DOI:
10.1039/D4NR01450B
(Paper)
Nanoscale, 2025,
17, 1260-1269
Cell specific mitochondria targeted metabolic alteration for precision medicine†‡
Received
2nd April 2024
, Accepted 2nd September 2024
First published on 23rd October 2024
Abstract
Mitochondria play important roles in the maintenance of cellular health. In cancer, these dynamic organelles undergo significant changes in terms of membrane hyperpolarization, altered metabolic functions, fusion–fission balance, and several other parameters. These alterations promote cancer growth, proliferation and spread, and the eventual development of metastatic disease and therapeutic resistance. Thus, routing therapeutics to the mitochondrial compartments can be one of the most promising methodologies for tackling such changes to achieve cancer control. Over the last decade, targeted cancer medicine has experienced tremendous growth, enabling the targeting of mitochondria for greater therapeutic specificity. Here, we demonstrate a feasibility method to specifically target the mitochondria of prostate cancer cells. We achieve such dual targeting by utilizing two functionalized polymers and constructing a single blended nanoparticle (NP). Such a targeting strategy was developed utilizing a polymeric platform that differed in terms of the length of the amphiphilic portions, the linker between the hydrophobic portions, and the attached targeting moieties. In doing this, we demonstrate prostate cancer specific mitochondrial delivery of a chemotherapeutic prodrug to create repair-resistant adducts within mitochondrial DNA promoting cellular death. This article documents the synthetic strategy, optimization of blended NPs for cell specific mitochondria targeting, and the utility of the proof-of-concept design was demonstrated using a combination of analytical and in vitro studies.
Introduction
Mitochondria play a major role in the cycle of carcinogenesis.1,2 The oxidative phosphorylation (OxPHOS) cycle involves a series of redox reactions that generate ATP and reactive oxygen species (ROS). The ROS contribute to normative cellular functions, but also potentiate damage to local mitochondrial DNA (mtDNA). Damage to mtDNA within the mitochondrial matrix is unlike nuclear DNA (nDNA) damage. A lack of excision repair machinery in the mitochondrial matrix greatly reduces, or essentially eliminates, the potential for repair of oxidative damage from adjacent ROS. The ROS-damaged mtDNA containing mutated genetic information and transcriptional ability will populate within tumors cells.3 The concentration of mutated mtDNA, demonstrating altered methylation markers throughout the DNA, can ultimately be deemed a causative factor for the propagation of cancerous masses.4 In particular, mutated mtDNA can put forward the following cancer-related mitochondrial markers: increased rate of glycolysis, hyperpolarization of the inner mitochondrial membrane (IMM), and alteration of the permeability pore complex of the membranes. The downstream effects of such mutation on the cell are (1) increased intracellular alkalinity, (2) increased extracellular acidity, and (3) endosomal vesicles in close proximity to the cellular periphery.5 The key action of mtDNA that puts it at the forefront as the prime target for the treatment of aggressive cancers is its contribution to apoptosis.6 Cisplatin, a Food and Drug Administration (FDA) approved chemotherapeutic drug, is highly efficacious in solid tumors, such as those of the ovary, breast, brain, kidney, and testicles. The basis of its efficacy stems from its ability to introduce intra- and inter-strand DNA cross-links with nDNA.7 However, drug-resistance and systemic toxicity are significant downsides to the use of cisplatin. The nDNA utilizes nuclear excision repair (NER) machinery to excise cross-linked regions, rendering the drug ineffective with prolonged use.7–9 Prostate cancer (PCa) is an example of a cisplatin-resistant tumor in which DNA adducts are repaired by NER mechanisms. Furthermore, it was estimated that between 84 and 90% of metastatic prostate cancer (mPCa) patients had metastasis to bone, and as a result of the US Preventive Services Task Force (USPSTF), diagnosis and identification of mPCa cases are on the rise.10–12
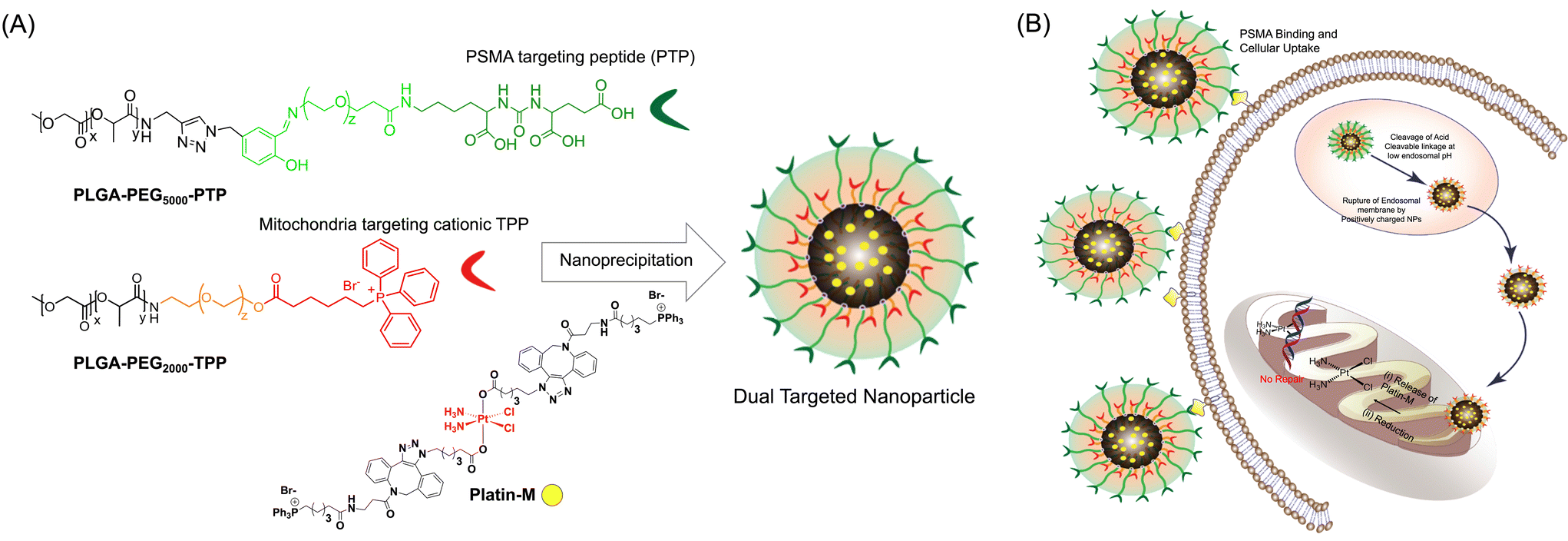
Mutated mtDNA continues to pose a threat towards the progression of the disease to an advanced stage. Thus, the targeting of mtDNA can be a promising tool for treating PCa and managing aggressive PCa in the metastatic stage. The absence of efficient mitochondrial DNA repair machinery and the role of mutated mtDNA in aggressive PCa provide compelling reasons for routing cisplatin to the mitochondrial lumen to develop alternative therapeutic options for metastatic PCa. The success of such a strategy would depend on the ability to target the mtDNA situated in the mitochondrial lumen with cisplatin and thus it requires a mitochondria-targeted cisplatin prodrug and a drug delivery system that can selectively deliver this prodrug to the mitochondrial matrix of PCa cells where the mitochondrial genome is located. In this work, we constructed a dual-targeted biodegradable polymeric nanoparticle (NP) system containing a cisplatin prodrug with mitochondria targeting ligands (Fig. 1) and demonstrated its in vitro efficacy. Polymeric NPs are extensively used to modulate the biodistribution (bioD), pharmacokinetics (PK), and safety profiles of drugs to achieve a better therapeutic window.13–23 We designed and constructed a biocompatible polymeric NP of PLGA-b-PEG functionalized with a terminal triphenylphosphonium (TPP) cation which has the ability to show mitochondrial association utilizing sequential pathways which include endosomal escape, mitochondrial outer membrane association via electrostatic interaction, intermembrane space crossing utilizing a hydrophobic surface, and finally recognition of a substantial negative mitochondrial membrane potential (ΔΨm) which exists across the inner mitochondrial membrane (IMM).24–29 We identified an optimized NP formulation of suitable diameter and surface charge for efficient endosomal escape and mitochondrial targeting and maximum mitochondrial matrix accumulation. The biological activity of Pt(IV) prodrugs involves the reduction and release of active cisplatin. Therefore, we designed and constructed a Pt(IV)-prodrug of cisplatin, Platin-M (Fig. 1), by appending lipophilic TPP cations in the axial positions of cisplatin using a combination of strain promoted cycloaddition based click reactions and traditional chemical approaches.26 Mitochondria of PCa cells maintain a hyperpolarized ΔΨm state compared to that of normal cells.30 Here, we demonstrate the introduction of Platin-M inside a dual targeted NP system to selectively deliver cisplatin inside the mitochondria of PCa cells. To deliver Platin-M with the highest efficiency inside the mitochondria of PCa cells, we encapsulated this prodrug inside a blended NP system containing a PLGA-b-PEG-prostate specific membrane antigen (PSMA)-targeting peptide (PLGA-b-PEG5000-PTP) where PLGA is connected to a longer PEG with an acid sensitive linker (Fig. 1A). The mitochondria-targeted polymer PLGA-b-PEG2000-TPP contains a shorter PEG. PSMA is a well-established tumor marker, which is up-regulated in PCa, particularly in advanced, hormone-independent, and metastatic disease.31 When the blended NPs are in circulation, cationic TPP molecules from PLGA-b-PEG2000-TPP with a shorter PEG linker are shielded by the long PEG-chains from PLGA-b-PEG5000-PTP to minimize TPP exposure to the cells of the mononuclear phagocyte system (MPS). These NPs undergo endocytosis by interaction of the PSMA targeting ligand on the NP surface with the antigens present on PCa cells. At the acidic pH of the endosomes, the longer PEG-chains with an acid cleavable linker are cleaved off and the mitochondria-targeted NP with Platin-M was released (Fig. 1B). Our recent studies with mitochondria-targeted NPs and early endosome marker EAA-1 indicated that these mito-targeted-NPs escape endosomes efficiently,24 and the released single-targeted NPs will disrupt the endosomal membrane and will accumulate inside the mitochondrial matrix efficiently to deliver Platin-M with the highest efficacy. The efficient endosomal escape of the targeted NPs can be attributed to the high buffering capacity of the TPP containing polymer in solution.32 In this paper, we summarize the proof-of-concept demonstration of the activity of such a dual targeted NP system (Fig. 1).
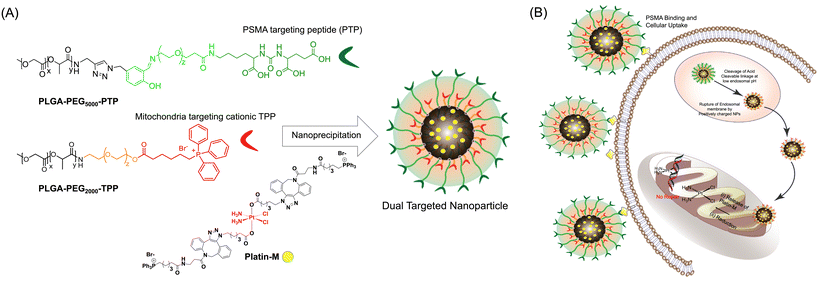 |
| | Fig. 1 Schematic representation of a dual targeting nanotherapeutic technology for accessing selectively the mitochondria of prostate cancer cells which express PSMA and utilization of such a strategy to deliver a cisplatin prodrug, Platin-M for attacking mtDNA. The Platin-M loaded dual targeted nanoparticle consists of the PSMA targeting ligand attached to an acid cleavable polymer backbone along with a mitochondria targeted polymer. Upon endosomal uptake, a longer PSMA targeted polymer was cleaved under the acidic conditions resulting in mitochondria-targeted NPs which can navigate to the mitochondria for Platin-M delivery to result in mt-DNA damage. (A) Structures of the polymers and depiction of the nanoparticle, (B) uptake of NPs in PSMA expressing PCa cells, cleavage of the acid sensitive linker in the endosomes, and uptake of the NPs in the mitochondria. | |
Results and discussion
Synthesis of a PSMA-targeted acid cleavable biodegradable polymer
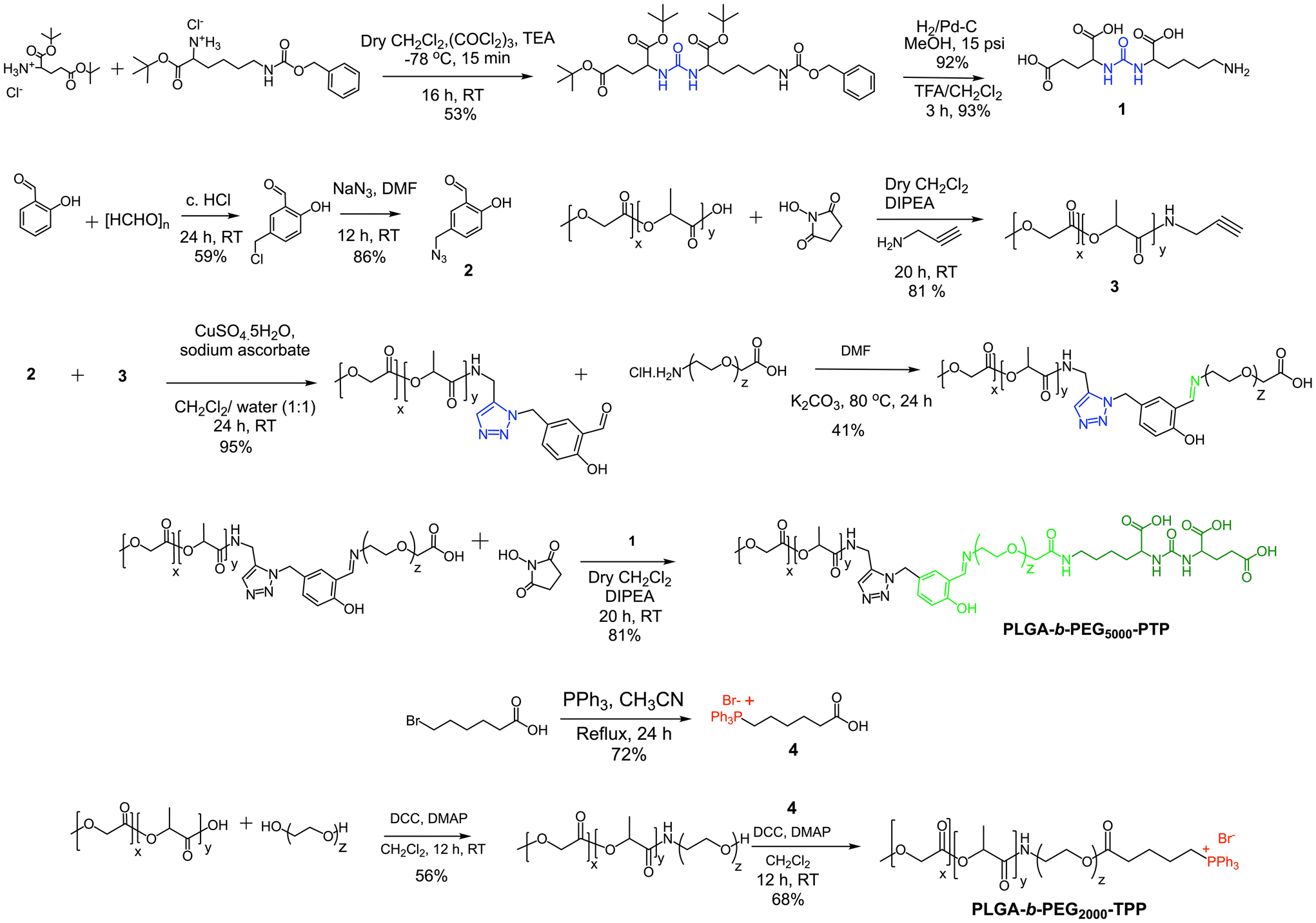
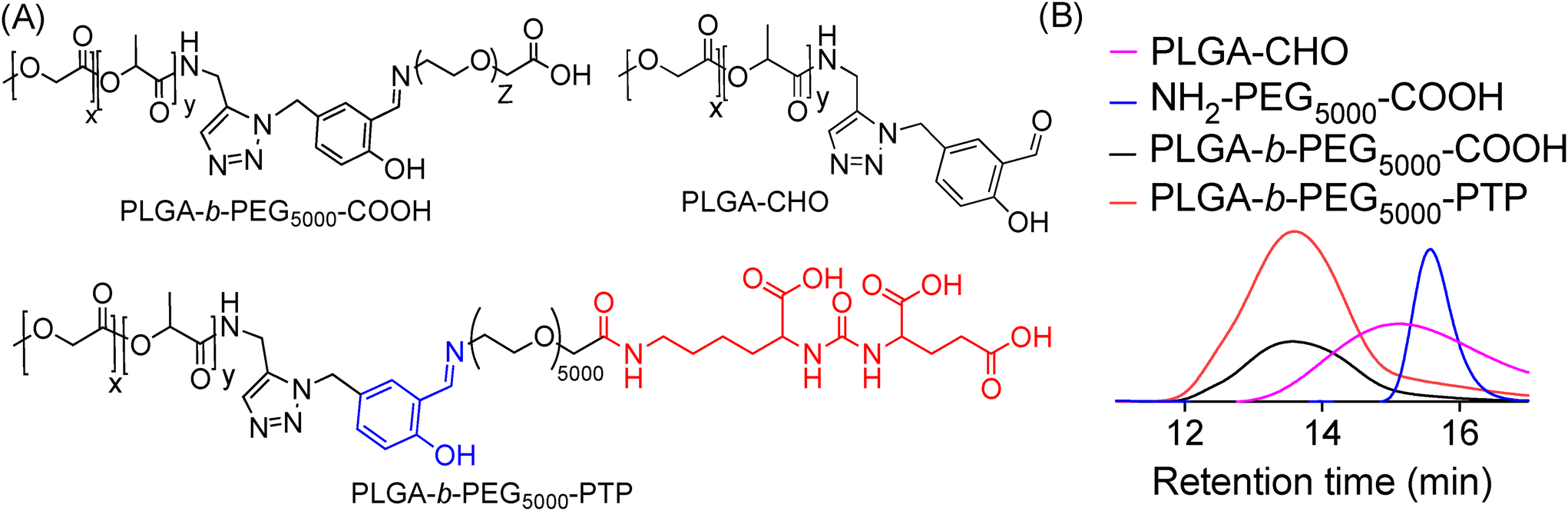
Our objective was to realize precise medicine tailored for prostate cancer by implementing strategies that enable PCa cell-specific mitochondria targeting followed by mitochondria and induce targeted alterations in metabolic processes within these cells. To accomplish this objective, it is imperative to develop a NP technology specifically designed for targeting PCa, which possesses acid-cleavable properties to be integrated with the mitochondria-targeting capabilities in the NP. This combined approach facilitates the delivery of prodrugs to the mitochondria of PCa cells, enhancing the overall therapeutic efficacy. In this approach, we exploited the significant binding affinity of the urea-based prostate-specific membrane antigen (PSMA) targeting peptide (PTP) analogue towards the antigens found on PCa cells. We synthesized a polymer by conjugating the PTP peptide to a PLGA-PEG, resulting in the formation of PLGA-b-PEG5000-PTP construct featuring an extended PEG chain and an acid-cleavable imine linker, achieved through a multistep synthetic route involving sequential reactions to achieve the desired structure (Fig. 2). To improve targeting precision and facilitate drug delivery to mitochondria, we developed a specialized small polymer named PLGA-b-PEG2000-TPP (Fig. 2). This polymer incorporates a triphenylphosphonium cation (TPP), enabling it to effectively target mitochondria and upon formulation into NPs, the positive charge of the TPP group enhances its affinity for the mitochondria, ensuring precise and efficient drug delivery to the organelle. The synthesis of the PSMA-targeted acid cleavable biodegradable polymer involved several steps aimed at incorporating a urea-based PSMA targeting peptide (PTP) analogue into the polymer structure. This peptide analog attached to the polymer backbone, as illustrated in Fig. 3. Throughout the synthesis process, each monomer and polymer formed at every step underwent thorough characterization using spectroscopic and analytical methods (ESI Fig. S1–S9‡). This rigorous characterization ensured the integrity and composition of the polymer at each stage of synthesis, confirming the successful incorporation of the PSMA targeting peptide analog and the acid-cleavable linker into the final polymer structure. The final PLGA-b-PEG-PTP polymer was characterized by NMR and GPC. The GPC of PLGA-b-PEG-PTP and a comparison of the retention time of the final polymer with the precursors confirmed its formation (Fig. 3). Overall, the synthesis strategy capitalized on the strong binding affinity of the urea based PSMA targeting peptide analog to design a biodegradable polymer capable of specifically targeting PCa cells. The inclusion of an acid cleavable linker enhances the polymer's functionality, allowing for the exposure of TPP ligands for targeting mitochondria, a final location of tumor for controlled release of therapeutic agents.
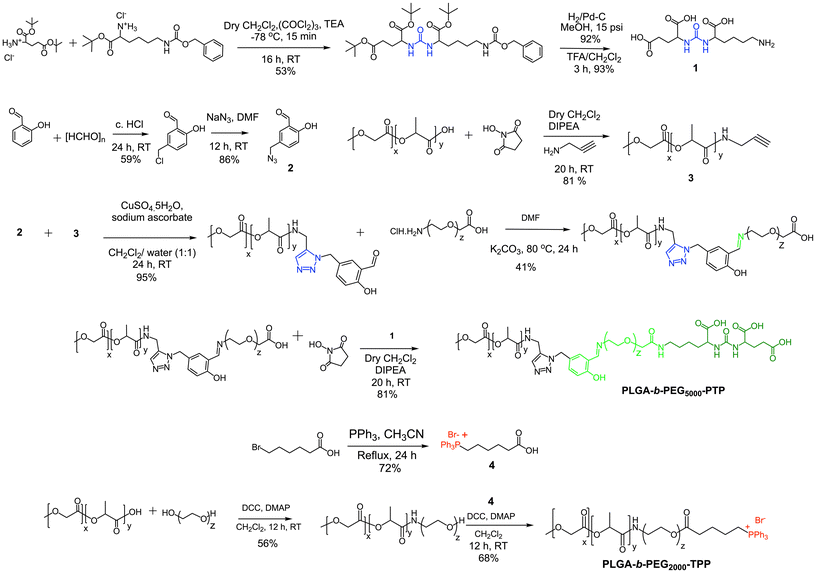 |
| | Fig. 2 Multistep synthetic strategy of PLGA-b-PEG5000-PTP and PLGA-b-PEG2000-TPP. L-Glutamic acid di-tert-butyl ester·HCl reacted with triphosgene followed by H-Lys(Z)-OtBu·HCl and subsequently deprotected the benzyl ether and tert-butyl protection to yield PSMA targeting peptide compound 1. Salicylaldehyde and paraformaldehyde reacted in the presence of hydrochloric acid, which was further reacted with sodium azide to form compound 2. Poly (D,L-lactide-co-glycolide)-carboxylic acid (PLGA3350-COOH) was reacted with propargyl amine after converting to its N-hydroxy succinimide (NHS) variant via the NHS coupling reaction to form compound 3. Compounds 2 and 3 were coupled via a click reaction to yield PLGA3350-CHO which is a PLGA molecule containing an aldehyde end group, which was further reacted with imien and carboxylic acid functionalized polyethylene glycol (H2N-PEG5000-COOH) to form an acid cleavable imine backbone containing a PLGA-PEG5000-COOH molecule. Finally, it was reacted with compound 1 to form the PSMA targeting peptide containing polymer PLGA-PEG5000-PTP. 6-Bromoheaxnoic acid was reacted with triphenyl phosphine to form TPP-hexanoic acid which contains the delocalized positive charge. PLGA3350-COOH was reacted with hydroxyl functionalized polyethylene glycol (HO-PEG2000-OH) using N,N′-dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP) resulting in PLGA3350-PEG2000-OH which was further reacted with TPP-hexanoic acid using DCC and DMAP coupling to form the mitochondria targeted molecule containing the polymer PLGA3350-PEG2000-TPP. | |
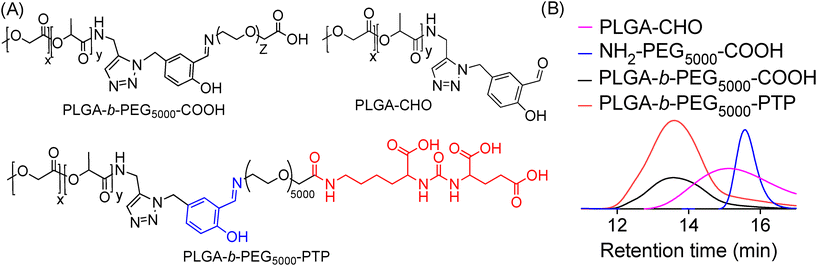 |
| | Fig. 3 (A) Structures of PSMA targeted PLGA-b-PEG-PTP with an acid cleavable linker and its precursors. (B) GPC chromatogram of PLGA-b-PEG-PTP and comparison with precursor polymers. | |
Synthesis and characterization of dual targeted NPs
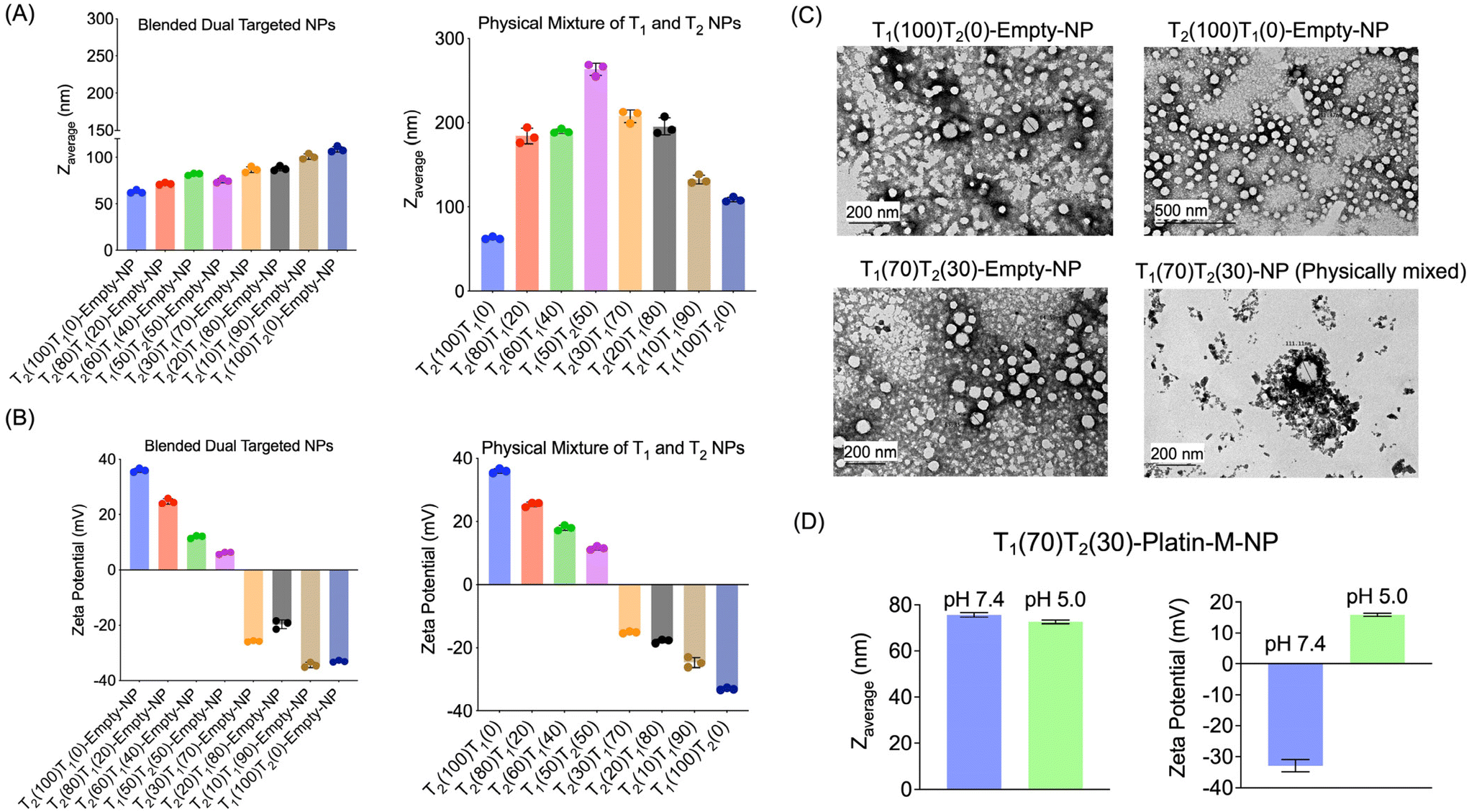
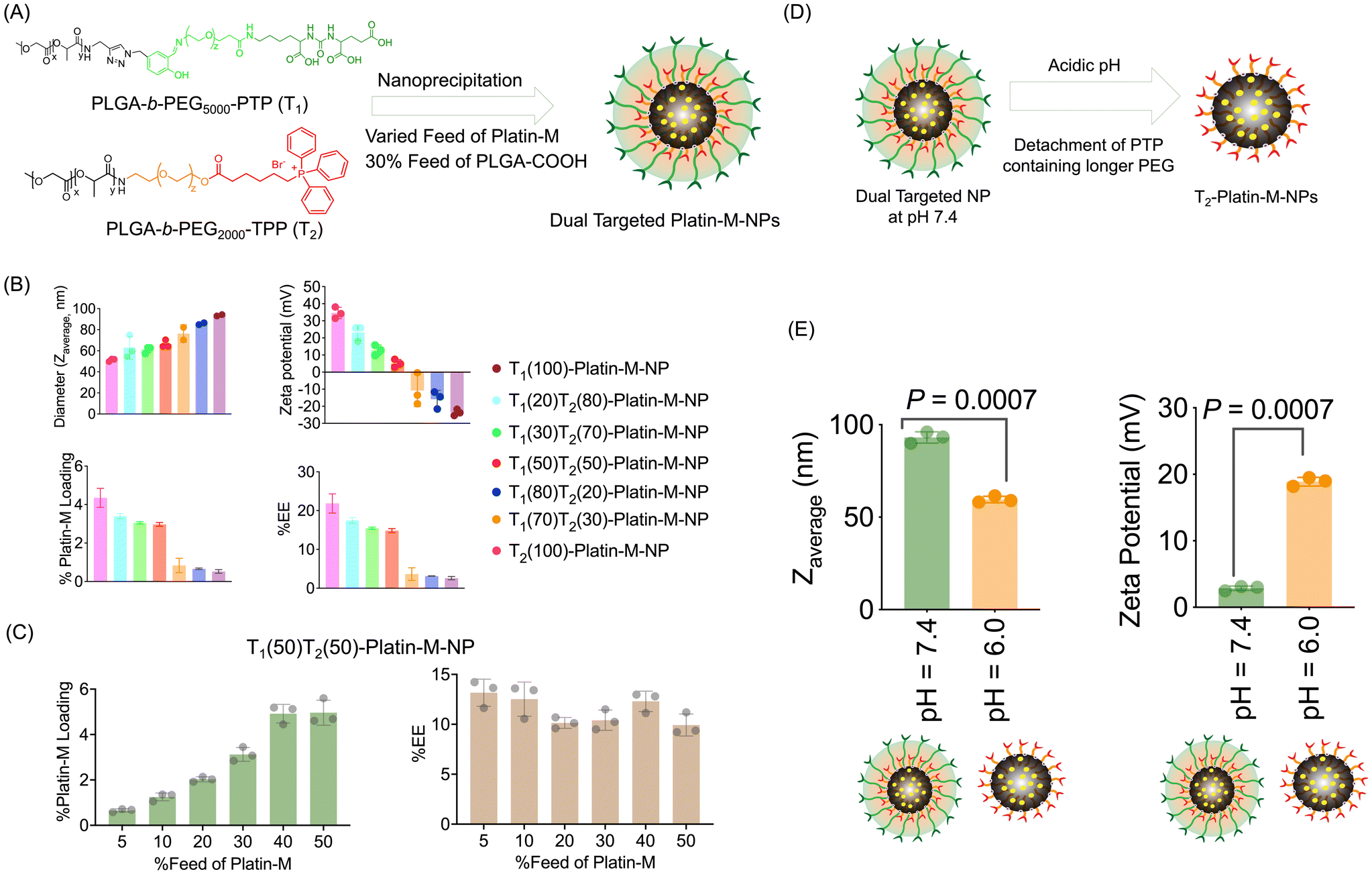
We synthesized drug free (empty) blended NPs consisting of PLGA-b-PEG5000-PTP and PLGA-b-PEG2000-TPP using a nanoprecipitation method by varying the percentages of both polymers from 0 to 100% (Fig. 4A). By varying the ratio of these two polymers, we aimed to understand their effects on the physicochemical properties such as the diameter and surface charge of the NPs. Characterization of these NPs was conducted using dynamic light scattering (DLS) to determine the average diameter, zeta potential, and polydispersity index (PDI) (Fig. 4A and B). We also synthesized individual NPs of PLGA-b-PEG5000-PTP and PLGA-b-PEG2000-TPP separately via the nanoprecipitation method and physically mixed two NPs at various ratios. Comparison of the zeta potentials of dual targeted NPs with single PTP and single TPP targeted NPs clearly demonstrated that the –TPP moieties are masked by the –PTP moieties as we predicted in the case of blended NPs. The physical mixture of NPs exhibited distinct characteristics, with the 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 physical mixture yielding larger NPs. This suggested that PLGA-b-PEG2000-TPP is not being masked but rather aggregated to form larger NPs. This control experiment demonstrated efficient blended NP formation. TEM images of these blended dual NPs (T1(30)T2(70)-empty-NP), physically mixed dual NPs, and single PTP and single TPP NPs revealed similar observations, with blended NPs displaying a defined structure, in contrast, physically mixed NPs aggregated. We further studied the effect of pH changes on dual targeted NPs and it was observed that these NPs are able to release the mitochondria-targeted NPs when the pH is changed to an acidic endosomal pH (Fig. 4D). Diameter analyses of dual targeted NPs at pH 7.4 and 5.0 demonstrated a decrease in the diameter when the pH was 5.0 and the zeta potential changed to a positive value (Fig. 4D). This study indicated that the dual targeted NPs can release the mitochondria targeted NPs at the acidic pH such as those observed in endosomes.
:50 physical mixture yielding larger NPs. This suggested that PLGA-b-PEG2000-TPP is not being masked but rather aggregated to form larger NPs. This control experiment demonstrated efficient blended NP formation. TEM images of these blended dual NPs (T1(30)T2(70)-empty-NP), physically mixed dual NPs, and single PTP and single TPP NPs revealed similar observations, with blended NPs displaying a defined structure, in contrast, physically mixed NPs aggregated. We further studied the effect of pH changes on dual targeted NPs and it was observed that these NPs are able to release the mitochondria-targeted NPs when the pH is changed to an acidic endosomal pH (Fig. 4D). Diameter analyses of dual targeted NPs at pH 7.4 and 5.0 demonstrated a decrease in the diameter when the pH was 5.0 and the zeta potential changed to a positive value (Fig. 4D). This study indicated that the dual targeted NPs can release the mitochondria targeted NPs at the acidic pH such as those observed in endosomes.
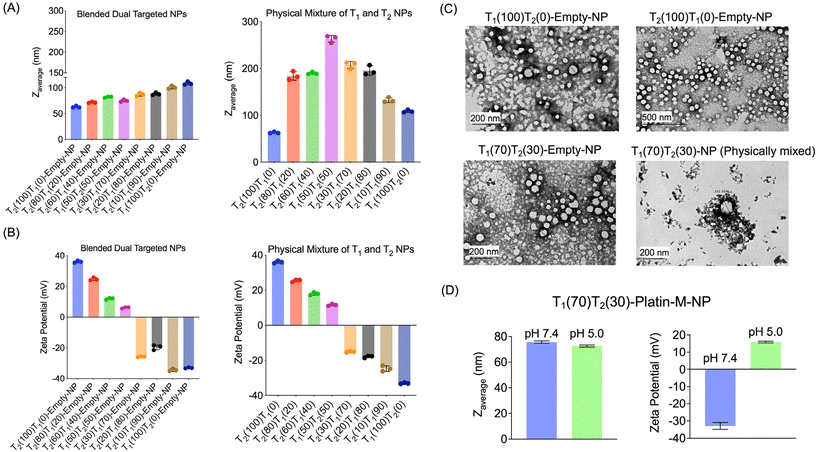 |
| | Fig. 4 Characterization of blended dual targeted nanoparticles and a physical mixture of two nanoparticles with varying ratios of polymers. (A) Diameter and (B) zeta potential of blended NPs and a physical mixture of NPs prepared using different ratios of –PTP (T1) and –TPP (T2) polymers. The ratio is indicated in parenthesis. (C) TEM images of single PTP and single TPP targeted NPs, blended dual targeted T1(30)T2(70)-empty-NPs and equivalent physically mixed dual NPs (T1(30)T2(70)-empty-NP). (D) Diameter and surface charge of blended dual targeted T1(70)T2(30)-empty-NPs at pH = 7.4 and pH = 5.0. | |
Dual targeted NPs containing Platin-M
We optimized the encapsulation of Platin-M inside the hydrophobic core of a blended NP composed of PLGA-b-PEG5000-PTP, PLGA-b-PEG2000-TPP, and PLGA-COOH using the nanoprecipitation method (Fig. 5A). The ratio of these two polymers was varied to understand the effects on the diameter and surface charge of NPs. An additional 30% PLGA-COOH was added along with 30% Platin-M to increase the loading of Platin-M inside the hydrophobic core of the NPs. All these NPs were characterized by DLS for size, zeta potential, and polydispersity. The platinum content was quantified by ICP-MS (Fig. 5A and B). Upon further investigating the Platin-M loading and encapsulation efficiency (EE), we found that the loading and EE drastically decreased beyond 50% of T1 (Fig. 5A and B). Based on these preliminary diameters, surface charge, and higher Platin-M prodrug loading, we decided to use a 50:50 blend of the two polymers to create dual targeted NPs containing Platin-M (dual targeted-Platin-M-NPs). We next studied the effects of percentage feed Platin-M variation on percentage loading and percentage encapsulation efficiency using the dual targeted 50:50 blend T1(50)T2(50) NPs. Analyses of NPs prepared by varying the Platin-M feed and determination of Platin-M loading by the Pt-based ICP-MS method indicated that the percentage loading can be increased by increasing the percentage feed up to 40% and then the encapsulation efficiency saturates (Fig. 5C). Thus, for all our subsequent studies, we used 30% Platin-M for NP synthesis. In all further studies, we used single targeted (PTP or T1)-Platin-M-NPs and single targeted (TPP or T2)-Platin-M-NPs as controls. We further studied the effect of a change in pH on dual targeted (T1, T2)-Platin-M-NPs and it was observed that these NPs can release the inner mitochondria-targeted NP when the pH is changed to an acidic endosomal pH (Fig. 5D and E). Analyses of dual targeted-Platin-M-NPs at pH 7.4 demonstrated a diameter of 93 nm and a zeta potential of 3 mV. However, when the pH was changed to 6.0, we observed a decrease in diameter (60 nm) and the zeta potential changed to a positive value of 19 mV (Fig. 5D). This study indicated that the dual targeted NP will be able to release the mitochondria targeted NP in the endosome in response to the change in pH to a more acidic value.
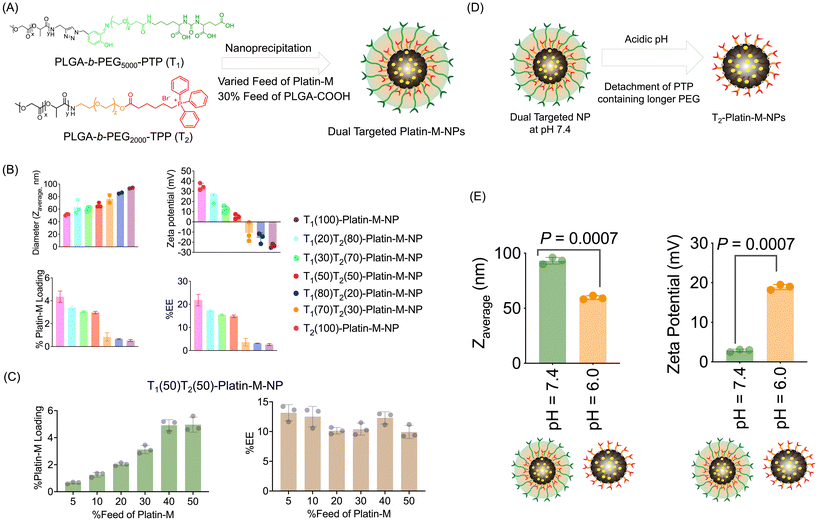 |
| | Fig. 5 Characterization of Platin-M encapsulated dual targeted NPs. (A) Blending of two polymers to generate dual targeted Platin-M loaded NPs. (B) Diameter and zeta potential of blended NPs prepared using different ratios of –PTP (T1) and –TPP (T2) polymers. The ratio is indicated in parenthesis. (C) Effects of the percentage Platin-M feed variation on the percentage loading and EE using T1(50)T2(50)-Platin-M-NPs. (D) A cartoon of pH driven cleavage of the outer shell of dual targeted NPs, and (E) demonstration of such cleavage by determining the diameter and surface charge at two pH values. | |
Uptake studies and mitochondrial effects of dual targeted NPs
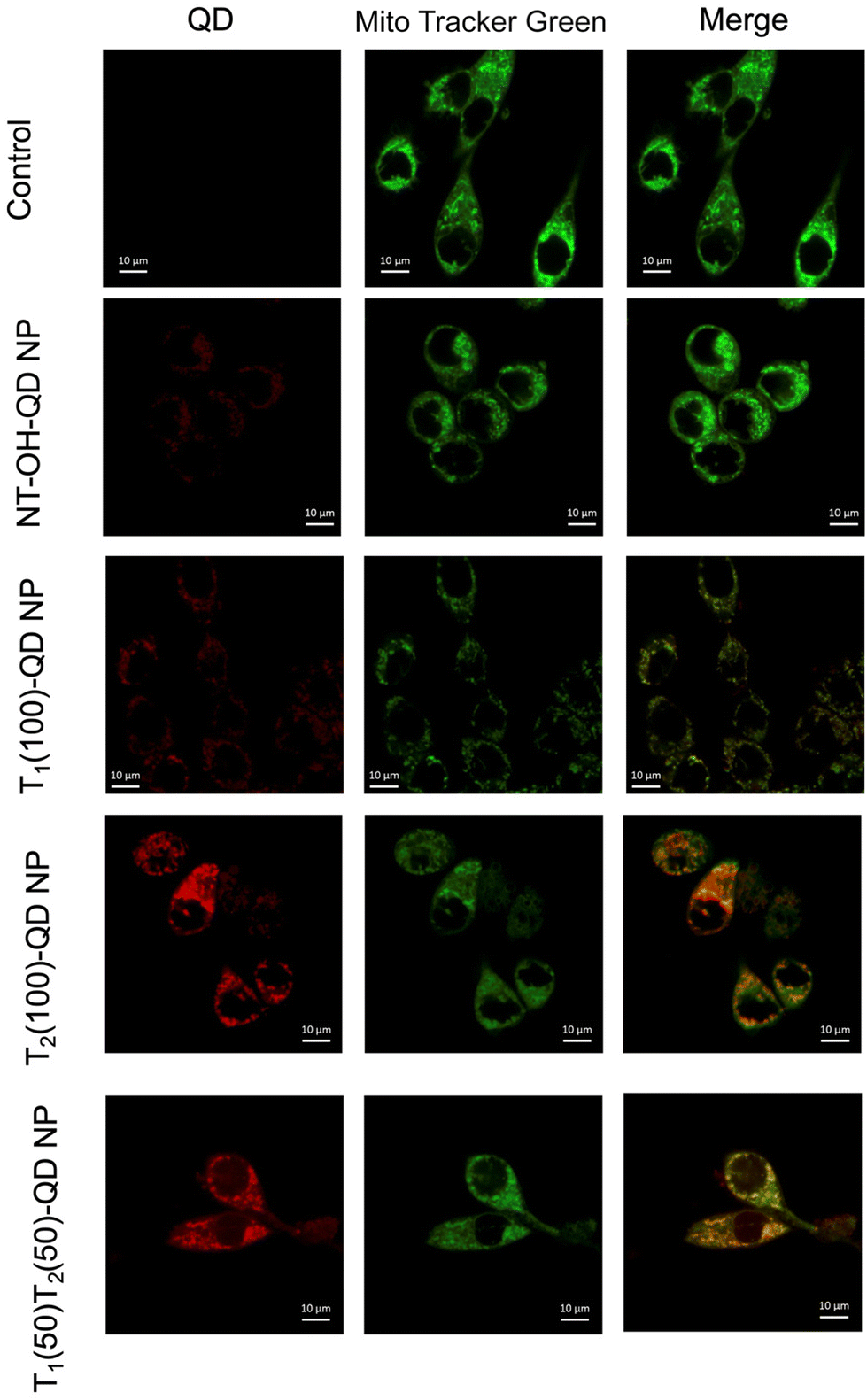
Mitochondrial delivery of the prodrug to the PSMA positive cells is another major objective of developing dual targeted NPs. To visualize the mitochondrial colocalization capability of the NPs, we synthesized red fluorescent quantum dot (QD) loaded NPs with different targeting strategies, including dual targeted, single targeted (PTP or T1), single targeted (TPP or T2), and completely non-targeted (NT) NPs. These NPs were formulated using PLGA-b-PEG5000-PTP, PLGA-b-PEG2000-TPP, PLGA-b-PEG2000-OH, and PLGA-PEG-QD. Specifically, PLGA-PEG-QD33,34 was used to incorporate red fluorescent quantum dots into the NPs. Preliminary uptake studies were conducted in PSMA-expressing LNCaP prostate cancer cells after treating them with 0.5 mg mL−1 NPs for 45 min. The results indicated the efficacy of the dual-targeted NPs in terms of cellular and mitochondrial association, as depicted in Fig. 6. While it was expected that single targeted TPP would exhibit higher colocalization with the mitochondria, single targeted PTP did not exhibit efficient colocalization. This suggests that the dual-targeted NPs have higher affinity for PSMA-expressing cells and exhibit enhanced cellular and mitochondrial uptake compared to the other formulations tested. These findings underscore the potential of the dual-targeted NPs for mitochondria targeted drug delivery to prostate cancer cells expressing PSMA, thereby highlighting their promising application in cancer therapy.
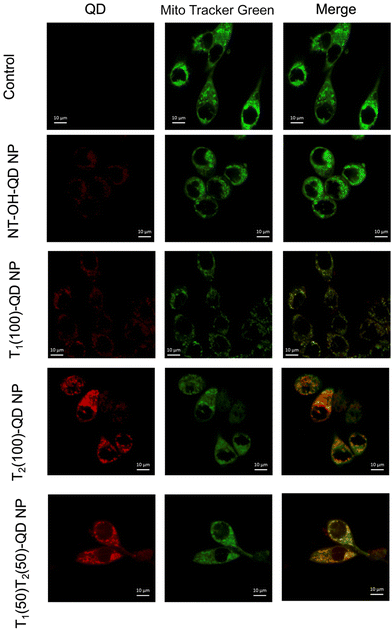 |
| | Fig. 6 Uptake studies and mitochondrial effects of dual targeted NPs. Uptake of dual-targeted NPs in LNCaP cells. [NPs]: 0.5 mg mL−1 (with respect to total NPs). Incubation time: 45 min at 37 °C. Live cell imaging was performed. | |
Activity of Platin-M-loaded dual targeted NPs
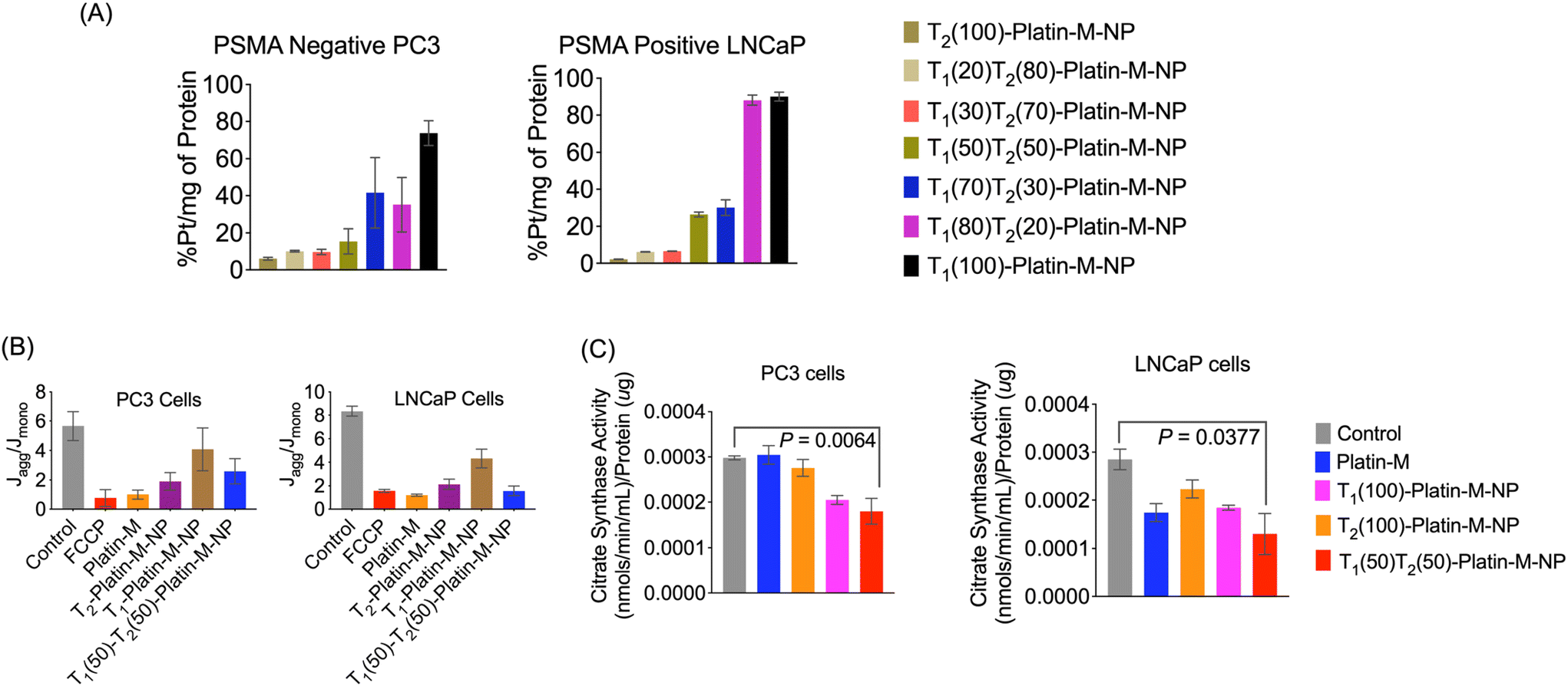
We investigated the overall cellular uptake of Platin-M loaded NPs in PSMA-positive LNCaP prostate cancer cells and compared their uptake in PSMA-negative PC3 prostate cancer cells. The completely TPP (T2) or PTP (T1) targeted NPs were used as controls and we treated the LNCaP and PC3 cells with a Platin-M loaded library of NPs at a concentration of 5 μM for 6 h, after which cell lysates were collected to determine the platinum content. ICP-MS based analyses of Pt treated cell lysates indicated that 50:50 blended dual targeted NPs showed significantly higher uptake in PSMA+ LNCaP cells (Fig. 7A). These studies indicated that we could generate and optimize a dual targeted NP containing Platin-M with the potential to deliver cisplatin in the mitochondria of PSMA+ PCa cells. The efficacy of dual targeted-Platin-M-NPs in inducing apoptosis in prostate cancer (PCa) cells was evaluated using the JC-1 assay to assess the state of ΔΨm, a key early marker of apoptosis. PC3 and LNCaP cells were treated with dual targeted-Platin-M-NPs at a concentration of 10 μM with respect to Platin-M for 24 h. The results revealed a greater induction of apoptosis in LNCaP cells, as evidenced by a decreased Jaggregate/Jmonomer ratio, indicating disruption of the mitochondrial membrane potential (Fig. 7B). Furthermore, the effect of Platin-M, when delivered with dual targeted NPs, on the mitochondrial total mass was determined using the citrate synthase activity assay. A decrease in the mtDNA content is typically associated with reduced citrate synthase activity and lower total mitochondrial mass. Treatment of LNCaP or PC3 cells with dual targeted-Platin-M-NPs at a concentration of 10 μM with respect to Platin-M for 24 h resulted in decreased citrate synthase activity, implying an impact of Platin-M on the mitochondria of PCa cells when delivered with a dual targeted-NP (Fig. 7C). These findings underscored the potential of dual targeted-Platin-M-NPs as effective inducers of apoptosis in PCa cells, particularly in LNCaP cells, and highlighted their ability to target and disrupt mitochondrial function, suggesting their promise for application in PCa therapy.35–38
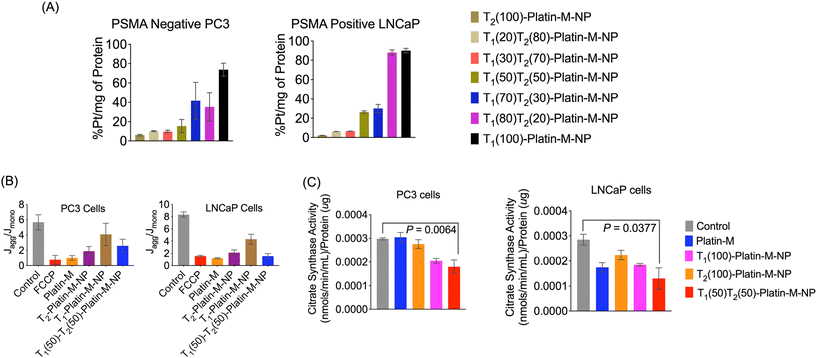 |
| | Fig. 7 Uptake and activity of Platin-M-loaded dual targeted NPs. (A) Demonstration of the targeted cellular uptake of the library of blended NPs in PSMA positive LNCaP compared to PSMA negative PC3 cells. Cells were treated with NPs at a concentration of 5 μM for 6 h and the cell lysates were used to quantify the NPs by ICP-MS by quantifying Pt. (B) Healthy vs. apoptotic cell upon treatment of LNCaP or PC3 cells with dual-targeted-Platin-M-NPs at a concentration of 10 μM with respect to Platin-M for 24 h by detecting the loss of ΔΨm using the lipophilic cationic dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide or JC-1. (C) A reduction in the citrate synthase activity in LNCaP and PC3 cells upon treatment of the cells with dual-targeted-Platin-M-NPs at a concentration of 10 μM with respect to Platin-M for 24 h. Citrate synthase is a TCA cycle enzyme which can be used as a marker of mitochondrial mass. | |
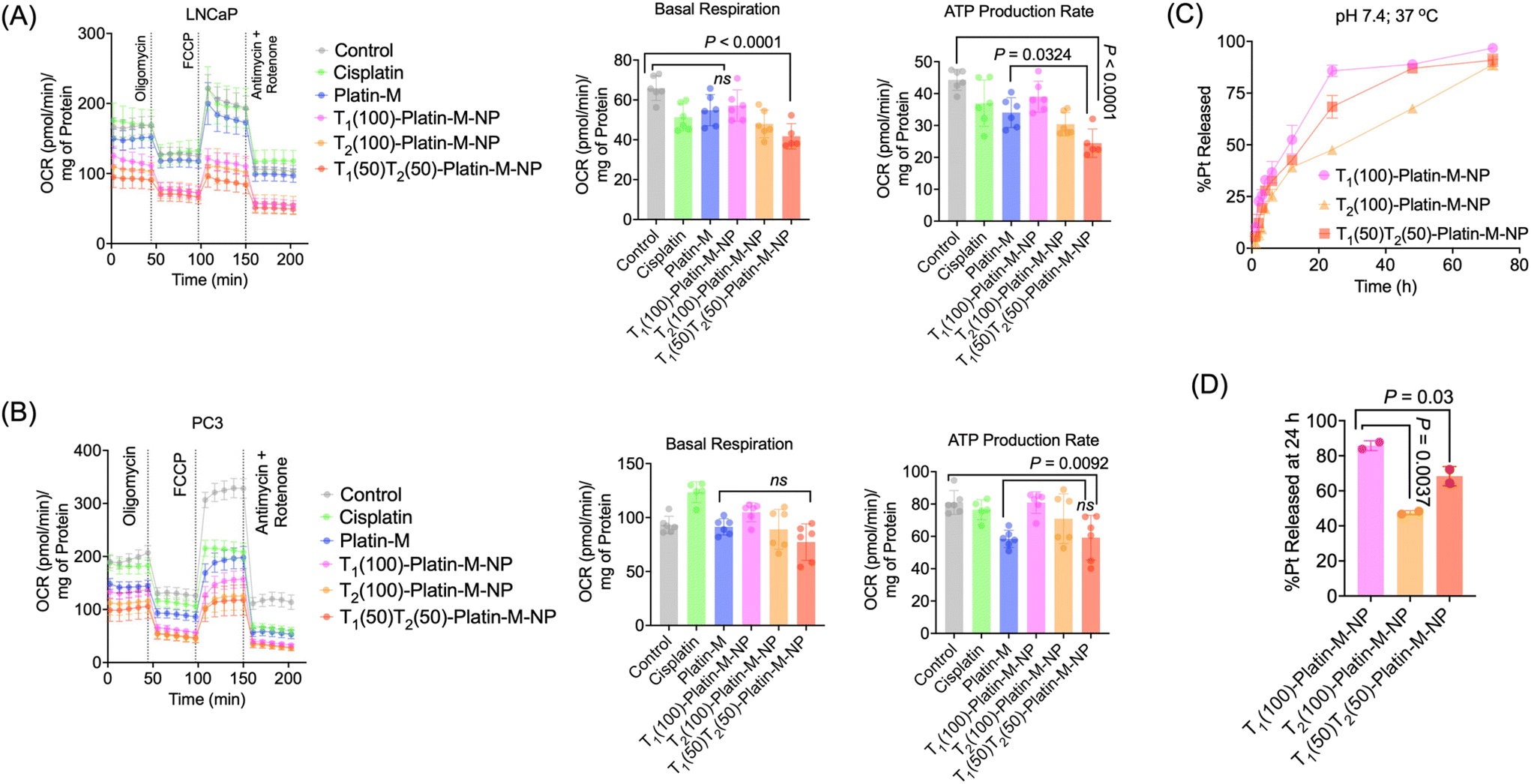
We further investigated the effects of Platin-M prodrug and Platin-M encapsulated in single targeted T1(100)-Platin-M, single targeted T2(100)-Platin-M, and dual targeted T1(50)T2(50)-Platin-M NPs on mitochondrial bioenergetics using Seahorse Mito-stress analyses in both PSMA expressing LNCaP and PSMA negative PC3 cells (Fig. 8A and B). We specifically assessed the basal respiration and ATP production rate of these cells after 24 h of treatment and found that Platin-M loaded dual targeted NPs are significantly more effective compared to control or free drug treatments in PMSA expressing LNCaP cells (Fig. 8A). In PSMA-negative PC3 cells, dual-targeted NPs notably reduced both basal respiration and ATP production rates compared to the control (Fig. 8B). Comparing the different NP treatments, it is evident that targeted delivery and mitochondrial release of the prodrug play a crucial role in impacting the cellular bioenergetics and enhancing efficacy. To support this further, we performed a time dependent release kinetics study using two single targeted and the dual targeted NPs against 1× PBS at a physiologically relevant pH of 7.4 at 37 °C (Fig. 8C). The NPs exhibited a controlled and gradual release of Platin-M from the core of the NPs over 72 h. After 24 h, the single targeted T1(100)-Platin-M-NPs released over 80% of the prodrug likely due to the easily cleavable imine bond present in the polymer; the mitochondria targeted T2(100)-Platin-M-NPs released over 47% during the same period (Fig. 7D). The enhanced mitochondrial targeting and delivery of Platin-M in T2(100)-Platin-M-NPs likely contributed to their superior efficacy compared with T1(100)-Platin-M-NPs. The dual-targeted NPs showed an intermediate release of around 68% at 37 °C and pH 7.4, indicating a balanced release profile. The combined benefits of higher prodrug release and more efficient mitochondrial targeting in PSMA expressing LNCaP cells accounted for the enhanced efficacy of dual targeted T1(50)T2(50)-Platin-M NPs over the free Platin-M prodrug and single-targeted NPs.
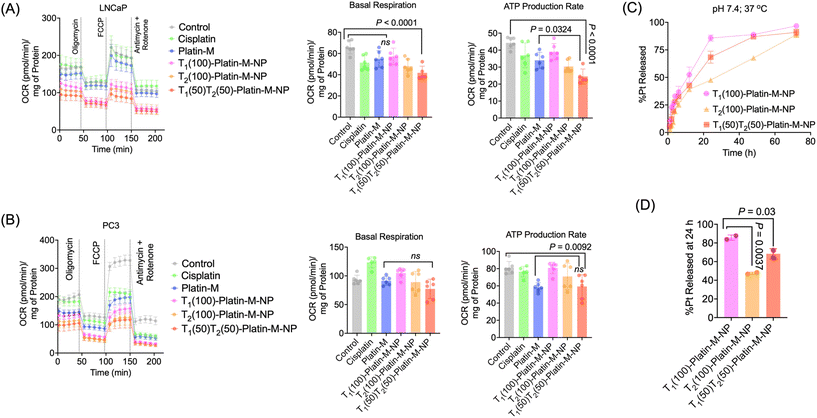 |
| | Fig. 8 Assessment of the impact of free Platin-M and Platin-M loaded single targeted T1(100)-Platin-M, single targeted T2(100)-Platin-M, and dual targeted T1(50)T2(50)-Platin-M NPs on the overall mitochondrial health of PCa cells. Analyses of mitochondrial OXPHOS by mito-stress test assays of (A) PSMA expressing LNCaP cells and (B) PSMA negative PC3 cells showing a significant reduction in basal respiration and ATP production rate upon treatment with T1(50)T2(50)-Platin-M-NPs. (C) Release kinetics of Platin-M from single targeted and dual targeted NPs. (D) Percentage of Pt released from single targeted and dual targeted NPs during a 72 h period at a physiological pH of 7.4 and a temperature of 37 °C. Statistical analyses were performed using ordinary one-way ANOVA. | |
Conclusions
In summary, to deliver a cisplatin prodrug inside the mitochondria of PCa cells with the highest efficiency, we designed and demonstrated encapsulation of the prodrug Platin-M inside a blended NP system containing a PLGA-b-PEG-prostate specific membrane antigen (PSMA)-targeting peptide (PLGA-b-PEG5000-PTP) where PLGA was connected to a longer PEG with an acid sensitive linker. The mitochondria-targeted polymer PLGA-b-PEG2000-TPP contained a shorter PEG. The dual-targeted NPs can release the single-targeted NPs at endosomal acidic pH. This approach enabled the creation of repair-resistant adducts within mitochondrial DNA, ultimately leading to cellular death. This innovative approach holds promise for enhancing the therapeutic effectiveness of cisplatin in PCa treatment by specifically targeting the mitochondria, thereby offering a potential strategy for improving outcomes in patients with advanced disease when studied further. We would like to emphasize that our innovation lies in the development of dual targeted NPs to selectively deliver the DNA damaging agent to the mitochondria of only cancer cells to form repair resistant DNA adducts using biodegradable components such as PLGA and PEG. Our findings highlight the power of synthetic strategies to realize precise medicine tailored for prostate cancer by implementing cell-specific and mitochondria targeting technologies.
Statistics
All data were expressed as mean ± SD (standard deviation). Statistical analyses were performed using GraphPad Prism® software v. 5.00.
Author contributions
S. D. conceptualized and designed the research, supervised experiments, and provided all resources. A. A., M. B., B. S., and U. B. synthesized monomers and polymers, and conducted troubleshooting of all synthetic methods. S. G. performed the synthesis of a few nanoparticles. A. A., M. B., and S. D. drafted the original manuscript. All authors discussed the results and commented on the manuscript.
Data availability
All data are presented in the manuscript and the ESI.‡ No additional data are available.
Conflicts of interest
There is no financial interest to disclose.
Acknowledgements
This work was supported, in whole or in part, by the Sylvester Comprehensive Cancer Center, the NCI funded Sylvester Comprehensive Cancer Center support grant 1P30CA240139 and a Bankhead-Coley Grant (8BC10) from the Florida Department of Health (to S. D.).
References
- M. A. Birch-Machin, The role of mitochondria in ageing and carcinogenesis, Clin. Exp. Dermatol., 2006, 31(4), 548–552 CrossRef CAS.
- P. Tokarz and J. Blasiak, Role of mitochondria in carcinogenesis, Acta Biochim. Pol., 2014, 61(4), 671–678 CrossRef.
- A. Chatterjee, E. Mambo and D. Sidransky, Mitochondrial DNA mutations in human cancer, Oncogene, 2006, 25(34), 4663–4674 CrossRef CAS.
- D. J. Smiraglia, M. Kulawiec, G. L. Bistulfi, S. G. Gupta and K. K. Singh, A novel role for mitochondria in regulating epigenetic modification in the nucleus, Cancer Biol. Ther., 2008, 7(8), 1182–1190 CrossRef CAS PubMed.
- J. Huotari and A. Helenius, Endosome maturation, EMBO J., 2011, 30(17), 3481–3500 CrossRef CAS PubMed.
- R. Wen, B. Banik, R. K. Pathak, A. Kumar, N. Kolishetti and S. Dhar, Nanotechnology inspired tools for mitochondrial dysfunction related diseases, Adv. Drug Delivery Rev., 2016, 99, 52–69 CrossRef CAS.
- S. Dasari and P. B. Tchounwou, Cisplatin in cancer therapy: Molecular mechanisms of action, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS.
- D. B. Zamble, D. Mu, J. T. Reardon, A. Sancar and S. J. Lippard, Repair of cisplatin−DNA adducts by the mammalian excision nuclease, Biochemistry, 1996, 35(31), 10004–10013 CrossRef CAS.
- J. G. Moggs, D. E. Szymkowski, M. Yamada, P. Karran and R. D. Wood, Differential human nucleotide excision repair of paired and mispaired cisplatin–DNA adducts, Nucleic Acids Res., 1997, 25(3), 480–491 CrossRef CAS PubMed.
- L. Bubendorf, A. Schöpfer, U. Wagner, G. Sauter, H. Moch, N. Willi, T. C. Gasser and M. J. Mihatsch, Metastatic patterns of prostate cancer: An autopsy study of 1,589 patients, Hum. Pathol., 2000, 31(5), 578–583 CrossRef CAS.
- G. Gandaglia, F. Abdollah, J. Schiffmann, V. Trudeau, S. F. Shariat, S. P. Kim, P. Perrotte, F. Montorsi, A. Briganti, Q.-D. Trinh, P. I. Karakiewicz and M. Sun, Distribution of metastatic sites in patients with prostate cancer: A population-based analysis, Prostate, 2014, 74(2), 210–216 CrossRef.
- M. M. Desai, G. E. Cacciamani, K. Gill, J. Zhang, L. Liu, A. Abreu and I. S. Gill, Trends in incidence of metastatic prostate cancer in the US, JAMA Network Open, 2022, 5(3), e222246 CrossRef PubMed.
- M. Ferrari, Cancer nanotechnology: Opportunities and challenges, Nat. Rev. Cancer, 2005, 5(3), 161–171 CrossRef CAS PubMed.
- R. Langer, Drug delivery and targeting, Nature, 1998, 392(6679), 5–10 CAS.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nanoparticle-mediated cellular response is size-dependent, Nat. Nanotechnol., 2008, 3(3), 145–150 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, Nanomedicine: Developing smarter therapeutic and diagnostic modalities, Adv. Drug Delivery Rev., 2006, 58(14), 1456–1459 CrossRef CAS.
- V. Wagner, A. Dullaart, A. K. Bock and A. Zweck, The emerging nanomedicine landscape, Nat. Biotechnol., 2006, 24(10), 1211–1217 CrossRef CAS PubMed.
- O. C. Farokhzad, J. J. Cheng, B. A. Teply, I. Sherifi, S. Jon, P. W. Kantoff, J. P. Richie and R. Langer, Targeted nanoparticle–aptamer bioconjugates for cancer chemotherapy in vivo, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(16), 6315–6320 CrossRef CAS.
- V. P. Torchilin, Micellar nanocarriers: Pharmaceutical perspectives, Pharm. Res., 2007, 24(1), 1–16 CAS.
- K. S. Soppimath, T. M. Aminabhavi, A. R. Kulkarni and W. E. Rudzinski, Biodegradable polymeric nanoparticles as drug delivery devices, J. Controlled Release, 2001, 70(1–2), 1–20 CrossRef CAS PubMed.
- O. C. Farokhzad, S. Y. Jon, A. Khadelmhosseini, T. N. T. Tran, D. A. LaVan and R. Langer, Nanopartide–aptamer bioconjugates: A new approach for targeting prostate cancer cells, Cancer Res., 2004, 64(21), 7668–7672 CrossRef CAS.
- R. Langer, Drug delivery. Drugs on target, Science, 2001, 293(5527), 58–59 CrossRef CAS.
- S. Dhar, N. Kolishetti, S. J. Lippard and O. C. Farokhzad, Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(5), 1850–1855 CrossRef CAS PubMed.
- S. Marrache and S. Dhar, Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(40), 16288–16293 CrossRef CAS PubMed.
- S. Marrache, S. Tundup, D. A. Harn and S. Dhar, Ex vivo programming of dendritic cells by mitochondria-targeted nanoparticles to produce interferon-gamma for cancer immunotherapy, ACS Nano, 2013, 7(8), 7392–7402 CrossRef CAS PubMed.
- S. Marrache, R. K. Pathak and S. Dhar, Detouring of cisplatin to access mitochondrial genome for overcoming resistance, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(29), 10444–10449 CrossRef CAS.
- S. Marrache, S. Tundup, D. A. Harn and S. Dhar, Ex vivo generation of functional immune cells by mitochondria-targeted photosensitization of cancer cells, Methods Mol. Biol., 2015, 1265, 113–122 CrossRef CAS PubMed.
- R. Wen and S. Dhar, Turn up the cellular power generator with vitamin E analogue formulation, Chem. Sci., 2016, 7(8), 5559–5567 RSC.
- R. K. Pathak, R. Wen, N. Kolishetti and S. Dhar, A prodrug of two approved drugs, cisplatin and chlorambucil, for chemo war against cancer, Mol. Cancer Ther., 2017, 16(4), 625–636 CrossRef CAS.
- I. C. Summerhayes, T. J. Lampidis, S. D. Bernal, J. J. Nadakavukaren, K. K. Nadakavukaren, E. L. Shepherd and L. B. Chen, Unusual retention of rhodamine 123 by mitochondria in muscle and carcinoma cells, Proc. Natl. Acad. Sci. U. S. A., 1982, 79(17), 5292–5296 CrossRef CAS.
- A. Ghosh and W. D. Heston, Tumor target prostate specific membrane antigen (PSMA) and its regulation in prostate cancer, J. Cell. Biochem., 2004, 91(3), 528–539 CrossRef CAS PubMed.
- N. D. Sonawane, F. C. Szoka and A. S. Verkman, Chloride accumulation and swelling in endosomes enhances DNA transfer by polyamine-DNA polyplexes, J. Biol. Chem., 2003, 278(45), 44826–44831 CrossRef CAS PubMed.
- S. Marrache, R. K. Pathak and S. Dhar, Formulation and optimization of mitochondria-targeted polymeric nanoparticles, Methods Mol. Biol., 2015, 1265, 103–112 CrossRef CAS PubMed.
- S. Marrache and S. Dhar, Biodegradable synthetic high-density lipoprotein nanoparticles for atherosclerosis, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(23), 9445–9450 CrossRef CAS.
- S. Guin, A. Ashokan, A. Pollack and S. Dhar, Lipid metabolism modulatory cisplatin prodrug sensitizes resistant prostate cancer towards androgen deprivation therapy, ACS Pharmacol. Transl. Sci., 2024, 7(9), 2820–2826 CrossRef CAS.
- A. A. Kalathil, S. Guin, A. Ashokan, U. Basu, B. Surnar, K. S. Delma, L. M. Lima, O. N. Kryvenko and S. Dhar, New Pathway for cisplatin prodrug to utilize metabolic substrate preference to overcome cancer intrinsic resistance, ACS Cent. Sci., 2023, 9, 1297–1312 CrossRef CAS.
- R. K. Pathak, U. Basu, A. Ahmad, S. Sarkar, A. Kumar, B. Surnar, S. Ansari, K. Wilczek, M. E. Ivan, B. Marples, N. Kolishetti and S. Dhar, A designer bow-tie combination therapeutic platform: An approach to resistant cancer treatment by simultaneous delivery of cytotoxic and anti-inflammatory agents and radiation, Biomaterials, 2018, 187, 117–129 CrossRef CAS.
- R. K. Pathak and S. Dhar, A nanoparticle cocktail: Temporal release of predefined drug combinations, J. Am. Chem. Soc., 2015, 137, 8324–8327 CrossRef CAS.
Footnotes |
| † Celebrating the contributions of Professor Santanu Bhattacharya. |
| ‡ Electronic supplementary information (ESI) available: Materials, instruments, and methods. See DOI: https://doi.org/10.1039/d4nr01450b |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Michael
Birnhak
a,
Bapurao
Surnar
ab,
Michael
Birnhak
a,
Bapurao
Surnar