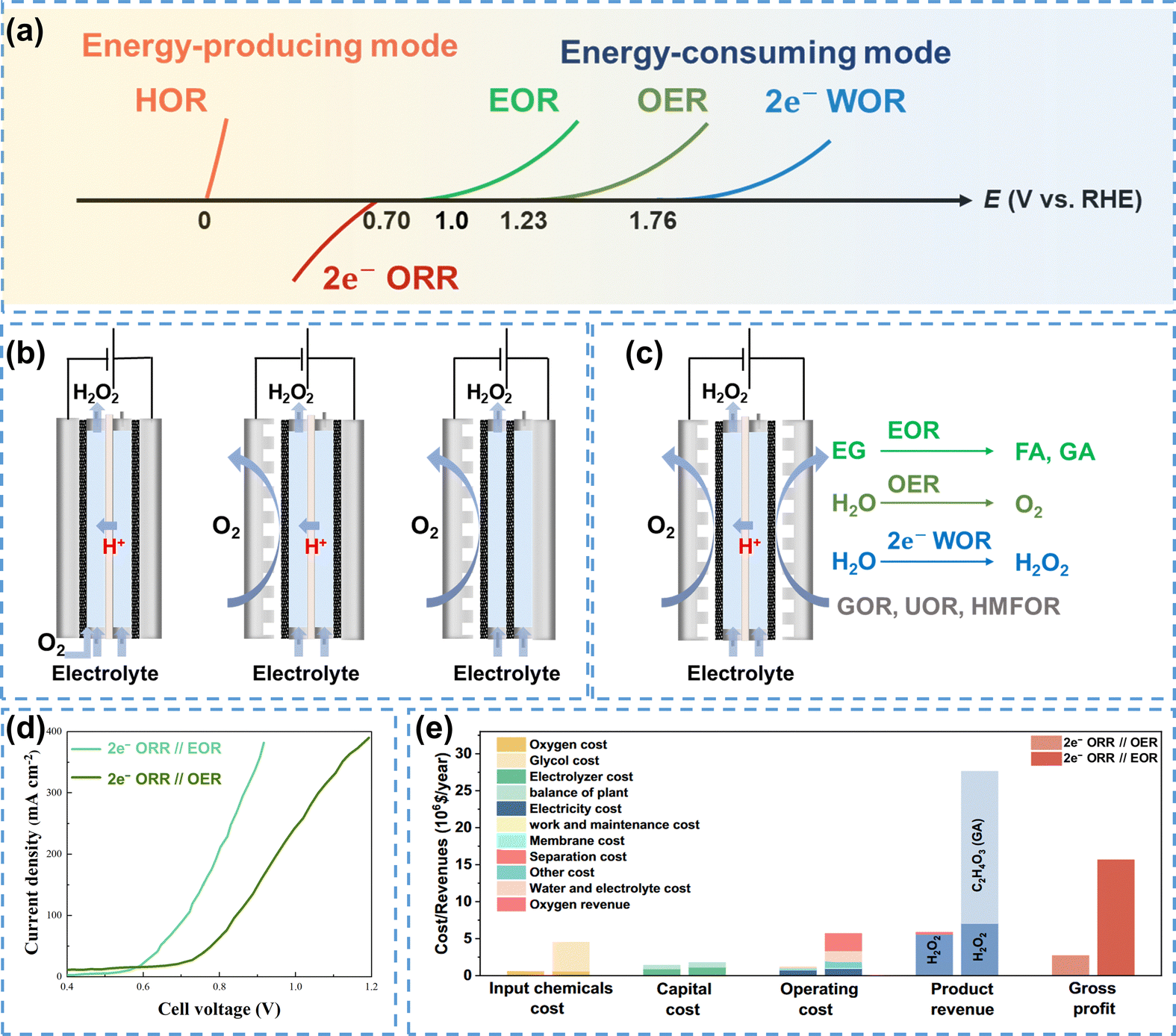 | ||
| Fig. 9 (a) Coupled systems of the 2e− ORR and possible anodic reactions; (b) the Huron–Dow process (left), adapted from ref. 128; conventional GDE flow-cell reactor (middle); membrane-free flow-cell reactor (right), adapted from ref. 129; (c) schematic of coupled systems for 2e− ORR production and high-value chemicals synthesis; (d) performance comparison of the 2e− ORR//EOR and 2e− ORR//OER systems, data were taken from ref. 130; (e) technoeconomic evaluation of the 2e− ORR//OER and 2e− ORR//EOR systems, reprinted with permission from ref. 131, copyright 2024, Springer Nature. | ||
In 1939, Berl proposed H2O2 electrosynthesis using activated carbon electrode as the cathode, which produced high concentrations of H2O2 and was anticipated to be commercially utilized.132 Initially, the commercial production of H2O2 through electrosynthesis relied on the Huron Dow process (on the left in Fig. 9b).128 This process used a strong alkaline solution as the electrolyte, wherein O2 was introduced into the electrolyte and reduced to HO2− at the cathode. The efficiency of this process was constrained by the low solubility of O2 in the electrolyte (∼9 ppm at 25 °C),133 as well as the inevitable decomposition of H2O2 in a strong alkaline solution.
The flow-cell using GDE as the cathode could significantly enhance the O2 mass transfer, and in turn increase the production rate of H2O2 (in the middle in Fig. 9b). However, the cation exchange membrane (CEM) used for separation would be gradually degraded by H2O2,134 which presented a great challenge to the long-term stability. A membrane-free reactor was then proposed by Chen et al.129 which could eliminate this issue ultimately. The reactor consisted of two carbon paper electrodes with the sides facing the catholyte coated with a hydrophobic polymer, which allowed the diffusion of gaseous O2 but prevented H2O2 from crossing to the anode (on the right in Fig. 9b). In addition, the membrane-free reactor exhibited improved H2O2 electrosynthesis due to the reduced internal resistance by removing the CEM.
The cathodic 2e− ORR is typically coupled with the anodic OER, which is kinetically sluggish and generates low-value O2.135 Replacing the OER with thermodynamically favorable organic oxidation reactions can improve the overall energy efficiency and economic viability of the system. Organic oxidation reactions such as the ethylene glycol oxidation reaction (EOR),130 glycerol oxidation reaction (GOR),136 urea oxidation reaction (UOR),137 5-hydroxymethylfurfural oxidation reaction (HMFOR),138 and furfural oxidation reaction (FuOR)139 have been used as the anodic reaction to couple with the 2e− ORR, as shown in Fig. 9c. Taking EOR as an example, the onset potential for the EOR is below 1 V, which is much lower than that required for the OER. In addition, ethylene glycol can be converted into value-added formic acid (FA) and glycolic acid (GA) through electrocatalytic reactions. As shown in Fig. 9d, the 2e− ORR//EOR coupled system required only 0.9 V to achieve an industrial level current density of 400 mA cm−2, while the 2e− ORR//OER system required a higher voltage of 1.2 V, which demonstrated the superior efficiency of the EOR-based system.130 Similarly, Sun et al.131 employed EOR as the anodic reaction and selectively oxidized ethylene glycol to high-value GA, which significantly increased the gross profit of the coupled system (Fig. 9e).
Theoretically, replacing the OER with the 2e− WOR could increase the H2O2 FE up to 200%, thus improving both the H2O2 production rate and the energy utilization efficiency.140,141 Xia et al.142 constructed the 2e− ORR//2e− WOR coupled system with oxygen-functionalized carbon nanotubes (O-CNTs) as the cathode and PTFE-coated carbon fiber paper (CFP-60%) as the anode, which enabled the simultaneous electrochemical synthesis of H2O2. This coupled system achieved an H2O2 FE exceeding 150% at 120 mA cm−2, highlighting the significant potential of carbon electrodes for the efficient production of H2O2 via 2e− ORR and 2e− WOR processes.
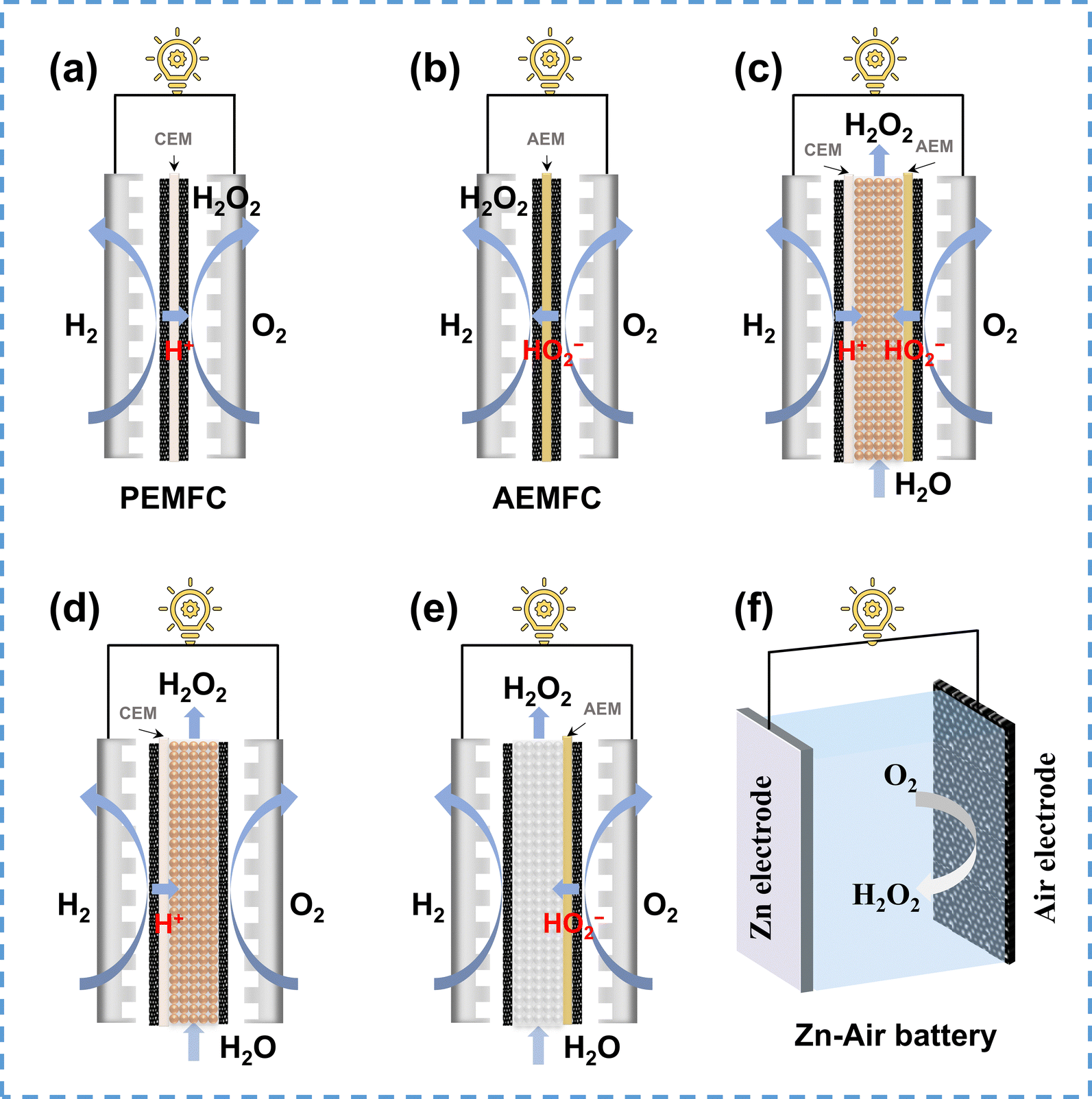 | ||
| Fig. 10 Schematic of various reactor configurations for H2O2 electrosynthesis in energy-producing mode: (a) proton exchange membrane fuel cell (PEMFC), adapted from ref. 143; (b) alkaline anion exchange membrane fuel cell (AEMFC), (c) dual membrane solid–electrolyte cell (SE-cell), adapted from ref. 147; (d) CEM solid–electrolyte cell, adapted from ref. 63; (e) AEM solid–electrolyte cell, and (f) Zn–air battery, adapted from ref. 148. | ||
The integrated PEMFC stack reactor is beneficial for large-scale H2O2 electrosynthesis. Nevertheless, the zero-gap structure of the MEA hinders the transportation and collection of produced H2O2, resulting in a high concentration of H2O2 within the MEA, which accelerates the H2O2RR and reduces the H2O2 selectivity. Various optimization strategies were adopted to mitigate this issue, including catalyst loading adjustment, MEA wettability modification, flow rate controlling, and reaction temperature regulation.149 Besides, the preparation methods of the MEA, including catalyst-coated membranes (CCM) and gas diffusion electrodes also influence the PEMFC performance for H2O2 electrosynthesis.150
In recent years, alkaline anion exchange membrane fuel cells (AEMFCs), which operate under milder conditions compared to PEMFCs, have garnered increasing attention. Carbon electrodes are well known for their excellent 2e− ORR performance under alkaline conditions, thus exhibiting great potential in AEMFC for H2O2 electrosynthesis.6 Unlike PEMFCs that produce H2O2 at the cathode, AMEFCs produce HO2− ions at the cathode, which pass through an anion exchange membrane (AEM) to the anode, and further combine with protons to form H2O2 (Fig. 10b). In addition to the H2O2 transfer hindrance in the MEA, another challenge arises in AEMFCs due to the strong decomposition ability of anodic catalysts towards H2O2, such as Pt/C, which inhibits the production of H2O2. The low ionic conductivity of AEM presents an additional challenge to the AEMFC performance,151 requiring more efforts to enhance the conductivity of AEM for better H2O2 electrosynthesis performance.
Using a solid electrolyte (SE) layer instead of conventional ion exchange membranes can effectively address the aforementioned issue of H2O2 transport in MEA, facilitating the co-generation of H2O2 and electricity. As illustrated in Fig. 10c, Xia et al.147 proposed a dual membrane solid–electrolyte cell consisting of AEM, CEM, and SE layers in between, which was composed of sulfonated styrene-divinylbenzene copolymer microspheres. In the reactor, H+ generated at the anode were conducted through the CEM and the HO2− generated at the cathode were conducted through the AEM, and they further combined to form H2O2 within the SE layer, ensuring high concentration H2O2 production (up to 20 wt%). Rawah et al.63 optimized the dual membrane reactor by removing the AEM and applying a hydrophobic treatment to the carbon electrode, which reduced the reactor internal resistance and enhanced the electrocatalytic performance (Fig. 10d). Following the same concept, the combination of anionic solid electrolyte and AEM could theoretically generate H2O2 in energy-producing mode, as depicted in Fig. 10e.
Apart from H2–O2 fuel cells, metal–air batteries such as zinc–air batteries (ZABs) are promising for H2O2 electrosynthesis under energy-producing mode. As shown in Fig. 10f, the typical structure of a ZAB consists of a zinc plate (for the oxidation reaction) and an air electrode (as the cathode for the 2e− ORR). Wang et al.148 first demonstrated the feasibility of H2O2 production using ZAB by employing a carbon electrode as the air electrode, achieving an output power of 36 mW cm−2 with a H2O2 yield of 0.593 mmol cm−2 h−1 at 0.8 V. Furthermore, Xie et al.152 improved the ZAB performance by using S-doped carbon as the air electrode, which increased the maximum output power to 82.7 mW cm−2 and maintained a high H2O2 production rate. Since H2O2 is unstable in strong alkaline electrolytes, which are commonly used in ZABs (6 M KOH), Lu et al.153 developed a neutral ZAB to avoid the decomposition of H2O2, which exhibited a high H2O2 selectivity of 91% and maintained a long-time stability up to 1500 hours in a low-temperature environment.
4.2 Considerations in practical-scale H2O2 electrosynthesis
Catalysts in a strongly oxidizing H2O2 environment could undergo dynamic evolutions, as the produced H2O2 may react with the catalysts, especially metal oxides,163,164 leading to changes in the electrochemical properties of the catalysts. For example, ZnO has been recognized as a promising catalyst for H2O2 production,165–167 however, this material could be converted to ZnO2 by H2O2,168 which inspires further investigations on the true active sites of ZnO electrocatalysts. Recently, Zhou et al.163 observed that using ZnO as the 2e− ORR catalyst, the H2O2 selectivity increased from 64.45% to nearly 100% within 6000 s (Fig. 11a). Raman spectroscopic studies of the ZnO pristine catalyst, activated at 0.2 V in O2-saturated electrolyte for different reaction times, confirmed the appearance of O22− signal (Fig. 11b), suggesting the in situ construction of a ZnO@ZnO2 heterogeneous structure. Density functional theory (DFT) calculations indicated that the heterogeneous interface between ZnO and the in situ constructed ZnO2 was likely the real active sites, which weakened the binding energies of both *OOH and *O2 and facilitated superior 2e− ORR performance (Fig. 11c). This hypothesis was further supported by the fact that ZnO@ZnO2 synthesized using 0.1 M H2O2 exhibited nearly identical selectivity as the ZnO@ZnO2 heterostructure evolved from ZnO under operando conditions. Similarly, using Ti2O3 as the initial 2e− ORR catalyst, the surface of Ti2O3 could transform into oxygen-deficient TiO2 during the reaction, which switched the ORR pathway from the 4e− to 2e− pathway (Fig. 11d), and this surface reconstruction from pristine Ti2O3 to TiO2 was subsequently confirmed by in situ Raman spectroscopy study (Fig. 11e) and XRD spectra collected after the ORR test (Fig. 11f).164
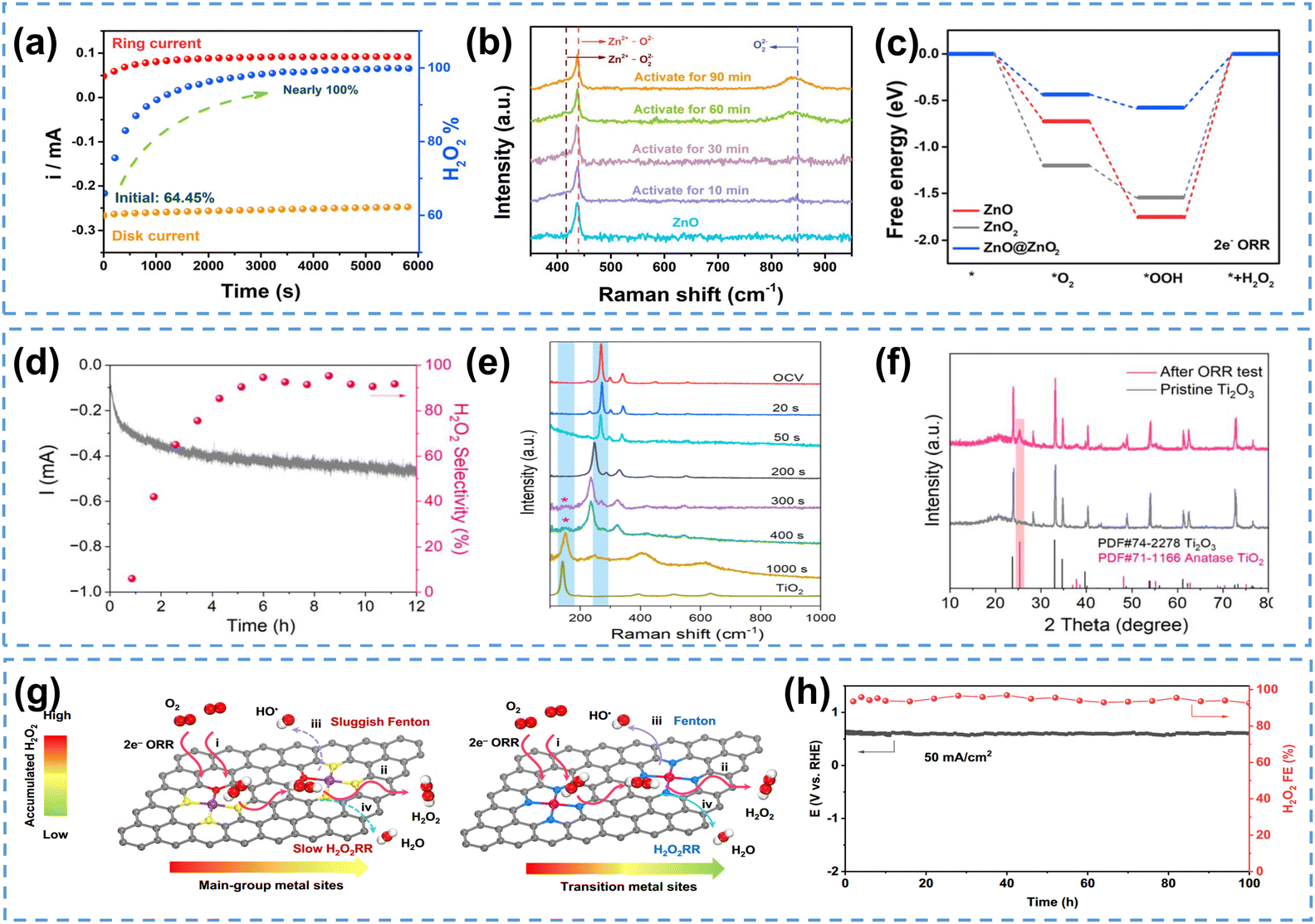 | ||
| Fig. 11 (a) Chronoamperometry measurement of the 2e− ORR on ZnO at 0.2 V, and H2O2 selectivity, (b) Raman spectra of ZnO electrochemically activated at 0.2 V for different periods of time, and (c) energy diagram of 2e− ORR intermediates on ZnO, ZnO2 and ZnO@ZnO2 (U = 0.7 V); (a)–(c) are reprinted with permission from ref. 163, copyright 2023, Royal Society of Chemistry; (d) chronoamperometry measurement of the 2e− ORR on Ti2O3 at 0.3 V and H2O2 selectivity, (e) in situ Raman spectra collected during the 2e− ORR on Ti2O3 at 0.2 V, and (f) XRD spectra of Ti2O3 before and after ORR electrocatalysis; (d) and (e) are reprinted with permission from ref. 164, copyright 2023, Royal Society of Chemistry; (g) schematics of Fenton reaction rate on the main-group metal catalysts (left) and transition metal catalysts (right), and (h) chronopotentiometry measurement on Pb SA/OSA at 50 mA cm−2, and H2O2 selectivity; (g) and (h) are reprinted with permission from ref. 169, copyright 2024, Springer Nature. | ||
The electrochemically generated H2O2 could also potentially induce the Fenton-like reactions, especially on transition metal catalysts,170 which produces strong oxidative ROS, raising concerns on the catalyst durability. Different from transition metals, main-group metals are Fenton-inactive for their fully occupied d-orbitals, and thus considered as an attractive option for mitigating Fenton-like reactions. Recently, Zhou et al.169 reported the main-group Pb SACs (Pb SA/OSC) catalyst for H2O2 electrosynthesis, achieving an industrial current density of 400 mA cm−2 with remarkable FE (∼90%). No ROS characteristic signals were observed during the 2e− ORR process, suggesting that Pb SACs had sluggish Fenton reaction rate compared to transition metal catalysts (Fig. 11g). This catalyst also exhibited excellent durability without degradation at 50 mA cm−2 (FE maintained over 90%) for at least 100 hours (Fig. 11h). It is also found that adding a radical scavenger (typically CeO2) could protect the catalyst from being attacked by radicals and improve the durability of ORR catalysts in PEMFC applications.171,172 This strategy could be potentially applied in 2e− ORR electrocatalyst design to rapidly eliminate ROS and improve the catalyst durability.
Joule heating is a universal phenomenon in electrolysis processes, which originates from the internal ohmic resistance in devices (eqn (15)).
| Q = i2Rt | (15) |
During electrolysis processes, Joule heating is mainly manifested by an increase in electrolyte temperature, severely affecting the H2O2 selectivity by promoting its self-decomposition reaction rate, especially in an alkaline environment under industrial current density.32 Recently, Ni et al.173 investigated the effect of current density on Joule heating by measuring the temperature of the electrolyte during electrolysis, and found that the temperature of the electrolyte gradually rose from 32.7 °C to 49.3 °C with increasing current density from 100 mA cm−2 to 300 mA cm−2 (Fig. 12a). Calculations showed that the H2O2 FE negatively correlated with the electrolyte temperature, consistent with experimental observations, indicating the promoting effect of Joule heating on H2O2 self-decomposition (Fig. 12b).
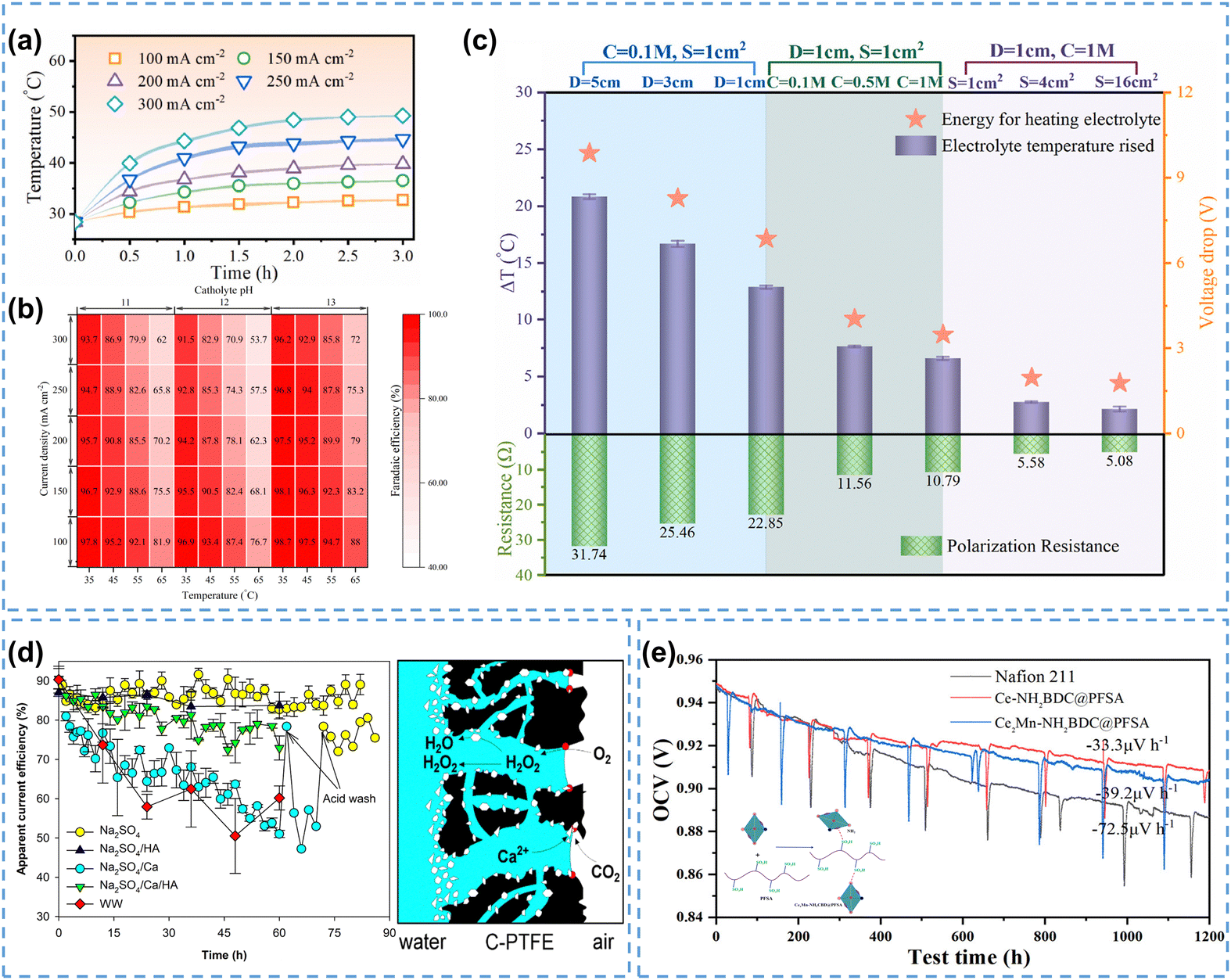 | ||
| Fig. 12 (a) Temperature of the electrolyte during the reaction at different current densities, (b) calculated H2O2 FE under different current densities and temperatures, (c) cell voltage drop, electrolyte temperature change, and cell resistance under different cell configurations; (a) and (b) are reprinted with permission from ref. 173, copyright 2024, American Chemical Society, and (c) is reprinted with permission from ref. 32, copyright 2024, Elsevier Ltd; (d) apparent current efficiency of H2O2 production in different natural waters (left), and schematic of CaCO3 precipitation process during H2O2 electrosynthesis (right), reprinted with permission from ref. 33, copyright 2023, Elsevier Ltd; (e) OCV holding test of Ce2Mn-NH2BDC@PFSA, CeNH2BDC@PFSA, and Nafion 211 membranes, and schematic of the MOF and PFSA composite membrane (inset), reprinted with permission from ref. 174, copyright 2024, American Chemical Society. | ||
The Joule heating effect not only reduces the H2O2 FE, but also brings additional energy loss of the entire reactor. The cell resistance is regarded as a determining factor in the Joule heating effect, and the mitigation measures include reducing the electrolyte compartment thickness and increasing electrolyte conductivity etc., which have been reviewed in Section 4.1.1. Researchers validated the effectiveness of these measures by reducing the electrode distance from 5 cm to 1 cm, increasing the electrolyte concentration from 0.1 M to 1 M, and expanding the electrode area from 1 cm2 to 16 cm2. Correspondingly, the cell resistance declined from 31.74 Ω to 5.08 Ω, and the temperature change (Δt) dropped from 20.9 °C to 2.2 °C, indicating a suppressed Joule heating effect (Fig. 12c).32 Considering that the scale-up of H2O2 electrosynthesis to industrial current density level would inevitably magnify the Joule heating effect, heat management becomes indispensable. Integrating the reactor with a cooling system may be an effective solution in terms of suppressing the Joule heating effect.
For household disinfection and/or waste water treatment, it is desirable to directly use tap water or natural water due to their accessibility, instead of the de-ionized water used in laboratories, in order to reduce the overall processing cost. However, the existing hardeners (such as Ca2+, Mg2+) and other dissolved organic matter in natural water usually cause fouling issues on electrodes during the electrocatalysis process, resulting in decreased electrode stability and selectivity. For example, Ma et al.33 found that the addition of Ca2+ in Na2SO4 solution led to a continuous decrease in the apparent current efficiency of H2O2 on carbon-based air diffusion electrodes (on the left in Fig. 12d). The CaCO3 precipitates negatively influenced the electrode by covering the active sites, and more severely, the hydrophilic CaCO3 precipitates could induce electrode flooding, resulting in sluggish mass transfer of O2 (on the right in Fig. 12d). The fouling issue caused by Ca2+/Mg2+ could be relieved by using complexing agents, which bond with Ca2+/Mg2+, significantly reducing their precipitation on the electrode surface.175
As one of the limiting factors for the long-term operation of H2O2 reactors, the stability of ion exchange membranes presents another challenge for the industrial production of H2O2. Currently, perfluorinated sulfonic acid (PFSA) based membranes (such as Nafion membranes) are commonly used for H2O2 electrosynthesis, however, they are prone to chemical degradation, which could be accelerated by various ROS species.176–179 Recently, novel stable membranes such as sulfonated polyphenylene membranes,180 hydrocarbon-based membranes,181 and sulfonated fluorene-based poly(phenyl ketone) membranes182 have been developed. Huang et al.174 utilized a self-assembly method to synthesize a molecular organic framework (MOF)-PFSA composite membrane (Fig. 12e inset), in which the MOF material (Ce2Mn-NH2BDC) could efficiently scavenge free radicals and mitigate the membrane degradation issue in PEMFCs. The open circuit voltage (OCV) decay rate of this composite membrane was 33.3 μV h−1 within a 1200-hour test, much smaller than 72.5 μV h−1 for the commercial Nafion 211 membrane, which indicated its excellent chemical stability (Fig. 12e). These novel ion exchange membranes have the potential to be applied in H2O2 electrosynthesis reactors. Recently, preliminary attempts have been made to develop membrane-free reactors in order to reduce the internal ohmic resistance and overall cost,129 which in fact could ultimately eliminate the membrane degradation issue.
5 2e− ORR integrated tandem systems for chemical synthesis
H2O2 is an important green oxidant and raw chemical material, with more than 30% of commercial H2O2 being used in traditional chemical synthesis.6 For example, H2O2 is widely used for the epoxidation of alkene,1 with titanium silicalite-1 (TS-1) serving as a highly active and selective catalyst when H2O2 is the primary oxidant.183 For the decarbonization of the traditional chemical manufacturing industry, integrating electrochemically produced H2O2 into chemical synthesis represents an innovative approach to the synthesis of high-value chemicals.184 In this section, we reviewed recent studies on the chemical synthesis using in situ generated H2O2.5.1 Ex-cell and in-cell modes of tandem systems
Tandem reactions, also known as cascade reactions, typically involve at least two consecutive chemical transformations, with each subsequent reaction relying on the product of the previous one.185 Integration of H2O2 electrosynthesis in tandem reactions allows on-site application of H2O2 or in situ generated ROS for subsequent chemical synthesis reactions, such as the oxidation of organic substrates.186In general, most reactors can be integrated in tandem reactions, including flow-cells, PEMFCs and SE cells. The SE cells are the most commonly used, as they can produce high concentrations of pure H2O2, which ensures the purity of synthesized chemicals. The 2e− ORR integrated tandem reactions can be implemented through two distinct modes: ex-cell mode and in-cell mode.187 As illustrated in Fig. 13a, the ex-cell mode consists of two separated reaction processes: the pure H2O2 electrosynthesis in the SE cell and the oxidation of organic substrates in the tower reactor, which facilitates the application of on-site produced H2O2. The ex-cell mode could be regarded as an ordinary production process with the electrochemical and chemical steps conducted independently, thereby minimizing mutual interference and improving the tandem system reliability. For example, Fan et al.127 developed a tandem reaction system using ex-cell mode for the synthesis of ethylene glycol under mild conditions. In this system, CNT-COOH served as the cathode to produce HO2−, and pure H2O2 was transferred into the subsequent tower reactor for the ethylene oxidation reaction. The ethylene glycol production rate correlated linearly with the H2O2 production rate with a slope of 0.75 (Fig. 13b), indicating that 75% of the electrochemically generated H2O2 had been utilized for ethylene glycol production. During the 100-hour stability test, the SE-cell voltage showed negligible decline, and the overall electron utilization efficiency was retained at ∼60%, exhibiting excellent stability (Fig. 13c).
