
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGas-flow activation of MOFs: unlocking efficient catalysis through dynamic bonding†
Mariana L.
Díaz-Ramírez‡
ab,
Sun Ho
Park‡
 a,
Marcos
Rivera-Almazo‡
c,
Erika
Medel‡
c,
Ricardo A.
Peralta
c,
Ilich A.
Ibarra
d,
Rubicelia
Vargas
*c,
Jorge
Garza
*c and
Nak Cheon
Jeong
*ab
a,
Marcos
Rivera-Almazo‡
c,
Erika
Medel‡
c,
Ricardo A.
Peralta
c,
Ilich A.
Ibarra
d,
Rubicelia
Vargas
*c,
Jorge
Garza
*c and
Nak Cheon
Jeong
*ab
aDepartment of Physics & Chemistry, DGIST, Daegu 42988, Korea. E-mail: nc@dgist.ac.kr
bCenter for Basic Science, DGIST, Daegu 42988, Korea
cDepartamento de Química, División de Ciencias Básicas e Ingeniería, Universidad Autónoma Metropolitana, Iztapalapa, CP 09340 CDMX, Mexico. E-mail: ruvf@xanum.uam.mx; jgo@xanum.uam.mx
dLaboratorio de Fisicoquímica y Reactividad de Superficies (LaFReS), Instituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior s/n, CU, Del. Coyoacán, 04510, Ciudad de México, Mexico
First published on 23rd December 2024
Abstract
Metal–organic frameworks (MOFs), characterized by dynamic metal–ligand coordination bonding, have pivotal roles in catalysis, gas storage, and separation processes, owing to their open metal sites (OMSs). These sites, however, are frequently occupied by Lewis-base solvent molecules, necessitating activation to expose the OMSs for practical applications. Traditional thermal activation methods involve harsh conditions, risking structural integrity. This study presents a novel ‘gas-flow activation’ technique using inert gases like nitrogen and argon to eliminate these coordinating solvent molecules at low temperatures, thereby maintaining the structural integrity of MOFs. We specifically explored this method with HKUST-1, demonstrating that gas-flow activation at mild temperatures is not only feasible but also superior in efficiency compared to the conventional thermal methods. This approach highlights the potential for safer, more efficient activation processes in MOF applications, making it a valuable addition to the repertoire of MOF activation techniques. This activation function of inert gas flow allows HKUST-1 as a catalyst for the hydrogenation of acetophenone even at room temperature. In addition, it is demonstrated that this ‘gas-flow activation’ is broadly applicable in other MOFs such as MOF-14 and UTSA-76. Furthermore, the findings reveal that dynamic coordination bonding, the repeating transient dissociation-association of solvent molecules at OMSs, are key mechanisms in facilitating this activation, pointing towards new directions for designing activation strategies that prevent structural damage.
Introduction
Metal–organic frameworks (MOFs) are crystalline porous materials composed of inorganic metal nodes connected by organic linkers. Their extensive structural diversity and the ability to finely tune physicochemical properties by modifying linkers or substituting metal nodes render MOFs promising for various applications, including catalysis,1–4 gas storage and separation,5–7 sensing,8–12 energy storage,13–16 and drug delivery.17–19 Notably, approximately 70% of MOFs feature open metal sites (OMSs) within the first coordination sphere of the metal centers, where labile ligands can dissociate, leaving vacant sites crucial for catalytic reactions and gas adsorption.20,21 These OMSs typically bind Lewis-basic (LB) solvent molecules in as-synthesized MOFs,20 reducing their efficiency in such applications, thus necessitating activation to expose the OMSs for practical use. However, developing a method to remove coordinating solvent molecules under mild conditions without damaging the framework remains challenging.The conventional activation of OMSs involves two steps: (i) solvent exchange—from a high-boiling-point solvent like water or N,N′-dimethylformamide (DMF) to a low-boiling-point solvent such as chloroform (trichloromethane, TCM), methylene chloride (dichloromethane, DCM), or methanol (MeOH); and (ii) heating the MOF typically between 120 to 200 °C under vacuum for extended periods (12 to 24 hours). Alternative methods such as freeze-drying22 and supercritical CO2 activation23–25 aim to mitigate damage from surface tension and capillary forces, yet their effectiveness in removing coordinating solvents from metal centers is limited. Consequently, novel room-temperature activation methods have been developed using TCM,26,27 DCM,28 dimethyl ether.29 These methods, especially so-called ‘chemical activation’ with such weakly coordinating solvents, effectively activate OMSs in Cu-HKUST-1 and Ni-MOF-74 by gradually replacing strongly coordinating solvents with weaker ones (see Fig. 1a).26,27 This activation also applies to unstable MOFs like MOF-143 (DUT-34),30 enhancing catalytic activity in processes such as the hydrogenation of acetophenone and acetone, outperforming MOFs with strongly coordinating solvents.31,32 Nevertheless, this chemical activation method has a drawback in that large amounts of TCM, DCM, or dimethyl ether are required to replace the strongly coordinating solvents.
