
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing acid–gas separations using free volume manipulation for microporous poly(arylene ether)s†
Taigyu
Joo‡
 a,
Yifan
Wu‡
b,
Tae Hoon
Lee
a,
Pablo A.
Dean
a,
Wan-Ni
Wu
a,
Timothy M.
Swager
b and
Zachary P.
Smith
a
a,
Yifan
Wu‡
b,
Tae Hoon
Lee
a,
Pablo A.
Dean
a,
Wan-Ni
Wu
a,
Timothy M.
Swager
b and
Zachary P.
Smith
a
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: zpsmith@mit.edu
bDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
First published on 27th January 2025
Abstract
To address global energy needs, traditional and renewable natural gas will likely be key energy sources for years to come. However, raw feeds require removal of impurities like hydrogen sulfide (H2S) and carbon dioxide (CO2) before use. In this study, we illustrate the key challenges of using traditional post-synthetic modification approaches to simultaneously enhance H2S/CH4 and CO2/CH4 selectivities in microporous polymer membranes, while also demonstrating how free volume manipulation (FVM) can overcome some of these challenges. By integrating tert-butoxycarbonyl-protected piperazinyl (PIP-tBOC) groups into a microporous poly(arylene ether) (PAE-1) and applying thermal treatment with oxygen to degrade the incorporated units in solid-state films, we successfully increased sorption capacity and diffusion selectivity. This modification enhanced the mixed-gas selectivity of H2S/CH4 and CO2/CH4 by 88% and 114%, respectively, compared to the original PAE-1 films. Consequently, the films achieved a combined acid gas (CAG) selectivity of 48, which approached the CAG upper bound for glassy polymers. The FVM process not only improved the selectivity of these membrane films but also markedly increased their resistance to plasticization, making them more suitable for industrial applications in acid–gas separation. This post-synthetic modification strategy, applicable to any glassy polymer containing a nucleophilic aromatic unit, provides a means to leverage the competitive sorption of H2S molecules and the molecular sieving properties of the polymer.
1. Introduction
Natural gas is a vital energy resource, playing a crucial role in meeting the world's energy demands. In 2022, global natural gas consumption increased to 150.6 trillion cubic feet, representing approximately 25% of the world's primary energy consumption.1 As a cleaner-burning fossil fuel compared to coal and oil, natural gas has been pivotal in the transition towards more sustainable energy systems.2 However, raw natural gas and biogas from renewable natural gas (RNG) typically contain a substantial amount of impurities, such as hydrogen sulfide (H2S), carbon dioxide (CO2), water vapor, and inert gases.3–5 Consequently, purification is essential to convert these gases into a usable energy source.A key component of this purification process is the removal of acid gases (i.e., H2S and CO2), a procedure also known as natural gas sweetening. Natural gas reservoirs in specific regions, such as the United States, China, Europe, and the Middle East, can contain high levels of H2S and CO2.6 For instance, natural gas feeds in China may comprise up to 17% H2S and 9% CO2,6 while those in the Middle East can contain up to 30% H2S and 10% CO2.7,8 Similarly, biogas derived from the anaerobic digestion of organic feed stocks can contain 15–60% CO2 and 0.1–2% H2S.4,5 The presence of acid gases is not just a health hazard due to their toxicity but also poses a risk to the lifetime of transportation pipelines through corrosion.9 Therefore, meeting pipeline specifications (e.g., <2% CO2, <4 ppm H2S in the US) ensures safe and efficient transport while simultaneously increasing the heating value of the fuel.3
Amine absorption is currently the predominant method for removing acid gases from natural gas.10,11 This process uses aqueous amine solutions to selectively bind H2S and CO2 through weak acid–base interactions. The captured acid gases are subsequently released by heating the saturated amines in a stripping column.11 Despite its widespread use, the amine absorption process is energy-intensive due to absorbent regeneration.11 In contrast, membrane technology, which employs semi-permeable membranes to separate gas mixtures, is a promising alternative. With lower energy requirements and a smaller operational footprint, membrane technology presents a more sustainable option for natural gas purification compared to conventional approaches.10,12
Polymeric membranes, such as cellulose acetate (CA) and polyimides, are currently the primary choice in the membrane-based gas separation market due to their solution processability and robust mechanical properties.12 However, their application is often limited by the fundamental tradeoff relationship between permeability and selectivity, a phenomenon graphically represented in classic upper bound plots.13,14 This tradeoff is a significant factor when comparing polymer membranes to other gas separation technologies, as polymer membranes often do not meet the performance levels required to replace conventional separation methods. In the theoretical analysis of upper bound plots by Freeman, he proposed that polymers must be engineered with either enhanced sorption selectivity or increased backbone stiffness along with wider intersegmental distances to surpass these performance limits and compete more effectively with other separation techniques.15 Following this theoretical structure–property guidance, a wide array of microporous organic polymer (MOP) membrane materials have been developed, including polymers of intrinsic microporosity (PIMs),16,17 ring-opening metathesis polymerization (ROMP)-based polymers,18,19 catalytic arene-norbornene annulation (CANAL)-based polymers,20,21 and poly(arylene ethers) (PAEs).22,23 MOPs are characterized by rigid and bulky monomer units that prevent tight packing of polymer segments, resulting in materials with high free volume when processed into films. These unique characteristics of MOPs result in high gas sorption capacity from their high free volume and good diffusion selectivity from their rigid monomer units, making them highly promising for gas separation applications.
Although recent advancements in polymer membrane materials have been significant, effectively separating both H2S and CO2 from natural gas using these materials presents distinct challenges, primarily due to the size and chemical properties of these gases. The kinetic diameter difference is notably larger between CO2 (3.30 Å) and CH4 (3.80 Å) than H2S (3.62 Å) and CH4, making CO2/CH4 separation relatively more dependent on diffusion selectivity.24 On the other hand, H2S/CH4 separation is largely governed by sorption selectivity, primarily due to considerably higher condensability of H2S (Tc = 373.5 K) compared to CH4 (Tc = 190.0 K).24 This sorption-dominant transport mechanism results in unique behaviors in H2S/CH4 separation not typically observed for the separation of other light gases in glassy polymers, which are usually controlled by diffusion selectivity and show downward upper bound slopes defined by the differences in gas kinetic diameters.15,25,26 For example, Liu et al. highlighted the unexpected benefits of membrane plasticization for the H2S/CH4 gas pair in mixed-gas permeation of a 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:60 H2S/CO2/CH4 gas mixture for 6FDA-DAM and 6FDA-DAM/DABA polyimide membranes.27 Typically, plasticization—the increase in polymer segmental mobility induced by strongly sorbing gases—leads to an increase in permeability and a decrease in selectivity.28 However, plasticization of these polyimide membranes resulted in an increase in both permeability and mixed-gas selectivity for H2S/CH4, while CO2/CH4 selectivity decreased due to a reduction in molecular sieving effects.27 This finding was attributed to plasticization creating additional free volume, thereby providing more sorption capacity for H2S molecules, while minimally affecting H2S/CH4 diffusion selectivity.27 In a related context, many post-synthetic modification strategies designed for CO2 separation, including the installation of polar functional groups, crosslinking, and thermal annealing, often do not suit H2S separations since these modifications tend to reduce free volume, thereby decreasing both permeability and selectivity.6,29,30 This issue of coupled functionality and densification poses a challenge in achieving high selectivity for both H2S/CH4 and CO2/CH4 separations simultaneously in mixed-gas conditions.
:20:60 H2S/CO2/CH4 gas mixture for 6FDA-DAM and 6FDA-DAM/DABA polyimide membranes.27 Typically, plasticization—the increase in polymer segmental mobility induced by strongly sorbing gases—leads to an increase in permeability and a decrease in selectivity.28 However, plasticization of these polyimide membranes resulted in an increase in both permeability and mixed-gas selectivity for H2S/CH4, while CO2/CH4 selectivity decreased due to a reduction in molecular sieving effects.27 This finding was attributed to plasticization creating additional free volume, thereby providing more sorption capacity for H2S molecules, while minimally affecting H2S/CH4 diffusion selectivity.27 In a related context, many post-synthetic modification strategies designed for CO2 separation, including the installation of polar functional groups, crosslinking, and thermal annealing, often do not suit H2S separations since these modifications tend to reduce free volume, thereby decreasing both permeability and selectivity.6,29,30 This issue of coupled functionality and densification poses a challenge in achieving high selectivity for both H2S/CH4 and CO2/CH4 separations simultaneously in mixed-gas conditions.
In this study, we first illustrate the complexity inherent in these acid–gas separation processes by examining H2S/CO2/CH4 ternary pure-gas and mixed-gas permeation data of glassy polymer membranes available in literature. In addressing the challenges of acid–gas separation, we also explore the potential of a post-synthetic modification technique known as free volume manipulation (FVM).31–34 FVM involves the sequential functionalization and removal of labile functional groups, such as tert-butoxycarbonyl (tBOC), to alter the physical packing structures and the gas transport properties of modified polymer membranes.31,32 The introduction of bulky functional groups into the polymer backbone expands the intersegmental distances when the polymer is processed into a solid-state film. By removing these labile groups through thermal treatment, free volume in the polymer matrix can be increased. Moreover, incorporating a small amount of oxygen during thermal treatment can further degrade the pendant functional groups into oxidative crosslinks, further enhancing diffusion selectivity.34 Given that larger free volume can provide more sorption capacity for H2S, while crosslinks can help maintain or increase the CO2/CH4 diffusion selectivity, FVM holds considerable potential as a post-synthetic modification strategy to improve acid–gas separations. Herein, we demonstrate the effectiveness of FVM for acid–gas separations using a microporous PAE that contains spirobifluorene and triptycene moieties (PAE-1).22,23 The PAE structure is modified by incorporating tBOC-protected piperazinyl (PIP-tBOC) groups, and FVM is applied under either inert conditions to retain piperazinyl functionality or in the presence of oxygen to induce crosslinking by degrading the entire PIP-tBOC unit. We evaluate the performance of these modified PAEs in separating binary and ternary acid–gas mixtures, providing valuable insights into the practical application and effectiveness of FVM-modified microporous polymers in conditions that closely mirror industrial settings for acid–gas separations.
2. Experimental section
2.1. Polymer synthesis
A detailed synthetic procedure is described in the ESI.† In short, tBOC-protected piperazinyl PAE-1 (PAE-PIP-tBOC) was synthesized in three steps. Pd-catalyzed C–O polycondensation and subsequent post-polymerization functionalization resulted in chloromethylated PAE-1 (PAE-Cl).23,35 The displacement of chloride by tBOC-protected piperazine as a nucleophile was used to install PIP-tBOC groups onto the polymer backbone.2.2. Film formation and free volume manipulation (FVM)
Membrane films were fabricated following a standard solution-casting procedure. PAE-1 and PAE-PIP-tBOC were dissolved in chloroform to create a 3 wt/v% polymer solution. This solution was subsequently filtered through a 0.45 μm syringe filter (VWR, 76479-008) into a STERIPLAN® glass Petri dish (DWK Life Sciences). The dish was covered and placed in a chemical fume hood for 72 h to allow complete solvent evaporation. Once the solvent had fully evaporated, the resultant film was separated from the Petri dish using a small amount of DI water, and then the films were dried in a vacuum oven at 90 °C for 12 h. Before conducting any experiments, all membrane samples were kept submerged in methanol to ensure consistent conditioning. Prior to any experiments, films were dried in a vacuum oven at 90 °C for 12 h under a dynamic vacuum to ensure thorough removal of any residual methanol.For FVM, PAE-PIP-tBOC films were placed on a Petri dish and transferred to a tube furnace (Carbolite Gero HST 12/900, USA). The furnace was purged with either pure N2 gas or a 3% O2 and balance N2 gas mixture at a rate of 0.5 L min−1 for at least 30 min. The O2 concentration in the furnace was continuously monitored using an O2 gas sensor (Rapidox 2100, Cambridge Sensotec, USA), which was directly connected to the outlet of the tube furnace. Subsequently, the gas flow was reduced to 0.3 L min−1, and the furnace was heated at a rate of 10 °C min−1 to 300 °C and maintained at 300 °C for 16 h. The furnace was then gradually cooled to room temperature.
2.3. Characterization
Thermogravimetric analysis (TGA) was conducted with a TA Instruments TGA550 (USA). For each experiment, samples were initially equilibrated at 50 °C for 10 min to stabilize their weight. Following this isothermal hold, the samples were heated to 700 °C at a rate of 10 °C min−1 under a nitrogen atmosphere.Attenuated total reflection Fourier transform infrared (ATR-FT-IR) spectroscopy measurements were carried out using a Bruker ALPHA Fourier transform infrared spectrometer (USA). For the analysis of each polymer film, a total of 64 scans were acquired across a spectral range of 400–4000 cm−1 with a resolution of 4 cm−1. The peaks in each spectrum were normalized relative to the maximum peak intensity observed.
13C magic angle spinning (MAS) solid-state nuclear magnetic resonance (SSNMR) spectroscopy was performed using a Bruker Avance Neo spectrometer (USA), operating at a frequency of 500.18 MHz and equipped with a 3.2 mm HX solids probe. To prepare the samples, polymer films were first finely cut, and then the films were carefully packed into 3.2 mm zirconia MAS rotors (Bruker, USA). The spectroscopic analysis was carried out at room temperature, collecting a total of 3060 scans at a MAS spinning rate of 20 kHz, a relaxation delay of 3 seconds, and a spectral window set to 200 ppm.
2.4. Permeation measurements
Pure-gas permeabilities of various light gases (He, H2, CH4, N2, O2, CO2, and H2S) were measured for each polymer film using a constant volume–variable pressure permeation testing apparatus purchased from Maxwell Robotics (USA). Each sample was carefully sealed onto brass supports using epoxy adhesive to ensure no leakage occurred between the upstream and downstream sides. Before conducting permeation tests, the entire system was evacuated under vacuum for 8 h. To prevent cross-contamination when switching gases, the upstream side was purged with high-pressure He, followed by holding the entire apparatus under vacuum for 0.5–1 h after testing each gas. All permeation tests were performed at a controlled temperature of 35 °C, maintaining an upstream pressure of approximately 15 psia (approximately 1 atm) and a downstream pressure below 9.5 torr.For mixed-gas experiments, a constant volume–variable pressure mixed-gas permeation apparatus from Maxwell Robotics (USA) was used. The feed gases comprising either a 50:50 CO2/CH4 or 20:20:60 H2S/CO2/CH4 mixture (mol%) were supplied at 35 °C under a total pressure of 32 psia (2.2 atm) or 115 psia (7.8 atm), respectively. Detailed experimental procedures are available in a previous publication from our group.36 Briefly, the entire system was degassed for at least 8 h, and the feed gas mixture was introduced at the upstream side of the film at a minimum flow rate of 0.24 L min−1 to reduce concentration polarization. The maximum stage cuts were 2.03 × 10−5 and 1.48 × 10−5 for the 50:50 CO2/CH4 and 20:20:60 H2S/CO2/CH4 mixtures, respectively. To ensure steady-state conditions for the mixed-gas tests, preliminary pure-gas permeation tests were performed for all gases that would be included in the mixed-gas experiments using identical partial pressures to determine the time lag. The permeated gas mixture was collected in a degassed downstream chamber and analyzed using a gas chromatograph (Agilent 7890B, USA). For permeation tests under humid conditions, a custom-built permeation system from Maxwell Robotics (USA) equipped with a dewpoint transmitter (Kahn Hygrometer SF82, USA) was used. For this system, the upstream and downstream of the membrane have equal relative humidity (i.e., 50%). Thus, there is no water vapor gradient in the membrane during testing. After reaching a steady-state, the downstream permeate mixture was collected and analyzed using a gas chromatograph (Agilent 8890, USA).
Pure-gas permeability (P) was calculated using the equation:
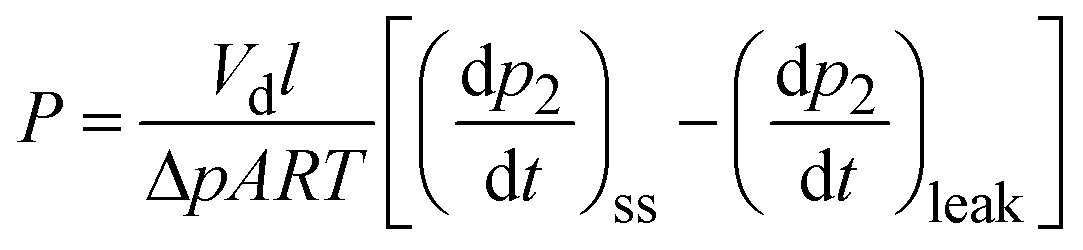 | (1) |
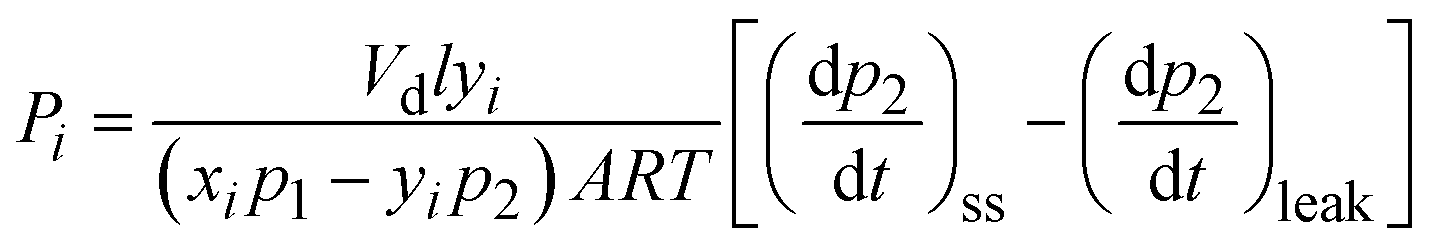 | (2) |
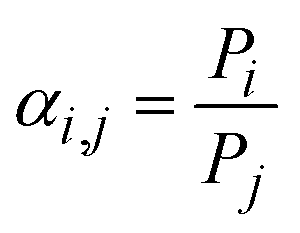 | (3) |
2.5. Pure-gas sorption measurements
Pure-gas sorption isotherms for H2S, CO2, and CH4 were conducted at 35 °C using a pressure decay sorption apparatus from Maxwell Robotics (USA). For each measurement, at least 0.1 g of polymer films were placed into a 3 cm3 sample cell. Before conducting sorption measurements, the entire apparatus was evacuated under full vacuum for at least 8 h. Each pressure was maintained for 0.3 h in the charge volume before injecting the gas into the sample chamber. Following the injection, the gas was allowed to equilibrate for 6 h for H2S, 1.5 h for CO2, and 2.5 h for CH4. Sorption isotherms were collected up to 167 psia (11.4 atm) for H2S and up to 700 psia (47.6 atm) for CO2 and CH4, and the results were fitted using the dual-mode sorption (DMS) model:36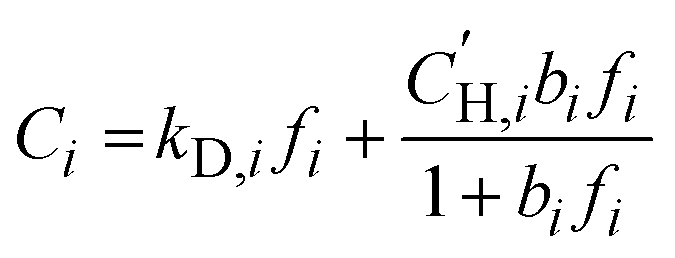 | (4) |
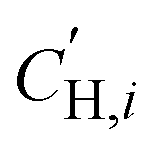 is the Langmuir sorption capacity (cmSTP3 cmpol−3), and bi is the Langmuir affinity constant (atm−1). The isotherm fits were constrained such that the slope of ln(kD) versus critical temperature (Tc) matched the slope of ln(Si) versus Tc at 8 atm (near the pressure at which the mixed-gas permeation was tested), where Si is the pure-gas sorption coefficient:37
is the Langmuir sorption capacity (cmSTP3 cmpol−3), and bi is the Langmuir affinity constant (atm−1). The isotherm fits were constrained such that the slope of ln(kD) versus critical temperature (Tc) matched the slope of ln(Si) versus Tc at 8 atm (near the pressure at which the mixed-gas permeation was tested), where Si is the pure-gas sorption coefficient:37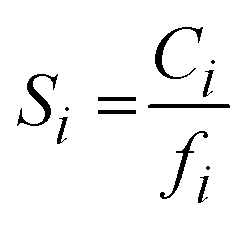 | (5) |
To model the competitive sorption effects on mixed-gas transport in a 20:20:60 H2S/CO2/CH4 ternary mixture, mixed-gas sorption isotherms were calculated based on the fitted parameters obtained from the pure-gas DMS model (eqn (4)),24 where j and k represent co-permeating gases:
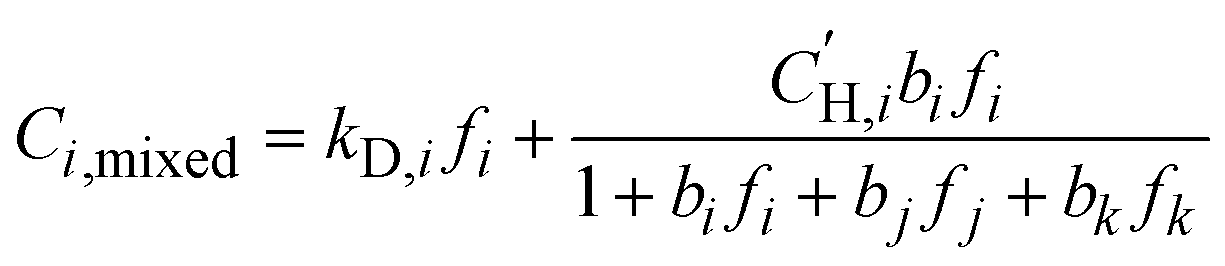 | (6) |
3. Results and discussion
3.1. Illustration of CO2/CH4 and H2S/CH4 trends
The upper bounds for common gas pairs, such as H2/CH4, CO2/CH4, and O2/N2, typically exhibit downward slopes.13,14 Within the sorption–diffusion framework, the permselectivity of these upper bounds can be separated into contributions from diffusion selectivity and sorption selectivity. Given that the activation energy of diffusion is directly proportional to the kinetic diameter squared from Brandt's model,38 and that diffusion selectivity correlates with the differences in kinetic diameters of gas pairs,39 larger differences in kinetic diameter tend to exhibit steeper downward slopes on the upper bound plots. For instance, the H2/CH4 pair, with a kinetic diameter difference of 0.91 Å, shows steeper slopes in comparison to O2/N2 (with a kinetic diameter difference of 0.18 Å) and CO2/CH4 (with a kinetic diameter difference of 0.5 Å).13,14 When considering light gases, Robeson et al. demonstrated that diffusion selectivity closely aligns with permselectivity,39 as would be expected for gases with weak transport contributions from sorption. Nevertheless, sorption selectivity still exhibits a small but notable downward slope as a function of increasing permeability or free volume, which is attributed to a molecular exclusion effect that more strongly impacts larger gases.39 Thus, the downward slopes of upper bound plots for light gas pairs commonly studied in literature are significantly influenced by diffusion selectivity with a modest contribution from sorption selectivity.39H2S and CH4, however, have unique physical properties compared to other light gas pairs studied in the upper bound relationships.13,14 The kinetic diameter difference between H2S and CH4 is only 0.18 Å, but H2S is far more polarizable than CH4, having a polarizability of 37.8 × 10−25 cm3 compared to 26.0 × 10−25 cm3 for CH4.40 Thus, sorption selectivity is anticipated to be more substantial in this separation relative to diffusion selectivity. In fact, annealed 6FDA-DAM:DABA (3:2), which has been frequently tested for H2S/CH4 separation, showed a H2S/CH4 diffusion selectivity of 1.0, whereas sorption selectivity was 8.0.29 Given the significantly higher polarizability of H2S compared to CH4 without a notable difference in their kinetic diameters, we anticipated that sorption of H2S would increase more rapidly with increasing free volume compared to sorption of CH4. In this case, the mechanism underlying H2S/CH4 separation differs fundamentally from that of CO2/CH4 separation, which predominantly depends on diffusion selectivity as the primary separation mechanism. This distinction presents a unique challenge in attaining high selectivity for both gas pairs simultaneously.
To understand the impact of the different separation mechanisms for H2S/CH4 and CO2/CH4 in acid gas separation involving H2S/CO2/CH4 ternary gas mixtures, literature data up to 2024 for these gases was collected to evaluate performance trends for conventional polymers and emerging microporous polymers. These trends are considered for H2S/CO2/CH4 ternary gas mixtures and the corresponding pure-gas tests in Fig. 1. In Fig. 1a–d, the data are categorized into conventional polymer structures (light blue color) and MOPs (taupe color), and the dashed lines represent linear fits for the data within each category. Conventional polymers include structures that are not classified as MOPs, such as 6FDA-based polyimides and CA films. It is important to note that mixture data in the literature cover a range of compositions, with H2S, CO2, and CH4 concentrations ranging from 0.5–20%, 3–45%, and 50–95%, respectively. Such variations can influence mixed-gas permeability and selectivity due to competitive sorption effects, whereby higher concentrations of condensable gases like H2S and CO2 molecules can exclude each other and CH4 molecules from the polymer.36 Although some studies have investigated mixtures with additional impurities like nitrogen and ethylene,41Fig. 1 focuses solely on ternary gas mixture data to avoid confounding interpretations of trends.
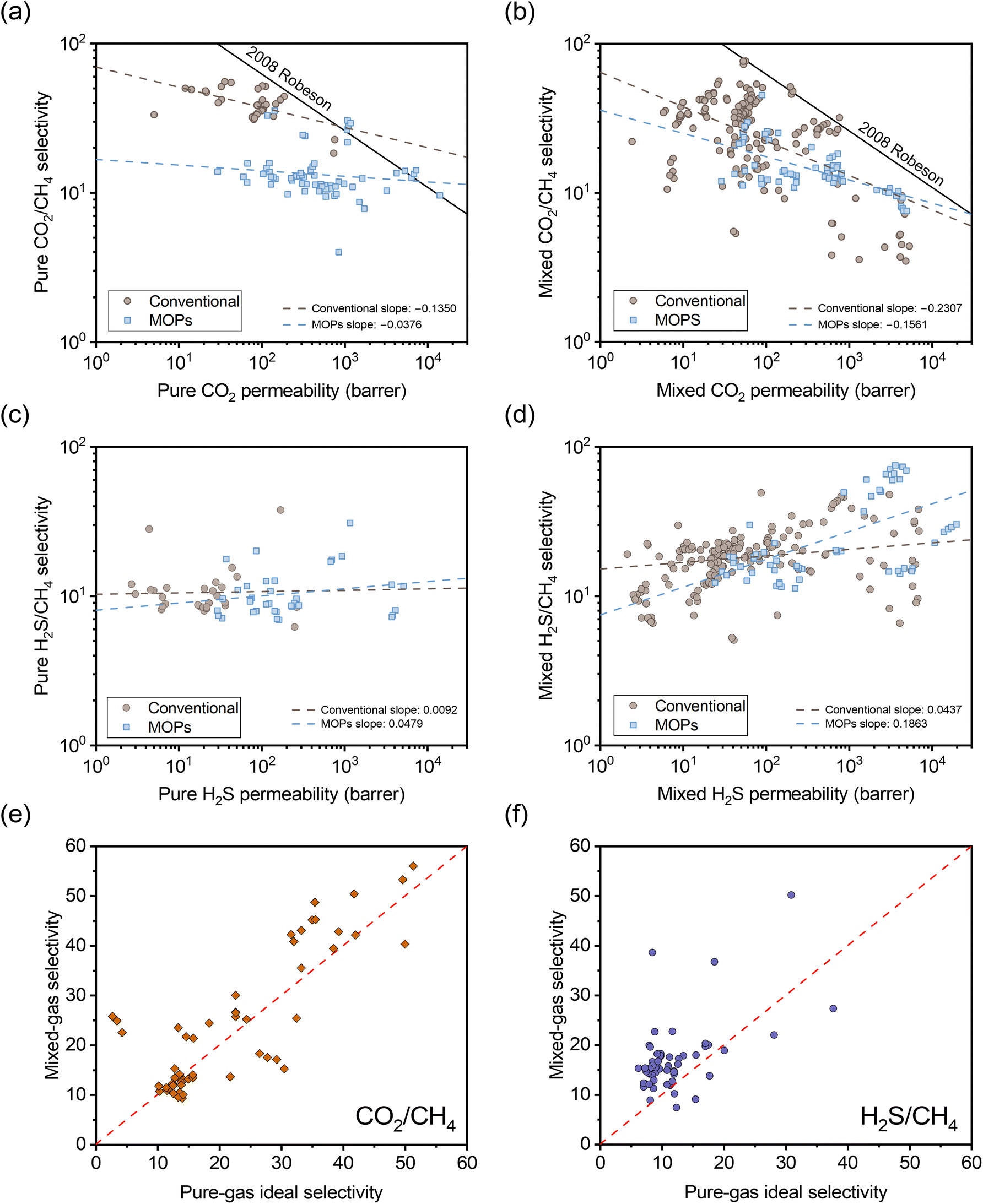 | ||
| Fig. 1 Comparison of selectivity versus permeability for (a) pure-gas CO2/CH4, (b) mixed-gas CO2/CH4, (c) pure-gas H2S/CH4, and (d) mixed-gas H2S/CH4 based on ternary H2S/CO2/CH4 permeation data available in the literature for glassy polymers.3,22,24,27,29,30,32,35,57–74 The dashed lines represent linear fits for conventional (non-MOP) glassy polymers (taupe circles) and MOPs (light blue squares). Comparison of mixed-gas selectivity for ternary H2S/CO2/CH4 mixtures and pure-gas selectivity for (e) CO2/CH4 and (f) H2S/CH4. The red dashed line represents a parity line. | ||
In Fig. 1a, both MOPs and conventional polymers exhibit a negative correlation between permeability and selectivity for CO2/CH4 separation. As previously discussed, this trend is typical for light gas pairs with relatively low sorption selectivity if diffusion selectivity primarily drives the separation, as is highlighted by the downward slopes in classic light gas upper bound plots.14 Although CO2 has higher sorption compared to CH4,39 the significant kinetic diameter difference allows diffusion selectivity to govern separation performance. This trend suggests that an increase in CO2 sorption capacity does not offset the reduction in diffusion selectivity as free volume is increased.
In the mixed-gas case depicted in Fig. 1b, the slopes for both conventional polymers and MOPs are noticeably steeper compared to the pure-gas case in Fig. 1a. Visually, the observed steeper slopes in mixed-gas tests result from selectivity reductions in membranes with higher permeability (i.e., higher free volume), whereas CO2/CH4 selectivity tends to be better maintained in membranes with lower permeability (i.e., lower free volume). Quantitatively, these data can be interpreted through the membrane “score” system, which quantifies the perpendicular distance of a data point from the upper bound line. The calculation of the score is formalized in the equation below, where a negative score signifies that the data point lies below the upper bound line:42
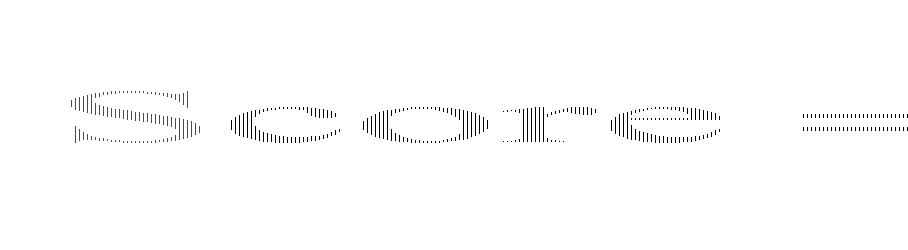 | (7) |
| Testing condition | Classification | CO2 permeability (barrer) | # of data | Average score |
|---|---|---|---|---|
| Pure-gas | Low free volume | <1000 | 80 | −0.889 ± 0.414 |
| High free volume | >1000 | 17 | −0.184 ± 0.359 | |
| H2S/CO2/CH4 mixed-gas | Low free volume | <1000 | 211 | −1.020 ± 0.573 |
| High free volume | >1000 | 24 | −0.775 ± 0.368 |
For H2S/CH4 in Fig. 1c, both MOPs and conventional polymers show a weak positive correlation between permeability and selectivity. As previously discussed, such trends are expected for the separation of gas pairs primarily driven by sorption mechanisms. For example, Lin et al. demonstrated that the upper bound for CO2/H2 separation exhibits a positive slope.44 This result showed that CO2/H2 separation, despite having unfavorable diffusion selectivity (CO2 being larger than H2), can still achieve a selectivity >1 due to its sorption-dominant separation mechanism.
Similarly, the positive slope for H2S/CH4 separation indicates that an increase in sorption selectivity with increase in free volume outweighs the decrease in diffusion selectivity. This behavior deviates from the typical tradeoff relationship between permeability and selectivity, demonstrating that greater free volume benefits both permeability and selectivity in H2S/CH4 separations. Current upper bound theory, which primarily relies on gas kinetic diameters to determine the upper bound slopes, appears insufficient in fully accounting for the separation of gas pairs when sorption selectivity plays a critical role. Developing more robust theory would require a more extensive dataset for diffusion and sorption coefficients for H2S/CH4. In the mixed-gas case presented in Fig. 1d, the slopes for both conventional polymers and MOPs become significantly steeper. Complementary to the CO2/CH4 case, the steeper slopes are attributed to a more significant increase in selectivity for membranes with higher permeability, highlighting the strong correlation between competitive sorption effects and free volume.
The opposing correlations between H2S/CH4 and CO2/CH4 present a unique challenge in achieving high selectivity for both gas pairs simultaneously, particularly for polymers with high free volume due to the more pronounced competitive sorption effects. Any change in free volume through post-synthetic modification can severely affect the selectivity for one of these gas pairs. Therefore, to optimize selectivity for both CO2/CH4 and H2S/CH4 in ternary mixed-gas permeation, a post-synthetic modification strategy should aim to maximize sorption (i.e., large free volume and favorable interactions with acid gases) while maintaining high size-sieving capability to preserve CO2/CH4 diffusion selectivity. Following these design principles, we have applied an FVM strategy to PAE-1.
3.2. FVM of PAE-1 membranes
As illustrated in Fig. 2, PAE-1 was first modified by functionalization with tBOC-protected piperazine to yield PAE-PIP-tBOC. Chloromethylation can occur on any aromatic unit within PAE-1 since the functionalization involves a nucleophilic attack by electron-rich aromatic groups on chloromethyl methyl ether. However, chloromethylation should occur predominantly on the more electron-rich ring of the triptycene moiety, specifically the aromatic unit without the tert-butyl (tBu) groups due to both the electron-donating effect from ether linkages and the minimal steric hindrance.45 A notable benefit of this synthesis route is the solution-processability of the resultant polymer, a feature that is not typically available in primary or secondary amine-functionalized polymers, as evidenced by PIM-NH2 and PAE-NH2.24,32,43 This solution-processability is crucial for scaling up the production of these microporous polymers for industrial applications. Furthermore, this synthesis route highlights the versatility of FVM, building upon our previous FVM applications where we used pendant polar groups that were either hydroxyl or amine groups. For example, tBOC groups have been installed on a 6FDA-HAB polyimide via the hydroxyl pendant group,31 while amines provided an installation site for tBOC on PIM-NH2.32 The incorporation of a labile moiety onto an aromatic unit in the present study suggests that FVM could potentially be applied to any polymer containing a nucleophilic aromatic unit that can be readily chloromethylated.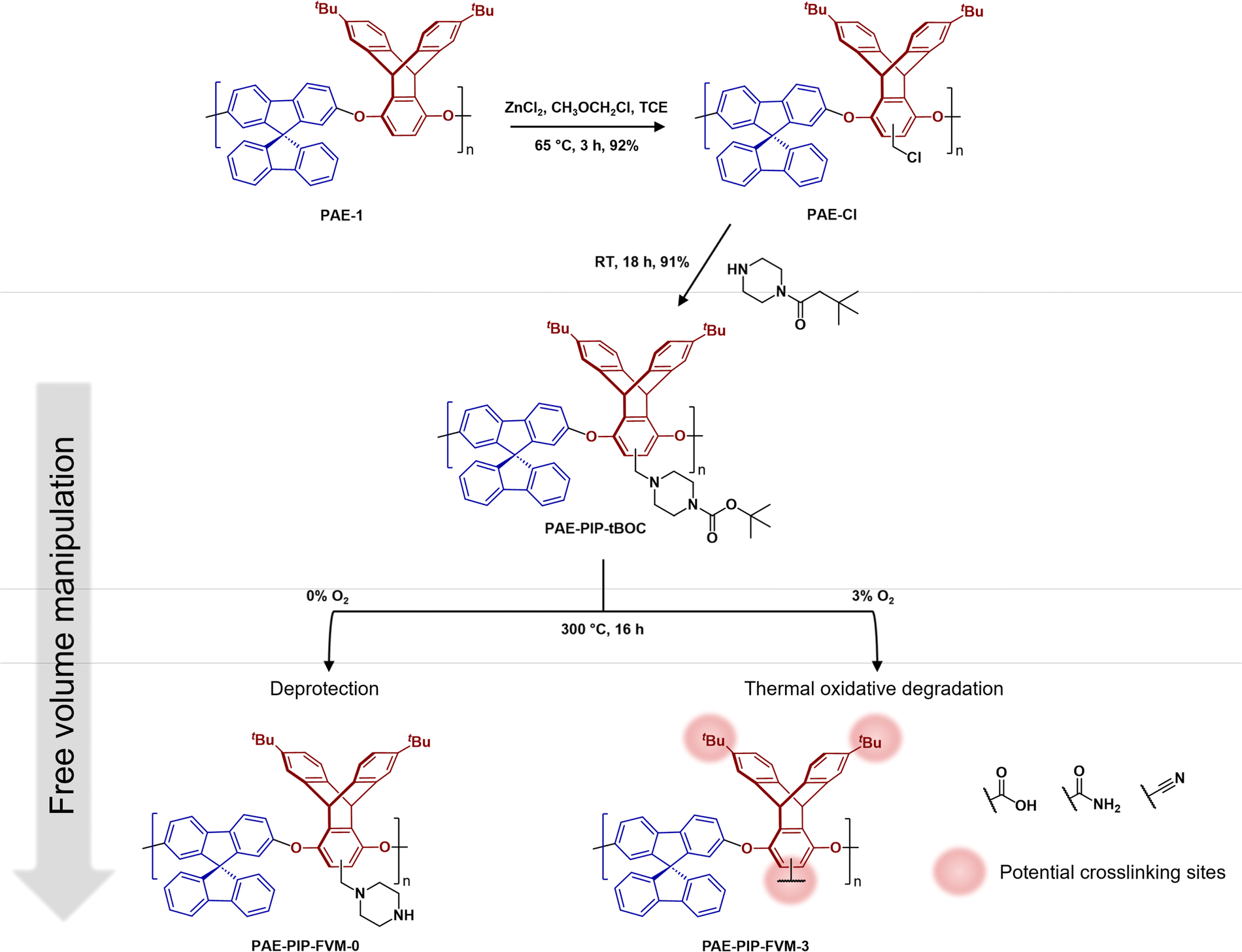 | ||
| Fig. 2 Functionalization routes for PAE-PIP-tBOC and a summary of FVM products. FVM is implemented in N2 atmosphere with either 0% or 3% O2 concentration. | ||
To implement FVM, PAE-PIP-tBOC films underwent thermal treatment at 300 °C for 16 h under an atmosphere of pure N2 or a gas mixture comprising 3% O2 and a balance of N2. The O2 concentration, deprotection temperature, and deprotection time were chosen based on optimization efforts shown in Fig. S9† and following our recent study on O2 deprotection chemistries for PIM-tBOC.34 The resulting samples are labeled as PAE-PIP-FVM-x, where x indicates the O2 concentration during FVM. As shown in Fig. 2, performing FVM in an inert atmosphere led to the removal of tBOC groups, yielding benzylic piperazinyl groups as pendant groups. Conversely, introducing a small amount of oxygen during the thermal treatment resulted in the degradation of the entire piperazyl unit, along with the formation of new crosslinks, as will be discussed later.
Fig. 3a presents TGA scans comparing the thermal degradation profiles of PAE-1 and PAE-PIP-tBOC films with the PAE-PIP-FVM films that already underwent thermal treatment at 300 °C. The PAE-PIP-tBOC sample showed a weight reduction of about 15% starting around 150 °C, which is attributed to the degradation of the tBOC groups. Detailed insights into the potential degradation mechanisms of tBOC groups are provided in previous FVM studies conducted by our group.32,34 If one tBOC group were attached for each repeat unit, the theoretical mass loss for complete deprotection of tBOC would be 11.0%. Thus, the higher observed weight loss is attributed to more than one chloromethylation site per repeating unit on the polymer, including potential chloromethylation on the spiro-bifluorine repeating units. For the thermally processed PAE-PIP-FVM samples, this initial mass loss was not observed, indicating that the tBOC groups were fully deprotected during the FVM treatment. Furthermore, the PAE-PIP-FVM-3 sample did not show similar mass loss profiles compared to those of PAE-PIP-tBOC and PAE-PIP-FVM-0 beyond 350 °C. This finding is attributed to the degradation of the piperazine group in the presence of oxygen. We hypothesize that this oxidative degradation likely involves the oxidation of methylene groups adjacent to the nitrogen atoms on the benzylic piperazinyl rings. This is analogous to the oxidative degradation pathways observed for PIM-tBOC in our previous study on FVM under oxygen,34 given the similarities in the side groups containing methylene adjacent to amines and the resulting functional groups after degradation, which will be discussed in detail.
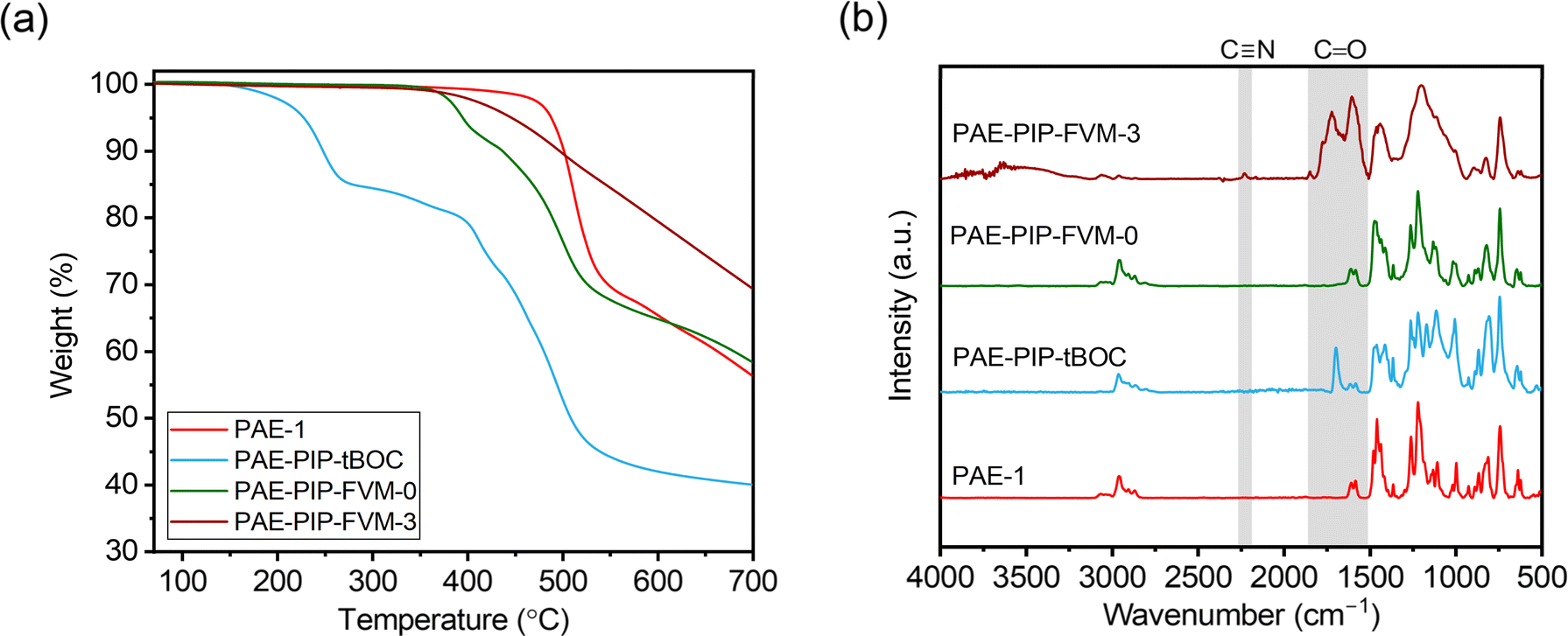 | ||
| Fig. 3 Comparisons of (a) TGA scans and (b) ATR-FT-IR spectra for PAE-1, PAE-PIP-tBOC, PAE-PIP-FVM-0, and PAE-PIP-FVM-3 films. | ||
ATR-FT-IR spectroscopy was used to identify the formation of new functional groups during the FVM process. As shown in Fig. 3b, the spectrum of the fresh PAE-1 film aligned with previously reported results for this polymer.22 After post-functionalization with PIP-tBOC groups, a distinct peak at 1695 cm−1 emerged, indicative of carbonyl groups in tBOC.32 However, after FVM of PAE-PIP-tBOC in N2 (i.e., PAE-PIP-FVM-0), this carbonyl peak completely disappeared. Unlike FVM of PIM-NH2 films, where a urea crosslinking peak at 1680 cm−1 was observed,32 this finding indicates that no observable urea crosslinking occurred in the FVM process for PAE-PIP-tBOC in an inert atmosphere. In the context of FVM of PIM-NH2, Mizrahi Rodriguez et al. proposed two mechanisms for urea crosslink formation, which involved a nucleophilic attack on either an in situ generated isocyanate or a carbamic acid intermediate by an adjacent deprotected amine group.32 With these mechanisms, primary amine groups need to be in proximity to the electron-deficient sites of either isocyanate or carbamic acid. Given that piperazinyl is bulkier compared to a primary amine, the imposed steric hindrance may prevent urea crosslinking during FVM of PAE-1 under inert conditions, particularly if the dominant mechanism involves carbamic acid.
The incorporation of oxygen during the FVM process led to several changes in the FT-IR spectrum. Notably, multiple peaks associated with carbonyl groups emerged in the PAE-PIP-FVM-3 sample, similar to those observed for PIM-tBOC films treated in the presence of oxygen.34 Specifically, a series of broad, overlapping peaks appeared at 1605 cm−1, 1655 cm−1, 1720 cm−1, and 1772 cm−1, along with a small peak at 1846 cm−1. These spectral changes are indicative of the formation of various carbonyl-containing species during thermal oxidative crosslinking. These species may include carboxylic acids, amides, ureas, imides, and anhydrides.32,34,46,47 The peak at 1720 cm−1, in particular, closely corresponds to the carboxylic acid peak noted in PIM-COOH.47 Additionally, a small peak at 2225 cm−1 was observed in the presence of oxygen. In our previous study investigating thermal oxidative crosslinks of PIM-tBOC films,34 this peak was ascribed to the formation of nitrile groups resulting from the dehydration of primary amides, which in turn are formed by the oxidation of amines.48 These findings underscore the complex chemical transformations that occur during FVM under oxidative conditions, leading to the formation of a range of functional groups.
The chemical differences among the PAE-1 film and the FVM derivatives were further investigated using MAS solid-state 13C NMR spectroscopy. As shown in Fig. 4a, the tBu carbons and carbonyl carbons associated with tBOC groups (peaks 1, 6, and 8) were absent in both FVM samples, suggesting the full deprotection of tBOC groups as observed in the TGA scans. In the case of PAE-PIP-FVM-3, the peaks corresponding to the methylene carbons in piperazinyl groups (peaks 3 and 5) were absent, corroborating observations from the TGA scan in Fig. 3a. As shown in the zoomed in scale of the spectra at higher chemical shifts in Fig. 4b, a distinct broad peak ranging from 162–174 ppm (peak 9) was observed when oxygen was present during FVM. This new peak is attributed to the formation of new species resulting from the thermal oxidative degradation of the piperazine group, as discussed previously. The chemical shift of this peak closely aligns with those of carboxylic acids and amides.34 In contrast, PAE-PIP-FVM-0 did not show any additional peak near this region. If urea crosslinking had occurred similar to the reported mechanism for PIM-tBOC films treated under inert conditions,32 a peak around 164 ppm would be expected. The lack of this urea peak coupled with the absence of the urea carbonyl peak at 1680 cm−1 in the FT-IR suggests that urea crosslinking did not occur in PAE-PIP-FVM-0 films. Interestingly, there was a notable reduction in the intensity of the peak at 31 ppm (peak 2) for PAE-PIP-FVM-3, which corresponds to the tBu carbons on the triptycene moiety. This observation is highlighted by the changing ratio between the peak intensities of the tBu carbon (peak 2) and the bridgehead carbon on the triptycene unit (peak 4): a decrease from 1.74 and 1.75 for PAE-1 and PAE-PIP-FVM-0, respectively, to 1.25 for PAE-PIP-FVM-3. We acknowledge that MAS solid-state NMR measurements are not entirely quantitative due to variations in peak intensity influenced by factors like contact time during cross-polarization and relaxation constants. Nevertheless, as our comparison focuses on identical carbons under consistent experimental parameters, the observed decrease in peak intensity suggests a change in the chemical environment surrounding the tBu groups. Although further investigation is required to confirm potential mechanisms, we hypothesize that the methyl groups in the tBu may serve as sites for hydrogen abstraction and could be involved in crosslinking through radical coupling.49
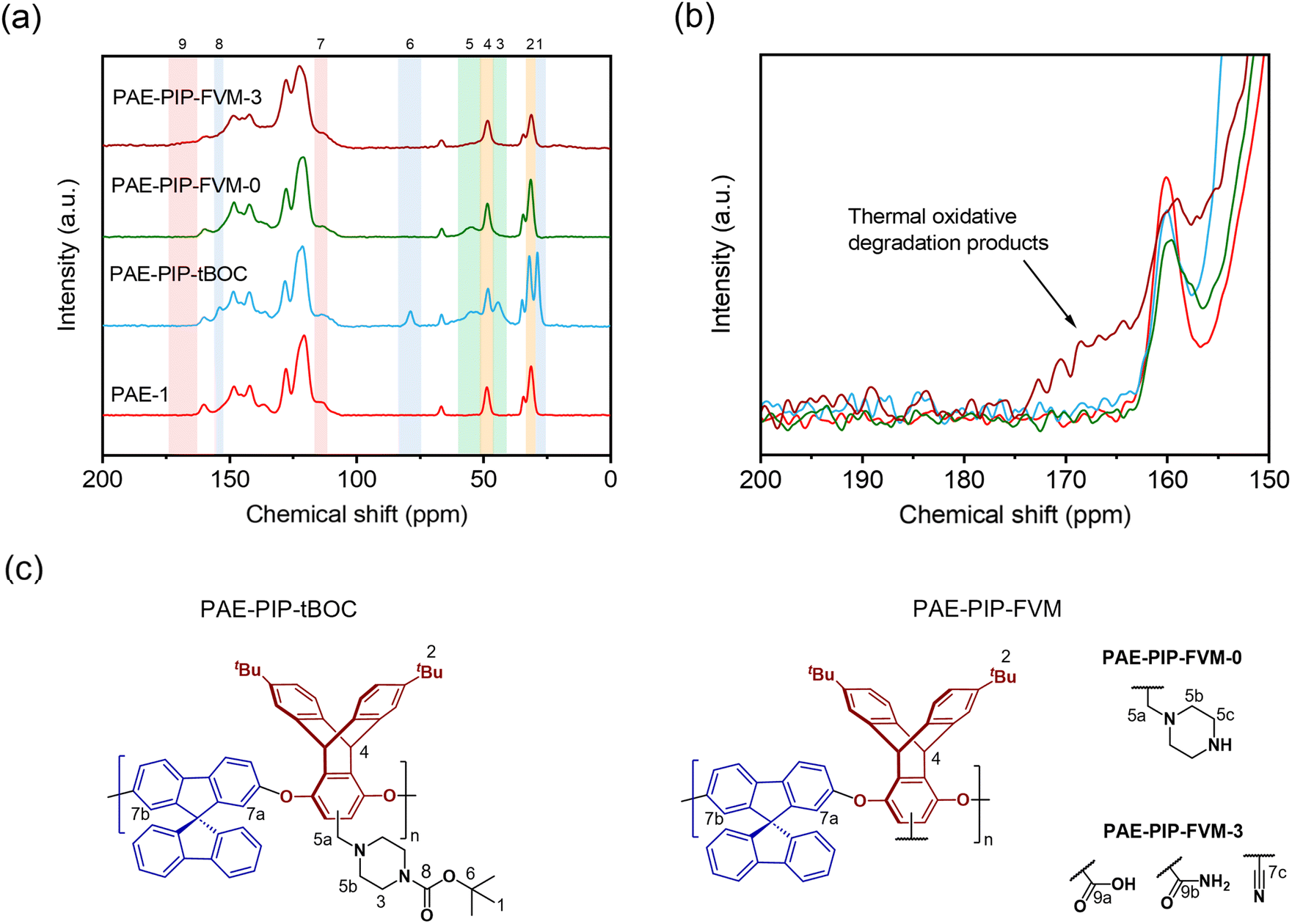 | ||
| Fig. 4 Comparison of (a) solid-state 13C NMR and (b) zoomed in scale of 13C NMR spectra for the PAE-1, PAE-PIP-tBOC, and FVM derivatives. The y-axis in (b) is zoomed in by a factor of 5 with all spectra superimposed for clarity. The peak assignments are shown on top of each plot. (c) The chemical structures of PAE-PIP-tBOC and PAE-PIP-FVM structures highlighting assigned peaks. | ||
3.3. Gas transport properties
The influence of FVM on pure-gas separation performance was examined using several gases. The results for CO2/CH4 separation are presented in Fig. 5a, while the results for other gases can be found in Table S5 and S6 and Fig. S8.† As shown in Fig. 5a, FVM treatment in the presence of oxygen led to a notable 113% increase in CO2/CH4 selectivity with a modest 16% increase in CO2 permeability compared to the pristine PAE-1 sample, allowing the sample to approach the 2008 upper bound.14 This finding indicates that the entire PIP-tBOC group effectively acts as a labile porogen to increase free volume. Simultaneously, crosslinks are formed through the oxidative degradation process to help maintain this modified free volume structure, as observed in the previous FVM studies.32,34 This increase in free volume is corroborated by other experiments. First, the BET surface area is increased after FVM in the presence of oxygen, which increased from 132.9 m2 g−1 for PAE-1 films to 161.3 m2 g−1 for PAE-PIP-FVM-3 films (Table S4†). It is noted that the BET surface area for PAE-1 films is significantly lower than the 668 m2 g−1 reported in a previous study22 because the BET measurements here were performed on films, while the previous study used powder samples. Second, Langmuir sorption capacity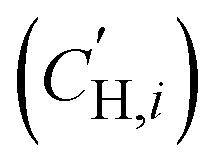 for all gases increased after FVM in the presence of oxygen (Table S10†). Finally, the pore size distributions calculated using nonlocal density functional theory (NLDFT) for CO2 adsorption at 273 K (Fig. S7b†) indicate higher pore volumes in both 3.5 Å and 5 Å ranges. Notably, PAE-PIP-FVM-3 showed a significantly higher volume of smaller pores compared to PAE-1. This finding aligns well with the previous characterization work of PIM-NH2-FVM using positron annihilation lifetime spectroscopy (PALS), which showed a 19% increase in the number of smaller free volume elements.32 It is also worth noting that PAE-PIP-FVM-3 exhibited larger standard deviations in both permeability and selectivity compared to other samples. This variation is attributed to the nature of the thermal oxidative crosslinking process, which involves a sequence of radical-mediated reactions at high temperatures.50,51 The formation of crosslinks can occur through the combination of radical sites on different polymer chains.51 This finding means that the kinetics of crosslink formation are influenced by the spatial arrangement of PIP-tBOC groups and the proximity of nascent radical sites on different chains. These solid-state reactions are naturally difficult to control, and hence, there is considerable variability in the separation performance of each sample subjected to FVM. This uncertainty was quantified by testing 5 different samples from distinct FVM implementations and reporting uncertainty in transport performance.
for all gases increased after FVM in the presence of oxygen (Table S10†). Finally, the pore size distributions calculated using nonlocal density functional theory (NLDFT) for CO2 adsorption at 273 K (Fig. S7b†) indicate higher pore volumes in both 3.5 Å and 5 Å ranges. Notably, PAE-PIP-FVM-3 showed a significantly higher volume of smaller pores compared to PAE-1. This finding aligns well with the previous characterization work of PIM-NH2-FVM using positron annihilation lifetime spectroscopy (PALS), which showed a 19% increase in the number of smaller free volume elements.32 It is also worth noting that PAE-PIP-FVM-3 exhibited larger standard deviations in both permeability and selectivity compared to other samples. This variation is attributed to the nature of the thermal oxidative crosslinking process, which involves a sequence of radical-mediated reactions at high temperatures.50,51 The formation of crosslinks can occur through the combination of radical sites on different polymer chains.51 This finding means that the kinetics of crosslink formation are influenced by the spatial arrangement of PIP-tBOC groups and the proximity of nascent radical sites on different chains. These solid-state reactions are naturally difficult to control, and hence, there is considerable variability in the separation performance of each sample subjected to FVM. This uncertainty was quantified by testing 5 different samples from distinct FVM implementations and reporting uncertainty in transport performance.
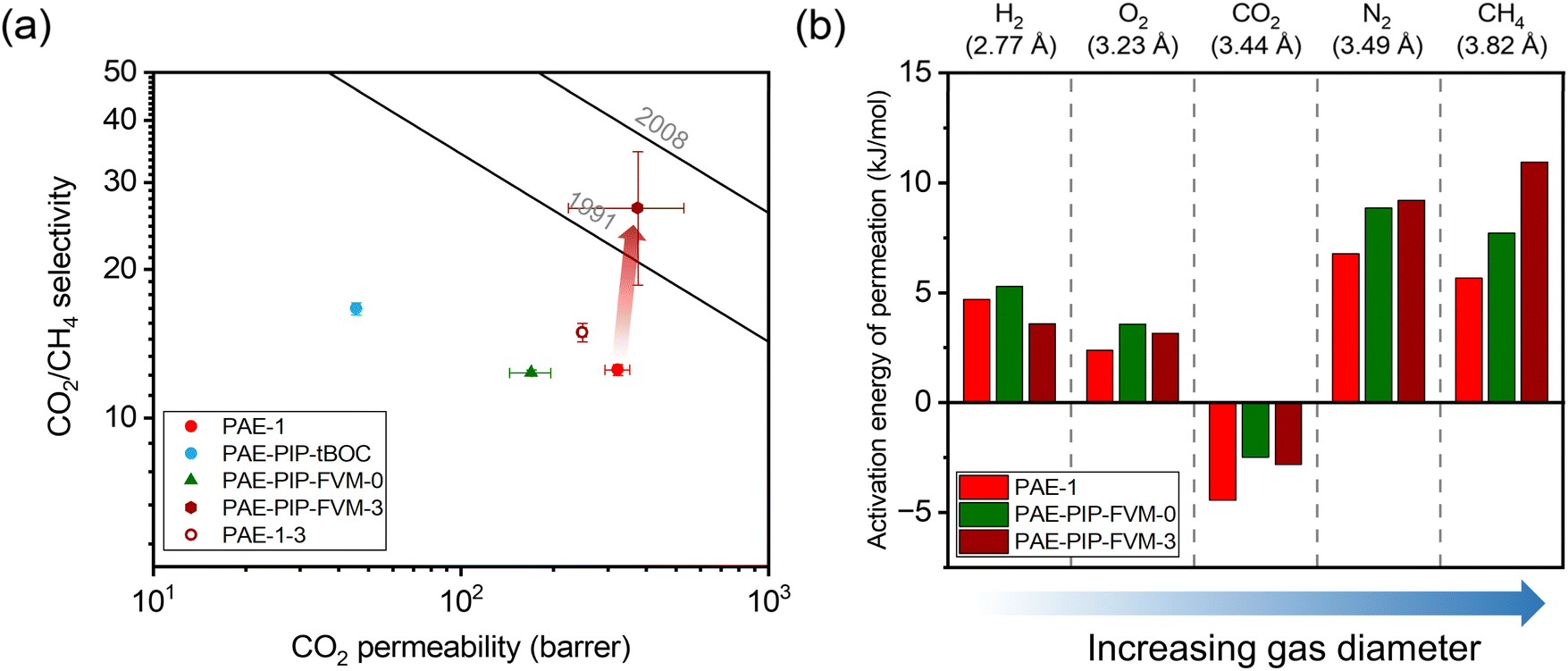 | ||
| Fig. 5 (a) Pure-gas CO2/CH4 permeability and selectivity for the PAE-1 and the FVM derivatives compared against the upper bounds. (b) Comparison of activation energy of permeation for the polymer samples studied in this work. The diffusion correlation gas diameters are shown above the plot.39 | ||
To further validate the effectiveness of the FVM approach, a control PAE-1 sample that was never chloromethylated or protected underwent the same thermal treatment as the FVM process (PAE-1-3 in Fig. 5a), as is represented by the unfilled red circle in Fig. 5a. The thermal treatment of PAE-1 under a small concentration of oxygen resulted in a 19% increase in CO2/CH4 selectivity, but with a 23% reduction in CO2 permeability, matching expectations for performance trends that would be expected from standard thermal annealing procedures. This comparison underscores that the enhancement in separation performance is not merely due to the thermal treatment under oxygen conditions but is significantly influenced by the changes in packing structure and crosslinking during FVM. For the sample that had undergone FVM under nitrogen (i.e., PAE-PIP-FVM-0), permeability and selectivity decreased by 48% and 2%, respectively. This observation closely aligns the findings from the previous work on amine functionalization of PIM-1, where CO2 separation performance decreased due to the partial immobilization of CO2 molecules by the amine groups.32 Regarding other non-CO2 gas pairs, as shown in Fig. S8,† a slight increase in selectivity was noted along with a decrease in permeability for PAE-PIP-FVM-0. This common permeability–selectivity tradeoff implies that the packing structures become more compact due to re-introduction of hydrogen bonding groups, as suggested by the reduced d-spacing shown in Fig. S4.† Thus, this sample does not benefit from the FVM process. In other words, the manipulated free volume is not effectively maintained without any crosslinks due to the lack of interchain rigidity. Similar observations have been reported in the FVM treatment of 6FDA-HAB polyimide, where no crosslinks were formed during the process.31 Such observations emphasize the vital role of crosslinks in preserving the modified free volume structure, especially considering that PAEs have a more flexible backbone due to their ether linkage compared to other microporous polymers like PIM-1.
To investigate the energetics of gas transport, we conducted variable-temperature permeation tests to determine the activation energy for gas permeation (Ep), which can be obtained by the following equation and can be decoupled into activation energies of diffusion (ED) and enthalpies of sorption (ΔHS):20
| P = P0e−Ep/RT = D0e−ED/RTS0e−ΔHS/RT | (8) |
To further investigate the gas transport properties under more industrially relevant conditions, we tested our membrane samples for mixed-gas permeation. Fig. 6a shows the binary mixed-gas permeation data for a 50:50 CO2/CH4 composition at a total feed pressure of 32 psia (approximately 2.2 atm). As shown in the figure, CO2/CH4 mixed-gas selectivity increased from the pure-gas case for all PAE samples due to the competitive sorption effect. This phenomenon contributed to a decrease in CH4 permeability under mixed-gas conditions, as detailed in Tables S11 and S12.† Particularly noteworthy is the performance of PAE-PIP-FVM-0, which showed a 33% increase in CO2/CH4 mixed-gas selectivity compared to an 8% and 21% increase for PAE-1 and PAE-PIP-FVM-3, respectively. This outcome underscores the influence of strong secondary interactions between the lone pairs of nitrogen in piperazinyl groups and the acidic gases.24,36 Compared to the mixed-gas performance for PAE-1, the FVM method for PAE-PIP-FVM-3 resulted in mixed-gas selectivities that were 121% higher, highlighting the effectiveness of FVM in enhancing CO2/CH4 separation. To better represent membrane performance under real-world conditions, mixed-gas permeation experiments must be conducted in the presence of humidity. Water vapor, which is common in natural gas reservoirs and RNG production processes, can significantly impact the performance of polymer membranes through effects such as plasticization and competitive sorption. This is an ongoing area of research in our group, and preliminary results for a CO2/CH4 mixture under humid conditions are presented in Table S14.†
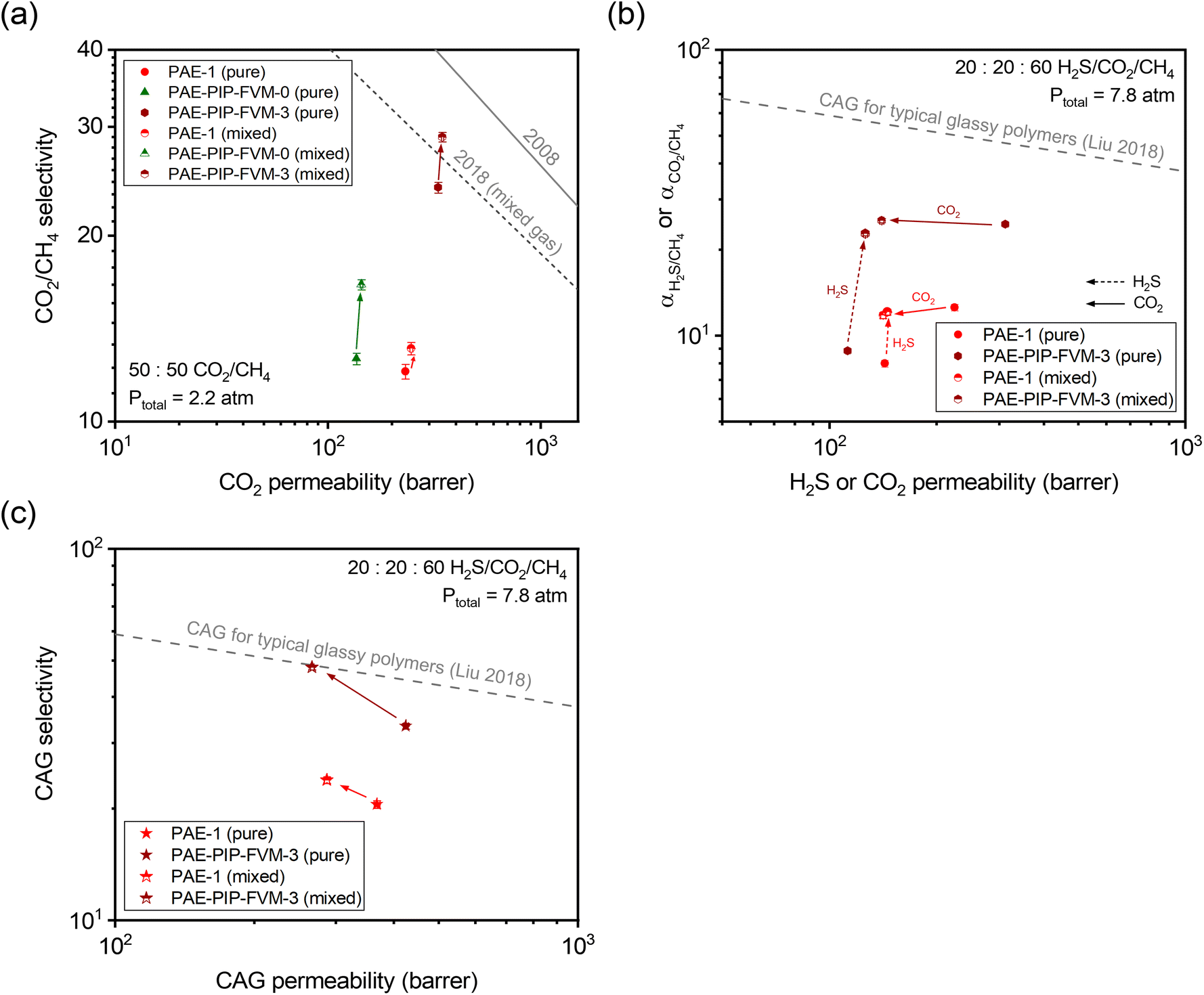 | ||
| Fig. 6 (a) Binary (50:50 CO2/CH4) mixed-gas separation performance compared against the pure- and mixed-gas upper bounds.14,75 (b) Comparison of ternary (20:20:60 H2S/CO2/CH4) mixed-gas separation performance and corresponding pure-gas data at equivalent partial pressures. (c) Comparison of combined acid gas (CAG) separation performance for results in (b). The dashed gray lines in (b) and (c) represent the CAG upper bound for typical glassy polymers.54 Filled symbols are pure-gas results, and half-filled symbols are mixed-gas results. The arrows indicate the direction of changes when going from pure-gas to mixed-gas conditions. | ||
As noted, the separation of acid gas mixtures containing both H2S and CO2 presents a challenge for polymer membranes based on free volume and free volume distribution effects. In this context, employing FVM in the presence of oxygen can be a promising strategy for enhancing acid–gas separation since it increases accessible free volume while simultaneously maintaining diffusion selectivity by inducing crosslinks. As such, we investigated permeation for ternary mixtures containing H2S for PAE-PIP-FVM-3 and compared the results with those of PAE-1. Fig. 6b shows the ternary mixed-gas permeation data for a 20:20:60 H2S/CO2/CH4 composition tested at a total feed pressure of 115 psia (approximately 7.8 atm) along with the corresponding pure-gas data at equivalent partial pressures. For CO2/CH4 pure-gas separation, PAE-PIP-FVM-3 showed increases in both permeability and selectivity following the FVM treatment of PAE-1, similar to the observation described in relation to Fig. 5a. However, there was a noticeable 21% decrease in H2S pure-gas permeability following FVM. This decrease in H2S permeability is attributed to the increased molecular-sieving properties of the FVM membranes. As shown in Table S13,† the diffusion coefficient of H2S decreased by 54% while sorption coefficient increased by 70% for PAE-PIP-FVM-3 compared to the pristine PAE-1. This observation aligns with expectations for a high free volume structure that contains a higher energetic barrier to larger gas molecules.
The advantage of FVM is particularly evident in the mixed-gas permeation case, where H2S molecules can significantly benefit from the increased free volume. As shown in Fig. 6b, H2S/CH4 mixed-gas selectivity for PAE-PIP-FVM-3 increased by 157% from the pure-gas selectivity, compared to only a 52% increase for PAE-1. This trend supports the concept that competitive sorption effects are more pronounced in membranes with higher free volume. In the ternary mixture, H2S molecules can effectively exclude both CO2 and CH4 molecules from the polymer matrix, leading to a notable decrease in CO2 permeability in the mixed-gas case. For the CAG separation shown in Fig. 6c, PAE-PIP-FVM-3 showed a 43% boost in mixed-gas CAG selectivity from its pure-gas counterpart and outperformed the PAE mixed-gas CAG selectivity by 102%, which only improved by 16% from the pure-gas case. This mixed-gas CAG performance for PAE-PIP-FVM-3 reached the CAG upper bound for typical glassy polymers,54 highlighting the effectiveness of FVM approach in enhancing the acid–gas separation performance.
To gain a deeper understanding of how FVM enhances ternary acid–gas separations, pure-gas DMS model parameters derived from fitting the pure-gas sorption isotherms in Fig. S13† were used to model the mixed-gas sorption isotherms for a 20:20:60 H2S/CO2/CH4 ternary mixture using eqn (6). The resulting mixed-gas sorption isotherms are depicted in Fig. S14.† By using these modeled mixed-gas sorption isotherms, we estimated mixed-gas sorption selectivity and calculated diffusion selectivity using the sorption–diffusion model:
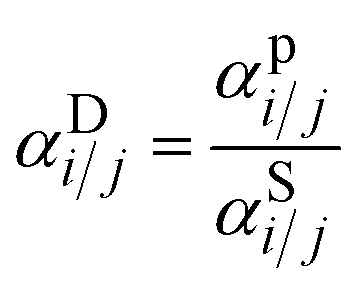 | (9) |
Fig. 7a illustrate the comparison between measured pure-gas and modelled mixed-gas sorption selectivities obtained from static equilibrium measurements (the secant slope approach). Mixture selectivities are for the composition tested for permeation in Fig. 6b. For all samples, CO2/CH4 sorption selectivity remained close to 1 in both pure- and mixed-gas cases. Although an increase in CO2/CH4 sorption selectivity may be anticipated based on competitive sorption effects, the negligible change in selectivity results from the relative exothermicity of H2S sorption, which suppresses CO2 sorption in a similar way to that of CH4 sorption for binary CO2/CH4 conditions. For H2S/CH4, the mixed-gas sorption selectivity slightly increased, as expected for the most condensable penetrant due to competitive effects. This increase was more pronounced in the PAE-PIP-FVM-3 sample, showing a 29% increase from pure-gas selectivity, compared to a 23% increase in the PAE-1 sample. This result aligns well with the hypothesis that FVM enhances sorption capacity, allowing H2S molecules to occupy more of the sorption domain compared to that for other gases. Fig. 7b illustrates a comparison of diffusion selectivity calculated using eqn (9). As previously discussed, the calculated diffusion selectivities may not represent accurate values during mixed-gas permeation, as they were derived from a combination of static equilibrium sorption measurements and dynamic permeation measurements. However, we can still derive some important insights under the assumptions of this analysis. As shown in Fig. 7b, PAE-PIP-FVM-3 exhibited notably higher diffusion selectivities in both pure- and mixed-gas cases for both gas pairs, indicating that FVM can be effective in enhancing the molecular sieving effect while also retaining an expanded free volume. For instance, CO2/CH4 mixed-gas diffusion selectivity increased from 12.1 to 20.4 after FVM. Notably, PAE-PIP-FVM-3 achieved a H2S/CH4 diffusion selectivity of 7.3 in mixed-gas tests. Achieving such high diffusion selectivity for H2S/CH4 is challenging, given their similar kinetic diameters.27 While further investigation with mixed-gas diffusivity measurements is necessary to validate our analysis based on equilibrium sorption measurements in eqn (9), the results suggest that FVM may enhance H2S/CH4 diffusion selectivity under mixed-gas conditions. These interesting findings may result from altered free volume architecture due to high H2S sorption in these functional microporous polymers during testing, but more experiments are required to qualify this possibility of emergent behavior due to penetrant sorption. This dual enhancement in selectivity for both H2S/CH4 and CO2/CH4 underlines the effectiveness of FVM in addressing the complexities of acid–gas separation in mixed-gas environments.
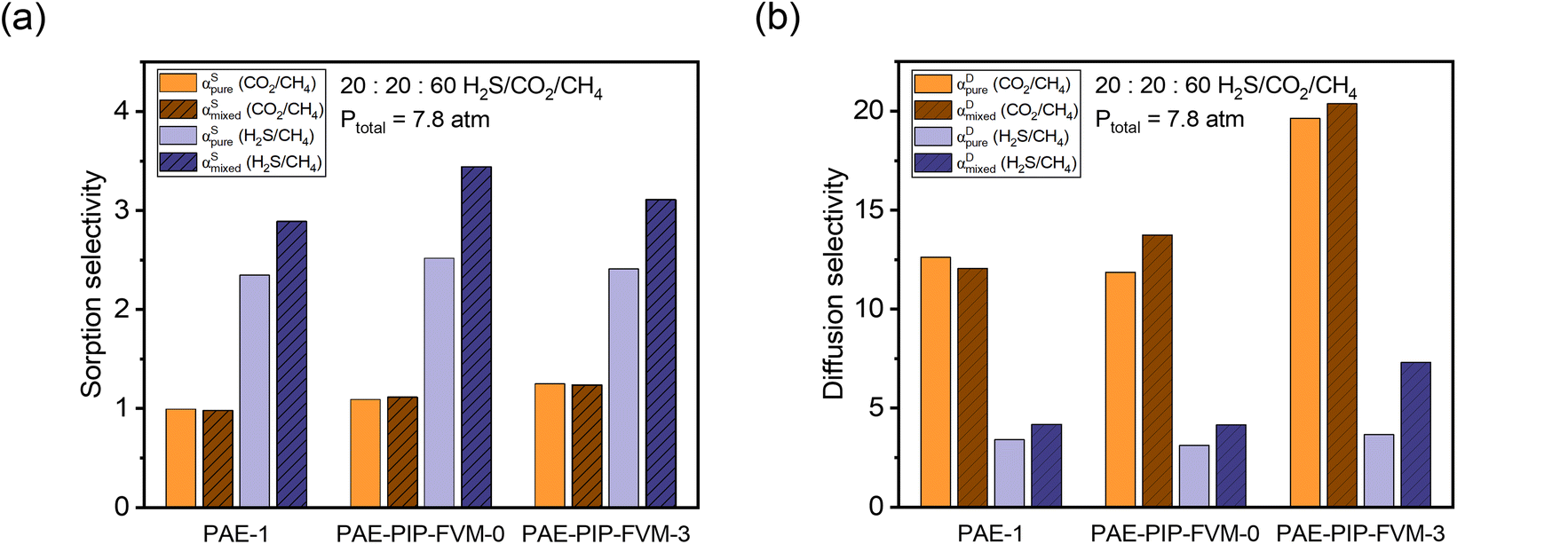 | ||
| Fig. 7 Comparison of pure-gas selectivities and modeled mixed-gas selectivities for a 20:20:60 H2S/CO2/CH4 mixture at 7.8 atm for (a) sorption and (b) diffusion. | ||
Penetrant-induced plasticization is a significant challenge for the deployment of membranes for various applications in the gas processing sector,28 so we investigated the influence of FVM on H2S- and CO2-induced plasticization. Under industrially relevant pressures, condensable penetrants like H2S and CO2 increase segmental and cooperative modes of motion in the polymer, leading to substantial losses in selectivity.6,36Fig. 8a and b depict the normalized H2S and CO2 permeabilities, respectively, for the PAE films at various feed fugacities. As shown in the figure, all PAE films demonstrated a decrease in permeability with increasing feed fugacity. This significant decrease in permeability at low fugacity is associated with the saturation of the Langmuir sorption mode.36 Notably, PAE-1 exhibited H2S plasticization pressure at 3 atm, while the PAE-PIP-FVM-3 and PAE-PIP-FVM-0 films showed enhanced resistance to plasticization, with plasticization pressures up to 5.7 atm and 6.4 atm, respectively. In the case of CO2, the pristine PAE-1 film showed plasticization pressure at 33.7 atm, whereas both FVM-treated samples did not exhibit a plasticization pressure up to the highest tested feed fugacity of 40 atm. These findings suggest that FVM treatment enhances membrane resistance to plasticization. A comparison of H2S-induced plasticization pressures from the literature, as illustrated in Fig. S16,† shows that PAE-PIP-FVM-0 has the highest H2S-induced plasticization resistance among polymers tested to date, along with PAE-PIP-FVM-3, which ranks third by the same comparison. These remarkable resistances may be attributed to the hydrogen bonds formed by the piperazinyl groups or the chemical functionality in the polymer matrix from thermal oxidative degradation. Secondary interactions, like hydrogen bonding, are known to be particularly effective in mitigating plasticization.28
 | ||
| Fig. 8 Comparison of normalized permeabilities at various feed fugacities containing (a) H2S and (b) CO2. Black arrows indicate the observed plasticization pressures. | ||
4. Conclusions
The challenge of natural gas and biogas sweetening using glassy polymers arises from opposing permeability–selectivity correlations for H2S/CH4 and CO2/CH4 separations. For H2S/CH4, separation is largely driven by sorption, with an increase in free volume significantly increasing the sorption domain for the highly condensable H2S gas molecules. This predominantly sorption-selective process overcomes the typical decrease in diffusion selectivity with increase in free volume. In this work, we have successfully implemented a FVM strategy on a microporous PAE polymer to effectively enhance the sorption capacity for H2S molecules while preserving CO2 selectivities through the enhancement of molecular sieving properties via thermal oxidative crosslinking. These crosslinks played a crucial role not only in preserving the CO2/CH4 selectivity but also in significantly improving the diffusion selectivity for H2S/CH4, which was evidenced by the results from our mixed-gas transport modeling. As a result, the FVM treatment with oxygen resulted in a remarkable 102% enhancement in mixed-gas selectivity for CAGs compared to the original PAE-1 films, demonstrating the effectiveness of this method in improving acid–gas separation. Based on chemical characterization, FVM in the presence of oxygen formed oxidative degradation species and new crosslinks. These modifications not only improved acid–gas separation efficiency but also significantly increased resistance to plasticization, making the resulting membrane from this post-synthetic modification approach of interest for industrial applications. Our study introduces a versatile post-synthetic modification approach that can be applied to any polymer containing a nucleophilic aromatic unit. Extending this strategy across various polymer compositions holds the promise of substantial enhancements in acid–gas separation performance and resistance to highly condensable gas feeds, a significant stride forward for gas separation membrane technologies.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Separation Science program under Award Number DE-SC0019087, the Office of Naval Research (ONR) under Award Numbers N00014-20-1-2418 and N00014-21-1-2666, and Eni S.p.A. through the MIT Energy Initiative. TMS is grateful for support from the National Science Foundation DMR-2207299. The authors would like to thank Walter Massefski and Sarah Willis at MIT's Department of Chemistry Instrumentation Facility (DCIF) for help with SSNMR characterization. The authors would also like to thank Sheng Guo for sharing the details of the polymer synthesis procedures.References
- U.S. Energy Information Administration, International Energy Outlook 2023, 2023 Search PubMed.
- A. Safari, N. Das, O. Langhelle, J. Roy and M. Assadi, Energy Sci. Eng., 2019, 7, 1075–1094 CrossRef.
- S. Yi, X. Ma, I. Pinnau and W. J. Koros, J. Mater. Chem. A, 2015, 3, 22794–22806 RSC.
- M. Syed, G. Soreanu, P. Falletta and M. Béland, Can. Agric. Eng., 2006, 48, 1–14 Search PubMed.
- L. R. C. Assunção, P. A. S. Mendes, S. Matos and S. Borschiver, Appl. Energy, 2021, 292, 116849 CrossRef.
- Y. Ma, H. Guo, R. Selyanchyn, B. Wang, L. Deng, Z. Dai and X. Jiang, J. Mater. Chem. A, 2021, 9, 20211–20240 RSC.
- D. J. Harrigan, J. A. Lawrence, H. W. Reid, J. B. Rivers, J. T. O'Brien, S. A. Sharber and B. J. Sundell, J. Membr. Sci., 2020, 602, 117947 CrossRef CAS.
- F. Hamad, M. Qahtani, A. Ameen, M. Vaidya, S. Duval, A. Bahamdan and F. Otaibi, Sep. Purif. Technol., 2020, 237, 116348 CrossRef CAS.
- M. Yadav, M. H. Sliem, A. M. Abdullah, K. M. Youssef and N. H. Al-Qahtani, Sustainability, 2022, 14, 8015 CrossRef CAS.
- D. F. Sanders, Z. P. Smith, R. Guo, L. M. Robeson, J. E. McGrath, D. R. Paul and B. D. Freeman, Polymer, 2013, 54, 4729–4761 CrossRef CAS.
- J. Hack, N. Maeda and D. M. Meier, ACS Omega, 2022, 7, 39520–39530 CrossRef CAS PubMed.
- M. Galizia, W. S. Chi, Z. P. Smith, T. C. Merkel, R. W. Baker and B. D. Freeman, Macromolecules, 2017, 50, 7809–7843 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- B. D. Freeman, Macromolecules, 1999, 32, 375–380 CrossRef CAS.
- P. M. Budd, E. S. Elabas, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib, C. E. Tattershall and D. Wang, Adv. Mater., 2004, 16, 456–459 CrossRef CAS.
- N. B. Mc Keown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- Y. He, F. M. Benedetti, S. Lin, C. Liu, Y. Zhao, H. Z. Ye, T. Van Voorhis, M. G. De Angelis, T. M. Swager and Z. P. Smith, Adv. Mater., 2019, 31, 1807871 CrossRef PubMed.
- S. Lin, K. R. Storme, Y. C. M. Wu, F. M. Benedetti, T. M. Swager and Z. P. Smith, J. Membr. Sci., 2023, 668, 121194 CrossRef CAS.
- H. W. H. Lai, F. M. Benedetti, Z. Jin, Y. C. Teo, A. X. Wu, M. G. De Angelis, Z. P. Smith and Y. Xia, Macromolecules, 2019, 52, 6294–6302 CrossRef CAS.
- H. W. H. Lai, F. M. Benedetti, J. M. Ahn, A. M. Robinson, Y. Wang, I. Pinnau, Z. P. Smith and Y. Xia, Science, 2022, 375, 1390–1392 CrossRef CAS PubMed.
- S. Guo, J. Y. Yeo, F. M. Benedetti, D. Syar, T. M. Swager and Z. P. Smith, Angew. Chem., Int. Ed., 2024, 63, e202315611 CrossRef CAS PubMed.
- S. Guo and T. M. Swager, J. Am. Chem. Soc., 2021, 143, 11828–11835 CrossRef CAS PubMed.
- K. Mizrahi Rodriguez, P. A. Dean, S. Guo, N. Roy, T. M. Swager and Z. P. Smith, J. Membr. Sci., 2024, 696, 122464 CrossRef CAS.
- L. M. Robeson, M. E. Dose, B. D. Freeman and D. R. Paul, J. Membr. Sci., 2017, 525, 18–24 CrossRef CAS.
- L. M. Robeson, Q. Liu, B. D. Freeman and D. R. Paul, J. Membr. Sci., 2015, 476, 421–431 CrossRef CAS.
- Y. Liu, Z. Liu, G. Liu, W. Qiu, N. Bhuwania, D. Chinn and W. J. Koros, J. Membr. Sci., 2020, 593, 117430 CrossRef CAS.
- K. Mizrahi Rodriguez, S. Lin, A. Wu, K. Storme, T. Joo, A. Grosz, N. Roy, D. Syar, F. M. Benedetti and Z. P. Smith, Chem. Soc. Rev., 2024, 53, 2435–3529 RSC.
- B. Kraftschik, W. J. Koros, J. R. Johnson and O. Karvan, J. Membr. Sci., 2013, 428, 608–619 CrossRef CAS.
- B. Kraftschik and W. J. Koros, Macromolecules, 2013, 46, 6908–6921 CrossRef CAS.
- S. Lin, T. Joo, F. M. Benedetti, L. C. Chen, A. X. Wu, K. Mizrahi Rodriguez, Q. Qian, C. M. Doherty and Z. P. Smith, Polymer, 2021, 212, 123121 CrossRef CAS.
- K. Mizrahi Rodriguez, S. Lin, A. X. Wu, G. Han, J. J. Teesdale, C. M. Doherty and Z. P. Smith, Angew. Chem., Int. Ed., 2021, 60, 6593–6599 CrossRef CAS PubMed.
- T. Joo, K. Mizrahi Rodriguez, H. Lee, D. P. Acharya, C. M. Doherty and Z. P. Smith, J. Mater. Chem. A, 2023, 11, 15943–15957 RSC.
- T. Joo, T. H. Lee, S. J. Kaser, W.-N. Wu, S. Wi, J. Y. Yeo and Z. P. Smith, Chem. Mater., 2024, 36, 4275–4290 CrossRef CAS.
- P. A. Dean, Y. Wu, S. Guo, T. M. Swager and Z. P. Smith, JACS Au, 2024, 4(10), 3848–3856 CrossRef CAS PubMed.
- K. Mizrahi Rodriguez, F. M. Benedetti, N. Roy, A. X. Wu and Z. P. Smith, J. Mater. Chem. A, 2021, 9, 23631–23642 RSC.
- Z. P. Smith, D. F. Sanders, C. P. Ribeiro, R. Guo, B. D. Freeman, D. R. Paul, J. E. McGrath and S. Swinnea, J. Membr. Sci., 2012, 415–416, 558–567 CrossRef CAS.
- W. W. Brandt, J. Phys. Chem., 1959, 63, 1080–1085 CrossRef CAS.
- L. M. Robeson, Z. P. Smith, B. D. Freeman and D. R. Paul, J. Membr. Sci., 2014, 453, 71–83 CrossRef CAS.
- S. Sircar, Ind. Eng. Chem. Res., 2006, 45, 5435–5448 CrossRef CAS.
- A. Hayek, A. Alsamah, Q. Saleem, R. H. Alhajry, A. A. Alsuwailem and F. I. Jassim, ACS Appl. Polym. Mater., 2022, 4, 9257–9271 CrossRef CAS.
- Q. Qian, P. A. Asinger, M. J. Lee, G. Han, K. Mizrahi Rodriguez, S. Lin, F. M. Benedetti, A. X. Wu, W. S. Chi and Z. P. Smith, Chem. Rev., 2020, 120, 8161–8266 CrossRef CAS PubMed.
- P. A. Dean, K. Mizrahi Rodriguez, S. Guo, N. Roy, T. M. Swager and Z. P. Smith, J. Membr. Sci., 2024, 696, 122465 CrossRef CAS.
- H. Lin, E. Van Wagner, B. D. Freeman, L. G. Toy and R. P. Gupta, Science, 2006, 311, 639–642 CrossRef CAS PubMed.
- E. V. Anslyn and D. A. Dougherty, in Modern Physical Organic Chemistry, University Science Books, Mill Valley, 2006, pp. 537–625 Search PubMed.
- Z. Xie, B. Dao, J. Hodgkin, M. Hoang, A. Hill and S. Gray, J. Polym. Res., 2011, 18, 965–973 CrossRef CAS.
- K. Mizrahi Rodriguez, A. X. Wu, Q. Qian, G. Han, S. Lin, F. M. Benedetti, H. Lee, W. S. Chi, C. M. Doherty and Z. P. Smith, Macromolecules, 2020, 53, 6220–6234 CrossRef CAS.
- M. Ganesan and P. Nagaraaj, Org. Chem. Front., 2020, 7, 3792–3814 RSC.
- Q. Song, S. Cao, R. H. Pritchard, B. Ghalei, S. A. Al-Muhtaseb, E. M. Terentjev, A. K. Cheetham and E. Sivaniah, Nat. Commun., 2014, 5, 4813 CrossRef PubMed.
- V. Kholodovych and W. J. Welsh, in Physical Properties of Polymers Handbook, ed. J. E. Mark, Springer, New York, 2nd edn, 2007 Search PubMed.
- K. Pielichowski, J. Njuguna and T. M. Majka, Thermal Degradation of Polymeric Materials, Elsevier Science, Amsterdam, 2nd edn, 2022 Search PubMed.
- B. D. Freeman and I. Pinnau, ACS Symp. Ser., 1999, 733, 1–27 CrossRef CAS.
- W. F. Yong, F. Y. Li, Y. C. Xiao, P. Li, K. P. Pramoda, Y. W. Tong and T. S. Chung, J. Membr. Sci., 2012, 407–408, 47–57 CrossRef CAS.
- G. Liu, V. Chernikova, Y. Liu, K. Zhang, Y. Belmabkhout, O. Shekhah, C. Zhang, S. Yi, M. Eddaoudi and W. J. Koros, Nat. Mater., 2018, 17, 283–289 CrossRef CAS PubMed.
- M. Lanč, K. Pilnáček, C. R. Mason, P. M. Budd, Y. Rogan, R. Malpass-Evans, M. Carta, B. C. Gándara, N. B. McKeown, J. C. Jansen, O. Vopička and K. Friess, J. Membr. Sci., 2019, 570–571, 522–536 CrossRef.
- D. R. Paul and W. J. Koros, J. Polym. Sci., Part B: Polym. Phys., 1976, 14, 675–685 CAS.
- S. Yi, B. Ghanem, Y. Liu, I. Pinnau and W. J. Koros, Sci. Adv., 2019, 5, eaaw5459 CrossRef PubMed.
- C. S. K. Achoundong, N. Bhuwania, S. K. Burgess, O. Karvan, J. R. Johnson and W. J. Koros, Macromolecules, 2013, 46, 5584–5594 CrossRef CAS.
- J. Vaughn and W. J. Koros, Macromolecules, 2012, 45, 7036–7049 CrossRef CAS.
- J. T. Vaughn and W. J. Koros, J. Membr. Sci., 2014, 465, 107–116 CrossRef CAS.
- G. O. Yahaya, M. S. Qahtani, A. Y. Ammar, A. A. Bahamdan, A. W. Ameen, R. H. Alhajry, M. M. B. Sultan and F. Hamad, Chem. Eng. J., 2016, 304, 1020–1030 CrossRef CAS.
- G. O. Yahaya, I. Mokhtari, A. A. Alghannam, S. H. Choi, H. Maab and A. A. Bahamdan, J. Membr. Sci., 2018, 550, 526–535 CrossRef CAS.
- A. Hayek, G. O. Yahaya, A. Alsamah, A. A. Alghannam, S. A. Jutaily and I. Mokhtari, Polymer, 2019, 166, 184–195 CrossRef CAS.
- A. Hayek, G. O. Yahaya, A. Alsamah and S. K. Panda, J. Appl. Polym. Sci., 2020, 137, 48336 CrossRef CAS.
- A. Hayek, A. Alsamah, G. O. Yahaya, E. A. Qasem and R. H. Alhajry, J. Mater. Chem. A, 2020, 8, 23354–23367 RSC.
- A. Hayek, A. Alsamah, E. A. Qasem, N. Alaslai, R. H. Alhajry and G. O. Yahaya, Sep. Purif. Technol., 2019, 227, 115713 CrossRef CAS.
- A. Hayek, A. Alsamah, N. Alaslai, H. Maab, E. A. Qasem, R. H. Alhajry and N. M. Alyami, ACS Appl. Polym. Mater., 2020, 2, 2199–2210 CrossRef CAS.
- J. A. Lawrence, D. J. Harrigan, C. R. Maroon, S. A. Sharber, B. K. Long and B. J. Sundell, J. Membr. Sci., 2020, 616, 118569 CrossRef CAS.
- G. Chatterjee, A. A. Houde and S. A. Stern, J. Membr. Sci., 1997, 135, 99–106 CrossRef CAS.
- G. Liu, A. Cadiau, Y. Liu, K. Adil, V. Chernikova, I.-D. Carja, Y. Belmabkhout, M. Karunakaran, O. Shekhah, C. Zhang, A. K. Itta, S. Yi, M. Eddaoudi and W. J. Koros, Angew. Chem., Int. Ed., 2018, 130, 15027–15032 CrossRef.
- S. J. Datta, A. Mayoral, N. M. Srivatsa Bettahalli, P. M. Bhatt, M. Karunakaran, I. D. Carja, D. Fan, P. G. M. Mileo, R. Semino, G. Maurin, O. Terasaki and M. Eddaoudi, Science, 2022, 376, 1080–1087 CrossRef CAS PubMed.
- W.-N. Wu, K. Mizrahi Rodriguez, N. Roy, J. J. Teesdale, G. Han, A. Liu and Z. P. Smith, ACS Appl. Mater. Interfaces, 2023, 15, 52893–52907 CAS.
- Z. Liu, Y. Liu, W. Qiu and W. J. Koros, Angew. Chem., Int. Ed., 2020, 59, 14877–14883 CrossRef CAS PubMed.
- M. Z. Ahmad, T. A. Peters, N. M. Konnertz, T. Visser, C. Téllez, J. Coronas, V. Fila, W. M. de Vos and N. E. Benes, Sep. Purif. Technol., 2020, 230, 115858 CrossRef CAS.
- Y. Wang, X. Ma, B. S. Ghanem, F. Alghunaimi, I. Pinnau and Y. Han, Mater. Today Nano, 2018, 3, 69–95 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta07738e |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |