DOI:
10.1039/D4TC04692G
(Paper)
J. Mater. Chem. C, 2025, Advance Article
AIEE active dual-state emissive tripodal pyridopyrazine derivatives as multi-stimuli responsive smart organic materials†
Received
5th November 2024
, Accepted 10th February 2025
First published on 11th February 2025
Abstract
This work presents the synthesis of five novel pyridopyrazine derivatives (PPDs) PP1–PP5. These PPDs have high quantum yields (Φ) up to 0.87 in solid and solution states. This dual-state emissive behavior was supported by AIEE properties. Single crystal X-ray diffraction (SCXRD) studies confirmed the absence of π–π stacking interactions in the unit cell. The SCXRD analysis of PP4 also revealed a twisted conformation and herringbone packing arrangement. All pyridopyrazine derivatives detected TFA reversibly in solution and the powdered state. Probe PP2 was also incorporated in the guar-gum borax hydrogel matrix and could detect TFA in the gel phase with a color change from blue to yellow. These PPDs were also exploited for their metal sensing abilities and could selectively sense Hg2+ ions with detection limits in ppm. Theoretical studies were also raised to support experimental findings. The changes in emissive properties with increasing temperature from 30 °C to 80 °C were also studied for these PPDs.
1. Introduction
Organic luminous materials have become important in recent years because of their intriguing photophysical characteristics. The use of these emissive organic compounds is currently being actively pursued in a wide range of applications, either in their solid form (such as optoelectronic devices or data encryption)1–7 or in solution (such as sensors or bioimaging).8–12 Recently, there has been a growing interest in designing emissive organic materials with substantial emissions in solution and solid states, also known as dual-state emission (DSE).13,14 DSE is important to fill the void between aggregation-caused quenching (ACQ) and aggregation-induced emission (AIE).15–17 To achieve DSE, the molecular design should incorporate a highly conjugated backbone with a rigid twisted structure.18,19 The rigid structure will restrict the intramolecular rotations and suppress non-emissive pathways, making the compound emissive in the solution state.20–23 The twisted scaffold is important for emission in the solid state as it prevents the possibility of π–π stacking, which is responsible for ACQ in the aggregated state.24 The conjugated backbone can be substituted with groups of different electronic natures to fine-tune intramolecular charge transfer (ICT), which is also helpful in achieving DSE.25–27
Pyridopyrazine is a well-known acceptor unit consisting of fused pyridine and pyrazine heterocyclic rings. Due to the presence of nitrogen atoms, it has the potential for various applications in the fields of medicine, sensing, and optics.28–30 To enhance the photophysical properties, these pyridopyrazine acceptor units can be substituted with donor groups to synthesize D–A push–pull molecules. Such molecules have the potential to show ICT or enhanced ICT on varying the substituents present in the donor and/or acceptor moieties.31 Moreover, the pyridopyrazine can undergo chelation, protonation, and hydrogen bond formation owing to the presence of three nitrogen atoms.29
Mercury (Hg) is a heavy metal and environmental contaminant.32,33 It bio-accumulates in the food chain and adversely affects the human neurological and renal systems.34 As per WHO guidelines, the acceptable limit for Hg2+ in drinking water must be lower than 6 ppb.35 So, the development of techniques for trace level Hg2+ detection is of great importance. The most effective techniques are chromogenic and fluorogenic sensing of mercuric ions, having the advantages of fast response, simple handling, and bio-imaging.36–38 These detection techniques also effectively sense volatile organic compounds such as trifluoroacetic acid (TFA). TFA is a corrosive and toxic volatile colorless acid that affects human health and the environment.39 It is widely used for industrial purposes; hence, its detection is also necessary to minimize its hazardous impacts.
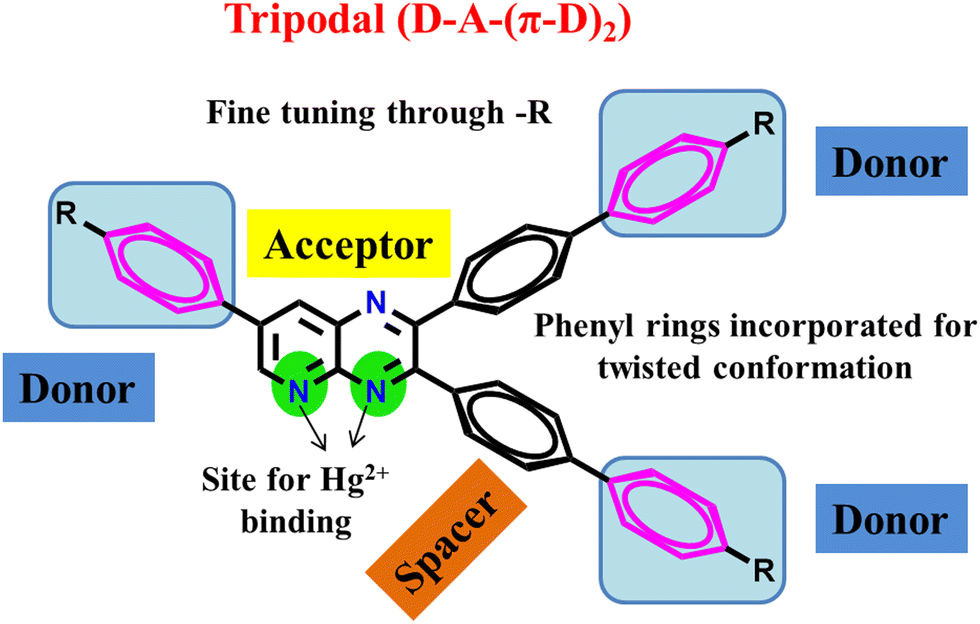
Previously, we reported the synthesis of quinoxaline and pyridopyrazine derivatives as donor–acceptor push–pull molecules.29,40–46 These compounds could efficiently act as sensors for Hg2+ ions, TFA, and moisture. In this work, we are reporting tripodal PPDs with a D–A–(π–D)2 arrangement (Fig. 1) with the ability to reversibly sense TFA in the solid and solution state, and Hg2+ ions in solution, showing solvatochromic properties with solvents of increasing polarity and AIEE in THF–H2O mixtures. Pyridopyrazine acts as an acceptor which is linked with substituted phenyl groups as donors. The donor groups at the 2- and 3-positions of pyridopyrazine are connected through 1,4-phenylene spacer groups. The phenyl spacer groups are introduced to provide twisting to the molecule to avoid the possibility of π–π stacking, making the compounds AIEE-active and emissive in the solid state. The core of the molecule is conjugated, and donor groups are varied based on their donation abilities to fine-tune the ICT character in these PPDs. The presence of three nitrogen atoms is further utilized in acidochromic applications. Also, the proximity of nitrogens, at 4- and 5-positions, offers a possible site for chelation of mercuric ions (Hg2+).43
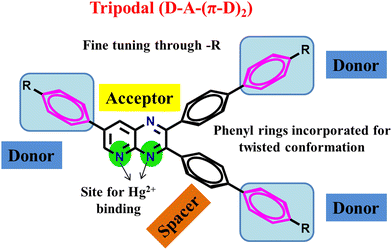 |
| | Fig. 1 Design of dual-state emissive PPDs as Hg2+ ion sensors. | |
2. Experimental details
2.1. Materials and measurement
The starting materials were purchased from Sigma-Aldrich and used as provided. All metal salts, AgNO3, Al(NO3)3·9H2O, Ba(NO3)2, Ca(NO3)2·4H2O, Co(NO3)2·6H2O, Cr(NO3)3·9H2O, Cu(NO3)2·3H2O, Fe(NO3)3·9H2O, Hg(NO3)2·H2O, KNO3, Mg(NO3)2·6H2O, NaNO3, Ni(NO3)2·6H2O, Pb(NO3)2, and Zn(NO3)2·6H2O were purchased from Sisco Research Laboratories Pvt. Ltd (SRL) – India. All the products were purified with column chromatography using 60–120 mesh silica as the stationary phase and a chloroform–methanol mixture as the mobile phase. The pure products were characterized with 1H-NMR, 13C-NMR, IR, and HRMS techniques. NMR was recorded on a Bruker Avance Neo 400 MHz, and spectra were analyzed using CDCl3 as a reference (7.26 ppm for 1H and 77.0 ppm for 13C). IR and HRMS spectra were recorded with a Shimadzu IRAffinity-1S and Agilent G6530AA (LC-HRMS-Q-TOF). Stock solutions of PP1–PP5 (2 × 10−3 M) were prepared in CHCl3, THF, CH3CN, DMSO and diluted accordingly. 0.015 M stock solutions of metal salts were prepared in HPLC water. For selectivity, sensitivity, and probe interaction with metal ion studies, 3 mL of PP1–PP5 (2 × 10−5 M) solutions were taken, and metal salt solutions were added accordingly. Absorption spectra were recorded on Lab India Analytical (UV 3092), and fluorescence on Hitachi F-4700 using HPLC solvents. The solid-state photoluminescence spectra and the corresponding quantum yields (integrated sphere method) were measured on a Fluorolog Horiba Jobinyvon spectrofluorometer and the fluorometer PTI Horiba QuantaMaster at excitation and emission slit widths of 5 nm. For the quantum yield measurement, a Quanta Phi integrating sphere equipped with a xenon lamp source is used. The experiment was performed at room temperature keeping the room light switched off. Density functional calculations are conducted to gain deeper insights for this project. All the structures (PP1–PP5) were fully optimized in THF solvent using the dispersion-corrected PBE0-D347,48 functional with the def2-TZVPP basis set49 using the Gaussian 16 program.50 The solvent effects were considered via the COSMO solvation model.51
2.2. General procedure for the synthesis of PP1–PP5
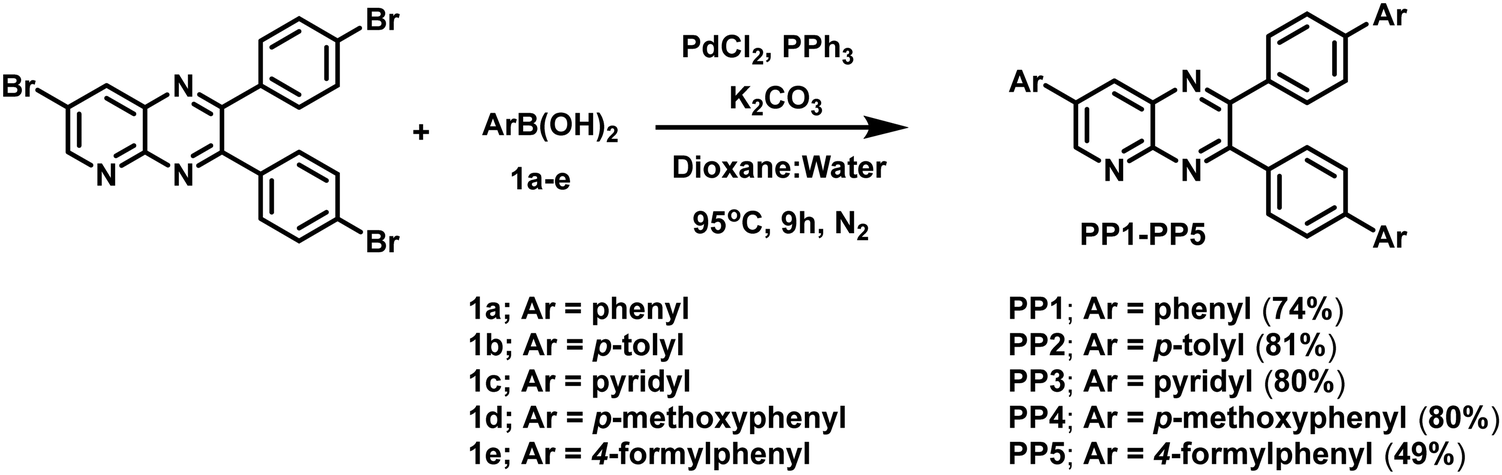
Products PP1–PP5 were obtained following the earlier reported procedure of the modified Suzuki coupling reaction.29,40–46 Palladium catalyst, Pd(PPh3)4, was generated in situ by the reaction of PdCl2 (0.06 mmol) and PPh3 (0.24 mmol) in 1,4-dioxane (16 mL) at 95 °C under a N2 atmosphere for 20 minutes. Furthermore, K2CO3 (6 mmol), 7-bromo-2,3-bis(4-bromophenyl)pyrido[2,3-b]pyrazine (1.0 mmol) and substituted boronic acid (2.4 mmol) and distilled water (8 mL) were added to the reaction mixture under a nitrogen atmosphere. Then, this reaction mixture was allowed to reflux for 9 h at 95 °C under a nitrogen atmosphere. The progress of the reaction was monitored using TLC. After completion, the crude product was extracted using a separating funnel with a chloroform–water mixture. The organic layer was dried over sodium sulfate and concentrated. It was further purified by column chromatography to obtain a pure product.
Synthesis of 2,3-di([1,1′-biphenyl]-4-yl)-7-phenylpyrido[2,3-b]pyrazine (PP1). The coupling reaction between 7-bromo-2,3-bis(4-bromophenyl)pyrido[2,3-b]pyrazine (520.02 mg, 1.0 mmol) and phenylboronic acid (1a) (292.63 mg, 2.4 mmol) resulted in the required PP1 as a bright-lemon yellow solid (378.6047 mg, 74%); m.p. = 250–252 °C (petroleum ether-CHCl3); λmax (CHCl3)/nm 267 (ε dm3 mol−1 cm−1 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 967), 286sh (47557) and 388 (33590); IR (cm−1): 3050, 3027, 1598, 1531, 1488, 1456, 1419,1396, 1338, 1263, 1223, 1179, 1111, 1064, 1038, 1007; δH (400 MHz, CDCl3) 9.46 (1H, d, J = 2.4 Hz), 8.69 (1H, d, J = 2.4 Hz), 7.84–7.80 (4H, m), 7.74–7.72 (2H, m), 7.66–7.57 (10H, m), 7.53–7.43 (5H, m), 7.40–7.34 (2H, m); δC (100 MHz, CDCl3) 155.5, 154.8, 153.5, 149.0, 142.2(2), 140.2(2), 138.2, 137.5, 137.0, 136.5, 136.1, 134.6, 130.9, 130.4, 129.5, 129.0, 128.9(2), 127.8(2), 127.6, 127.2, 127.1, 126.9; HRMS (ESI-TOF) m/z calcd for C37H26N3, [M + H]+ 512.2121; found: 512.2130.
967), 286sh (47557) and 388 (33590); IR (cm−1): 3050, 3027, 1598, 1531, 1488, 1456, 1419,1396, 1338, 1263, 1223, 1179, 1111, 1064, 1038, 1007; δH (400 MHz, CDCl3) 9.46 (1H, d, J = 2.4 Hz), 8.69 (1H, d, J = 2.4 Hz), 7.84–7.80 (4H, m), 7.74–7.72 (2H, m), 7.66–7.57 (10H, m), 7.53–7.43 (5H, m), 7.40–7.34 (2H, m); δC (100 MHz, CDCl3) 155.5, 154.8, 153.5, 149.0, 142.2(2), 140.2(2), 138.2, 137.5, 137.0, 136.5, 136.1, 134.6, 130.9, 130.4, 129.5, 129.0, 128.9(2), 127.8(2), 127.6, 127.2, 127.1, 126.9; HRMS (ESI-TOF) m/z calcd for C37H26N3, [M + H]+ 512.2121; found: 512.2130.
Synthesis of 2,3-bis(4′-methyl-[1,1′-biphenyl]-4-yl)-7-(p-tolyl)pyrido[2,3-b]pyrazine (PP2). The coupling reaction between 7-bromo-2,3-bis(4-bromophenyl)pyrido[2,3-b]pyrazine (520.02 mg, 1.0 mmol) and p-tolylboronic acid (1b) (326.30 mg, 2.4 mmol) gave the required PP2 as a bright lemon-yellow solid (448.40 mg, 81%); m.p. = 190–192 °C (petroleum ether–CHCl3); λmax (CHCl3)/nm 272 (ε dm3 mol−1 cm−1 71410), 293sh (57885) and 395 (44557); IR (cm−1): 3034, 2974, 2920, 1741, 1603, 1533, 1492, 1430, 1374, 1215, 1118, 1062, 1001; δH (400 MHz, CDCl3) 9.44 (1H, d, J = 2.4 Hz), 8.65 (1H, d, J = 2.8 Hz), 7.79 (2H, d, J = 8.3 Hz), 7.73–7.69 (4H, m), 7.64–7.59 (4H, m), 7.54 (4H, d, J = 8.0 Hz), 7.38 (2H, d, J = 8.0 Hz), 7.28–7.25 (5H, m, merged with CHCl3 peak), 2.46 (3H, s), 2.40 (6H, d, J = 1.6 Hz); δC (100 MHz, CDCl3) 155.4, 154.8, 153.4, 148.8, 142.1, 142.0, 139.1, 138.0, 137.7, 137.6, 137.3, 136.7, 136.1, 134.1, 133.6, 130.8, 130.3, 130.2, 129.6(2), 127.4, 127.0, 126.6, 21.3, 21.2; HRMS (ESI-TOF) m/z calcd for C40H32N3, [M + H]+ 554.2591; found: 554.2596.
Synthesis of 7-(pyridin-4-yl)-2,3-bis(4-(pyridin-4-yl)phenyl)pyrido[2,3-b]pyrazine (PP3). The coupling reaction between 7-bromo-2,3-bis(4-bromophenyl)pyrido[2,3-b]pyrazine (520.02 mg, 1.0 mmol) and 4-pyridylboronic acid (1c) (295.0 mg, 2.4 mmol) resulted in the required PP3 as a mustard-yellow solid (411.50 mg, 80%); m.p. = 284–288 °C (petroleum ether–CHCl3); λmax (CHCl3)/nm 266 (ε dm3 mol−1 cm−1 81197), 308sh (31475) and 374 (41311); IR (cm−1): 3028, 2965, 2924, 1741, 1589, 1533, 1464, 1430, 1361, 1270, 1215, 1062, 1028; δH (400 MHz, CDCl3): 9.49 (1H, d, J = 2.4 Hz), 8.83 (2H, d, J = 5.6 Hz), 8.77 (1H, d, J = 2.8 Hz), 8.68 (4H, d, J = 0.8 Hz), 7.86–7.84 (2H, m), 7.78–7.67 (9H, m), 7.56–7.55 (4H, m); δC (100 MHz, CDCl3) 155.8, 154.6, 153.1, 150.9, 150.3, 150.2, 149.8, 147.4, 147.3, 143.7, 139.4, 139.3, 138.9, 138.5, 135.9, 135.6, 131.1, 130.7, 127.2, 127.0, 121.8, 121.6; HRMS (ESI-TOF) m/z calcd for C34H23N6, [M + H]+ 515.1979; found: 515.1988.
Synthesis of 2,3-bis(4′-methoxy-[1,1′-biphenyl]-4-yl)-7-(4-methoxyphenyl)pyrido[2,3-b]pyrazine (PP4). The coupling reaction between 7-bromo-2,3-bis(4-bromophenyl)pyrido[2,3-b]pyrazine (520.02 mg, 1.0 mmol) and p-methoxyphenylboronic acid (1d) (364.69 mg, 2.4 mmol) resulted in the required PP4 as a gold-yellow solid (481.39 mg, 80%); m.p. = 270–272 °C (petroleum ether–CHCl3); λmax (CHCl3)/nm 282 (ε dm3 mol−1 cm−1 82524), 309sh (68360) and 405 (55918); IR (cm−1): 3048, 2924, 2827, 2730, 2357, 2163, 1693, 1603, 1575, 1443, 1388, 1346, 105, 1208, 1166 1062, 1007; δH (400 MHz, CDCl3) 9.42 (1H, d, J = 2.4 Hz), 8.61 (1H, d, J = 2.0 Hz), 7.77 (4H, t, J = 7.4 Hz), 7.69 (2H, d, J = 8.0 Hz), 7.60–7.55 (8H, m), 7.10 (2H, d, J = 8.4 Hz), 7.00–6.97 (4H, m), 3.90 (3H, s), 3.85 (6H, s); δC (100 MHz, CDCl3) 160.5, 159.6(2), 155.2, 154.8, 153.2, 148.6, 141.7, 141.6, 137.6, 137.0, 136.4, 136.1, 133.5, 132.7, 130.8, 130.3, 128.8, 128.7, 128.2, 126.6, 126.3, 115.0, 114.3(2), 55.5, 55.4; HRMS (ESI-TOF) m/z calcd for C40H32N3O3, [M + H]+ 602.2438; found: 602.2443.
Synthesis of 4′,4′′′-(7-(4-formylphenyl)pyrido[2,3-b]pyrazine-2,3-diyl)bis(([1,1′-biphenyl] – 4-carbaldehyde)) (PP5). The coupling reaction between 7-bromo-2,3-bis(4-bromophenyl)pyrido[2,3-b]pyrazine (520.02 mg, 1.0 mmol) and 4-formylphenylboronic acid (1e) (359.86 mg, 2.4 mmol) resulted in the required PP5 as a gold-yellow solid (292.0 mg, 49%); m.p. = 220 °C (petroleum ether–CHCl3); λmax (CHCl3)/nm 299 (ε dm3 mol−1 cm−1 81934), 331sh (40918) and 386 (44754); IR (cm−1): 3041, 2927, 2826, 2725, 1696, 1602, 1535, 1440, 1379, 1339, 1306, 1211, 1171, 1104, 1070; δH (400 MHz, CDCl3) 10.14 (1H, s), 10.07 (2H, d, J = 1.9 Hz), 9.51 (1H, d, J = 1.9 Hz), 8.76 (1H, d, J = 2.4 Hz), 8.10 (2H, d, J = 8.1 Hz), 8.01–7.96 (6H, m), 7.85 (2H, d, J = 8.3 Hz), 7.81–7.77 (6H, m), 7.70 (4H, dd, J = 10.9, 8.4 Hz); δC (100 MHz, CDCl3) 191.8(2), 191.5, 155.7, 154.7, 153.4, 149.4, 145.9, 142.1, 141.0, 140.9, 138.4, 137.9, 137.0, 136.4, 136.0, 135.6(2), 135.4, 131.0, 130.7, 130.6, 130.4, 130.3, 128.2, 127.7, 127.6, 127.3; HRMS (ESI-TOF) m/z calcd for C40H26N3O3, [M + H]+ 596.1969; found: 596.1967.
2.3. Incorporation of PP2 in guar-gum borax hydrogel
Guar gum (375 mg) was dissolved in 20 mL deionized water at 60 °C under 500 rpm stirring for 2 h.52 To the guar gum solution, THF solution of probe PP2 (20 μM) was added and stirred for 30 minutes. Then, the cross-linker borax (200 μL, 10% borax solution in water) was added slowly to the guar gum suspension of PP2 under stirring. The prepared hydrogel was left aside for 3–4 h to attain maximum stability. Then, the hydrogel is washed thoroughly with deionized water to remove excess unreacted borax solution. Parafilm was used to seal the hydrogel and was stored in a refrigerator for further studies.
3. Results and discussion
3.1 Synthesis and structural characterization
Suzuki coupling reaction was employed to couple 7-bromo-2,3-bis(4-bromophenyl)pyrido[2,3-b]pyrazine and arylboronic acids in dioxane:water in the presence of in situ prepared Pd(PPh3)4 catalyst and K2CO3 as the base at 95 °C under a N2 atmosphere to give novel pyridopyrazine derivatives (PP1–PP5) in moderate yields (Scheme 1). The compounds were fully characterized via a comprehensive analysis of the 1H-NMR, 13C-NMR, and HRMS spectra (Fig. S1–S15, ESI†). These non-hygroscopic, and air-stable solid compounds were soluble in solvents such as chloroform, dichloromethane, acetonitrile, dimethyl sulfoxide, toluene, and tetrahydrofuran, but are water insoluble.
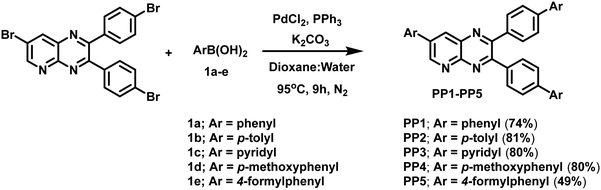 |
| | Scheme 1 Synthesis of tripodal pyridopyrazine derivatives PP1–PP5. | |
3.2. Single crystal X-ray analysis
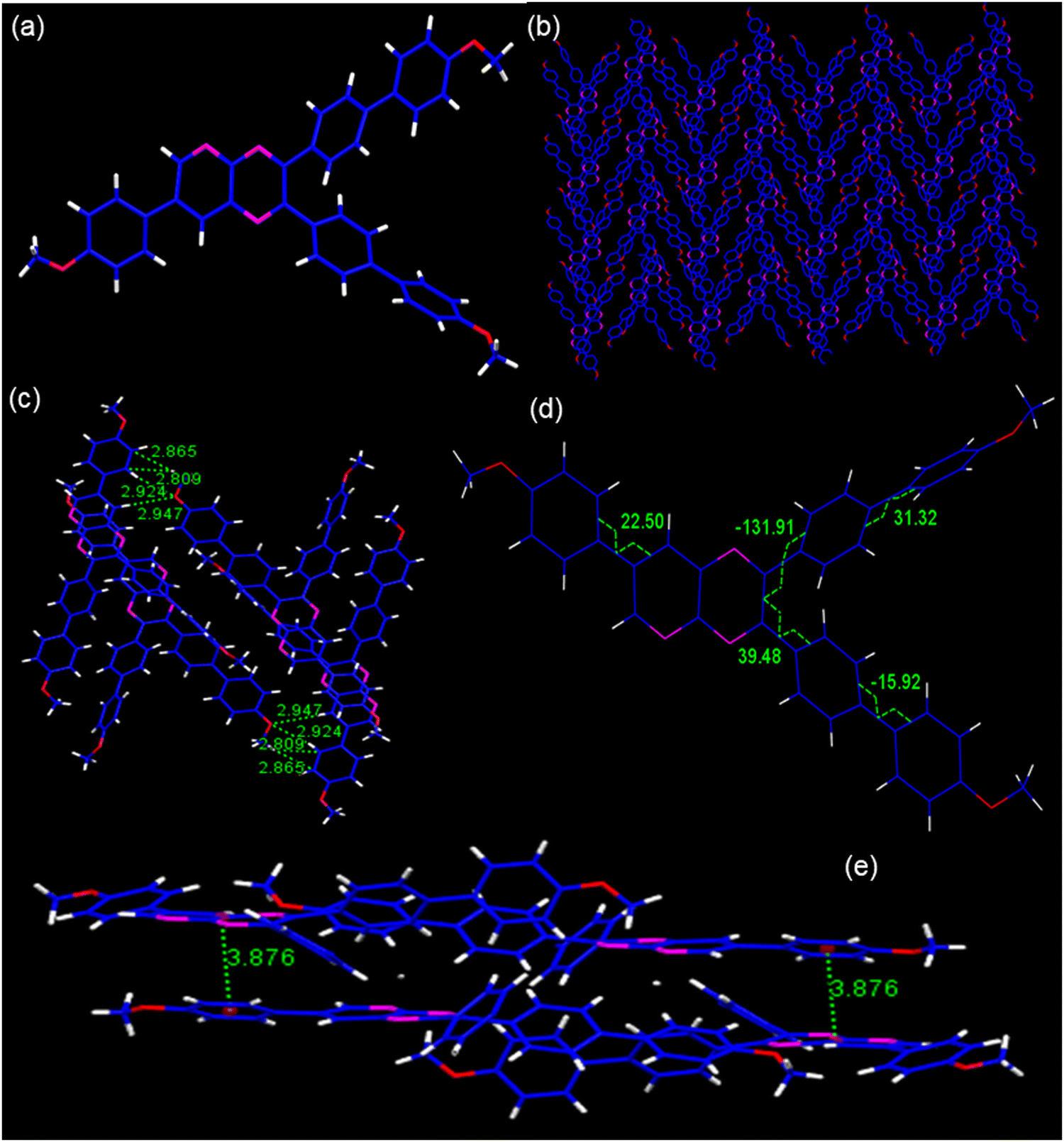
Single crystals of PP4 (CCDC 2301058) were grown by slow evaporation of the chloroform–hexane mixture of the compound. As shown, the PP4 (Fig. 2a) crystal belongs to a monoclinic crystal system and crystallized in the P21/c space group with four molecules of PP4 in one unit cell with cell parameters as a = 13.2222(5), b = 21.5979(8) and c = 12.2263(4) Å. (Table S1, ESI†). The crystal packing arrangement of PP4 showed a herringbone arrangement (Fig. 2b) with no π–π interactions and the existence of only weak C–H interactions at 2.865 Å and 2.809 Å distance (Fig. 2c) between different layers of the molecules. PP4 has a planar pyridopyrazine unit and twisted biphenyl segments on its 2- and 3-positions. The torsion angles were measured to be 39.48, −131.91, and 22.50 between pyridopyrazine and 1,4-phenylene rings, which indicated a distorted conformation (Fig. 2d). In a unit cell, the molecules showed donor (D) to acceptor (A) arrangement with interactions between electron donating p-methoxyphenyl and electron accepting pyridopyrazine at a distance of 3.876 Å (Fig. 2e), typical of push–pull molecules.53,54 There are also H-bonding interactions between O of –OCH3 and phenyl H at 2.924 Å and 2.947 Å55 (Fig. 2c). The highly twisted conformation of PP4, herringbone packing arrangement and absence of π–π interactions suggest the possibility of AIEE in these PPDs.43,44,56
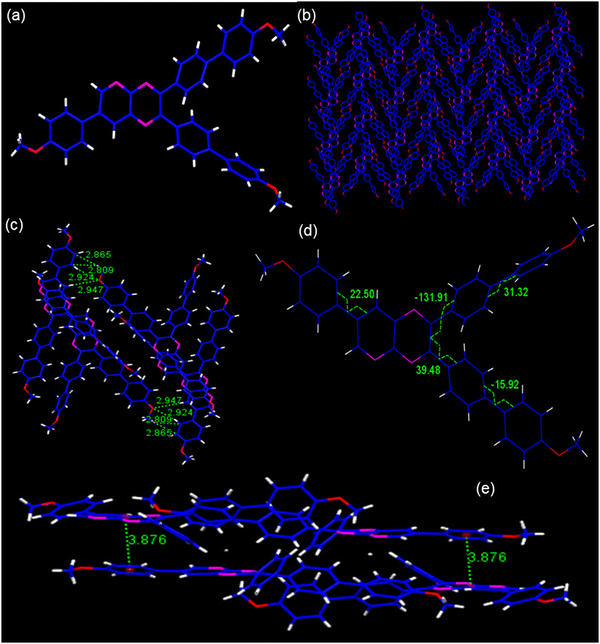 |
| | Fig. 2 (a) The molecular structure; (b) packing arrangements; (c) H-bonding and C–H interactions; (d) torsion angles, and (e) D–A interactions in PP4. | |
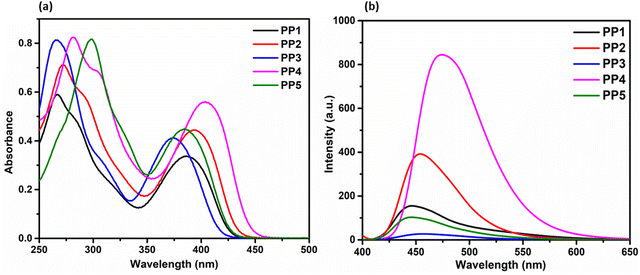 |
| | Fig. 3 (a) The absorption (1 × 10−5 M) and (b) emission (2 × 10−5 M) spectra of PP1–PP5 in CHCl3. | |
3.3. Photophysical analysis
3.3.1. Absorption studies. The UV-visible absorption properties for PP1–PP5 were investigated at a concentration of 1 × 10−5 M in CHCl3 solvent (Fig. 3a). PP1 with the phenyl group as a donor displayed absorption bands at 267 nm and 388 nm with a shoulder at 286 nm. The lower energy absorption (higher wavelength) corresponds to ICT transition, while the higher energy (lower wavelength) peak corresponds to π–π* transition.57,58 The shoulder is due to the electronic transitions between different vibrational states. On substituting the phenyl donor with electron donating p-tolyl in PP2, absorption bands were red shifted to 272 nm and 395 nm with a shoulder at 293 nm. The spectra were further red-shifted on replacing phenyl group with an electron donating p-methoxyphenyl donor in PP4, showing absorption bands at 282 nm and 405 nm with a shoulder at 309 nm. When the electron donating groups are replaced with an electron withdrawing pyridine in PP3, the bands were blue-shifted to 266 nm and 374 nm with a shoulder at 308 nm. On substituting with an electron withdrawing 4-formylphenyl group in PP5, the absorption corresponding to ICT was blue-shifted due to the electron withdrawing nature. However as expected, the band corresponding to π–π* transition was red-shifted to 299 nm as the conjugation of the molecule was increased (Table 1).
Table 1 Absorption and emission data of PP1–PP5
| Compound |
λabsb (nm) |
λemc (nm) |
Stokes shift cm−1 (nm) |
Quantum yieldd |
ε (M−1 cm−1) |
λeme (nm) |
Quantum yieldf |
| Shoulder. The absorption spectra are measured in CHCl3 at 1 × 10−5 M. The emission spectra are measured in CHCl3 at 2 × 10−5 M. Quantum yield in solution with reference to quinine sulfate (10−6 M in 0.1 M H2SO4). The emission wavelengths in the solid state. Quantum yield in the solid state with reference to BaSO4. |
| PP1 |
267, 286a, 388 |
447 |
3401 (59) |
0.21 |
47032 |
474 |
0.87 |
| PP2 |
272, 293a, 395 |
455 |
3338 (60) |
0.34 |
62188 |
485 |
0.65 |
| PP3 |
266, 308a, 374 |
456 |
4808 (82) |
0.03 |
57756 |
480 |
0.07 |
| PP4 |
282, 309a, 405 |
475 |
3639 (70) |
0.70 |
78238 |
535 |
0.84 |
| PP5 |
299, 331a, 386 |
446 |
3535 (61) |
0.10 |
62486 |
472 |
0.12 |
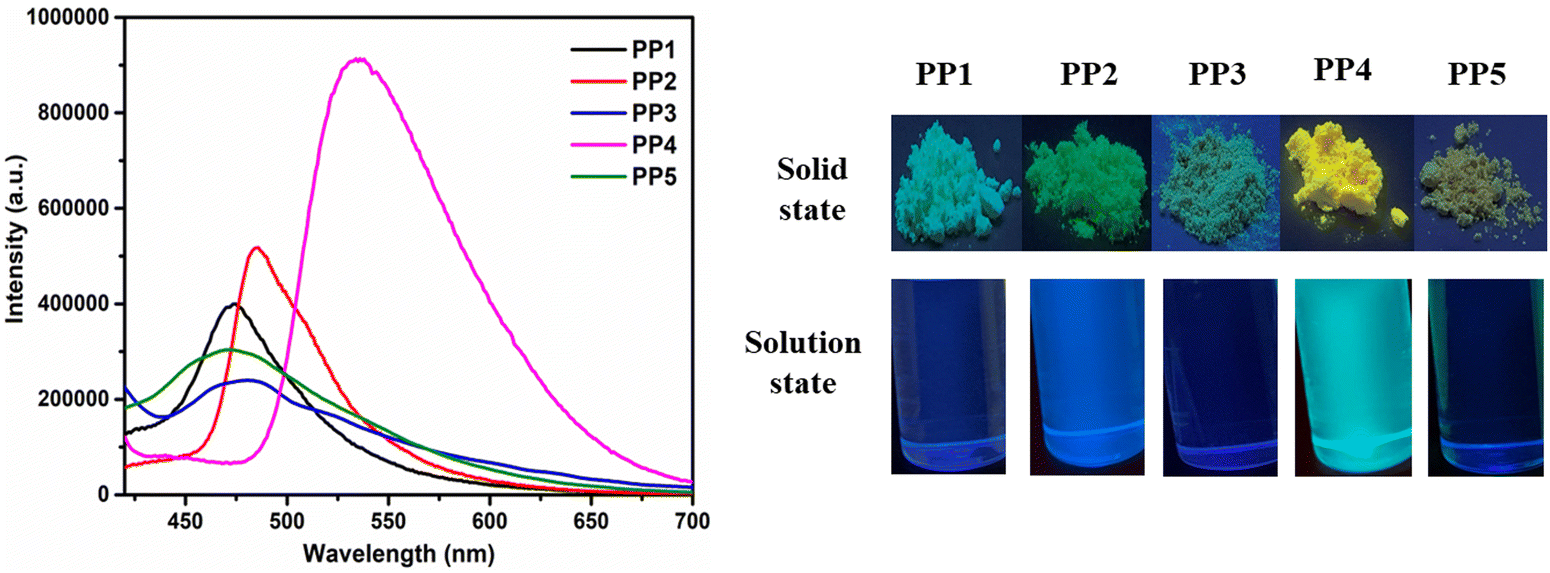
3.3.2. Fluorescence studies in the solution and solid state. The emission spectra of PP1–PP5 were recorded in 2 × 10−5 M CHCl3 solution and in the solid state (Fig. 3b and Fig. 4). All the compounds were excited at their respective absorption maxima. PP1–PP5 were emissive in both the solution and the solid states. They displayed blue color emission in CHCl3 (Fig. S16, ESI†), which shifted to higher wavelength emission in the solid state, with PP4 showing a change in emission color from blue to yellow on moving from solution to the solid state (Fig. 4). The pyridopyrazine appended with phenyl donor PP1 showed λem = 447 nm and λem = 474 nm in solution and the solid state, respectively. When the phenyl donor was replaced with an electron withdrawing 4-formylphenyl group in PP5, an almost negligible blue-shift of 1 nm and 2 nm was observed in the solution and solid state, respectively. The emission maximum was red-shifted on substituting phenyl with electron donating p-tolyl in PP2, and p-methoxyphenyl in PP4 in both the solution and solid state, suggesting enhanced ICT between the acceptor pyridopyrazine and donor groups.59 The emission data in solution and the solid state, along with Stokes shifts, have been included in Table 1. The quantum yields were also calculated in both states and are presented in Table 1. These photophysical studies show that compounds PP1–PP5 are emissive in the solid and solution states with high quantum yields and have π-conjugated twisted conformation critical to DSEgens. So, PP1–PP5 can be termed as DSEgens.
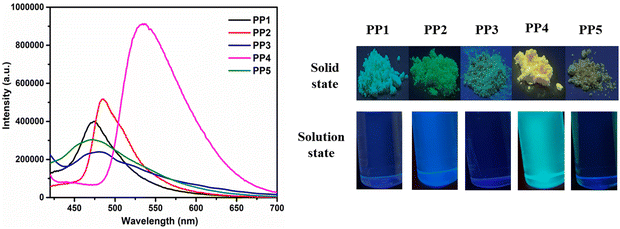 |
| | Fig. 4 The emission spectra of PP1–PP5 in the solid state and the pictures showing the emission colors of PP1–PP5 in the solid and solution (CHCl3) state under a UV lamp (365 nm). | |
3.4. Solvatochromism
It is well known that pyridopyrazine is an electron acceptor, and the substitution of electron donating groups on it generates intramolecular charge transfer (ICT) transitions.30 It is expected that PP1–PP5 behave as D–A–(π–D)2 type donor–acceptor molecules. The optical characteristics of such molecules in solution are known to be significantly influenced by the varying polarity of the solvents. Hence, we studied the emission behavior of PPDs in solvents with varied polarity such as toluene, chloroform (CHCl3), tetrahydrofuran (THF), acetonitrile (CH3CN), and dimethyl sulfoxide (DMSO). The pyridopyrazine derivatives PP1, PP2, and PP4, which are substituted with electron donating groups phenyl, p-tolyl, and p-methoxyphenyl, respectively, showed the solvatochromic behavior on varying the solvent polarity (Fig. S17, ESI†). PP1, PP2, and PP4 showed red shifts in the range of 10–63 nm (Table S2, ESI†), on varying the solvent polarity from toluene to DMSO, giving rise to positive solvatochromism. This positive behavior is caused by the reorientation of the solvent molecules, which stabilizes and increases the dipole moments of the excited states of molecules in the polar solvents.16,60 The pyridopyrazine derivatives PP3 and PP5 substituted with pyridyl and 4-formylphenyl groups, respectively, did not show a patterned shift in wavelength on increasing the solvent polarity. This observation shows that substituting acceptor pyridopyrazine rings with EDGs exhibits enhanced ICT due to increased donor–acceptor interactions compared to substituting them with EWGs.
3.5. Aggregation-induced emission enhancement (AIEE)
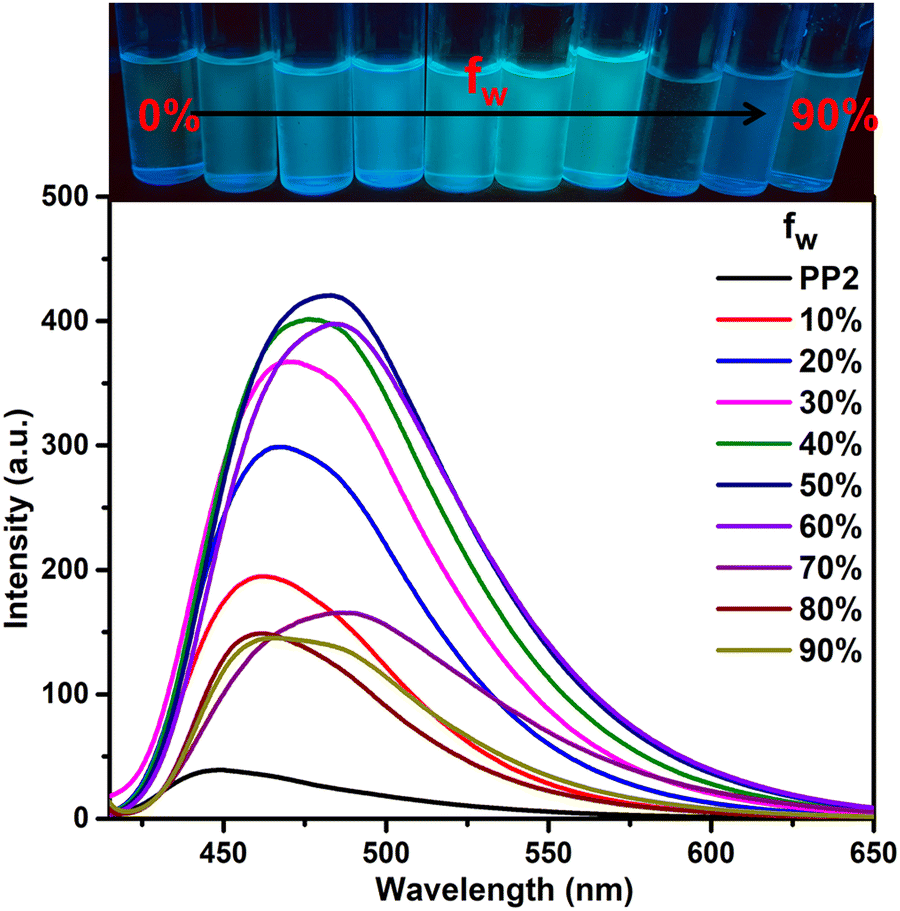
A design rationale for dual-state emitters is established only after evaluating the photophysical properties in aggregated and solid states. After performing emission studies in the solid state (Section 3.3.2), we further recorded the photophysical properties of PP1–PP5 in the aggregated state. The single crystal structural analysis of PP4 showed a twisted conformation and a herringbone packing arrangement with no intermolecular π–π interactions. These characteristics in the SCXRD indicate the possibility of AIEE properties on aggregation.61 So, the AIEE characteristics for PP1–PP5 are evaluated in THF–water (THF–H2O) solvent mixtures. PP1–PP5 are soluble and weakly emissive in THF but insoluble in water. The increase in water fractions can lead to aggregate formation of the hydrophobic molecules. In THF–H2O mixtures, all the PPDs showed AIEE with slight red-shifts and then blue shifts in emission maxima (Fig. 5 and Fig. S18, ESI†). PP1 showed a red-shift of 24 nm with a 33-fold enhancement in emission intensity at 70% fw (water fraction in THF). PP2 exhibited an 11-fold increase in emission intensity with a red-shift of 34 nm at fw = 50%. PP4 and PP5 also showed red-shifted emission maxima at fw = 80% and 70%, respectively. PP3 showed a slight blue-shift of 3 nm with a 2-fold increase in emission intensity at fw = 90%. These emission shifts are the result of the combined effects of aggregation and quantum effects.62 The bathochromic shifts with increasing water fraction are the result of enhanced ICT due to increased solvent polarity and/or J-type stacking on aggregation.63 The blue shifts at higher water fractions in PP3 are due to stronger quantum effects than the red shifts caused by aggregation.62 To analyze the particle size distribution of molecular aggregates, a dynamic light scattering (DLS) experiment was performed at different THF–H2O fractions. The average particle size of the aggregates of PP1–PP5 in 0% H2O, fw when emission is maximum and in 90% H2O were recorded and are summarized in Table S3 (ESI†). The findings show that the particle size increases on adding water, showing that aggregation is responsible for the enhanced emission.
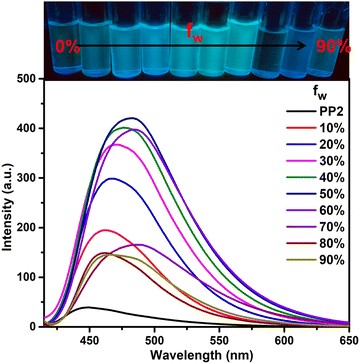 |
| | Fig. 5 Emission spectra of PP2 (2 × 10−5 M) in THF–H2O fractions. | |
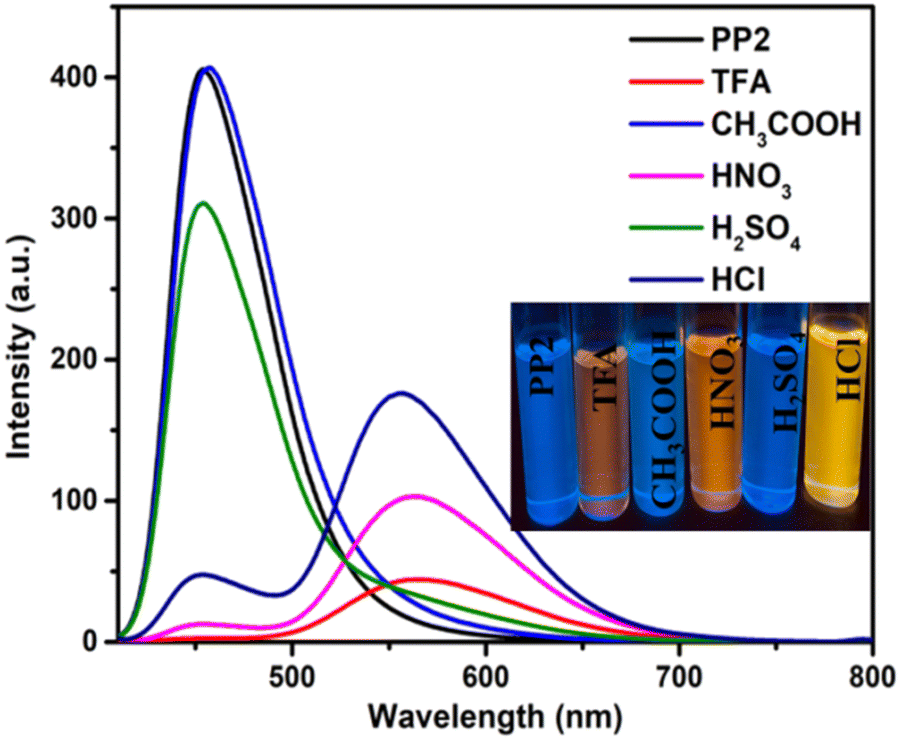
3.6. Acidofluorochromism
Because the pyridopyrazine units in DSEgens (PP1–PP5) are easily protonated by acid, PP1–PP5 were investigated as a dual-state fluorescence sensor for acidic solution and vapor in the solution and solid states, respectively. Different acids, such as CH3COOH, HCl, H2SO4, and HNO3, were studied for sensitivity. Adding 0.01 mL of these acids (except CH3COOH) to CHCl3 solutions of PP1–PP5 resulted in red-shifts and color changes (Fig. 6 and Fig. S19, ESI†). CH3COOH, being a weak acid, could not protonate the pyridopyrazine derivatives, and hence, no significant changes were observed. TFA and HNO3 addition showed the maximum changes. However, since TFA is more volatile and an organic acid, our further investigation was conducted only on TFA.
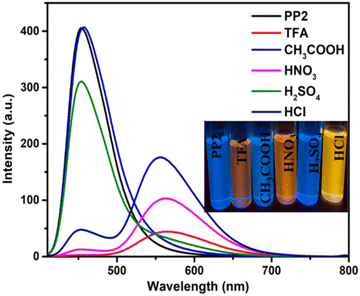 |
| | Fig. 6 The emission spectra of PP2 (2 × 10−5 M) in the presence of 0.02 mL of different acids. | |
3.6.1. In solution.
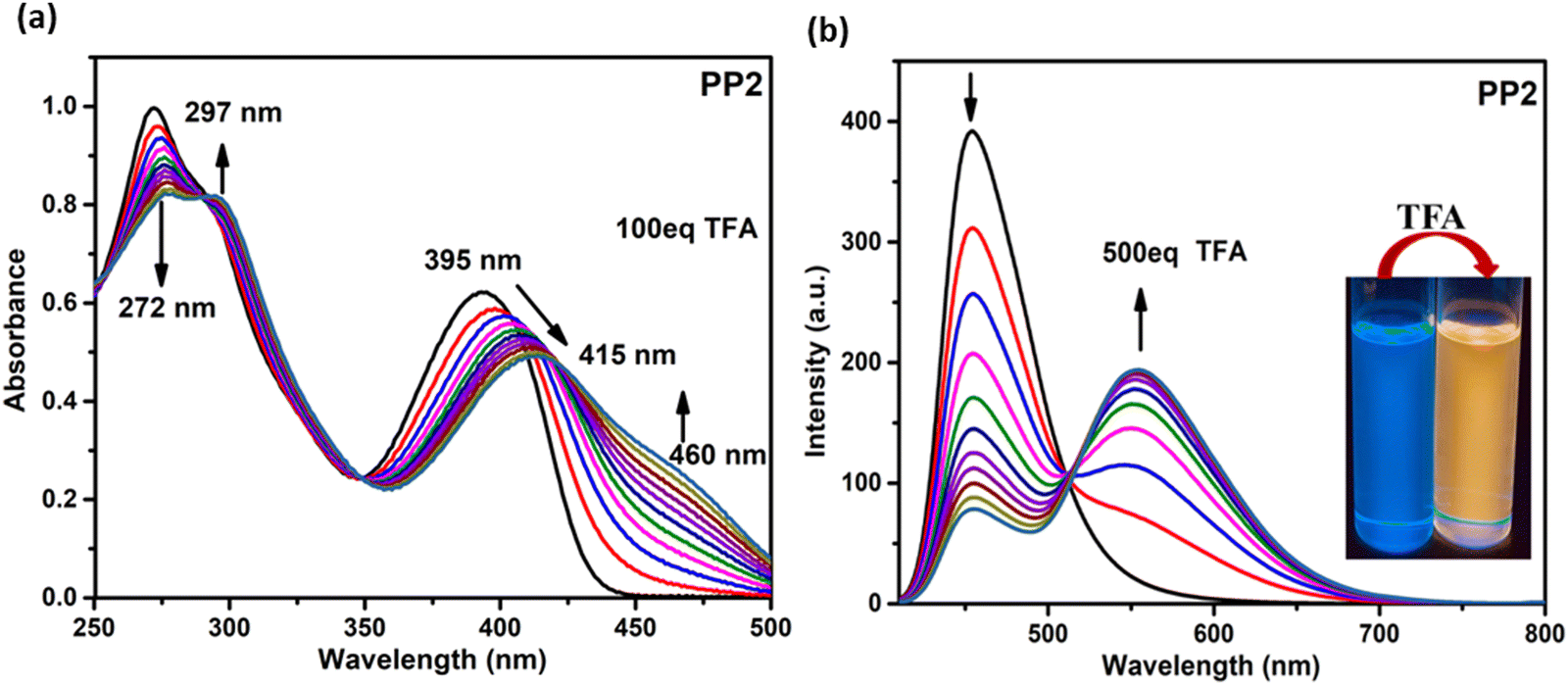
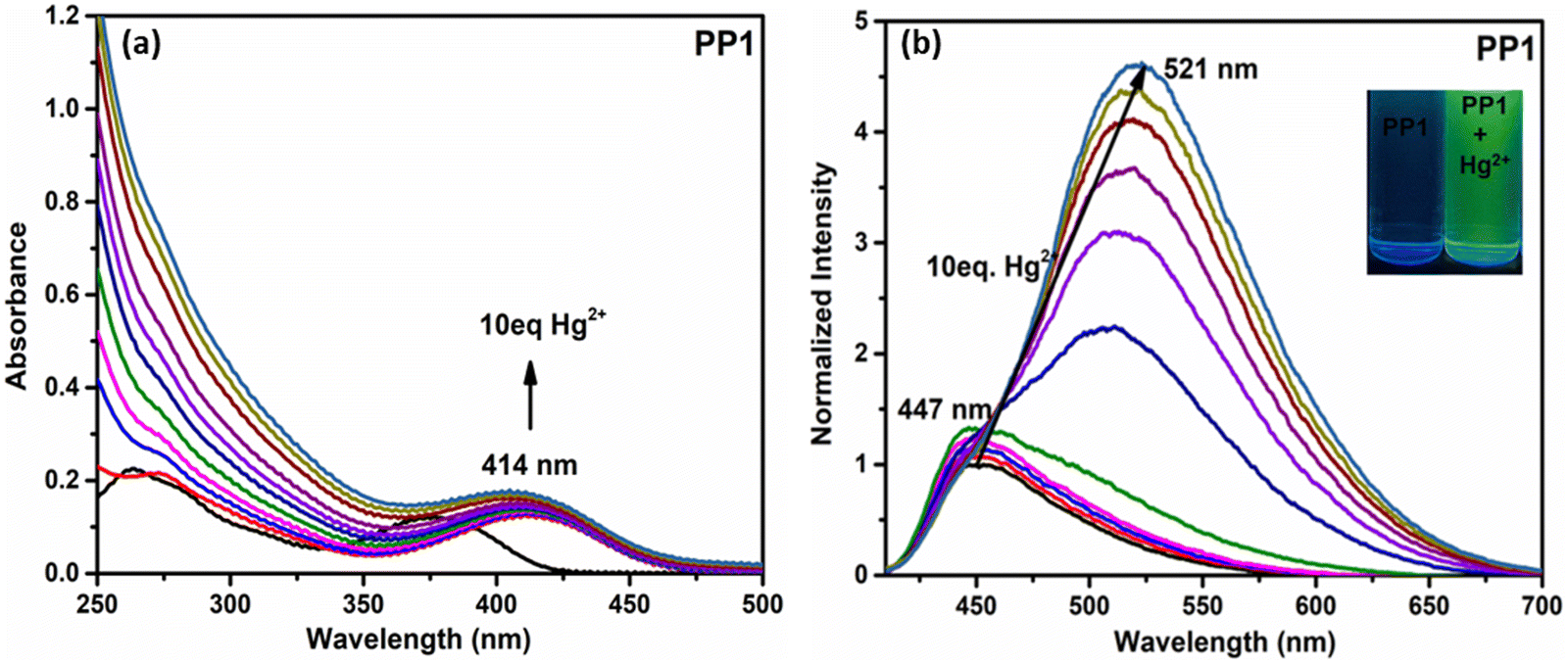
3.6.1.1. Absorption studies. On adding TFA to the CHCl3 solutions of PP1–PP5 (1 × 10−5 M), the colorless solution turned light yellow. The absorption titration experiments were performed to get insights into the binding mechanisms of PP1–PP5 with TFA (Fig. 7a and Fig. S20, ESI†). The absorption maxima band of PP2 at 272 nm showed a decrease in absorbance. In comparison, the band at 395 nm exhibited a red-shift of 20 nm and a new shoulder around 460 nm with the gradual addition of TFA (from 0 to 100 eq.) to the CHCl3 solution (Fig. 7). A red-shift of 20 nm was observed along with three isosbestic points in the absorption titration curves of PP2. Similarly, three isosbestic points were observed for PP1 and PP4, showing red shifts of 16 nm and 27 nm with new peaks at 460 nm and 479 nm, respectively. PP3 and PP5 also showed shifts in absorbance spectra when 100 eq. of TFA was added gradually, but no distinct isosbestic points were seen due to the co-existence of more than one species.29 These red shifts could be protonation-induced and attributed to an enhanced ICT between the donor and acceptor of these D–A–(π–D)2 systems.64 Three isosbestic points indicate the presence of three protonation sites in these DSEgens. Thus, the binding stoichiometry between PPDs and TFA is 1:3.40
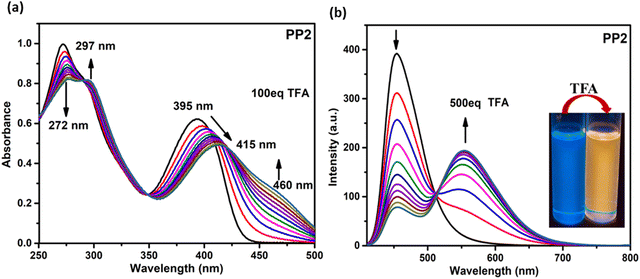 |
| | Fig. 7 (a) The absorption (1 × 10−5 M, CHCl3) and (b) emission (2 × 10−5 M, CHCl3) spectra of PP2 as a function of the concentration of TFA added. Inset: Photographs of PP2 solution before and after adding TFA under UV light (365 nm). | |
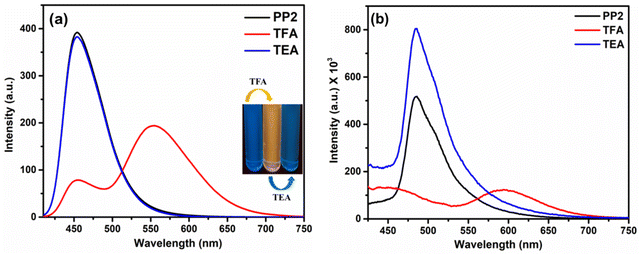 |
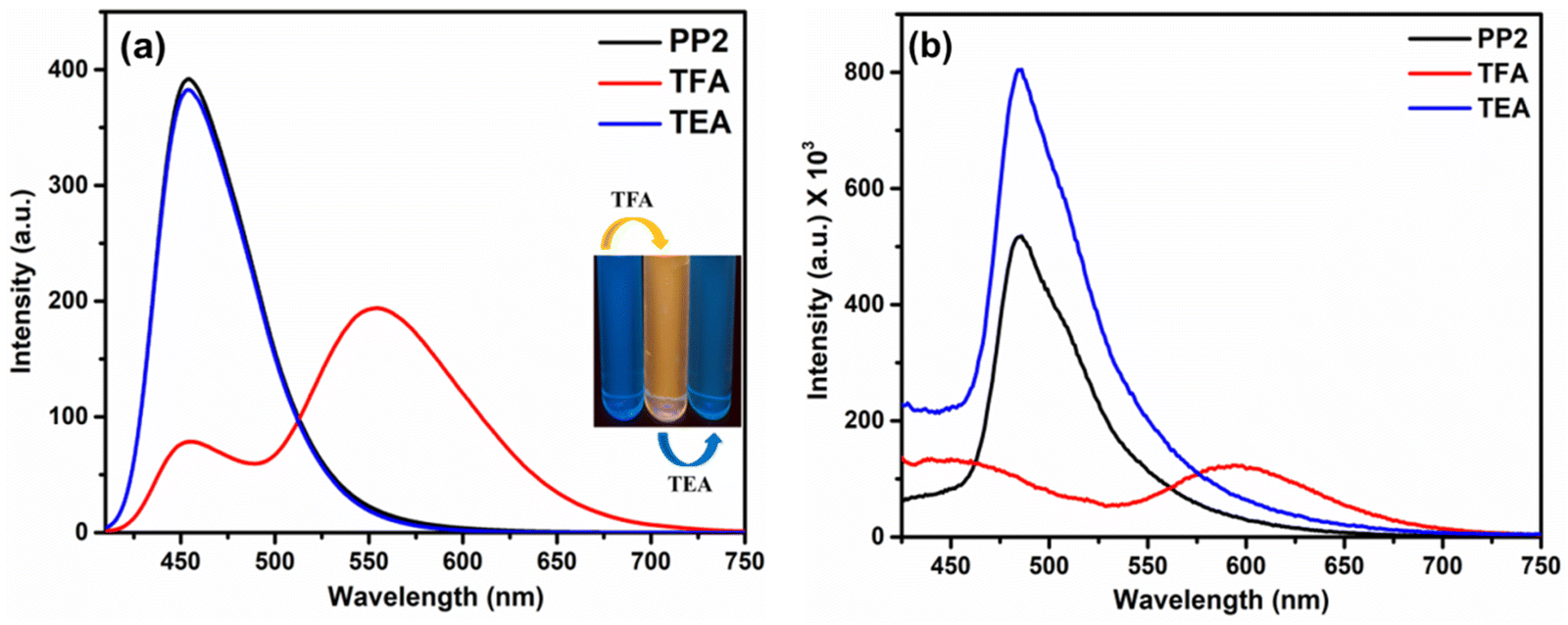
| | Fig. 8 Emission spectra of PP2 after sequential addition of TFA and TEA in (a) CHCl3 (inset: Image after sequential addition of TFA and TEA in CHCl3 under UV light) and (b) the solid state. | |
3.6.2. In the solid state. As discussed in Section 3.3.2, the solid state emission studies showed that PP1–PP5 are emissive in the solid state. To explore their ability as solid state probes, we examined the photoluminescence of the probes after fuming them with TFA. In the solid state, the pristine compound PP2 emitted at 485 nm. The emission was quenched and a new red-shifted peak was observed at 594 nm on fuming the solid sample with TFA vapors (Fig. 8b). Also, a color change was seen from pale to bright orange (Fig. 9). Similarly, PP1 and PP4–PP5 also showed new-red shifted λem at 581 nm, 585 nm, and 577 nm, respectively, with a color change from yellow to orange (Fig. 9 and Fig. S22, ESI†). PP3 showed a red shift of 16 nm in the emission wavelength along with pale to fluorescent yellow transformation (Fig. S22, ESI†). These changes were reversible as a response to TEA exposure of the TFA-fumed sample (Fig. 8 and 9). Furthermore, to explore the sensitivity of our probe in a portable matrix, we incorporated PP2 in a guar gum-borax hydrogel matrix.66 In the gel phase, PP2 could sense TFA with a color change from blue to yellow under UV light and yellow to red under daylight (Fig. 10). Here also, the changes were reversible on adding TEA.
 |
| | Fig. 9 Photos of PP1–PP5 upon fuming with TFA and TEA vapors under (i) daylight and (ii) UV light (365 nm). | |
 |
| | Fig. 10 PP2 incorporated hydrogel matrix showing a color change on adding TFA under UV light (365 nm). | |
Furthermore, to confirm the protonation of nitrogens of the pyridopyrazine ring, the 1H-NMR titrations were performed with probe PP1. The gradual addition of 3 equivalents of TFA resulted in downfield shifts in the NMR signals, but the maximum impact was seen on pyridopyrazine protons (Fig. S23, ESI†). The HA proton was shifted from 9.46 ppm to 9.52 ppm to 9.57 ppm to 9.63 ppm, and the HB proton shifted from 8.69 ppm to 8.78 ppm to 8.88 ppm to 9.08 ppm on sequential addition of 3 equivalents of TFA. These substantial shifts in pyridopyrazine protons as compared to other protons support the fact that protonation is occurring on the pyridopyrazine ring nitrogens, and the binding stoichiometry is 1:3. These changes in the NMR spectrum of PP1 were reverted on adding equal amounts of TEA (Fig. S23, ESI†). The binding stoichiometry between the probes and TFA was also determined through Job's plot and was found to be 1:3 (Fig. S24, ESI†).
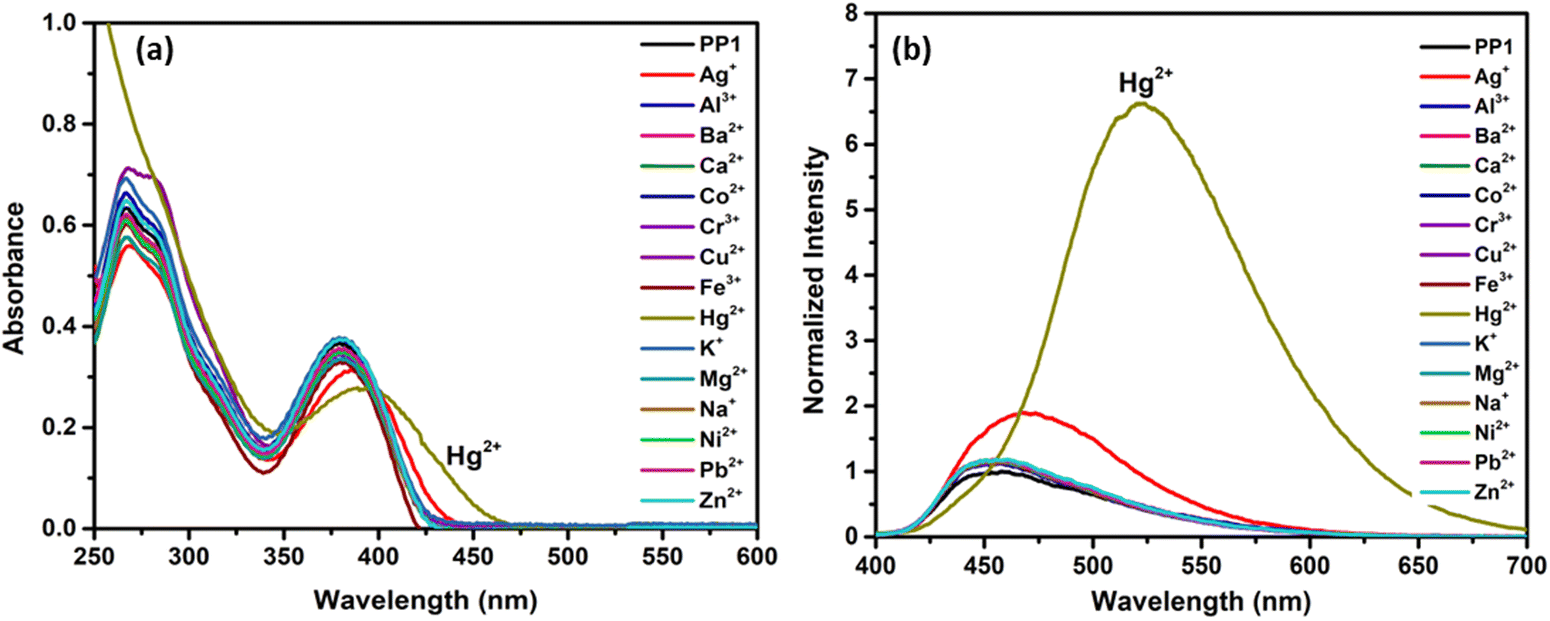
3.7. Metal ion recognition studies
These PPDs contain adjacent nitrogens with a lone pair of electrons that can act as ligands to bind metal ions. So, the effect of the addition of 10 equivalents of various metal ion solutions such as Na+, K+, Ag+, Ca2+, Ba2+, Co2+, Ni2+, Cu2+, Zn2+, Hg2+, Pb2+, Al3+, Cr3+ and Fe3+ on the synthesized PPDs PP1–PP5 was studied in THF–H2O mixtures. The THF–H2O fractions were selected as per the AIEE studies. For PP1, the maximum emission enhancement was observed at 70% H2O in THF, so the metal sensing studies were done at water fraction (fw) = 70%. As per AIEE, maximum emission for PP2–PP5 was obtained at fw = 50%, fw = 90%, fw = 80% and fw = 70%, respectively. Interaction of Hg2+ ions with PP1–PP5 except PP4 induced AIE and showed a “turn-on” response along with red shifts in the absorption and emission spectra67 (Fig. 11 and Fig. S25, ESI†). A “turn-off” response with a slight red-shift in emission and absorption wavelength was observed in PP4 (Fig. S25, ESI†). A slight emission enhancement was also observed with Ag+ ions, but it was not as pronounced as that observed in the presence of Hg2+ ions, so we carried out further experiments with Hg2+ metal ions only.
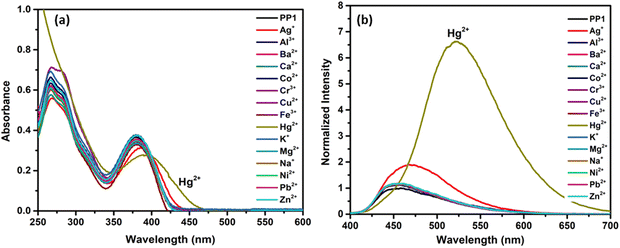 |
| | Fig. 11 (a) Absorption (1 × 10−5 M) and (b) emission (2 × 10−5 M) spectra of PP1 showing the selectivity of the probes towards Hg2+ ions in the presence of other competitive metal ions in a THF–H2O mixture. | |
To further understand the effect of Hg2+ ions on probes PP1–PP5, titration experiments with increasing concentrations of Hg2+ ions were performed in the respective THF–H2O mixtures. The absorption spectra of PP1–PP5 showed a red shift in the range of 26–44 nm (Fig. 12a and Fig. S26, ESI†). The emission titrations for PP1 exhibited increased emission intensity along with a red-shift in the wavelength from 447 nm to 521 nm on the addition of 10 equivalents of Hg2+ ions (Fig. 12b). Similarly, PP2, PP3, and PP5 showed enhanced emission accompanied by red-shifts of 80 nm, 18 nm, and 58 nm on gradually adding 10 eq. of Hg2+ ions (Fig. S26, ESI†). The probe PP4 showed a fluorescence quenching along with the red shift of 33 nm in the emission maximum (Fig. S26, ESI†). The emission spectral changes were accompanied by color changes from blue to yellow for PP1, PP2, and PP4. At the same time, PP3 and PP5 showed color changes from yellow to orange under UV light (Fig. 13). The red shifts observed in the absorption and emission spectra can be attributed to the greater charge transfer from the donor groups to the Hg2+ ion-bound pyridopyrazine acceptor. The increase in fluorescence intensity on adding Hg2+ ion solution to PP1–PP3 and PP5 is due to chelation enhancement of fluorescence (CHEF).68 The fluorescence quenching observed in PP4 on adding Hg2+ ions can be attributed to PET in the chelated form.69 There could be a photo-induced electron transfer taking place from the excited state HOMO of ligand PP4 to the LUMO of the metal ion.
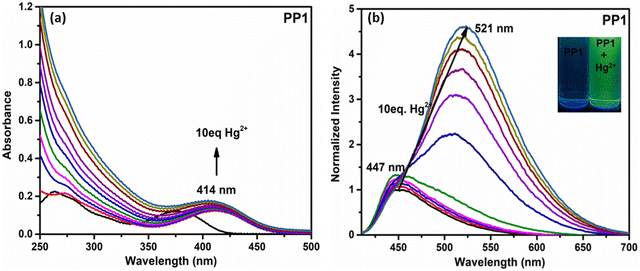 |
| | Fig. 12 The (a) absorption and (b) emission spectra of PP1 as a function of Hg equivalents. | |
 |
| | Fig. 13 Color changes observed under UV light (365 nm) in PP1–PP5 on adding 10 eq. Hg2+ ions. | |
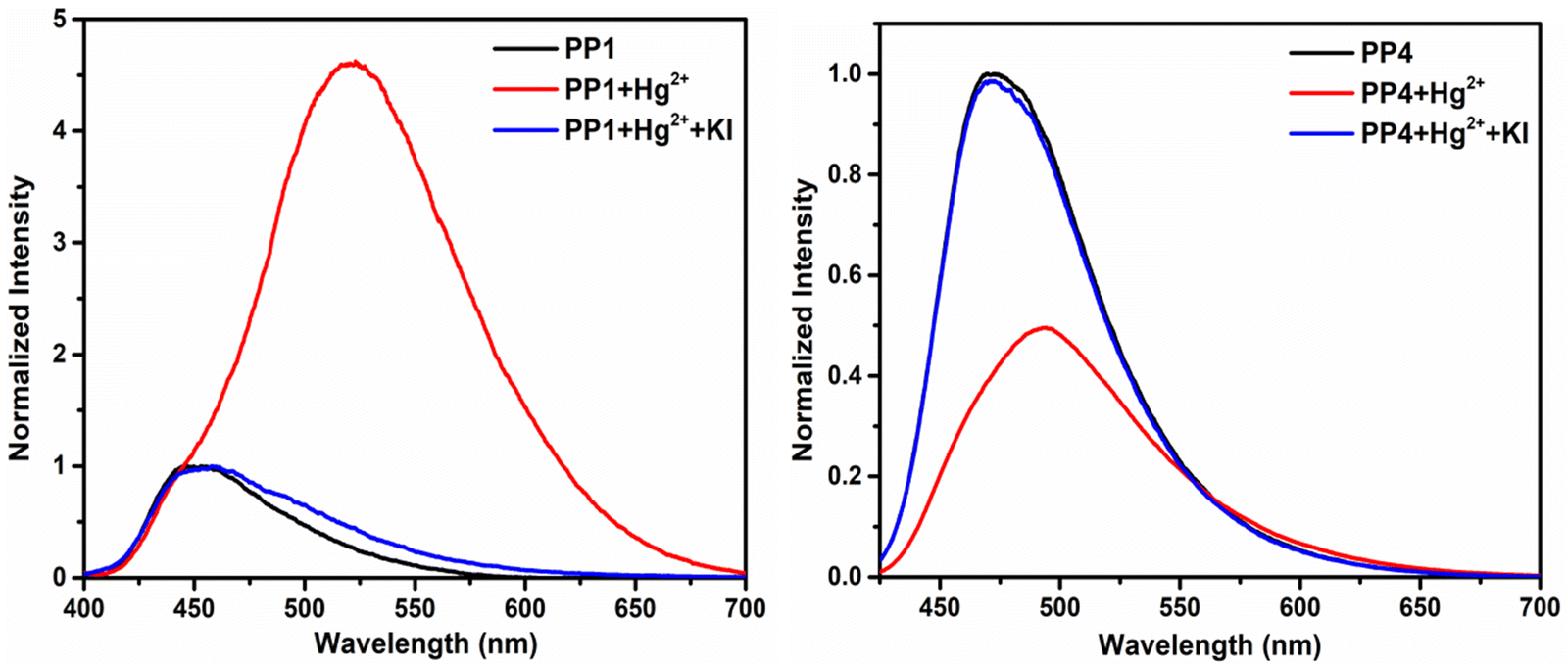
All fluorochromic changes observed in PP1–PP5 on adding Hg2+ ions were reversed on adding equal amounts of KI solution in water (Fig. 14). In order to determine the binding mechanism of Hg2+ ions with the probes, a 1H-NMR titration experiment was performed by adding excess Hg2+ ions to the CDCl3 solution of PP4. Six protons of the –OMe group appended on the pyrazine ring showed an upfield shift, while three protons of the –OMe group appended on the pyridine ring showed a downfield shift. Similar upfield and downfield shifts were also observed in the respective aromatic protons of the phenyl rings. A downfield shift of the Ha proton and upfield shift of the Hb proton of the pyridopyrazine ring suggests that the chelation is taking place at the nitrogen atoms at positions 4- and 5- of the pyridopyrazine ring (Fig. 15).43 The binding stoichiometry was evaluated using Job's method of continuous variation. The Job's plots revealed a 1:1 binding ratio between the probes and Hg2+ ions (Fig. S27, ESI†). The detection limits were calculated using D.L. = 3σ/s, where σ stands for the standard deviation of the probe and s is the slope of the intensity vs. amount of Hg2+ ion plot. The detection limits were found to be 2.74–26.28 nM (Table S4, ESI†).
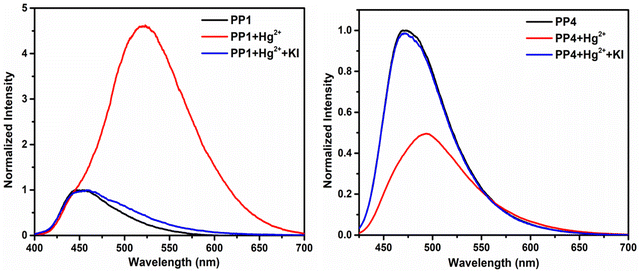 |
| | Fig. 14 Fluorescence spectra of PP1 in THF–H2O (3:7 v/v) and PP4 in THF–H2O (2:8 v/v) showing reversible Hg2+ ion sensing in the presence of KI. | |
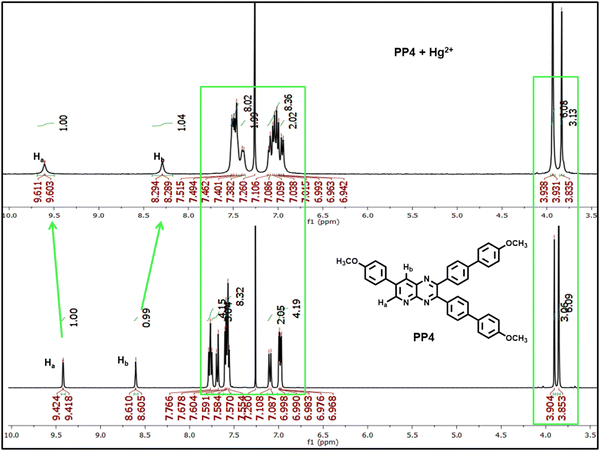 |
| | Fig. 15 1H-NMR spectrum of PP4 on adding excess Hg2+ ions in CDCl3 solvent. | |
3.8. Theoretical calculations
3.8.1. Molecular structure. Density functional calculations are conducted to gain deeper insights into the relationship between molecular structure and properties. All the structures (PP1–PP5) were fully optimized in THF solvent. The optimized structures are depicted in Fig. S28 (ESI†). The DFT-optimized structure is in good agreement with the single crystal X-ray structure available for PP4. In all compounds, the pyridopyrazine unit is planar, while the biphenyl segments are twisted. The calculated HOMO–LUMO gaps are presented in Table 2. The HOMO and LUMO orbitals for PP1–PP5 are plotted using the Charmol 1.3 program70 and are presented in Fig. S29 and S30 (ESI†), respectively. In all the compounds studied, the HOMOs are primarily localized on the biphenyl rings that are connected to the pyridopyrazine moiety, while the LUMOs are predominantly situated on the pyridopyrazine structure. The absorption and emission spectra for PP1–PP5 are calculated using time-dependent density functional theory (TDDFT) with the same exchange–correlation functional used for the DFT optimization. Chloroform solvent was used for the TDDFT calculations, via the COSMO solvation model. TDDFT excitation and emission wavelengths (nm), oscillator strengths, and major composition in terms of MO contribution are depicted in Table S5 (ESI†). For all the compounds the first major excitation is the transition from HOMO to LUMO.
Table 2 HOMO–LUMO energies and the gaps in eV
| Compound |
Pristine ligand |
Hg2+–ligand |
Protonated ligand |
| EHOMO |
ELUMO |
ΔELUMO–HOMO |
EHOMO |
ELUMO |
ΔELUMO–HOMO |
EHOMO |
ELUMO |
ΔELUMO–HOMO |
| PP1 |
−6.35 |
−2.51 |
3.85 |
−7.40 |
−6.78 |
0.62 |
−7.96 |
−6.23 |
1.73 |
| PP2 |
−6.36 |
−2.84 |
3.51 |
−7.23 |
−6.64 |
0.60 |
−7.69 |
−6.13 |
1.56 |
| PP3 |
−6.95 |
−3.05 |
3.90 |
−7.78 |
−7.10 |
0.68 |
−8.52 |
−6.48 |
2.04 |
| PP4 |
−6.47 |
−2.88 |
3.59 |
−6.89 |
−6.44 |
0.46 |
−7.31 |
−6.00 |
1.31 |
| PP5 |
−6.67 |
−2.97 |
3.69 |
−7.62 |
−6.99 |
0.63 |
−8.22 |
−6.40 |
1.83 |
These theoretical findings support our experimental results that PP1–PP5 have twisted molecular structures and a donor–acceptor relationship between the substituted biphenyls and pyridopyrazine moiety, giving rise to ICT character in these compounds.
3.8.2. Interaction with Hg2+ and TFA. To find out the probable binding sites for electron deficient Hg2+ and protons, the electrostatic potential maps (ESP) of PP1–PP5 were investigated through DFT, presented in Fig. S31 (ESI†). The ESPs indicate that the nitrogens of the pyridopyrazine moiety are the electron-rich centers that can potentially bind with the Hg2+ and protons from TFA. Optimizing the Hg2+–ligand complexes and protonated ligands results in stable complexes. Interaction energies (ΔEint) are calculated for the following reactions with eqn (1). The values are reported in Table 3. High negative ΔEint values indicate the formation of highly stable complexes (Table 3). The HOMO and LUMO orbitals for all these complexes are depicted in Fig. S32–S35 (ESI†). For the Hg2+–ligand complexes, the LUMOs are located mainly on the metal center. These theoretical findings are in line with NMR studies, which show that the Hg2+ and protonation takes place at the pyridopyrazine nitrogens as there is availability of electron density due to the presence of lone pairs on the nitrogens.
| Hg2+ + PPD ligand = Hg2+ − ligand or 3H+ + PPD ligand = protonated ligand |
| | |
ΔEint = Eproducts − Ereactants
| (1) |
Table 3 Interaction energies (ΔEint in kcal mol−1) of all the complexes
| Compound |
Hg2+–ligand |
Protonated ligand |
| PP1 |
−123.39 |
−742.48 |
| PP2 |
−125.87 |
−746.52 |
| PP3 |
−118.63 |
−731.32 |
| PP4 |
−142.04 |
−751.16 |
| PP5 |
−118.86 |
−733.87 |
3.9. Temperature sensing
We also investigated the effect of temperature on these PPDs by gradually increasing the temperature from 30 °C to 80 °C. All the probes PP1–PP5 showed quenching of fluorescence intensity accompanied by blue-shift in the emission wavelength (Fig. S36, ESI†). The blue shifts in emission wavelength with increasing temperature might be due to the increase in the rate of non-emissive de-excitation from the upper vibrational levels.71
3.10. Thermogravimetric analysis
Thermograms are specific to each molecule and communicate information about the material's moisture and volatile content, longevity, multi-component composition, thermal stability, and oxidative stability.72 Here, we performed thermo-gravimetric analysis to get an insight into the thermal stabilities of the dual-state emissive PPDs. High thermal stabilities are crucial for emissive molecules to be used in device fabricating applications. These PPDs are thermally stable. The decomposition temperatures (Td) range from 182 °C to 389 °C (Fig. S37, ESI†). Thus, these synthesized PPDs are potential candidates for material applications.
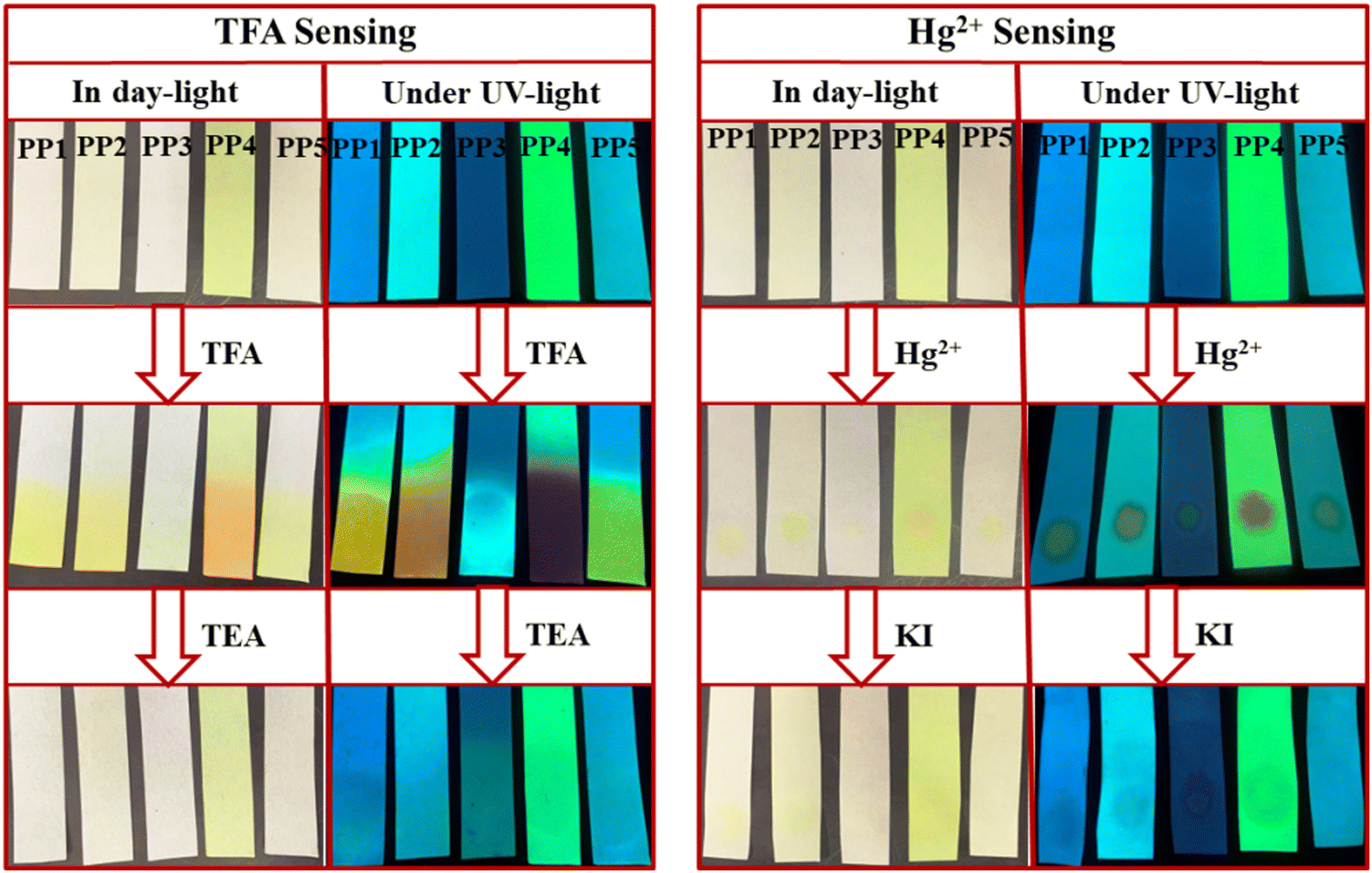
3.11. Practical applications
The practical application of these PPDs was studied by making portable paper strips. Whatman filter paper strips were loaded with ligands PP1–PP5 by dipping them in the respective solutions and drying them (Fig. 16). The paper strips of PP1–PP5 displayed visible color changes, from colorless to yellow/orange in daylight upon exposure to TFA fumes. The effect of exposure was also evident under UV lamp turning blue/yellow fluorescence to deep yellow (PP1), orange (PP2), blue (PP3), red (PP4) and light yellow (PP5) fluorescence. These changes can be reverted after fuming with TEA. Similarly, for Hg2+ detection, paper strips were prepared by coating the filter paper strips with a solution of PP1–PP5 (2 × 10−3 M) in THF solvent (Fig. 16). A yellow color spot was visible on the ligand loaded test strips after spotting with Hg2+ ion-containing water solution (1 × 10−3 M), under daylight and a UV lamp. This effect of Hg2+ ions can also be reversed by dipping the test strips in KI solution (1 × 10−3 M).
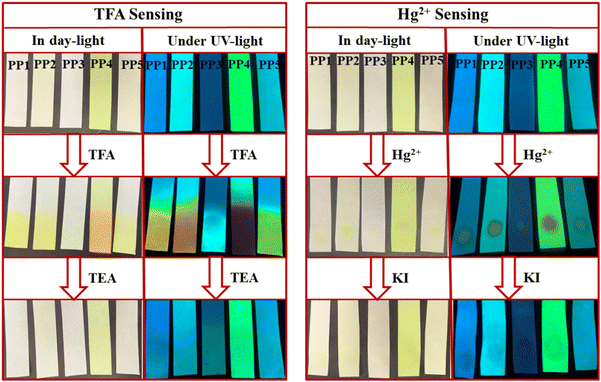 |
| | Fig. 16 Paper strips of PP1–PP5 employed to sense TFA and Hg2+ ions in daylight and under UV light (365 nm). | |
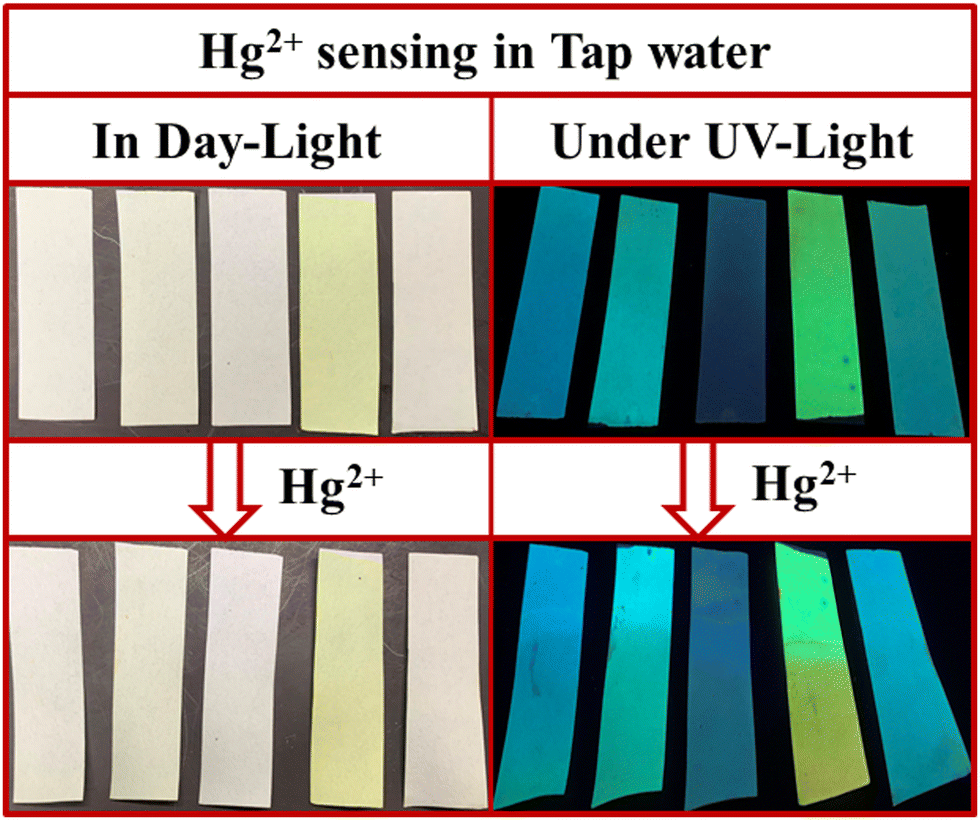
We also studied the real-life application of these PPDs by testing the tap water sample using test strips. The 9 mL tap water at Department of Chemistry, University of Delhi was spiked with 1 mL of 2 × 10−3 M Hg2+ ions. The PPD loaded paper strips were dipped in this tap water. The strips showed a slight color change under daylight but exhibited noticeable changes in fluorescence under UV light (Fig. 17). So, these PPDs have the potential to act as portable sensors for Hg2+ and TFA analytes.
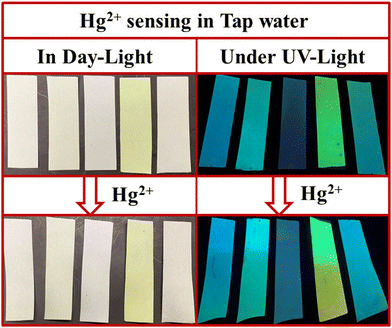 |
| | Fig. 17 Hg2+ ions sensing with paper strips of PP1–PP5 in tap water in daylight and under UV light (365 nm). | |
4. Conclusion
To summarize, a series of novel tripodal pyridopyrazine derivatives PP1–PP5 were designed, synthesized, and characterized. The push–pull nature of the compounds was tuned by varying the donor abilities of substituents on the acceptor pyridopyrazine unit. The single crystal XRD of PP4 revealed that it attains a twisted conformation, forming a herringbone packing arrangement. These compounds are AIEE-active in THF–H2O mixtures and are emissive in both the solution and solid state. Only PP1, PP2, and PP5 substituted with electron donor groups exhibited positive solvatochromism on moving from non-polar to polar solvents. All the PPDs exhibited reversible acidofluorochromism in solution and the solid state. Probe PP2 was also incorporated in a guar-gum borax hydrogel matrix for fabricating a portable TFA sensor. These PPDs were also sensitive towards mercuric ions and showed a “turn on” response for PP1–PP3 and PP5 with detection limits of 2.74 nM to 26.28 nM. These responses with Hg2+ ions could be reversed by adding KI. These experimental findings of PP1–PP5 having a twisted structure, ICT character and pyridopyrazine nitrogens acting as binding sites for Hg2+ and TFA are supported by theoretical studies. These PPDs also have good thermal stabilities and the potential to act as fluorometric thermometers in the temperature range of 30 °C to 80 °C. These results demonstrate that PP1–PP5 are dual-state emitting, dual chemosensors for TFA and Hg2+ and have great potential as organic materials.
Author contributions
Monika Lamoria: writing – original draft, visualization, validation, methodology, investigation, formal analysis. Debashree Manna: investigation, software, writing – original draft. Marilyn Daisy Milton: writing – review & editing, supervision, project administration, funding acquisition, conceptualization.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
MDM gratefully acknowledges financial support from the Institution of Eminence, University of Delhi (Faculty Research Project grants IoE/2024-25/12/FRP). The Council for Scientific and Industrial Research (CSIR) India is also accredited for awarding Junior and Senior Research Fellowships to M. L. This work was supported by the Ministry of Education, Youth and Sports of the Czech Republic through the e-INFRA CZ (ID:90254). We also acknowledge the Department of Chemistry, University of Delhi for IR, TGA, absorption and fluorescence facilities and the University Science Information Centre (USIC), the University of Delhi for NMR, HRMS, Solid PL, dynamic light scattering (DLS) and single crystal X-ray facilities.
References
- N. Sivakumar, K. Tashiro, A. Alsulmi and R. Jayavel, Opt. Mater., 2023, 143, 114173, DOI:10.1016/j.optmat.2023.114173.
- S. E. Jadhav, P. S. Singh, B. R. Madje, S. Chacko and R. M. Kamble, J. Lumin., 2023, 263, 120160, DOI:10.1016/j.jlumin.2023.120160.
- R. Iftikhar, F. Z. Khan and N. Naeem, Mol. Diversity, 2023, 28, 271–307, DOI:10.1007/s11030-022-10597-0.
- J. Hu, T. Han, Y. Liu, X. Zhang, Y. Duan, Z. Li and T. Han, Spectrochim. Acta, Part A, 2020, 239, 118486, DOI:10.1016/j.saa.2020.118486.
- Q. Wang, B. Lin, M. Chen, C. Zhao, H. Tian and D. H. Qu, Nat. Commun., 2022, 13, 4185, DOI:10.1038/s41467-022-31978-x.
- J. Wu, C. Li, Q. Chen, L. Xu, M. Jian and J. Zhao, J. Mater. Chem. C, 2022, 10, 10595–10608, 10.1039/D2TC01332K.
- P. Meti, H. H. Park and Y. D. Gong, J. Mater. Chem. C, 2020, 8, 352–379, 10.1039/C9TC05014K.
- M. R. Miah, M. Yang, S. Khandaker, M. M. Bashar, A. K. Alsukaibi, H. M. Hassan, H. Znad and M. R. Awual, Sens. Actuators, A, 2022, 347, 113933, DOI:10.1016/j.sna.2022.113933.
- T. Sachdeva, S. Gupta and M. D. Milton, Current Org. Chem., 2020, 24, 1976–1998, DOI:10.2174/1385272824999200729132853.
- J. H. Park, H. Kim, D. Y. Kim and S. D. Yang, Bull. Korean Chem. Soc., 2015, 36, 1778–1783, DOI:10.1002/bkcs.10335.
- P. Meti, J. W. Yang and Y. D. Gong, Dyes Pigm., 2018, 156, 233–242, DOI:10.1016/j.dyepig.2018.03.006.
- S. Zeng, S. J. Li, X. J. Sun, T. T. Liu and Z. Y. Xing, Dyes Pigm., 2019, 170, 107642, DOI:10.1016/j.dyepig.2019.107642.
- Z. Meng, Y. Li, K. Liao, Y. Sun, W. Zhang, B. Song, X. Zhu and X. Q. Hao, Dyes Pigm., 2024, 222, 111882, DOI:10.1016/j.dyepig.2023.111882.
- Y. Qi, W. Liu, J. Wang, Y. Wu, F. Liu, L. Ye and B. Lei, Dyes Pigm., 2023, 220, 111745, DOI:10.1016/j.dyepig.2023.111745.
- Y. Yin, A. Ding, F. He, C. Wang, L. Kong and J. Yang, J. Phys. Chem. B, 2022, 126, 2921–2929, DOI:10.1021/acs.jpcb.2c01387.
- J. Xue, F. Tang, A. Ding, F. He, J. Huang, L. Kong and J. Yang, J. Lumin., 2022, 250, 119119, DOI:10.1016/j.jlumin.2022.119119.
- L. A. Rodríguez-Cortés, F. J. Hernández, M. Rodríguez, R. A. Toscano, A. Jiménez-Sánchez, R. Crespo-Otero and B. Rodríguez-Molina, Matter, 2023, 6, 1140–1159, DOI:10.1016/j.matt.2023.01.015.
- V. D. Singh, A. K. Kushwaha and R. S. Singh, Dyes Pigm., 2021, 187, 109117, DOI:10.1016/j.dyepig.2020.109117.
- Y. Yin, A. Ding, L. Yang, L. Kong and J. Yang, Mater. Chem. Front., 2022, 6, 1261–1268, 10.1039/D2QM00067A.
- S. H. Chen, S. H. Luo, L. J. Xing, K. Jiang, Y. P. Huo, Q. Chen and Z. Y. Wang, Chem. Eur. J., 2022, 28, e202103478, DOI:10.1002/chem.202103478.
- K. Ohno, F. Narita, H. Yokobori, N. Iiduka, T. Sugaya, A. Nagasawa and T. Fujihara, Dyes Pigm., 2021, 187, 109081, DOI:10.1016/j.dyepig.2020.109081.
- Y. Zhang, A. Ding, S. Guo, F. He, L. Kong, R. H. H. Duan and J. Yang, J. Mater. Chem. C, 2022, 10, 6078–6084, 10.1039/D2TC00350C.
- M. Li, Y. Niu, X. Zhu, Q. Peng, H. Y. Lu, A. Xia and C. F. Chen, Chem. Commun., 2014, 50, 2993–2995, 10.1039/C3CC49680E.
- S. Kumar, P. Singh, P. Kumar, R. Sivastava, S. K. Pal and S. Ghosh, J. Phys. Chem. C, 2016, 120, 12723–12733, DOI:10.1021/acs.jpcc.6b01351.
- L. Zou, S. Guo, H. Lv, F. Chen, L. Wei, Y. Gong, Y. Liu and C. Wei, Dyes Pigm., 2022, 198, 109958, DOI:10.1016/j.dyepig.2021.109958.
- X. Zhang, Y. Zhou, M. Wang, Y. Chen, Y. Zhou, W. Gao, M. Liu, X. Huang and H. Wu, Chem. – Asian J., 2020, 15, 1692–1700, DOI:10.1002/asia.202000322.
- Y. Xu, L. Ren, D. Dang, Y. Zhi, X. Wang and L. Meng, Chem. – Eur. J., 2018, 24, 10383–10389, DOI:10.1002/chem.201802201.
- M. Pettersson, D. S. Johnson, C. Subramanyam, K. R. Bales, C. W. Ende, B. A. Fish, M. E. Green, G. W. Kauffman, P. B. Mullins, T. Navaratnam and S. M. Sakya, J. Med. Chem., 2014, 57, 1046–1062, DOI:10.1021/jm401782h.
- M. Lamoria and M. D. Milton, Dyes Pigm., 2023, 213, 111182, DOI:10.1016/j.dyepig.2023.111182.
- S. S. Mahadik, D. R. Garud, A. P. Ware, S. S. Pingale and R. M. Kamble, Dyes Pigm., 2021, 184, 108742, DOI:10.1016/j.dyepig.2020.108742.
- S. K. Panja, N. Dwivedi and S. Saha, RSC Adv., 2016, 6, 105786–105794, 10.1039/C6RA17521J.
- A. L. Li, Z. L. Wang, W. Y. Wang, Q. S. Liu, Y. Sun, S. F. Wang and W. Gu, Microchem. J., 2021, 160, 105682, DOI:10.1016/j.microc.2020.105682.
- J. Wang, Y. Liu, Z. Dong, J. Chao, H. Wang, Y. Wang and S. Shao, J. Hazard. Mater., 2020, 382, 121056, DOI:10.1016/j.jhazmat.2019.121056.
- F. Lafzi, A. S. Hussein, H. Kilic and S. Bayindir, J. Photochem. Photobiol., A, 2023, 444, 114958, DOI:10.1016/j.jphotochem.2023.114958.
- Q. S. Liu, Z. H. Yang, Z. L. Wang, Y. Sun, L. L. Chen, L. Sun, X. B. Sun and W. Gu, J. Photochem. Photobiol., A, 2022, 423, 113597, DOI:10.1016/j.jphotochem.2021.113597.
- S. H. Chen, K. Jiang, Y. H. Liang, J. P. He, B. J. Xu, Z. H. Chen and Z. Y. Wang, Food Chem., 2023, 428, 136800, DOI:10.1016/j.foodchem.2023.136800.
- P. Purushothaman and S. Karpagam, Spectrochim. Acta, Part A, 2024, 305, 123518, DOI:10.1016/j.saa.2023.123518.
- M. Tripathy, U. Subuddhi and S. Patel, Dyes Pigm., 2020, 174, 108054, DOI:10.1016/j.dyepig.2019.108054.
- X. Niu, H. Zhang, X. Wu, S. Zhu, H. Feng and W. Liu, J. Mol. Struct., 2022, 1259, 132754, DOI:10.1016/j.molstruc.2022.132754.
- S. Gupta and M. D. Milton, Dyes Pigm., 2019, 165, 474–487, DOI:10.1016/j.dyepig.2019.02.038.
- S. Gupta and M. D. Milton, J. Mol. Struct., 2022, 1264, 133275, DOI:10.1016/j.molstruc.2022.133275.
- S. Gupta and M. D. Milton, J. Photochem. Photobiol., A, 2022, 424, 113630, DOI:10.1016/j.jphotochem.2021.113630.
- S. Gupta and M. D. Milton, New J. Chem., 2018, 42, 2838–2849, 10.1039/C7NJ04573E.
- S. Gupta and M. D. Milton, J. Photochem. Photobiol., A, 2021, 419, 113444, DOI:10.1016/j.jphotochem.2021.113444.
- M. Lamoria and M. D. Milton, Dyes Pigm., 2024, 229, 112303, DOI:10.1016/j.dyepig.2024.112303.
- M. Lamoria, D. Manna and M. D. Milton, J. Mol. Struct., 2024, 1319, 139385, DOI:10.1016/j.molstruc.2024.139385.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170, DOI:10.1063/1.478522.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104, DOI:10.1063/1.3382344.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305, 10.1039/B508541A.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsuji, et al., Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 1, 1993, 799–805, 10.1039/P29930000799.
- D. Ghosh, M. Basak, D. Deka and G. Das, Int. J. Biol. Macromol., 2023, 229, 615–623, DOI:10.1016/j.ijbiomac.2022.12.310.
- T. Sachdeva and M. D. Milton, Dyes Pigm., 2020, 181, 108539, DOI:10.1016/j.dyepig.2020.108539.
- S. Garain, S. N. Ansari, A. A. Kongasseri, B. C. Garain, S. K. Pati and S. J. George, Chem. Sci., 2022, 13, 10011–10019, 10.1039/D2SC03343G.
- L. Schaeffer, The Practice of Medicinal Chemistry, Academic Press, 2008, pp. 464–480. DOI:10.1016/B978-0-12-417205-0.00014-6.
- Y. Sun, Z. Lei and H. Ma, J. Mater. Chem. C, 2022, 10, 14834–14867, 10.1039/D2TC02512D.
- D. M. Kapse, P. S. Singh, M. Ghadiyali, S. Chacko and R. M. Kamble, RSC Adv., 2022, 12, 6888–6905, 10.1039/d2ra00128d.
- P. Thirumurugan, D. Muralidharan and P. T. Perumal, Dyes Pigm., 2009, 81, 245–253, DOI:10.1016/j.dyepig.2008.10.018.
- S. E. Jadhav, P. S. Singh, B. R. Madje, S. Chacko and R. M. Kamble, J. Lumin., 2023, 263, 120160, DOI:10.1016/j.jlumin.2023.120160.
- K. Prabakaran, R. Manivannan and Y. A. Son, Spectrochim. Acta, Part A, 2023, 285, 121874, DOI:10.1016/j.saa.2022.121874.
- S. Bhuin, P. Chakraborty, P. Sivasakthi, P. K. Samanta, P. Yogeeswari and M. Chakravarty, J. Mater. Chem. C, 2023, 11, 11270–11282, 10.1039/d3tc01793a.
- C. Zeng, T. Yang, Z. Wang, K. Chen, Q. Ge, W. Peng, J. Zhang, J. Liu, T. Zhang and M. Sun, J. Lumin., 2024, 269, 120452, DOI:10.1016/j.jlumin.2024.120452.
- H. Sun, X. X. Tang, B. X. Miao, Y. Yang and Z. Ni, Sens. Actuators, B, 2018, 267, 448–456, DOI:10.1016/j.snb.2018.04.022.
- S. Gupta and M. D. Milton, Dyes Pigm., 2021, 195, 109690, DOI:10.1016/j.dyepig.2021.109690.
- Y. Zhan, Dyes Pigm., 2020, 173, 108002, DOI:10.1016/j.dyepig.2019.108002.
- D. Ghosh, M. Basak, D. Deka and G. Das, J. Mol. Liq., 2022, 363, 119816, DOI:10.1016/j.molliq.2022.119816.
- Y. Gu, R. Jia, Y. Yu, S. Li, J. Zhu, X. Feng and Y. Lu, ACS Appl. Mater. Interfaces, 2024, 16, 10805–10812, DOI:10.1021/acsami.3c19521.
- B. Zhang, Q. Suo, Q. Li, J. Hu, Y. Zhu, Y. Gao and Y. Wang, Tetrahedron, 2022, 120, 132878, DOI:10.1016/j.tet.2022.132878.
- D. Udhayakumari, J. Inclusion Phenom. Macrocyclic Chem., 2022, 102, 451–476, DOI:10.1007/s10847-022-01138-1.
- J. Chalupsky, Charmol - program for molecular graphics, Prague, Czech Republic, 2012.
- C. Cao, X. Liu, Q. Qiao, M. Zhao, W. Yin, D. Mao, H. Zhang and Z. Xu, Chem. Commun., 2014, 50, 15811–15814, 10.1039/C4CC08010F.
- N. Saadatkhah, A. C. Garcia, S. Ackermann, P. Leclerc, M. Latifi, S. Samih, G. S. Patience and J. Chaouki, Can. J. Chem. Eng., 2020, 98, 34–43, DOI:10.1002/cjce.23673.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Debashree Manna
a,
Debashree Manna