
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Origins of intrinsic p-type conductivity, p–n transition and substoichiometry in SrO†
Taifeng
Liu‡
 *ab,
Xingfan
Zhang‡
b,
Jingcheng
Guan
b,
Xuebo
Chen
*ac,
You
Lu
d,
Thomas W.
Keal
d,
John
Buckeridge
e,
C. Richard A.
Catlow
bf and
Alexey A.
Sokol
*b
*ab,
Xingfan
Zhang‡
b,
Jingcheng
Guan
b,
Xuebo
Chen
*ac,
You
Lu
d,
Thomas W.
Keal
d,
John
Buckeridge
e,
C. Richard A.
Catlow
bf and
Alexey A.
Sokol
*b
aNational & Local Joint Engineering Research Center for Applied Technology of Hybrid Nanomaterials, College of Chemistry and Molecular Sciences, Henan University, Kaifeng 475004, P. R. China. E-mail: tfliu@vip.henu.edu.cn
bKathleen Lonsdale Materials Chemistry, Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: a.sokol@ucl.ac.uk
cKey Laboratory of Theoretical and Computational Photochemistry of Ministry of Education, Department of Chemistry, Beijing Normal University, Beijing 100875, P. R. China. E-mail: xuebochen@bnu.edu.cn
dScientific Computing Department, STFC Daresbury Laboratory, Warrington WA4 4AD, Cheshire, UK
eSchool of Engineering, London South Bank University, 103 Borough Road, London SE1 OAA, UK
fSchool of Chemistry, Cardiff University, Park Place, Cardiff, CF10 1AT, UK
First published on 4th February 2025
Abstract
To understand the complex electrical behaviour and deviations from ideal stoichiometry in strontium oxide we have investigated its defect chemistry using a hybrid quantum mechanical/molecular mechanical (QM/MM) embedded-cluster approach. Depending on the temperature and oxygen partial pressure, oxygen interstitials or strontium and oxygen vacancies are found to dominate, while strontium interstitials are rare. Notably, charge-neutral oxygen interstitials form a peroxy-like closed-shell configuration, which is the commonest native point defect in SrO under normal conditions explaining the Sr substoichiometry which is not electrically active. Formally charged double acceptors strontium vacancies prove to be the primary source of the hole excess over negative carriers supplied by donor species, contributing to the material's p-type conductivity. Based on our calculations, we predict that at ultralow oxygen partial pressure (P = 1.0 × 10−15 bar) and high temperatures (>1100 K) in SrO, the electron concentration surpasses the hole concentration, which has previously been reported in pure BaO (also at about 1100 K) and the double barium–strontium oxide (at 850 K) by D. W. Wright, (Nature, 1949, 4173, 714) with the oxygen split interstitial acting as a donor. On increasing the oxygen partial pressure, the hole concentration exceeds the electron concentration, resulting in effective p-type conductivity. Only under low oxygen pressures (e.g., 10−8 bar) and high extrinsic donor concentrations (>1017 cm−3) might SrO switch to n-type conductivity at high temperatures (>1250 K). This study provides essential insights into intrinsic defects and mechanisms of SrO's p-type conductivity, aiding in understanding and predicting other p-type materials.
1. Introduction
Strontium oxide (SrO) belongs to the alkaline-earth oxides family, characterized by its highly symmetrical rock salt structure. It is a wide band gap semiconductor (5.9 eV) with a high exciton binding energy and electrochemical stability,1 making it suitable for diverse applications including catalysis and microelectronics.2,3 SrO could potentially serve as a candidate for diluted half-metallic ferromagnetic semiconductors (DMSs) when doped with non-magnetic elements like carbon4 and nitrogen5 or transition metals.6 Intrinsic defects in SrO play a crucial role in all these applications. The p-type conductivity of SrO observed in experiments7 may be attributed to the intrinsic defects.Intrinsic defects in SrO have been widely explored experimentally. Kappers8 reported observation of the intrinsic V− (a positive hole trapped at a cation vacancy) and V0 centers (two holes trapped at a cation vacancy) in single-crystal SrO on a sample x-irradiated at 77 K using electron spin resonance (ESR) spectroscopy. Tench et al.9 studied the formation of cation and anion vacancies in SrO after irradiation by 20 MeV protons, again using ESR. They found that the rate of formation of cation vacancies fits a model of interstitial-vacancy recombination. The efficiency of vacancy production is an order of magnitude less than that predicted for primary displacements confirming the importance of recombination between vacancies and those interstitials formed within a critical volume around the vacancy. Seeman et al.10 found by ESR a new type of V center in SrO, which consists of an O− ion (localized hole) near a cation vacancy surrounded by two OH− groups. They also found that there are several types of superoxide (O2− centers) in SrO in x-irradiated SrO single crystals using ESR. All these centers consist of an O2− molecule stabilized near various defects.11 Johnson and Hensley12 reported that the observation of the F centers (oxygen vacancies containing two electrons) at room temperature peaked at 2.5 eV and the F+ centers (oxygen vacancies containing one electron) peaked at about 3.1 eV. Feldott and Summers13 observed that the F+ band appears to be carried by electrons and has a thermal activation energy of 0.1 eV.
Intrinsic defects in SrO have also been widely studied using theoretical calculations. Yamamoto et al.14 investigated the Vo in SrO using first-principles PAW calculations with the Perdew–Burke–Ernzerhof (PBE) generalised gradient approximation (GGA) functional. The main conclusion was that the energy state of the neutral oxygen vacancy is located at 1.02 eV below the conduction band minimum (CBM). They also found that the formation of the oxygen vacancy is enhanced under a reducing atmosphere. Sun et al.15 also investigated the Sr vacancy in SrO using the GGA + U approach with U applied on O 2p orbitals. They found that the neutral and singly charged Sr vacancy could introduce a magnetic moment into the SrO crystal which leads to the material showing half-metallicity. The same authors16 reported that the optical absorption band of F and F+ color centers peak at 2.49 eV and 3.06 eV, respectively by using hybrid HSE calculations. Middleburgh et al.17 reported using GGA-PAW pseudopotentials that the accommodation of excess oxygen in SrO results in the formation of a peroxide ion centred on an oxygen site, rather than forming a single oxygen species.
Despite this extensive range of studies, there is no comprehensive understanding of all types of intrinsic defects in SrO nor of the reason for its native p-type conductivity. Previous theoretical studies have primarily utilized supercell approaches, which have limitations in modeling charged defects.18,19 In this work, we investigated intrinsic defects in SrO using the hybrid quantum mechanical/molecular mechanical (QM/MM) embedded-cluster method,20–23 which avoids spurious interactions between periodic images of charged defects and thus overcomes the disadvantage of the supercell approach. We examine oxygen and strontium vacancies, single oxygen and strontium interstitials, and a peroxide-like oxygen interstitial using the PBE0 (ref. 24) and BB1K25 hybrid functionals. Formation energies of various defects are calculated, and the origin of the p-type conductivity related to these defects is investigated.
2. Computational details
2.1 QM/MM calculations
The hybrid QM/MM embedded-cluster method21 has been employed to model the relevant point defects in SrO within the ChemShell package.23,26,27 In this approach, a Sr-centered 129-atom QM cluster was embedded within a large cluster with 7599 atoms in total. The QM cluster is treated by hybrid density functional theory (DFT) and the surrounding region is treated at the MM level of theory. The interface region between the QM and MM regions serves as the buffer layer to minimize the mismatch of the QM and MM levels of theory. Here, we use a specially designed local embedding pseudopotential on the cationic sites in the form of a linear combination of three Gaussian functions19 with the fitted parameters in atomic units as given below:r2Up(r) = −53.9247r![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e−20.5435r2 + 19.9965r2e−1.3535r2 +1.973r2e−0.762r2 e−20.5435r2 + 19.9965r2e−1.3535r2 +1.973r2e−0.762r2 | (1) |
For the QM part of the calculations, we use Density Functional Theory (DFT) with triple-zeta plus polarization Gaussian basis set28 on oxygen and strontium, including a 28-electron effective core potential (ECP) on Sr,29 as implemented in the NWCHEM30 software. To reduce the computational load, we have removed the f function from the oxygen basis set and some of the highly diffuse functions from the strontium basis sets, which do not contribute to ionic bonding. As mentioned, for electron exchange and correlation, the PBE0 (ref. 24 and 31) functional with 25% exact exchange, and BB1K25 functional with 42% exact exchange are used.
For the MM part of the simulation, pairwise Buckingham interatomic potentials are used in the context of the shell model:32
 | (2) |
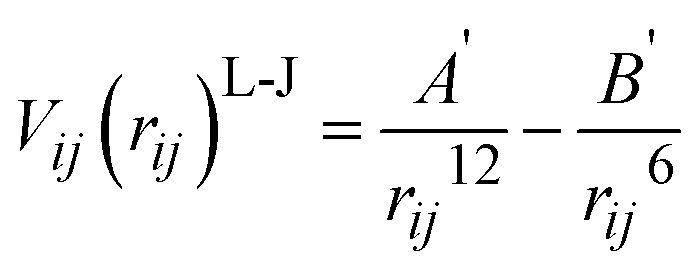 | (3) |
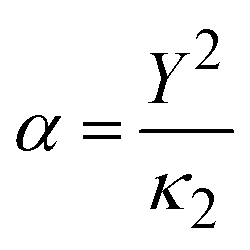 , where Y is the shell charge. The electrostatic Coulomb interaction is calculated by
, where Y is the shell charge. The electrostatic Coulomb interaction is calculated by 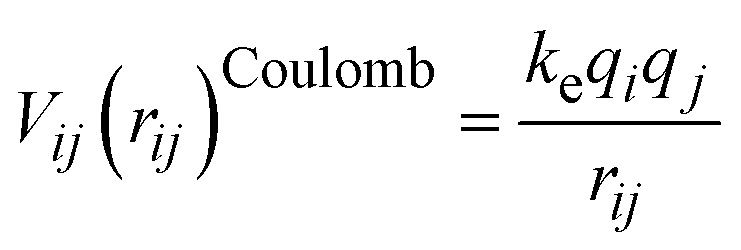 with ke, the dimensional Coulomb constant (14.3996 eV Å e−2).
with ke, the dimensional Coulomb constant (14.3996 eV Å e−2).
The General Utility Lattice Package (GULP) code33,34 is employed for the MM part of the calculation, which is divided into two regions. One is the MM-active region where all the atoms are allowed to relax, while the other is the MM-frozen region where all the atoms are kept fixed to simulate the bulk environment. The outermost layer of the whole cluster is fitted point charges, and its role is to eliminate the effects of surface termination and reproduce the Madelung potential of bulk SrO.
We employed the GULP code to develop a new shell-model interatomic potential for SrO. Our previous work has demonstrated the importance of explicit incorporation of relative ionic polarisabilities in fitting the shell-model potential for metal oxides.18 This approach significantly enhances the capability to accurately model the electrostatics and polarisation in defect and surface calculations.18,35 To calculate the ionic polarisabilities of Sr2+ and O2− in the SrO lattice, we employed an embedded-cluster model, where a single QM ion was surrounded by MM ions as the environment. DFT calculations were performed at the PBE0 level of theory with the same basis set as one used in defect QM/MM calculations using NWChem.30 Computational details can be found in our previous work.18 Curiously, a consistent polarisability of 5.71 a.u. was obtained for both Sr2+ and O2− ions. Therefore, we imposed a constraint to equalise the k2 values on both ions in the potential fitting. Parameters of oxygen–oxygen repulsion potential were taken from ref. 36. By adjusting the shell charges and fitting other short-range parameters against experimental data, an optimised potential was determined for SrO, as shown in Table 1.
| (a) Short range potentials | |||||||
|---|---|---|---|---|---|---|---|
| Interaction | A (eV) | ρ (Å) | C 6 (eV Å6) | A′ (eV Å12) | B′ (eV Å6) | r min (Å) | r max (Å) |
| O2− shell–Sr2+ core | 1865.3676 | 0.348774 | 0.0 | 10.0 | 43.0 | 0 | 15 |
| O2− shell–O2− shell | 22764.3 |
0.149 | 0.0 | 10.0 | 43.0 | 0 | 15 |
| (b) Shell model | ||
|---|---|---|
| Species | Y (e) | k 2 (eV Å−2) |
| Sr2+ shell | −2.45 | 45.235768 |
| O2− shell | −2.45 | 45.235768 |
The performance of the new potential set in modelling bulk SrO is shown in Table 2 in comparison to experiment and DFT results. In general, the new potential is able to accurately reproduce the lattice constant and dielectric constants, which are critical to QM/MM modelling. The calculated elastic properties, phonon frequencies, and lattice energy are also in good agreement with experimental and theoretical data.
| Observable | Shell model | Expt. | DFT |
|---|---|---|---|
| a 0 (Å) | 5.1602 | 5.1615 (ref. 37) | 5.17 (PBE0),38 5.12 (PBEsol0)38 |
| ε 0 | 13.09 | 13.1 (ref. 39) | |
| ε ∞ | 3.11 | 3.46 (ref. 39) | 3.03 (PBE0),38 3.09 (PBEsol0)38 |
| C 11 | 198.59 | 165,40 175 (ref. 41) | 170 (GGA)42 |
| C 12 | 70.82 | 49 (ref. 41) | 48 (GGA)42 |
| C 44 | 70.82 | 56 (ref. 40 and 41) | 59 (GGA)42 |
| B 0 (GPa) | 113.41 | 86,40 89 (ref. 41) | 89 (GGA)42 |
| TOΓ (cm−1) | 244.93 | 224.2,43 234.2 (ref. 44) | 217.85 (LDA)45 |
| LOΓ (cm−1) | 502.91 | 485.0,43 482.7 (ref. 44) | 474.13 (LDA)45 |
| ΔHL (eV) | −34.16 | −33.40 (ref. 46) |
2.2 Calculations of the intrinsic defects formation energies
The formation energy of a defect in the charge state q is defined as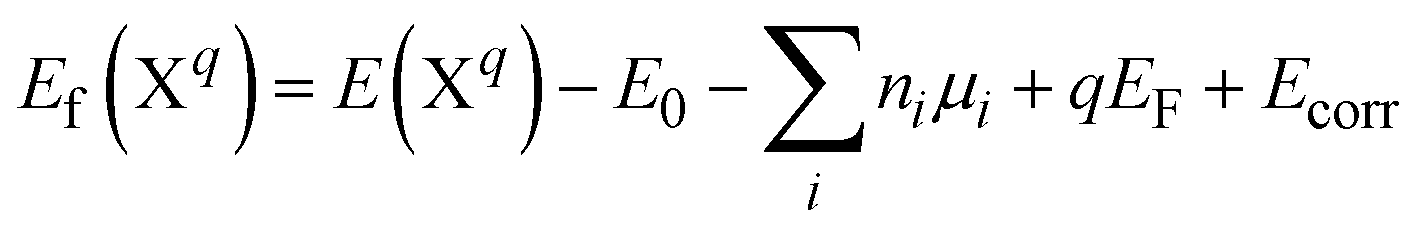 | (4) |
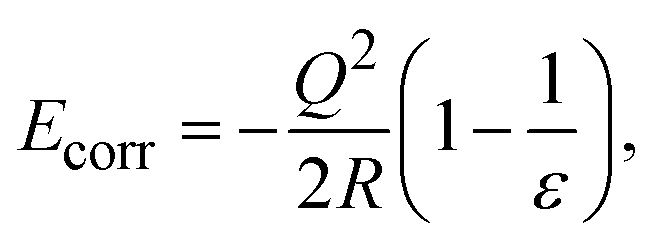 | (5) |
The formation of charged defects also depends on the growth conditions of the materials. In the O-rich/Sr-poor conditions, the upper limit is determined by the formation of O2 molecules, in which μO = 0 eV and μSr = ΔHf(SrO). In the O-poor/Sr-rich conditions, the lower limit of μO is determined by the SrO. In the QM/MM calculations, the reference energies for an O2 molecule and a single Sr2+ ion are calculated using the NWCHEM with the corresponding basis set and functional. The sum of the first two ionization potentials of Sr (5.094 eV and 11.030 eV (ref. 46)) and the sublimation enthalpy of Sr46 (ΔHsubl(Sr), 1.70 eV) are then used to obtain the reference energy of Sr(s). The SrO formation enthalpy (−6.182 eV) is calculated using the SrO cohesive enthalpy (10.45 eV (ref. 47)), the sublimation enthalpy of Sr, and the O2 dissociation energy (5.136 eV (ref. 46)).
2.3 Oxygen chemical potential
As shown in eqn (4), the formation energy of a point defect is a function of the Fermi level and respective chemical potentials, which are therefore affected by the experimental conditions, in particular, the oxygen partial pressure P and temperature T. Reuter and Scheffler48 give the relationship between the oxygen chemical potential μO(T,P) and the oxygen partial pressure P as following: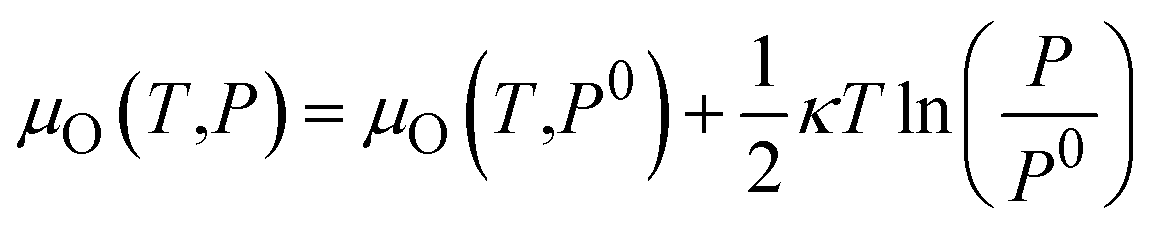 | (6) |
| μO(T,P0) = ½[H(T,P0,O2) − H(0 K,P0,O2)] − ½T[S(T,P0,O2) − S(0 K,P0,O2)] | (7) |
2.4 Charge carrier and defect concentrations
The self-consistent Fermi energy level and the equivalent defect and carrier concentrations can be obtained using the defects formation energy as shown in eqn (4). The concentration of a defect X in charge state q is calculated by: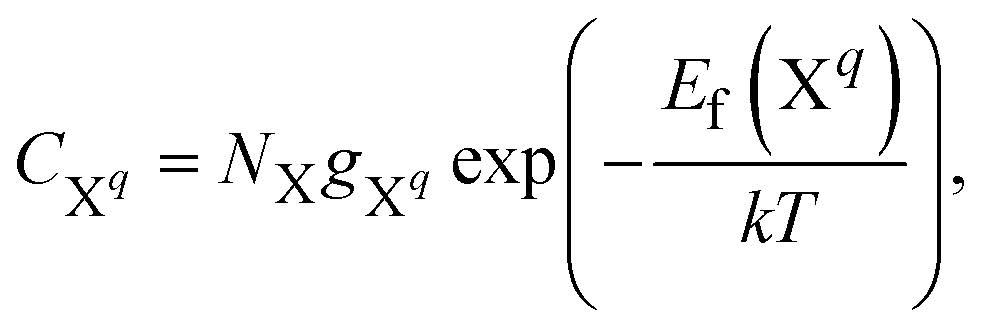 | (8) |
The electron n0 and hole p0 concentrations can be calculated as:
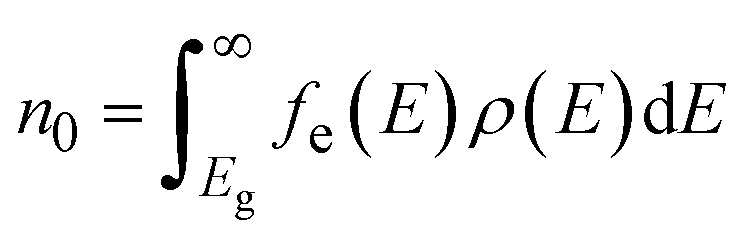 | (9) |
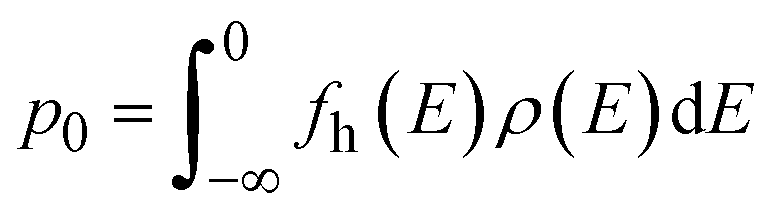 | (10) |
 | (11) |
| fh(E) = 1 − fe(E) | (12) |
To calculate ρ(E) of SrO, the Vienna Ab initio Simulation Package (VASP)51–54 code is employed using the PBE0 functional with the projector augmented wave (PAW)55 method to model the interactions between the core and active, or valence electrons. There are ten and six valence electrons in Sr and O atoms. The crystallographic unit cell containing eight atoms is used in the DOS calculations. The plane wave cutoff energy is 650 eV and 9 × 9 × 9 Monkhorst–Pack56k-points mesh is used. The convergence of the total energy and total force are 1 × 10−6 eV and 0.001 eV Å−1, respectively.
3. Results and discussion
SrO is an archetypal rock-salt structured binary oxide with well-known basic defects, i.e., cation and anion vacancies and interstitials.57,58 In the following sections, we focus on the results of our calculations of the energy of formation for these defects in their main charge states before we turn our attention to the problem of thermodynamic equilibrium between different defects and charge carriers in this system.3.1 Formation energies of oxygen and strontium vacancies
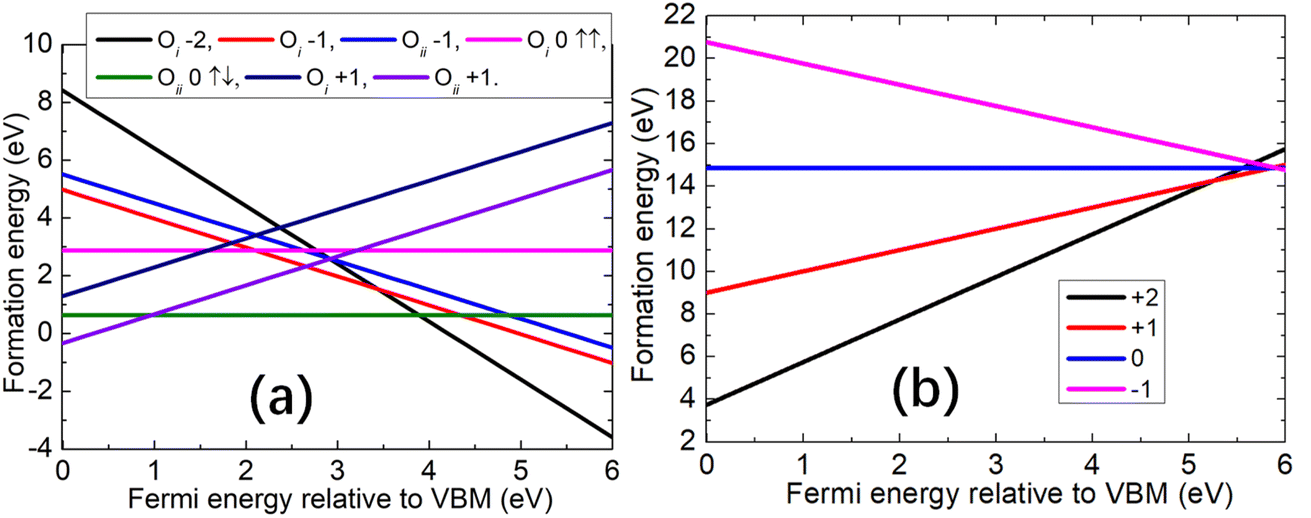
We first consider the formation of the oxygen vacancy (Vo) and strontium vacancy (VSr). For Vo, the formation energies of the charge states of +2, +1, 0, and −1 are calculated. The charge neutral state can have both singlet and triplet configurations, for which calculations have been performed; for the −1 charge state, both the low and high spin configurations are investigated. For VSr, the formation energies of the charge states of −2, −1, 0, +1, and +2 are considered. For the neutral VSr, we found that the difference in energy between the singlet and triplet is 82 (121) meV using the PBE0 (BB1K) functional. For the charge state of +1, the high spin state is 19 (14) meV lower than that of the low spin state. For the charge state of +2, the spin configurations are singlet, triplet, and quintuplet. The energy of the quintuplet is 24 (6) meV lower than the triplet state, and 38 (7) meV lower than the singlet state. Thus, all the energies of different spin configurations in the VSr are close, so we did not include them in the formation energy diagram where only the one with the lowest energy is shown. The formation energies were calculated using the PBE0 and BB1K functionals, but there is little difference between the results for the two functionals as shown in the ESI.† Taking the neutral case of Vo and VSr, we found the formation energy difference of these two defects is only 0.2 eV. Therefore, we only show the results calculated using the PBE0 functional.For the calculations of vacancies, we consider both the O-rich and O-poor conditions. In the O-poor conditions, the formation energy of Vo will shift down the enthalpy of formation of SrO which is measured at about 6.182 eV.59 The oxygen vacancy has a lower formation energy under O-poor conditions indicating it will be easier to form.
The structure of the charge neutral Vo is shown in Fig. 1(a), and we found that there is little distortion around the oxygen vacancy. The formation energies of Vo as a function of the Fermi energy level relative to the VBM under O-rich are shown in Fig. 1(b). Importantly, for the Vo, the formation energy of the low spin configuration is more than 1 eV lower compared to the high spin configuration in both charge states 0 and −1. From Fig. 1(b), we also find that if the Fermi level lies between the VBM and the middle of the band gap, Vo with charge +2 has the lowest formation energy. Almost 1 eV above the middle of the band gap, the charge +1 Vo dominates. We use (n/m) to denote the charge of Vo changing from q = n to q = m, and for (+2/+1), the transition energy level is ∼2.85 eV above the VBM. For the remaining range close to the CBM, charge-neutral oxygen vacancies are favoured, and the transition energy level for (+1/0) is ∼4.4 eV above the VBM.
 | ||
| Fig. 1 (a) Structures of charge neutral oxygen vacancy, and (b) formation energies of oxygen vacancies, and (c) structures of the charge neutral strontium vacancies, and (d) formation energies of strontium vacancies, with the crossing point shown in (e). All the formation energies are plotted as a function of the Fermi energy level relative to the valence band maximum (VBM) under O-rich conditions. | ||
The structure of the charge neutral VSr is shown in Fig. 1(c) and there is a some distortion of the oxygen atoms that originally bonded to the strontium. The formation energy diagrams for VSr under O-rich conditions are shown in Fig. 1(d) with the crossing point shown in Fig. 1(e). In contrast to Vo, the formation energies of VSr under O-poor conditions shifts up 6.182 eV (ref. 59) indicating it is favored under O-rich conditions. Close to the VBM, the charge +2 states of VSr have the lowest formation energy, and for the larger part of the band gap, the charge state −2 predominates. The energy transition level of (+2/−2) of VSr lies at ∼2.80 eV. Around 2.80 eV above VBM, there is a small region where all charge states (from −2 to +2) have similar formation energies.
3.2 Formation energies of oxygen and strontium interstitials
As noted, SrO is a cubic rock-salt structured material, and interstitial defects might be expected to occupy a special high-symmetry cubic site with fractional coordinates (0.25, 0.25, 0.25). For the charge neutral interstitial oxygen case, we found the geometry of the singlet and triplet configurations are quite different. In the triplet configuration, the interstitial oxygen sits in the center of the distorted cube as shown in Fig. 2(a). In contrast, for the singlet configuration, the interstitial oxygen atom forms an O–O bond with an oxygen ion of the cube as shown in Fig. 2(b) with a bond length of 1.40 Å and ∼2.2 eV lower in energy than the triplet.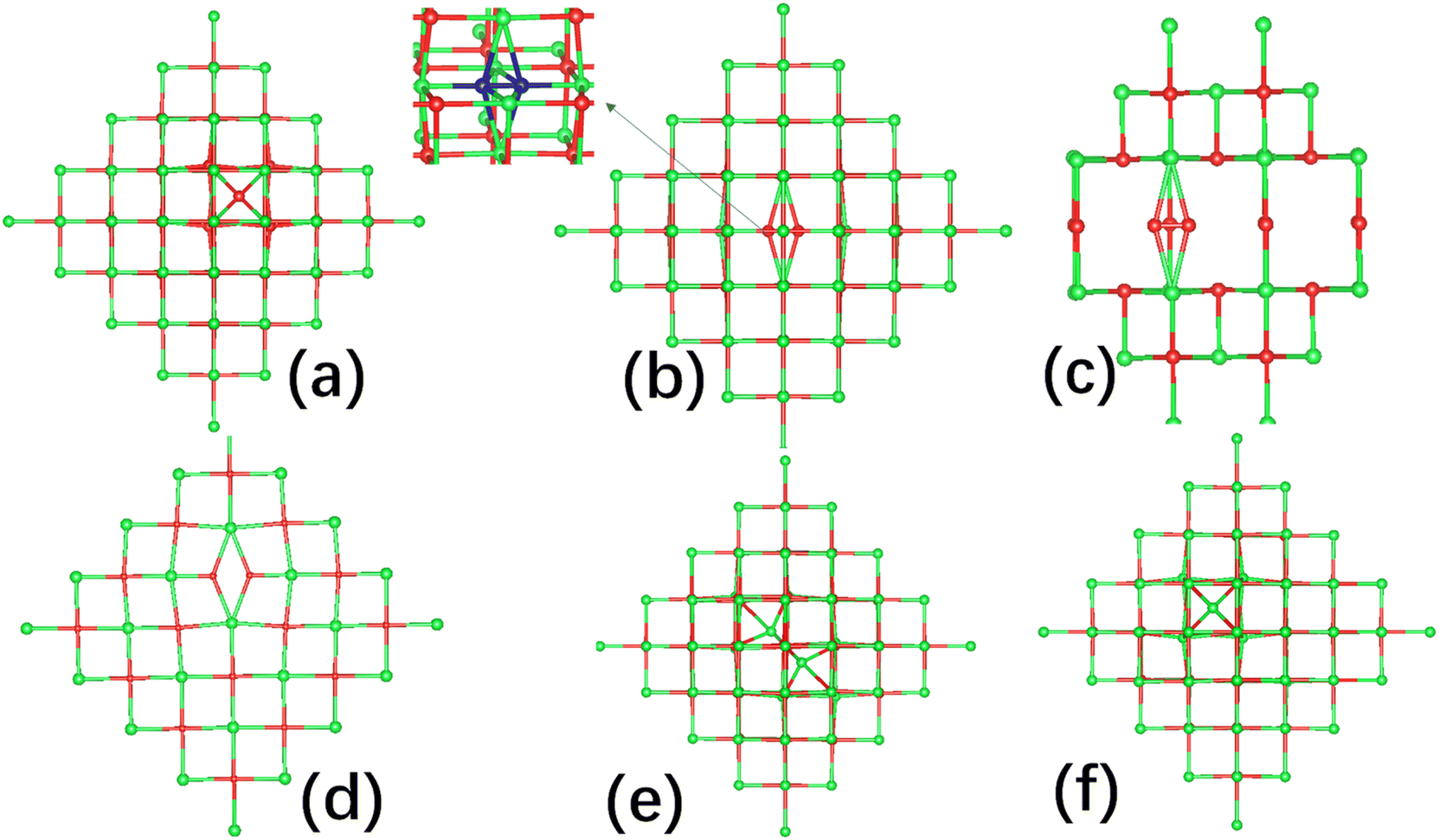 | ||
| Fig. 2 The geometry of interstitial defects in SrO. (a) Cubic interstitial oxygen, (b) the charge neutral split interstitial oxygen, (c) the split interstitial oxygen with charge state +1, (d) the split interstitial oxygen with charge state −1, (e) unstable split interstitial strontium, and (f) cubic interstitial strontium, Sri sitting in the center of the cube (in the ball and stick molecular diagram, green color is reserved for Sr, red for O, and the split interstitial O, peroxide ion is highlighted in blue). | ||
Thus we identify two types of oxygen interstitial which are cubic interstitial (denoted as Oi) and split interstitial (denoted as Oii). For the cubic oxygen interstitial Oi, charge states −2, −1, 0, and +1 were considered. For the split interstitial Oii, except the charge neutral case, we also investigated the charge state +1 and −1. Interestingly, in the charge state +1, we found that the O–O points two neighbouring oxygen atoms with a bond length of 1.31 Å as shown in Fig. 2(c). This is reasonable, as this species carries a +1 charge and interacts with the oxygen atoms which take negative charges. Similarly, in charge −1, we found the O–O is aligned along [100] direction pointing towards strontium atoms as they take positive charge with the O–O bond length 1.87 Å shown in Fig. 2(d).
For the strontium interstitial Sri defect,60 we did not observe a defect species with a Sr–Sr bond formed on the optimization. In order not to miss this kind of defect, we explore it in more detail as follows. Nine configurations are constructed by fixing the Sr–Sr bond length within the range of 2.4 to 3.3 Å. After optimizing these configurations, we obtained an energy curve which was fitted with a fourth-order polynomial, as shown in Fig. S1 in the ESI.† We found that the lowest energy position occurs when the Sr–Sr bond length is approximately 2.9 Å, as illustrated in Fig. 2(e). The total energy of this configuration is 0.56 eV higher than that of the normal interstitial structure. We then fully optimized this configuration. Unsurprisingly, it reverted to the normal interstitial structure as shown in Fig. 2(f). We conclude that the configuration with the Sr–Sr bond is unstable and may represent a transition state towards the normal interstitial structure. Therefore, we only have one type of cubic strontium interstitial which is shown in Fig. 2(f). Similarly to the strontium vacancies, in the charge neutral case, the singlet configuration is only 0.15 eV lower than that of the triplet configuration.
The formation energy diagram for oxygen interstitial under O-rich conditions is shown in Fig. 3(a) while for the O-poor conditions the energies will shift up 6.182 eV.59 Under O-rich conditions, the formation energy of oxygen interstitial has a lower energy with the same charge state than that in O-poor conditions. For the cubic interstitial Oi in SrO, we found that the charge states of −2 have the lowest formation energies in the upper parts of the band gap. In the lowest part of the band gap, the split interstitial Oii with charge +1 has the lowest formation energy. In the middle area of the band gap, the charge neutral split interstitial Oii dominates. The corresponding transition energy level (+1/0) is ∼1.0 eV above the VBM, and above this level, the singlet charge neutral configuration Oii is preferentially formed. The transition energy level (0/−2) is ∼3.9 eV above the VBM, and above this level, the cubic interstitial configuration Oi with charge −2 will dominate. For the charge state −1, it prefers to form close to the CBM. At this charge state, the formation energy of cubic interstitial Oi is 0.53 eV lower than that of the split interstitial Oii. For the cubic interstitial Oi with charge state −1, the transition energy level (0/−1) lies at 4.3 eV. But for the split interstitial Oii, the transition energy level (0/−1) is 4.9 eV. This indicates these species could transfer to each other under photoirradiation.
 | ||
| Fig. 3 Formation energies of (a) oxygen interstitial Oi and (b) strontium interstitial Sri as a function of the Fermi energy level relative to the VBM under O-rich conditions. | ||
For Sri, from the formation energy diagram under O-rich conditions (for O-poor conditions, the energies will shift down 6.182 eV) shown in Fig. 3(b), we find across almost the whole band gap, the charge state of +2 is the most stable. There is only a small energy range below the CBM, where the charge state of +1 of Sri has a lower formation energy. The energy transition level of (+2/+1) is ∼5.3 eV above the VBM, which is close to the CBM, as Sr in a +1 charge state is too large to fit in the interstitial site. Only by losing the additional valence shell electron, as in the charge states +2, the ion can be stabilized at the cube site.
3.3 Charge carrier and defect concentrations
The equilibrium-state concentrations of charge carriers are determined by intrinsic defects in SrO which are also influenced by the oxygen partial pressure (P) and temperature. In this section, various realistic oxygen partial pressures P = 1.0 × 10−15, 1.0 × 10−8, 1.0 × 10−3, and 1 atm are considered. The equilibrium carrier and defect concentrations calculated under these conditions are shown in Fig. 4 where the code SC-FERMI50,61 is used. We note that, alternative software exploiting similar methodological approaches like the DefAP code developed by Murphy et al.62 have also been reported in the literature. | ||
| Fig. 4 The equilibrium concentrations of holes p0, electrons n0, Vo, VSr, Oi and Sri under different oxygen partial pressure P, (a) P = 1.0 × 10−15 atm, (b) P = 1.0 × 10−8 atm, (c) P = 1.0 × 10−3 atm, and (d) P = 1 atm. | ||
At lower oxygen partial pressure (P = 1.0 × 10−15 atm), we observe significant concentrations of three types of intrinsic defects (Vo, VSr, and Oi). The concentrations of Vo and VSr are comparable across a wide range of temperatures. However, below approximately 700 K, Oi exhibits the highest concentration. Between 700 K and 1100 K, the concentrations of defects Vo and VSr surpass that of Oi, leading to a rapid increase in the n0 concentration with temperature. At 1100 K, the n0 and p0 concentrations are balanced. Above 1100 K, the electron concentrations exceed those of the holes, changing the system behavior to n-type. With an increase in oxygen partial pressure, such as at 10−8 atm, a similar trend is observed as with 10−15 atm. However, the intersection point of the electron and hole carrier concentrations is reached at around ∼1500 K. Upon further increasing the oxygen partial pressure to 10−3 atm, we observed that the concentration of Oi is highest below ∼1300 K, and the hole concentrations exceed the electron concentrations from 0 to 1500 K. Similarly, at P = 1 atm, a comparable trend is observed as with 10−3 atm. Under these four oxygen partial pressures, we observed that the concentration of Sri is very low due to its high formation energy, indicating its formation to be unfavorable in SrO.
Based on these calculated results, we confirm that SrO exhibits higher hole concentrations than the electron concentration under normal experimental conditions which leads to its p-type behavior. The origin of its p-type conductivity stems from the VSr with charge −2. The doubly charged oxygen vacancy at lower Fermi levels overtakes from the singly charged which successfully competes with holes as a charge compensating species for Sr vacancies. This leads to the concentrations of holes reduced and in turn, reduced the p-type conductivity.
3.4 Charge carrier concentrations under different acceptors and donors concentrations
We also studied whether the intrinsic defects respond to the presence of ionized impurities, both acceptors and donors. In SrO, silver (Ag) substituting on a Sr site commonly acts as an acceptor, while indium (In) substituting for Sr acts as a donor. The singly ionized acceptors and donors are denoted as AgSr−, and InSr+, respectively. The concentrations of acceptors and donors chosen were 109, 1011, 1013, and 1017 cm−3. At an oxygen partial pressure P = 1 atm, the electron and hole concentrations under different acceptor and donor concentrations are shown in Fig. 5(a) and (b). We found that with higher concentrations of acceptors AgSr−, the hole concentrations are higher while the electron concentrations are lower. For the donors InSr+, it is the opposite behaviour, as expected. The hole concentrations are, however, always higher than the electron concentrations regardless of the presence of acceptors AgSr− and donors InSr+ in the system at oxygen partial pressure 1 atm which indicates that the p-type conductivity is not suppressed or inverted by the ionized impurities. When the oxygen partial pressure is reduced to 10−3 atm, similar results are found.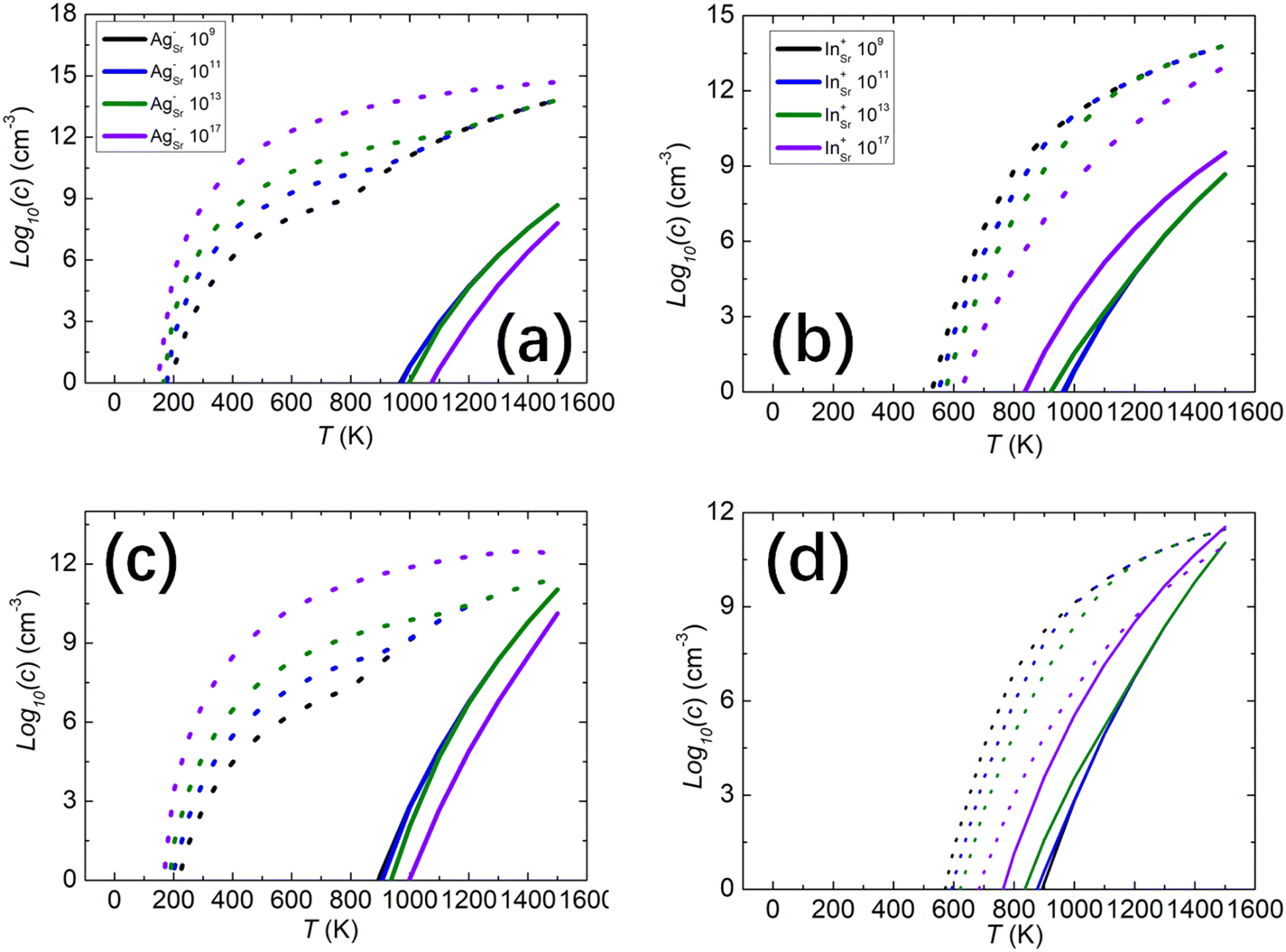 | ||
| Fig. 5 Electron (solid lines) and hole (dotted lines) concentrations in the presence of different acceptor and donor concentrations in unit with different oxygen partial pressures, (a) different AgSr− acceptor concentrations at P = 1 atm, (b) different InSr+ donor concentrations at P = 1 atm, (c) different AgSr− acceptor concentrations at P = 10−8 atm, and (d) different InSr+ donor concentrations at P = 10−8 atm. | ||
On further reducing the oxygen partial pressure to 10−8 atm, the concentration of electrons and holes begin to show differences, as illustrated in Fig. 5(c) and (d). With acceptor concentrations AgSr− of 109, 1011 and 1013 cm−3, the hole concentration is higher than the electron concentration from 200 to 1400 K. At temperatures higher than 1400 K, the electron concentration approaches the hole concentration, indicating that the system may transition from p-type to n-type conductivity. However, at high acceptor concentrations, such as 1017 cm−3, the acceptors will accept more electrons and produce more holes. The hole concentration remains higher than the electron concentration, indicating the system maintains and enhances its p-type conductivity. For low concentrations of donors InSr+, such as 109, 1011 and 1013 cm−3, the effect on electron and hole concentration is small. Only above 1400 K does the electron concentration start to exceed the hole concentration. For the rest of the temperature range, the hole concentration remains higher than the electron concentration. At higher donor concentrations, such as 1017 cm−3, the donors produce more electrons, significantly affecting the carrier concentration. Around 1100 K, the electron concentration surpasses the hole concentration, indicating a change in the system's conductivity. We note that there is a change in the trend of hole concentration around T > 800 K in the presence of acceptors in Fig. 5(a) and (c), and the reason is that the concentration of VSr becomes higher at that temperature which contributes to the hole concentrations. All these results indicate that modifying the p-type conductivity of SrO is challenging. At normal oxygen partial pressure and low donor concentrations, the p-type conductivity remains dominant. Only under low oxygen partial pressure, such as 10−8 atm, and high donor concentrations, exceeding 1017 cm−3, might its p-type conductivity switch to n-type.
3.5 Discussion
To evaluate our calculated results, we compared them with experimental data and other theoretical studies. In our calculations, the Schottky defect pair (VO2+ + VSr2−) was determined to have a formation energy of 4.47 eV using the PBE0 functional. The Frenkel defect pairs were calculated to have formation energies of 7.68 eV (VO2+ + Oi2−) and 8.25 eV (Sri2+ + VSr2−). Similar values were obtained using the BB1K functional, as detailed in the ESI.† Our results indicate that Schottky defects are the dominant defect type. This is consistent with the work of Murarka and Swalin,7,63 who reported that Schottky defects are predominant at high temperatures in conductivity experiments, with estimated formation energies of 3.7–4.0 eV, aligning well with our calculations. Ramani and Rao64 reported the Schottky defect formation energy of 4.23 eV from Mott and Littleton model calculations employing two parameter repulsive potentials which is consistent with our calculated result. Mackrodt and Stewart65 also confirmed that the Schottky pair is the most stable defect in SrO, but with a higher formation energy of 6.0 eV using their shell model potential calculations. Richardson66 reported a similar higher value of 7.5 eV using a three-body potential. However, our calculations suggest that the charge-neutral oxygen interstitial (Oi) is the dominant defect in SrO under usual conditions, except under very low oxygen partial pressures. This type of defect, however, cannot be detected by conductivity measurements. A piece of possible experimental evidence could be referred to the 2.1 eV slopes reported in conductivity measurements by Copeland and Swalin7 at T > 1000 K and P = 10−12 atm, attributed to the formation of Oi1− and a hole. Under such conditions, the chemical potential of oxygen (μO) is estimated to be at least −2.3 eV below the O-rich limit (μO = 0 eV),18 which increases the formation enthalpy of the oxygen split interstitial to at least 2.8 eV according to our PBE0 calculation. The increased entropy effect at high temperatures could account for the higher defect energies in our calculations compared to experimental measurements.4. Conclusions
In summary, our calculations on intrinsic defects in SrO, using a hybrid QM/MM embedded-cluster approach have shown that oxygen vacancies exhibit the lowest formation energies in the +2, +1, and 0 charge states, with charge-neutral vacancies having the lowest energy in the singlet state. Strontium vacancies display the lowest formation energy in the −2 and +2 charge states. Similarly, oxygen interstitials with charge states of −2 and 0 exhibit the lowest formation energies. In the neutral singlet state, a peroxide-like oxygen interstitial emerges with the lowest formation energy, characterized by the interstitial oxygen forming an O–O bond with a lattice oxygen atom. In interstitial defects, the geometry involves the interstitial oxygen atom positioned at the center of the SrO cube. Strontium interstitials are predominantly present in the +2 charge state, exhibiting high formation energies, indicating them to be unfavorable in SrO. Our analysis suggests that dominant defects in SrO primarily consist of oxygen vacancies, strontium vacancies, and oxygen interstitials, and in particular the peroxide-like oxygen interstitial. Confirmation of these findings is evident in the charge carrier and defect concentration results. Under the lowest oxygen partial pressure (P = 1.0 × 10−15 atm), the peroxide-like oxygen interstitial exhibits the highest concentration, while oxygen vacancies and strontium vacancies remain balanced. The hole concentration stems from strontium vacancies with charge −2, and it is the origin of the p-type conductivity of SrO. Our results align well with experimental observations of SrO exhibiting p-type conductivity, with strontium vacancies with charge −2 being identified as the origin. Besides, the neutral oxygen interstitial is readily formed in SrO and has the largest concentration under normal oxygen partial pressure. In general, transparent conducting oxides of p-type character are rare compared to n-type, and understanding the fundamental limitations for their existence is important. Here the physics of SrO provides a valuable insight and indeed there are successful modifications of basic SrO which already led to new p-type conducting materials such as SrCuO.67 This study offers valuable insights into the design of p-type conducting materials based on defect engineering.Data availability
The data supporting this article has been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by The Key Technologies R&D Program of Henan Province (no. 242102521002) and the National Natural Science Foundation of China (grant # 22173026, 21703054). The authors acknowledge the use of the THOMAS, YOUNG, and ARCHER2 UK National Supercomputing Service (http://www.archer2.ac.uk) via membership of U.K.'s HEC Materials Chemistry Consortium, which is funded by EPSRC (Grant No. EP/P020194, EP/T022213, EP/R029431, EP/W014378 and EP/W014580).References
- G. P. Summers, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 20, 5275–5279 CrossRef CAS.
- F. El-Sayed, M. S. A. Hussien, T. H. AlAbdulaal, A.-H. Abdel-Aty, H. Y. Zahran, I. S. Yahia, M. S. Abdel-wahab, E. H. Ibrahim, M. A. Ibrahim and H. Elhaes, J. Mater. Res. Technol., 2022, 20, 959–975 CrossRef CAS.
- Y. Wei, X. Hu, Y. Liang, D. C. Jordan, B. Craigo, R. Droopad, Z. Yu, A. Demkov, J. L. Edwards Jr and W. J. Ooms, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2002, 20, 1402–1405 CAS.
- V. An Dinh, M. Toyoda, K. Sato and H. Katayama-Yoshida, J. Phys. Soc. Jpn., 2006, 75, 093705 CrossRef.
- M. Seike, V. A. Dinh, K. Sato and H. K. Yoshida, Phys. B, 2012, 407, 2875–2878 CrossRef CAS.
- S. Berri, A. Kouriche, D. Maouche, F. Zerarga and M. Attallah, Mater. Sci. Semicond. Process., 2015, 38, 101–106 CrossRef CAS.
- W. D. Copeland and R. A. Swalin, J. Phys. Chem. Solids, 1968, 29, 313–325 CrossRef CAS.
- L. A. Kappers, Solid State Commun., 1977, 21, 883–885 CrossRef CAS.
- A. J. Tench and M. J. Duck, J. Phys. C: Solid State Phys., 1975, 8, 257 CrossRef CAS.
- V. Seeman, S. Reifman and T. Lehto, Phys. Status Solidi B, 1981, 105, 409–412 CrossRef CAS.
- V. Seeman, S. Reifman, T. Lehto and Ü. Haldre, Phys. Status Solidi B, 1980, 102, 459–465 CrossRef CAS.
- B. P. Johnson and E. B. Hensley, Phys. Rev., 1969, 180, 931–934 CrossRef CAS.
- S. J. Feldott and G. P. Summers, Solid State Commun., 1976, 18, 347–349 CrossRef.
- T. Yamamoto and T. Mizoguchi, Ceram. Int., 2013, 39, S287–S292 CrossRef CAS.
- R.-X. Sun, T.-Y. Liu, K.-L. Wu, C.-Y. Shi and J.-M. Song, J. Magn. Magn. Mater., 2021, 522, 167524 CrossRef CAS.
- R.-X. Sun, T.-Y. Liu, C.-Y. Shi, J.-M. Song and K.-L. Wu, Mater. Sci. Semicond. Process., 2021, 133, 105940 CrossRef CAS.
- S. C. Middleburgh, K. P. D. Lagerlof and R. W. Grimes, J. Am. Ceram. Soc., 2013, 96, 308–311 CrossRef CAS.
- X. Zhang, L. Zhu, Q. Hou, J. Guan, Y. Lu, T. W. Keal, J. Buckeridge, C. R. A. Catlow and A. A. Sokol, Chem. Mater., 2023, 35, 207–227 CrossRef CAS PubMed.
- J. Buckeridge, C. R. A. Catlow, M. R. Farrow, A. J. Logsdail, D. O. Scanlon, T. W. Keal, P. Sherwood, S. M. Woodley, A. A. Sokol and A. Walsh, Phys. Rev. Mater., 2018, 2, 054604 CrossRef CAS.
- P. Sherwood, A. H. de Vries, M. F. Guest, G. Schreckenbach, C. R. A. Catlow, S. A. French, A. A. Sokol, S. T. Bromley, W. Thiel, A. J. Turner, S. Billeter, F. Terstegen, S. Thiel, J. Kendrick, S. C. Rogers, J. Casci, M. Watson, F. King, E. Karlsen, M. Sjøvoll, A. Fahmi, A. Schäfer and C. Lennartz, J. Mol. Struct. THEOCHEM, 2003, 632, 1–28 CrossRef CAS.
- A. A. Sokol, S. T. Bromley, S. A. French, C. R. A. Catlow and P. Sherwood, Int. J. Quantum Chem., 2004, 99, 695–712 CrossRef CAS.
- C. R. A. Catlow, J. Buckeridge, M. R. Farrow, A. J. Logsdail and A. A. Sokol, Quantum Mechanical/Molecular Mechanical (QM/MM) Approaches, in Handbook of Solid State Chemistry, ed. R. Dronskowski, S. Kikkawa and A. Stein, Wiley-VCH, 2017, ch. 17, vol. 5, (Part 5) Theoretical Description, DOI:10.1002/9783527691036.hsscvol5012.
- Y. Lu, K. Sen, C. Yong, D. S. D. Gunn, J. A. Purton, J. Guan, A. Desmoutier, J. Abdul Nasir, X. Zhang, L. Zhu, Q. Hou, J. Jackson-Masters, S. Watts, R. Hanson, H. N. Thomas, O. Jayawardena, A. J. Logsdail, S. M. Woodley, H. M. Senn, P. Sherwood, C. R. A. Catlow, A. A. Sokol and T. W. Keal, Phys. Chem. Chem. Phys., 2023, 25, 21816–21835 RSC.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- Y. Zhao, B. J. Lynch and D. G. Truhlar, J. Phys. Chem. A, 2004, 108, 2715–2719 CrossRef CAS.
- Y. Lu, M. R. Farrow, P. Fayon, A. J. Logsdail, A. A. Sokol, C. R. A. Catlow, P. Sherwood and T. W. Keal, J. Chem. Theory Comput., 2019, 15, 1317–1328 CrossRef CAS PubMed.
- J. Kästner, J. M. Carr, T. W. Keal, W. Thiel, A. Wander and P. Sherwood, J. Phys. Chem. A, 2009, 113, 11856–11865 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. Kaupp, P. v. R. Schleyer, H. Stoll and H. Preuss, J. Chem. Phys., 1991, 94, 1360–1366 CrossRef CAS.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. Van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. de Jong, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- B. G. Dick and A. W. Overhauser, Phys. Rev., 1958, 112, 90–103 CrossRef.
- J. D. Gale, J. Chem. Soc., Faraday Trans., 1997, 93, 629–637 RSC.
- J. D. Gale and A. L. Rohl, Mol. Simul., 2003, 29, 291–341 CrossRef CAS.
- X. Zhang, T. Liu, L. Zhu, J. Guan, Y. Lu, T. W. Keal, J. Buckeridge, C. R. A. Catlow and A. A. Sokol, Angew. Chem., Int. Ed., 2023, 62, e202308411 CrossRef CAS PubMed.
- C. R. A. Catlow and J. S. Anderson, Proc. R. Soc. London, Ser. A, 1977, 353, 533–561 CAS.
- J. Bashir, R. T. A. Khan, N. M. Butt and G. Heger, Powder Diffr., 2002, 17, 222–224 CrossRef CAS.
- A. J. Logsdail, D. O. Scanlon, C. R. A. Catlow and A. A. Sokol, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 155106 CrossRef.
- J. L. Jacobson and E. R. Nixon, J. Phys. Chem. Solids, 1968, 29, 967–976 CrossRef CAS.
- P. R. Son and R. A. Bartels, J. Phys. Chem. Solids, 1972, 33, 819–828 CrossRef CAS.
- Z. P. Chang and E. K. Graham, J. Phys. Chem. Solids, 1977, 38, 1355–1362 CrossRef CAS.
- T. Tsuchiya and K. Kawamura, J. Chem. Phys., 2001, 114, 10086–10093 CrossRef CAS.
- K. H. Rieder, R. Migoni and B. Renker, Phys. Rev. B, 1975, 12, 3374–3379 CrossRef CAS.
- W. Martienssen and H. P. D. Warlimont, Springer Handbook of Condensed Matter and Materials Data, ed. W. Martienssen and H. Warlimont, Springer Berlin, Heidelberg, 2005, vol. XVIII, p. 1121, DOI:10.1007/3-540-30437-1.
- M. Souadkia, B. Bennecer and F. Kalarasse, J. Phys. Chem. Solids, 2012, 73, 129–135 CrossRef CAS.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC press, 2014 Search PubMed.
- C. Giancarlo, B.-R. Sophie, A. Bernard, N. Claudine and F. Fabio, J. Phys.: Condens. Matter, 2000, 12, 3671 CrossRef.
- K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 65, 035406 CrossRef.
- D. R. Stull and H. Prophet, JANAF thermochemical tables, National Bureau of Standards, Gaithersburg, MD, 2nd edn, 1971, DOI:10.6028/NBS.NSRDS.37.
- J. Buckeridge, D. Jevdokimovs, C. R. A. Catlow and A. A. Sokol, Phys. Rev. B, 2016, 94, 180101 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- A. Stoneham, Contemp. Phys., 1979, 20, 535–545 CrossRef CAS.
- A. M. Stoneham, Theory of Defects in Solids: Electronic Structure of Defects in Insulators and Semiconductors, Oxford University Press, 2001 Search PubMed.
- I. J. Brink and C. E. Holley, J. Chem. Thermodyn., 1978, 10, 259–266 CrossRef CAS.
- P. V. Kovtunenko and L. K. Ya, Russ. Chem. Rev., 1979, 48, 243 CrossRef.
- J. Buckeridge, Comput. Phys. Commun., 2019, 244, 329–342 CrossRef CAS.
- W. D. Neilson and S. T. Murphy, Comput. Mater. Sci., 2022, 210, 111434 CrossRef CAS.
- S. P. Murarka and R. A. Swalin, J. Phys. Chem. Solids, 1971, 32, 1277–1285 CrossRef CAS.
- G. Ramani and K. J. Rao, J. Solid State Chem., 1976, 16, 63–71 CrossRef CAS.
- W. C. Mackrodt and R. F. Stewart, J. Phys. C: Solid State Phys., 1979, 12, 5015 CrossRef CAS.
- D. D. Richardson, Phys. Status Solidi B, 1981, 106, 223–227 CrossRef CAS.
- A. Mariya Thomas, S. Salam, M. C. Santhoshkumar, S. Devasia and E. I. Anila, J. Phys.: Conf. Ser., 2019, 1172, 012008 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: (1) Energies curves as a function of the Sr–Sr bond length; (2) the data of formation energies of all the defect species using PBE0 and BB1K functionals. See DOI: https://doi.org/10.1039/d4ta07690g |
| ‡ T. Liu and X. Zhang contributed equally to this work as co-first authors. |
| This journal is © The Royal Society of Chemistry 2025 |