
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrode engineering considerations for high energy efficiency Li–CO2 batteries†
Jingzhao
Wang
a,
Xin
Chen
a,
Xiangming
Cui
a,
Mi
Zhou
a,
Jianan
Wang
 *a,
Wenbiao
Liu
c,
Hang
Ma
c,
José V.
Anguita
b,
S. Ravi P.
Silva
b,
Kai
Yang
*b and
Wei
Yan
a
*a,
Wenbiao
Liu
c,
Hang
Ma
c,
José V.
Anguita
b,
S. Ravi P.
Silva
b,
Kai
Yang
*b and
Wei
Yan
a
aDepartment of Environmental Science and Engineering, State Key Laboratory of Multiphase Flow in Power Engineering, School of Energy and Power Engineering, Xi'an Jiaotong University, Xi'an 710049, China. E-mail: wangjn116@xjtu.edu.cn
bAdvanced Technology Institute, University of Surrey, Guildford, Surrey GU2 7XH, UK. E-mail: kai.yang@surrey.ac.uk
cCenter of Research & Development, Yunnan Yuntianhua Co., Ltd, 1417 Dianchi Road, Kunming 650228, China
First published on 20th January 2025
Abstract
Li–CO2 batteries (LCBs) offer significant potential for high energy storage and efficient CO2 utilization. However, their practical application is hindered by challenges such as low energy efficiency, poor rate performance, and limited cycle life. To address these issues, it is crucial to develop gas electrodes with a highly conductive, catalytic, and robust network to facilitate rapid and reversible CO2 conversion. In this work, a comprehensive design for high-performance gas electrodes in LCBs is presented. The critical structure–property relationships of gas electrodes have been investigated with a focus on optimal substrate and catalytic site construction. The developed self-supporting electrodes, featuring ultrafine nanocatalyst decoration within a hierarchical porous and conductive structure, exhibited superior electrochemical performance, including ultrahigh areal capacity (over 10 mA h cm−2), excellent reversibility, and high energy efficiency (over 80%) under practical operating conditions. Furthermore, flexible Li–CO2 pouch cells were successfully fabricated, showing stable operation and high tolerance to mechanical stress, indicating significant potential for large-scale applications in high-energy-density flexible power devices. The principles and guidelines established for gas electrode design are expected to advance the development of superior LCBs and other catalyst-based energy systems.
1 Introduction
Li–CO2 batteries (LCBs), with CO2 as the direct reactant, have been regarded as an emerging sustainable energy technology with the dual function of CO2 conversion and utilization. Based on the unique CO2 conversion process (3CO2 + 4Li ↔ 2Li2CO3 + C), LCBs can deliver a discharge voltage of ∼2.8 V (vs. Li/Li+) and a high theoretical energy density of 1876 W h kg−1 (three times higher than that of conventional lithium-ion batteries).1 The great potential to utilize CO2 in high-density energy storage devices makes the LCBs a promising technology for future Mars and deep-sea exploration.2,3The development of LCBs is still relatively new with several major research limitations such as high polarization and low energy efficiency, poor recyclability and short cycle life, and poor rate performance.4,5 To address the above challenges, it is imperative to develop an efficient electrocatalyst to facilitate fast and reversible reactant conversion in both CO2 reduction reaction (CO2RR) and CO2 evolution reaction (CO2ER) processes.6 Extensive research has been conducted to produce suitable electrocatalysts that optimise the performance of LCBs,7,8 using a wide range of materials. Carbon nanomaterials (graphene, porous activated carbon, carbon nanotubes, etc.) to transition metal compounds (oxides, carbides, etc.) and noble metals/alloys (Pt, Ir, Ru, etc.) have been widely researched.9–11 However, despite recent advancements, practical LCB performance parameters such as specific areal capacity and operating current density are still unable to fulfil the requirements for realistic application scenarios.12 There is therefore a need to investigate and overhaul the systematic design of gas electrodes for performance optimization.
The typical gas electrode configuration is composed of several key components, including the porous supporting substrate (e.g., carbon paper, carbon fibres, copper foam, etc.), electrocatalyst, conductive agent (e.g., acetylene black) and binder (e.g., polyvinylidene difluoride).13 This conventional gas electrode structure has several drawbacks.14 The binder, as an inactive ingredient, may trigger undesirable side reactions or passivate catalytic sites. The gradual detachment of the electrocatalyst from the substrate due to weak adhesion15 could lead to diminished electrochemical performance such as fast capacity decay and reduced cycle life. In order to address these issues, an effective strategy is to design self-supporting gas cathodes to improve catalytic efficiency and maintain electrode integrity.16,17 Ideally, the self-supporting electrodes must provide abundant catalytic sites with optimized electrocatalyst utilization, hierarchical pore size distribution and interfacial affinity for rapid mass diffusion, sufficient conductive networks and a robust mechanical support. The various physicochemical properties (e.g., porosity, catalytic, gas and electrolyte permeability, electrical conductivity, etc.) of gas electrodes significantly affect the electrochemical performance of LCBs.18 It is therefore crucial to correlate LCB performance with physicochemical properties of gas electrodes and achieve a more informed and efficient practical optimization.
Herein, we provide a systematic evaluation of gas electrodes and demonstrate fast electrode engineering for high energy efficiency LCBs. Key parameters in the gas electrode design have been elucidated. With the ultrafast and controllable electrocatalyst engineering strategy, an ultrafine nanoparticle electrocatalyst has been effectively loaded onto the robust substrate. Optimized graphene/carbon nanotube aerogel-based gas electrodes with a three-dimensional conductive and sufficient catalytic framework have been constructed. Superior LCB performance can be achieved with ultrahigh areal discharge capacity (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 247 μA h cm−2) at a current density of 20 μA cm−2, excellent reversibility and energy efficiency (82%), and high stability. We believe that the demonstrated electrode design principle and regulation strategy will inspire broad electrocatalyst-based energy applications.
247 μA h cm−2) at a current density of 20 μA cm−2, excellent reversibility and energy efficiency (82%), and high stability. We believe that the demonstrated electrode design principle and regulation strategy will inspire broad electrocatalyst-based energy applications.