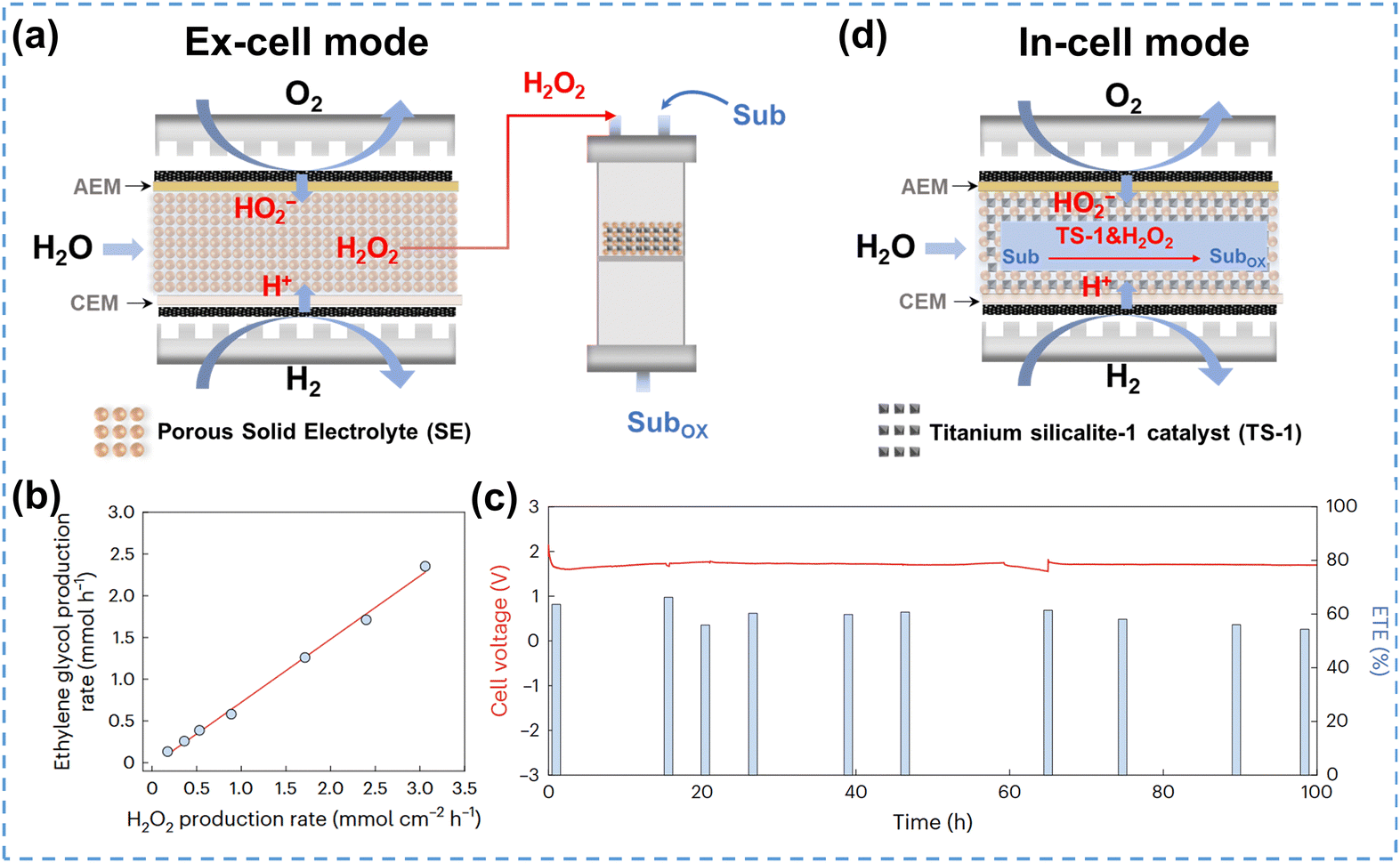 | ||
| Fig. 13 (a) Schematic of ex-cell mode for 2e− ORR integrated tandem reactions, adapted from ref. 127; (b) ethylene glycol production rate vs. H2O2 production rate, and (c) stability test for ethylene glycol production; (b) and (c) are reprinted with permission from ref. 127, copyright 2023, Springer Nature; (d) schematic of the in-cell mode for the 2e− ORR integrated tandem reactions, adapted from ref. 188. | ||
However, tandem reactions using the ex-cell mode diminish the benefit of electrochemical in situ production of highly active H2O2 due to the separation between the electrochemical and chemical reaction steps. Moreover, the ex-cell mode is unable to make full use of the high concentration of H2O2 accumulated near the electrode surface to accelerate the subsequent reactions, as the highly concentrated H2O2 is usually diluted into the bulk solution before flowing to the downstream tower reactor. Zhang et al.188 proposed an innovative in-cell mode based on SE cells, which incorporated the H2O2 electrosynthesis and the oxidation of organic substrates in a single integrated reactor. As illustrated in Fig. 13d, the middle layer of the reactor was filled with SE and TS-1 catalyst, which enabled multiple catalytic steps to proceed simultaneously. The feasibility of this novel in-cell tandem system was verified by the phenol oxidation reaction, which achieved 94.68% selectivity to target products (catechol and hydroquinone) under optimized conditions. This excellent catalytic performance was attributed to the full utilization of the in situ generated H2O2 and the direct activation of TS-1.
Although non-conductive TS-1 may increase the internal resistance of the reactor, the in-cell mode remains highly effective for chemical synthesis using in situ generated H2O2. We have summarized the applications of 2e− ORR integrated tandem systems for chemical synthesis in the past three years in Table S4 (ESI†) for readers’ reference. In the following sections, we will discuss the H2O2 concentration and ROS effects in this efficient tandem system mode with cutting-edge examples, and reveal its unique application values.
5.2 H2O2 concentration effect in the in-cell mode
During the 2e− ORR at the electrode, the produced H2O2 will accumulate near the electrode surface region, leading to a significantly higher H2O2 concentration compared to the bulk solution.34,189 In batch H2O2 electrosynthesis, the elevated concentration near the electrode surface region can initiate undesired H2O2RRs.101 However, H2O2 in tandem reaction systems is directly used as an intermediate oxidizing agent, therefore the high H2O2 concentration near the electrode surface will facilitate subsequent organic substrate oxidation reactions.The H2O2 concentration effect in the SE cell was first demonstrated by Zhang et al.,34 by visualization of the H2O2 concentration gradient between the electrode surface region and the bulk region using finite element simulations. Apparently, a sharp increase in H2O2 concentration up to 2.06 wt% was observed near the electrode, which was 25 times higher than the average concentration in the bulk solution (0.08 wt%), as shown in Fig. 14a. Using the ethylene epoxidation reaction as the model reaction, the in-cell mode SE reactor (Fig. 14b) significantly enhanced ethylene epoxidation rate, and tripled glycol yield compared to the ex-cell mode. This reactor maintained a 99.7% selectivity for ethylene glycol production over 200 hours, demonstrating excellent stability and high application potential.
 | ||
| Fig. 14 (a) Finite element simulations of the interfacial H2O2 concentration and the bulk solution concentration, and (b) schematic of the integrated SE reactor; (a) and (b) are reprinted with permission from ref. 34, copyright 2023, Elsevier; (c) schematic of the flow-cell with a bifunctional electrode for the propylene oxidation reaction, reprinted with permission from ref. 190, copyright 2023, Elsevier; (d) schematic of direct electrochemical oxidation of alkane (left), and a tandem reaction process for alkane oxidation in PEMFCs (right), reprinted with permission from ref. 191, copyright 2023, Wiley-VCH. | ||
To maximize the utilization of the H2O2 concentration effect for high-value chemical synthesis, the organic oxidation catalysts (e.g. TS-1) should be closely distributed on the electrode surface region. Liu et al.190 prepared a bifunctional electrode by mixing nanoscale TS-1 with oxygen-doped carbon catalyst (C–O), which ensured both effective H2O2 electrosynthesis and the activation of TS-1 by H2O2 near the electrode. Fig. 14c illustrated a simple flow-cell reactor for ethylene epoxidation reaction using the bifunctional electrode, which produced propylene glycol with 80% selectivity and achieved a yield of 4.28 mmol h−1 gcatalyst−1 when the mass ratio of C–O:TS-1 was adjusted to 1:3.
PEMFCs have been increasingly explored for the production of high-value chemicals, and current research studies primarily focus on the direct electrocatalytic reactions involving precious metal catalysts.192 As discussed in Section 4.1.3, the electrosynthesis of H2O2 via PEMFCs faces the dilemma of H2O2 transport within the zero-gap MEA, which hinders the batch H2O2 electrosynthesis. Luckily, the H2O2 concentration effect can turn this disadvantage into an advantage, as the high concentration of H2O2 accumulated by MEA can be directly utilized in tandem reactions to produce high-value chemicals. As depicted in Fig. 14d, Yang et al.191 developed a new strategy to oxidize propane by utilizing the high concentration of H2O2 in PEMFCs. The cathode was constructed by a 2e− ORR catalyst and propane oxidation catalyst MIL-53 (Al, Fe). Under mild reaction conditions (80 °C), the propane oxidation was significantly enhanced by the high concentration of H2O2 in the zero-gap MEA, achieving a yield of 11.4 mmol L−1 h−1 of C3 oxygenates, which was approximately ten times higher than that reported in direct alkane oxidization at the PEMFC anode.
5.3 ROS effect in the in-cell mode
The in situ generated H2O2 could also decompose to produce ROS, such as superoxide radicals (˙O2−), hydroxyl radicals (˙OH), and hydroperoxyl radicals (˙OOH),193 which typically exhibit stronger oxidizing ability compared to H2O2, thus accelerating organic substrate oxidation reactions. The ROS effect in the in-cell mode will bring more opportunities for chemical synthesis, especially for reactants that are difficult to activate, such as methane (CH4) and nitrogen (N2).Taking the CH4 oxidation reaction as an example, converting CH4 into high-value liquid oxygenates typically requires harsh conditions of high temperatures and pressures.194 Kim et al.195 proposed a strategy for the partial oxidation of CH4 using ROS generated by electro-synthesized H2O2 under ambient conditions. As illustrated in Fig. 15a, H2O2 was synthesized on a carbon electrode and subsequently activated to generate ROS to participate in the CH4 oxidation reaction. The further mechanistic study revealed that H2O2 could not react with CH4 directly, whereas ROS (e.g. ˙OH and ˙OOH radicals) generated by H2O2 significantly activated CH4, forming a series of liquid oxygenates such as CH3OH, CH3OOH, and HCOOH. This system achieved a high selectivity (80.7%) and production rate (18.9 μmol h−1) for converting CH4 to HCOOH under ambient conditions (Fig. 15b), offering a new method for the direct partial oxidation of CH4.
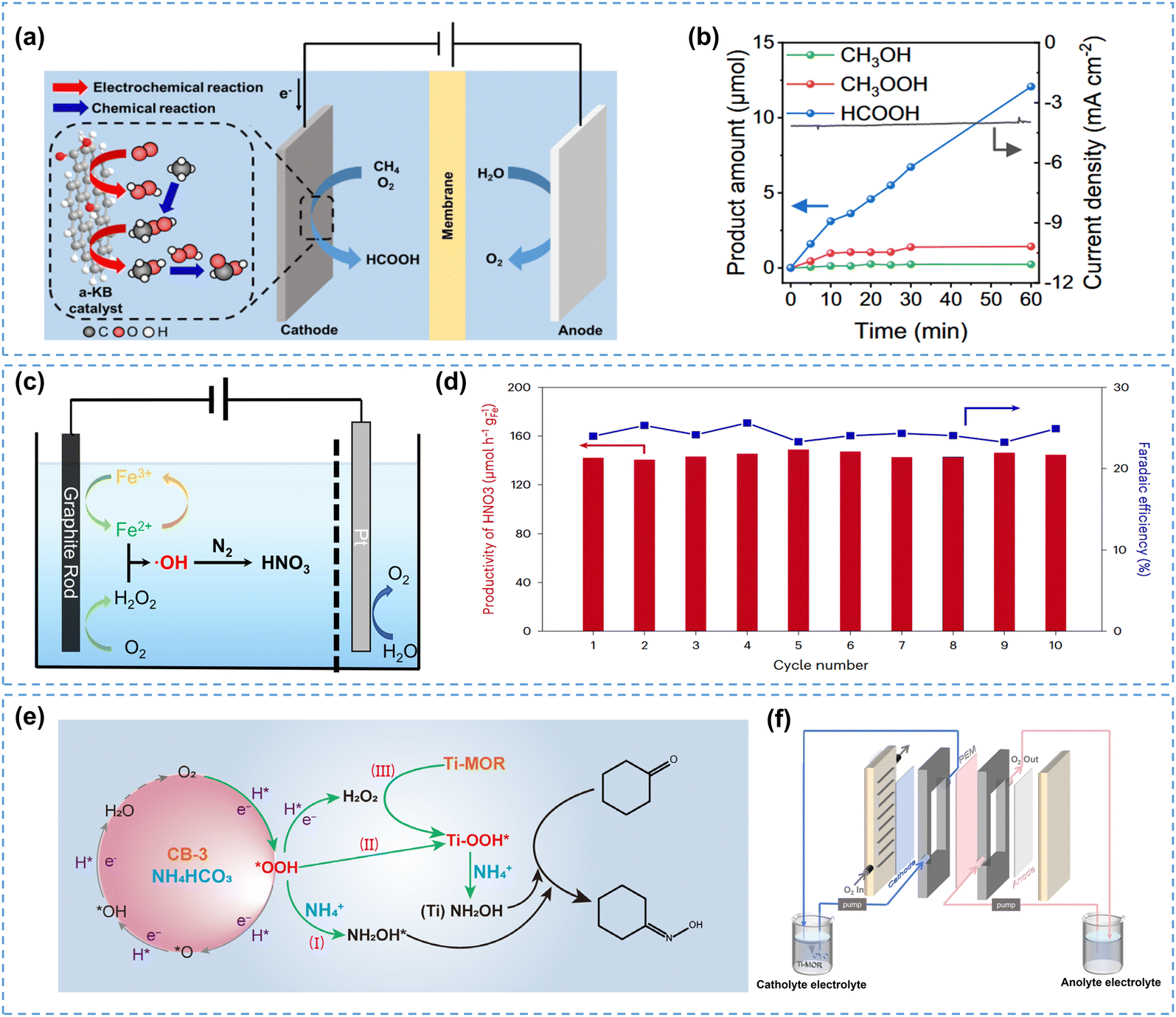 | ||
| Fig. 15 (a) Schematic of the electro-assisted CH4 partial oxidation process, and (b) production rates of liquid products; (a) and (b) are reprinted with permission from ref. 195, copyright 2023, Springer Nature; (c) schematic of N2 oxidation using electro-Fenton generated ˙OH, and (d) performance of the graphite rod cathode over ten reaction cycles; (c) and (d) are reprinted with permission from ref. 196, copyright 2023, Springer Nature; (e) schematic of the proposed pathways for the CYC oximation in the 2e− ORR integrated system, and (f) flow-cell reactor for CYC oximation; (e) and (f) are reprinted with permission from ref. 197, copyright 2024, American Association for the Advancement of Science. | ||
It has been demonstrated that oxygen functional groups in carbon materials favor the generation of ˙OH from H2O2 in acidic media.198,199 Lin et al.200 developed an oxygen-rich carbon electrode, which facilitated the oxidation of acetic acid to peroxyacetic acid due to abundant ˙OH radicals activated from H2O2.
The electro-Fenton reaction is a well-known process that can effectively activate H2O2 to generate ROS. In this process, Fe2+ reacts with H2O2 to produce ˙OH and Fe3+, and the Fe3+ is reduced back to Fe2+ at the cathode, forming a continuous cyclic conversion of the Fe2+/Fe3+ redox pair.201 Chen et al.196 successfully oxidized N2 to HNO3 using ˙OH generated by the electro-Fenton reaction. As illustrated in Fig. 15c, O2 was reduced to H2O2 on the graphite electrode and further converted into ˙OH by Fe2+. The ˙OH is a strong nucleophile reagent, which activated N2 and produced HNO3. This tandem reaction system demonstrated good stability over ten reaction cycles, maintaining a faradaic efficiency of ∼25% and a HNO3 yield of 140 μmol h−1 gFe−1 (Fig. 15d).
In the above examples, ROS directly interact with organic substrates to produce high-value chemicals, while in most cases, catalysts such as TS-1 are indispensable to catalyze the organic substrate oxidation reactions. Therefore, it is essential to explore the interaction between the catalysts and ROS in tandem reactions. The ROS directly transferred to near the TS-1 catalyst, forming the active Ti-OOH species, which accelerated the reaction kinetics of the ethylene oxidation reaction.202 Yuan et al.197 proposed that the direct generation of active Ti-OOH species from the interaction between ˙OOH radicals and the Ti-MOR catalyst was the primary pathway (pathway II) in the tandem system (Fig. 15e). Further application using the GDE reactor, equipped with external catholyte containing Ti-MOR catalyst (Fig. 15f), converted CYC (60 mM) to oxime (57 mM) with a yield of 95% at a current of 60 mA.
6 Summaries and prospects
The decentralized production of H2O2 via the 2e− ORR is an important method for clean and sustainable synthesis of H2O2, offering a green, safe and energy-efficient alternative to the anthraquinone process. The aspects of H2O2 electrosynthesis beyond catalysts have been discussed in this review, with a particular focus on the reactor design and its integration into tandem systems.Various aspects in reactor design have been discussed, including electrode surface/interface modifications and designs for cell configurations. Firstly, by discussing the surface wettability of carbon electrodes, it is suggested that an electrode surface with appropriate hydrophobicity is preferred to enhance the overall performance. Subsequently, the strategies for controlling the carbon electrode–electrolyte interface microenvironment have been reviewed. By reducing the proton concentration at the electrode interface or slowing the proton transfer rate, the H2O2RR can be effectively inhibited, thus enhancing the H2O2 production. Additionally, enhancing the electric field can improve the stability of 2e− ORR intermediates, and boost H2O2 electrosynthesis. Furthermore, the optimizations of key reactor components, as well as the challenges in practical-scale H2O2 electrosynthesis, have been summarized, revealing that optimized FFP and EC structures can enhance the overall performance. In the last part, we focus on the applications of the 2e− ORR by integrating it into tandem systems for value-added chemical synthesis. The H2O2 concentration and ROS effects in 2e− ORR integrated tandem reactions have been discussed in detail. Despite recent advances in electrodes and reactors for improved H2O2 electrosynthesis, challenges still remain in achieving large-scale H2O2 production, and future studies may focus on the following aspects:
(1) Investigate the dynamic evolution of three-phase interfaces (TPIs). The stable TPIs are determining factors on the electrode activity, stability and H2O2 selectivity in the application of GDE-based reactors. Currently, the states of the TPIs are generally represented by the three wetting states on the electrode, which are simply evaluated by measuring the contact angle before and post reaction. 3D images and dynamic evolution of TPIs are greatly needed for a better understanding of the construction of TPIs. The utilization of confocal laser scanning microscopy, micro-computed tomography 3D imaging, and environmental scanning electron microscopy could provide valuable insights into the reaction mechanisms and construction of durable TPIs.
(2) Investigate the cation effect on the 2e− ORR comprehensively. Although previous studies suggest that cations create an alkaline-like microenvironment, which is beneficial for H2O2 electrosynthesis, researchers indicate that a localized alkaline-like microenvironment can be formed without cation addition,203,204 particularly at higher current densities due to significant proton consumption. Other researchers ascribe the enhanced H2O2 electrosynthesis performance to the AMC-induced electric field modification,106 however, this effect has not been validated at larger current densities. The effect of AMC on H2O2 electrosynthesis can be rather complicated, and future studies should explore the comprehensive effects of cations, including their influences on interface electric fields and H-bond networks, and further verify the universality of the cation effects on different types of catalysts.
(3) Investigate the dynamic evolution of catalysts under operando conditions. The harsh oxidative conditions during the 2e− ORR can potentially alter catalyst structure and accelerate the degradation process. Designing and developing novel catalysts require a thorough evaluation of their resistance to oxidative conditions, as well as their evolution mechanisms. Advanced operando characterizations are needed, such as in situ Raman spectroscopy and operando EXAFS characterization, which enable real-time monitoring of catalyst structures. Understanding of the catalyst evolution under operando conditions will provide valuable insights into the fundamental aspects in material science and guide the design and development of efficient and durable electrocatalysts.
(4) Explore more application scenarios for in situ generated H2O2. A particularly promising direction is to incorporate chemical feedstocks into 2e− ORR integrated tandem systems to synthesize high-value chemicals, which represents sustainable and environmentally-friendly pathways. Integrating H2O2 electrosynthesis with other chemical reaction systems not only enhances energy efficiency but also enables the production of more complex chemical compounds under mild conditions. Recently, Kong et al.205 demonstrated a groundbreaking approach by integrating three electrolysis systems—CO2RR, nitrogen reduction reaction (NRR), and 2e− ORR—to achieve the green synthesis of glycine, a key amino acid. Moving forward, reactor designs should focus on tailoring systems for specific applications, thereby maximizing the utilization efficiency of H2O2 and contributing to sustainable chemical processes.
(5) Assess the economic feasibility and perform lifecycle analyses of H2O2 electrosynthesis and 2e− ORR integrated tandem systems. A comprehensive techno-economic analysis should evaluate product efficiency, scalability, and lifecycle impacts, and ultimately guide the development of cost-effective, resource-efficient, and sustainable chemical production processes. The analyses should include not only direct economic costs, such as pre-investments and electricity consumption, but also include the environmental costs, with a particular emphasis on carbon emissions and their environmental impacts.
Data availability
No primary research results, software or code have been included and no new data have been generated or analyzed in this review.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are thankful for the financial support from the National Natural Science Foundation of China (grant number 22172112) and the Fundamental Research Funds for the Central Universities.References
- R. L. Myers, The 100 Most Important Chemical Compounds: A Reference Guide, Greenwood Press, 2007 Search PubMed.
- X. Wen, X. Zhang, M. Wang, C. Yuan, J. Lang, X. Li, H. Wei, D. Mandler and M. Long, Appl. Catal., B, 2024, 342, 123437 CrossRef.
- C. Xia, Y. Xia, P. Zhu, L. Fan and H. Wang, Science, 2019, 366, 226–231 CrossRef.
- M. Wang, J. Cheng, W. Xu, D. Zhu, W. Zhang, Y. Wen, W. Guan, J. Jia and Z. Lu, Nat. Nanotechnol., 2024, 20, 67–74 CrossRef PubMed.
- M. Lu, X. Liu, C. Jing, X. Wang, L. Ding, F. Gao, L. Ren, S. Dai, X. Zhong and J. Wang, Adv. Funct. Mater., 2024, 2412170 Search PubMed.
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Čolić, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, ACS Catal., 2018, 8, 4064–4081 CrossRef.
- S. C. Perry, D. Pangotra, L. Vieira, L.-I. Csepei, V. Sieber, L. Wang, C. Ponce de León and F. C. Walsh, Nat. Rev. Chem., 2019, 3, 442–458 CrossRef.
- A. Byeon, W. C. Yun, J. M. Kim and J. W. Lee, ChemElectroChem, 2023, 10, e202300234 CrossRef.
- Y. Tian, D. Deng, L. Xu, M. Li, H. Chen, Z. Wu and S. Zhang, Nano-Micro Lett., 2023, 15, 122 CrossRef CAS.
- D. Zhang, E. Mitchell, X. Lu, D. Chu, L. Shang, T. Zhang, R. Amal and Z. Han, Mater. Today, 2023, 63, 339–359 CrossRef CAS.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. L. Stephens and J. Rossmeisl, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS.
- M. Huang, Z. Cui, Z. Li and W. Sheng, ACS Catal., 2024, 14, 2095–2106 CrossRef CAS.
- Z. Yu, H. Deng, Q. Yao, L. Zhao, F. Xue, T. He, Z. Hu, W.-H. Huang, C.-W. Pao, L.-M. Yang and X. Huang, Nat. Commun., 2024, 15, 9346 CrossRef CAS.
- H. Li, P. Wen, D. S. Itanze, Z. D. Hood, S. Adhikari, C. Lu, X. Ma, C. Dun, L. Jiang, D. L. Carroll, Y. Qiu and S. M. Geyer, Nat. Commun., 2020, 11, 3928 CrossRef CAS PubMed.
- Q. Sun, G. Xu, B. Xiong, L. Chen and J. Shi, Nano Res., 2023, 16, 4729–4735 CrossRef CAS.
- X.-L. Zhang, X. Su, Y.-R. Zheng, S.-J. Hu, L. Shi, F.-Y. Gao, P.-P. Yang, Z.-Z. Niu, Z.-Z. Wu, S. Qin, R. Wu, Y. Duan, C. Gu, X.-S. Zheng, J.-F. Zhu and M.-R. Gao, Angew. Chem., Int. Ed., 2021, 60, 26922–26931 CrossRef CAS PubMed.
- W. Liu, R. Chen, Z. Sang, Z. Li, J. Nie, L. Yin, F. Hou and J. Liang, Adv. Mater., 2024, 36, 2406403 CrossRef CAS.
- S. Chen, T. Luo, J. Wang, J. Xiang, X. Li, C. Ma, C.-W. Kao, T.-S. Chan, Y.-N. Liu and M. Liu, Angew. Chem., Int. Ed., 2024, e202418713 Search PubMed.
- H. Huang, M. Sun, S. Li, S. Zhang, Y. Lee, Z. Li, J. Fang, C. Chen, Y.-X. Zhang, Y. Wu, Y. Che, S. Qian, W. Zhu, C. Tang, Z. Zhuang, L. Zhang and Z. Niu, J. Am. Chem. Soc., 2024, 146, 9434–9443 CrossRef CAS PubMed.
- J. Du, G. Han, W. Zhang, L. Li, Y. Yan, Y. Shi, X. Zhang, L. Geng, Z. Wang, Y. Xiong, G. Yin and C. Du, Nat. Commun., 2023, 14, 4766 CrossRef CAS PubMed.
- C. Zhang, W. Shen, K. Guo, M. Xiong, J. Zhang and X. Lu, J. Am. Chem. Soc., 2023, 145, 11589–11598 CrossRef CAS.
- Y. Long, J. Lin, F. Ye, W. Liu, D. Wang, Q. Cheng, R. Paul, D. Cheng, B. Mao, R. Yan, L. Zhao, D. Liu, F. Liu and C. Hu, Adv. Mater., 2023, 35, e2303905 CrossRef PubMed.
- Z. Lin, C. Han, G. E. P. O'Connell and X. Lu, Angew. Chem., Int. Ed., 2023, 62, e202301435 CrossRef CAS PubMed.
- Y. Wen, T. Zhang, J. Wang, Z. Pan, T. Wang, H. Yamashita, X. Qian and Y. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202205972 CrossRef CAS.
- N. Li, C. Huang, X. Wang, Y. Feng and J. An, Chem. Eng. J., 2022, 450, 138246 CrossRef CAS.
- X. Zhang, X. Zhao, P. Zhu, Z. Adler, Z.-Y. Wu, Y. Liu and H. Wang, Nat. Commun., 2022, 13, 2880 CrossRef CAS.
- J. L. Hübner, L. E. B. Lucchetti, H. N. Nong, D. I. Sharapa, B. Paul, M. Kroschel, J. Kang, D. Teschner, S. Behrens, F. Studt, A. Knop-Gericke, S. Siahrostami and P. Strasser, ACS Energy Lett., 2024, 9, 1331–1338 CrossRef.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- L. Cui, B. Chen, D. Chen, C. He, Y. Liu, H. Zhang, J. Qiu, L. Liu, W. Jing and Z. Zhang, Nat. Commun., 2024, 15, 10632 CrossRef CAS PubMed.
- B. Ni, P. Shen, G. Zhang, J. Zhao, H. Ding, Y. Ye, Z. Yue, H. Yang, H. Wei and K. Jiang, J. Am. Chem. Soc., 2024, 146, 11181–11192 CAS.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12, 136 CrossRef CAS.
- C. Ni, W. Xu, N. Deng and X. Huang, J. Cleaner Prod., 2024, 480, 144066 CrossRef CAS.
- Y. Ma, E. Zhao, G. Xia, J. Zhan, G. Yu and Y. Wang, Water Res., 2023, 229, 119503 CrossRef CAS PubMed.
- S.-K. Zhang, Y. Feng, A. Elgazzar, Y. Xia, C. Qiu, Z. Adler, C. Sellers and H. Wang, Joule, 2023, 7, 1887–1901 CrossRef CAS.
- J. Kim, J. H. Kim, C. Oh, H. Yun, E. Lee, H.-S. Oh, J. H. Park and Y. J. Hwang, Nat. Commun., 2023, 14, 4704 CrossRef CAS.
- S. Chen, S. Liang, R. Huang, M. Zhang, Y. Song, Y. Zhang, S. Tao, L. Yu and D. Deng, Nat. Synth., 2024, 3, 76–84 CrossRef CAS.
- Z. Lu, G. Chen, S. Siahrostami, Z. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. Lin, Y. Liu, T. F. Jaramillo, J. K. Nørskov and Y. Cui, Nat. Catal., 2018, 1, 156–162 CrossRef CAS.
- C. Xia, J. Y. Kim and H. Wang, Nat. Catal., 2020, 3, 605–607 CrossRef CAS.
- J. S. Choi, G. V. Fortunato, D. C. Jung, J. C. Lourenco, M. R. V. Lanza and M. Ledendecker, Nanoscale Horiz., 2024, 9, 1250–1261 RSC.
- R. Zhou, Y. Zheng, M. Jaroniec and S.-Z. Qiao, ACS Catal., 2016, 6, 4720–4728 Search PubMed.
- P. Wang, T. Hayashi, Q. Meng, Q. Wang, H. Liu, K. Hashimoto and L. Jiang, Small, 2017, 13, 1601250 CrossRef.
- Y. Wang, Y. Zou, L. Tao, Y. Wang, G. Huang, S. Du and S. Wang, Nano Res., 2019, 12, 2055–2066 CrossRef CAS.
- Y. He, S. Liu, M. Wang, Q. Cheng, T. Qian and C. Yan, J. Energy Chem., 2023, 80, 302–323 CrossRef CAS.
- K. Shi, Z. Ren, Z. Meng and X. Feng, ChemCatChem, 2024, 16, e202301308 CrossRef CAS.
- G. Xia, Y. Tian, X. Yin, W. Yuan, X. Wu, Z. Yang, G. Yu, Y. Wang and M. Wu, Appl. Catal., B, 2021, 299, 120655 CrossRef CAS.
- Z. Xing, K. Shi, Z. S. Parsons and X. Feng, ACS Catal., 2023, 13, 2780–2789 CrossRef CAS.
- J. Ryu and D. W. Lee, J. Mater. Chem. A, 2024, 12, 10012–10043 RSC.
- C. Zhang, Z. Xu, N. Han, Y. Tian, T. Kallio, C. Yu and L. Jiang, Sci. Adv., 2024, 9, eadd6978 Search PubMed.
- H. Yang, Y. Liu, X. Liu, X. Wang, H. Tian, G. I. N. Waterhouse, P. E. Kruger, S. G. Telfer and S. Ma, eScience, 2022, 2, 227–234 CrossRef.
- K. Dong, J. Liang, Y. Wang, Z. Xu, Q. Liu, Y. Luo, T. Li, L. Li, X. Shi, A. M. Asiri, Q. Li, D. Ma and X. Sun, Angew. Chem., Int. Ed., 2021, 60, 10583–10587 CrossRef CAS.
- X. You, F. Hou, T. Xie, A. Cai, H. He, G. Li, F. Zhang, W. Peng, X. Fan and Y. Li, J. Colloid Interface Sci., 2023, 639, 333–342 CrossRef CAS PubMed.
- R. T. Ferrell and D. M. Himmelblau, J. Chem. Eng. Data, 1967, 12, 111–115 CrossRef CAS.
- B. Carnes and N. Djilali, J. Power Sources, 2005, 144, 83–93 CrossRef CAS.
- Z. Liu, K. Li, L. Liu, H. Song, Y. Zhang, M. Tebyetekerwa, X. Zhang, K. Wang, L. Xu and J. Wang, Appl. Catal., B, 2024, 357, 124311 CrossRef CAS.
- J. Wang, C. Li, M. Rauf and W. Wang, ACS Sustainable Chem. Eng., 2022, 11, 436–443 CrossRef.
- K. Wang and M. Pera-Titus, Sci. Adv., 2024, 10, eado5448 CrossRef CAS PubMed.
- Q. Zhang, M. Zhou, G. Ren, Y. Li, Y. Li and X. Du, Nat. Commun., 2020, 11, 1731 CrossRef CAS.
- B. Hu, M. Wang, D. Li, J. Chen, C. Li and L. Wang, Adv. Energy Sustainability Res., 2023, 4, 230014 Search PubMed.
- J. Chen, H. Qiu, Y. Zhao, H. Yang, L. Fan, Z. Liu, S. Xi, G. Zheng, J. Chen, L. Chen, Y. Liu, L. Guo and L. Wang, Nat. Commun., 2024, 15, 5893 CrossRef CAS PubMed.
- P. Xia, L. Zhao, X. Chen, Z. Ye, Z. Zheng, Q. He and I. Sirés, Appl. Catal., B, 2024, 343, 123467 CrossRef CAS.
- F. Yu, M. Zhou and X. Yu, Electrochim. Acta, 2015, 163, 182–189 CrossRef CAS.
- Y. Qiao, N. Ren, X. Li, J. An, X. Wang and N. Li, Chem. Eng. J., 2023, 463, 142417 CrossRef CAS.
- B. Sabri Rawah, M. Albloushi and W. Li, Chem. Eng. J., 2023, 466, 143282 CrossRef CAS.
- H. Wang, S. Jiang, H. Yu, K. Deng, Z. Wang, X. Li, Y. Xu and L. Wang, J. Mater. Chem. A, 2023, 11, 13633–13639 RSC.
- P. Cao, X. Quan, K. Zhao, X. Zhao, S. Chen and H. Yu, ACS Catal., 2021, 11, 13797–13808 CrossRef CAS.
- H. Xu, X. H. Lv, H. Y. Wang, J. Y. Ye, J. Yuan, Y. C. Wang, Z. Y. Zhou and S. G. Sun, ChemSusChem, 2022, 15, e202102587 Search PubMed.
- B. L. French, J. J. Wang, M. Y. Zhu and B. C. Holloway, Thin Solid Films, 2006, 494, 105–109 CrossRef CAS.
- J. Wang, M. Zhu, R. A. Outlaw, X. Zhao, D. M. Manos and B. C. Holloway, Carbon, 2004, 42, 2867–2872 CrossRef CAS.
- L. X. Zhang, Z. Sun, J. L. Qi, J. M. Shi, T. D. Hao and J. C. Feng, Carbon, 2016, 103, 339–345 CrossRef CAS.
- Y. Wang, R. Shi, L. Shang, L. Peng, D. Chu, Z. Han, G. I. N. Waterhouse, R. Zhang and T. Zhang, Nano Energy, 2022, 96, 107046 CrossRef CAS.
- D. Zhang, C. Tsounis, Z. Ma, D. Djaidiguna, N. M. Bedford, L. Thomsen, X. Lu, D. Chu, R. Amal and Z. Han, Small, 2022, 18, e2105082 CrossRef.
- A. Kozbial, F. Zhou, Z. Li, H. Liu and L. Li, Acc. Chem. Res., 2016, 49, 2765–2773 CrossRef CAS PubMed.
- D. Zhang, C. Tsounis, Z. Ma, L. Peng, Z. Lin, H. Yin, F. Hussain, C. Cazorla, D. Chu, R. Amal and Z. Han, Cell Rep. Phys. Sci., 2023, 4, 101643 CrossRef CAS.
- F. Mugele and J.-C. Baret, J. Phys.: Condens. Matter, 2005, 17, R705 CrossRef CAS.
- L. Cui, B. Chen, L. Zhang, C. He, C. Shu, H. Kang, J. Qiu, W. Jing, K. Ostrikov and Z. Zhang, Energy Environ. Sci., 2024, 17, 655–667 RSC.
- J. Chen, H. Qiu, Y. Zhao, H. Yang, L. Fan, Z. Liu, S. Xi, G. Zheng, J. Chen, L. Chen, Y. Liu, L. Guo and L. Wang, Nat. Commun., 2024, 15, 5893 Search PubMed.
- R. Haaring, P. W. Kang, J. W. Lee, J. Lee and H. Lee, ACS Appl. Mater. Interfaces, 2024, 16, 28731–28741 CrossRef CAS.
- Y. Sheng, Y. Zhao, X. Wang, R. Wang and T. Tang, Electrochim. Acta, 2014, 133, 414–421 CrossRef CAS.
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 33–40 CrossRef CAS.
- Y. J. Sa, C. W. Lee, S. Y. Lee, J. Na, U. Lee and Y. J. Hwang, Chem. Soc. Rev., 2020, 49, 6632–6665 RSC.
- J. Wu, Chem. Rev., 2022, 122, 10821–10859 CrossRef CAS PubMed.
- T. Kumeda and K. Sakaushi, Curr. Opin. Electrochem., 2022, 36, 101121 CrossRef CAS.
- P. Sebastián-Pascual, Y. Shao-Horn and M. Escudero-Escribano, Curr. Opin. Electrochem., 2022, 32, 100918 CrossRef.
- M. M. Waegele, C. M. Gunathunge, J. Li and X. Li, J. Chem. Phys., 2019, 151, 160902 CrossRef PubMed.
- J. L. Hubner, L. E. B. Lucchetti, H. N. Nong, D. I. Sharapa, B. Paul, M. Kroschel, J. Kang, D. Teschner, S. Behrens, F. Studt, A. Knop-Gericke, S. Siahrostami and P. Strasser, ACS Energy Lett., 2024, 9, 1331–1338 CrossRef CAS PubMed.
- X. Zhang, X. Zhao, P. Zhu, Z. Adler, Z. Y. Wu, Y. Liu and H. Wang, Nat. Commun., 2022, 13, 2880 CrossRef CAS PubMed.
- D. Park and Y. Jung, Chem Catal., 2024, 4, 100823 CrossRef CAS.
- X. Y. Li, T. Wang, Y. C. Cai, Z. D. Meng, J. W. Nan, J. Y. Ye, J. Yi, D. P. Zhan, N. Tian, Z. Y. Zhou and S. G. Sun, Angew. Chem., Int. Ed., 2023, 62, e202218669 CrossRef CAS PubMed.
- P. Cao, X. Zhao, Y. Liu, H. Zhang, K. Zhao, S. Chen, H. Yu, F. Dong, N. N. Nichols, J. G. Chen and X. Quan, Angew. Chem., Int. Ed., 2024, 63, e202406452 CrossRef CAS.
- Y. Fan, Y. Chen, W. Ge, L. Dong, Y. Qi, C. Lian, X. Zhou, H. Liu, Z. Liu, H. Jiang and C. Li, J. Am. Chem. Soc., 2024, 146, 7575–7583 CrossRef CAS PubMed.
- Y. Fang, Y. Fan, K. Xie, W. Ge, Y. Zhu, Z. Qi, Z. Song, H. Jiang and C. Li, Angew. Chem., Int. Ed., 2023, 62, e202304413 CrossRef CAS PubMed.
- E. L. Gyenge and C. W. Oloman, J. Appl. Electrochem., 2001, 31, 233–243 Search PubMed.
- G. A. Kolyagin and V. L. Kornienko, Russ. J. Appl. Chem., 2006, 79, 746–751 CrossRef CAS.
- P. Li, Y. Jiang, Y. Hu, Y. Men, Y. Liu, W. Cai and S. Chen, Nat. Catal., 2022, 5, 900–911 CrossRef CAS.
- Q. Nian, X. Zhang, Y. Feng, S. Liu, T. Sun, S. Zheng, X. Ren, Z. Tao, D. Zhang and J. Chen, ACS Energy Lett., 2021, 6, 2174–2180 Search PubMed.
- Y. Fang, Y. Fan, K. Xie, W. Ge, Y. Zhu, Z. Qi, Z. Song, H. Jiang and C. Li, Angew. Chem., Int. Ed., 2023, 62, e202304413 CrossRef CAS PubMed.
- C. Yang, F. Sun, Z. Qu, X. Li, W. Zhou and J. Gao, ACS Energy Lett., 2022, 7, 4398–4407 CrossRef CAS.
- L. Jing, Q. Tian, W. Wang, X. Li, Q. Hu, H. Yang and C. He, Adv. Energy Mater., 2024, 14, 2304418 CrossRef CAS.
- Q. Tian, L. Jing, Y. Yin, Z. Liang, H. Du, L. Yang, X. Cheng, D. Zuo, C. Tang, Z. Liu, J. Liu, J. Wan and J. Yang, Nano Lett., 2024, 24, 1650–1659 CrossRef CAS PubMed.
- Q. Tian, L. Jing, H. Du, Y. Yin, X. Cheng, J. Xu, J. Chen, Z. Liu, J. Wan, J. Liu and J. Yang, Nat. Commun., 2024, 15, 983 CrossRef CAS PubMed.
- K.-H. Wu, D. Wang, X. Lu, X. Zhang, Z. Xie, Y. Liu, B.-J. Su, J.-M. Chen, D.-S. Su, W. Qi and S. Guo, Chem, 2020, 6, 1443–1458 CAS.
- S. Zhao, Y. Wang, Y. Hao, L. Yin, C. H. Kuo, H. Y. Chen, L. Li and S. Peng, Adv. Mater., 2024, 36, e2308925 CrossRef PubMed.
- J. Guo, Y. Zheng, Z. Hu, C. Zheng, J. Mao, K. Du, M. Jaroniec, S.-Z. Qiao and T. Ling, Nat. Energy, 2023, 8, 264–272 CAS.
- H. Li, P. Cao, H. Zhang, K. Wang, S. Chen, H. Yu and X. Quan, Chem. Eng. J., 2024, 497, 154523 CrossRef CAS.
- S. R. Kelly, C. Kirk, K. Chan and J. K. Nørskov, J. Phys. Chem. C, 2020, 124, 14581–14591 CrossRef CAS.
- J. Lee, J. S. Lim, G. Yim, H. Jang, S. H. Joo and Y. J. Sa, ACS Appl. Mater. Interfaces, 2021, 13, 59904–59914 CrossRef CAS PubMed.
- Y. Ding, W. Zhou, L. Xie, S. Chen, J. Gao, F. Sun, G. Zhao and Y. Qin, J. Mater. Chem. A, 2021, 9, 15948–15954 RSC.
- J. Yu, J. Yin, R. Li, Y. Ma and Z. Fan, Chem Catal., 2022, 2, 2229–2252 CrossRef CAS.
- B. Pan, Y. Wang and Y. Li, Chem Catal., 2022, 2, 1267–1276 CrossRef CAS.
- J. Chen, Y. Zhao, H. Yang, T. Zhang, L. Fan, C. Li and L. Wang, Nanoscale, 2023, 15, 3832–3840 RSC.
- K. Zhou and L. Li, J. Alloys Compd., 2024, 1002, 175549 CrossRef CAS.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- Y. Ding, W. Zhou, J. Li, J. Wang, L. Xie, X. Meng, J. Gao, F. Sun, G. Zhao and Y. Qin, ACS Energy Lett., 2023, 8, 3122–3130 CrossRef CAS.
- W. Shen, C. Zhang, M. Alomar, Z. Du, Z. Yang, J. Wang, G. Xu, J. Zhang, J. Lv and X. Lu, Nano Res., 2023, 17, 1217–1224 CrossRef.
- W. Shen, C. Zhang, X. Wang, Y. Huang, Z. Du, M. Alomar, J. Wang, J. Lv, J. Zhang and X. Lu, ACS Mater. Lett., 2023, 6, 17–26 CrossRef.
- Z. Mou, Y. Mu, L. Liu, D. Cao, S. Chen, W. Yan, H. Zhou, T. S. Chan, L. Y. Chang and X. Fan, Small, 2024, 20, e2400564 CrossRef PubMed.
- F. She, Z. Guo, F. Liu, Z. Yu, J. Chen, Y. Fan, Y. Lei, Y. Chen, H. Li and L. Wei, ACS Catal., 2024, 14, 10928–10938 CrossRef CAS.
- P. Cao, X. Quan, X. Nie, K. Zhao, Y. Liu, S. Chen, H. Yu and J. G. Chen, Nat. Commun., 2023, 14, 172 CrossRef CAS PubMed.
- Z. Deng, S. J. Choi, G. Li and X. Wang, Chem. Soc. Rev., 2024, 53, 8137–8181 RSC.
- M. R. Gerhardt, A. A. Wong and M. J. Aziz, J. Electrochem. Soc., 2018, 165, A2625–A2643 Search PubMed.
- Y. Chen, C. Zhen, Y. Chen, H. Zhao, Y. Wang, Z. Yue, Q. Wang, J. Li, M. D. Gu, Q. Cheng and H. Yang, Angew. Chem., Int. Ed., 2024, 63, e202407163 Search PubMed.
- S. Subramanian, K. Yang, M. Li, M. Sassenburg, M. Abdinejad, E. Irtem, J. Middelkoop and T. Burdyny, ACS Energy Lett., 2023, 8, 222–229 CrossRef CAS PubMed.
- D. Corral, J. T. Feaster, S. Sobhani, J. R. DeOtte, D. U. Lee, A. A. Wong, J. Hamilton, V. A. Beck, A. Sarkar, C. Hahn, T. F. Jaramillo, S. E. Baker and E. B. Duoss, Energy Environ. Sci., 2021, 14, 3064–3074 RSC.
- J. Li, Z. Wang, S. Liu, Z. Chen, J. Yang, Z. Chen, A. Li, Q. Wen, L. Wang, S. Qiu, C. Cui, H. Deng and F. Deng, Chem. Eng. J., 2024, 481, 148452 CrossRef CAS.
- S. Jia, H. Yu, J. Na, Z. Liu, K. Lv, Z. Ren, S. Sun and Z. Shao, ACS Appl. Mater. Interfaces, 2024, 16, 23099–23108 CAS.
- B. N. Ruggiero, X. K. Lu, K. Adonteng, J. Dong, J. M. Notestein and L. C. Seitz, Chem. Eng. J., 2024, 486, 150246 CrossRef CAS.
- L. Fan, Y. Zhao, L. Chen, J. Chen, J. Chen, H. Yang, Y. Xiao, T. Zhang, J. Chen and L. Wang, Nat. Catal., 2023, 6, 585–595 CrossRef CAS.
- P. C. Foller and R. T. Bombard, J. Appl. Electrochem., 1995, 25, 613–627 CrossRef CAS.
- Z. Chen, S. Chen, S. Siahrostami, P. Chakthranont, C. Hahn, D. Nordlund, S. Dimosthenis, J. K. Nørskov, Z. Bao and T. F. Jaramillo, React. Chem. Eng., 2017, 2, 239–245 RSC.
- J. Qi, Y. Du, Q. Yang, N. Jiang, J. Li, Y. Ma, Y. Ma, X. Zhao and J. Qiu, Nat. Commun., 2023, 14, 6263 CrossRef CAS PubMed.
- Y. Sun, K. Fan, J. Li, L. Wang, Y. Yang, Z. Li, M. Shao and X. Duan, Nat. Commun., 2024, 15, 6098 CrossRef CAS PubMed.
- E. Berl, Trans. Electrochem. Soc., 1939, 76, 359 CrossRef.
- G. A. Truesdale and A. L. Downing, Nature, 1954, 173, 1236 CrossRef CAS.
- T. Kinumoto, M. Inaba, Y. Nakayama, K. Ogata, R. Umebayashi, A. Tasaka, Y. Iriyama, T. Abe and Z. Ogumi, J. Power Sources, 2006, 158, 1222–1228 CrossRef CAS.
- J. Li and H. Duan, Chem, 2024, 10, 1–32 Search PubMed.
- H. Sheng, A. N. Janes, R. D. Ross, H. Hofstetter, K. Lee, J. R. Schmidt and S. Jin, Nat. Catal., 2022, 5, 716–725 CrossRef CAS.
- S.-K. Geng, Y. Zheng, S.-Q. Li, H. Su, X. Zhao, J. Hu, H.-B. Shu, M. Jaroniec, P. Chen, Q.-H. Liu and S.-Z. Qiao, Nat. Energy, 2021, 6, 904–912 CrossRef CAS.
- J. Wang, X. Liu, C. Ma, H. Fu, S. Chen, N. Li, Y. Li, X. Fan and W. Peng, Chem Catal., 2024, 4, 101090 CrossRef CAS.
- L. Li, C. Tang, Y. Zheng, B. Xia, X. Zhou, H. Xu and S. Z. Qiao, Adv. Energy Mater., 2020, 10, 2000789 CrossRef CAS.
- K. Dong, J. Liang, Y. Wang, Y. Ren, Z. Xu, H. Zhou, L. Li, Q. Liu, Y. Luo, T. Li, A. M. Asiri, Q. Li, D. Ma and X. Sun, Chem Catal., 2021, 1, 1437–1448 CrossRef CAS.
- Z. Wang, X. Duan, M. G. Sendeku, W. Xu, S. Chen, B. Tian, W. Gao, F. Wang, Y. Kuang and X. Sun, Chem Catal., 2023, 3, 100672 CrossRef CAS.
- C. Xia, S. Back, S. Ringe, K. Jiang, F. Chen, X. Sun, S. Siahrostami, K. Chan and H. Wang, Nat. Catal., 2020, 3, 125–134 CrossRef CAS.
- K. Otsuka and I. Yamanaka, Electrochim. Acta, 1990, 35, 319–322 CrossRef CAS.