 | ||
| Fig. 1 (a) Schematic illustration of the coordination states of Cu(II) centers with various solvents: strong coordination (DMF, DMSO), intermediate coordination (MeOH, EtOH, H2O), and weak coordination (DCM, TCM). The Cu–Cu paddlewheel node is depicted, showing the dynamic coordination changes. (b) Schematic representation of the dynamic dissociation processes of coordinating H2O molecule at Cu(II) centers, highlighting the role of collision kinetics and electronic interaction with nitrogen. | ||
To overcome these MOF activation techniques, we explored ‘gas-flow activation’, employing inert gases like nitrogen (N2) and argon (Ar) to remove coordinating solvents at mild temperatures, thereby maintaining MOFs' structural integrity and operational efficiency. We hypothesized that this method facilitates the dynamic coordinative bonding of LB solvent molecules, which are in a state of transient dissociation and re-association. In other words, these coordinating solvent molecules in the dynamic state instantaneously dissociate and are subsequently removed by expulsion through inert gas collisions (see Fig. 1b). We confirmed the effectiveness of this process in OMS-possessing MOFs, including HKUST-1, MOF-14, and UTSA-76. Notably, ‘Ar-flow activation’ has proven particularly effective for the catalytic hydrogenation of acetophenone in HKUST-1. This method highlights the potential for safer, more efficient MOF activation processes, emphasizing the dynamic coordination bonding behavior of solvent molecules as a crucial mechanism, thereby pointing towards innovative directions for designing non-destructive activation strategies.
Results and discussion
The dynamicity of coordinating molecules led us to investigate whether these molecules could be dissociated and subsequently removed from the OMSs using a dry inert gas flow at a temperature lower than that used in conventional thermal activation. Our initial approach was to determine whether introducing dry N2 gas could reduce the activation temperature compared to a static atmosphere (i.e., conditions without applying N2 flow or vacuum). For this purpose, we selected HKUST-1 due to its ideal structural characteristics: its distinctive Cu–Cu paddlewheel nodes, where two oxygen atoms from each benzenetricarboxylate (btc) ligand bridge two copper centers, as depicted in Fig. 1a. Furthermore, each Cu center is linked to four oxygen atoms, each from a btc ligand. This specific coordination geometry ensures that all OMSs at Cu centers, where LB solvent molecules can bind, face the large open pore and are thus readily accessible to guest molecules. This structural arrangement results in a strong Raman scattering at the Cu–Cu node, with the band appearing approximately between 165–230 cm−1, varying according to the chemical environment around the Cu centers, such as the presence or absence of coordinating LB solvent, its type, its coordination strength, and its interactions with pore-filling solvent (i.e., hydrogen bonding and dipole interactions). Consequently, the Raman band distinguishes between the open (activated state) and coordinating state of OMSs.Considering this spectroscopic feature, we prepared H2O-loaded HKUST-1 crystals (hereafter referred to as H2O-HK; also see the scanning electron microscope (SEM) image in Fig. S1†), following the methods outlined in Section S1 of ESI.† After verifying the phase and chemical purity of the samples by the powder X-ray diffraction (PXRD) pattern and 1H nuclear magnetic resonance (NMR) spectrum of H2O-HK (Fig. S2†), we measured the Raman spectrum of the H2O-HK sample. Subsequently, we monitored changes in the Cu state (H2O-coordination or coordination-free state) at varying temperatures, continuously supplying N2 at a rate of 50 mL min−1 (Fig. S3†). Initially, the H2O-HK exhibited a characteristic Raman band at 175 cm−1, indicative of the presence of both coordinating and pore-filling H2O molecules. After 30 minutes of N2 flow at 25 °C, the Raman band shifted to 166 cm−1, suggesting the evacuation of pore-filling H2O molecules while leaving only the coordinating H2O molecules. After continuing the N2 flow for 120 minutes at 25 °C, the band shifted to 230 cm−1, indicating complete OMS activation. It is noteworthy that while static thermal activation without N2 flow required 45 min at 90 °C (Fig. S4†), N2-flow activation achieved the same result in just 5 min at a temperature of 40 °C (Fig. S3†). This result indicates that the N2-flow method can conserve the energy with a gain of approximately 40 J g−1 compared to the static thermal method (Section S5†).
