
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed aerobic homocoupling of aliphatic olefins to dienes: evidence for rate-limiting concerted metalation–deprotonation†
Sam
Van Minnebruggen
 a,
Harry
Poels-Ryckeboer
a,
Hendrik
Van Dessel
a,
Frederick
Martens
a,
Wouter
Stuyck
a,
Tom
Nelis
a,
Igor
Beckers
a,
Aram
Bugaev
b and
Dirk
De Vos
*a
a,
Harry
Poels-Ryckeboer
a,
Hendrik
Van Dessel
a,
Frederick
Martens
a,
Wouter
Stuyck
a,
Tom
Nelis
a,
Igor
Beckers
a,
Aram
Bugaev
b and
Dirk
De Vos
*a
aCentre for Membrane Separations, Adsorption, Catalysis and Spectroscopy for Sustainable Solutions (cMACS), KU Leuven, 3001 Leuven, Belgium. E-mail: Dirk.Devos@kuleuven.be
bSuperXAS Beamline, Paul Scherrer Institute, 5232 Villigen, Switzerland
First published on 9th December 2024
Abstract
Palladium(II)-catalyzed dehydrogenative coupling of aliphatic olefins would enable an efficient route to (conjugated) dienes, but remains scarcely investigated. Here, 2-hydroxypyridine (2-OH-pyridine) was found to be an effective ligand for Pd(II) in the activation of vinylic C(sp2)–H bonds. While reoxidation of Pd(0) is challenging in many catalytic oxidations, one can avoid in this reaction that the reoxidation becomes rate-limiting, even under ambient O2 pressure, by working in coordinating solvents. Via kinetic studies the elementary steps governing this reaction were elucidated, resulting in enhanced performance (turnover frequency) of the Pd(II)/2-OH-pyridine system. The diene product is formed via a consecutive activation of two olefins on the same Pd atom, followed by a β-hydride elimination. The first olefin activation, viz. the C–H activation, determines the overall reaction rate under these conditions. The catalytic complex was studied by ESI-MS and X-ray absorption spectroscopy, revealing that the coordination sphere of the working palladium complex contains two 2-OH-pyridine ligands.
Introduction
Dienes are vital in the chemical industry, with applications ranging from polymer building blocks (e.g. for polybutadiene)1 to fine chemicals2–7 (e.g. for vitamin A synthesis). The simplest diene, 1,3-butadiene, is a petrochemical building block produced in large scale (ca. 20.5 Mton annually in 2023).8 Traditionally, its demand was largely met by naphtha-fed steam crackers,9 but lately on-purpose C4-dehydrogenation has come to prominence to sustain the growth in butadiene demand.10 However, as the chain length of the aliphatic alkane increases, the dehydrogenation increasingly produces coke rather than dienes.Dehydrogenative homocoupling of aliphatic olefins requires an oxidant to thermodynamically drive the catalytic cycle. Using O2 as oxidant to couple identical olefins to dienes is a largely unexplored challenge.11 Previous studies have focused mainly on the dehydrogenative coupling of two different olefins (Fig. 1), in particular combinations of olefins with electronically activating substituents (e.g. styrene-like) and with metal-coordinating, directing groups (e.g. acrylates or acrylamides).12–16 Intramolecular coupling of olefin moieties has been reported as well, driven by stabilization of the formed aromatic product.17 Finally, homocoupling of olefins has been described, but these reports remained limited to activated styrene-type reactants.18,19 All previous cases also rely on specific terminal oxidants (Cu salts, benzoquinones…) which negatively affects process economics of such reactions.20 An alternative approach that has been reported, used ethylene not only as reactant, but also as terminal oxidant.21,22
 | ||
| Fig. 1 Overview of different pathways to obtain diene moieties via coupling reactions. | ||
Like other oxidative Pd(II)-catalyzed reactions, such as the Fujiwara–Moritani reaction (dehydrogenative alkenylation of arenes),23,24 the allylic acetoxylation25 and the Wacker oxidation,26 the herein reported dehydrogenative homocoupling of aliphatic olefins leaves the metal in a reduced, zerovalent state after product formation. In these known reactions, various concepts have been developed to attain high activities/yields, even under mild reoxidation conditions (e.g. 1 bar O2, in absence of redox co-catalysts).27–31 Rapid reoxidation is necessary to prevent formation of inactive Pd(0)-species.32 Additionally, the use of strong acids should be avoided in order to suppress the formation of oligomers and olefin isomerization.33–35 The present work system uncovers a Pd-(2-OH-pyridine) catalyst that effects selective homocoupling of α-olefins to dienes.
