
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing solid-state battery performance with spray-deposited gradient composite cathodes†
Matt P.
Tudball
 a,
Will J.
Dawson
ab,
Joshua H.
Cruddos
ab,
Francesco
Iacoviello
a,
Andrew R. T.
Morrison
ab,
Alexander J. E.
Rettie
abc and
Thomas S.
Miller
*abc
a,
Will J.
Dawson
ab,
Joshua H.
Cruddos
ab,
Francesco
Iacoviello
a,
Andrew R. T.
Morrison
ab,
Alexander J. E.
Rettie
abc and
Thomas S.
Miller
*abc
aElectrochemical Innovation Lab, Department of Chemical Engineering, University College London, London, WC1E 7JE, UK. E-mail: t.miller@ucl.ac.uk
bThe Faraday Institution, Quad One, Harwell Science and Innovation Campus, Didcot, OX11 0RA, UK
cAdvanced Propulsion Lab, Marshgate, University College London, London, E20 2AE, UK
First published on 28th January 2025
Abstract
Solid state electrolytes, which replace flammable liquid ones, are seen as being key to deployment of safe and high capacity batteries based on lithium metal anodes. Yet these materials often suffer from poor electrode/electrolyte contact which limits Li+ transport and active material utilisation. To overcome these barriers, effective methods to intimately connect active materials and electrolytes must be developed and demonstrated. In this work gradient composite cathodes of lithium iron phosphate (LFP) and polyethylene oxide (PEO) were manufactured using spray deposition to remove the planar electrode/electrolyte interface in solid-state batteries with polymeric electrolytes. These graded cathodes achieved ten times lower resistance and superior cycle life and rate testing performance compared to ungraded cathodes made in the same way. Graded composite cathodes maintained stable capacity after 80 cycles and functioned well at rates up to 2C, whereas ungraded composite cathodes failed to deliver any useable capacity after 80 cycles and at rates higher than C/5. Hence, this work acts as a demonstration that simple electrode structuring can have a significant impact on cell performance, offering a route towards the stabilization of solid-state batteries in real-world applications.
1 Introduction
Solid state batteries (SSBs) represent a significant advancement in energy storage technologies, enabling the use of high-capacity lithium metal anodes without the associated safety risk of a flammable liquid electrolyte. However, whilst inorganic solid-state electrolytes (SSEs) such as lithium lanthanum zirconium oxide (LLZO) and lithium phosphorous oxynitride (LiPON) boast high Li+ conductivity and stability, they suffer from complexities in manufacturing, including high processing costs and difficulties in device integration.1 Polymer based electrolytes or solid polymer electrolytes (SPEs), such as polyethylene oxide (PEO), can benefit from cheap slurry based manufacturing methods but generally have limited electrochemical stability windows and poorer Li+ conductivity.2–6A major issue that affects all solid state electrolyte materials is the ability to ‘wet’ the electrode; poor contact between electrolyte and active material increases resistance and leads to decreased capacity.7 In order to rectify the poor wetting in the electrode, a composite electrode consisting of a mixture of electrolyte and active material is commonly used, thus providing ionic connection to the bulk of the electrode. Despite this, the planar interface that exists between the composite cathode and the bulk solid electrolyte is still a site of significant degradation of solid state batteries and this can worsen with volume expansion caused by routine cycling.8 Furthermore, poor connection of the composite cathode with the solid electrolyte increases resistance leading to decreased Li+ ion conductivity which can worsen performance. Full separation of the composite cathode from the solid electrolyte, which can occur with cell aging, produces areas of inactive material thus reducing overall capacity and shortening the battery lifetime.7 The clear planar interface between composite cathode and solid electrolyte is a source of resistivity, degradation and a cause of capacity loss with prolonged cycling. A method of removing this planar solid boundary would be to grade the composite cathode and electrolyte into each other, thus providing the necessary ionic connection without an obvious interface from which resistivity or disconnections can easily occur.
Gradients have previously been manufactured into battery electrodes in order to optimise the ionic and electronic conductivity by adjusting the ratio of active material or conductive carbon throughout the thickness, thereby influencing the through-thickness porosity.9–12 For example, by increasing the ratio of conductive carbon near the current collector and active material near the surface of the electrode, rate performance of a lithium iron phosphate (LiFePO4, LFP) battery was improved by 520% at 3C.9 This concept has also been applied to SSB's using starch-templated LLZO pellets as a porous substrate for cathode deposition, which resulted in good contact between electrode and electrolyte which significantly decreased interfacial resistance when compared to non-porous LLZO.13 Gradients therefore provide a promising method of improving SSB performance, prioritising Li+ movement through the electrode thickness, while also removing the planar electrode/electrolyte boundary. Manufacturing true gradients can be easily achieved using a layer-by-layer deposition technique in which the material ratio in the electrode can be altered through-thickness. In this work, layer-by-layer spray deposition is used in a one-step manufacturing process to implement true gradients in an LFP-based composite cathode, creating a gradient of PEO from the base to the surface of the electrode. When paired with a spray deposited PEO-based SPE layer, the planar boundary between electrode and electrolyte is effectively removed. These novel graduated composite cathodes were then tested for resistance, rate capability, and cycle life. The gradient composite cathodes showed significantly lower resistance and performed better in cycle-life and rate testing compared to homogenous ungraded cathodes.
2 Methodology
Materials used for the manufacture of composite cathodes were LFP (lithium iron phosphate, 1.5 μm, MSE supplies), carbon black (Super C65, C-NERGY, Imerys), PEO (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MV, Sigma-Aldrich), LiTFSi (Bis(trifluoromethane)sulfonimide lithium salt, 99.95%, Sigma-Aldrich), LLZO (Al-Doped Lithium Lanthanum Zirconate Garnet, Li6.25Al0.25La3Zr2O12, 99.9%, 500 nm, Ampcera) and IPA (isopropyl alcohol, ≥99%, Sigma-Aldrich).
000 MV, Sigma-Aldrich), LiTFSi (Bis(trifluoromethane)sulfonimide lithium salt, 99.95%, Sigma-Aldrich), LLZO (Al-Doped Lithium Lanthanum Zirconate Garnet, Li6.25Al0.25La3Zr2O12, 99.9%, 500 nm, Ampcera) and IPA (isopropyl alcohol, ≥99%, Sigma-Aldrich).
To manufacture the composite cathodes, LFP and PEO solutions were spray deposited through two AccuMist (SonoTek) nozzles and associated stirring syringe inputs using a SonoTek Exactacoat ultrasonic spray coater. The heated bed was set at 60 °C and the films were sprayed onto 1 mm thick 14 mm diameter stainless steel disks at a combined nozzle flow rate of 0.25 ml min−1. The flow rates of the LFP and PEO solutions were kept constant at 0.125 ml min−1 for the ungraded samples and a program to vary the flow rates between 0 and 0.25 ml min−1 was used for the graded samples, see Fig. 1(b–d). The shaping air was limited to 2 psi to ensure the stainless steel disks did not move and to improve atomisation; 80 sets of 4 spray passes comprised each composite cathode which were 30 μm thick and ∼7 mg cm−2. The 50 μm thick SPE layer was sprayed directly onto the previously coated composite cathode at the same conditions using one AccuMist nozzle over 80 sets of 4 spray passes. After spray deposition, the films were cooled and dried under vacuum at room temperature overnight before being transferred to an Ar-filled glovebox.
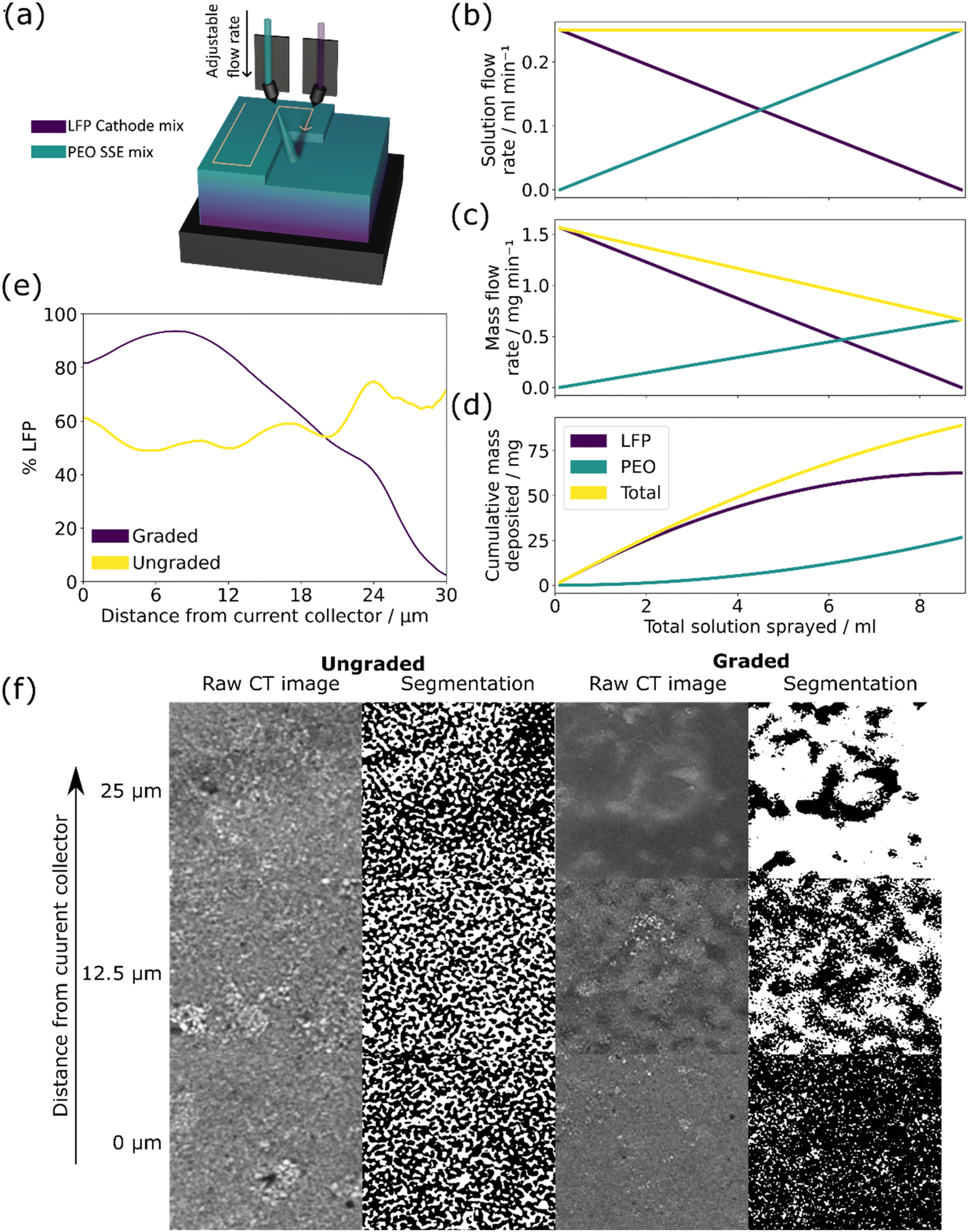 | ||
| Fig. 1 The manufacturing methodology and characterisation of graded and ungraded LFP/PEO composite cathodes. (a) Schematic of the rastering two-spray nozzle set-up and the subsequent gradient film. (b–d) Showing the calculated spray parameters and estimated solution and mass flow rates of the LFP and PEO solutions with (b) the solution flow rates through the spray coater throughout the full spray, (c) the associated mass flow of the two different concentrations solutions and (d) the cumulative deposited mass of each material. (e and f) The characterisation of graded and ungraded composite cathodes using X-ray CT with (e) experimental results showing the % of LFP measured throughout the thickness of the electrodes as calculated using the segmentations, (f) shows three of the raw X-ray CT slices and corresponding segmentations at different distances from the current collector, in the segmentation column white indicates PEO and black indicates LFP. | ||
Composite cathodes were assembled into coin cells with a lithium metal anode and tested on a Biologic VMP-300 (Biologic) at 60 °C in a Binder KB23 cooling incubator (Binder) at C-rates equivalent to the theoretical capacity of LFP (170 mA h g−1) and the theoretical capacity of the composite cathodes (∼1.2 mA h). All cycling was performed between 2.5 V and 3.65 V and no voltage hold was used. EIS measurements were taken after every charge and discharge post a one-hour rest period and was performed between 10 mHz and 7 MHz with 20 points per decade and an amplitude of 10 mV. The DRT (Distribution of Relaxation Times) transformation and analysis of the real and imaginary impedance was performed using RelaxIS 3 (RHD Instruments). A Gaussian function was applied for data discretization, with a second-order regularization fitting parameter (λ) of 0.0001, and a shape factor of 0.5 used. The software facilitated the determination of peak positions and shifts across cycles during charge and discharge.
X-ray computed tomography (X-ray CT) of the graded and ungraded composite cathodes was performed on a lab-based micro-CT X-ray instrument (Zeiss Xradia 620 Versa, Carl Zeiss). 1601 projections were taken with an exposure time of 5 seconds, magnification of 20×, a source operating voltage of 80 kV, and source current of 125 μA with a tungsten emission and a pixel size of 381 nm. Reconstruction was done using a cone beam filtered back projection algorithm using commercial software (Reconstructor Scout-and-Scan, Zeiss, Carl Zeiss). The data was cropped using Avizo3D 2023.2 software (ThermoFischer) and segmented using ilastik software using the methodology from previous work in Dawson et al.14,15
3 Results
Graded and ungraded composite cathodes were manufactured by spray depositing LFP and PEO solutions simultaneously, changing the flow rate of the two solutions to build a through-thickness gradient. The compositions of the LFP, PEO, and SPE solutions used are shown in Table 1. The two spray nozzles were rotated to −24° and 24° respectively at a height of 60 cm to ensure overlap of the two spray shapes and the nozzles moved in a raster pattern over the substrate to build up the film layer by layer, see Fig. 1(a). The two syringe inputs were programmed to increase/decrease the flow rate of the LFP and PEO solutions between 0 and 0.25 ml min−1 throughout the spray as shown in Fig. 1(b), with the associated mass flow rate in Fig. 1(c). Both graded and ungraded cathodes were manufactured to be 66.8 wt% LFP/carbon mix which required the use of different concentration precursor solutions; the impact of this can be seen in (d) where the plot of cumulative mass deposited shows the gradient of each material through the thickness of the film is curved rather than linear. This effect could be resolved by reprogramming the flow rates using a curved program rather than a linear one, although this was deemed to be unnecessary as the actual change in structure would be minimal.
LFP/carbon (14.04 mg ml−
1
) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 DI water:IPA by volume :1 DI water:IPA by volume
|
|
| LFP | 95.15% |
| Carbon black | 4.85% |
|
|
|
PEO/LiTFSI (5.96 mg ml−
1
) 1:1 DI water:IPA by volume
|
|
| PEO (100000 MV) |
60.4% |
| LiTFSI | 39.6% |
|
|
| SPE–PEO/LiTFSI/LLZO (13.15 mg ml− 1 ) in DI water | |
| PEO (10000 MV) |
76% |
| LiTFSI | 16% |
| Al-doped LLZO | 8% |
Fig. 1(e) shows the measured through-thickness LFP % of the ungraded and graded composite cathodes from the current collector to the surface of the electrode as calculated from the segmented X-ray CT scans in Fig. 1(f), using image analysis of the X-ray CT slices to determine the LFP ratio. There is a steady drop in LFP % in the graded cathode from ∼100% to 0% over the 30 μm thickness, whereas the LFP % in the ungraded cathode remains relatively stable at ∼60%. The slight increase in LFP % in the graded cathode between 0 and 10 μm is thought to be due to the settling of the molten PEO during spraying which displaced some LFP. The raw X-ray CT slices and associated segmentations shown in Fig. 1(f) demonstrate the density difference of the materials at distances from the current collector of 0 μm, 12.5 μm and 25 μm in an electrode of ∼30 μm thickness. The segmentation was based on the greyscale value of the raw slices, with highly dense LFP appearing lighter in the raw slices and darker in the segmented ones. The change in density throughout the graded electrode is clear with the 0 μm slice of the graded cathode appearing as a dense LFP structure and the 25 μm slice containing mostly polymer. By contrast, the ungraded electrode shows the same ratio and structure of LFP and PEO at 0 μm, 12.5 μm, and 25 μm, demonstrating the effectiveness of spray deposition in manufacturing true gradients in deposited films.
3.1 Cycle-life performance
The graded and ungraded composite cathodes were tested for capacity loss during long-term cycling at a rate of C/10 in which graded composite cathodes performed better over 80 cycles than ungraded ones. Fig. 2(a) displays the typical capacity per cycle results of the graded and ungraded composite cathodes. Here and across multiple repeats, see ESI Fig. 1(a and b),† the graded electrodes maintained capacity after extensive cycling significantly better than the ungraded despite the fact that the ungraded electrodes had higher initial discharge capacity (∼100–120 mA h g−1) than the graded ones (∼80 mA h g−1). The initial lower capacity for the graded composite cathode is thought to be due to the reduced amount of PEO near the current collector which limits ionic conductivity near the base of the electrode and creates areas of less active LFP. Capacity loss in the ungraded cathode is quite significant, decreasing from ∼100 mA h g−1 to almost zero after 80 cycles; by contrast, the graded cathodes retained high capacity remaining at ∼80 mA h g−1 after 80 cycles, thus negating the impact of the lower initial capacity.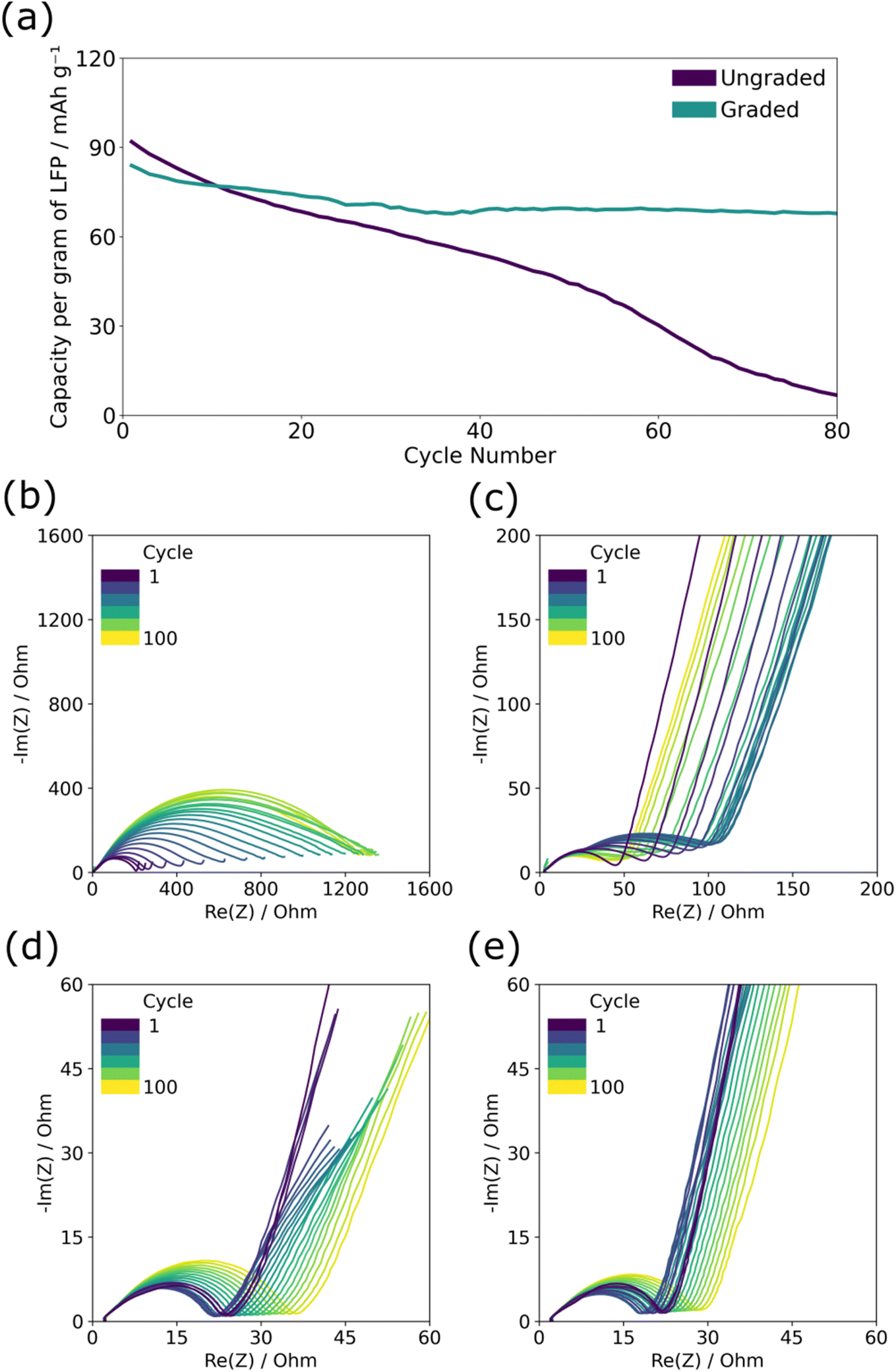 | ||
| Fig. 2 The cycle life performance and charge and discharge EIS of graded and ungraded composite LFP/PEO cathodes against lithium metal. (a) The discharge capacity per gram of LFP at C/10. (b–e) EIS spectra of graded and ungraded cathodes measured every 10th cycle in either the charged or discharged state with (b) ungraded charged, (c) ungraded discharged, (d) graded charged, (e) graded discharged. | ||
The electrochemical impedance spectroscopy shown in Fig. 2(b–e) demonstrates the difference in the charge transfer resistance across the cell for the two different structures throughout cycle life testing. The total resistance can be observed by the size of the semicircle along the x-axis, and is made up of overlapping semicircles of the bulk PEO ionic resistance and two main planar interface resistances, one for the cathode/electrolyte interface and the other for the anode/electrolyte interface, whereas the straight diagonal line following the semicircle provides information about diffusion processes.7 The graded cathode had a lower initial resistance on the first cycle (∼20 Ω in both states) compared to the ungraded cathode (160 Ω in the charged state and 50 Ω discharged). The impedance of the ungraded cathode after the first charge, Fig. 2(b), of ∼160 Ω is much higher than that of the discharge (c) ∼50 Ω. It has been shown that the resistance of the discharged state should be lower than that of the charged, partially due to the intrinsic differences in lithium-ion diffusion coefficient in LiFePO4 and FePO4.7,16 Furthermore, throughout the 80 cycles, the graded cathode has a smaller increase in resistance than the ungraded one, particularly in the charged state, with resistance after the final charge of ∼1400 Ω for the ungraded cathode and ∼40 Ω for the graded. The initial lower resistance alongside the smaller increase in resistance of the graded cathode suggests that the charge transfer in either the bulk PEO or the planar interfaces of the graded cathode is improved and suffers less damage with cycling.
To resolve greater detail in the EIS analysis, distribution of relaxation times (DRT) analysis of the spectra was performed, allowing overlapping relaxation processes to be resolved into a series of local maxima linked to specific time constants (τ).17 In the DRT analysis, the charge transfer peak in the charged ungraded composite cathode, ESI Fig. 2a,† can be seen to have grown in size and shifted from ∼10−3 s−1 to ∼10−1 s−1 over 80 cycles, suggesting that charge transfer both increased in resistance and became slower with cycle induced degradation; this would account for the distinct loss in capacity. By contrast, there was very little change in the charge transfer peak over the course of cycling in the graded composite cathode, ESI Fig. 2c.† As the material composition of the bulk PEO is the same in both SPE layers, it is assumed the charge transfer resistance between the composite cathode and the bulk PEO SPE is the primary cause of degradation. With the graded composite cathode, capacity loss or resistance increase during cycling is more unlikely to occur as there is no clear plane between the two layers in which disconnection can take place.
3.2 Rate performance
Graded and ungraded composite cathodes were tested for rate performance between C/10 and 2C and the corresponding discharge capacity and EIS plots are shown in Fig. 3. While ungraded cathodes had higher capacity at C/10, similar to cycle life testing, at all rates above C/10 the graded composite cathodes performed better. The capacity of the ungraded composite cathodes dropped from ∼100–120 mA h g−1 in the initial C/10 to 50 mA h g−1 at C/5 and then to ∼0 mA h g−1 at C/2, 1C, and 2C. Based on the recovery of the capacity in the final C/10 cycles back to ∼80 mA h g−1, this significant capacity loss can be attributed to poor rate performance and not solely to cycle life degradation. The graded composite cathodes maintained a relatively stable capacity up to C/2 of between 50 and 80 mA h g−1, dropping to ∼30–40 mA h g−1 at 1C and, then to ∼10 mA h g−1 at 2C. Graded cathodes consistently achieved higher capacity than the ungraded cathodes at rates above C/10 across repeats, see ESI Fig. 1(c and d),† demonstrating improved rate performance despite the initially lower capacity.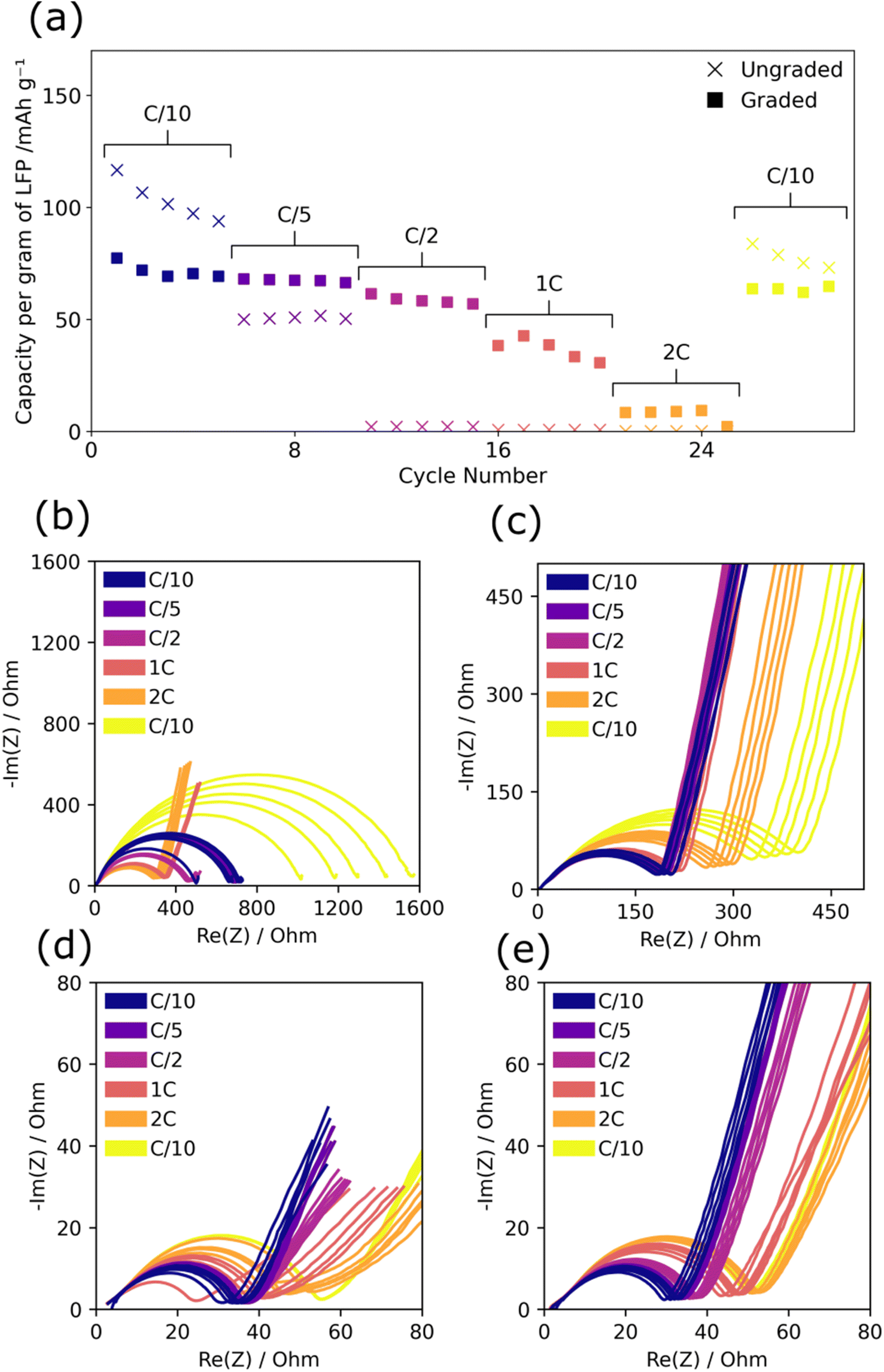 | ||
| Fig. 3 The rate performance and charge and discharge EIS of graded and ungraded composite LFP/PEO cathodes against lithium metal. (a) Voltage vs. capacity plot at increasing rates of both graded and ungraded cathodes. (b–d) EIS of graded and ungraded cathodes per cycle in either the charged or discharged state with (b) ungraded charged, (c) ungraded discharged, (d) graded charged, (e) graded discharged. | ||
Similarly to cycle life testing, the total resistance of the graded cathodes was substantially lower than ungraded cathodes at all points (graded between 20 Ω and 60 Ω and ungraded between 170 Ω and 1600 Ω), see Fig. 3(b–e). In all cases except for the ungraded charged EIS (b), the measured resistance increased with rate and the final C/10 cycles show around twice the resistance of the initial C/10 cycles. This suggests that there was some permanent degradation that occurred with high-rate cycling. The DRT analysis of the charged states in ESI Fig. 3 (a and c)† shows that the charge transfer resistance peak for the final C/10 cycles was higher than for the initial C/10 cycles with a higher increase in resistance for the ungraded composite cathode which further suggests permanent degradation of the PEO SPE/composite cathode interface.
The ungraded charged EIS, Fig. 3(b) showed slightly different behaviour as the resistance decreased with rate from C/2 onwards and then increased by a significantly larger proportion for the final C/10 going from ∼300 Ω to ∼1600 Ω. The 2C resistance value of ∼400 Ω and EIS profile showing a longer diffusion tail was similar to that of the discharged state which suggested that at high C-rates full lithiation had not taken place and that as rate was increased the charged EIS profile more resembled the discharged state. The DRT analysis in ESI Fig. 3(a)† shows a high diffusion peak (∼102 s−1) during 1C and 2C testing for the ungraded charged state which confirms that there was minimal lithiation due to the lack of diffusion throughout the PEO structure. This was confirmed by the low capacity recorded for the ungraded cells between C/2 and 2C and may also explain the high capacity seen in the final C/10 cycles. At high rates, limited lithiation has occurred therefore there is minimal degradation and the capacity remains abnormally high, although in repeats the recorded capacity of the final C/10 cycles varies greatly, see ESI Fig. 1(c).† This phenomenon was not present in the graded cathodes which indicates that a more complete lithiation has occurred at high C-rates. The DRT peak at ∼102 s−1 associated with lithium diffusion in the charged graded composite cathode, ESI Fig. 3(c),† follows an opposite trend to the charged ungraded composite cathode as peak height decreased with increased C-rate. This demonstrates that in the graded composite cathode, diffusion does not contribute as significant a proportion of impedance at higher rates, unlike the ungraded composite cathode in which the increase in diffusion-based impedance prevents any real lithiation from occurring. The improved lithium diffusion in the graded composite cathodes is therefore thought to be a major factor in their superior rate performance and must be due to the changes in the PEO SPE/composite cathode interface.
Overall, the graded cathodes have lower resistance than the ungraded cathodes with smaller resistance increases at higher rates demonstrating the stability of the graded composite cathode structure to maintain good lithium mobility under high current densities. This could be due to the improved electrolyte pathways through the cathode created by the gradient which are less constricted with lower tortuosity and therefore promote fast lithium movement and reduce charge transfer resistance which manifests as superior rate performance and the ability to pass higher current loads. Alongside this, the lack of a distinct electrolyte/electrode boundary promotes ionic contact and reduces rate-based degradation of the interface.
3.3 PEO oxidation
PEO oxidation was observed during cycle life and rate testing for the graded composite cathodes but not for the ungraded. This oxidation manifests as a noisy or infinite charge profile starting after ∼40 cycles in the cycle life testing and from C/2 onwards in rate testing, see Fig. 4. PEO oxidation can also account for the slight instability in recorded capacity of the graded composite cathode between cycles 20 and 40 in Fig. 2(a). The EIS analysis discussed in Fig. 2(d) show that during these cycles the shape of the diffusion tail changes; the DRT analysis in ESI Fig. 2(c)† also shows that the diffusion peak reduces in size for this period. PEO oxidation and the subsequent impact on lithium diffusion may have caused the capacity fluctuation.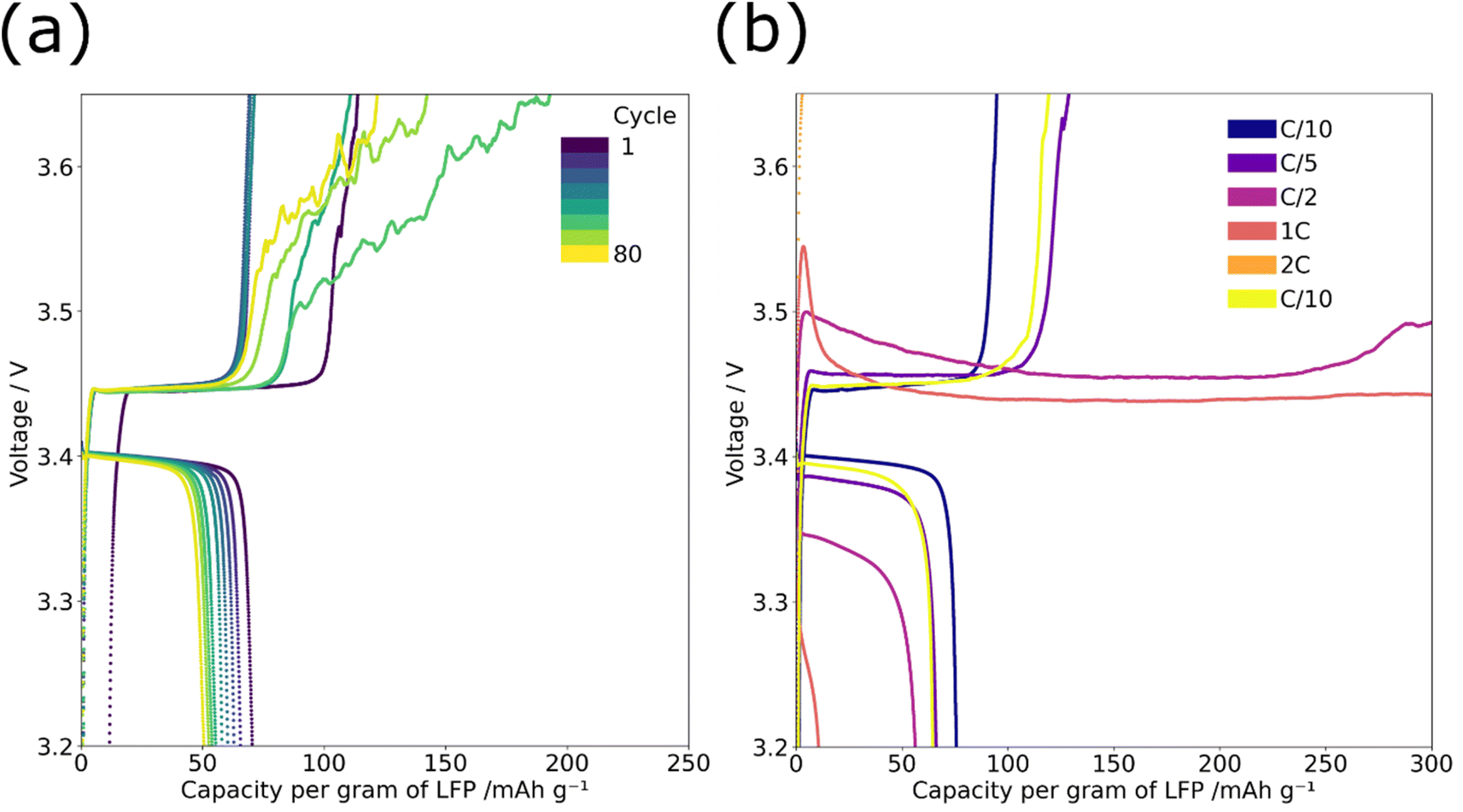 | ||
| Fig. 4 The voltage capacity curves of graded composite cathodes showing charge and discharge for (a) cycle life and (b) rate capability for every 5 cycles demonstrating the noisy/infinite charge profiles of PEO oxidation. | ||
PEO oxidation is a phenomenon that is discussed often in the literature with various oxidation voltages proposed for the decomposition of the PEO chain and terminal groups. Seidl et al. suggest that PEO oxidation of terminal OH groups occurs at 3.2 V vs. Li/Li+ and the oxidation of the PEO chain occurs at 3.6 V vs. Li/Li+;3 while there is a degradation of the PEO chain above 3.6 V vs. Li+ it is thought there is no significant loss of Li+ conductivity below 3.9 V vs. Li/Li+.4 It is likely that the oxidation of PEO is exacerbated here due to the use of shorter-chain PEO (100000 MV) which has a higher ratio of terminal OH groups; short-chain PEO was used because ultrasonic spray deposition is sensitive to the viscosity of the material.
PEO oxidation was only seen with the graded cathodes; the gradient structure places fewer LFP particles on the surface of the electrode and this may impact the spatial distribution of the electrochemical reactions within the electrode. It is known that there is a reaction rate gradient in an electrode, based on lithium ion diffusion, with the surface of the electrode closest to the electrolyte experiencing faster reaction rates than the material adjacent to the current collector.18,19 When this effect is combined with the gradient of active material within the cathode it likely places a large current density on the few LFP particles near the surface which raises the potential difference in these localised areas to the point at which the oxidation of PEO occurs. This effect, which is closely linked to current density, therefore would be amplified at higher rates; this is evident in Fig. 4(b) where the effect did not appear in the cycling behaviour until C/5 when a noisy elongated voltage profile was seen. At rates above C/2 the voltage profile changed further and an infinite charge was recorded, this suggests that oxidation is interminable with higher current densities, likely due to greater localised potential differences.
In ungraded cathodes the high Li+ flux at the electrode surface was more evenly distributed across more LFP particles therefore the localised potential difference remains below the oxidation limit and no noisy or infinite charge profiles were seen. While this remains an issue with PEO as a solid electrolyte material there are many suggested methods2–6,19–22 to prevent oxidation at lower voltages such as polymer blending,2 adding fillers,20 sacrificial electrolyte additives21 and cathode coating.22 To keep this work as widely relevant to other literature studies as possible, these methods to prevent PEO oxidation were not attempted here. While this is a clear challenge associated with PEO-based electrolytes this work shows the viability of graded composite cathodes in improving charge transfer at electrode electrolyte interfaces thereby increasing cycle life and rate performance. This technique is relevant to other solid-state batteries and spray-deposited gradient structures provide a method for more electrode design alongside simple manufacturing techniques.
4 Conclusions
Bi-nozzle spray deposition was used to manufacture graded LFP/PEO composite cathodes. EIS analysis showed that graded LFP/PEO/Li cells had 10 times lower resistance than ungraded cathodes and maintained stable capacity after 80 cycles and continued to provide capacity at a rate of 2C. By contrast, ungraded composite cathodes manufactured by the same method retained almost no capacity after 80 cycles and did not function at rates higher than C/5. It is thought that the graded composite cathode structure provides better lithium-ion pathways throughout the electrode and improves the electrode/electrolyte contact by removing the distinct planar boundary thus preventing degradation of the interface. Despite lower initial cycle capacity at low rates, graded composite cathodes have longer cycle life and better rate performance than ungraded electrodes. Spray deposition therefore represents a one-step manufacturing method for composite cathodes which allows for precise through-thickness structures to be designed and implemented, in contrast to traditional homogenous slurry-based approaches. This approach is not limited to PEO or even polymer-based solid electrolytes and could be utilised to prevent cathode/electrolyte interface degradation in a range of solid-state battery materials.Data availability
Data for this article, including raw electrochemical cycling data and electrochemical impedance spectroscopy are available at the UCL Research Data Repository at https://doi.org/10.5522/04/27908829.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TSM acknowledges the Faraday Battery Challenge through the High Silicon Content Anodes for a Solid-State Battery project (HISTORY, project number 10040711). TSM, MPT, WJD, ARTM also acknowledge the Faraday Institution through the Lithium Sulfur Technology Accelerator (LiSTAr) program (EP/S003053/1, Grant FIRG014, FIRG058) and Nextrode (Grant FIRG015, FIRG066).References
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- W. Hou, Y. Ou and K. Liu, Chem. Res. Chin. Univ., 2022, 38, 735–743 CrossRef CAS.
- L. Seidl, R. Grissa, L. Zhang, S. Trabesinger and C. Battaglia, Adv. Mater. Interfaces, 2022, 9, 2100704 CrossRef CAS.
- J. Qiu, X. Liu, R. Chen, Q. Li, Y. Wang, P. Chen, L. Gan, S.-J. Lee, D. Nordlund, Y. Liu, X. Yu, X. Bai, H. Li and L. Chen, Adv. Funct. Mater., 2020, 30, 1909392 Search PubMed.
- G. Homann, L. Stolz, J. Nair, I. C. Laskovic, M. Winter and J. Kasnatscheew, Sci. Rep., 2020, 10, 4390 CrossRef CAS PubMed.
- Y. Xia, T. Fujieda, K. Tatsumi, P. P. Prosini and T. Sakai, J. Power Sources, 2001, 92, 234–243 CrossRef CAS.
- V. Wurster, C. Engel, H. Graebe, T. Ferber, W. Jaegermann and R. Hausbrand, J. Electrochem. Soc., 2019, 166, A5410 Search PubMed.
- Y. Zhu, X. He and Y. Mo, J. Mater. Chem. A, 2016, 4, 3253–3266 RSC.
- C. Cheng, R. Drummond, S. R. Duncan and P. S. Grant, J. Power Sources, 2020, 448, 227376 Search PubMed.
- W. Deng, W. Shi, Q. Liu, J. Jiang, X. Li and X. Feng, ACS Sustain. Chem. Eng., 2020, 8, 17062–17068 CrossRef CAS.
- C. Huang, M. Dontigny, K. Zaghib and P. S. Grant, J. Mater. Chem. A, 2019, 7, 21421–21431 RSC.
- E. G. Sukenik, L. Kasaei and G. G. Amatucci, J. Power Sources, 2023, 579, 233327 CrossRef CAS.
- J. van den Broek, S. Afyon and J. L. M. Rupp, Adv. Energy Mater., 2016, 6, 1600736 CrossRef.
- S. Berg, D. Kutra, T. Kroeger, C. N. Straehle, B. X. Kausler, C. Haubold, M. Schiegg, J. Ales, T. Beier, M. Rudy, K. Eren, J. I. Cervantes, B. Xu, F. Beuttenmueller, A. Wolny, C. Zhang, U. Koethe, F. A. Hamprecht and A. Kreshuk, Nat. Methods, 2019, 16, 1226–1232 CrossRef CAS PubMed.
- W. J. Dawson, A. R. T. Morrison, F. Iacoviello, A. M. Boyce, G. Giri, J. Li, T. S. Miller and P. Shearing, Batteries Supercaps, 2024, e202400260 CrossRef CAS.
- Z.-Y. Mao, Y.-P. Sun and K. Scott, J. Electroanal. Chem., 2016, 766, 107–119 CrossRef CAS.
- M. Hahn, D. Rosenbach, A. Krimalowski, T. Nazarenus, R. Moos, M. Thelakkat and M. A. Danzer, Electrochim. Acta, 2020, 344, 136060 CrossRef CAS.
- A. J. Pearse, E. Gillette, S. B. Lee and G. W. Rubloff, Phys. Chem. Chem. Phys., 2016, 18, 19093–19102 RSC.
- O. J. Borkiewicz, K. W. Chapman and P. J. Chupas, Phys. Chem. Chem. Phys., 2013, 15, 8466 RSC.
- B. Chen, H. Huang, Y. Wang, Z. Shen, L. Li, Y. Wang, X. Wang, X. Li and Y. Wang, ChemElectroChem, 2022, 9, e202101277 CrossRef CAS.
- J. Tan, X. Li, Q. Li, Z. Wang, H. Guo, G. Yan, J. Wang and G. Li, Ionics, 2022, 28, 3233–3241 CrossRef CAS.
- K. Nie, X. Wang, J. Qiu, Y. Wang, Q. Yang, J. Xu, X. Yu, H. Li, X. Huang and L. Chen, ACS Energy Lett., 2020, 5, 826–832 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se01736f |
| This journal is © The Royal Society of Chemistry 2025 |