To demonstrate that this N2-flow activation could be applied to other solvent coordinations, we prepared HKUST-1 crystals coordinating with TCM, DCM, MeOH, MeCN, EtOH, DMF, and DMSO (hereafter TCM-HK, DCM-HK, MeOH-HK, MeCN-HK, EtOH-HK, DMF-HK, and DMSO-HK, respectively). We then monitored the changes in the Raman band of each sample at different temperatures, applying dry N2 flow (Fig. S5–S11†). Our findings revealed that the lowest activation temperatures for each sample correlate strongly with the coordination strength of the LB solvent, as observed in previous studies, where the Cu–Cu vibrational Raman band closely relates to the solvent's coordination strength in the order: TCM ≤ DCM < H2O < MeOH ≈ MeCN ≤ EtOH < DMF (Fig. 2).32,33 Thus, TCM and DCM are the most labile solvents for dissociating from Cu centers, while DMF and DMSO pose the greatest challenge (DMSO-HK were not fully activated even at 180 °C; Fig. S11†). To gain a deeper understanding of the coordination strength, we calculated the interaction energies of the solvents at Cu(II) OMS using the DFT method (Fig. 3, S12 and Tables S1–S5†). The DFT results closely agree with our experimental findings, except for MeOH, which displays an exceptionally high interaction energy due to non-conventional hydrogen bonding between the framework oxygen and methanol's C–H hydrogen. Although such hydrogen bonding does not significantly influence the actual dissociation ability, the relatively high activation temperatures of EtOH-HK and DMF-HK compared to their coordination energies can be attributed to their large molecular sizes, which impact their diffusion through the pores. To corroborate these DFT findings and further explore non-conventional hydrogen bondings, we also calculated the electron density (ρBCP) and density energy (HBCP) at the bond critical point between the coordinating atom in the solvent molecule and the OMS using the theoretical package, Atoms in Molecules (AIM) (Tables S6–S13†). The AIM results generally align with the DFT results, although some calculated values appear somewhat erratic.
 | ||
| Fig. 2 (a) Schematic illustration of the Raman spectra change showing the Cu–Cu stretching vibration at 230 cm−1 and 175 cm−1 for different coordination states of Cu(II) induced by the activation. (b) Activation temperatures determined by in situ Raman spectroscopy under N2 flow (blue columns) and static conditions (green column) for various solvents. The graph highlights the transition of activation temperature for H2O-coordinating HKUST-1 from static (90 °C) to N2 flow conditions (40 °C). | ||
 | ||
| Fig. 3 DFT-calculated interaction energies for the coordination of various solvent molecules at Cu(II) OMS per unit cell of HKUST-1, indicating the coordination strength. | ||
To test our hypothesis that another inert gas can facilitate the removal of coordinating solvents by collisional expulsion, we conducted gas-flow activation using argon (Ar, Mm = 39.948 g mol−1). Like the N2-flow experiments, we monitored the dissociation of solvent molecules under dry Ar flow using Raman analysis with samples of H2O-HK, EtOH-HK, DMF-HK, and DMSO-HK (Fig. S13–S16†). Compared to N2-flow activation, Ar-flow activation significantly reduced the activation temperature—from 40 to 25 °C for H2O-HK and from 90 to 60 °C for EtOH-HK (Fig. 4)—attributable to the 42% higher kinetic energy of Ar (4.41 × 10−11 J s−1) than that of N2 (3.09 × 10−11 J s−1) (Section S9†), along with their similar polarizabilities (1.6411 and 1.7403 × 10−24 cm3 for Ar and N2, respectively).34 However, due to their high boiling points and strong coordination strengths, neither DMF-HK nor DMSO-HK samples were effectively activated using Ar-flow.
 | ||
| Fig. 4 (a) Schematic illustrations of the dynamic coordination bonding of H2O molecule and its dissociation process under N2 (left) and Ar flow (right) conditions. (b) Activation temperatures of H2O-, EtOH-, and DMF-coordinated to HKUST-1 under N2 (blue columns) and Ar (pink columns) flow conditions, showing the impact of different inert gases on the activation temperature. | ||
We also considered whether gas-flow activation might be achieved through coordination exchange, where N2 or Ar molecules instantaneously coordinate at the OMSs after displacing pre-coordinating solvent molecules. To investigate this possibility, we conducted Raman spectroscopy with a coordination-free activated HKUST-1 sample while infusing N2 and Ar gas into the quartz holder (Fig. S17†). We also performed control experiments under vacuum conditions (0.1 mbar) for comparison. Although we hypothesized that N2 or Ar coordination at the OMS might cause a redshift in the Cu–Cu vibration Raman band from 230 cm−1, the band remained at 230 cm−1 in all tests, indicating that neither N2 nor Ar coordinates at the OMS, thus supporting the hypothesis that the transportation of transiently dissociated solvent molecules by collision is the most plausible mechanism, rather than coordination exchange.