2 Experimental section
2.1 Cathode preparation
2.2 Characterization
Field emission scanning electron microscopy (FE-SEM) images were obtained using a GeminiSEM 500 (China) microscope. Field Emission Transmission Electron Microscopy (FE-TEM) and energy dispersive spectroscopy (EDS) mapping were conducted on a JEOL JEM-F200 (Japan) instrument. Time of flight secondary ion mass spectrometry (TOF-SIMS) surface scanning images were taken on a TOF-SIMS-M6 (Germany). Fourier transform infrared (FTIR) spectra were collected on a BRUKER TENSOR 37 FTIR (Germany, ATR Kit) spectrometer. X-ray diffraction (XRD) patterns were collected on a SHIMADZU XRD-6000 diffractometer using Cu-Kα radiation. Brunauer–Emmett–Teller (BET) specific surface area analysis and Barrett–Joyner–Halenda (BJH) pore size and volume analysis were performed on a JW-BK200B JWGB (China) instrument. X-ray photoelectron spectroscopy (XPS) measurements were carried out on a Kratos Axis Ultra (England) instrument using monochromatic Al Kα radiation (150 W, 15 kV and 1486.6 eV) at 10−9 torr. Raman measurements were taken on a Renishaw InVia Raman microscope with a 532 nm laser.2.3 Li–CO2 battery assembling and electrochemical measurements
2032-type coin batteries were assembled with the self-supported gas electrode as the cathode (cathode cases have several holes for CO2 diffusion), Whatman® glass microfiber membranes (Sigma-Aldrich) as the separator, 1 M LiTFSI in TEGDME as the electrolyte (DoDoChem, SuZhou), and lithium foil (16 mm) as the anode. The prepared cathode electrodes were directly used as the self-supporting cathode without any binder. The batteries were assembled in an Ar-filled glove box (O2 < 0.1 ppm; H2O < 0.1 ppm) and measured in a homemade atmosphere control chamber filled with high-purity CO2.The Li–CO2 pouch cell was assembled with a Pt@GCA cathode (2 × 3 cm), with lithium foil pressed on the copper mesh as the anode, and finally sealed with aluminium–plastic films with six punched holes at the cathode side.
The galvanostatic charge/discharge (GCD) tests were carried out on a LAND-CT2001A (Wuhan, China) battery test system. A CHI 660D electrochemical workstation (Shanghai, China) was used to record cyclic voltammograms (CV) with a scan rate of 0.1 mV s−1 and for electrochemical impedance spectroscopy (EIS) experiments with an impedance frequency range of 0.01–100 kHz. All batteries were rested in a pure CO2 atmosphere for 10 h prior to testing.
3 Results and discussion
3.1 Self-supporting substrate evaluation
The working principle of LCBs is much more intricate than the intercalation reaction in conventional LIBs. The operation of LCBs involves multiple processes such as gas–solid conversion reaction and discharge product evolution, with distinct electrode morphology changes. As the main location of multi-interface (CO2, electrolyte, solid catalyst) reactions, the optimization of gas electrodes with sufficient catalytic sites and suitable pore structures is crucial for improving the electrochemical performance of LCBs (Fig. 1). An ideal gas electrode for practical LCBs is expected to possess the following characteristics: (1) abundant pore structure to ensure rapid mass transfer and adequate surface area to accommodate discharging products; (2) high conductivity to promote fast electron transfer under high current operation; (3) excellent mechanical stability to maintain the electrode integrity during manufacturing and in harsh operational environments; and (4) highly efficient catalytic sites to accelerate both CO2 reduction reaction (CO2RR) and CO2 evolution reaction (CO2ER) processes. Self-supporting electrodes (e.g., carbon aerogels, carbon nanotube films, foam metals, etc.) have been widely developed as promising gas electrodes for metal–gas batteries.19–21 A typical configuration for self-supporting electrodes includes the conductive substrate and electrocatalyst loading. Physicochemical properties of the substrate, such as morphology, porosity, conductivity, and wettability, significantly affect the electrochemical performance of LCBs.22 Based on the design principles of an ideal gas electrode, we started by evaluating the suitable conductive substrate for the self-supporting electrodes. Key evaluation parameters are summarized in Table S1.†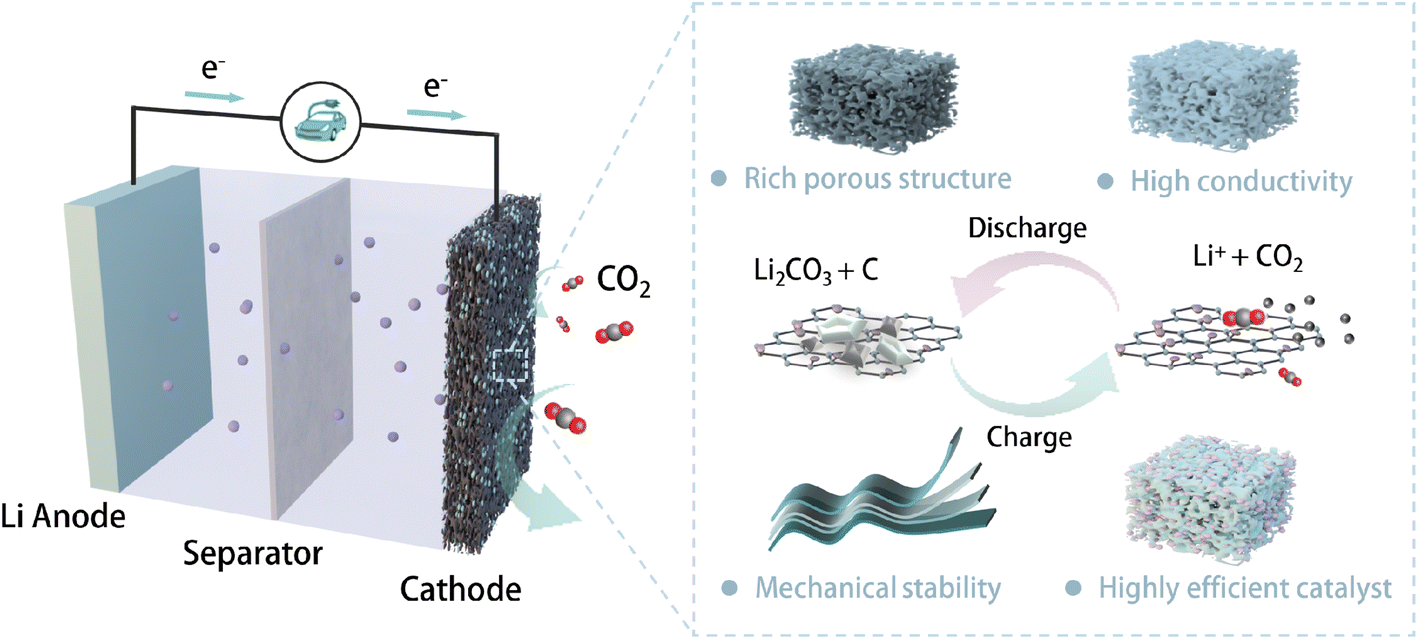 | ||
| Fig. 1 Schematic illustration of a typical reversible Li–CO2 battery structure and desirable properties for an ideal gas electrode. | ||