- I. Yamanaka, S. Tazawa, T. Murayama, R. Ichihashi and N. Hanaizumi, ChemSusChem, 2008, 1, 988–992 CrossRef CAS PubMed.
- I. Yamanaka, S. Tazawa, T. Murayama, T. Iwasaki and S. Takenaka, ChemSusChem, 2010, 3, 59–62 CrossRef CAS PubMed.
- W. Li, A. Bonakdarpour, E. Gyenge and D. P. Wilkinson, ChemSusChem, 2013, 6, 2137–2143 CrossRef CAS PubMed.
- C. Xia, Y. Xia, P. Zhu, L. Fan and H. Wang, Science, 2019, 366, 226–231 CrossRef CAS PubMed.
- W. Wang, Y. Hu, Y. Liu, Z. Zheng and S. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 31855–31859 CrossRef CAS PubMed.
- H. Li, P. Wen, D. S. Itanze, Z. D. Hood, S. Adhikari, C. Lu, X. Ma, C. Dun, L. Jiang, D. L. Carroll, Y. Qiu and S. M. Geyer, Nat. Commun., 2020, 11, 3928 CrossRef CAS PubMed.
- J. Yang, R. Ding, C. Liu, Q. Xu, S. Liu and X. Yin, J. Phys.: Energy, 2024, 6, 015022 CAS.
- G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377, 1–35 CrossRef CAS.
- R. Xie, C. Cheng, R. Wang, J. Li, E. Zhao, Y. Zhao, Y. Liu, J. Guo, P. Yin and T. Ling, ACS Catal., 2024, 14, 4471–4477 CrossRef CAS.
- T. Lu, M. Sun, F. Wang, S. Chen, Y. Li, J. Chen, X. Liao, X. Sun, Y. Liu, F. Wang, B. Huang and H. Wang, ACS Nano, 2024, 18, 15035–15045 CrossRef CAS PubMed.
- H. Sheng, R. D. Ross, J. R. Schmidt and S. Jin, ACS Energy Lett., 2023, 8, 196–212 CrossRef CAS.
- Y. Wang, C. Han, L. Ma, T. Duan, Y. Du, J. Wu, J.-J. Zou, J. Gao, X.-D. Zhu and Y.-C. Zhang, Small, 2024, 20, 2309448 CrossRef CAS PubMed.
- Y. Bu, R. Ma, Y. Wang, Y. Zhao, F. Li, G.-F. Han and J.-B. Baek, Adv. Mater., 2024, 36, 2412670 CrossRef CAS PubMed.
- C. He, C. Xia, F.-M. Li, J. Zhang, W. Guo and B. Y. Xia, Adv. Energy Mater., 2024, 14, 2303233 CrossRef CAS.
- Y. Li, D. Luan and X. W. Lou, Adv. Mater., 2024, 36, 2412386 CrossRef CAS PubMed.
- M. Deng, D. Wang and Y. Li, Adv. Mater., 2024, 36, 2314340 CrossRef CAS PubMed.
- Y.-Y. Yan, W.-J. Niu, W.-W. Zhao, R.-J. Li, E.-P. Feng, B.-X. Yu, B.-K. Chu and C.-Y. Cai, Adv. Energy Mater., 2024, 14, 2303506 CrossRef CAS.
- L. Zhao, R. Yan, B. Mao, R. Paul, W. Duan, L. Dai and C. Hu, Small, 2024, 20, 2403029 CrossRef CAS PubMed.
- M. Mazzucato, A. Facchin, M. Parnigotto and C. Durante, ACS Catal., 2024, 14, 6369–6403 CrossRef CAS.
- Y. Zhou, L. Xu, J. Wu, W. Zhu, T. He, H. Yang, H. Huang, T. Cheng, Y. Liu and Z. Kang, Energy Environ. Sci., 2023, 16, 3526–3533 RSC.
- Y. Yao, H. Wang, K. Dong, H. Li, J. Liang, R. Li, S. Sun, Z. Cai, X. He, D. Zheng, Y. Luo, S. Alfaifi, D. Ma, W. Hu and X. Sun, J. Mater. Chem. A, 2023, 11, 22154–22160 RSC.
- S. Ding, B. Xia, M. Li, F. Lou, C. Cheng, T. Gao, Y. Zhang, K. Yang, L. Jiang, Z. Nie, H. Guan, J. Duan and S. Chen, Energy Environ. Sci., 2023, 16, 3363–3372 RSC.
- Q. Huang, B. Xia, M. Li, H. Guan, M. Antonietti and S. Chen, Nat. Commun., 2024, 15, 4157 CrossRef CAS PubMed.
- S. Ding, Y. Zhang, F. Lou, M. Li, Q. Huang, K. Yang, B. Xia, C. Tang, J. Duan, M. Antonietti and S. Chen, Mater. Today Energy, 2023, 38, 101430 CrossRef CAS.
- V. Alvarado-Pérez, L. I. Cabrera-Lara, G. López-Téllez, D. Mendoza-Anaya, S. Hernández-López and M. Camacho-López, Mater. Chem. Phys., 2019, 233, 180–184 CrossRef.
- X. Zhou, Y. Min, C. Zhao, C. Chen, M.-K. Ke, S.-L. Xu, J.-J. Chen, Y. Wu and H.-Q. Yu, Nat. Commun., 2024, 15, 193 CrossRef PubMed.
- J. Xu, X. Zheng, Z. Feng, Z. Lu, Z. Zhang, W. Huang, Y. Li, D. Vuckovic, Y. Li, S. Dai, G. Chen, K. Wang, H. Wang, J. K. Chen, W. Mitch and Y. Cui, Nat. Sustainability, 2021, 4, 233–241 CrossRef PubMed.
- X. Cheng, X. Jiang, S. Yin, L. Ji, Y. Yan, G. Li, R. Huang, C. Wang, H. Liao, Y. Jiang and S. Sun, Angew. Chem., Int. Ed., 2023, 62, e202306166 CrossRef CAS PubMed.
- H. Xie, X. Xie, G. Hu, V. Prabhakaran, S. Saha, L. Gonzalez-Lopez, A. H. Phakatkar, M. Hong, M. Wu, R. Shahbazian-Yassar, V. Ramani, M. I. Al-Sheikhly, D.-E. Jiang, Y. Shao and L. Hu, Nat. Energy, 2022, 7, 281–289 CrossRef CAS.
- C. Ni, Z. Fan, N. Deng and X. Huang, ACS Appl. Energy Mater., 2024, 7, 3116–3124 CrossRef CAS.
- H. Huang, Z. Zhong, J. Li and H. Li, ACS Appl. Energy Mater., 2024, 7, 10804–10814 CrossRef CAS.
- M. S. Enstrup, J. Steinmann, F. G. Daragan, B. Dangpiaei and U. Kunz, ChemSusChem, 2024, 17, e202400491 CrossRef CAS PubMed.
- H. Nguyen, C. Klose, L. Metzler, S. Vierrath and M. Breitwieser, Adv. Energy Mater., 2022, 12, 2103559 CrossRef CAS.
- H. Zhang and P. K. Shen, Chem. Rev., 2012, 112, 2780–2832 CrossRef CAS PubMed.
- D. W. Shin, M. D. Guiver and Y. M. Lee, Chem. Rev., 2017, 117, 4759–4805 CrossRef CAS PubMed.
- M. Adamski, N. Peressin and S. Holdcroft, Mater. Adv., 2021, 2, 4966–5005 RSC.
- F. Liu and K. Miyatake, J. Mater. Chem. A, 2022, 10, 7660–7667 RSC.
- F. Akinori, S. Hironori and T. Takashi, Comput. Theor. Chem., 2017, 1121, 44–48 CrossRef CAS.
- W. Li, Y. Xie, L. Chen, Z. Lin, Z. Zhao, G. Chen, J. Pang and Z. Jiang, J. Membr. Sci., 2024, 706, 122952 CrossRef CAS.
- C. P. Gordon, H. Engler, A. S. Tragl, M. Plodinec, T. Lunkenbein, A. Berkessel, J. H. Teles, A.-N. Parvulescu and C. Copéret, Nature, 2020, 586, 708–713 CrossRef CAS PubMed.
- Q. Jiang, Y. Ji, T. Zheng, X. Li and C. Xia, ACS Mater. Au, 2024, 4, 133–147 CrossRef CAS PubMed.
- X. Li, K. Wu, S. Chen, B. Yuan, J. Wang, C. Tang and Q. Zhang, Chem Catal., 2024, 4, 100997 CrossRef CAS.
- M. H. Guan, T. Wu and A. H. Lu, ChemCatChem, 2023, 15, e202301311 CrossRef CAS.
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC.
- H. Zhang, S. Wu, X. Huang, L. Li, Q. Liao and Z. Wei, Chem. Eng. J., 2022, 428, 131534 CrossRef CAS.
- Q. Jiang, Y. Ji, T. Zheng, X. Li and C. Xia, ACS Mater. Au, 2024, 4, 133–147 CrossRef CAS PubMed.
- X.-C. Liu, W.-K. Yao, B.-Y. Su, Y.-H. Hong, T. Wang, Z.-Y. Zhou and S.-G. Sun, Electrochem. Commun., 2023, 151, 107510 CrossRef CAS.
- C. H. Yang, X. C. Liu, Y. Li, S. Yuan, T. Wang, Z. Y. Zhou and S. G. Sun, ChemSusChem, 2023, 16, e202300699 CrossRef CAS PubMed.
- X. Jiang, R. Chen, Y.-X. Chen and C.-Z. Lu, Chem. Synth., 2024, 4, 4–6 Search PubMed.
- C. Y. Chen, C. Tang, H. F. Wang, C. M. Chen, X. Zhang, X. Huang and Q. Zhang, ChemSusChem, 2016, 9, 1194–1199 CrossRef CAS PubMed.
- X. Meng, X. Cui, N. P. Rajan, L. Yu, D. Deng and X. Bao, Chem, 2019, 5, 2296–2325 CAS.
- J. Kim, J. H. Kim, C. Oh, H. Yun, E. Lee, H. S. Oh, J. H. Park and Y. J. Hwang, Nat. Commun., 2023, 14, 4704 CrossRef CAS PubMed.
- S. Chen, S. Liang, R. Huang, M. Zhang, Y. Song, Y. Zhang, S. Tao, L. Yu and D. Deng, Nat. Synth., 2023, 3, 76–84 CrossRef.
- Y. Yuan, L. Chen, Z. Wan, K. Shi, X. Teng, H. Xu, P. Wu and J. Shi, Sci. Adv., 2024, 10, eado1755 CrossRef CAS PubMed.
- Y. Li, W. Cao and X. Zuo, Environ. Res., 2022, 212, 113508 CrossRef CAS PubMed.
- X. Qin, K. Zhao, X. Quan, P. Cao, S. Chen and H. Yu, J. Hazard. Mater., 2021, 416, 125859 CrossRef CAS PubMed.
- Z. Lin, Felice, G. E. P. O'Connell, T. Wan, D. Zhang, L. Peng, D. Chu, X. Lu and Z. Han, Chem. Eng. J., 2024, 481, 148736 CrossRef CAS.
- E. Brillas, I. Sirés and M. A. Oturan, Chem. Rev., 2009, 109, 6570–6631 CrossRef CAS PubMed.
- M. H. Guan, L. Y. Dong, T. Wu, W. C. Li, G. P. Hao and A. H. Lu, Angew. Chem., Int. Ed., 2023, 62, e202302466 CrossRef CAS PubMed.
- B. N. Ruggiero, A. R. Weidner, J. M. Notestein and L. C. Seitz, J. Phys. Chem. C, 2023, 127, 20640–20651 CrossRef CAS.
- B. N. Ruggiero, K. M. Sanroman Gutierrez, J. D. George, N. M. Mangan, J. M. Notestein and L. C. Seitz, J. Catal., 2022, 414, 33–43 CrossRef CAS.
- X. Kong, C. Liu, Z. Xu, J. Zhao, J. Ni, H. Li, T. Zheng, C. Xia, Z. Geng and J. Zeng, Angew. Chem., Int. Ed., 2024, e202411160, DOI:10.1002/anie.202411160.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00232f |
| This journal is © The Royal Society of Chemistry 2025 |