Based on the above understanding and considering the two places where solvent molecules reside (the crystal exterior and interior), we propose two distinct mechanisms for the gas-flow activation process. The removal of non-coordinating solvent molecules on the crystal exterior occurs through direct collision with inert gas molecules, which possess high translational speed due to convection. In contrast, the removal of coordinating solvent molecules within the crystal interior proceeds in two steps: (1) dissociation of coordinating molecules by the collision with inert gas molecules and (2) diffusion of the dissociated molecules to the crystal surface, driven by a decreasing concentration gradient, after exterior drying.
Additionally, we explored the loss of coordinating solvent molecules from HKUST-1 using the thermogravimetric analysis (TGA), maintaining the sample at a stable lower temperature until weight loss stabilized, then incrementally raising the temperature to 25, 40, 50, 60, 90, 120, 150, and 180 °C. As depicted in Fig. S18,† the weight loss of DCM-HK, TCM-HK, H2O-HK, and MeOH-HK samples reached a plateau at 90 °C, while that of MeCN-HK stabilized at 120 °C, EtOH-HK at 150 °C, DMF-HK at over 180 °C, and DMSO-HK at over 180 °C. Although this thermal analysis does not precisely distinguish between pore-filling and coordinating solvents, we calculated the stoichiometric amounts of each solvent associated with the Cu centers based on their final masses (Tables S14 and S15†). Accordingly, the temperatures at which solvents began dissociating from the OMSs—25 °C for DCM and TCM, 40 °C for H2O, 50 °C for MeOH and MeCN, and 60 °C for EtOH—correspond closely to those observed in the Raman experiments.
While these results confirm the efficacy of gas-flow activation at the Cu centers, such experiments have thus far been conducted on single crystals or small milligram-scale samples. Therefore, we aimed to examine larger, bulk samples of more than 200 mg of HKUST-1 by employing N2 adsorption isotherms and testing BET surface areas with EtOH-HK samples activated thermally at 180 °C and by Ar-flow at rates of 50 mL min−1 (Thermal-Act_EtOH-HK and Ar-Act_EtOH-HK, respectively). For the Ar-Act_EtOH-HK samples, we controlled the activation temperature at 25, 40, and 90 °C to compare the degree of activation. The results showed that Thermal-Act_EtOH-HK (180 °C) and Ar-Act_EtOH-HK (90 °C) yielded very similar BET-specific surface areas of 1992 and 1907 m2 g−1, respectively, as illustrated in Fig. 5a. However, the Ar-Act_EtOH-HK samples activated at 40 and 25 °C exhibited significantly reduced surface areas of 1350 and 948 m2 g−1, respectively, which are 29% and 50% lower, respectively, than the Ar-Act_EtOH-HK sample activated at 90 °C. In these Ar-activated samples, the total pore volume increased from 0.358 to 0.520 and up to 0.775 cm3 g−1 depending on the activation temperature (25, 40, or 90 °C, respectively), as depicted in Fig. 5b. As corroborated by other studies, these lower BET surface areas and total pore volumes undoubtedly resulted from remaining EtOH molecules in the sample after Ar-flow at the lower temperatures (25 and 40 °C).35,36 For instance, Ar-Act_EtOH-HK (25 and 40 °C) contained 1.13 and 0.93 EtOH molecules per Cu center, respectively, while Ar-Act_EtOH-HK (90 °C) contained only 0.14 EtOH molecules per Cu center, as confirmed by 1H NMR (Fig. S19a†). Nonetheless, after N2 sorption measurements, all samples retained their crystallinity (Fig. S19b†).
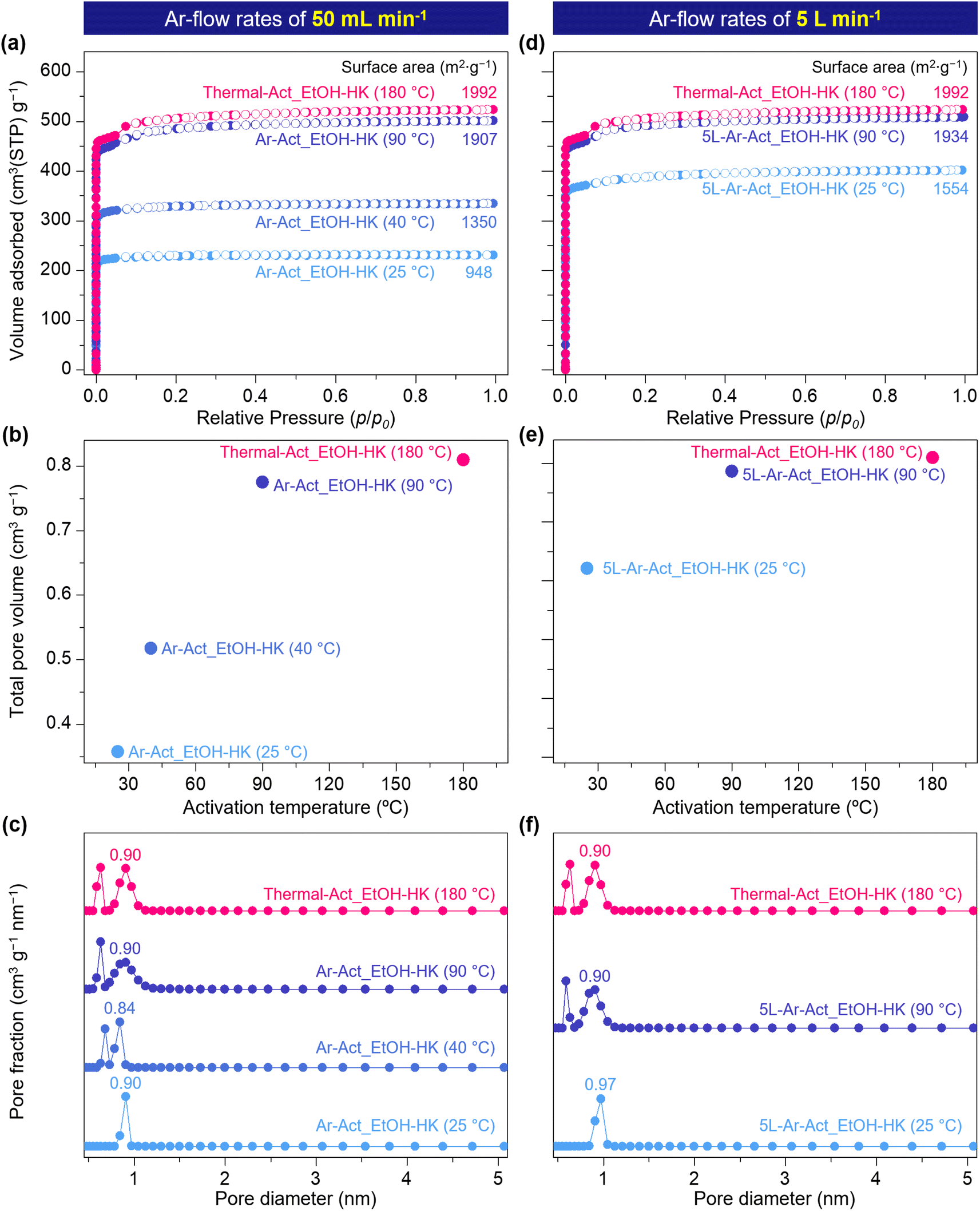 | ||
| Fig. 5 (a and d) N2 adsorption/desorption isotherms, (b and e) plot of total pore volume as a function of activation temperature, and (c and f) NLDFT calculation for the pore size distribution of EtOH-HKUST-1 under different activation conditions: thermal activation at 180 °C (pink), Ar activation at 90 °C (dark blue), Ar activation at 40 °C (blue), and Ar activation at 25 °C (light blue) after (a–c) 50 mL min−1 and (d–f) 5 L min−1 Ar-flow activation. | ||
To further evaluate the effect of the kinetic energy of the gas flow on the activation of HKUST-1, we conducted bulk activation experiments using an Ar flow rate of 5 L min−1 at 25 and 90 °C [5L-Ar-Act_EtOH-HK (25 °C) and 5L-Ar-Act_EtOH-HK (90 °C)]. At the higher flow rate (higher kinetic energy), the BET surface area and total pore volume of 5L-Ar-Act_EtOH-HK at 25 °C increased substantially by 64% and 79%, respectively (1554 m2 g−1 and 0.62 cm3 g−1, as shown in Fig. 5d–f), compared to Ar-Act_EtOH-HK, which was tested under a lower flow rate. Despite these improvements, the BET surface area and total pore volume for 5L-Ar-Act_EtOH-HK (25 °C) remained significantly lower than those of Thermal-Act_EtOH-HK (180 °C), indicating that some EtOH molecules were still coordinated to Cu centers. According to 1H NMR results, 0.37 EtOH molecules per Cu center remained in the sample after flowing Ar at 5 L min−1 for 24 hours (Fig. S20†). Moreover, 5L-Ar-Act_EtOH-HK (25 °C) exhibited only pores with a size of around 0.97 nm, with no narrower pores present (Fig. 5f), similar to the case with Ar-Act_EtOH-HK (25 °C). By contrast, 5L-Ar-Act_EtOH-HK (90 °C) showed the closest BET surface area and total pore volume values (1934 m2 g−1 and 0.78 cm3 g−1), along with a pore size distribution very similar to Thermal-Act_EtOH-HK (180 °C), highlighting the complete removal of EtOH molecules, as confirmed by 1H NMR (Fig. S20†). These results indicate that a flow of dry Ar at a moderate temperature (90 °C) effectively activates the OMSs in HKUST-1 without causing structural damage. Additionally, a comparable BET surface area to Thermal-Act_EtOH-HK (180 °C) can only be achieved with a high Ar flow rate (5 L min−1) due to its greater efficacy in dissociating EtOH molecules from Cu centers as a result of its higher kinetic energy (4.91 × 10−5 J s−1) compared to the lower flow rate (50 mL min−1, 4.41 × 10−11 J s−1).
