
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of inner hydrophobicity of dendrimer nanomicelles on biodistribution: a PET imaging study†
Tom
Roussel‡
a,
Twiany
Cruz-Dubois‡
bc,
Beatrice
Louis
bc,
Erik
Laurini
d,
Ling
Ding
a,
Laure
Balasse
c,
Vincent
Nail
bc,
Françoise
Dignat-George
b,
Suzanne
Giorgio
a,
Sabrina
Pricl
 de,
Benjamin
Guillet
bc,
Philippe
Garrigue
bc and
Ling
Peng
*a
de,
Benjamin
Guillet
bc,
Philippe
Garrigue
bc and
Ling
Peng
*a
aAix Marseille University, CNRS, Centre Interdisciplinaire de Nanoscience de Marseille (CINaM), UMR 7325, Equipe Labellisée Ligue Contre le Cancer, Marseille, France. E-mail: ling.peng@univ-amu.fr
bAix Marseille University, INSERM, INRAE, C2VN, Marseille, France
cAix Marseille University, CNRS, CERIMED, Marseille, France
dMolecular Biology and Nanotechnology Laboratory, Department of Engineering and Architectures, University of Trieste, Trieste 34127, Italy
eDepartment of General Biophysics, Faculty of Biology and Environmental Protection, University of Lodz, Lodz 90-136, Poland
First published on 17th December 2024
Abstract
Self-assembly is a powerful strategy for building nanosystems for biomedical applications. We have recently developed small amphiphilic dendrimers capable of self-assembling into nanomicelles for tumor imaging. In this context, we studied the impact of increased hydrophobicity of the amphiphilic dendrimer on hydrophilic/hydrophobic balance and consequently on the self-assembly and subsequent biodistribution. Remarkably, despite maintaining the exact same surface chemistry, similar zeta potential, and small size, the altered and enlarged hydrophobic component within the amphiphilic dendrimer led to enhanced stability of the self-assembled nanomicelles, with prolonged circulation time and massive accumulation in the liver. This study reveals that even structural alteration within the interior of nanomicelles can dramatically impact biodistribution profiles. This finding highlights the deeper complexity of rational design for nanomedicine and the need to consider factors other than surface charge and chemistry, as well as size, all of which significantly impact the biodistribution of self-assembling nanosystems.
Introduction
Bioimaging has revolutionized modern medicine both in diagnosis, by providing accurate disease detection, grading and staging, as well as in monitoring response and efficacy of treatment.1–3 The increasing number of applications with different imaging modalities has led to high demand for imaging agents capable of improving imaging sensitivity, specificity, and spatial resolution. Nanotechnology-based systems are expected to meet the needs for bioimaging by virtue of their ability to accommodate different imaging agents in abundance within nanoscale dimensions, allowing increased local concentrations of imaging reporters for multimodal imaging with superior sensitivity and specificity.3–7 As such, a wide range of nanosystems, including liposomes, polymers, quantum dots, iron oxide nanoparticles, gold nanoparticles, and carbon nanodots, have been explored and studied as bioimaging probes.3–7Among the different nanomaterials studied, a unique class of synthetic macromolecules called dendrimers are particularly attractive for the construction of imaging probes by virtue of their well-defined dendritic structure, multivalent cooperativity and nanosize per se.8–13 Recently, we developed small amphiphilic dendrimers 1–3 (Fig. 1) that self-assemble into supramolecular nanomicelles, allowing effective tumor imaging by using single-photon emission computed tomography (SPECT) and positron emission tomography (PET).14–16 These amphiphilic dendrimers 1–3 consist of a long hydrophobic alkyl chain and a hydrophilic poly(amidoamine) (PAMAM) dendron bearing four macrocyclic terminals for chelating radionuclides such as 111In3+ for SPECT imaging and 68Ga3+ for PET imaging (Fig. 1B). Importantly, all three dendrimers 1–3 self-assemble into nanosized micelles (denoted as 1@, 2@ and 3@, respectively) each carrying a much higher number of radionuclide reporters than in the non-assembled state. Owing to their nanoscale size, these micelles can passively target tumors by exploiting the “enhanced permeability and retention” (EPR) effect (Fig. 1)14–16 that results from the leaky vasculature and dysfunctional lymphatic drainage in the tumor microenvironment.17–21 The accumulation of the nanosized agents and their increased local concentrations within the tumor lesions improve imaging sensitivity and specificity for effective cancer detection.
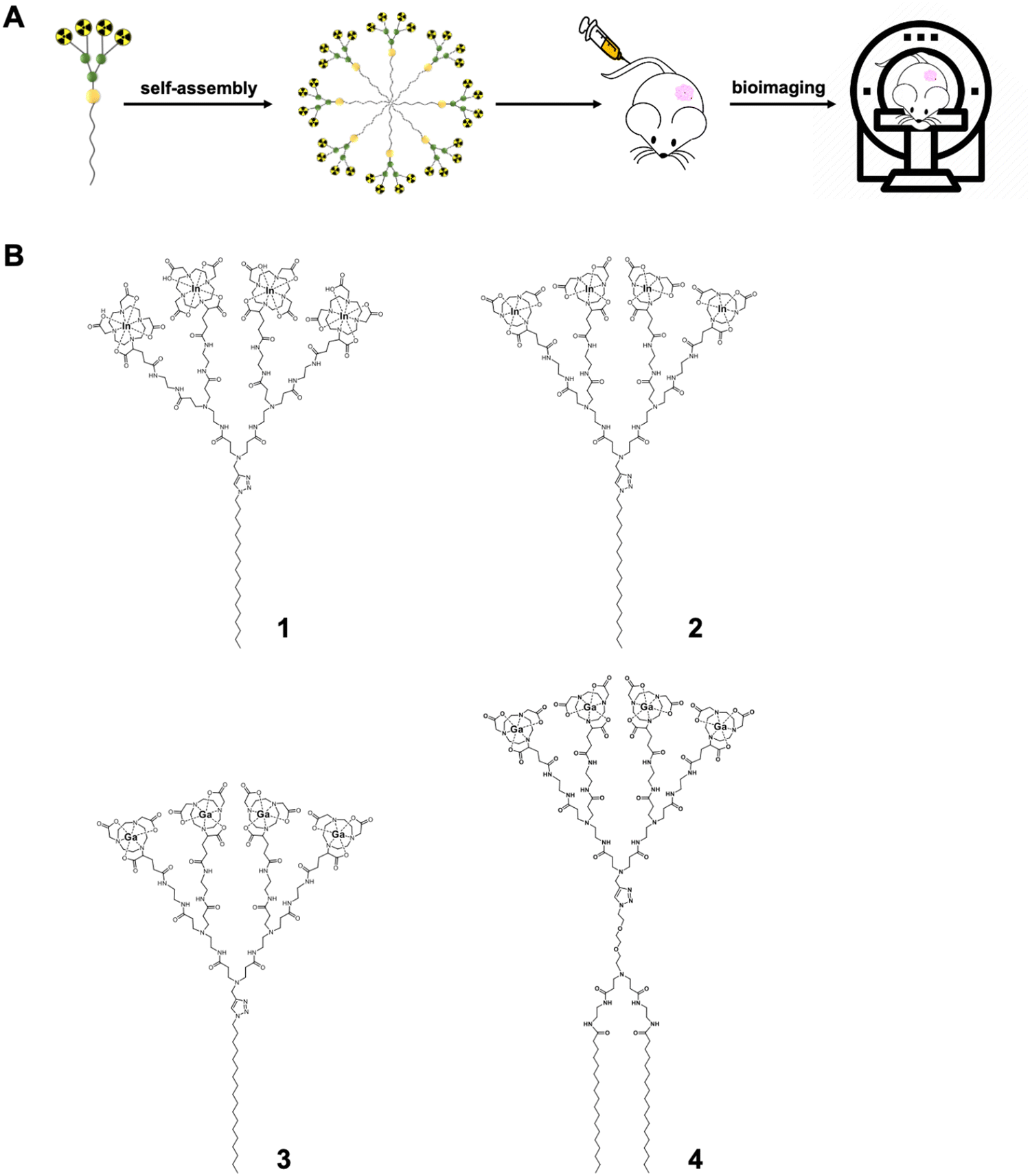 | ||
| Fig. 1 Amphiphilic dendrimers self-assemble into nanomicelles for bioimaging. (A) Schematic illustration of the supramolecular dendrimer nanosystem based on the self-assembling amphiphilic dendrimer for bioimaging. (B) Chemical structures of the amphiphilic dendrimers 1–4 developed for nuclear imaging, with 1–2 for single-photon emission computed tomography (SPECT) and 3–4 for positron emission tomography (PET), respectively. | ||
Notably, although the imaging probe 1@ was effective for tumor imaging using SPECT, it showed a distinctly high liver retention.15 By substituting the larger macrocyclic chelating DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) ring with the smaller NOTA (1,4,7-triazacyclononane-1,4,7-triacetic acid) cage, the resulting nanotracer 2@ had considerably reduced liver uptake.16 This difference can be attributed to the significant, yet different impact that each chelator has on the complex size, charge, geometry, and lipophilicity when chelated with radionuclide metal ions.22,23 Indeed, while the zeta potential of 1@ bearing the negatively charged In3+/DOTA-caged terminals was negative, it was positive in 2@ with the neutral In3+/NOTA-complex terminals. This finding of the positive charge of 2@ drastically reducing its uptake in the liver contrasts with the majority of reports in the literature, highlighting that positively charged nanoparticles preferentially accumulate in this organ. Thus, our study provides new perspectives for improving the biodistribution of positively charged nanosystems for biomedical applications.
It should be mentioned that 3@ exhibited excellent performance in tumor imaging using PET.14 By exploiting both dendritic multivalence and passive tumor targeting mediated by the EPR effect, the nanotracer 3@ demonstrated superior imaging sensitivity and specificity, with significantly higher PET signal ratios compared to the clinical gold standard [18F]FDG (2-fluorodeoxyglucose). Most importantly, 3@ even allowed the detection of tumors that are otherwise undetectable using [18F]FDG. However, 3@ still accumulated in the liver, which may constitute a limitation for its future clinical translation.
We were intrigued by the potential for reducing liver accumulation through structural modification of amphiphilic dendrimers. Differently from the change introduced in 2 involving the hydrophilic dendron bearing the NOTA-chelating terminals, we were curious as to whether modification of the hydrophobic component of dendrimer 3 could obviate liver uptake. The hydrophobic section of an amphiphilic molecule not only affects the balance between hydrophilicity and hydrophobicity, but also influences molecular geometry.24,25 Thus, the structural change in the hydrophobic region would be expected to have an impact on the packing and stability of the resulting self-assembled nanostructure.26–29 We therefore wanted to investigate the impact of changing the hydrophobic component of amphiphilic dendrimer 3 on its self-assembly stability and, subsequently, its biodistribution and liver accumulation. We hypothesized that increasing the size of the hydrophobic portion in dendrimer 3 would enhance the overall hydrophobicity of the amphiphilic dendrimer and shift the hydrophilic–hydrophobic balance to improve self-assembling stability and subsequently alter its biodistribution.30
Accordingly, we designed and synthesized the amphiphilic dendrimer 4 with two hydrophobic C18 chains (Fig. 1B), resulting in a larger hydrophobic entity compared to dendrimer 3, while retaining the same hydrophilic PAMAM dendron as 3. In particular, 4 exhibited enhanced self-assembly into nanomicelles 4@ that were also more stable than those formed by 3. However, PET/CT imaging revealed that 4@ had significantly higher liver accumulation compared to 3@, despite their same surface chemistry, spherical morphology and similar surface charge, as well as their small size. Although studies have reported that stable nanoparticles are preferable for imaging probes,31–33 our results clearly show that nanoparticle stability is not necessarily conducive to reduced hepatic uptake. This finding highlights a deeper level of complexity that requires consideration in the rational design of self-assembling nanosystems as imaging agents or therapeutics in the field of nanomedicine.
Results and discussion
To synthesize 4 we first used a click reaction to couple the hydrophilic PAMAM dendron AD-1 with the hydrophobic entity AD-2 (Fig. 2). We then proceeded with amidation, followed by conjugation of the macrocyclic cages at the dendrimer terminals, with subsequent deprotection and finally chelation with gallium ions (Fig. 2). Specifically, we prepared the hydrophilic PAMAM dendron AD-1 and the hydrophobic component AD-2 according to well-established protocols.34–36 The click reaction between AD-1 and AD-2 was successful and yielded AD-3 effectively (Fig. 2). However, similar migration of AD-3 and AD-2 on silica gel made purification of AD-3via column separation tedious and laborious. Furthermore, AD-3 showed strong adsorption on silica gel with eluent systems even containing ammonia, resulting in a moderate isolation yield of 55%. Gratifyingly, the amidation of AD-3 with ethylenediamine advanced efficiently, producing AD-4 with an excellent yield of 95% (Fig. 2). It should be mentioned that AD-4 exhibited low solubility in water, although its purification by dialysis proved to be effective. Additionally, AD-4 showed poor solubility in various organic solvents such as DMF, DMSO, CH2Cl2, THF, MeOH, acetonitrile, acetone, and ethyl acetate. This impeded the coupling reaction between AD-4 and the NOTA reagent of NODA-GA(tBu)3 in DMF under the conditions previously validated for the synthesis of 3.14 Consequently, we opted for mixed solvents of DMF and dichloromethane, which enabled the successful conjugation of NODA-GA(tBu)3 to all the four dendrimer terminals. Subsequent deprotection of the tBu group using trifluoroacetic acid (TFA), followed by dialysis, yielded the desired NOTA-conjugated dendrimer AD-NOTA (Fig. 2).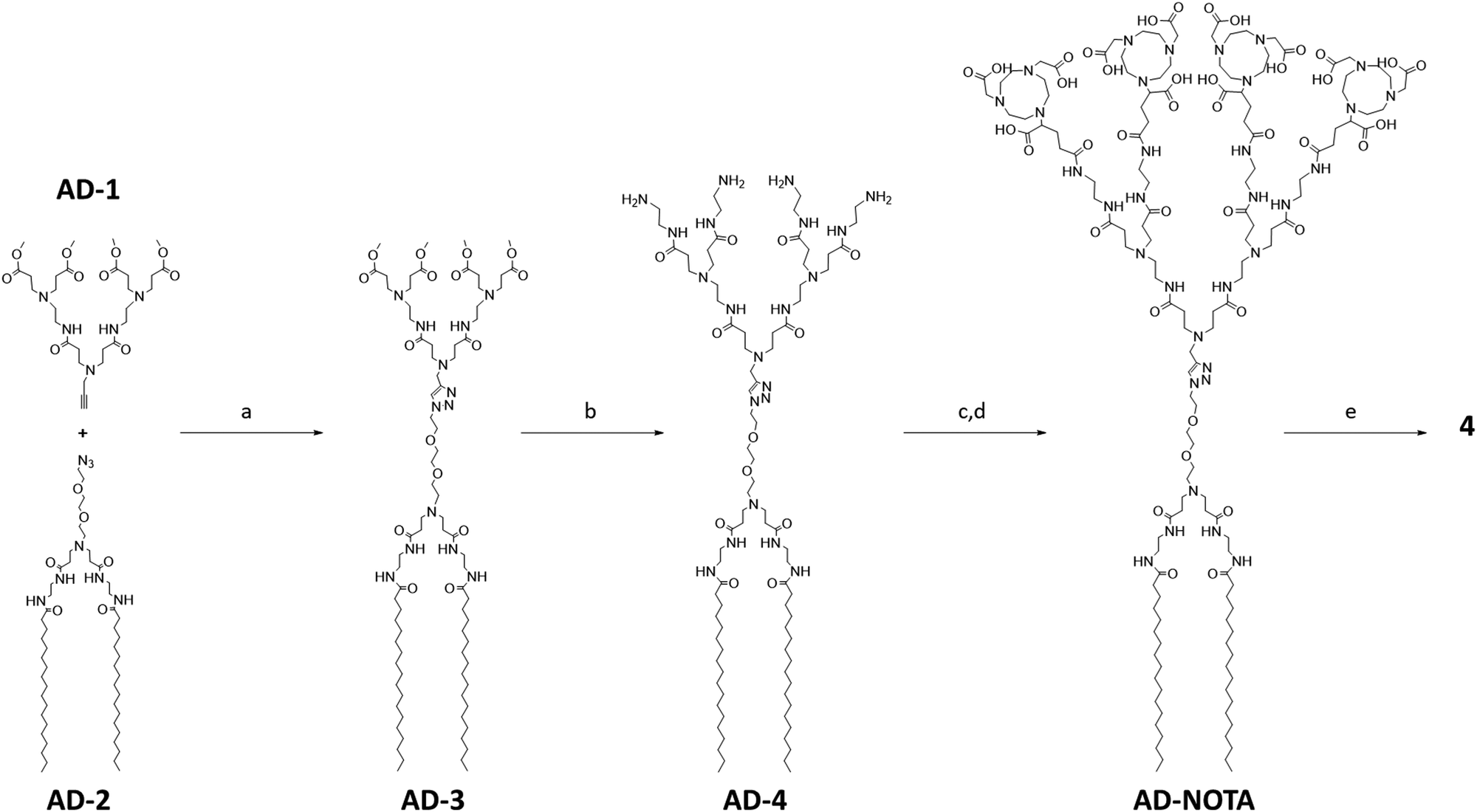 | ||
| Fig. 2 Synthesis of the amphiphilic dendrimer 4. Synthesis scheme: (a) CuI, DBU, DMF, 65 °C, 3 h; (b) EDA, MeOH, 30 °C, 3 days; (c) NODA-GA(tBu)3, PyBOP, NMM, DCM/DMF, 30 °C, 3 days; (d) TFA, DCM, 30 °C, 16 h; (e) [69Ga]GaCl3, 1.0 mM HCl, 25 °C, 15 min. | ||
Further chelation of the dendrimer AD-NOTA with the stable isotope of gallium (69Ga) using GaCl3 gave 4 in 90% yield, even after purification using dialysis. Remarkably, the chelating reaction proceeded rapidly and efficiently, even at room temperature and within 15 minutes. This can be ascribed to the perfect match of size, geometry and denticity between the NOTA ring and the small Ga3+ ion, allowing the formation of the chelating complex with excellent thermodynamic stability and kinetic inertness.23,37,38 Four Ga3+ ions successfully chelated within 4, as confirmed by high-resolution mass spectroscopy. The observed isotopic pattern was characteristic of the quadruple charged species [4 + 4H]4+ of the expected molecular structure (Fig. S2, ESI†). The experimental mass obtained matched the theoretical molecular weight within an error of less than 0.3 ppm.
With dendrimer 4 synthesized and in hand, we first studied its self-assembly in aqueous solution using dynamic light scattering (DLS).39 The DLS analysis revealed the presence of two populations of nanoparticles: one measuring 16 nm characteristic of nanomicelles and a larger one measuring 173 nm (Fig. 3A). The intensity ratio of the signals from the smaller and larger NPs was 4.9, indicating an over 1 million folds greater number of smaller NPs than larger NPs, highlighting a hugely predominant formation of small NPs for 4@ and a negligible population of larger nanoassemblies. The larger NPs, fortunately negligible in number, are possibly the result of small nanomicelle aggregation. DLS analysis of 3@ revealed similar small nanoparticles of 13 nm and larger aggregates of 309 nm (Fig. 3F), with the number of smaller to larger NPs also far exceeding one million. Both 4@ and 3@ therefore self-assembled in aqueous solution with a predominance of small nanomicelles and negligible population of large aggregates.
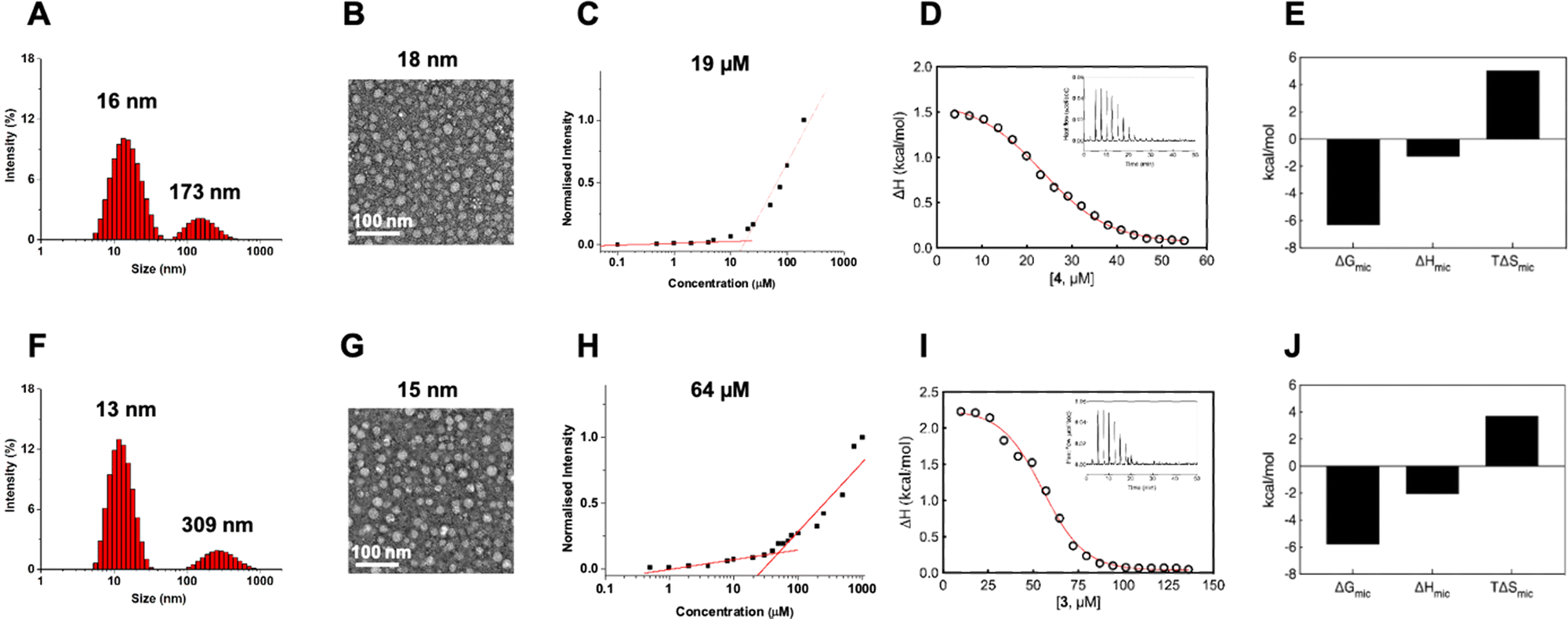 | ||
| Fig. 3 Self-assembly of the amphiphilic dendrimers 4 (top row) and 3 (bottom row) into small and uniform nanomicelles in water. (A) and (F) Dynamic light scattering analysis of 4@ and 3@, (B) and (G) transmission electron microscopy of 4@ and 3@, (C) and (H) critical micelle concentration measured using the Nile Red method for 4@ and 3@, respectively. (D) and (I) Representative integrated experimental isothermal titration calorimetry (ITC) profiles (circles) and data fitting with a sigmoidal function (red line) for 4@ and 3@, respectively. The inset shows the corresponding ITC raw data. (E) and (J) Thermodynamic parameters (i.e., Gibbs free energy of micellization (ΔGmic), micellization enthalpy (ΔHmic) and micellization entropy (TΔSmic)) as determined using ITC for the self-assembling process of 4 and 3, respectively. | ||
Further examination of size and morphology of 4@ using transmission electron microscopy (TEM) confirmed the DLS result, revealing an abundance of small and spherical nanoparticles measuring 18 ± 4 nm yet no visible larger aggregates at all (Fig. 3B). Similarly, TEM also confirmed the formation of small and spherical nanomicelles of 15 ± 4 nm for 3@ with no notable formation of large aggregates (Fig. 3G). Collectively, these results support the favorable chemical structure and geometry of the amphiphilic dendrimer 4 for assembly into nanomicelles. The difference in size of 4@ (16–18 nm) compared to 3@ (13–15 nm) can be reasonably ascribed to the difference in the chemical structures of the two dendrimers and the potential steric hindrance created by the enlarged hydrophobic entity in 4 during the self-assembly and packing process.
Interestingly, examination of the surface charge of both 4@ and 3@ exhibited similar zeta potentials of +10 mV (ESI,† Fig. S3). The positive zeta potentials can be attributed to the protonation of tertiary amine groups present in the dendrimer branches, with the dendrimer terminals remaining neutral in charge owing to the Ga/NOTA complexation. Atomistic molecular dynamics (MD) simulations of the nanomicelles 3@ and 4@ support this hypothesis, revealing the confinement of hydrophobic tails within the micellar cores of both nanoassemblies, with only the Ga/NOTA terminals distributed on the micellar surfaces (ESI,† Fig. S4A and B). The corresponding radial distribution functions (RDFs) indicate that no amphiphile tails reach the surface in either micelle, highlighting a clear space segregation between the hydrophobic cores and the surface-located Ga/NOTA terminals (ESI,† Fig. S4C and D). Moreover, the absence of hydrophobic tails on both micellar surfaces suggests they do not contribute toward the surface potential, which is therefore likely determined solely by inner protonated tertiary amines.
We further evaluated the critical micelle concentration (CMC) of 4 using a fluorescent spectroscopic assay with Nile red. 4 exhibited a significantly lower CMC (approximately 19 μM) compared to 3 (64 μM) (Fig. 3C and H), indicating the ability of 4 to readily self-assemble to form nanomicelles at very low concentrations, lower than required for 3. To better understand the self-assembly process of 3 and 4 into nanomicelles, we studied their demicellization process using isothermal titration calorimetry (ITC) by following a well-established protocol.40–42 Notably, the ITC experiments (Fig. 3D and I) provided CMC values of 23 μM for 4 and 57 μM for 3, which closely matched the CMC values obtained using fluorescence spectroscopy.
From a thermodynamic perspective, when a concentrated solution of 4@ or 3@ was injected into water, predominantly endothermic signals were observed during the demicellization process (Fig. 3D and I). Consequently, for the reverse process of dendrimer micellization, negative (exothermic) enthalpy values of ΔHmic = −1.29 ± 0.11 kcal mol−1 for 4 and ΔHmic = −2.06 ± 0.09 kcal mol−1 for 3 were determined using integrated data fitting, representing the difference between final and initial heat in the titration curve (Fig. 3D and I). The negative enthalpic component can be attributed to favorable hydrogen bonding and polar interactions among the dendron branches; consequently, in a smaller and more compact structure like 3@, these stabilizing forces are higher. The adopted ITC protocol also allowed measurement of the Gibbs free energy of micellization (ΔGmic) and its entropic component (TΔSmic). As indicated by the negative sign of ΔGmic, both self-assembly processes were thermodynamically spontaneous in water. However, as expected due to the lower CMC value obtained for dendrimer 4, 4@ had a more favorable ΔGmic (−6.33 kcal mol−1) compared to 3@ (−5.79 kcal mol−1) (Fig. 3E and J). Interestingly, the derived entropic contribution led to defining the self-assembly processes for both as driven by entropy, with TΔSmic values of +5.04 kcal mol−1 for 4@ and +3.73 kcal mol−1 for 3@ being higher in magnitude than the corresponding ΔHmic components (Fig. 3E and J). The entropic contribution is largely provided by a strong network of hydrophobic interactions among the hydrocarbon tails and is related to the release of water molecules in the hydration shells around the hydrophobic parts of the dendrimer monomeric units. Consequently, the double-alkyl portion of 4 has a more stabilizing effect than the mono-alkyl tail of 3 in forming the nanomicellar structure. Finally, the aggregation number (Nagg) for both amphiphilic systems estimated using ITC was 20 ± 1 for 4@ and 15 ± 1 for 3@, consistent with the dimensions obtained using TEM and DLS. The higher number of monomers in 4@ could also explain the larger average size.
Encouraged by all the evidence favoring efficient self-assembly and stability of 4@, we then wanted to study its biodistribution in a mouse model, using micro-PET/CT imaging. To this end, we first synthesized the radioactive dendrimer [68Ga]Ga-4 using [68Ga]Ga3+ in ammonium acetate buffer (Fig. 4A), successfully reaching an excellent level of radiochemical purity (RCP) of 95 ± 2%, as confirmed by radio-thin-layer chromatography. Additionally, [68Ga]Ga-4 demonstrated remarkable stability, maintaining its RCP for at least 4 hours at 37 °C, both in 0.9% NaCl solution and in human serum (Fig. 4B). These data validated the high radiochemical purity and stability essential for PET imaging.
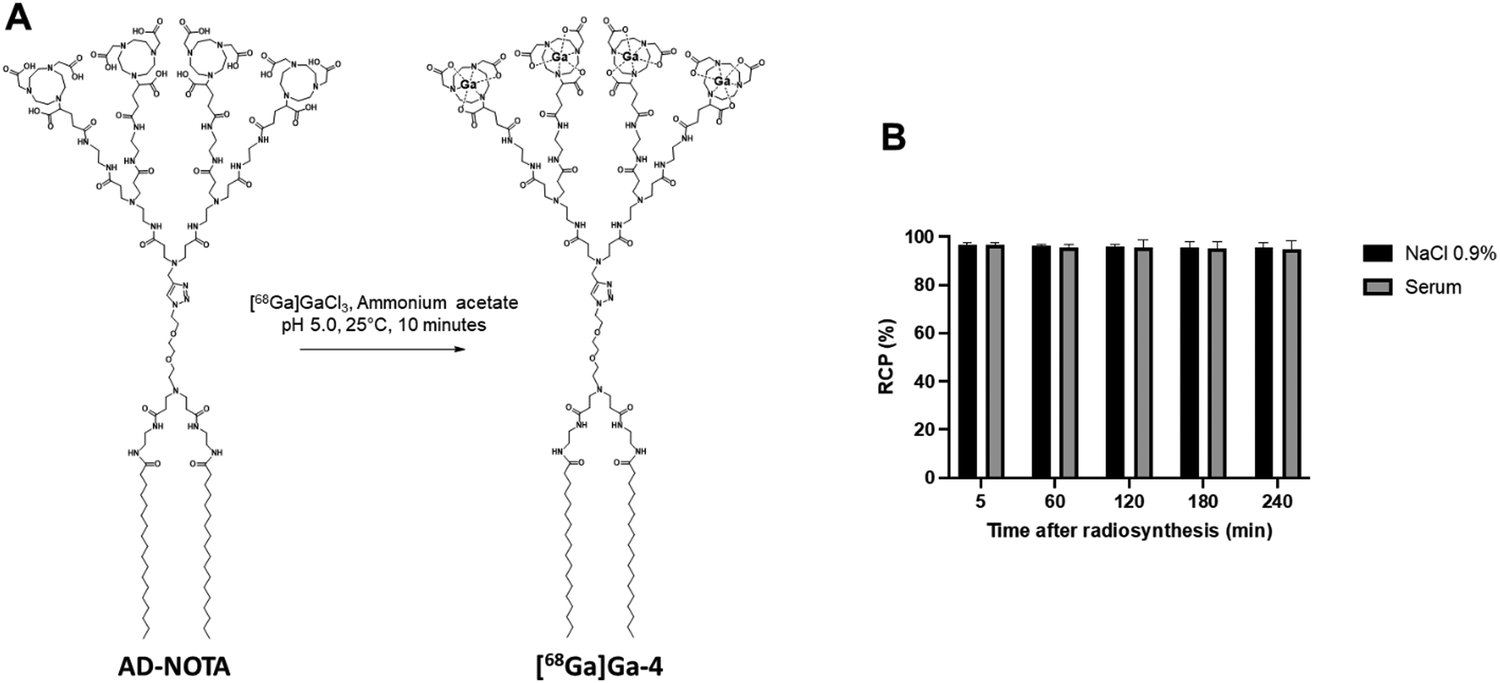 | ||
| Fig. 4 Radiolabeling and stability of dendrimer 4. (A) Fast and reliable radiolabeling of dendrimer 4 with gallium-68. (B) Radiochemical purity and stability of [68Ga]Ga-4 in NaCl 0.9% and human serum at 37 °C from 5 to 240 minutes after radiosynthesis, as assessed by instant radio-thin-layer chromatography. | ||
With the radiolabeled dendrimers in hand, we then carried out an in vivo biodistribution study on [68Ga]Ga-4@ in healthy CD1/Swiss mice (n = 4) using PET/CT imaging. Surprisingly, [68Ga]Ga-4@ exhibited an extremely high and time-dependent accumulation in the liver, in sharp contrast to [68Ga]Ga-3@ (Fig. 5). [68Ga]Ga-4@ also showed a less systemic PET background signal at 2 hours post-injection, as reflected by the quantified activities in a representative muscle (Fig. 5). [68Ga]Ga-3@ was predominantly eliminated in the bladder (Fig. 5B) through the renal system as its primary excretion route, and showed higher retention in the bladder and kidney than [68Ga]Ga-4@ (Fig. 5B and Table S1, ESI†). Although both nanoparticles are small, the larger size of 4@ may in part explain the above discrepancy, as size is an important factor for the biodistribution of nanoparticles. For example, quantum dots coated with short PEG chains measuring less than 5.5 nm were rapidly taken up by the liver, while larger quantum dots (>6.5 nm) distributed to organs such as lymph nodes and pancreas.43 Also, iron oxide magnetic nanoparticles measuring 10 nm showed high uptake by the liver, whereas the larger nanoparticles of 20 nm, 30 nm and 40 nm showed greater uptake by the spleen.44 Earlier studies using covalently-constructed dendrimer-based magnetic resonance contrast agents have already demonstrated the accumulation in the liver of dendrimers with hydrophobic chemical entities in their interior, and highlighted the dramatic impact of small size variations of 1 or 2 nm on their biodistribution in liver, kidney and lymph nodes.45,46 Our results using self-assembled dendrimer nanoparticles reflect trends for biodistribution similar to those of conventional dendrimer agents. It is important to note that, in this study, the rapid renal elimination favors the use of [68Ga]Ga-3@ as a radiotracer for bioimaging, while the excessive uptake of [68Ga]Ga-4@ in the liver and the increased accumulation in this organ over time (Fig. 5) exclude its further use in biomedical applications.
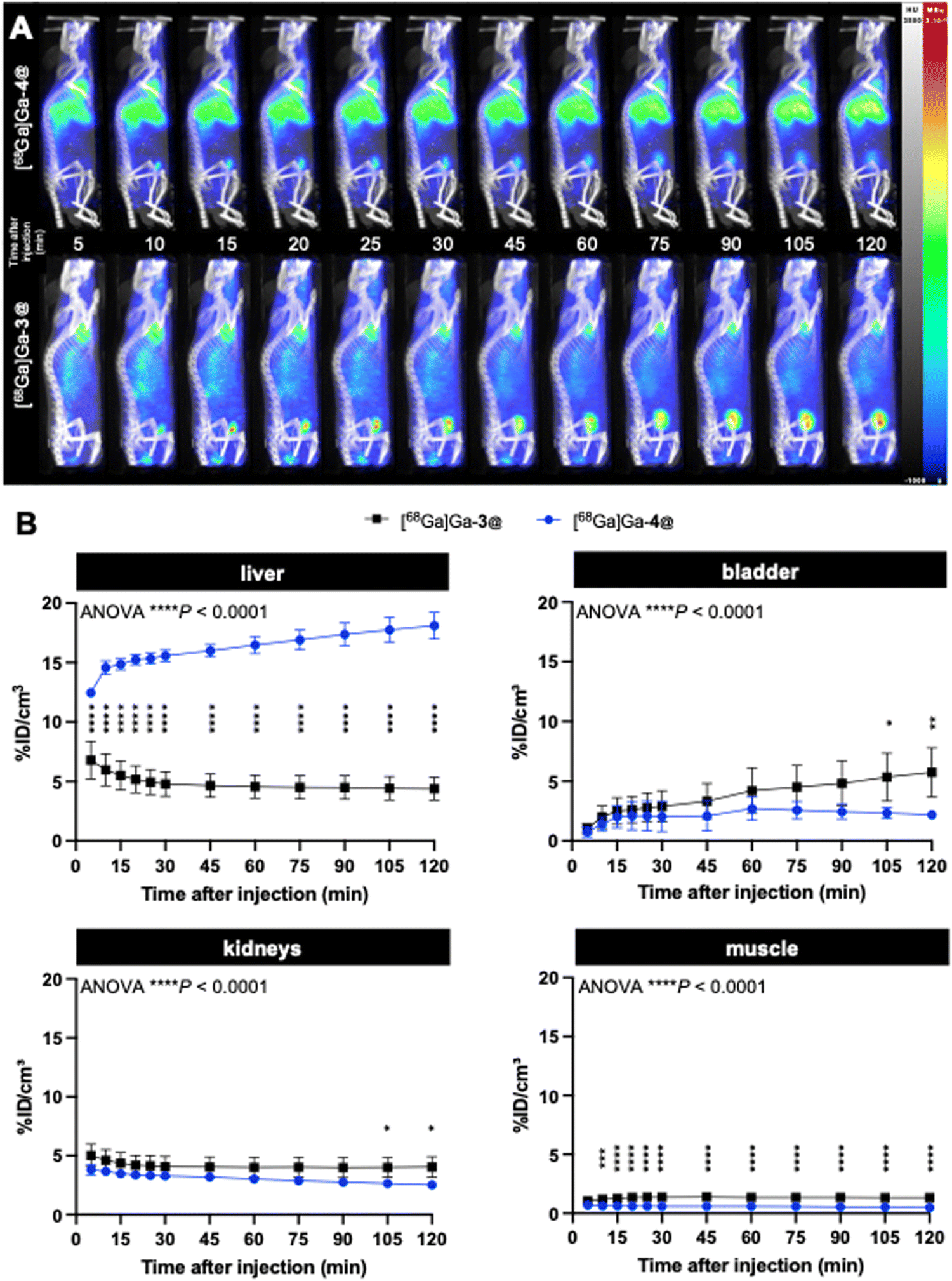 | ||
| Fig. 5 Dynamic PET/CT biodistribution of [68Ga]Ga-4@ and [68Ga]Ga-3@ up to 2 h after intravenous injection in healthy mice (n = 4): (A) representative images and (B) PET quantifications of uptake in the liver, bladder, kidney and muscle over time, analyzed using a 2-way ANOVA followed by Šídák's post-hoc test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. | ||
The imaging results on liver uptake of [68Ga]Ga-4@ were further confirmed using ex vivo gamma counting, which showed a significantly high retention in the liver but a much lower retention in all other organs (Fig. 6A and Table S2, ESI†). It should be noted that discrepancies were found between PET imaging quantifications and ex vivo gamma-counting quantifications of [68Ga]Ga-4@ biodistribution in the heart, kidneys, and lungs (Fig. 5B and 6A and Tables S1 and S2, ESI†). This could be ascribed to the elevated plasmatic contribution to the quantified activity, as these organs are highly vascularized. In fact, the calculated plasmatic half-life of [68Ga]Ga-4@ (577.8 min) showed a more than 2-fold increase in circulation time compared to that of [68Ga]Ga-3@ (247.0 min) with a significantly different kinetic profile (Fig. 6B and Table S3, ESI†). The longer half-life of [68Ga]Ga-4@ may also be partially responsible for its higher liver uptake, as prolonged circulation time in the bloodstream promotes accumulation in the liver. Consequently, although [68Ga]Ga-4@ and [68Ga]Ga-3@ have exactly the same hydrophilic dendron entity, the hydrophobic part of these amphiphilic dendrimers plays a crucial role in their self-assembling stability, circulation time, and biodistribution profile in vivo. Our findings underscore the importance of fine-tuning the amphiphilic dendrimer structure to achieve the desired safe biodistribution profile with minimal retention in the liver and/or other vital organs.
 | ||
| Fig. 6 Ex vivo gamma counting of main organs 2 h after [68Ga]Ga-4@ and [68Ga]Ga-3@ intravenous injection in healthy mice (A). Blood kinetics of [68Ga]Ga-4@ or [68Ga]Ga-3@ up to 2 h after intravenous injection in healthy mice (B). Data were analyzed using a 2-way ANOVA test followed by Šídák's post-hoc test. ****P < 0.0001. | ||
Although a detailed and insightful understanding of the significant difference in biodistribution between [68Ga]Ga-4@ and [68Ga]Ga-3@ requires further study, it is highly likely that the disparity in their chemical structures and the consequential differences in stability and size could also influence their interactions with proteins in biofluids and tissues. This in turn could affect their fate during systemic circulation, resulting in distinct differences in biodistribution and liver accumulation. Increasing evidence shows that, upon entry into biological tissues or fluids, nanoparticles often spontaneously adsorb proteins and biomolecules, hence forming protein coronas on their surface. The type, amount, and conformation of these biomolecules in the biocoronas critically dictate the fate and biodistribution of the nanoparticles, as well as their biocompatibility and safety.47–49 Therefore, we hypothesized that [68Ga]Ga-4@ might form a biocorona different from that formed by [68Ga]Ga-3@, resulting in their disparate biodistributions. This warrants further investigation and detailed analysis falling outside the scope of the present study.
Conclusion
In summary, we studied the impact of increased hydrophobicity and the resulting altered hydrophilic/hydrophobic balance on the self-assembling of amphiphilic dendrimer 4 and its subsequent biodistribution. We observed that this structural modification not only led to an enhanced stability of the self-assembled nanomicelles, an enlarged size, and a prolonged circulation time, but also resulted in massive accumulation in the liver. This study emphasizes the importance of finding the right balance between hydrophobicity and hydrophilicity in amphiphilic dendrimers in order to ensure optimal self-assembling stability and achieve desirable biodistribution patterns. It highlights that nanoparticles even with the same chemical entities on the surface, similar surface charge, and small size can exhibit dramatically different biodistribution profiles as a result of different hydrophobic components buried within their interior core for different compactivity. Further research is needed to refine these parameters and develop nanoparticles with improved biodistribution profiles for safer and more effective biomedical applications.Author contributions
LP coordinated the project; TR, LD synthesized the agents; TR, TD and BL performed the characterization; TD, BL, LB and VN performed the radiosynthesis, in vivo imaging acquisitions and image treatments; EL, SP performed the ITC experiments and the computer simulations; TR, TD, LB, VN, PG, SG, SP and LP analyzed the data; TR, TD, PG, EL, FDG, BG, SP and LP wrote the paper. All authors proofed the manuscript.Data availability
All data generated in this study are included in the article and the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank Mrs Sandrine Pons, Mrs Nadhumati Abdou, Mr Michel Skandalovski and Mr Samy Vigier for their technical support. This work was supported by the Ligue Nationale Contre le Cancer (EL2016, EL2021 LNCCLiP, LP; doctoral fellowship grant, TD), the French National Research Agency under the framework of the ERA-NET EURONANOMED European Research project ‘NAN-4-TUM’ (LP) and under the framework of the generic call 2022 ‘THERAnanoSTIC’ project (PG), the EU H2020 Research and Innovation program NMBP “SAFE-N-MEDTECH” (2019-2023) (grant agreement No. 814607, TR, LP), the EU Horizon Europe Mission Cancer program “HIT-GLIO” (2023-2027) (grant agreement No. 101136835, LP) and China Scholarship Council (LD). This work was partially performed by a platform member of the France Life Imaging network (grant ANR-11-INBS-0006, CERIMED, BG). E. L. and S. P. acknowledge access to supercomputing resources and financial support from ICSC-Centro Nazionale di Ricerca in high-performance computing, big data, and quantum computing (Spoke 7: WP4 (Pilot applications), T.2.8 (Development and optimization of HPC-based integrated workflows based on flagship codes for personalized (nano)medicine); WP5 (Materials Foundry), T.2.3 (Development of computational workflows based on atomistic molecular simulations for the prediction of key properties of molecular system and high-performance (nano)materials for biological, pharmaceutical and industrial application)).References
- M. L. James and S. S. Gambhir, A Molecular Imaging Primer: Modalities, Imaging Agents, and Applications, Phys. Rev., 2012, 92(2), 897–965, DOI:10.1152/physrev.00049.2010.
- C. Li, A Targeted Approach to Cancer Imaging and Therapy, Nat. Mater., 2014, 13(2), 110–115, DOI:10.1038/nmat3877.
- C. Andreou, R. Weissleder and M. F. Kircher, Multiplexed Imaging in Oncology, Nat. Biomed. Eng., 2022, 6(5), 527–540, DOI:10.1038/s41551-022-00891-5.
- J. Sun, Z. Huangfu, J. Yang, G. Wang, K. Hu, M. Gao and Z. Zhong, Imaging-Guided Targeted Radionuclide Tumor Therapy: From Concept to Clinical Translation, Adv. Drug Delivery Rev., 2022, 190, 114538, DOI:10.1016/j.addr.2022.114538.
- A. S. Thakor, J. V. Jokerst, P. Ghanouni, J. L. Campbell, E. Mittra and S. S. Gambhir, Clinically Approved Nanoparticle Imaging Agents, J. Nucl. Med., 2016, 57(12), 1833–1837, DOI:10.2967/jnumed.116.181362.
- E.-K. Lim, T. Kim, S. Paik, S. Haam, Y.-M. Huh and K. Lee, Nanomaterials for Theranostics: Recent Advances and Future Challenges, Chem. Rev., 2015, 115(1), 327–394, DOI:10.1021/cr300213b.
- H. Chen, W. Zhang, G. Zhu, J. Xie and X. Chen, Rethinking Cancer Nanotheranostics, Nat. Rev. Mater., 2017, 2(7), 17024, DOI:10.1038/natrevmats.2017.24.
- A.-M. Caminade, A. Hameau, C.-O. Turrin, R. Laurent and J.-P. Majoral, Dendritic Metal Complexes for Bioimaging. Recent Advances, Coord. Chem. Rev., 2021, 430, 213739, DOI:10.1016/j.ccr.2020.213739.
- Z. Lyu, L. Ding, A. Tintaru and L. Peng, Self-Assembling Supramolecular Dendrimers for Biomedical Applications: Lessons Learned from Poly(Amidoamine) Dendrimers, Acc. Chem. Res., 2020, 53(12), 2936–2949, DOI:10.1021/acs.accounts.0c00589.
- L. Ding, Z. Lyu, D. Dhumal, C.-L. Kao, M. Bernard and L. Peng, Dendrimer-Based Magnetic Resonance Imaging Agents for Brain Cancer, Sci. China Mater., 2018, 61(11), 1420–1443, DOI:10.1007/s40843-018-9323-6.
- R. M. Kannan, E. Nance, S. Kannan and D. A. Tomalia, Emerging Concepts in Dendrimer-Based Nanomedicine: From Design Principles to Clinical Applications, J. Intern. Med., 2014, 276(6), 579–617, DOI:10.1111/joim.12280.
- A. R. Menjoge, R. M. Kannan and D. A. Tomalia, Dendrimer-Based Drug and Imaging Conjugates: Design Considerations for Nanomedical Applications, Drug Discovery Today, 2010, 15(5–6), 171–185, DOI:10.1016/j.drudis.2010.01.009.
- T. Barrett, G. Ravizzini, P. Choyke and H. Kobayashi, Dendrimers in Medical Nanotechnology, IEEE Eng. Med. Biol. Mag., 2009, 28(1), 12–22, DOI:10.1109/MEMB.2008.931012.
- P. Garrigue, J. Tang, L. Ding, A. Bouhlel, A. Tintaru, E. Laurini, Y. Huang, Z. Lyu, M. Zhang, S. Fernandez, L. Balasse, W. Lan, E. Mas, D. Marson, Y. Weng, X. Liu, S. Giorgio, J. Iovanna, S. Pricl, B. Guillet and L. Peng, Self-Assembling Supramolecular Dendrimer Nanosystem for PET Imaging of Tumors, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(45), 11454–11459, DOI:10.1073/pnas.1812938115.
- L. Ding, Z. Lyu, A. Tintaru, E. Laurini, D. Marson, B. Louis, A. Bouhlel, L. Balasse, S. Fernandez, P. Garrigue, E. Mas, S. Giorgio, S. Pricl, B. Guillet and L. Peng, A Self-Assembling Amphiphilic Dendrimer Nanotracer for SPECT Imaging, Chem. Commun., 2020, 56(2), 301–304, 10.1039/C9CC07750B.
- L. Ding, Z. Lyu, B. Louis, A. Tintaru, E. Laurini, D. Marson, M. Zhang, W. Shao, Y. Jiang, A. Bouhlel, L. Balasse, P. Garrigue, E. Mas, S. Giorgio, J. Iovanna, Y. Huang, S. Pricl, B. Guillet and L. Peng, Surface Charge of Supramolecular Nanosystems for In Vivo Biodistribution: A MicroSPECT/CT Imaging Study, Small, 2020, 16(37), 2003290, DOI:10.1002/smll.202003290.
- R. Sun, J. Xiang, Q. Zhou, Y. Piao, J. Tang, S. Shao, Z. Zhou, Y. H. Bae and Y. Shen, The Tumor EPR Effect for Cancer Drug Delivery: Current Status, Limitations, and Alternatives, Adv. Drug Delivery Rev., 2022, 191, 114614, DOI:10.1016/j.addr.2022.114614.
- Y. Matsumura and H. Maeda, A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs Search PubMed.
- J. Fang, H. Nakamura and H. Maeda, The EPR Effect: Unique Features of Tumor Blood Vessels for Drug Delivery, Factors Involved, and Limitations and Augmentation of the Effect, Adv. Drug Delivery Rev., 2011, 63(3), 136–151, DOI:10.1016/j.addr.2010.04.009.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, Tumor Vascular Permeability and the EPR Effect in Macromolecular Therapeutics: A Review, J. Controlled Release, 2000, 65(1–2), 271–284, DOI:10.1016/S0168-3659(99)00248-5.
- Y. Shi, R. Van Der Meel, X. Chen and T. Lammers, The EPR Effect and beyond: Strategies to Improve Tumor Targeting and Cancer Nanomedicine Treatment Efficacy, Theranostics, 2020, 10(17), 7921–7924, DOI:10.7150/thno.49577.
- R. van der Meel, E. Sulheim, Y. Shi, F. Kiessling, W. J. M. Mulder and T. Lammers, Smart Cancer Nanomedicine, Nat. Nanotechnol., 2019, 14(11), 1007–1017, DOI:10.1038/s41565-019-0567-y.
- T. I. Kostelnik and C. Orvig, Radioactive Main Group and Rare Earth Metals for Imaging and Therapy, Chem. Rev., 2019, 119(2), 902–956, DOI:10.1021/acs.chemrev.8b00294.
- A.-M. Caminade, R. Laurent, B. Delavaux-Nicot and J.-P. Majoral, “Janus” Dendrimers: Syntheses and Properties, New J. Chem., 2012, 36(2), 217–226, 10.1039/C1NJ20458K.
- V. Percec, D. A. Wilson, P. Leowanawat, C. J. Wilson, A. D. Hughes, M. S. Kaucher, D. A. Hammer, D. H. Levine, A. J. Kim, F. S. Bates, K. P. Davis, T. P. Lodge, M. L. Klein, R. H. DeVane, E. Aqad, B. M. Rosen, A. O. Argintaru, M. J. Sienkowska, K. Rissanen, S. Nummelin and J. Ropponen, Self-Assembly of Janus Dendrimers into Uniform Dendrimersomes and Other Complex Architectures, Science, 2010, 328(5981), 1009–1014, DOI:10.1126/science.1185547.
- F. Zeng and S. C. Zimmerman, Dendrimers in Supramolecular Chemistry: From Molecular Recognition to Self-Assembly, Chem. Rev., 1997, 97(5), 1681–1712 CrossRef CAS PubMed.
- J.-M. Lehn, Toward Self-Organization and Complex Matter, Science, 2002, 295(5564), 2400–2403, DOI:10.1126/science.1071063.
- A. C. Mendes, E. T. Baran, R. L. Reis and H. S. Azevedo, Self-Assembly in Nature: Using the Principles of Nature to Create Complex Nanobiomaterials: Self-Assembling in Nature, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 5(6), 582–612, DOI:10.1002/wnan.1238.
- E. Apartsin and A. Caminade, Supramolecular Self-Associations of Amphiphilic Dendrons and Their Properties, Chem. – Eur. J., 2021, 27(72), 17976–17998, DOI:10.1002/chem.202102589.
- D. Zhang, E. N. Atochina-Vasserman, J. Lu, D. S. Maurya, Q. Xiao, M. Liu, J. Adamson, N. Ona, E. K. Reagan, H. Ni, D. Weissman and V. Percec, The Unexpected Importance of the Primary Structure of the Hydrophobic Part of One-Component Ionizable Amphiphilic Janus Dendrimers in Targeted mRNA Delivery Activity, J. Am. Chem. Soc., 2022, 144(11), 4746–4753, DOI:10.1021/jacs.2c00273.
- K. Chen and X. Chen, Design and Development of Molecular Imaging Probes, Curr. Top. Med. Chem., 2010, 10(12), 1227–1236, DOI:10.2174/156802610791384225.
- S. Goel, C. G. England, F. Chen and W. Cai, Positron Emission Tomography and Nanotechnology: A Dynamic Duo for Cancer Theranostics, Adv. Drug Delivery Rev., 2017, 113, 157–176, DOI:10.1016/j.addr.2016.08.001.
- L. Cheng, K. Yang, Q. Chen and Z. Liu, Organic Stealth Nanoparticles for Highly Effective in Vivo Near-Infrared Photothermal Therapy of Cancer, ACS Nano, 2012, 6(6), 5605–5613, DOI:10.1021/nn301539m.
- J. Chen, A. Ellert-Miklaszewska, S. Garofalo, A. K. Dey, J. Tang, Y. Jiang, F. Clément, P. N. Marche, X. Liu, B. Kaminska, A. Santoni, C. Limatola, J. J. Rossi, J. Zhou and L. Peng, Synthesis and Use of an Amphiphilic Dendrimer for siRNA Delivery into Primary Immune Cells, Nat. Protoc., 2021, 16(1), 327–351, DOI:10.1038/s41596-020-00418-9.
- X. Liu, J. Zhou, T. Yu, C. Chen, Q. Cheng, K. Sengupta, Y. Huang, H. Li, C. Liu, Y. Wang, P. Posocco, M. Wang, Q. Cui, S. Giorgio, M. Fermeglia, F. Qu, S. Pricl, Y. Shi, Z. Liang, P. Rocchi, J. J. Rossi and L. Peng, Adaptive Amphiphilic Dendrimer Based Nanoassemblies as Robust and Versatile siRNA Delivery Systems, Angew. Chem., Int. Ed., 2014, 53(44), 11822–11827, DOI:10.1002/anie.201406764.
- T. Yu, X. Liu, A. Bolcato-Bellemin, Y. Wang, C. Liu, P. Erbacher, F. Qu, P. Rocchi, J. Behr and L. Peng, An Amphiphilic Dendrimer for Effective Delivery of Small Interfering RNA and Gene Silencing In Vitro and In Vivo, Angew. Chem., Int. Ed., 2012, 51(34), 8478–8484, DOI:10.1002/anie.201203920.
- M. D. Bartholomä, Recent Developments in the Design of Bifunctional Chelators for Metal-Based Radiopharmaceuticals Used in Positron Emission Tomography, Inorg. Chim. Acta, 2012, 389, 36–51, DOI:10.1016/j.ica.2012.01.061.
- E. W. Price and C. Orvig, Matching Chelators to Radiometals for Radiopharmaceuticals, Chem. Soc. Rev., 2014, 43(1), 260–290, 10.1039/C3CS60304K.
- S. Bhattacharjee, DLS and Zeta Potential – What They Are and What They Are Not, J. Controlled Release, 2016, 235, 337–351, DOI:10.1016/j.jconrel.2016.06.017.
- Design and Delivery of SiRNA Therapeutics, ed. H. J. Ditzel, M. Tuttolomondo, S. Kauppinen, Methods in Molecular Biology, Springer, US: New York, NY, 2021; vol. 2282 DOI:10.1007/978-1-0716-1298-9.
- A. C. Rodrigo, S. M. Bromfield, E. Laurini, P. Posocco, S. Pricl and D. K. Smith, Morphological Control of Self-Assembled Multivalent (SAMul) Heparin Binding in Highly Competitive Media, Chem. Commun., 2017, 53(47), 6335–6338, 10.1039/C7CC02990J.
- M. Russi, R. Valeri, D. Marson, C. Danielli, F. Felluga, A. Tintaru, N. Skoko, S. Aulic, E. Laurini and S. Pricl, Some Things Old, New and Borrowed: Delivery of Dabrafenib and Vemurafenib to Melanoma Cells via Self-Assembled Nanomicelles Based on an Amphiphilic Dendrimer, Eur. J. Pharm. Sci., 2023, 180, 106311, DOI:10.1016/j.ejps.2022.106311.
- H. S. Choi, B. I. Ipe, P. Misra, J. H. Lee, M. G. Bawendi and J. V. Frangioni, Tissue- and Organ-Selective Biodistribution of NIR Fluorescent Quantum Dots, Nano Lett., 2009, 9(6), 2354–2359, DOI:10.1021/nl900872r.
- L. Yang, H. Kuang, W. Zhang, Z. P. Aguilar, Y. Xiong, W. Lai, H. Xu and H. Wei, Size Dependent Biodistribution and Toxicokinetics of Iron Oxide Magnetic Nanoparticles in Mice, Nanoscale, 2014, 7(2), 625–636, 10.1039/C4NR05061D.
- H. Kobayashi and M. W. Brechbiel, Nano-Sized MRI Contrast Agents with Dendrimer Cores, Adv. Drug Delivery Rev., 2005, 57(15), 2271–2286, DOI:10.1016/j.addr.2005.09.016.
- M. R. Longmire, M. Ogawa, P. L. Choyke and H. Kobayashi, Dendrimers as High Relaxivity MR Contrast Agents, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 6(2), 155–162, DOI:10.1002/wnan.1250.
- M. Mahmoudi, The Need for Robust Characterization of Nanomaterials for Nanomedicine Applications, Nat. Commun., 2021, 12(1), 5246, DOI:10.1038/s41467-021-25584-6.
- M. Mahmoudi, The Need for Improved Methodology in Protein Corona Analysis, Nat. Commun., 2022, 13(1), 49, DOI:10.1038/s41467-021-27643-4.
- M. Mahmoudi, M. P. Landry, A. Moore and R. Coreas, The Protein Corona from Nanomedicine to Environmental Science, Nat. Rev. Mater., 2023, 8, 422–438, DOI:10.1038/s41578-023-00552-2.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01266f |
| ‡ These authors contributed equally to the study. |
| This journal is © The Royal Society of Chemistry 2025 |