Results & discussion
In a first series of experiments on the dehydrogenative homocoupling of 1-octene, different functionalized pyridine ligands were evaluated, as these are known to facilitate the activation of C(sp2)–H bonds in both arenes and olefins.29,36 The considered reaction times are short (typically <1 h) to probe kinetic behaviour. Productivity of the system is expressed in turnover numbers (TON), as is usual for other, similar homocoupling reactions.25,32,37 Table S1 (ESI)† summarizes the results obtained for different ligands, revealing that pyridine moieties bearing a hydroxy functionality in the ortho position facilitate the dehydrogenative coupling of 1-octene to yield hexadecadiene isomers (Fig. 2; identification of product isomers in Fig. S1(a)†). 1H-NMR showed that predominantly linear C16 compounds were formed; no C8 ketones or acetoxylated products were observed. Besides the thermodynamically most stable conjugated diene (68%), also non-conjugated dienes were formed in significant amounts (32%). Notably, the rate of reaction was influenced by the functional groups on the ligand, as illustrated in Fig. 2, with electron-withdrawing groups enhancing the activity. To obtain a stable catalytic activity under atmospheric O2 pressure, it was found essential to use coordinating solvents like dimethylacetamide (DMA) (Fig. S3†). As observed before in other C–H functionalization reactions, such coordinating solvents help in suppressing catalyst deactivation by avoiding agglomeration of metallic Pd.36,38 | ||
| Fig. 2 Product selectivity (left) and linear free-energy relationship between reaction rate and σ-parameter of substituents on the 2-OH-pyridine ligand (right). Reaction conditions: 15 μmol Pd(OAc)2, 7 eq. ligand, 1.0 mL 1-octene, 1.4 mL dimethylacetamide (DMA) and 50 μL tetradecane at 90 °C under O2 atmosphere (sparged) after 10 min. The ligands used in this screening and the corresponding σ-values are shown in Table S2† (σpara is used for the substituent in the para position with respect to the OH-group, as in e.g. 5-NO2-2-OH-pyridine). | ||
Having identified a suitable ligand and solvent for the dehydrogenative olefin homocoupling, the influence of individual reaction parameters was assessed. To ensure that kinetics are not governed by the reoxidation, different O2 pressures were screened (Fig. 3(a)). This shows clearly that from atmospheric O2 pressure onwards, reoxidation is no longer rate limiting when working in DMA with 2-OH-pyridine as ligand. In such conditions, the effect of the 2-OH-pyridine/Pd ratio was investigated (Fig. 3(b)). In the absence of ligand, negligible product formation was observed. Increasing the amount of ligand led to an optimum ligand/metal ratio centered around 2. Saturation of the complex hindered the reaction at large ligand excesses, thus demonstrating the importance of this ratio. Additionally, carboxylic acids with varying pKa were evaluated as additives (Fig. 3(c)). Carboxylic acids can play various roles in oxidative Pd chemistry; for instance, carboxylates can assist in C–H activation by concerted metalation–deprotonation (CMD). Interestingly, stronger acids negatively affected the reactivity, while at more moderate pKa values, an activity plateau was reached. Strong Brønsted acids may hamper the deprotonation of the 2-hydroxypyridine (pKa = 11.6) to form an anionic ligand; this anionic character can play an important role in accepting protons from the olefin substrate, facilitating the cleavage of the C(sp2)–H bond through a CMD mechanism.39,40 With the strongest acids, even the 2-pyridone ring itself may be protonated (pKa = 0.8), thus impeding coordination of the Pd. This ligand protonation might also explain the poor performance of 2-OH-pyridines with electron-withdrawing substituents over long reaction times upon addition of carboxylic acid (Fig. 2 and S2†).41,42
 | ||
| Fig. 3 Influence of individual reaction parameters on the kinetics. (a) Variation of O2 pressure; (b) effect of the ratio of 2-OH-pyridine ligand to Pd; (c) effect of the pKa of the added carboxylic acid; (d) effect of the amount of acetic acid. Reaction conditions: 15 μmol Pd(OAc)2, 7 eq. 2-OH-pyridine (varied for (b)), 100 eq. acetic acid (or 0.7 M AcOH) (amount varied in (d), other acids (R-COOH) used in (c) are shown in Table S3†), 1.4 mL DMA, 0.6 mL 1-octene and 50 μL tetradecane at 100 °C under O2 atmosphere (sparged, varied for (a)) after 60 min. | ||
Moreover, varying the amount of carboxylic acid had significant impact on the system's activity (Fig. 3(d)). While the system is active even without carboxylic acid, a 4-fold activity increase was achieved by raising the acetic acid concentration to 3 M. In such large concentrations, the acid acts rather as a polar, protic solvent than as a ligand. An increased proticity of the solvent enhances the dissociation of bidentate coordinated 2-OH-pyridine by interacting via hydrogen bonding interactions.43,44 Interestingly, no difference in activity was observed when AcOH was substituted with its deuterated equivalent (Fig. S4†), implying that a protonation is not the slow step in the cycle. Moreover, considering only potassium acetate in absence of ligand did not yield any product.
With optimal conditions established for the ancillary ligands and solvent, the focus shifted towards the mechanism and the effect of the reaction parameters. Either one, or both of the olefin activation steps might be rate-limiting. In these respective cases, the rate would exhibit a first (or higher) order dependence on the substrate concentration. Fig. 4 illustrates an initial linear increase in rate with an increasing concentration of 1-octene. This linear relationship indicates a first order dependence in olefin concentration; thus the rate-limiting step involves the activation of only one olefin reactant. As the olefin concentration is further increased, the rate reaches a plateau, indicating that the olefin activation is no longer rate-limiting. As the olefin concentration was found to be important, reactions were also conducted in pure 1-octene. Although minor traces of product were observed, palladium black formation indicates that reoxidation becomes the limiting step at this point, either due to lack of coordinating solvent, or due to the limited solubility of 2-OH-pyridine in 1-octene.
 | ||
| Fig. 4 Effect of the olefin concentration on turnover frequency. Reaction conditions: 15 μmol Pd(OAc)2, 7 eq. 2-OH-pyridine, 100 eq. AcOH, 1.4 mL DMA, x M 1-octene and 50 μL tetradecane at 100 °C under O2 atmosphere (sparged) after 30 min. | ||
A plausible reaction mechanism comprises a consecutive C–H activation, migratory insertion and finally a β-hydride elimination, which is in agreement with the observed first order in olefin.28 In a typical monometallic pathway, one Pd atom consecutively performs both olefin activation steps. However, a bimetallic pathway is also possible, in which two Pd(II)-vinyl fragments undergo transmetallation to yield a diene product.43 Both options can be distinguished by determining the order of the reaction in Pd. In the monometallic pathway, a first-order dependence is expected as only one Pd atom is involved in the mechanism. In contrast, the bimetallic pathway involves two Pd atoms throughout the catalytic cycle, resulting in a second-order dependence on Pd concentration.
The results observed in Fig. 5 confirm a first-order dependence on [Pd], supporting the proposed monometallic migratory insertion and β-hydride elimination steps, shown in Scheme 1. Now the question arises, which step is rate-limiting: the C–H activation or the migratory insertion of the olefin. A first approach to distinguish these options is based on the activation energies. By comparing initial rates between 70 and 110 °C, an apparent activation energy (Ea,app) of 80 kJ mol−1 was found (Fig. 6). Such values are typically associated with the activation of C(sp2)–H bonds,39,44 whereas for the migratory insertion lower values have been reported in literature, as this only involves cleaving the vinylic π-bond.45
 | ||
| Fig. 5 Mono- (left) and bimetallic (right) pathway for both olefin activation steps in the dehydrogenative olefin homocoupling (top) and the effect of the Pd(OAc)2 concentration on product formation (bottom). Reaction conditions: x μmol Pd(OAc)2, 7 eq. 2-OH-pyridine, 100 eq. AcOH, 2.5 mL 1-octene, 1.4 mL DMA and 50 μL tetradecane at 100 °C under O2 atmosphere (sparged) after 30 min. | ||
 | ||
| Scheme 1 Proposed mechanism for the Pd-catalyzed dehydrogenative homocoupling of simple olefins to (non-) conjugated dienes. | ||
 | ||
| Fig. 6 Effect of the temperature on reaction rates. Reaction conditions: 15 μmol Pd(OAc)2, 7 eq. 2-OH-pyridine, 100 eq. AcOH, 1.4 mL DMA, 0.6 mL 1-octene and 50 μL tetradecane at 70–130 °C under O2 atmosphere (sparged) after 30 min. | ||
Increasing the temperature beyond 110 °C resulted in rapid Pd(0) precipitation and degradation of the (conjugated) product, yielding heavier compounds (Fig. S1(a) and S6†). Prolonged reaction times had similar effects, limiting the per-pass productivity of the system. Accumulation of the diene product is in that sense unwanted, as the diene is likely to undergo consecutive reaction, deteriorating the product yield after long reaction times. Although this can be partly suppressed by working in solvent-like quantities of olefin, the increased reactivity of the (conjugated) diene with respect to the olefin reactant hampers the per-pass conversion. This contrasts with the findings for arene homocoupling, in which the product exhibits largely similar reactivity as the original arene reactant. To address this issue, a solution was proposed involving intermittent product removal. The reaction was periodically halted and cooled down, leading to phase separation. The apolar layer contained 1-octene and the products, while the polar layer contained DMA and the catalyst. As such, a cumulative turnover number of 130 mol diene product per mol Pd was achieved, as demonstrated in Fig. S5.† The obtained apparent activation energy, corresponding to the activation of the C(sp2)–H bond, suggests that a kinetic isotope effect (KIE) should be observed upon switching to a deuterated equivalent of the substrate. Indeed, when comparing styrene and styrene-d8, a KIE of 4.7 was obtained (see product identification Fig. S1(f)† and 7 (left)). Considering that the C–H activation is rate-limiting, the influence of electronic properties of substituents on the reaction rates of substituted styrenes was studied. The resulting structure–activity relationship is shown in Fig. 7 (right). Electron-donating groups, such as –OCH3, were found to enhance rates, whereas electron-withdrawing groups, like –Cl, diminished reactivity. Additionally, the –Cl group did not lead to side reactions. Bulky substituents, like t-butyl groups, were found to cause deviations from this structure–activity relationship, presumably due to steric effects. The ρ-value obtained from the linear free-energy relationship (−0.96) is mildly negative, compared to literature values for electrophilic aromatic substitutions (e.g. nitration of benzene; ρ = −5.9).47 Both the ρ-value as well as the occurrence of a KIE support that the first olefin activation indeed follows a CMD mechanism.48 A similar behaviour was described for the electrophilic CMD in the oxidative coupling of thiophenes.49
 | ||
| Fig. 7 Kinetic isotope effect for the dehydrogenative homocoupling of styrene vs. styrene-d8 (left). Linear free-energy relationship between reaction rate and σ-parameter of substituted functionalized styrene reactants (right). Reaction conditions: 15 μmol Pd(OAc)2, 3 eq. 2-OH-pyridine, 100 eq. AcOH, 1.4 mL DMA, 2 mL olefin, and 50 μL tetradecane at 80 °C under O2 atmosphere (sparged) after 10 min. σp values were obtained from literature.46 | ||
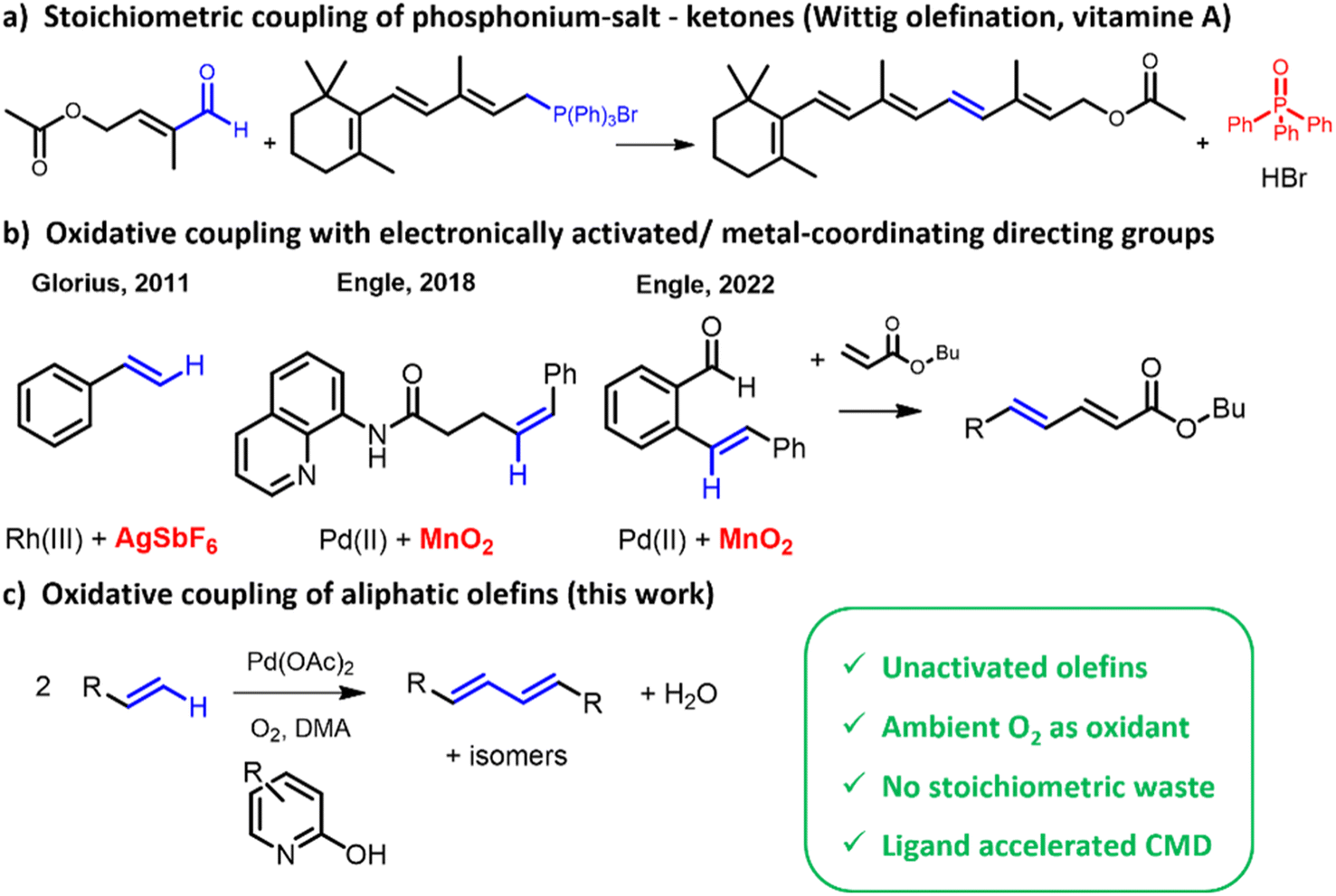
Having clarified the impact of the electronic properties in aromatic olefins, the reaction of a series of aliphatic olefins was studied (Fig. 8). In line with its larger number of protons in terminal methylidene groups, 1,7-octadiene is converted faster than 1-octene, but if the reaction rates are normalized per number of vinylic protons, similar turnover frequencies (TOF) are found for both reactants. Given the large amounts of 1,7-octadiene used, the product mixture is dominated by intermolecular coupling product. Notably, the obtained GC chromatogram as well as the 1H-NMR spectrum resemble strongly that of 1-octene, indicating that conjugated and non-conjugated products are formed in similar ratios, depending on the kinetically controlled β-hydride elimination (Scheme 1). In the mechanistically similar oxidative alkenylation of benzene with 1-pentene, β-hydride elimination likewise controls the ratio of conjugated vs. non-conjugated products.28 The reaction with internal olefins like trans-2-octene and cyclooctene is much slower, due to the lack of terminal vinylic protons and an increased steric hindrance. Upon comparing 1- and trans-2-octene, the relative reactivity of an individual proton of ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 was found to be four times higher than that of the CH– internal vinylic proton. Additionally, the number of different products formed increased for 2-octene (see product identification, Fig. S1†). This can be attributed to the different numbers of (non-equivalent) allylic protons compared to 1-octene (5 vs. 2 allylic protons). The presence of methyl groups on the olefin (as in 2-Me-1-heptene) was also found to hamper the activation of the vinylic C–H bonds. If a methyl group is present further down the aliphatic chain, the steric hindrance is reduced (as in 4-Me-1-hexene and vinylcyclohexane), yielding a comparable result as with 1-octene. In case fewer or no allylic protons are present, like for vinylcyclohexane and t-butylethylene, the product mixture shifts significantly towards trans,trans-coupled products, as the formation of non-conjugated dienes is severely hindered. Allylcyclohexane behaves in that sense again very similar to 1-octene. Lastly, testing styrene in the same conditions shows that the reactivity of this aromatic olefin is ten times higher than that of 1-octene.
CH2 was found to be four times higher than that of the CH– internal vinylic proton. Additionally, the number of different products formed increased for 2-octene (see product identification, Fig. S1†). This can be attributed to the different numbers of (non-equivalent) allylic protons compared to 1-octene (5 vs. 2 allylic protons). The presence of methyl groups on the olefin (as in 2-Me-1-heptene) was also found to hamper the activation of the vinylic C–H bonds. If a methyl group is present further down the aliphatic chain, the steric hindrance is reduced (as in 4-Me-1-hexene and vinylcyclohexane), yielding a comparable result as with 1-octene. In case fewer or no allylic protons are present, like for vinylcyclohexane and t-butylethylene, the product mixture shifts significantly towards trans,trans-coupled products, as the formation of non-conjugated dienes is severely hindered. Allylcyclohexane behaves in that sense again very similar to 1-octene. Lastly, testing styrene in the same conditions shows that the reactivity of this aromatic olefin is ten times higher than that of 1-octene.
 | ||
| Fig. 8 Effect of vinyl substitution in the olefin substrate on the turnover frequency (TOF), normalized for the number of vinylic protons (e.g. 3 for 1-octene). Reaction conditions: 15 μmol Pd(OAc)2, 3 eq. 2-OH-pyridine, 492 μL AcOH, 1 mL olefin, 1.4 mL dimethylacetamide and 50 μL tetradecane at 90 °C under O2 atmosphere (sparged) after 10 min. | ||
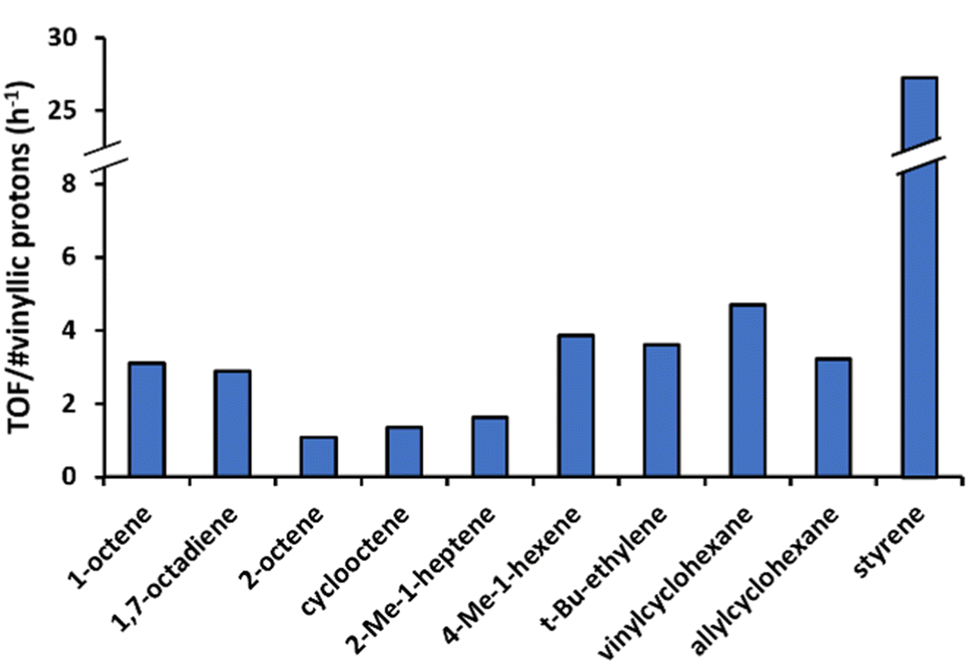
For the cross-coupling of two different olefins, the productivity is expected to depend on the choice of the electron-rich olefin, since the cycle starts with a C–H activation of the most electron-rich olefin reactant. Depending on the respective tendencies to undergo migratory insertion, the product distribution between homo- and cross-coupled diene is expected to be strongly shifted to cross-coupled product. Specifically, olefins bearing electron-withdrawing groups are preferentially inserted, as the electron-withdrawing group induces a partial positive charge on the β-vinylic carbon atoms.46Fig. 9 illustrates that similar reaction rates are obtained in the cross-coupling of an acrylate and 1-octene, as in the homocoupling of 1-octene. This confirms that the C–H activation of the electron-rich 1-octene is rate determining. Despite employing a 2.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar excess of 1-octene to t-butyl acrylate, the cross-coupled product dominates the obtained product mixture by 60%. This highlights that even though the first step is rate-limiting, selectivity in the migratory insertion can still be tuned to a large extent, depending on the choice of the second olefin and the ratio of both olefins. Notably, no homocoupled hexenedioate esters were detected, underscoring the challenging nature of C–H activation in electron-deficient olefins and the distinct electronic preferences of each individual olefin activation step. Similarly, when considering the cross-coupling of 1-octene with styrene (equimolar amounts), negligible amounts of hexadecadienes were obtained (7%, Fig. S1k†). This finding corresponds to earlier results showing limited homocoupling of 1-octene at 80 °C (Fig. 6). In that sense, the overall productivity of the catalyst is largely determined by its ability to activate the styrene C–H bonds, as indicated by the similar activity in pure styrene and the 1-octene-styrene mixture (Fig. 7 and 8). More specifically, 45% of cross-coupled products are formed, together with 48% of styrene homocoupled products. This indicates that there is no preference to incorporate either styrene or 1-octene in the migratory insertion.
:1 molar excess of 1-octene to t-butyl acrylate, the cross-coupled product dominates the obtained product mixture by 60%. This highlights that even though the first step is rate-limiting, selectivity in the migratory insertion can still be tuned to a large extent, depending on the choice of the second olefin and the ratio of both olefins. Notably, no homocoupled hexenedioate esters were detected, underscoring the challenging nature of C–H activation in electron-deficient olefins and the distinct electronic preferences of each individual olefin activation step. Similarly, when considering the cross-coupling of 1-octene with styrene (equimolar amounts), negligible amounts of hexadecadienes were obtained (7%, Fig. S1k†). This finding corresponds to earlier results showing limited homocoupling of 1-octene at 80 °C (Fig. 6). In that sense, the overall productivity of the catalyst is largely determined by its ability to activate the styrene C–H bonds, as indicated by the similar activity in pure styrene and the 1-octene-styrene mixture (Fig. 7 and 8). More specifically, 45% of cross-coupled products are formed, together with 48% of styrene homocoupled products. This indicates that there is no preference to incorporate either styrene or 1-octene in the migratory insertion.
 | ||
| Fig. 9 Olefin competition experiment. Reaction conditions: 15 μmol Pd(OAc)2, 7 eq. 2-OH-pyridine, 500 eq. AcOH, 1.4 mL DMA, 0 or 2.5 mL 1-octene, 0 or 1 mL t-butyl acrylate and 50 μL tetradecane at 100 °C under O2 atmosphere (sparged) after 45 min. | ||
As a full mechanistic understanding has been achieved, the active complex was characterized by electrospray ionization-mass spectroscopy (ESI-MS)51 and X-ray absorption spectroscopy (XAS).
In Fig. 10, the ESI-MS of Pd(OAc)2 dissolved in acetonitrile clearly shows palladium complexes (with the characteristic six isotopes). Significant changes were found upon addition of 2-OH-pyridine. More specifically, the addition of the ligand yields an active complex coordinated with two 2-OH-pyridine ligands, which corresponds to the optimum ligand/catalyst ratio determined earlier (Fig. 3(b)). Moreover, also the ex situ X-ray absorption spectroscopy results are shown in Fig. 9. From the edge jump in X-ray near edge structure (XANES), it can indeed be concluded that the Pd-complex is in its divalent state (see Fig. S7†). Additionally, the extended X-ray absorption fine structure (EXAFS) analysis revealed that the coordination sphere of the palladium complex with 2-OH-pyridine ligands differs distinctly from the Pd(OAc)2 reference. Additionally, in situ measurements reveal a swift transition of the Pd(OAc)2 precursor towards the active complex, featuring Pd–N interactions (Fig. S9†). Notably, in both cases of Pd(OAc)2 (with and without 2-OH-pyridine), no Pd–Pd interactions were observed, highlighting that the coordinating properties of DMA inhibit the formation of Pd(0) particles. In case ligand is present, the oxidation state of the Pd-species decreases slightly, suggesting that a portion of the Pd is in the zerovalent state due to ongoing reaction. These findings provide a comprehensive mechanistic understanding of the reaction and shed light on the nature of the active species considered herein.
 | ||
| Fig. 10 Characterization of the active complex by ESI-MS and XAS. ESI-MS measurement of Pd(OAc)2 dissolved in CH3CN (a) and with additional 2-OH-pyridine (b). Conditions: 0.89 μmol Pd(OAc)2, 0 or 10 eq. 2-OH-pyridine and 1 mL CH3CN; positive mode.50 (c) XANES spectra for the Pd-(2-hydroxypyridine) and Pd(OAc)2 compared to some reference Pd precursors. (d) Experimental κ2-weighted Fourier transformed EXAFS for the Pd-(2-hydroxypyridine) and Pd(OAc)2 complexes compared to some reference Pd-precursors. Conditions: Pd(OAc)2 dissolved in DMA (with and without ligand) benchmarked with reference Pd-precursors (PdO and metallic Pd foil). | ||
In summary, this study introduces a novel approach for the dehydrogenative coupling of aliphatic olefins, resulting in the formation of dienes. Detailed mechanistic investigations revealed the importance of maintaining a high olefin concentration in order to achieve significant activities. The rate-limiting step was identified to be the initial C–H activation of the olefin. To address product degradation, intermittent product removal by phase separation was found to improve overall productivity, resulting in a cumulative TON of 130. These findings have significant implications, offering a pathway for synthesizing long-chain internal dienes, which have been challenging to obtain thus far. This breakthrough opens up new possibilities for the economically viable production of dienes and their potential applications in various fields.
Abbreviations
| 2-OH-pyridine | 2-Hydroxypyridine |
| AcOH | Acetic acid |
| DMA | Dimethylacetamide |
| E a,app | Apparent activation energy |
| ESI-MS | Electrospray injection mass spectroscopy |
| EXAFS | Extended X-ray absorption fine structure |
| KIE | Kinetic isotope effect |
| Pd(OAc)2 | Palladium diacetate |
| TOF | Turnover frequency (h−1) |
| TON | Turnover number (mol product per mol Pd) |
| XANES | X-ray absorption near-edge structure |
| XAS | X-ray absorption spectroscopy |
Data availability
The data that support the findings of this study are available from the corresponding author, D. D. V., upon reasonable request.Author contributions
Under the supervision of D. D. V., S. V. M. was responsible for the conception, design and interpretation of the experiments. H. P. R. and T. N. performed additional experiments. H. V. D., F. M., W. S. and I. B. gave their critical input on the manuscript. W. S. and A. B. helped with the XAS measurements. A. B. interpreted the results of the XAS measurements.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. V. M., H. P. R., H. V. D., F. M., I. B. and A. B. express their gratitude to the FWO for funding (1SA0921N, 1SH8L24N, 1S31822N, 1SHA524N, 1291724N and G0781118N) and to Dr Kirill Lomachenko and Dr Kwinten Janssens for his support during the XAS measurements (ESRF, CH7275). W. S. and D. D. V. thank VLAIO via the Flemish spearhead cluster Catalisti for financial support (Moonshot cluster SBO project ECAP and FF-PFAS).References
- J. Grub and E. Löser, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2011, pp. 381–396 Search PubMed.
- J. Panten and H. Surburg, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2015, pp. 1–55 Search PubMed.
- R. Metcalf and A. Horowitz, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2014, pp. 1–94 Search PubMed.
- E. Breitmaier, Terpenes: Flavors, Fragrances, Pharmaca, Pheromones, Wiley, 2006 Search PubMed.
- X. Zhang, M. Wang, M. Zhang, Y. Xu and T. P. Loh, Org. Lett., 2013, 15(21), 5531–5533 CrossRef CAS PubMed.
- R. Rapagnani, R. Dunscomb, A. Fresh and I. Tonks, Nat. Chem., 2022, 14(8), 877–883 CrossRef CAS PubMed.
- J. Luca Schwarz, H. Huang, T. Paulisch and F. Glorius, ACS Catal., 2019, 10(2), 1621–1627 CrossRef.
- Butadiene Industry Capacity and Capital Expenditure Forecasts with Details of All Active and Planned Plants to 2028, available at https://www.globaldata.com/store/report/butadiene-market-analysis/#:%7E:text=Thebutadienecapacitywas20.47,growthduringtheoutlookperiod. (accessed September 2024).
- J. Moulijn, M. Makkee and A. Van Diepen, Chemical Process Technology, Wiley, 2013, pp. 1–576 Search PubMed.
- K. Weissermel and H. Arpe, in Industrial Organic Chemistry, Wiley, 2003, pp. 107–126 Search PubMed.
- M. Lu, J. Goh, M. Maraswami, Z. Jia, J. Tian and T. Loh, Chem. Rev., 2022, 122, 17479–17646 CrossRef CAS PubMed.
- G. Meng, L. Hu, H. Chan, J. Qiao and J. Yu, J. Am. Chem. Soc., 2023, 145(24), 13003–13007 CrossRef CAS PubMed.
- M. Liu, J. Sun, T. Erbay, H. Ni, R. Martin-Montero, P. Liu and K. Engle, Angew. Chem., 2022, 61(25), e202203624 CrossRef CAS PubMed.
- Y. Xu, J. Lu and T. Loh, J. Am. Chem. Soc., 2009, 131(4), 1372–1373 CrossRef CAS.
- M. Liu, P. Yang, M. Karunananda, Y. Wang, P. Liu and K. Engle, J. Am. Chem. Soc., 2018, 140(17), 5805–5813 CrossRef CAS PubMed.
- T. Besset, N. Kuhl, F. Patureau and F. Glorius, Chem.–Eur. J., 2011, 17(26), 7167–7171 CrossRef CAS PubMed.
- M. Maraswami, T. Diggins, J. Goh, R. Tio, W. Ong, H. Hirao and T. Loh, ACS Catal., 2021, 11(18), 11494–11500 CrossRef CAS.
- Y. Wen, J. Xie, C. Deng and Y. Wu, Synlett, 2015, 26(12), 1755–1758 CrossRef CAS.
- T. Zhu, Z. Li, F. Xiao and W. Duan, Tetrahedron Lett., 2018, 59(34), 3238–3241 CrossRef CAS.
- T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7(2), 245–261 CrossRef CAS PubMed.
- Y. Gao, T. Emge, K. Krogh-Jespersen and A. Goldman, J. Am. Chem. Soc., 2018, 140, 2260–2264 CrossRef CAS PubMed.
- M. Wilklow-Marnell, B. Li, T. Zhou, K. Krogh-Jespersen, W. Brennessel, T. Emge, A. Goldman and W. Jones, J. Am. Chem. Soc., 2017, 139(26), 8977–8989 CrossRef CAS PubMed.
- C. Jia, W. Lu, T. Kitamura and Y. Fujiwara, Org. Lett., 1999, 1(13), 2097–2100 CrossRef CAS.
- H. Huang, P. Bellotti, P. Chen, K. Houk and F. Glorius, Nat. Synth., 2022, 1, 59–68 CrossRef CAS.
- A. Campbell, P. White, I. Guzei and S. Stahl, J. Am. Chem. Soc., 2010, 132(43), 15116–15119 CrossRef CAS PubMed.
- J. Keith and P. Henry, Angew. Chem., 2009, 48(48), 9038–9049 CrossRef CAS.
- X. Jia, L. Frye, W. Zhu, S. Gu and B. Gunnoe, J. Am. Chem. Soc., 2020, 142(23), 10534–10543 CrossRef CAS PubMed.
- W. Zhu and B. Gunnoe, ACS Catal., 2020, 10(19), 11519–11531 CrossRef CAS.
- P. Wang, P. Verma, G. Xia, J. Shi, J. Qiao, S. Tao, P. Cheng, M. Poss, M. Farmer, K. Yeung and J. Yu, Nature, 2017, 551, 489–493 CrossRef CAS PubMed.
- S. Van Minnebruggen, C. Marquez, B. Krasniqi, K. Janssens, N. Van Velthoven, J. Vercammen, B. De Soete, A. Bugaev and D. De Vos, Chem. Commun., 2023, 59(16), 2319–2322 RSC.
- L. Van Emelen, V. Lemmens, C. Marquez, S. Van Minnebruggen, O. Usoltsev, A. Bugaev, K. Janssens, K. Cheung, N. Van Velthoven and D. De Vos, ACS Appl. Mater. interfaces, 2022, 14(46), 51867–51880 CrossRef CAS.
- N. Van Velthoven, S. Waitschat, S. Chavan, P. Liu, S. Smolders, J. Vercammen, B. Bueken, S. Bals, K. Lillerud, N. Stock and D. De Vos, Chem. Sci., 2019, 10, 3616–3622 RSC.
- R. Matsuura, M. Karunananda, M. Liu, N. Nguyen, D. Blackmond and K. Engle, ACS Catal., 2021, 11, 4239–4246 CrossRef CAS PubMed.
- S. Van Minnebruggen, T. De Baerdemaeker, K. Cheung, A. Parvulescu, U. Müller, P. Tomkins, R. de Oliveira-Silva, X. Meng, F. Xiao, T. Yokoi, W. Zhang, D. Sakellariou and D. De Vos, J. Catal., 2022, 406, 206–212 CrossRef.
- K. Cheung, S. Schouterden, S. Van Minnebruggen, P. Tomkins, T. De Baerdemaeker, A. Parvulescu, T. Yokoi and D. De Vos, ChemCatChem, 2023, 15(6), e202300070 CrossRef CAS.
- I. Beckers, A. Bugaev and D. De Vos, Chem. Sci., 2023, 14, 1176–1183 RSC.
- Y. Izawa and S. Stahl, Adv. Synth. Catal., 2010, 352(18), 3223–3229 CrossRef CAS PubMed.
- A. Trzeciak and A. Augustyniak, Coord. Chem. Rev., 2019, 384, 1–20 CrossRef CAS.
- J. Vercammen, M. Bocus, S. Neale, A. Bugaev, P. Tomkins, J. Hajek, S. Van Minnebruggen, A. Soldatov, A. Krajnc, G. Mali, V. Van Speybroeck and D. De Vos, Nat. Catal., 2020, 3(12), 1002–1009 CrossRef CAS.
- I. Beckers, M. Henrion and D. De Vos, ChemCatChem, 2019, 12(1), 90–94 CrossRef.
- S. Gorelsky, D. Lapointe and K. Fagnou, J. Org. Chem., 2012, 77(1), 658–668 CrossRef CAS PubMed.
- J. Vana, J. Bartacek, J. Hanusek, J. Roithova and M. Sedlak, J. Org. Chem., 2019, 84(20), 12746–12754 CrossRef CAS PubMed.
- D. Wang, Y. Izawa and S. Stahl, J. Am. Chem. Soc., 2014, 136(28), 9914–9917 CrossRef CAS PubMed.
- I. Beckers and D. De Vos, iScience, 2023, 26(1), 105790 CrossRef CAS.
- P. Hanley and J. Hartwig, J. Am. Chem. Soc., 2011, 133(39), 15661–15673 CrossRef CAS.
- M. Smith and J. March, March's Advanced Organic Chemistry, Wiley, 2007, pp. 395–417 Search PubMed.
- H. Jaffé, Chem. Rev., 1953, 53(2), 191–261 CrossRef.
- E. Simmons and J. Hartwig, Angew. Chem., 2012, 51(13), 3066–3072 CrossRef CAS PubMed.
- L. Wang and B. Carrow, ACS Catal., 2019, 9(8), 6821–6836 CrossRef CAS PubMed.
- J. Alexander, in The Metal-Carbon Bond, Wiley, 1985, pp. 339–400 Search PubMed.
- P. Liigand, K. Kaupmees, K. Haav, J. Liigand, I. Leito, M. Girod, R. Antoine and A. Kruve, Anal. Chem., 2017, 89(11), 5665–5668 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06686c |
| This journal is © The Royal Society of Chemistry 2025 |