Meanwhile, we hypothesized that Ar-Act_EtOH-HK (90 °C) would show high catalytic activity as effectively as Thermal-Act_EtOH-HK (180 °C) in the catalytic hydrogenation of acetophenone at room temperature, given that both had fully activated OMSs. As a result, Ar-Act_EtOH-HK (90 °C) exhibited high conversion efficiencies of 95% within 60 min, with a very similar profile to Thermal-Act_EtOH-HK (180 °C), whereas MeCN-HK, with Cu sites occupied by MeCN coordination, yielded a low conversion efficiency (∼10%, Fig. 6). However, we observed that the framework of the HKUST-1 completely collapsed after the reaction as reported in literature,37 indicating its limitation in recycling the catalyst (see PXRD analysis in Fig. S21†). To expand the above results, we examined additional experiments using MeCN-MOF-14 and MeCN-UTSA-76 that possess crystal sizes similar to HKUST-1 (Fig. S1 and S22†). As a result, Ar-Act_MeCN-MOF-14 exhibited higher catalytic activity compared to non-activated MeCN-MOF-14, demonstrating that Ar-flow activation enhances the catalytic performance of MOF-14 effectively. However, we found that MeCN-UTSA-76 was challenging due to its intrinsic low catalytic efficiency. These results support our hypothesis that the gas-flow activation can expose a significant number of OMSs under mild conditions; thus, it is a pretty effective activation method. These findings are promising, suggesting potential applications in an integrated experimental setup where in situ activation under inert gas flow followed by gas phase reactions can be conducted efficiently.
 | ||
| Fig. 6 (a) Schematic representation of the catalytic hydrogenation of acetophenone to 1-phenylethanol using HKUST-1. (b) Conversion rates of acetophenone to 1-phenylethanol over time for MeCN-activated HKUST-1 under different activation conditions: thermal activation at 180 °C (purple), Ar activation at 90 °C (pink), and non-activated MeCN-HKUST-1 (black). The graph demonstrates the enhanced catalytic performance of the Ar-flow-activated HKUST-1 sample. | ||
To explore whether this gas-flow activation method could broadly apply to other MOFs, we further extended our investigation to include MOF-14 and UTSA-76. In alignment with our hypotheses, when subjected to an Ar-flow activation protocol, both MOFs demonstrated complete activation at significantly lower temperatures of 40 °C, compared to the higher temperature required under static air conditions (90 °C for MOF-14 and 60 °C for UTSA-76) (Fig. 7 and S23–S27†). This experiment underscores the potential of the gas-flow activation techniques in enhancing the activation efficiency, suggesting a promising strategy for optimizing the performance of these materials in various applications.
 | ||
| Fig. 7 Activation temperatures of EtOH-coordinated HKUST-1 (EtOH-HK), MOF-14 (EtOH-MOF-14), and UTSA-76 (EtOH-UTSA-76) under static conditions (green columns) and Ar-flow conditions (pink columns). The graph highlights the differences in activation temperatures for each MOF, indicating the efficiency of Ar flow in lowering the activation temperature compared to static conditions. | ||
Conclusions
This study introduces the dynamic coordinative bonding behavior of OMS-coordinating solvent molecules despite their different coordinative strength and a subsequent innovative inert gas-flow activation method for MOFs using N2 and Ar, which significantly enhances activation efficacy under milder conditions compared to traditional methods. By employing inert gases to remove coordinating solvents at lower temperatures, this technique preserves the structural integrity of MOFs and expedites activation processes without the need for high temperatures or vacuum conditions. Our findings demonstrate that this method can rapidly activate OMSs, as shown through successful applications in HKUST-1, MOF-14, and UTSA-76. The technique's effectiveness across various copper-based MOFs suggests its broad applicability for improving MOF performance in catalysis, gas storage, and separation. Future research will explore extending this method to other MOF types and assessing its industrial scalability. The potential for this gas-flow activation to revolutionize MOF preparation and utilization offers promising avenues for advanced environmental and energy applications.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
M. L. D.-R. and S. H. P. designed and conducted all experiments under the supervision of N. C. J., M. R.-A., E. M., R. A. P., I. A. I., R. V., and J. G. performed the theoretical study. M. L. D.-R. and S. H. P. initially drafted the manuscript, and it was subsequently revised by N. C. J. All authors approved the final version of the manuscript. M. L. D.-R., S. H. P., M. R.-A., and E. M. contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministry of Science and ICT (MSIT) of Korea under the auspices of the Basic Science Research Program sponsored by the National Research Foundation (NRF-2023R1A2C2004838) and by the DGIST R&D Program (25-HRHR+-01). M. L. D.-R. thanks F. B. Z. for the scientific discussions. The authors thank the facilities provided by the Laboratorio de Supercómputo y Visualización en Paralelo at the Universidad Autónoma Metropolitana-Iztapalapa. EM and M. R.-A. thanks CONAHCyT, México for PhD grants 709721 and 567379.Notes and references
- H. Han, X. Zheng, C. Qiao, Z. Xia, Q. Yang, L. Di, Y. Xing, G. Xie, C. Zhou, W. Wang and S. Chen, ACS Catal., 2022, 12, 10668–10679 CrossRef CAS.
- D. Mukherjee, B. Van der Bruggen and B. Mandal, Chemosphere, 2022, 295, 133835 CrossRef CAS PubMed.
- T. K. Pal, D. De and P. K. Bharadwaj, Fuel, 2022, 320, 123904 CrossRef CAS.
- L. Liu, S. Du, Y. Xiao, X. Guo, S. Jin, G. Shao and F. Zhang, Appl. Catal., B, 2023, 338, 123094 CrossRef CAS.
- A. N. Hong, H. Yang, X. Bu and P. Feng, EnergyChem, 2022, 4, 100080 CrossRef CAS.
- J. Pei, X. W. Gu, C. C. Liang, B. Chen, B. Li and G. Qian, J. Am. Chem. Soc., 2022, 144, 3200–3209 CrossRef CAS PubMed.
- K. Qu, K. Huang, J. Xu, L. Dai, Y. Wang, H. Cao, Y. Xia, Y. Wu, W. Xu, Z. Yao, X. Guo, C. Lian and Z. Xu, Angew. Chem., Int. Ed., 2022, 61, e202213333 CrossRef CAS PubMed.
- P. Qin, B. A. Day, S. Okur, C. Li, A. Chandresh, C. E. Wilmer and L. Heinke, ACS Sens., 2022, 7, 1666–1675 CrossRef CAS.
- Y. Shen, A. Tissot and C. Serre, Chem. Sci., 2022, 13, 13978–14007 RSC.
- T. Wu, X. J. Gao, F. Ge and H. G. Zheng, CrystEngComm, 2022, 46, 7881–7901 RSC.
- S. Kamal, M. Khalid, M. S. Khan and M. Shahid, Coord. Chem. Rev., 2023, 474, 214859 CrossRef CAS.
- X. Su, Z. Zhong, X. Yan, T. Zhang, C. Wang, Y. X. Wang, G. Xu and L. Chen, Angew. Chem., Int. Ed., 2023, 62, e202302645 CrossRef CAS PubMed.
- P. E. Lokhande, S. Kulkarni, S. Chakrabarti, H. M. Pathan, M. Sindhu, D. Kumar, J. Singh, A. Kumar, Y. Kumar Mishra, D. C. Toncu, M. Syväjärvi, A. Sharma and A. Tiwari, Coord. Chem. Rev., 2022, 473, 214771 CrossRef CAS.
- F. Shahbazi Farahani, M. S. Rahmanifar, A. Noori, M. F. El-Kady, N. Hassani, M. Neek-Amal, R. B. Kaner and M. F. Mousavi, J. Am. Chem. Soc., 2022, 144, 3411–3428 CrossRef CAS PubMed.
- G. Xu, C. Zhu and G. Gao, Small, 2022, 18, 2203140 CrossRef CAS.
- J. Yin, N. Li, M. Liu, Z. Li, X. Wang, M. Cheng, M. Zhong, W. Li, Y. Xu and X. H. Bu, Adv. Funct. Mater., 2023, 33, 2211950 CrossRef CAS.
- E. Linnane, S. Haddad, F. Melle, Z. Mei and D. Fairen-Jimenez, Chem. Soc. Rev., 2022, 51, 6065–6086 RSC.
- N. Rabiee, M. Atarod, M. Tavakolizadeh, S. Asgari, M. Rezaei, O. Akhavan, A. Pourjavadi, M. Jouyandeh, E. C. Lima, A. Hamed Mashhadzadeh, A. Ehsani, S. Ahmadi and M. R. Saeb, Microporous Mesoporous Mater., 2022, 335, 111670 CrossRef CAS.
- T. Cui, Y. Zhang, G. Qin, Y. Wei, J. Yang, Y. Huang, J. Ren and X. Qu, Nat. Commun., 2023, 14, 1974 CrossRef CAS PubMed.
- J. N. Hall and P. Bollini, React. Chem. Eng., 2019, 4, 207–222 RSC.
- U. Kökçam-Demir, A. Goldman, L. Esrafili, M. Gharib, A. Morsali, O. Weingart and C. Janiak, Chem. Soc. Rev., 2020, 49, 2751–2798 RSC.
- L. Ma, A. Jin, Z. Xie and W. Lin, Angew. Chem., Int. Ed., 2009, 48, 9905–9908 CrossRef CAS PubMed.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. O. Yazaydin and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS PubMed.
- O. K. Farha, C. E. Wilmer, I. Eryazici, B. G. Hauser, P. A. Parilla, K. O'Neill, A. A. Sarjeant, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 9860–9863 CrossRef CAS.
- T. C. Wang, W. Bury, D. A. Gomez-Gualdron, N. A. Vermeulen, J. E. Mondloch, P. Deria, K. Zhang, P. Z. Moghadam, A. A. Sarjeant, R. Q. Snurr, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 3585–3591 CrossRef CAS.
- J. Bae, J. S. Choi, S. Hwang, W. S. Yun, D. Song, J. Lee and N. C. Jeong, ACS Appl. Mater. Interfaces, 2017, 9, 24743–24752 CrossRef CAS PubMed.
- J. S. Choi, J. Bae, E. J. Lee and N. C. Jeong, Inorg. Chem., 2018, 57, 5225–5231 CrossRef CAS PubMed.
- H. K. Kim, W. S. Yun, M. B. Kim, J. Y. Kim, Y. S. Bae, J. Lee and N. C. Jeong, J. Am. Chem. Soc., 2015, 137, 10009–10015 CrossRef CAS PubMed.
- K. R. Wright, K. Nath and A. J. Matzger, Angew. Chem., Int. Ed., 2022, e202213190 CAS.
- J. Bae, C. Y. Lee and N. C. Jeong, Bull. Kor. Chem. Soc., 2021, 42, 658–666 CrossRef CAS.
- J. Bae, S. H. Park, D. Moon and N. C. Jeong, Commun. Chem., 2022, 5, 51 CrossRef CAS PubMed.
- S. H. Park, R. A. Peralta, D. Moon and N. C. Jeong, J. Mater. Chem. A, 2022, 10, 23499–23508 RSC.
- J. Bae, E. J. Lee and N. C. Jeong, Chem. Commun., 2018, 54, 6458–6471 RSC.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, 2016 Search PubMed.
- W. Qiu, Y. Wang, C. Li, Z. Zhan, X. Zi, G. Zhang, R. Wang and H. He, Chin. J. Catal., 2012, 33, 986–992 CrossRef CAS.
- J. Bentley, G. S. Foo, M. Rungta, N. Sangar, C. Sievers, D. S. Sholl and S. Nair, Ind. Eng. Chem. Res., 2016, 55, 5043–5053 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Catal. Commun., 2017, 97, 74–78 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07011a |
| ‡ M. L. D.-R., S. H. P., M. R.-A., and E. M. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |