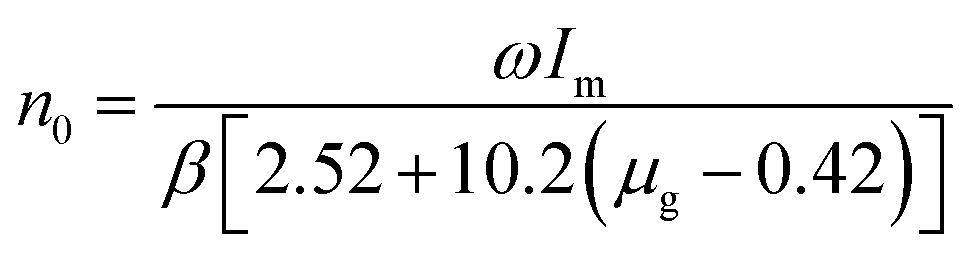DOI:
10.1039/D5TC00486A
(Paper)
J. Mater. Chem. C, 2025, Advance Article
Enhanced photodegradation performance based on the surface plasmon resonance effect of Ag/Ca2Al2SiO7:Pr3+ ultraviolet long afterglow driven in a photo-Fenton system†
Received
5th February 2025
, Accepted 19th February 2025
First published on 20th February 2025
Abstract
Long afterglow photocatalysts have demonstrated the potential to support all-weather photocatalytic reactions. In this work, an Ag/Ca2Al2SiO7:Pr3+ (Ag/CASO:Pr3+) composite was synthesized to enhance afterglow-driven photocatalytic activity. Ag loading introduced the surface plasmon resonance (SPR) effect, which increased light absorption capacity. It extended the afterglow duration and improved afterglow intensity by storing and releasing hot electrons. The Schottky junction between Ag and CASO:Pr3+ inhibited the recombination of photogenerated electrons and holes, increasing the active sites in photocatalytic reactions. The photodegradation performance of Ag/CASO:Pr3+ increased by up to 6.6-fold compared to CASO:Pr3+ under afterglow-driven conditions. The concentration of active species was further increased by adding Fe3+ and H2O2 to construct the Fenton system. Ag/CASO:Pr3+ achieved a 55% removal rate of TC and a 60% removal rate of NFX under its own afterglow-driven conditions within 1 h, following 10 min of ultraviolet light irradiation. This work expands the use of long afterglow luminescent materials and advances the development of all-weather photocatalytic technology.
1. Introduction
Photocatalytic technology demonstrates remarkable potential in the treatment of organic pollutants in wastewater and sewage.1,2 This technology uses specific photocatalysts to generate free radicals with strong oxidizing power under ultraviolet or visible light irradiation, typically hydroxyl radicals (˙OH).3 These radicals are highly efficient in oxidizing and decomposing organic pollutants into harmless substances, such as H2O and CO2.4,5 Defect engineering, doping strategies, surface modification and heterostructure construction are widely used to enhance the performance of photocatalysts.6–9 Although these photocatalysts exhibit outstanding degradation performance, their activity is highly dependent on continuous light irradiation.10,11 This limitation restricts their practical application due to factors such as the alternation of day and night, energy consumption, technical challenges and environmental factors of the equipment.
Photocatalytic technology adeptly resolves the aforementioned challenges using long afterglow phosphors.12–14 Long afterglow phosphors achieve sustained luminescence through their internal trap energy levels.15,16 The key feature of this material lies in its ability to store photolytic energy and release the stored energy in the dark to support continuous catalytic processes.17,18 However, the primary challenge faced by this technology is the significant competition between the catalytic process and the long afterglow luminescence process. The afterglow release occurs due to the radiative recombination of photogenerated electrons after the light source is removed. When the afterglow material is used as a photocatalyst, the stored photogenerated carriers are required both to produce afterglow to drive the photocatalytic reaction and to participate in the photocatalytic process to form active species. This means that there is a competitive utilization of photogenerated carriers between the afterglow emission process and the photocatalytic reaction process. In addition, the consumption of photogenerated electrons during the photocatalytic reaction leads to the accumulation of holes, further resulting in the rapid recombination of photogenerated carriers. This significantly reduces the photocatalytic efficiency of the afterglow photocatalyst and limits the development of long afterglow photocatalytic technology in practical applications.10,16 Therefore, optimizing the utilization efficiency of photogenerated carriers is a key challenge for the further development of long afterglow photocatalysts.
The photo-Fenton method is an advanced chemical oxidation technology.19–21 It centers on the reaction of Fe2+ with H2O2 to generate ˙OH and the light-driven Fe2+/Fe3+ cycles.22,23 The generation pathways of ˙OH in the photolytic Fenton reaction primarily include the following three mechanisms: (1) the reaction between ferrous ions (Fe2+) and hydrogen peroxide (H2O2) under the irradiation of ultraviolet (UV) or visible light to produce ˙OH.24 (2) The direct activation of H2O2 by light energy for the formation of ˙OH.22 (3) The reaction of photogenerated holes with H2O to generate ˙OH.25 The photo-Fenton reaction can generate a higher concentration of reactive species. It maximizes the utilization of photogenerated charge carriers within the limited afterglow duration, enabling efficient degradation of organic pollutants. The photo-Fenton reaction has received much attention for its ability to efficiently remove organic pollutants from water.26–30 Wu et al. degraded tetracycline hydrochloride (TC) using a MIL-101(Fe)/photo-/H2O2 system and demonstrated that visible light can accelerate the Fe2+/Fe3+ cycle in the photo-Fenton system.31 Zhao et al. used a ZnFe2O4/SrWO4 catalytic material to effectively degrade 99.18% of RhB dye in the photo-Fenton process in only 20 min with remarkable cycling stability.32 Currently, although this process can effectively utilize electrons and holes, the issue of rapid consumption of photogenerated carriers has not yet been overcome. The photocatalysts need to be optimally designed to enhance their photogenerated carrier generation and utilization efficiency. The surface plasmon resonance (SPR) effect of Ag can improve the light absorption of the photocatalyst and increase the number of photogenerated carriers, thereby playing an important role in the photocatalytic reaction.33–35 Therefore, the integration of Ag nanoparticles with the photo-Fenton reaction process is used to enhance the degradation performance of long afterglow photocatalysts. Ca2Al2SiO7 (CASO) possesses sufficient structural rigidity and a wide bandgap. It enables the CASO to have energy level traps near the conduction band edge, which can capture the photogenerated electrons.36 The combination of Ag with a low work function and CASO enhances the electron storage capacity. It has the potential to extend the afterglow duration and intensity. It is crucial for enhancing the performance of afterglow photocatalysts.
In this study, the CASO:Pr3+ phosphors with a UV long afterglow emission were prepared by a high-temperature solid-state process. The Ag nanoparticles were successfully loaded onto the CASO:Pr3+ by photoreduction deposition to create Ag/CASO:Pr3+. The low power function of the Ag gives Ag/CASO:Pr3+ a higher electron storage capacity. The light absorption properties of the CASO:Pr3+ are improved through the SPR effect of Ag loading. The degradation efficiency of the Ag/CASO:Pr3+ composite is enhanced by the generation of hot electrons and the separation of photogenerated carriers. The photo-Fenton system further enhanced the utilization of photogenerated carriers. The removal rate of RhB, TC and NFX driven by afterglow reached 82%, 55% and 60% within 1 h, respectively. These findings will help to broaden the long afterglow photocatalytic technology development and applications.
2. Experimental
2.1 Chemicals
CaCO3 and H2O2 were purchased from Sinopharm Chemical Reagent Co., Ltd. AgNO3 was purchased from Tianjin Windship Chemical Reagent Technology Co., Ltd. Al2O3, SiO2, isopropanol (IPA), p-benzoquinone (BQ), ethylenediamine tetraacetic acid (EDTA), rhodamine B (RhB), tetracycline hydrochloride (TC), norfloxacin (NFX) and ferric chloride hexahydrate (FeCl3·6H2O) were purchased from Macklin Reagent Co, Ltd. Pr6O11was purchased from Aladdin Reagent Co, Ltd. All materials were used without further processing.
2.2 Preparation of CASO:Pr3+
The CASO:0.01Pr3+ phosphor was synthesized by a high-temperature solid-state method.37–41 First, the raw materials, CaCO3 (0.9959 g), Al2O3 (0.5843 g), SiO2 (0.3004 g), and Pr6O11 (0.0085 g), were weighed according to the stoichiometric ratios. They were thoroughly mixed and ground for 30 min in an agate mortar and pestle. Then, the mixture was transferred to an alumina crucible and placed in a tube furnace sintering at 1300 °C for 6 h in the air (Fig. S1, ESI†). The agglomerate was ground at room temperature and in an agate mortar again. The obtained powder is the CASO:Pr3+ phosphor.
2.3 Preparation of Ag/CASO:Pr3+
The Ag/CASO:0.01Pr3+ phosphor was prepared by a photoreduction deposition method. First, 0.0001 g, 0.0002 g, 0.0003 g, 0.0004 g, 0.0005 g and 0.0006 g of AgNO3 powder were accurately weighed and dissolved each in 100 mL of H2O. Subsequently, 0.5 g of CASO:Pr3+ powder was added to the solution. The suspension was continuously stirred for 15 min under a 300 W Xe lamp irradiation. The precipitate was collected by centrifugation and washed several times with deionized water. The product was then dried at 60 °C for 12 h. Finally, the Ag/CASO:Pr3+ solid powder was obtained. The obtained samples were labeled as Ag/CASO:Pr3+-1, Ag/CASO:Pr3+-2, Ag/CASO:Pr3+-3, Ag/CASO:Pr3+-4, Ag/CASO:Pr3+-5 and Ag/CASO:Pr3+-6, respectively.
2.4 Photocatalytic degradation experiment
The photocatalytic activity of CASO:Pr3+ and Ag/CASO:Pr3+ were evaluated by degrading RhB in a dark environment after 10 min of irradiation under a 254 nm UV lamp. The photocatalyst (10 mg) and RhB solution (50 mL, 10 mg L−1) were added to a beaker. The Fenton reaction was employed to degrade RhB using 1 mL of H2O2 and 0.1 mg of FeCl3·6H2O. The aliquots (2 mL) were taken at regular intervals and filtered through filter paper to remove the excess powder. The absorbance of the aliquot was measured using a UV-vis spectrophotometer.
2.5 Characterization
The morphology of CASO:Pr3+ and Ag/CASO:Pr3+ were characterized by scanning electron microscopy (SEM, Hitachi, Regulus, 8100) and transmission electron microscopy (TEM, Talos F200X). The elemental compositions of the samples were measured by energy-dispersive X-ray spectroscopy (EDS, Ultim Max 65, Oxford). The crystal phase of all the phosphor samples was identified using X-ray powder diffraction (XRD) analysis with a Rigaku D/MAX-RB diffractometer (Tongda TD-3500 automatic X-ray diffractometer system). The emission and excitation spectra were measured by the fluorescence spectrometer (Hitachi, F-7000) equipped with a 150 W Xe lamp. Long afterglow decay curve measurements were performed with a fluorescence spectrometer after the CASO:Pr3+ and Ag/CASO:Pr3+ were irradiated by UV light (254 nm) for about 10 min. Electrochemical impedance spectra (EIS) and Mott–Schottky plots were recorded with an electrochemical workstation (ParStat 2273). Thermoluminescence (TL) curves were measured with an FJ-427 A TL meter (Beijing Nuclear Instrument Factory). X-ray photoelectron spectra (XPS) of CASO and Ag/CASO:Pr3+ were characterized by a ThermoFisher 250xi X-ray spectrometer. The electron spin resonance (ESR) spectra were measured using an ESR spectrometer (Bruker A-300). Brunauer–Emmett–Teller (BET) surface area measurements of the CASO:Pr3+ and Ag/CASO:Pr3+ were performed by N2 adsorption (Autosorb-iQ).
3. Results and discussion
3.1 Structure and surface analysis of CASO:Pr3+ and Ag/CASO:Pr3+
The morphologies of CASO:