
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydride-mediated chemoselective C–H bond formation during benzoic acid hydrodeoxygenation on anatase TiO2†
Mikyung Hwang‡
,
Jeremy Hu‡,
Michael J. Janik and
Konstantinos Alexopoulos *
*
Department of Chemical Engineering, The Pennsylvania State University, University Park, Pennsylvania 16801, USA. E-mail: kxa5325@psu.edu
First published on 4th March 2025
Abstract
The chemoselective hydrogenation of benzoic acid to aromatic products such as benzaldehyde, benzyl alcohol, toluene, and benzene is studied on anatase TiO2 (001) using density functional theory (DFT) and microkinetic modeling (MKM). Oxygen vacancy (Ovac) sites are more active for benzoic acid hydrogenation than fully oxidized surface regions, with a nearly 2.5 eV lower energy for the first C–H bond formation reaction to occur near Ovac. This favorable C–H bond formation mechanism on anatase TiO2 occurs between hydrides (H−) and monoanionic intermediates coadsorbed in Ovac. A steady-state microkinetic model is constructed using computed reaction energies and activation barriers to determine rates of product formation at different reaction temperatures. Product selectivities differ in distinct temperature ranges, caused by competition among reaction and product desorption steps. Sensitivity analysis shows which elementary steps significantly affect the overall benzoic acid hydrogenation activity and selectivity.
1. Introduction
The production of aromatic products such as benzaldehyde, benzyl alcohol, toluene, and benzene1–3 has been of interest due to their versatility as commodity chemicals and intermediates in chemical and pharmaceutical industries.4 The chemoselective hydrogenation of benzoic acid has previously been identified as an attractive route for the formation of these aromatic products.1,2,5,6 Benzoic acid is a relatively inexpensive feedstock that can be sourced from a variety of waste products. For example, polyethylene terephthalate (PET) plastic waste pyrolysis produced benzoic acid as the major product.7,8 Thus, finding methods to catalytically upgrade benzoic acid to value-added products could make diverting PET plastic waste from landfills economically more feasible.Selectivity is a challenging issue for aromatic carboxylic acid hydrogenation,9 since hydrogenating the aromatic ring is typically easier than preserving its aromaticity and exclusively hydrogenating the carboxyl group.1,6,10,11 Although some studies have demonstrated promising results in achieving aromatic product selectivity, there remains considerable variation in both the distribution of products obtained and the proposed reaction mechanisms.2,5,12,13 In principle, benzoic acid hydrogenation can produce aromatic products such as benzaldehyde, benzyl alcohol, benzene, and toluene (Fig. 1).1 Notably, the carbon of the carboxyl group undergoes several C–H bond formation steps upon the consecutive hydrogenation of benzoic acid towards toluene. Meanwhile, benzene can be produced through the decarboxylation of benzoic acid12,14 or the decarbonylation of benzaldehyde.15,16 Benzene production via the demethylation of toluene appears unlikely as only CO and CO2 (i.e., no CH4) are detected under relevant conditions.17,18 Among the possible aromatic products of benzoic acid, hydrogenated products such as toluene and benzyl alcohol are often preferred over benzaldehyde due to their value-added demand as solvents and ester precursors.14,19–21
 | ||
| Fig. 1 Reaction scheme for the chemoselective hydrogenation of benzoic acid to aromatic products. | ||
The development of a superior catalyst for the selective hydrogenation of benzoic acid, capable of achieving high selectivity for specific aromatic products, remains an active area of research.6,9 Numerous studies focused on ring hydrogenation for cyclohexanecarboxylic acid production.22–26 While some studies have reported supported metals such as Au1,16,27 and Pt2 to be selective for aromatic products, the specific role of oxide catalysts or supports in benzoic acid hydrogenation remains unclear. Early studies have reported high yields and selectivity for benzaldehyde on ZrO2 (ref. 5 and 6) and CeO2,28 albeit at higher temperatures than on extended metal clusters. For example, benzoic acid hydrogenation on Au/CeO2 was reported to occur at 573 K,1 while activity on CeO2 occurs between 523–723 K.28 The only study of benzoic acid hydrogenation on TiO2 reported lower selectivity for benzaldehyde and higher selectivity for toluene and benzene (i.e., relative to HfO2 and ZrO2).6 Ponec et al. hypothesized from their experiments on various metal oxides that the M–O bond strength and, by extension, the concentration of surface oxygen vacancy (Ovac) sites dictates the selectivity for benzaldehyde.6,9,28 The same authors suggested mechanisms such as a reverse Mars and van Krevelen or radical-like benzoate decomposition as responsible for benzaldehyde formation.9,28,29 To date, no ab initio studies have been published to provide atomistic confirmation of elementary reaction mechanisms or selectivity determining steps in benzoic acid hydrodeoxygenation (HDO) on metal oxides.
The search for highly selective catalysts for the chemoselective hydrogenation of complex aromatics is not exclusive to benzoic acid hydrogenation. Single-atom catalysts (SACs), with metals atomically dispersed on a support,30,31 have emerged as remarkably selective catalysts for similar reactions. For example, Liu et al. reported highly active and selective Ag SACs on anatase TiO2 for phenolic products from guaiacol, a biomass-derived aromatic (i.e., aromatic products preferred over ring hydrogenation).32 Both experimental and DFT studies confirm that the Ag single atoms serve as sites for H2 activation and a source of continuous hydrogen spillover to the TiO2 support.32–34 Since single atom catalysts have the metal atomically dispersed on the surface at extremely low coverages35,36 and because Ag is typically not active for hydrogenation,32 the vast majority of hydrogenation reactions, such as C–H bond formation, is expected to occur on the TiO2 surface (i.e., whereas the metal mainly acts as a source of spillover H atoms).
Since hydrogenation conditions over metal oxides require temperatures above 523 K,28 the presence of Ovac is expected on the oxide surface.37,38 Nuclear magnetic resonance (NMR) results39 as well as our previous work33 on anatase TiO2 have identified hydrides in Ovac as stable species of spillover H atoms when the surface is exposed to high temperature reducing conditions. Thus, in this paper, we use DFT and MKM to achieve two goals: 1) determine the mechanisms and selectivity of benzoic acid hydrodeoxygenation on anatase TiO2, and 2) examine the roles and effects that Ovac-adsorbed hydrides have in the hydrodeoxygenation reaction in general and the C–H bond formation steps in particular.
2. Methods
2.1. Development of surface models
The (001) facet of anatase TiO2, a low index facet of anatase considered to be the most active for catalytic reactions,40–42 was used for this study. Periodic 2 × 2 supercells of (001) anatase were constructed with a vacuum region of 15 Å above the slab to minimize periodic dipole interactions normal to the surface. The slab is two layers thick, with each layer defined as the minimum depth of atoms to achieve TinO2n stoichiometry in the 2 × 2 supercell; each layer is comprised of three atomic sublayers (i.e., six atomic layers in total to form a Ti24O48 unit cell). The three uppermost atomic layers are allowed to freely relax during geometric optimizations, while the bottom three layers are fixed in position to model the bulk anatase support (Fig. S1†). Atomic figures shown in this publication were constructed using the Amsterdam modeling suite (AMS).43Surface oxygen atoms are characterized based on their local coordination, with oxygens coordinated to two and three Ti atoms referred to as O2C and O3C, respectively (Fig. 2a). Ovac readily form on the surface as a result of TiO2 reduction (i.e., two spillover H atoms combine with a surface O to desorb as H2O and leave an Ovac).44,45 Previous studies have established that bridging O2C atoms require the least amount of energy to form a vacancy,33,46 so models of the reduced TiO2 (001) surface have an O2C vacant (Fig. 2b).
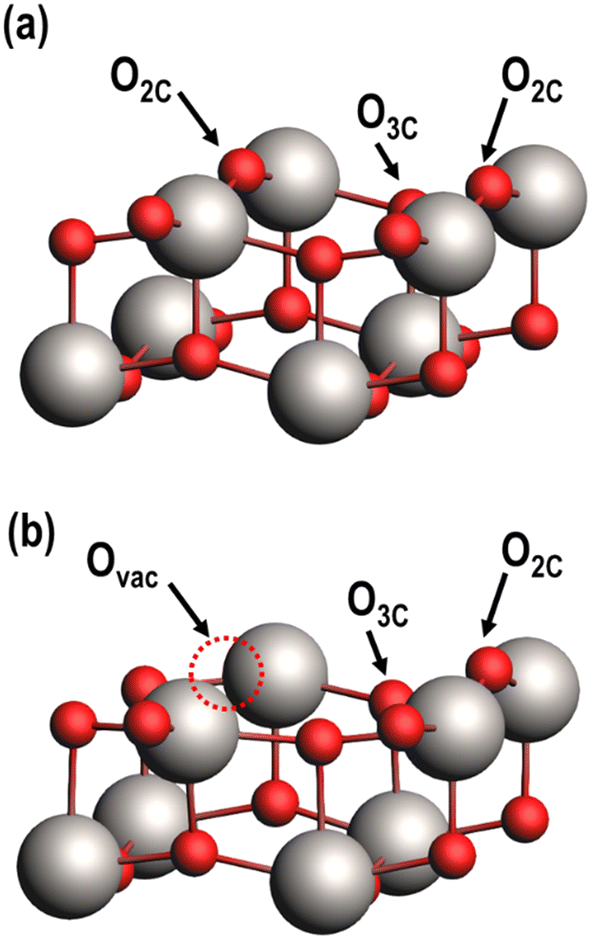 | ||
| Fig. 2 The anatase TiO2 (001) surface with (a) the fully oxidized surface having one pair each of O2C and O3C, and (b) a reduced surface with an O2C substituted with an Ovac. Only the two uppermost atomic layers are shown for clarity. Ti (gray), O (red), and an Ovac (dashed circle) are depicted. | ||
2.2. Electronic structure methods
DFT was implemented through the Vienna ab initio simulation package (VASP)47 using the Perdew–Burke–Ernzerhof (PBE) exchange correlation functional.48 The projector augmented-wave (PAW) method was used to take into account core–valence interactions49 and PBE + D3 was used to correct for dispersion interactions.50,51 The self-interaction error from strongly correlated d-orbital electrons on Ti requires the use of DFT+U corrections on Ti.52 The Hubbard's U parameter was set to 3 eV, as it was confirmed to give reasonable results for the energy of reduction of TiO2 to Ti2O3 and was proposed for computing redox cycles on TiO2 by Hu and Metiu.53 During geometric optimizations, iterative calculations were considered converged when the forces on atoms reached less than 0.05 eV Å−1. The self-consistent field tolerance was set to 10−5 eV. All calculations were spin-polarized. The Monkhorst–Pack k-point mesh of 3 × 3 × 1 and a plane-wave basis set cutoff energy of 450 eV were used.54,55 Valence electrons considered for each atom type were Ti (3s2 3p6 4s1 3d3), O (2s2 2p4), C (2s2 2p2), and H (1s1). Transition state searches were performed using the climbing image nudged elastic band method (CI-NEB), with the search considered converged when the transition state has a tangent force less than 0.05 eV Å−1 and one imaginary vibrational frequency.Adsorption energy of an absorbate to the TiO2 surface (ΔEads) was calculated as:
| ΔEads = Eadsorbate_on_surface − Eadsorbate − Ebare_surface | (1) |
 | (2) |
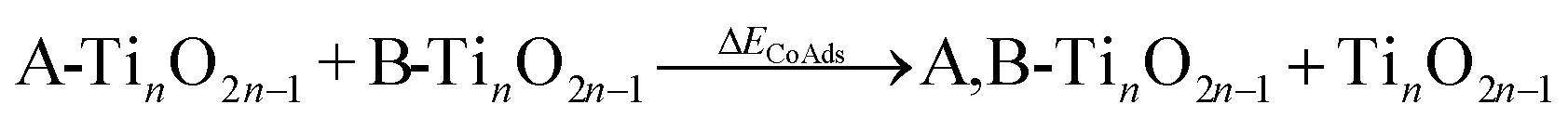 | (3) |
2.3. Kinetic model development
A dual site microkinetic model (MKM) was constructed to predict the hydrogenation performance of anatase TiO2 (001) at reaction conditions. Reduced TiO2 sites were used to facilitate the C–H bond formation reactions occurring at Ovac (*), while spillover H atoms are sourced from fully oxidized regions away from the C–H bond formation sites (**). When H atoms are needed for each C–H bond formation step, the spillover H atoms are assumed to be continuously sourced from (**). The coordinates of all surface intermediates and transition states included in our MKM are provided in the Appendix within the ESI.†A gas-phase reference adjustment is made for DFT adsorption and desorption energies in the MKM to match the overall thermodynamics to the NIST gas-phase thermodynamic values (see eqn (S1), Table S1†).56,57 Entropy changes corresponding to adsorption or desorption of gas phase species were calculated using correlations available in the literature for standard gas phase absolute entropies as a function of temperature58,59 and approximations of their entropies as adsorbed species developed by Campbell et al. (eqn (4)).60 More specifically,
| S0ads(T) = 0.70S0gas(T) − 3.3R | (4) |
| S0TS,des(T) = S0ads(T)/0.68 | (5) |
Equilibrium constants were calculated for each elementary step i using standard free energy changes (ΔG0) that combine the re-referenced DFT energetics with the aforementioned approximations for the entropy changes (ΔS0):63,64
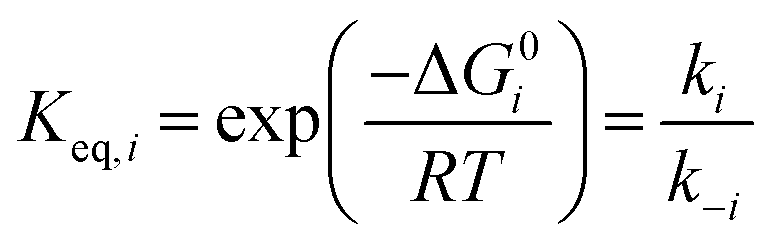 | (6) |
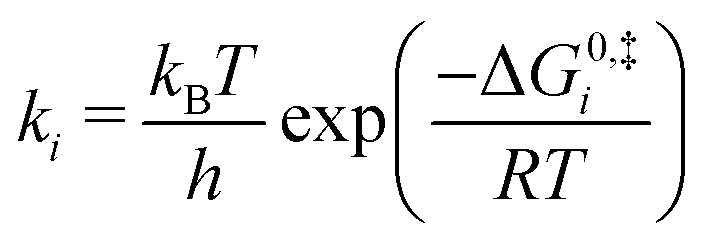 | (7) |
To determine surface coverages and reaction rates, a set of ordinary differential equations is setup (see eqn (S3) in ESI†) and integrated with time until steady state is reached, namely:
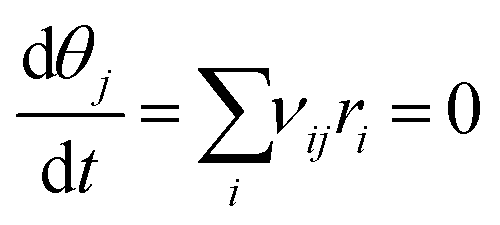 | (8) |
 | (9) |
Sensitivity analysis of the product formation rates was performed with respect to individual kinetic rate constants using the degree of rate control (eqn (10)),63,70,71 with each reaction rate constant perturbed by 2% and the rate of reactant consumption (i.e., benzoic acid) taken to be the rate of interest.
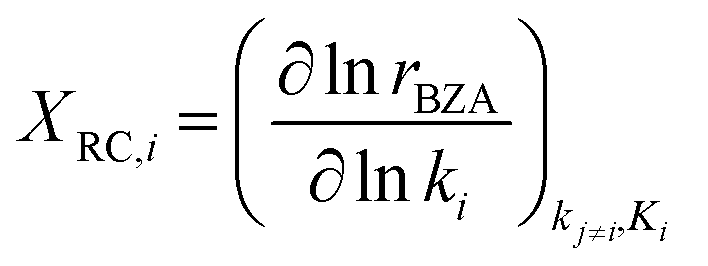 | (10) |
The partial equilibrium index (PEI) was calculated for each elementary reaction to gauge which steps are near equilibrium, thus not likely to be the rate determining step (eqn (11)).72,73 A reaction step with a PEI between 0.45 and 0.55 is generally considered partially equilibrated.
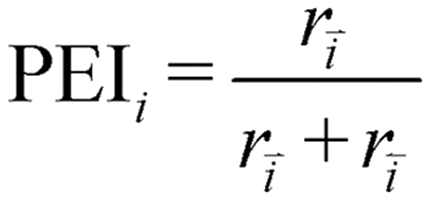 | (11) |
3. Results and discussion
3.1. Adsorption of benzoic acid on fully oxidized and reduced TiO2
Both fully oxidized and reduced sites on anatase (001) will be present at high temperature hydrogenation conditions. This is evidenced by H2O desorption and, by extension, Ovac formation during temperature programmed reduction (TPR)6,32 and the presence of reduced Ti states using resonant photoemission spectroscopy.74 DFT results quantified an Ovac formation energy of ΔEvac = 1.57 eV on this (001) surface, while this energy decreases as low as ΔEvac = 1.03 eV near Ag single atoms.33 Thus, stable adsorption structures were determined for benzoic acid on both the fully oxidized and reduced (i.e., with an Ovac) surfaces of anatase (001) (Fig. 3).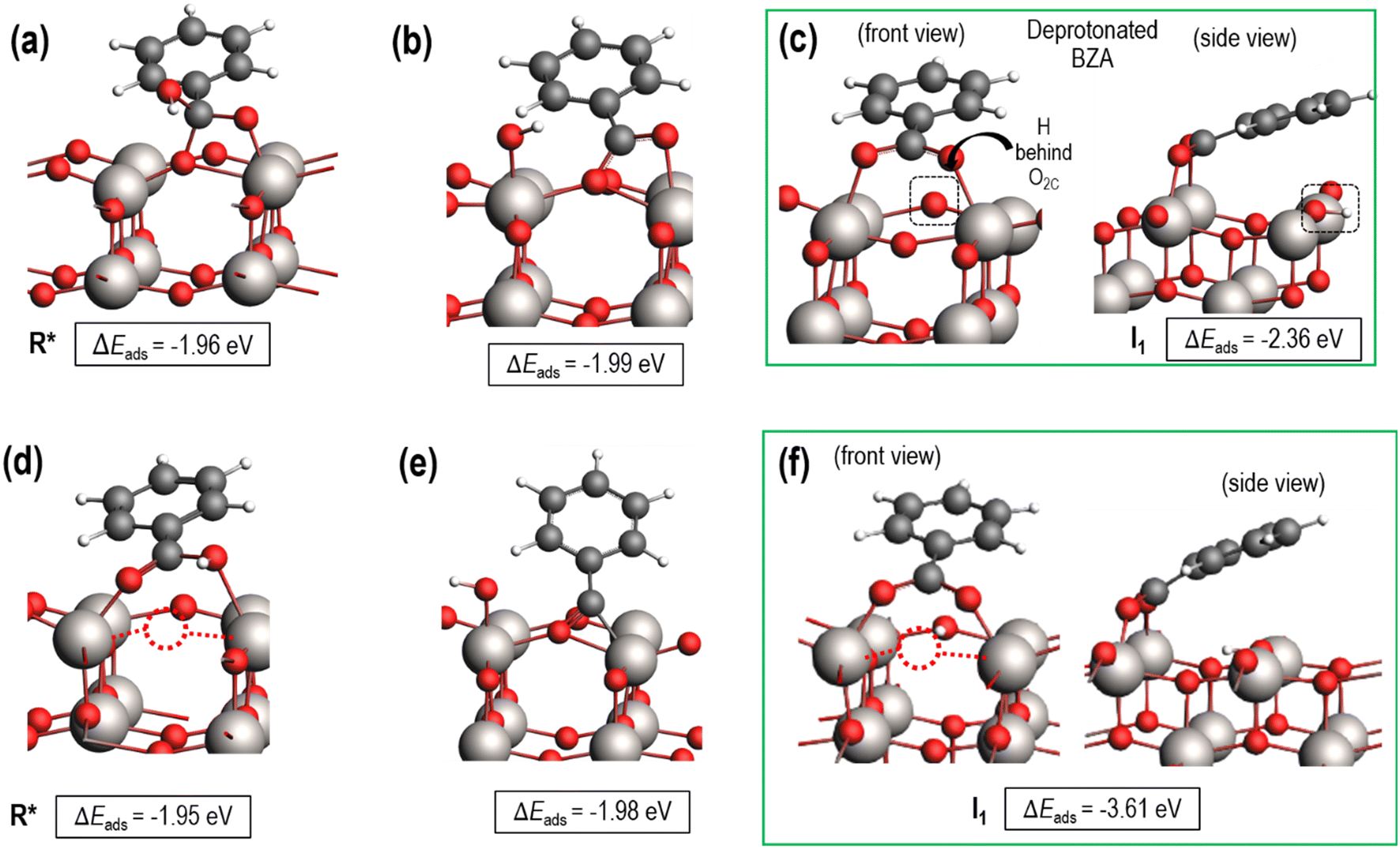 | ||
| Fig. 3 Benzoic acid adsorption on fully oxidized and reduced anatase TiO2 (001). Top row (a–c): Benzoic acid adsorbs on the fully oxidized surface either (a) molecularly, (b) dissociatively with the –OH group on a Ti atom, and (c) dissociatively by deprotonation. Bottom row (d–f): Benzoic acid adsorption on the reduced anatase TiO2 (001) surface with an Ovac of an O2C site. Stable structures included benzoic acid (d) molecular adsorption, (e) dissociative adsorption with –OH on a Ti atom, and (f) dissociative adsorption by deprotonation. R* and I1 labels in a), c), d), and f) are referenced in Fig. 4. Ti (gray), O (red), C (black), H (white), and an Ovac (dashed circle) are depicted. | ||
Benzoic acid molecular adsorption is thermodynamically favorable on both the fully oxidized (Fig. 3a) and reduced (Fig. 3d) surfaces, with ΔEads around −2 eV for both. On the Ovac surface, benzoic acid adsorbs into the vacancy with Ti–O bond formation between the O atoms of benzoic acid and each of the Ti atoms near the Ovac. This favorable adsorption state is 0.6 eV more stable than with a single O atom from benzoic acid bound in approximately the Ovac position (not shown). Dissociation of the hydroxyl group from benzoic acid is marginally more favorable on the fully oxidized (Fig. 3b) and reduced surfaces (Fig. 3e). However, dissociative adsorption of benzoic acid via deprotonation into an Ovac-bound benzoate anion and a proton on an O2C was thermodynamically more favorable by 0.4 eV on the fully oxidized surface (Fig. 3c) and more than 1.5 eV on the reduced surface (Fig. 3f). These results agree well with studies which used infrared reflection-adsorption spectroscopy (IRRAS),75 as well as scanning tunneling microscopy (STM) and low energy electron diffraction (LEED)76 to confirm the spontaneous dissociative adsorption of benzoic acid to benzoate and hydroxyl groups on TiO2. The deprotonated adsorption structure is consistent with previous DFT studies of benzoic acid adsorption.77–79 Furthermore, carboxylic acid deprotonation on anatase TiO2 is not only thermodynamically favorable but also essentially barrierless, with an experimental study reporting a barrier of around 0.05 eV for formic acid deprotonation on anatase (101) at 300 K.80 Thus, further steps of hydrodeoxygenation are assumed to proceed from deprotonated, adsorbed benzoic acid on both the fully oxidized and reduced TiO2 surfaces.
3.2. Benzoic acid hydrogenation to benzaldehyde on fully oxidized TiO2 (001)
Stable structures were compiled along a plausible path of the first C–H bond formation of benzoic acid to benzaldehyde on the fully oxidized TiO2 (001) surface. These structures and their relative energies were used to build a reaction energy diagram (Fig. 4). Starting with gas phase benzoic acid and the bare TiO2 (001) surface, benzoic acid dissociatively adsorbs on the surface in its deprotonated form, as a benzoate anion and a H+ (I1, Fig. 3c). The H atom then migrates to the support O2C near to the benzoate anion (I2); I2 is the most stable state compared to other structures of H adsorbed on atoms near the benzoate species. The first C–H bond formed (I3) by H migration from O2C to the carboxyl C proceeds uphill in energy by 3.98 eV. To close the catalytic cycle, a series of elementary steps follows, where Ovac is formed on O2C next to the deprotonated phenylmethanediol (I3), whose O atom refills the vacancy as a final step, producing benzaldehyde as a result of that step; the stepwise addition of 2 H atoms and desorption of O as H2O (i.e., I3 + 1/2H2 → I4 and subsequently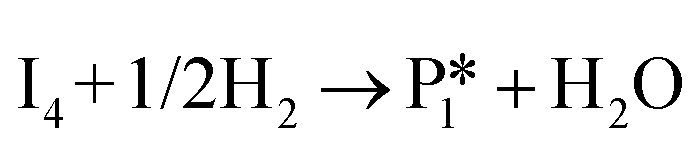 ) occur favorably. The final adsorbed state is essentially an oxidized precursor of benzaldehyde
) occur favorably. The final adsorbed state is essentially an oxidized precursor of benzaldehyde 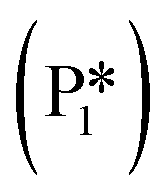 . The desorption of benzaldehyde into the gas phase (P1) reforms the fully oxidized TiO2 surface and requires a reaction energy of 2.76 eV. Based on the reaction energy diagram of Fig. 4, at least 3.98 eV energy is required to form benzaldehyde from benzoic acid on the fully oxidized surface even without any kinetic barriers, indicating that this mechanism is too unfavorable to occur at relevant reaction temperatures.
. The desorption of benzaldehyde into the gas phase (P1) reforms the fully oxidized TiO2 surface and requires a reaction energy of 2.76 eV. Based on the reaction energy diagram of Fig. 4, at least 3.98 eV energy is required to form benzaldehyde from benzoic acid on the fully oxidized surface even without any kinetic barriers, indicating that this mechanism is too unfavorable to occur at relevant reaction temperatures.
3.3. Benzoic acid hydrogenation to benzaldehyde and decarboxylation to benzene on reduced TiO2 (001)
The reaction energy profile of benzoic acid hydrogenation to benzaldehyde and decarboxylation to benzene on the reduced TiO2 (001) surface with an Ovac was determined (Fig. 5). The relative energies are referenced to gas phase benzoic acid and the reduced TiO2 (001) surface with an Ovac. Dissociative adsorption of benzoic acid into the benzoate anion (on the Ovac) and H+ (on the O2C opposite of the Ovac, I1, Fig. 3f) is enthalpically favorable by 3.6 eV. The proton adsorbed on O2C then migrates into the Ovac, forming a hydride species (I2).33,39 In effect, this state has two (−1) anions co-adsorbed in an Ovac, benzoate and H−. The migration of H into Ovac to form two anionic species (I1 → I2) proceeds uphill by 0.79 eV. The benzoate and hydride co-adsorption energy using eqn (3) is 0.58 eV, showing there is a repulsive interaction to placing two anionic ligands on the same Ovac site. From intermediate I2, either C–H bond formation leading to benzaldehyde or decarboxylation leading to benzene can occur.We first examine how intermediate I2 may proceed to form benzaldehyde. The reaction between the hydride and benzoate in the vacancy results in a C–H bond formation (I2 → I3), which proceeds downhill in reaction energy by 0.89 eV with a barrier of 0.64 eV. An alternate mechanism to form this first C–H bond was considered where H directly migrates from the O on benzoic acid to the C atom (R*, depicted in Fig. 3a, to I3), forgoing both benzoic acid deprotonation and C–H bond formation via the anion-hydride mechanism (Fig. S3†). The activation barrier for this alternate mechanism is greater than 1 eV, suggesting that the hydride-mediated mechanism between the hydride and benzoate anion will be kinetically more favorable for C–H bond formation. Comparing relative rate constants for this competing path at a reaction temperature of 575 K (ref. 6) results in benzoic acid undergoing deprotonation nine orders of magnitude faster than C–H bond formation from the non-hydride route (eqn (S2)†). We also considered routes to C–H bond formation from which the H is passed to C from a surface O atom (not shown); however, these all proceed through either the hydride intermediate or by first placing the H onto the O atom of benzoate (i.e., going back to molecularly adsorbed benzoic acid).
Once the C–H bond is formed through the hydride-mediated mechanism, structure I3 can be considered as deprotonated phenylmethanediol. I3 can further undergo hydrogenation to form benzaldehyde or decarboxylation to form benzene. In the case of hydrogenation, two H atoms are successively added on one of the O atoms of the deprotonated diol to form H2O that desorbs. This leads to benzaldehyde adsorbed on Ovac  , which proceeds thermodynamically uphill in energy by 0.99 eV. Finally, desorption of benzaldehyde from the Ovac surface requires more than 2.8 eV. Alternatively, benzaldehyde can first desorb from I3 (oxidized precursor of benzaldehyde) by leaving one O in Ovac (refilling Ovac) followed by the reformation of Ovac (not shown).
, which proceeds thermodynamically uphill in energy by 0.99 eV. Finally, desorption of benzaldehyde from the Ovac surface requires more than 2.8 eV. Alternatively, benzaldehyde can first desorb from I3 (oxidized precursor of benzaldehyde) by leaving one O in Ovac (refilling Ovac) followed by the reformation of Ovac (not shown).
Notably for the first C–H bond formation in benzoic acid, the required energy is at least 2.5 eV lower on the reduced surface than the fully oxidized surface (i.e., compare 1.43 to 3.98 eV in Fig. 5 and 4, respectively). We conclude that carboxylic acid reduction on the TiO2 surface will occur preferentially at Ovac sites. Therefore, further mechanistic steps (i.e., hydrogenation of benzaldehyde, etc.) were considered on the TiO2 surface with Ovac. The involvement of vacancy sites in the hydrodeoxygenation mechanism is supported by earlier experimental hypotheses that benzoic acid hydrogenation activity on reducible metal oxides increases with decreasing M-O bond strength (i.e., higher Ovac concentrations).6,28
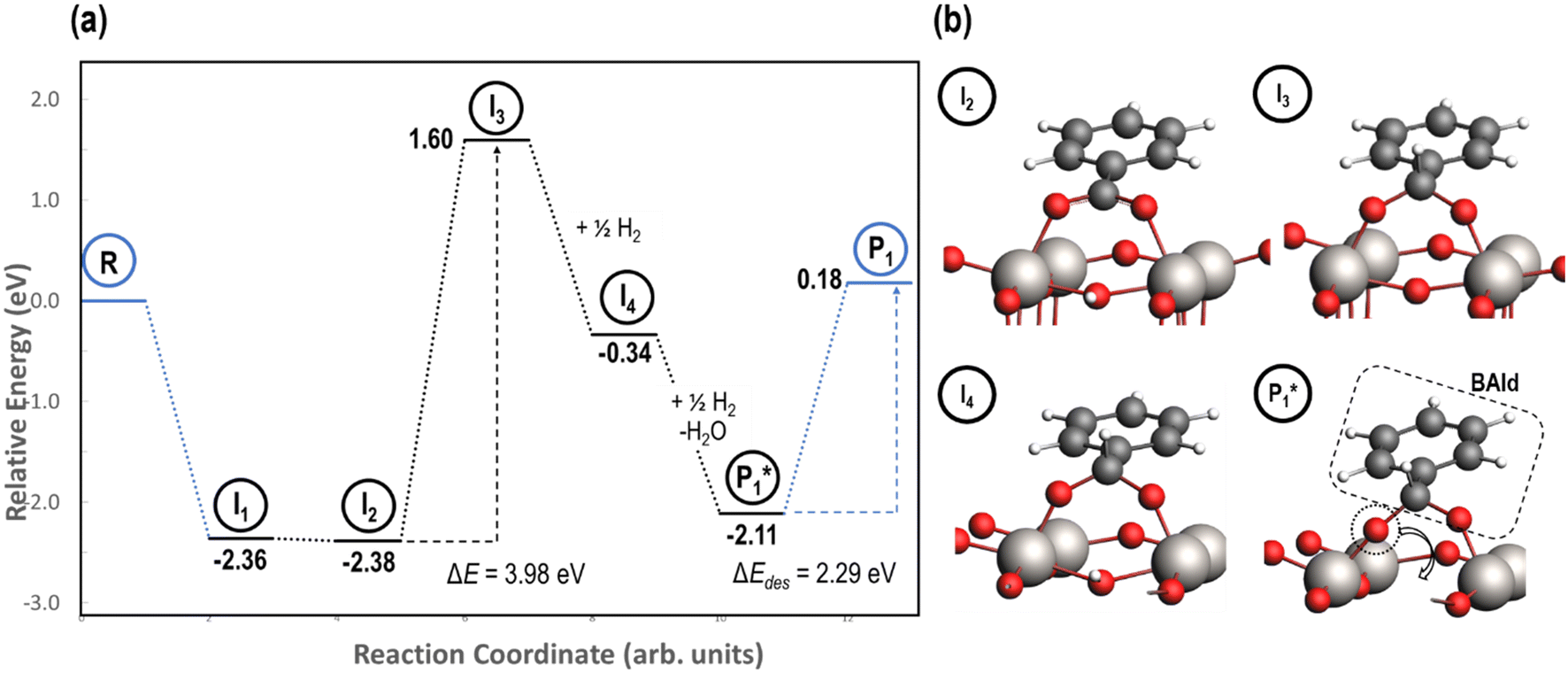 | ||
| Fig. 4 Reaction energy diagram (a) with optimized intermediate structures (b) of benzoic acid hydrogenation to benzaldehyde on fully oxidized anatase TiO2 (001). Stable intermediate I1 corresponds to Fig. 3c. The energies of stable intermediates are referenced to gas phase benzoic acid and the fully oxidized TiO2 (001) surface. Structures with gas phase molecules are circled in blue and adsorbed species in black. Only the uppermost atomic layer is shown for the sake of clarity. Ti (gray), O (red), C (black), and H (white) are depicted. | ||
Instead of hydrogenation to benzaldehyde, decarboxylation to benzene can occur from benzoate intermediates.81–83 Decarboxylation reactions have been well-documented on metal oxides and confirmed experimentally using IR spectroscopy and CO2 desorption.17 Starting with I2, CO2 can be eliminated from the adsorbed benzoate, resulting in a phenyl group co-adsorbed with a hydride in the Ovac (I2 → I10 + CO2). Following the release of CO2, a 0.46 eV activation barrier is required to form the C–H bond and produce benzene adsorbed on the Ovac (I10 to 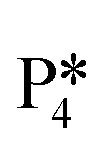 ). This C–H bond formation step to form benzene is thermodynamically favorable by 0.51 eV, with the desorption energy of benzene being 1.21 eV. Nonetheless, the energy barrier required to decarboxylate benzoate (at least 2.22 eV for I2 to I10) is higher than that required to hydrogenate benzoate (1.43 eV for I2 to I3) on the reduced anatase TiO2 (001) surface. Hence, we also considered a decarboxylation pathway from I3.
). This C–H bond formation step to form benzene is thermodynamically favorable by 0.51 eV, with the desorption energy of benzene being 1.21 eV. Nonetheless, the energy barrier required to decarboxylate benzoate (at least 2.22 eV for I2 to I10) is higher than that required to hydrogenate benzoate (1.43 eV for I2 to I3) on the reduced anatase TiO2 (001) surface. Hence, we also considered a decarboxylation pathway from I3.
This intermediate may undergo decarboxylation by shifting the hydrogen bound to the carbon of the carboxyl group to the carbon in the aromatic ring (I3 → I11).69 This hydrogen shift is an endothermic reaction with a barrier of 2.32 eV. Subsequently, the C–C bond between the carbon of the carboxyl group and the carbon in the aromatic ring can be broken, forming CO2 and benzene adsorbed on the surface (I11 → I12). The energy barrier for this C–C bond breaking is 0.26 eV. The desorption energies for CO2 and benzene are 0.65 eV and 1.21 eV, respectively. Nonetheless, I3 needs to surpass a much higher energy barrier (2.32 eV for I3 to I11) on its way to decarboxylation than what is required for its further hydrogenation (0.99 eV for I3 to 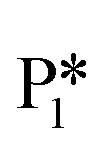 ). Overall, these results indicate that it is energetically more favorable to hydrogenate benzoic acid to benzaldehyde on the reduced anatase TiO2 (001) surface.
). Overall, these results indicate that it is energetically more favorable to hydrogenate benzoic acid to benzaldehyde on the reduced anatase TiO2 (001) surface.
3.4. Benzaldehyde hydrogenation to benzyl alcohol and decarbonylation to benzene on reduced TiO2 (001)
Next, the reaction path of benzaldehyde reduction to benzyl alcohol or benzene on the surface with an Ovac was considered (Fig. 6). The decarbonylation of benzaldehyde to form benzene is examined through a mechanism similar to a 1,2-hydrogen shift,69,84,85 where the H atom migrates to an adjacent C atom ( → I13). The migration of H in this step requires an energy barrier of 2.04 eV. Subsequent C–C bond breaking, resulting in adsorbed CO and benzene molecules (I13 → I14), has a 0.42 eV energy barrier. Desorption of CO requires 0.86 eV, leaving benzene adsorbed on the Ovac
→ I13). The migration of H in this step requires an energy barrier of 2.04 eV. Subsequent C–C bond breaking, resulting in adsorbed CO and benzene molecules (I13 → I14), has a 0.42 eV energy barrier. Desorption of CO requires 0.86 eV, leaving benzene adsorbed on the Ovac 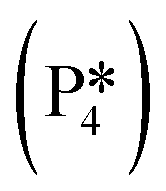 . As a final step, benzene desorbs from the reduced surface, as already described in the decarboxylation pathways of the previous section.
. As a final step, benzene desorbs from the reduced surface, as already described in the decarboxylation pathways of the previous section.
Alternatively, benzaldehyde hydrogenation to benzyl alcohol requires additional O–H formation and C–H bond formation. The addition of H to form the O–H bond proceeds uphill in reaction energy by 0.75 eV (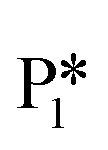 → I5), which ultimately forms a monoanionic ligand adsorbed on the Ovac (i.e., benzyl alcohol missing a hydrogen on C). The subsequent co-adsorption of a hydride on the Ovac with the negatively charged ligand proceeds uphill by an additional 0.75 eV (I5 → I6). However, the C–H bond formation from the co-adsorbed species, resulting in adsorbed benzyl alcohol (I6 →
→ I5), which ultimately forms a monoanionic ligand adsorbed on the Ovac (i.e., benzyl alcohol missing a hydrogen on C). The subsequent co-adsorption of a hydride on the Ovac with the negatively charged ligand proceeds uphill by an additional 0.75 eV (I5 → I6). However, the C–H bond formation from the co-adsorbed species, resulting in adsorbed benzyl alcohol (I6 → 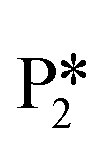 ), is exothermic and has the same barrier (i.e., 0.64 eV) as the one reported in the previous section for the first C–H bond formation (I2 → I3) via the hydride-mediated mechanism. Finally, benzyl alcohol has a desorption energy of 1.53 eV, which is 1.36 eV lower than the desorption energy of benzaldehyde. Nevertheless, an energy barrier of 2.14 eV (see Fig. 6) needs to be surpassed to hydrogenate adsorbed benzaldehyde
), is exothermic and has the same barrier (i.e., 0.64 eV) as the one reported in the previous section for the first C–H bond formation (I2 → I3) via the hydride-mediated mechanism. Finally, benzyl alcohol has a desorption energy of 1.53 eV, which is 1.36 eV lower than the desorption energy of benzaldehyde. Nevertheless, an energy barrier of 2.14 eV (see Fig. 6) needs to be surpassed to hydrogenate adsorbed benzaldehyde  and form adsorbed benzyl alcohol
and form adsorbed benzyl alcohol  , which is larger than the 1.43 eV hydrogenation energy barrier reported for benzaldehyde formation from benzoate (see Fig. 5). This is largely attributed to a higher co-adsorption energy of the monoanionic ligand (I6) with the hydride in Ovac (ΔECoAds = 0.84 eV).
, which is larger than the 1.43 eV hydrogenation energy barrier reported for benzaldehyde formation from benzoate (see Fig. 5). This is largely attributed to a higher co-adsorption energy of the monoanionic ligand (I6) with the hydride in Ovac (ΔECoAds = 0.84 eV).
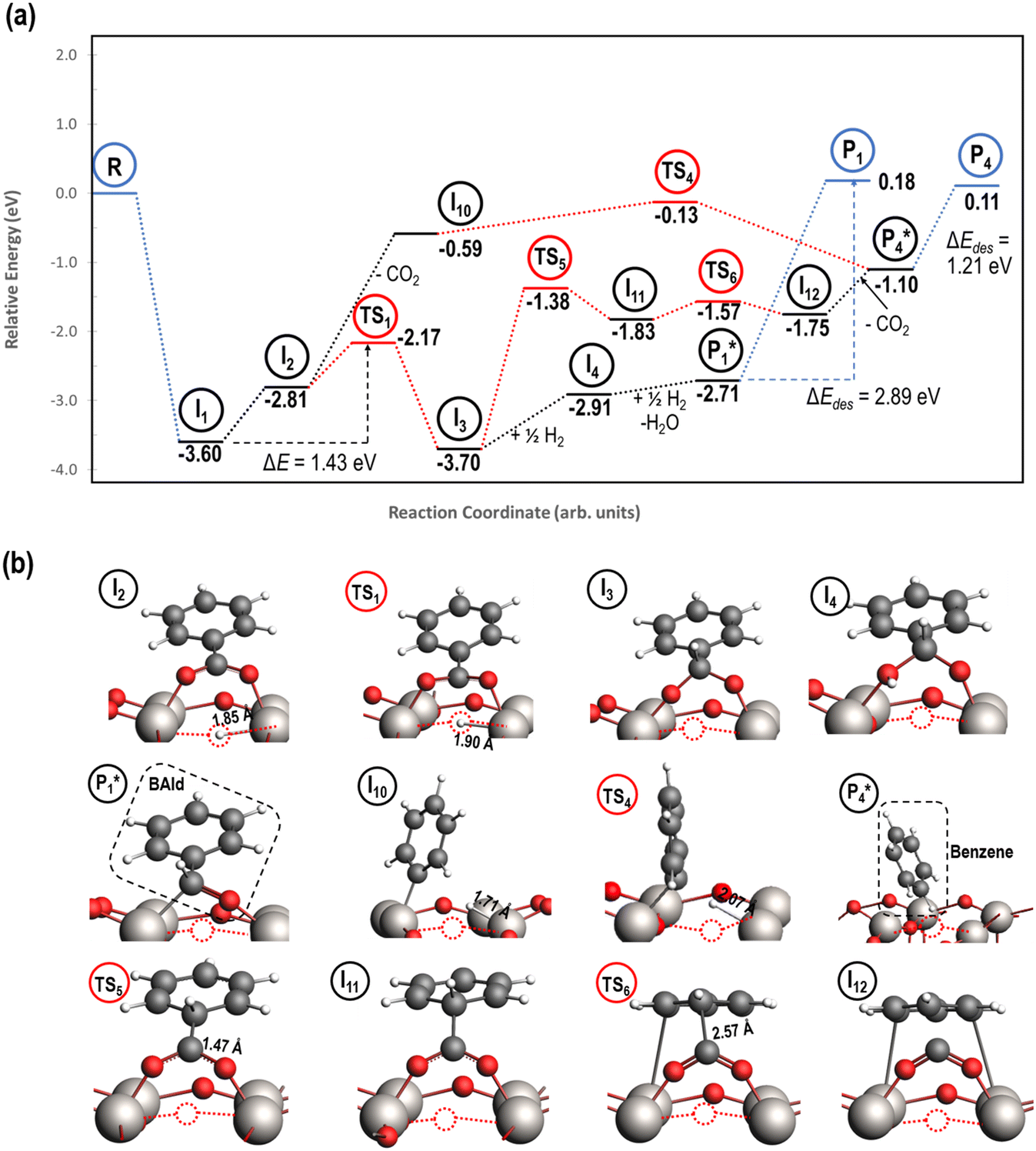 | ||
| Fig. 5 Reaction energy diagram (a) with optimized intermediate structures (b) of benzoic acid hydrogenation to benzaldehyde and decarboxylation to benzene on reduced anatase TiO2 (001) with an Ovac. Stable intermediate I1 corresponds to Fig. 3f. All energies are referenced to gas phase benzoic acid and the reduced TiO2 (001) surface. Structures with gas phase molecules are circled in blue, adsorbed species in black, and transition states in red. Only the uppermost atomic layer is shown for the sake of clarity. Ti (gray), O (red), C (black), H (white), and Ovac (dashed circle) are depicted. | ||
An alternate mechanism was explored for benzyl alcohol formation, reversing the order of H atom addition. The C–H bond is formed first, followed by the O–H bond to form benzyl alcohol (Fig. S4†). A hydride-mediated mechanism for C–H formation is no longer possible in this path, as the ligand co-adsorbed in the Ovac is neutral in charge (i.e., benzaldehyde). Co-adsorption of a hydride with benzaldehyde in Ovac is significantly unfavorable (not shown), with a co-adsorption energy of more than 2.60 eV. Instead, the alternative mechanism forms the first C–H bond from a proton adsorbed on the O3C of TiO2 with a hydrogenation energy barrier (from S1 to TSS1) that is 0.21 eV lower than the one depicted in Fig. 6 (from 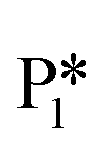 to TS2). This result highlights that alternative mechanisms may be competitive with the hydride-mediated mechanism if anionic ligand repulsion leads to a high co-adsorption energy.
to TS2). This result highlights that alternative mechanisms may be competitive with the hydride-mediated mechanism if anionic ligand repulsion leads to a high co-adsorption energy.
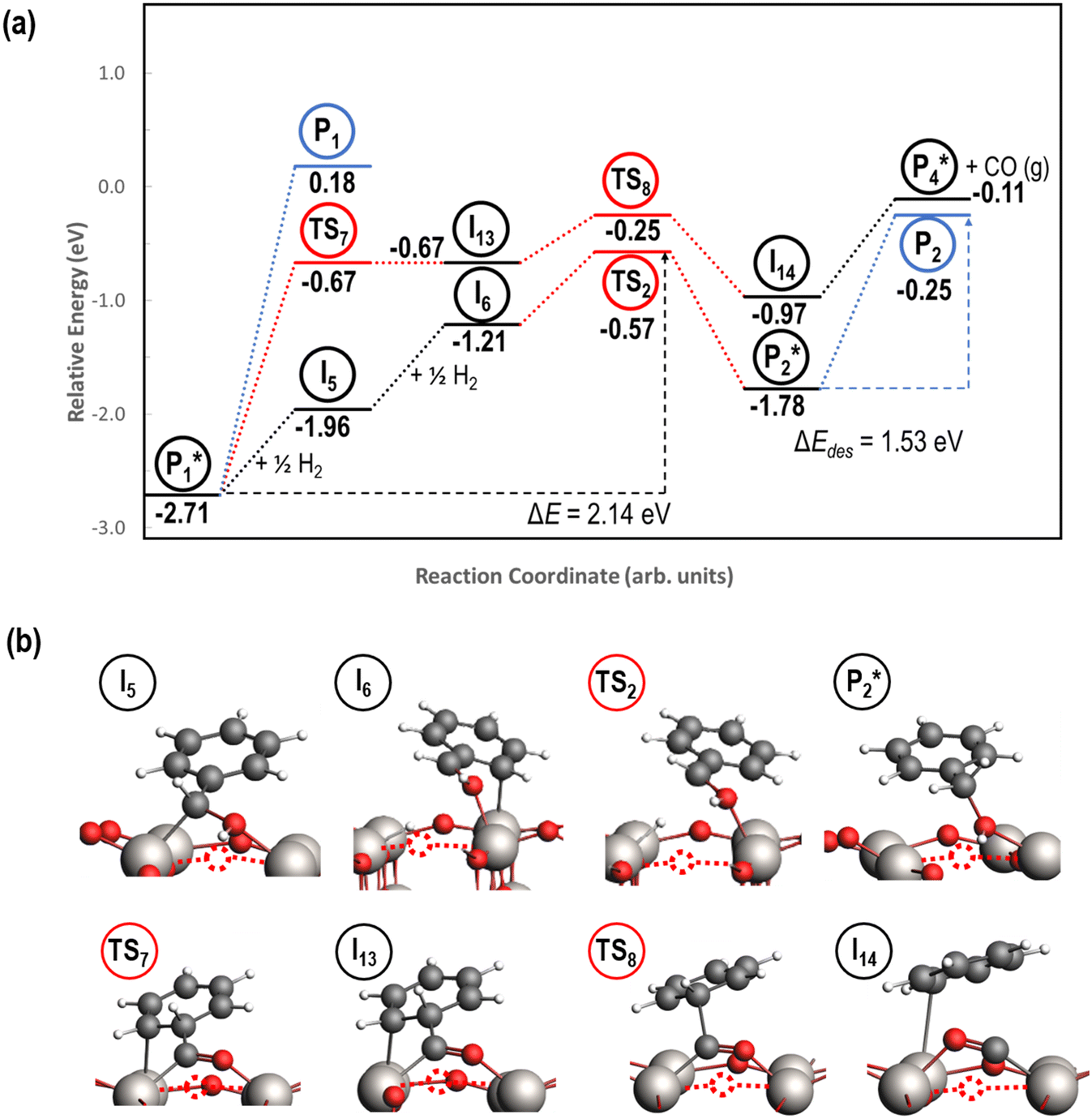 | ||
| Fig. 6 Reaction energy diagram (a) with optimized intermediate structures (b) of benzaldehyde hydrogenation to benzyl alcohol on reduced anatase TiO2 (001) with an Ovac. Structures with gas phase molecules are circled in blue, adsorbed species in black, and transition states in red. Only the uppermost atomic layer is shown for the sake of clarity. Ti (gray), O (red), C (black), H (white), and Ovac (dashed circle) are depicted. | ||
3.5. Benzyl alcohol hydrogenation to toluene on reduced TiO2 (001)
A mechanism of benzyl alcohol hydrogenation to toluene is proposed in Fig. 7. The most energetically viable mechanism of toluene production is directly from hydrogenating benzyl alcohol
is directly from hydrogenating benzyl alcohol 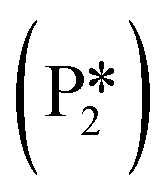 . The removal of the hydroxyl group from benzyl alcohol via the desorption of H2O (
. The removal of the hydroxyl group from benzyl alcohol via the desorption of H2O (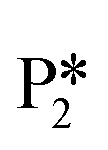 → I7) is thermodynamically favorable by 0.37 eV, leaving a benzyl ligand (I7) adsorbed in the vacancy (i.e., a monoanionic ligand). The addition of a hydride co-adsorbed into the vacancy (I7 → I8) proceeds slightly uphill in reaction energy, attributed to a low co-adsorption energy of ΔECoAds = 0.28 eV between the two ligands (i.e., only slightly net repulsive). Once again, the C–H bond formation to form toluene proceeds through a thermodynamically favorable reaction mechanism between a hydride and an electrophilic carbon center in a monoanionic ligand on an Ovac, with the reaction proceeding downhill by 0.18 eV (I8 →
→ I7) is thermodynamically favorable by 0.37 eV, leaving a benzyl ligand (I7) adsorbed in the vacancy (i.e., a monoanionic ligand). The addition of a hydride co-adsorbed into the vacancy (I7 → I8) proceeds slightly uphill in reaction energy, attributed to a low co-adsorption energy of ΔECoAds = 0.28 eV between the two ligands (i.e., only slightly net repulsive). Once again, the C–H bond formation to form toluene proceeds through a thermodynamically favorable reaction mechanism between a hydride and an electrophilic carbon center in a monoanionic ligand on an Ovac, with the reaction proceeding downhill by 0.18 eV (I8 → 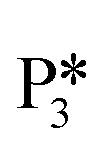 ). The energy barrier required to hydrogenate benzyl alcohol to toluene, 1.22 eV (Fig. 7), is the lowest compared to those of benzaldehyde and benzyl alcohol formation (Fig. 5 and 6, respectively). Finally, toluene has the lowest desorption energy compared to previous hydrogenation intermediates, suggesting that toluene may be a prominent gas phase product. Notably, the direct formation of toluene from benzaldehyde by bypassing the formation of benzyl alcohol is very unfavorable, since an energy barrier of 4.89 eV (see Fig. S5†) needs to be surpassed to hydrogenate adsorbed benzaldehyde
). The energy barrier required to hydrogenate benzyl alcohol to toluene, 1.22 eV (Fig. 7), is the lowest compared to those of benzaldehyde and benzyl alcohol formation (Fig. 5 and 6, respectively). Finally, toluene has the lowest desorption energy compared to previous hydrogenation intermediates, suggesting that toluene may be a prominent gas phase product. Notably, the direct formation of toluene from benzaldehyde by bypassing the formation of benzyl alcohol is very unfavorable, since an energy barrier of 4.89 eV (see Fig. S5†) needs to be surpassed to hydrogenate adsorbed benzaldehyde 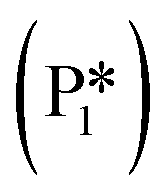 and form adsorbed benzyl (I7).
and form adsorbed benzyl (I7).
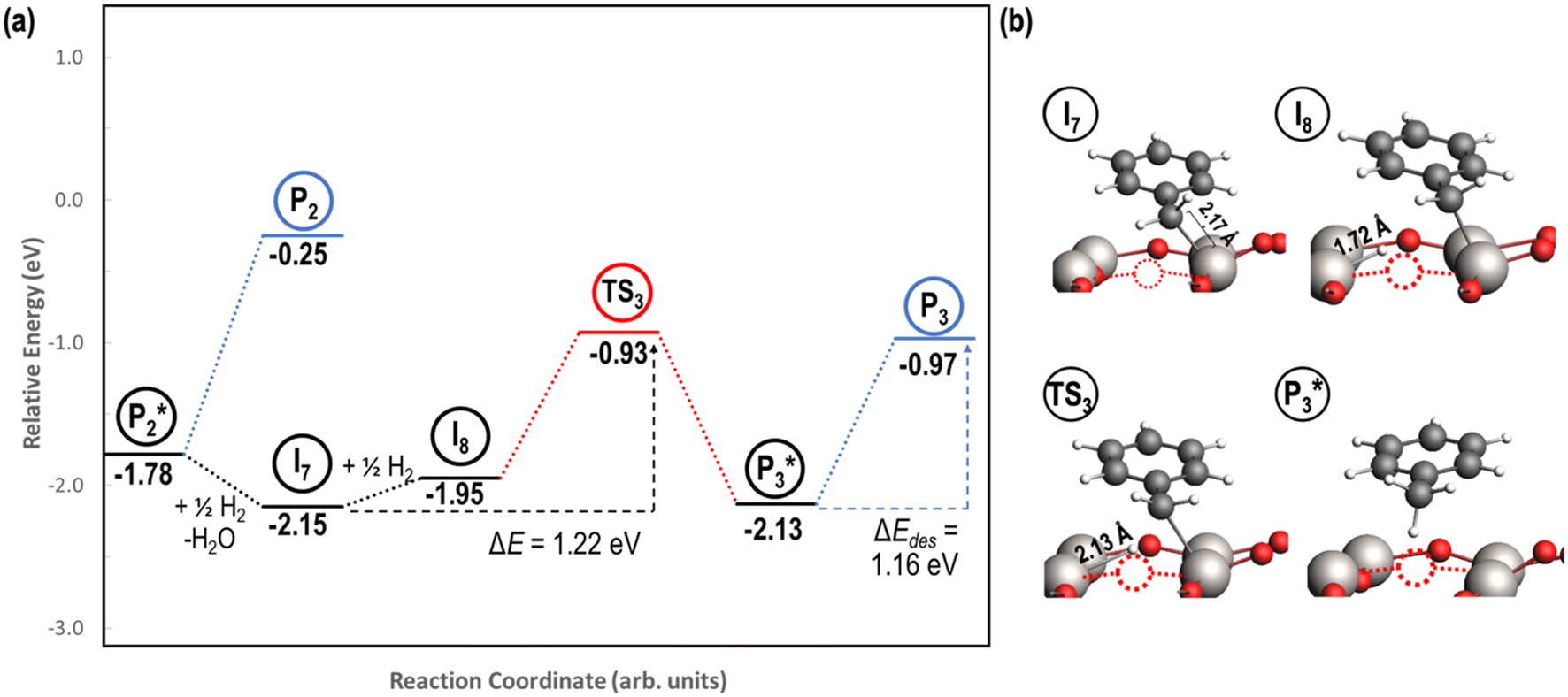 | ||
Fig. 7 Reaction energy diagram (a) with optimized intermediate structures (b) of benzyl alcohol hydrogenation to toluene  on reduced anatase TiO2 (001) with an Ovac. Structures with gas phase molecules are circled in blue, adsorbed species in black, and transition states in red. Only the uppermost atomic layer is shown for the sake of clarity. Ti (gray), O (red), C (black), H (white), and Ovac (dashed circle) are depicted. on reduced anatase TiO2 (001) with an Ovac. Structures with gas phase molecules are circled in blue, adsorbed species in black, and transition states in red. Only the uppermost atomic layer is shown for the sake of clarity. Ti (gray), O (red), C (black), H (white), and Ovac (dashed circle) are depicted. | ||
Thus, C–H bonds consistently form through co-adsorbed hydrides and ligands that have −1 formal charges (i.e., monoanionic ligands) bound in Ovac, with reasonably low kinetic barriers around or below 2 eV. The reaction energies of C–H bond formation between a hydride and the monoanionic species of interest (e.g., benzoate, phenyl, benzyl) in an Ovac on TiO2 are also consistently thermodynamically favorable. However, high reaction temperatures may still be necessary to form Ovac and to overcome barriers associated with the co-adsorption of two anionic species in Ovac (i.e., ΔECoAds). Partial pressures of hydrogen and, by extension, surface H coverages under reduction conditions will also affect the rates of C–H bond formation.
3.6. Microkinetic model of benzoic acid hydrogenation in an Ovac site
A microkinetic model (MKM) that included reactions of H2 dissociation, benzoic acid adsorption, sequential hydrogenation, decarboxylation or decarbonylation, and desorption of products, consisting of 24 elementary steps (Table 1), was constructed to predict the relative rates of product formation under reaction conditions using the DFT reaction energies and barriers (as summarized in Fig. S6a† and corrected by the values reported in Table S1†). The availability of H adsorbed on O sites (H**) is derived from H2 heterolytic dissociative adsorption on the fully oxidized TiO2 (001) surface (Fig. S7†), with a reaction energy of −0.33 eV and an energy barrier of 0.23 eV.| Product | Benz | BAld | BnOH | Tol | |||
|---|---|---|---|---|---|---|---|
| Path | D1 | D2 | D3 | H1 | H2 | H3 | |
| 0 | 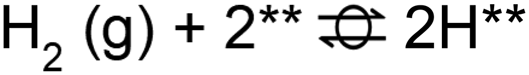 |
0 | 0 | 1 | 1 | 1 | 1 |
| 1 | 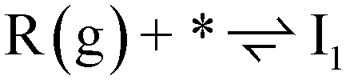 |
1 | 1 | 1 | 1 | 1 | 1 |
| 2 | 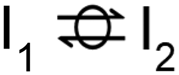 |
1 | 1 | 1 | 1 | 1 | 1 |
| 3 | 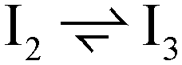 |
0 | 1 | 1 | 1 | 1 | 1 |
| 4 | 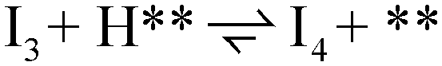 |
0 | 0 | 1 | 1 | 1 | 1 |
| 5 | 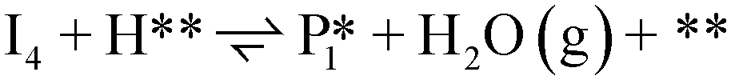 |
0 | 0 | 1 | 1 | 1 | 1 |
| 6 |  |
0 | 0 | 0 | 1 | 0 | 0 |
| 7 | 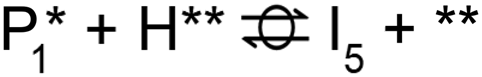 |
0 | 0 | 0 | 0 | 1 | 1 |
| 8 | 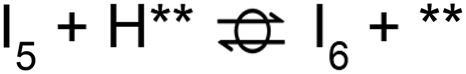 |
0 | 0 | 0 | 0 | 1 | 1 |
| 9 | 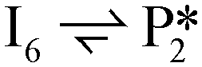 |
0 | 0 | 0 | 0 | 1 | 1 |
| 10 | 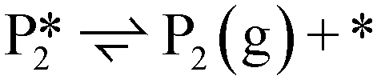 |
0 | 0 | 0 | 0 | 1 | 0 |
| 11 | 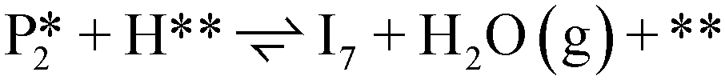 |
0 | 0 | 0 | 0 | 0 | 1 |
| 12 | 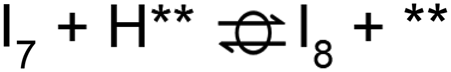 |
0 | 0 | 0 | 0 | 0 | 1 |
| 13 | 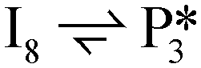 |
0 | 0 | 0 | 0 | 0 | 1 |
| 14 | 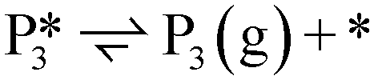 |
0 | 0 | 0 | 0 | 0 | 1 |
| 15 | 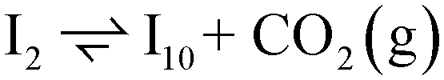 |
1 | 0 | 0 | 0 | 0 | 0 |
| 16 | 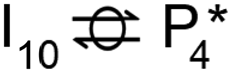 |
1 | 0 | 0 | 0 | 0 | 0 |
| 17 | 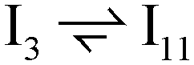 |
0 | 1 | 0 | 0 | 0 | 0 |
| 18 | 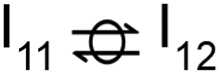 |
0 | 1 | 0 | 0 | 0 | 0 |
| 19 | 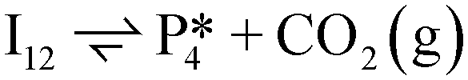 |
0 | 1 | 0 | 0 | 0 | 0 |
| 20 | 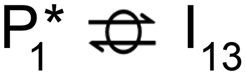 |
0 | 0 | 1 | 0 | 0 | 0 |
| 21 | 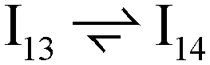 |
0 | 0 | 1 | 0 | 0 | 0 |
| 22 | 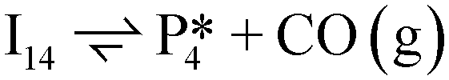 |
0 | 0 | 1 | 0 | 0 | 0 |
| 23 | 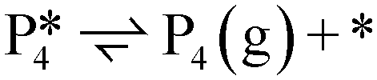 |
1 | 1 | 1 | 0 | 0 | 0 |
| Path D1/D2 (decarboxylation to Benz): R (g) ⇌ P4 (g) + CO2 (g) | |||||||
| Path D3 (decarbonylation to Benz): R (g) + H2 (g) ⇌ P4 (g) + H2O (g) + CO (g) | |||||||
| Path H1 (hydrogenation to BAld): R (g) + H2 (g) ⇌ P1 (g) + H2O (g) | |||||||
| Path H2 (hydrogenation to BnOH): R (g) + 2 H2 (g) ⇌ P2 (g) + H2O (g) | |||||||
| Path H3 (hydrogenation to Tol): R (g) + 3 H2 (g) ⇌ P3 (g) + 2 H2O (g) | |||||||
The detailed rate equations are described in eqn (S3).† The coupled system of 19 differential equations relating the rates of all intermediate surface species was solved simultaneously in the MKM. Gas phase concentrations were held constant, i.e., PH2 = PBZA = 1 bar. Note that the presence of surface hydroxyls was not considered on the reduced surface with Ovac under the dry conditions in which we performed MKM, since the formation of these species is not favorable under these conditions according to the thermodynamic analysis shown in Fig. S8.† For reaction temperatures varying from 450 to 900 K, the formation rate of each product was calculated using the steady state concentrations of the surface intermediates (i.e., Fig. S9b†). The steady state rates of benzaldehyde, benzyl alcohol, toluene, and benzene formation are shown as a function of reaction temperature in Fig. 8a. While the rates of product formation are negligible at low temperatures, an increase in T to 650 K brings the rates within an order of a magnitude difference from hydrogenation turnover frequencies on single atom catalysts.86 For example, the experimental turnover frequency of guaiacol hydrodeoxygenation on Ag1/TiO2 was determined to be near 10−3 s−1 at 573 K.32
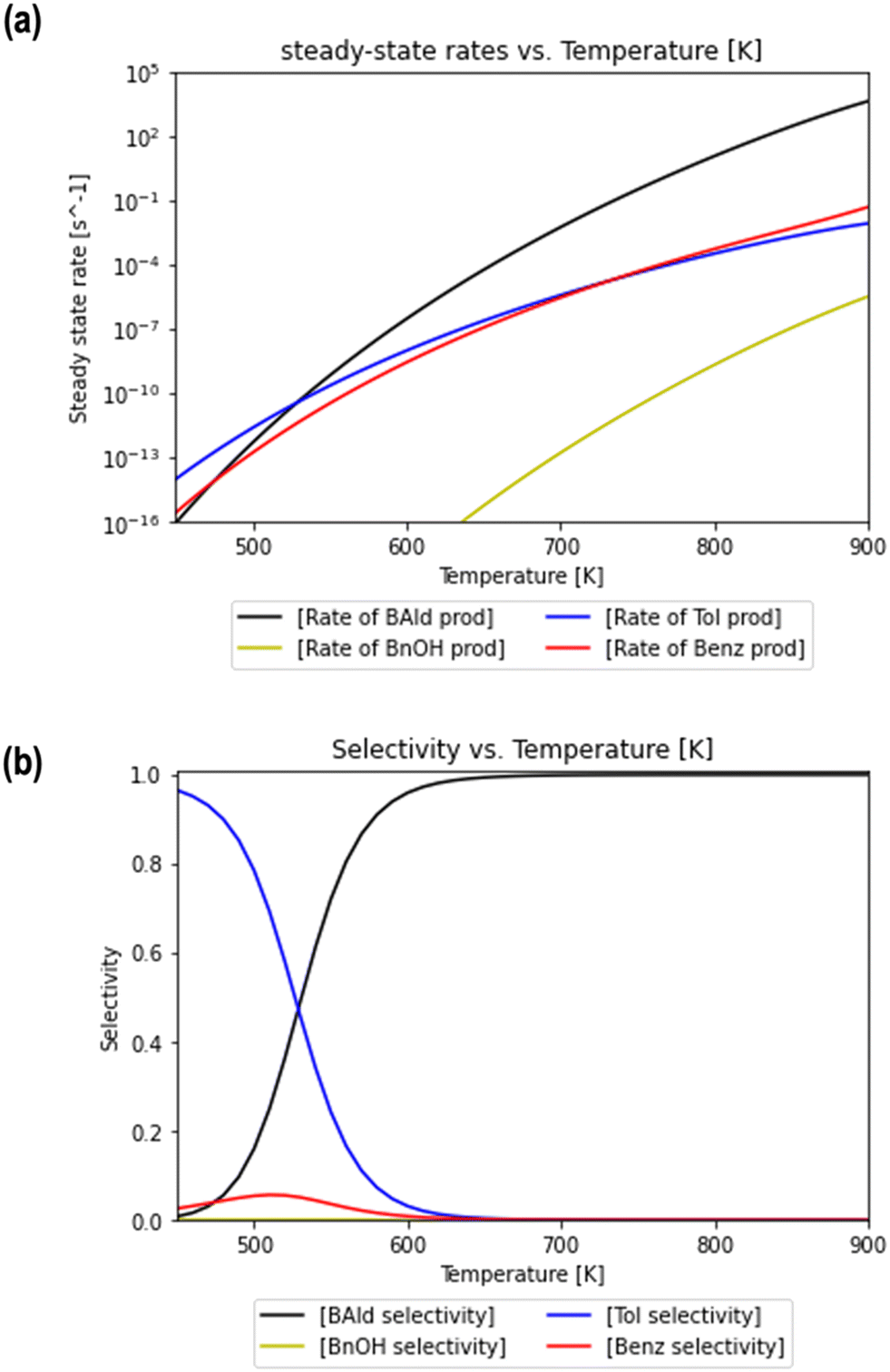 | ||
| Fig. 8 Rates of formation and selectivity of products predicted through the microkinetic model. Steady state rates (a) of benzaldehyde (black), benzyl alcohol (blue circles), toluene (dashed red), and benzene (dotted green) production are plotted as a function of reaction temperature. The relative selectivity of each product (b) is calculated using rates of formation at steady state at various temperatures. The coverages of select surface intermediates at different temperatures are included in Fig. S9b.† | ||
Steady state rates of production were also used to calculate the relative selectivity of each product for the same temperature range (Fig. 8b). Two distinct temperature ranges are observed in the MKM, each of which has a specific product rate dominate. At low temperatures, the selectivity to toluene dominates over the selectivity to benzene and benzaldehyde. The small amount of benzene is formed through decarbonylation and not decarboxylation at these low T conditions as shown by a detailed rate analysis (Fig. S9a†). At higher temperatures, the selectivity to benzaldehyde consistently increases and is the highest amongst all products at temperatures of 540 K and above. Meanwhile, selectivity to benzene and toluene drops to zero as the temperature increases above 540 K. Over the complete temperature range, no selectivity to benzyl alcohol is observed.
Thus, the MKM predicts that the relative selectivity for toluene is the highest compared to those for all four products at low temperatures till 540 K, while the relative selectivity of benzaldehyde dominates at temperatures of 540 K and higher. With the Ovac (*) completely covered with adsorbed benzaldehyde 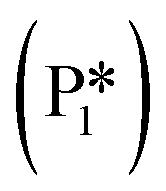 for the most part of the investigated temperature range (see Fig. S9b†), this switch in selectivity from toluene to benzaldehyde as the temperature increases is a direct consequence of benzaldehyde desorption being more activated than its further hydrogenation (see Fig. 6). In agreement with the current MKM study, the limited experimental data available for benzoic acid HDO on metal oxides (i.e., ZrO2, HfO2, TiO2) also report the production of toluene, benzene, and benzaldehyde at low temperatures and a steep increase in benzaldehyde selectivity as temperature increases.6 Potential modifications in the entropy treatment and using an alternative pathway for benzaldehyde hydrogenation (as depicted in Fig. S4†) may affect the selectivity of benzene and toluene at low temperatures where the production rates are lower than 10−10 s−1 and the onset temperature where benzaldehyde becomes dominant (Fig. S10†). Nevertheless, the overall trend of increase in benzaldehyde selectivity with increasing temperature remains consistent.
for the most part of the investigated temperature range (see Fig. S9b†), this switch in selectivity from toluene to benzaldehyde as the temperature increases is a direct consequence of benzaldehyde desorption being more activated than its further hydrogenation (see Fig. 6). In agreement with the current MKM study, the limited experimental data available for benzoic acid HDO on metal oxides (i.e., ZrO2, HfO2, TiO2) also report the production of toluene, benzene, and benzaldehyde at low temperatures and a steep increase in benzaldehyde selectivity as temperature increases.6 Potential modifications in the entropy treatment and using an alternative pathway for benzaldehyde hydrogenation (as depicted in Fig. S4†) may affect the selectivity of benzene and toluene at low temperatures where the production rates are lower than 10−10 s−1 and the onset temperature where benzaldehyde becomes dominant (Fig. S10†). Nevertheless, the overall trend of increase in benzaldehyde selectivity with increasing temperature remains consistent.
We further analyze the MKM using the degree of rate control at the crucial temperature of 540 K (Table 1, Fig. S11a†), where several products are observed. Almost all elementary steps contribute insignificantly to the overall reaction rate when perturbated, with a degree of rate control near zero. The reaction steps with the degree of rate control higher than 0.1 involve benzaldehyde desorption, decarbonylation via C–C bond cleavage and further hydrogenation to benzyl alcohol via hydride-mediated C–H bond formation. The results suggest that these elementary steps have significant impacts on the overall benzoic acid consumption. Additionally, the degree of selectivity control71 quantifies how significantly each elementary step contributes to the relative increase in selectivity with respect to each product (Fig. S11b†).
The partial equilibrium index (PEI) is also calculated for each elementary step at 540 K (Table 1, Fig. S11c†). A majority of the elementary steps are not near equilibrium, which makes it difficult with PEI alone to pinpoint any one reaction that is the rate-determining step of the system. Eight elementary steps can be considered near-equilibrated with a PEI between 0.45 and 0.55. Contrarily, no steps have PEIs near zero, which would generally correspond to reactions with very low forward reaction rates. The PEI also confirms the results from the degree of rate control, namely that the reactions with a high degree of rate control are also far from equilibrium (i.e., benzaldehyde desorption, decarbonylation, and further hydrogenation). In summary, the PEIs and degrees of rate control give a comprehensive overview of the microkinetic model and suggest which elementary steps should be targeted through rational catalyst design. For example, modifications to the catalyst that can alter the desorption, decarbonylation, or further hydrogenation of benzaldehyde (i.e., the non-equilibrated elementary steps with the highest degree of rate control) are expected to have significant impacts on the overall reaction according to the microkinetic model. Though not represented mathematically in the MKM that predicts rate per vacancy site, promoting vacancy formation would also generate more active sites and enhance the rate.
4. Conclusions
C–H bond formation to four aromatic products (i.e., benzaldehyde, benzyl alcohol, toluene, and benzene) on anatase TiO2 (001) can occur through a hydride-mediated mechanism on surface Ovac sites. In this mechanism, a negatively charged monoanionic intermediate ligand reacts with a favorable reaction energy with a hydride coadsorbed in Ovac. The overall reaction energy diagram of benzoic acid hydrodeoxygenation was constructed using plausible intermediates and kinetic barriers related to C–H bond formation as well as decarboxylation and decarbonylation. Finally, a microkinetic model was constructed using the overall mechanism and DFT energetics. The MKM suggests that selectivity to toluene is favored over the other three products at low temperatures, while benzaldehyde selectivity dominates at moderate-to-high temperatures. Competing mechanisms such as those for benzaldehyde hydrogenation, decarbonylation, and desorption result in the model predicting different product formation rates and selectivities. MKM analysis through degree of rate control and partial equilibrium index calculations shows several elementary steps that are far from equilibrium and thus can be targeted in the future for potentially significant impacts on the overall rate and selectivity of benzoic acid hydrodeoxygenation.Data availability
Data are available within the article and its ESI.†Disclaimer
The views expressed herein do not necessarily represent the views of the U.S. Department of Energy or the United States Government.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the U.S. Department of Energy's Office of Energy Efficiency and Renewable Energy (EERE) under the Advanced Manufacturing Office Award Number DE-EE0007897 awarded to the REMADE Institute, a division of Sustainable Manufacturing Innovation Alliance Corp. Computational resources were provided through the Extreme Science and Engineering Discovery Environment (XSEDE), supported by the National Science Foundation (grant no. ACI-1548562).References
- N. Perret, X. Wang, J. J. Delgado, G. Blanco, X. Chen, C. M. Olmos, S. Bernal and M. A. Keane, Selective Hydrogenation of Benzoic Acid over Au Supported on CeO2 and Ce0.62Zr0.38O2: Formation of Benzyl Alcohol, J. Catal., 2014, 317, 114–125, DOI:10.1016/j.jcat.2014.06.010.
- X. Chen, Z. Wang, H. Daly, R. Morgan, H. Manyar, C. Byrne, A. S. Walton, S. F. R. Taylor, M. Smith, R. Burch, P. Hu and C. Hardacre, Hydrogenation of Benzoic Acid to Benzyl Alcohol over Pt/SnO2, Appl. Catal., A, 2020, 593, 117420, DOI:10.1016/j.apcata.2020.117420.
- J. Pritchard, G. A. Filonenko, R. Van Putten, E. J. M. Hensen and E. A. Pidko, Heterogeneous and Homogeneous Catalysis for the Hydrogenation of Carboxylic Acid Derivatives: History, Advances and Future Directions, Chem. Soc. Rev., 2015, 3808–3833, 10.1039/c5cs00038f.
- B. Chen, U. Dingerdissen, J. G. E. Krauter, H. G. J. Lansink Rotgerink, K. Möbus, D. J. Ostgard, P. Panster, T. H. Riermeier, S. Seebald, T. Tacke and H. Trauthwein, New Developments in Hydrogenation Catalysis Particularly in Synthesis of Fine and Intermediate Chemicals, Appl. Catal., A, 2005, 280(1), 17–46, DOI:10.1016/j.apcata.2004.08.025.
- M. W. De Lange, J. G. Van Ommen and L. Lefferts, Deoxygenation of Benzoic Acid on Metal Oxides: 1. The Selective Pathway to Benzaldehyde, Appl. Catal., A, 2001, 220(1–2), 41–49 CrossRef.
- C. A. Koutstaal and V. Ponec, New Aspects of the Selective Reduction of Aromatic Carboxylic Acids to the Aldehydes, Science and technology in catalysis, 1994, pp. 105–110 Search PubMed.
- C. Ozdemir, S. Şahinkaya, E. Kalıpcı, M. K. Oden, Ö. Çepelioğullar and A. E. Pütün, Utilization of Two Different Types of Plastic Wastes from Daily and Industrial Life, Journal of Selcuk University Natural and Applied Science, 2013, 2(2), 694–706 Search PubMed.
- S. D. Anuar Sharuddin, F. Abnisa, W. M. A. Wan Daud and M. K. Aroua, A Review on Pyrolysis of Plastic Wastes, Energy Convers. Manage., 2016, 308–326, DOI:10.1016/j.enconman.2016.02.037.
- Y. Sakata, C. A. Van Tol-Koutstaal and V. Ponec, Selectivity Problems in the Catalytic Deoxygenation of Benzoic Acid, J. Catal., 1997, 169(1), 13–21 CrossRef CAS.
- J. Li, J. Zhang, S. Wang, G. Xu, H. Wang and D. G. Vlachos, Chemoselective Hydrodeoxygenation of Carboxylic Acids to Hydrocarbons over Nitrogen-Doped Carbon-Alumina Hybrid Supported Iron Catalysts, ACS Catal., 2019, 9(2), 1564–1577, DOI:10.1021/acscatal.8b04967.
- J. Ullrich and B. Breit, Selective Hydrogenation of Carboxylic Acids to Alcohols or Alkanes Employing a Heterogeneous Catalyst, ACS Catal., 2018, 8(2), 785–789, DOI:10.1021/acscatal.7b03484.
- T. Yokoyama, T. Setoyama, N. Fujita, M. Nakajima, T. Maki and K. Fujii, Novel Direct Hydrogenation Process of Aromatic Carboxylic Acids to the Corresponding Aldehydes with Zirconia Catalyst, Appl. Catal., A, 1992, 88(2), 149–161 CrossRef CAS.
- D. D. Poondi, The Influence of Metal-Support Interactions on the Hydrogenation Activity and Selectivity of Benzene-Toluene Mixtures, Benzaldehyde, and Phenylacetaldehyde, PhD thesis, The Pennsylvania State University, 1996 Search PubMed.
- D. Haffad, U. Kameswari, M. M. Bettahar, A. Chambellan and J. C. Lavalley, Reduction of Benzaldehyde on Metal Oxides, J. Catal., 1997, 172(1), 85–92 CrossRef CAS.
- A. Ausavasukhi, T. Sooknoi and D. E. Resasco, Catalytic Deoxygenation of Benzaldehyde over Gallium-Modified ZSM-5 Zeolite, J. Catal., 2009, 268(1), 68–78, DOI:10.1016/j.jcat.2009.09.002.
- Y. Hao, C. Pischetola, F. Cárdenas-Lizana and M. A. Keane, Selective Liquid Phase Hydrogenation of Benzaldehyde to Benzyl Alcohol Over Alumina Supported Gold, Catal. Lett., 2020, 150(3), 881–887, DOI:10.1007/s10562-019-02944-y.
- S. T. King and E. J. Strojny, An in Situ Study of Methyl Benzoate and Benzoic Acid Reduction on Yttrium Oxide by Infrared Spectroscopic Flow Reactor, J. Catal., 1982, 76(2), 274–284 CrossRef CAS.
- M. A. Vannice and D. Poondi, The Effect of Metal-Support Interactions on the Hydrogenation of Benzaldehyde and Benzyl Alcohol, J. Catal., 1997, 169(1), 166–175 CrossRef CAS.
- J. T. Bhanushali, I. Kainthla, R. S. Keri and B. M. Nagaraja, Catalytic Hydrogenation of Benzaldehyde for Selective Synthesis of Benzyl Alcohol: A Review, ChemistrySelect, 2016, 3839–3853, DOI:10.1002/slct.201600712.
- B. Nair, Final Report on the Safety Assessment of Benzyl Alcohol, Benzoic Acid, and Sodium Benzoate, Int. J. Toxicol., 2001, 20(3), 23–50, DOI:10.1080/10915810152630729.
- F. Pinna, F. Menegazzo, M. Signoretto, P. Canton, G. Fagherazzi and N. Pernicone, Consecutive Hydrogenation of Benzaldehyde over Pd Catalysts Influence of Supports and Sulfur Poisoning, Appl. Catal., A, 2001, 219(1–2), 195–200 CrossRef CAS.
- M. Tang, S. Mao, M. Li, Z. Wei, F. Xu, H. Li and Y. Wang, RuPd Alloy Nanoparticles Supported on N-Doped Carbon as an Efficient and Stable Catalyst for Benzoic Acid Hydrogenation, ACS Catal., 2015, 5(5), 3100–3107, DOI:10.1021/acscatal.5b00037.
- X. Xu, M. Tang, M. Li, H. Li and Y. Wang, Hydrogenation of Benzoic Acid and Derivatives over Pd Nanoparticles Supported on N-Doped Carbon Derived from Glucosamine Hydrochloride, ACS Catal., 2014, 4(9), 3132–3135, DOI:10.1021/cs500859n.
- M. Guo, X. Kong, C. Li and Q. Yang, Hydrogenation of Benzoic Acid Derivatives over Pt/TiO2 under Mild Conditions, Commun. Chem., 2021, 4(1), 54, DOI:10.1038/s42004-021-00489-z.
- H. Zhang, J. Dong, X. Qiao, J. Qin, H. Sun, A. Wang, L. Niu and G. Bai, In-Situ Generated Highly Dispersed Nickel Nanoclusters Confined in MgAl Mixed Metal Oxide Platelets for Benzoic Acid Hydrogenation, J. Catal., 2019, 372, 258–265, DOI:10.1016/j.jcat.2019.03.012.
- S. B. Shinde and R. M. Deshpande, Catalytic Hydrogenation of Benzoic Acid, in New Advances in Hydrogenation Processes - Fundamentals and Applications, InTech, 2017, DOI:10.5772/66428.
- N. Perret, F. Cárdenas-Lizana and M. A. Keane, Selective Hydrogenation of Benzaldehyde to Benzyl Alcohol over Au/Al 2O3, Catal. Commun., 2011, 16(1), 159–164, DOI:10.1016/j.catcom.2011.09.017.
- Y. Sakata and V. Ponec, Reduction of Benzoic Acid on CeO2 and, the Effect of Additives, Appl. Catal., A, 1998, 166, 173–184 CrossRef.
- P. Mars and D. W. Van Krevelen, Oxidations Carried out by Means of Vanadium Oxide Catalysts, Chem. Eng. Sci., 1954, 3, 41–59 CrossRef.
- A. Wang, J. Li and T. Zhang, Heterogeneous Single-Atom Catalysis, Nat. Rev. Chem., 2018, 65–81, DOI:10.1038/s41570-018-0010-1.
- M. T. Darby, M. Stamatakis, A. Michaelides and E. C. H. Sykes, Lonely Atoms with Special Gifts: Breaking Linear Scaling Relationships in Heterogeneous Catalysis with Single-Atom Alloys, J. Phys. Chem. Lett., 2018, 5636–5646, DOI:10.1021/acs.jpclett.8b01888.
- K. Liu, P. Yan, H. Jiang, Z. Xia, Z. Xu, S. Bai and Z. C. Zhang, Silver Initiated Hydrogen Spillover on Anatase TiO2 Creates Active Sites for Selective Hydrodeoxygenation of Guaiacol, J. Catal., 2019, 369, 396–404, DOI:10.1016/j.jcat.2018.11.033.
- J. Hu, E. M. Kim, M. J. Janik and K. Alexopoulos, Hydrogen Activation and Spillover on Anatase TiO2-Supported Ag Single-Atom Catalysts, J. Phys. Chem. C, 2022, 126(17), 7482–7491, DOI:10.1021/acs.jpcc.2c01670.
- W. C. Conner and J. L. Falconer, Spillover in Heterogeneous Catalysis, Chem. Rev., 1995, 95, 759–788 CrossRef CAS.
- X. He, Q. He, Y. Deng, M. Peng, H. Chen, Y. Zhang, S. Yao, M. Zhang, D. Xiao, D. Ma, B. Ge and H. Ji, A Versatile Route to Fabricate Single Atom Catalysts with High Chemoselectivity and Regioselectivity in Hydrogenation, Nat. Commun., 2019, 10(1), 3663, DOI:10.1038/s41467-019-11619-6.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Isolated Metal Atom Geometries as a Strategy for Selective Heterogeneous Hydrogenations, Science, 2012, 335(6073), 1209–1212 CrossRef CAS.
- X. Pan, M. Q. Yang, X. Fu, N. Zhang and Y. J. Xu, Defective TiO2 with Oxygen Vacancies: Synthesis, Properties and Photocatalytic Applications, Nanoscale, 2013, 3601–3614, 10.1039/c3nr00476g.
- R. Schaub, E. Wahlström, A. Rønnau, E. Lægsgaard, I. Stensgaard and F. Besenbacher, Oxygen-Mediated Diffusion of Oxygen Vacancies on the TiO2(110) Surface, Science, 2003, 299, 377–379, DOI:10.1126/science.1059133.
- K. Liu, G. Hou, P. Gao, X. Nie, S. Bai, M. J. Janik and Z. C. Zhang, Evolution of Multiple Spillover Hydrogen Species on Anatase Titanium Dioxide, Cell Rep. Phys. Sci., 2022, 3(12), 101190, DOI:10.1016/j.xcrp.2022.101190.
- X. Han, Q. Kuang, M. Jin, Z. Xie and L. Zheng, Synthesis of Titania Nanosheets with a High Percentage of Exposed (001) Facets and Related Photocatalytic Properties, J. Am. Chem. Soc., 2009, 131(9), 3152–3153, DOI:10.1021/ja8092373.
- O. Carp, C. L. Huisman and A. Reller, Photoinduced Reactivity of Titanium Dioxide, Prog. Solid State Chem., 2004, 33–177, DOI:10.1016/j.progsolidstchem.2004.08.001.
- U. Diebold, The Surface Science of Titanium Dioxide, Surf. Sci. Rep., 2003, 48(5–8), 53–229 CrossRef CAS.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, Chemistry with ADF, J. Comput. Chem., 2001, 22(9), 931–967, DOI:10.1002/jcc.1056.
- G. Pacchioni, Oxygen Vacancy: The Invisible Agent on Oxide Surfaces, ChemPhysChem, 2003, 1041–1047, DOI:10.1002/cphc.200300835.
- T. L. Thompson and J. T. Yates, TiO2-Based Photocatalysis: Surface Defects, Oxygen and Charge Transfer, Top. Catal., 2005, 197–210, DOI:10.1007/s11244-005-3825-1.
- A. Roldán, M. Boronat, A. Corma and F. Illas, Theoretical Confirmation of the Enhanced Facility to Increase Oxygen Vacancy Concentration in TiO2 by Iron Doping, J. Phys. Chem. C, 2010, 114(14), 6511–6517, DOI:10.1021/jp911851h.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B, 1996, 54(16), 11169 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed.
- P. E. Blochl, Projector Augmented-Wave Method, Phys. Rev. B, 1994, 50(24), 17953 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104, DOI:10.1063/1.3382344.
- W. Heckel, T. Würger, S. Müller and G. Feldbauer, Van Der Waals Interaction Really Matters: Energetics of Benzoic Acid on TiO2 Rutile Surfaces, J. Phys. Chem. C, 2017, 121(32), 17207–17214, DOI:10.1021/acs.jpcc.7b03149.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Band Theory and Mott Insulators: Hubbard U Instead of Stoner I, Phys. Rev. B, 1991, 44(3), 943 CrossRef CAS PubMed.
- Z. Hu and H. Metiu, Choice of U for DFT+U Calculations for Titanium Oxides, J. Phys. Chem. C, 2011, 115(13), 5841–5845, DOI:10.1021/jp111350u.
- H. J. Monkhorst and J. D. Pack, Special Points for Brillonin-Zone Integrations, Phys. Rev. B: Solid State, 1976, 13(12), 5188–5192 CrossRef.
- H. V. Thang, G. Pacchioni, L. DeRita and P. Christopher, Nature of Stable Single Atom Pt Catalysts Dispersed on Anatase TiO2, J. Catal., 2018, 367, 104–114, DOI:10.1016/j.jcat.2018.08.025.
- V. Vorotnikov, S. Wang and D. G. Vlachos, Group Additivity for Estimating Thermochemical Properties of Furanic Compounds on Pd(111), Ind. Eng. Chem. Res., 2014, 53(30), 11929–11938, DOI:10.1021/ie502049a.
- P. J. Linstrom and W. G. Mallard, The NIST Chemistry WebBook: A Chemical Data Resource on the Internet, J. Chem. Eng. Data, 2001, 46(5), 1059–1063, DOI:10.1021/je000236i.
- V. Diky, C. Muzny, A. Smolyanitsky, A. Bazyleva, R. D. Chirico, J. W. Magee, E. Paulechka, A. F. Kazakov, S. Townsend, E. Lemmon, M. D. Frenkel and K. Kroenlein, ThermoData Engine (TDE) Version 10 (Pure Compounds, Binary Mixtures, Ternary Mixtures, and Chemical Reactions): NIST Standard Reference Database 103b, National Institute of Standards and Technology, Gaithersburg, MD, 2015 Search PubMed.
- C. L. Yaws, Yaws' Handbook of Thermodynamic and Physical Properties of Chemical Compounds, Knovel, 2003 Search PubMed.
- C. T. Campbell and J. R. V. Sellers, The Entropies of Adsorbed Molecules, J. Am. Chem. Soc., 2012, 134(43), 18109–18115, DOI:10.1021/ja3080117.
- Y. P. Li, J. Gomes, S. M. Sharada, A. T. Bell and M. Head-Gordon, Improved Force-Field Parameters for QM/MM Simulations of the Energies of Adsorption for Molecules in Zeolites and a Free Rotor Correction to the Rigid Rotor Harmonic Oscillator Model for Adsorption Enthalpies, J. Phys. Chem. C, 2015, 119(4), 1840–1850, DOI:10.1021/jp509921r.
- M. A. Vannice, S. H. Hyun, B. Kalparci and W. C. Liauh, Entropies of Adsorption in Heterogeneous Catalytic Reactions, J. Catal., 1979, 56(3), 358–362 CrossRef CAS.
- A. H. Motagamwala and J. A. Dumesic, Microkinetic Modeling: A Tool for Rational Catalyst Design, Chem. Rev., 2021, 1049–1076, DOI:10.1021/acs.chemrev.0c00394.
- A. A. Gokhale, S. Kandoi, J. P. Greeley, M. Mavrikakis and J. A. Dumesic, Molecular-Level Descriptions of Surface Chemistry in Kinetic Models Using Density Functional Theory, Chem. Eng. Sci., 2004, 59, 4679–4691, DOI:10.1016/j.ces.2004.09.038.
- H. Eyring, The Activated Complex in Chemical Reactions, J. Chem. Phys., 1935, 3(2), 63–71, DOI:10.1063/1.1749604.
- H. Eyring, The Activated Complex And The Absolute Rate Of Chemical Reactions, Chem. Rev., 1935, 17(1), 65–77 CrossRef CAS.
- J. D. Kammert, A. Chemburkar, N. Miyake, M. Neurock and R. J. Davis, Reaction Kinetics and Mechanism for the Catalytic Reduction of Propionic Acid over Supported ReO XPromoted by Pd, ACS Catal., 2021, 11(3), 1435–1455, DOI:10.1021/acscatal.0c04328.
- L. A. Gomez, C. Q. Bavlnka, T. E. Zhang, D. E. Resasco and S. P. Crossley, Revealing the Mechanistic Details for the Selective Deoxygenation of Carboxylic Acids over Dynamic MoO3 Catalysts, ACS Catal., 2023, 13(13), 8455–8466, DOI:10.1021/acscatal.3c01053.
- E. A. Solano Espinoz and W. E. Vallejo Narváez, Density Functional Theory and RRKM Calculations of Decompositions of the Metastable E-2,4-pentadienalmolecular Ions, J. Mass Spectrom., 2010, 45(7), 722–733, DOI:10.1002/jms.1760.
- C. T. Campbell, The Degree of Rate Control: A Powerful Tool for Catalysis Research, ACS Catal., 2017, 2770–2779, DOI:10.1021/acscatal.7b00115.
- C. T. Campbell, Micro- and Macro-kinetics: Their Relationship in Heterogeneous Catalysis, Top. Catal., 1994, 1, 353–366 CrossRef CAS.
- G. R. Wittreich, K. Alexopoulos and D. G. Vlachos, Microkinetic Modeling of Surface Catalysis, in Handbook of Materials Modeling: Applications: Current and Emerging Materials, Springer International Publishing, 2nd edn, 2020, pp. 1377–1404, DOI:10.1007/978-3-319-44680-6_5.
- A. Samant and D. G. Vlachos, Overcoming Stiffness in Stochastic Simulation Stemming from Partial Equilibrium: A Multiscale Monte Carlo Algorithm, J. Chem. Phys., 2005, 123(14), 144114, DOI:10.1063/1.2046628.
- A. G. Thomas, W. R. Flavell, A. K. Mallick, A. R. Kumarasinghe, D. Tsoutsou, N. Khan, C. Chatwin, S. Rayner, G. C. Smith, R. L. Stockbauer, S. Warren, T. K. Johal, S. Patel, D. Holland, A. Taleb and F. Wiame, Comparison of the Electronic Structure of Anatase and Rutile TiO2 Single-Crystal Surfaces Using Resonant Photoemission and X-Ray Absorption Spectroscopy, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75(3), 035105, DOI:10.1103/PhysRevB.75.035105.
- M. Buchholz, M. Xu, H. Noei, P. Weidler, A. Nefedov, K. Fink, Y. Wang and C. Wöll, Interaction of Carboxylic Acids with Rutile TiO2(110): IR-Investigations of Terephthalic and Benzoic Acid Adsorbed on a Single Crystal Substrate, Surf. Sci., 2016, 643, 117–123, DOI:10.1016/j.susc.2015.08.006.
- Q. Guo, I. Cocks and E. M. Williams, The Adsorption of Benzoic Acid on a TiO2(110) Surface Studied Using STM, ESDIAD and LEED, Surf. Sci., 1997, 393(1–3), 1–11 CAS.
- N. Martsinovich, D. R. Jones and A. Troisi, Electronic Structure of TiO2 Surfaces and Effect of Molecular Adsorbates Using Different DFT Implementations, J. Phys. Chem. C, 2010, 114(51), 22659–22670, DOI:10.1021/jp109756g.
- S. P. Bates, G. Kresse and M. J. Gillan, The Adsorption and Dissociation of ROH Molecules on TiO2(110), Surf. Sci., 1998, 409(2), 336–349 CrossRef CAS.
- P. Persson, R. Bergstrom and S. Lunell, Quantum Chemical Study of Photoinjection Processes in Dye-Sensitized TiO2 Nanoparticles, J. Phys. Chem. B, 2000, 104(44), 10348–10351, DOI:10.1021/jp002550p.
- G. Tabacchi, M. Fabbiani, L. Mino, G. Martra and E. Fois, The Case of Formic Acid on Anatase TiO2(101): Where Is the Acid Proton?, Angew. Chem., Int. Ed., 2019, 58(36), 12431–12434, DOI:10.1002/anie.201906709.
- L. Xue, W. Su and Z. Lin, Mechanism of Silver- and Copper-Catalyzed Decarboxylation Reactions of Aryl Carboxylic Acids, Dalton Trans., 2011, 40(44), 11926–11936, 10.1039/c1dt10771b.
- L. J. Gooßen, N. Rodríguez, C. Linder, P. P. Lange and A. Fromm, Comparative Study of Copper- and Silver-Catalyzed Protodecarboxylations of Carboxylic Acids, ChemCatChem, 2010, 2(4), 430–442, DOI:10.1002/cctc.200900277.
- Q. Jin, J. Li, A. Ariafard, A. J. Canty and R. A. J. O'Hair, Substituent Effects in the Decarboxylation Reactions of Coordinated Arylcarboxylates in Dinuclear Copper Complexes, [(Napy)Cu2(O2CC6H4X)]+†, Eur. J. Mass Spectrom., 2017, 23(6), 351–358, DOI:10.1177/1469066717729067.
- M. John, K. Alexopoulos, M. F. Reyniers and G. B. Marin, Mechanistic Insights into the Formation of Butene Isomers from 1-Butanol in H-ZSM-5: DFT Based Microkinetic Modelling, Catal. Sci. Technol., 2017, 7(5), 1055–1072, 10.1039/c6cy02474b.
- M. Saunders and M. R. Kates, Rates of Degenerate 1,2-Hydride and 1,2-Methide Shifts from the Carbon-13 Nuclear Magnetic Resonance Spectra of Tertiary Alkyl Cations, J. Am. Chem. Soc., 1978, 100(2), 7082–7083 CrossRef CAS.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Selective Hydrogenation over Supported Metal Catalysts: From Nanoparticles to Single Atoms, Chem. Rev., 2020, 683–733, DOI:10.1021/acs.chemrev.9b00230.
Footnotes |
| † Electronic supplementary information (ESI) available: Computational methods and details, alternative mechanisms of product formation, additional geometric details and configurations of reaction intermediates, overall reaction energy diagram, kinetic model parameters and differential equations, surface coverages, and detailed rate analysis. See DOI: https://doi.org/10.1039/d4re00561a |
| ‡ Denotes equal authorship. |
| This journal is © The Royal Society of Chemistry 2025 |