The graphene–carbon nanotube aerogel (GCA) with a three-dimensional (3D) porous structure was synthesized with hydrothermal and freeze-drying methods. As the pure graphene-based aerogel obtained through hydrothermal reduction may suffer from severe self-aggregation and result in a less stable internal 3D porous structure, 1D carbon nanotubes were added during the hydrothermal process to enhance the long-distance conductivity and mechanical strength.23 Commonly used flexible substrates, including CNTs and electrospun PAN-based CNFs, were also compared. Fig. S3† shows the digital photographs of the three substrates. All of them displayed favourable flexibility and mechanical stability. The microstructure of the three substrates was characterized using a scanning electron microscope (SEM). An interconnected 3D porous structure was created in the GCA substrate as shown in Fig. 2a. Carbon nanotubes cross-linked the wrinkled graphene and mitigated agglomeration of graphene sheets, hence constructing a 3D porous conductive network. By contrast, CNT and CNF films were constituted of 3D interconnected fibre structures as shown in Fig. 2b and c. Differently, the diameter of a single carbon nanotube fibre was only about 50 nm, while the diameter was around 200 nm for the CNF. The nitrogen adsorption–desorption test indicated that the GCA has the largest Brunauer–Emmett–Teller (BET) surface area of 92.4 m2 g−1 (Fig. 2d and S4†). The pore size distribution in Fig. S5† shows that the GCA exhibited a hierarchical porous structure with the pore size concentrated below 10 nm. The hierarchical porosity is beneficial for providing both rapid mass transfer and sufficient reaction surface. Electrolyte wettability significantly affects the interfacial interaction and charge transfer resistance for metal–gas batteries.24,25 Contact angle analysis was conducted to evaluate the electrolyte wettability of various substrates (Fig. S6†). The GCA demonstrates the lowest contact angle (9.5°), which is much smaller than those of CNT (27.7°) and CNF films (24.4°). This high electrode/electrolyte affinity of the GCA substrate is expected to facilitate interfacial charge transfer. Fig. 2e shows the Raman spectra of the GCA, CNT and CNF. All samples show the D (∼1340 cm−1) and G (∼1580 cm−1) Raman bands, which are characteristic of carbon. The transition that gives rise to the D-band is Raman inactive in highly ordered graphitic planes; however, it becomes Raman active in the presence of defects, hence this band is attributed to defects or disordered carbon within graphitic domains. The G-band arises from the E2g stretching vibrations of the graphite lattice.21,26 For the CNFs, the Raman spectrum shows G and D bands of roughly equal intensities (IG and ID, respectively) and also, both these band are broad, with full width at half maximum (FWHM) values of 85 cm−1 and 90 cm—1, respectively. These are characteristic features consistent with significant amounts of disordered carbon within the CNFs.27 The spectrum of the CNTs shows a significantly more prominent and sharper G-peak, with an ID/IG ratio of ∼0.22 and a FWHM value of 15 cm−1, which indicates a graphitic structure with significantly fewer defects, compared to the CNFs. Also, a weak D′ peak is observed at 1610 cm−1 which is also activated by defects indicative of remnant defects within the graphitic structure.28 The G-peak of the GCA has the lowest ID/IG ratio of 0.08 and a FWHM of only 5 cm−1, indicating a high quality of the graphene lattice. Such low intensity ratios, with the absence of the D′ peak indicate that the D-peak arises from defects that are located at the edge boundary of the material rather than within the bulk, which underscores the excellent quality of the graphitic structure.29,30 Furthermore, a low-energy shoulder at 1580 cm−1 corresponding to the G− split is observed. This splitting of the G-peak into the G− and G+ (1595 cm−1) peaks occurs due to the different curvature of the nanotubes along the radial and longitudinal axes, which highlights structures with narrow curvature and a high degree of structural quality.28,30 These are necessary for the integration of the base materials involved in forming the GCA and potentially higher conductivity.31
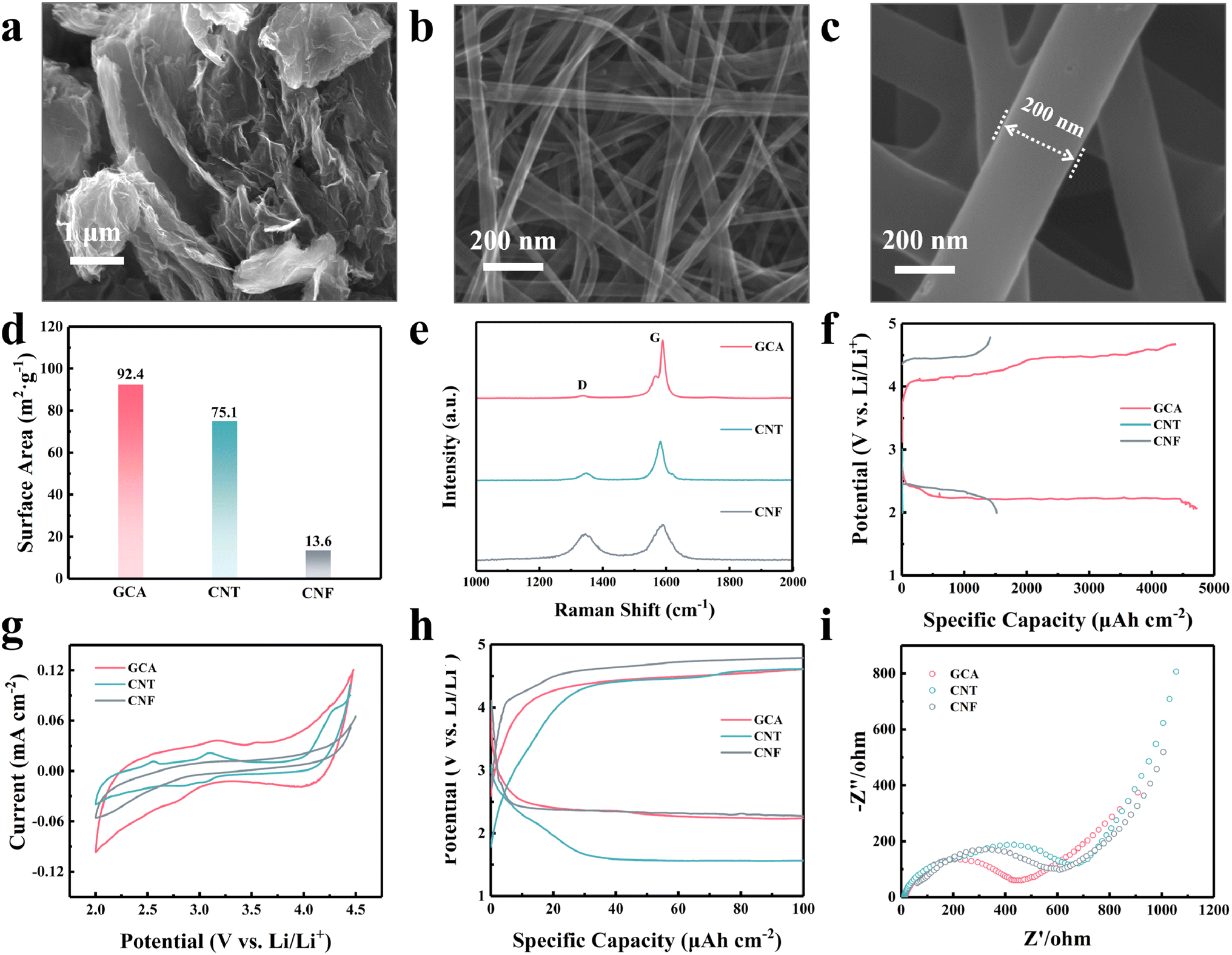 | ||
| Fig. 2 SEM characterization of different substrates: (a) GCA, (b) CNT and (c) CNF. (d) Comparison of surface area. (e) Raman spectra of the three cathodes. (f) The full discharge curves at a cut-off voltage of 2 V. (g) CV curves using different substrates with a scanning rate of 0.1 mV s−1. (h) The discharge–charge profile comparison of LCBs. (i) EIS spectra of the three cathodes at open circuit potential (OCP). | ||
To better evaluate their electrochemical properties, GCA, CNT and CNF films were directly utilized as self-supporting cathodes for LCBs. The full discharge results (Fig. 2f) confirmed that the GCA provides the highest discharge areal capacity (4720 μA h cm−2) owing to the sufficient reaction interface created by the unique porous structure. Fig. 2g depicts the cyclic voltammetry (CV) curves of the three substrates in the voltage range from 2.0 to 4.5 V (vs. Li/Li+). GCA showed higher reaction current and integral area during both CO2RR and CO2ER processes indicating that the GCA possesses excellent catalytic kinetics in LCBs. The galvanostatic discharge and charge tests were performed at a current density of 20 μA cm−2 with a cut-off areal capacity of 100 μA h cm−2. The GCA exhibited the lowest overpotential of 2.15 V compared with CNT (2.87 V) and CNF (2.35 V) films as shown in Fig. 2h. Electrochemical impedance spectra (EIS) revealed that GCA electrodes present the lowest charge transfer resistance (Rct) at open circuit potential as shown in Fig. 2i. This could be ascribed to the facilitated mass diffusion and better electrolyte affinity in the hierarchical porous electrode structure. In general, the GCA delivered much better electrochemical performance than CNT and CNF films, making it a more appropriate substrate among them.
3.2 Efficient catalytic electrode construction
Highly efficient cathode electrocatalysts are crucial to promote more practical LCB performance. After screening the suitable substrate, we further explored rapid and controllable electrocatalyst loading onto the substrates by the fast Joule heating approach. The Joule heating method has been widely used and explored thanks to its high efficiency, energy saving, wide applicability and uniform particle size distribution of the synthesised nanoparticles (NPs).32,33 Pt NPs with restricted electrocatalyst loading were deposited on the GCA substrate by fast Joule heating (denoted as Pt@GCA). Notably, this approach allows us to effectively optimize catalyst utilisation and tailor the catalytic sites. The overall preparation process of Pt@GCA is described in Fig. 3a. The Pt precursor was dissolved in deionised water (25 mM) and then added dropwise to the GCA at 20 μL cm−2. The Pt-decorated GCA was then thermally treated in the Joule heating system. The thermal shock effect enabled the fast conversion of the precursor into Pt NPs (Fig. S7, see the video in the ESI† for details).34 The detailed microstructure of Pt@GCA is shown in the SEM images (Fig. 3b). The GCA substrate retained a well-defined three-dimensional structure with no signs of aggregation. Uniformly distributed NPs can be observed on the GCA surface, with more detailed images in Fig. S8.† To determine the mass loading of Pt NPs on GCA, Pt@GCA was heated to 900 °C in air to remove the carbonaceous GCA. Based on the thermogravimetry data (Fig. S9†), the Pt content in Pt@GCA was calculated to be 1.25 wt%. Pt NPs were also introduced into the CNT and CNF films with the same Joule heating treatment (Fig. S10†). The optical photographs in Fig. S11† show that the heat-treated free-standing Pt@GCA cathode still has good resilience. The BET surface of the Pt@GCA cathode slightly increased (148.9 m2 g−1) compared with GCA electrodes (Fig. S12†) because of the thermal decomposition of impurities during the thermal shock process.35 | ||
| Fig. 3 (a) Schematic illustration of the preparation processes of the self-supporting Pt@GCA electrode. Morphological and compositional characterization: (b) SEM images, (c) HRTEM images and the inset shows the SAED image, (d) XRD patterns of GCA, Pt@CNT, Pt@CNF and Pt@GCA electrodes. (e) ToF-SIMS observation of the Pt@GCA electrode. | ||
High resolution transmission electron microscopy (HRTEM) was used to characterize the Pt nanoparticles. A uniform particle size was observed, as shown in Fig. S13.† The elemental mapping image revealed the uniform distribution of Pt NPs on graphene sheets (Fig. S14†). The average Pt nanoparticle size was measured to be approximately 6.27 nm (Fig. S15†). Lattice spacing measurements of 0.224 nm and 0.196 nm corresponding to the (111) and (200) faces of Pt, respectively, were obtained (Fig. 3c and S16†).36,37 This was further confirmed using the selected area electron diffraction (SAED) pattern as shown in the inset of Fig. 3c, implying the high crystallization degree of Pt NPs. Structural information of different gas electrodes was observed with XRD. Pt (111) peaks at 40.2° were identified according to the PDF standard card (PDF#87-0647). Among the three electrodes, Pt@GCA displayed an intensified Pt (111) peak compared to Pt@CNT and Pt@CNF (Fig. 3d), indicating a higher Pt (111) composition which is expected to enhance the intrinsic catalytic performance of the Pt-based catalysts for LCBs.38,39 The areal distribution of Pt NPs in the GCA was characterized by ToF-SIMS. The Pt element was observed to be uniformly distributed as shown in Fig. 3e.
The compositional characteristics of Pt@GCA were explored by X-ray photoelectron spectroscopy (XPS) measurements (Fig. S17†). As shown in Fig. S18,† the high-resolution spectra of Pt 4f split into two peaks at 71.1 and 74.5 eV, which can be attributed to Pt 4f7/2 and Pt 4f5/2. No other Pt impurity phase can be found. In addition, Fig. S19† exhibits a similar ID/IG value existing in both samples, indicating that the extremely fast heating process does not generate obvious carbon defects.
3.3 Electrochemical performance evaluation with self-supporting cathodes
Pt@GCA cathodes were used as self-supporting cathodes for the LCB coin cell testing. The electrochemical performance was initially evaluated through CV testing. When cycled at 0.1 mV s−1 in the potential range from 2.0 V to 4.5 V, Pt@GCA exhibited the lowest onset potentials and the highest reaction currents in both discharging and charging processes compared to other electrodes (Fig. 4a). These CV results indicate that Pt@GCA enhances the CO2ER and CO2RR catalytic kinetics, potentially improving CO2 utilisation and energy efficiency of LCBs. Notably, Pt@GCA demonstrated the largest cathode peak integral area, suggesting abundant active sites on the electrode surface and the highest discharge capacity among the samples tested. More GCD tests were conducted to evaluate capacity-related performance. Without CO2 injection, negligible capacity could be generated (Fig. S20†). Benefiting from the facilitated CO2 conversion, Pt@GCA was able to deliver a superior discharge areal capacity of 11247 μA h cm−2 as shown in Fig. 4b (tested at a current density of 20 μA cm−2 with the cut-off voltage set at 2.0 V) exceeding those of most reported cathode electrocatalysts (Fig. S21†). The ultrafine Pt nanoparticles anchored in the GCA substrate create highly efficient and sufficient catalytic sites to convert CO2 and deliver high areal discharge capacity. Rate performance was evaluated with various current densities in the range of 0.02–0.16 mA cm−2 (Fig. 4c). Stable discharging and charging plateaus can be observed for Pt@GCA with varied current densities. The overpotentials are 0.71, 0.72, 0.85, 0.93 and 1.06 V at 0.02, 0.04, 0.08, 0.10 and 0.16 mA cm−2, respectively, which correspond to energy efficiencies of 81.30%, 79.88%, 76.27%, 72.58% and 69.93%, respectively. In sharp contrast, the overpotential of pure GCA was reached even at 3.18 V (Fig. S22†) while the final charge potential of the Pt@GCA was still below 3.5 V even when operated under 0.16 mA cm−2.
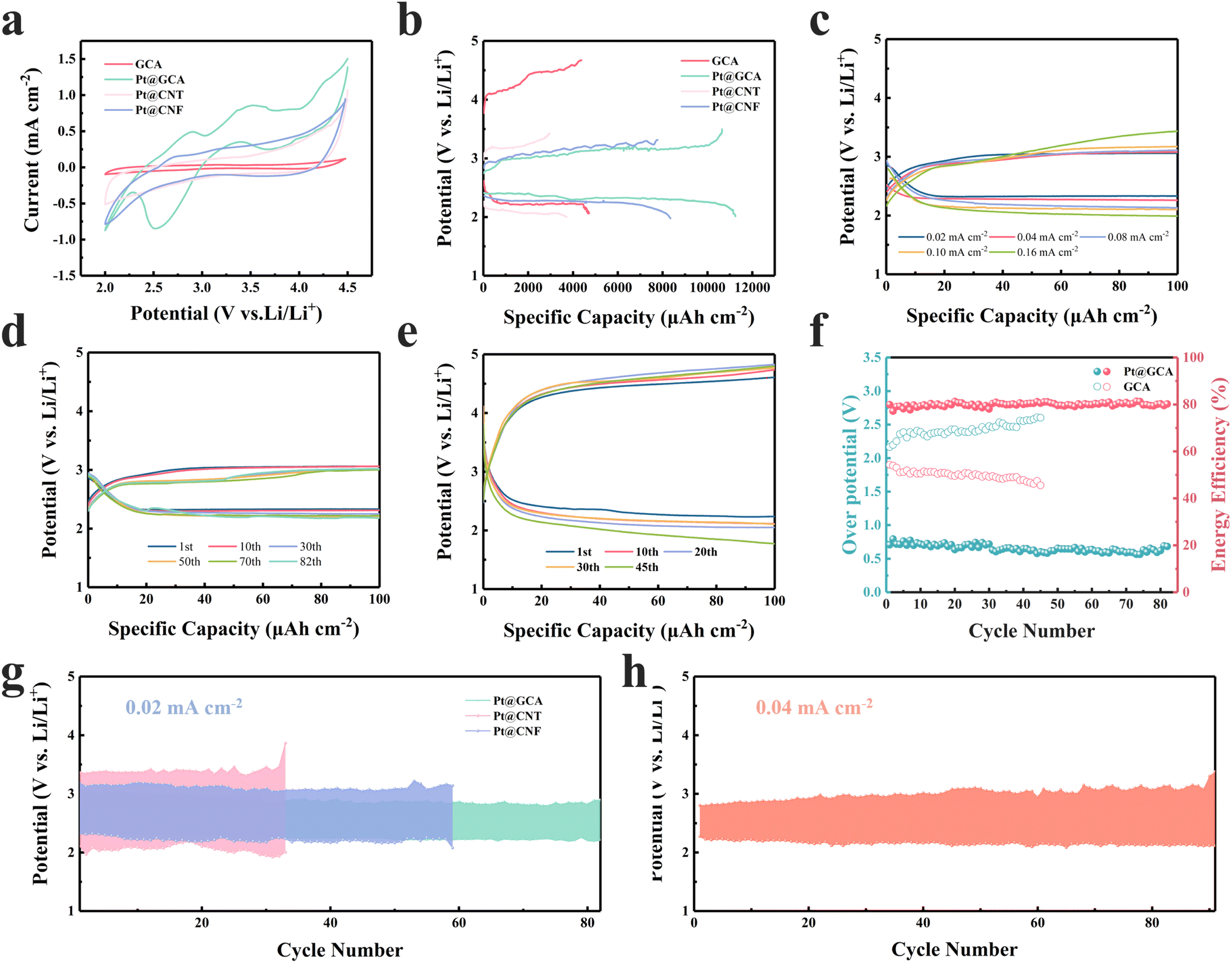 | ||
| Fig. 4 Electrochemical performance evaluation. (a) CV performance comparison using different electrodes. (b) The full discharging capacity curves. (c) Rate performance of Pt@GCA based LCBs. Long cycle performance of (d) Pt@GCA and (e) GCA cathodes. (f) Energy efficiency and overpotential comparison of Pt@GCA and GCA. (g) Cycle performance of Pt@GCA, Pt@CNT and Pt@CNF cathodes at 0.02 mA cm−2. (h) Cycle performance of Pt@GCA at 0.04 mA cm−2. | ||
At a current density of 20 μA cm−2 and a cut-off capacity of 100 μA h cm−2 (equal to 1000 mA h gcatalyst−1 based on the catalyst loading on the electrode), Pt@GCA exhibits a 3.05 V charging plateau and an overpotential of merely 0.71 V at the first cycle (Fig. 4d). In comparison, the overpotentials of other cathodes have all increased, with the overpotential of the GCA cathode reaching as high as 2.15 V under the same test conditions (Fig. S23† and 4e). In the subsequent cycle, the LCBs with the Pt@GCA cathode displayed low overpotential cycling performance, where the overpotential was below 0.75 V, energy efficiency was above 80% (Fig. 4f) and the charging plateau was at ∼3 V even after 82 cycles. However, the GCA can only be used for 45 cycles with the unstable discharging and charging plateaus, gradually increased overpotentials and corresponding reduced energy efficiency (Fig. 4f). The significant improvement in overpotential and stable cycle performance is mainly attributed to the sufficient catalytic sites and high reversibility of Pt@GCA. The outstanding electrochemical performance is superior to many currently reported cathodic catalysts as summarized in Table S2.† Compared with the reported cathodes, the LCBs with the Pt@GCA cathode achieve a high areal discharge capacity, low overpotential and competitive energy efficiency.
Cycle performance of gas electrodes with different substrates is compared as shown in Fig. 4g. LCBs based on Pt@CNT displayed the lowest and unstable discharging plateaus as well as the highest overpotential. The charging voltage sharply increased to over 3.5 V after only 33 cycles in Pt@CNT based LCBs. Overpotential of Pt@CNF based LCBs could be lowered compared with Pt@CNT. However, the cycle life ended within 60 cycles. Remarkably, the Pt@GCA cathode based LCBs exhibit the most stable long-term cycle stability with the lowest overpotential. Furthermore, Pt@GCA showed stability after more than 91 cycles, exhibiting a terminal charging voltage lower than 3.5 V at greater current densities of 0.04 mA cm−2 (Fig. 4h). A low charging potential results in effective inhibition of significant detrimental side reactions such as electrolyte and carbon substrate decomposition to ensure stable cycle performance.4,40
3.4 Reversibility investigation and degradation analysis
Here we combine morphological and compositional characterization experiments to reveal the reaction mechanism in the Pt@GCA cathode. The morphology characteristic of the discharging product and electrode surface evolution could greatly affect the reversibility of LCBs.41 The electrode surface changes after the discharging and charging processes were first observed by SEM. The electrode surface of Pt@GCA was covered with the thin-film discharging product after discharging (Fig. 5a). Upon charging, the discharging product fully decomposed and a clean electrode surface can be observed (Fig. 5b). Generally, the thin-film morphology discharging product formed following the Frank–van der Merwe model and decomposed more readily.42 However, the discharging product turned out to be large particles on the GCA electrode surface due to the lack of sufficient active catalytic sites (Fig. S24†). The Partial discharging product remained on the GCA surface at the fully charged state, which implies a poorer reversibility than Pt@GCA. EIS measurements before and after cycling were conducted to further investigate the overall reversibility of different cathodes (Fig. S25 and S26†). According to Fig. S25a,† Pt@GCA delivers the lowest Rct value before cycling among all cathodes. Upon discharging, the Rct values of the four cathodes increase as the reaction product gradually cover the electrode surface. However, after the subsequent charging process, the Rct of Pt@GCA recovered to its initial level before cycling, indicating the effective decomposition of the discharging product and demonstrating high reversibility (Fig. S25b†). In contrast, other cathodes show unrecoverable Rct at the fully charged state with a poorer reversibility (Fig. S26†). | ||
| Fig. 5 SEM images of Pt@GCA after (a) discharging and (b) recharging processes. Compositional characterization of Pt@GCA electrodes at different operation states: (c) Raman spectra, XPS spectra of (d) C 1s and (e) Li 1s, and (f) XRD spectra. SEM images of cycled electrodes, (g) Pt@GCA, (h) Pt@CNT and (i) Pt@CNF. | ||
The chemical composition evolution on the Pt@GCA cathode surface was further investigated by ex situ interfacial spectroscopic characterization. Raman spectra in Fig. 5c display the evolution of representative chemicals of Li2CO3. It is usually difficult to detect the evolution of discharging carbon products as their spectra could be covered by strong carbonaceous substrate signals. The Li2CO3 stretching peak can be identified at ∼1090 cm−1 during the discharging process. This peak disappears upon charging, evidencing reversibility.43,44 However in the GCA based LCBs, the Li2CO3 Raman signal could still be detected at the fully charged state, indicating a poorer reversibility (Fig. S27†). The XPS spectra of C 1s and Li 1s in Fig. 5d and e show significant Li2CO3 generation signals at 290.5 eV and at 55.5 eV, respectively, after discharging.45,46 In the subsequent charging process, the composition signal belonging to Li2CO3 disappeared completely indicating high reversibility. Furthermore, the ex situ XRD (Fig. 5f) and FTIR (Fig. S28†) characterization of the Pt@GCA cathode in different states also presented the reversible evolution of Li2CO3 during cycling. The combination of various component characterization studies reveals the highly reversible reaction in Pt@GCA based LCBs.
Electrode integrity reflected the mechanical stability of the electrodes. The cycled gas electrodes were disassembled for SEM observation (charged state). The Pt@GCA cathode can retain the 3D porous structure and stable loading of Pt NPs after long-term operation (>800 h) as shown in Fig. 4g. More importantly, the absence of discharging product residues on the electrode surface indicates complete decomposition and high reversibility. The morphology of the Li anode from the same battery after disassembling was also observed. After long-term cycling of over 800 h, the Li foil was almost completely corroded (Fig. S29†). The corresponding XRD spectra of the cycled lithium metal anode presented significant generation of LiOH and Li2CO3 which is assumed to be the side reaction of the Li anode with dissolved CO2 during battery operation (Fig. S30†). Therefore, we believe that the final suspension of Pt@GCA based LCBs could be attributed to the Li anode failure. However in the Pt@CNT based LCBs, a large amount of irregularly shaped discharging product was observed on the cycled electrode surface (Fig. 5h). It is proposed that the insulating discharging products would gradually passivate the active catalytic sites and further inhibit the reversible conversion of CO2. This irreversible generation of insulating Li2CO3 on the electrocatalyst surface results in high overpotentials and shortens the cycle lifetime. Worse still, the reaction stress produced during repeated catalytic processes caused the detachment of Pt NPs (Fig. S31†) away from the CNT substrate.15,47 The Pt@CNF electrodes also suffered severe structural damage after long-term cycling (Fig. 5i). The carbon nanofibers possess a large proportion of inactive and defect-rich carbon surface, which is vulnerable to potential superoxide attack, leading to catalyst structural collapse.31,48,49
3.5 Flexible Li–CO2 battery demonstration
To verify the promising application of Pt@GCA cathodes in future wearable electronics, flexible Li–CO2 pouch cells were fabricated. Due to the excellent flexibility and mechanical strength of Pt@GCA cathodes, the as-prepared pouch cell was able to reliably power LED strips under various bending conditions (Fig. 6a). Also, the as-prepared Li–CO2 pouch cell was able to still provide a stable open circuit voltage after repeated bending (Fig. 6b). Further cycling performance testing showed that the as-developed Li–CO2 pouch cell was stable after cycles for longer than 200 h at 20 μA cm−2, demonstrating promising practical application of the flexible LCBs (Fig. 6c). | ||
| Fig. 6 Pt@GCB based flexible LCB testing, (a) powering LEDs at different bending angles. (b) Open-circuit potential under different bending conditions. (c) Cycle performance of the flexible LCBs at 20 μA cm−2. | ||
4 Conclusion
In summary, a comprehensive and systematic evaluation of gas electrode design and engineering was conducted in our work. The three-dimensional graphene/carbon nanotube aerogel with a hierarchical porous structure was identified as a promising conductive substrate for LCBs. Ultrafine Pt NPs with a restricted mass loading were successfully loaded on the substrate via an efficient Joule heating technique. A highly conductive and catalytic network was constructed in the optimal Pt@GCA electrodes. Charge transfer resistance can be significantly reduced owing to the 3D porous and affiliative electrode structure. Additionally, abundant active catalytic sites were uniformly distributed on the robust conductive substrate enabling highly reversible CO2 conversion. Benefiting from the optimized gas electrode properties, Pt@GCA delivers superior electrochemical performance as a LCB cathode. Outstanding areal discharge capacity of over 10 mA h cm−2 at 20 μA cm−2 was obtained. Low overpotential under high current densities can be achieved with excellent energy efficiencies of over 80%. The reversible CO2RR and CO2ER processes were investigated by the combination of various morphological and compositional characterisation experiments. Flexible Li–CO2 pouch cells were also fabricated that can provide practical operation with high mechanical tolerance. This work emphasizes the importance of systematic electrode engineering considerations for high-performance LCBs. The proposed electrode structure regulation further indicates an effective way to enhance the electrochemical performance of metal–gas batteries, fuel cells and broad electrocatalyst-related applications.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors greatly acknowledge the financial support from the National Natural Science Foundation of China (52172097), New Energy Material Innovation Consortium Projects of Yunnan Province (202302AB080018), and the Royal Society International Exchanges 2021 Cost Share (NSFC) scheme (IEC\NSFC\211074).References
- Y. Qiao, J. Yi, S. Wu, Y. Liu, S. Yang, P. He and H. Zhou, Joule, 2017, 1, 359–370 CrossRef CAS
.
- B. Lu, B. Chen, D. Wang, C. Li, R. Gao, Y. Liu, R. Mao, J. Yang and G. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2216933120 CrossRef CAS PubMed
- Y. Liu, R. Mao, B. Chen, B. Lu, Z. Piao, Y. Song, G. Zhou and H.-M. Cheng, Mater. Today, 2023, 63, 120–136 CrossRef CAS
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng and P. G. Bruce, J. Am. Chem. Soc., 2013, 135, 494–500 CrossRef CAS PubMed
- Y. Jiao, J. Qin, H. M. K. Sari, D. Li, X. Li and X. Sun, Energy Storage Mater., 2021, 34, 148–170 CrossRef
- J. Lin, W. Song, C. Xiao, J. Ding, Z. Huang, C. Zhong, J. Ding and W. Hu, Carbon Energy, 2023, 5, e313 CrossRef CAS
- A. Sarkar, V. R. Dharmaraj, C. H. Yi, K. Iputera, S. Y. Huang, R. J. Chung, S. F. Hu and R. S. Liu, Chem. Rev., 2023, 123, 9497–9564 CrossRef CAS PubMed
- J. Chen, X.-Y. Chen, Y. Liu, Y. Qiao, S.-Y. Guan, L. Li and S.-L. Chou, Energy Environ. Sci., 2023, 16, 792–829 RSC
- Z. Zhang, Q. Zhang, Y. Chen, J. Bao, X. Zhou, Z. Xie, J. Wei and Z. Zhou, Angew Chem. Int. Ed. Engl., 2015, 54, 6550–6553 CrossRef CAS PubMed
- L. Liu, Y. Qin, K. Wang, H. Mao, H. Wu, W. Yu, D. Zhang, H. Zhao, H. Wang, J. Wang, C. Xiao, Y. Su and S. Ding, Adv. Energy Mater., 2022, 12, 2103681 CrossRef CAS
- L. Chen, J. Zhou, J. Zhang, G. Qi, B. Wang and J. Cheng, Energy Environ. Mater., 2023, 6, e12415 CrossRef CAS
- F. Wang, Y. Li, X. Xia, W. Cai, Q. Chen and M. Chen, Adv. Energy Mater., 2021, 11, 2100667 CrossRef CAS
- J.-L. Liao, S. Zhang, T.-S. Bai, F.-J. Ji, D.-P. Li, J. Cheng, H.-Q. Zhang, J.-Y. Lu, Q. Gao and L.-J. Ci, Rare Met., 2023, 42, 1969–1982 CrossRef CAS
- J.-T. Li, Z.-Y. Wu, Y.-Q. Lu, Y. Zhou, Q.-S. Huang, L. Huang and S.-G. Sun, Adv. Energy Mater., 2017, 7, 1701185 CrossRef
- Z.-Z. Shen, Y.-Z. Zhang, C. Zhou, R. Wen and L.-J. Wan, J. Am. Chem. Soc., 2021, 143, 21604–21612 CrossRef CAS PubMed
- A. Hu, C. Shu, C. Xu, R. Liang, J. Li, R. Zheng, M. Li and J. Long, J. Mater. Chem. A, 2019, 7, 21605–21633 RSC
- Y. Xiao, S. Hu, Y. Miao, F. Gong, J. Chen, M. Wu, W. Liu and S. Chen, Small, 2024, 20, 2305009 CrossRef CAS PubMed
- Y. Xiao, F. Du, C. Hu, Y. Ding, Z. L. Wang, A. Roy and L. Dai, ACS Energy Lett., 2020, 5, 916–921 CrossRef CAS
- H. Zhao, D. Li, H. Li, A. G. Tamirat, X. Song, Z. Zhang, Y. Wang, Z. Guo, L. Wang and S. Feng, Electrochim. Acta, 2019, 299, 592–599 CrossRef CAS
- J. Zhou, X. Li, C. Yang, Y. Li, K. Guo, J. Cheng, D. Yuan, C. Song, J. Lu and B. Wang, Adv. Mater., 2019, 31, 1804439 CrossRef PubMed
- W. Yu, L. Liu, Y. Yang, N. Li, Y. Chen, X. Yin, J. Niu, J. Wang and S. Ding, Chem. Eng. J., 2023, 465, 142787 CrossRef CAS
- J. Du, Q.-Y. Lin, J.-Q. Zhang, S.-L. Hou and A.-B. Chen, Rare Met., 2023, 42, 2284–2293 CrossRef CAS
- T. Yang, L. Li, T. Zhao, Y. Ye, Z. Ye, S. Xu, F. Wu and R. Chen, Adv. Energy Mater., 2021, 11, 2102454 CrossRef CAS
- H. Gao and B. M. Gallant, Nat. Rev. Chem, 2020, 4, 566–583 CrossRef
- E. R. Ezeigwe, L. Dong, R. Manjunatha, Y. Zuo, S.-Q. Deng, M. Tan, W. Yan, J. Zhang and D. P. Wilkinson, Nano Energy, 2022, 95, 106964 CrossRef CAS
- S. A. Chernyak, A. S. Ivanov, K. I. Maslakov, A. V. Egorov, Z. Shen, S. S. Savilov and V. V. Lunin, Phys. Chem. Chem. Phys., 2017, 19, 2276–2285 RSC
- B. O. Boskovic, V. Stolojan, R. U. A. Khan, S. Haq and S. R. P. Silva, Nat. Mater., 2003, 2, 126 CrossRef CAS
- J. H. Lehman, M. Terrones, E. Mansfield, K. E. Hurst and V. Meunier, Carbon, 2011, 49, 2581–2602 CrossRef CAS
- J. V. Anguita, T. R. Pozegic, M. Ahmad and S. R. P. Silva, ACS Appl. Nano Mater., 2021, 4, 5211–5219 CrossRef CAS
- M. S. Dresselhaus, G. Dresselhaus, A. Jorio, A. G. Souza Filho and R. Saito, Carbon, 2002, 40, 2043–2061 CrossRef CAS
- D. M. Itkis, D. A. Semenenko, E. Y. Kataev, A. I. Belova, V. S. Neudachina, A. P. Sirotina, M. Havecker, D. Teschner, A. Knop-Gericke, P. Dudin, A. Barinov, E. A. Goodilin, Y. Shao-Horn and L. V. Yashina, Nano Lett., 2013, 13, 4697–4701 CrossRef CAS PubMed
- Y. Yao, Z. Huang, P. Xie, S. D. Lacey, R. J. Jacob, H. Xie, F. Chen, A. Nie, T. Pu, M. Rehwoldt, D. Yu, M. R. Zachariah, C. Wang, R. Shahbazian-Yassar, J. Li and L. Hu, Science, 2018, 359, 1489–1494 CrossRef CAS PubMed
- Y. Qiao, S. Xu, Y. Liu, J. Dai, H. Xie, Y. Yao, X. Mu, C. Chen, D. J. Kline, E. M. Hitz, B. Liu, J. Song, P. He, M. R. Zachariah and L. Hu, Energy Environ. Sci., 2019, 12, 1100–1107 RSC
- S. Liu, Y. Shen, Y. Zhang, B. Cui, S. Xi, J. Zhang, L. Xu, S. Zhu, Y. Chen, Y. Deng and W. Hu, Adv. Mater., 2022, 34, 2106973 CrossRef CAS PubMed
- D. Xia, J. Mannering, P. Huang, Y. Xu, Q. Li, H. Li, Y. Qin, A. N. Kulak and R. Menzel, J. Am. Chem. Soc., 2024, 146, 159–169 CrossRef CAS PubMed
- W. Wang, Z. Wang, M. Yang, C.-J. Zhong and C.-J. Liu, Nano Energy, 2016, 25, 26–33 CrossRef CAS
- M. Wang, H. Liu, J. Ma and G. Lu, Appl. Catal., B, 2020, 266, 118647 CrossRef CAS
- C. Li, N. Clament Sagaya Selvam and J. Fang, Nano-Micro Lett., 2023, 15, 83 CrossRef CAS PubMed
- S. Chen, K. Yang, H. Zhu, J. Wang, Y. Gong, H. Li, M. Wang, W. Zhao, Y. Ji, F. Pan, S. R. P. Silva, Y. Zhao and L. Yang, Nano Energy, 2023, 117, 108872 CrossRef CAS
- S. A. Freunberger, Y. Chen, N. E. Drewett, L. J. Hardwick, F. Bardé and P. G. Bruce, Angew. Chem., Int. Ed., 2011, 50, 8609–8613 CrossRef CAS PubMed
- J. Zou, G. Liang, F. Zhang, S. Zhang, K. Davey and Z. Guo, Adv. Mater., 2023, 35, 2210671 CrossRef CAS PubMed
- J. Zhou, T. Wang, L. Chen, L. Liao, Y. Wang, S. Xi, B. Chen, T. Lin, Q. Zhang, C. Ye, X. Zhou, Z. Guan, L. Zhai, Z. He, G. Wang, J. Wang, J. Yu, Y. Ma, P. Lu, Y. Xiong, S. Lu, Y. Chen, B. Wang, C.-S. Lee, J. Cheng, L. Gu, T. Zhao and Z. Fan, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2204666119 CrossRef CAS PubMed
- Y. Jin, F. Chen and J. Wang, ACS Sustain. Chem. Eng., 2020, 8, 2783–2792 CrossRef CAS
- X. Zhang, T. Wang, Y. Yang, X. Zhang, Z. Lu, J. Wang, C. Sun, Y. Diao, X. Wang and J. Yao, ACS Energy Lett., 2021, 6, 3503–3510 CrossRef CAS
- K. Wang, D. Liu, L. Liu, X. Li, H. Wu, Z. Sun, M. Li, A. S. Vasenko, S. Ding, F. Wang and C. Xiao, Adv. Sci., 2023, 10, 2205959 CrossRef CAS PubMed
- J. Zhang, F. Wang, G. Qi, J. Cheng, L. Chen, H. Liu and B. Wang, Adv. Funct. Mater., 2021, 31, 2101423 CrossRef CAS
- O. Keisar, Y. Ein-Eli, Y. Alfi and Y. Cohen, J. Power Sources, 2020, 450, 227545 CrossRef CAS
- Z. Zhang, X. G. Wang, X. Zhang, Z. Xie, Y. N. Chen, L. Ma, Z. Peng and Z. Zhou, Adv. Sci., 2018, 5, 1700567 CrossRef PubMed
- N. Mahne, S. E. Renfrew, B. D. McCloskey and S. A. Freunberger, Angew. Chem., Int. Ed., 2018, 57, 5529–5533 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se01582g |
| This journal is © The Royal Society of Chemistry 2025 |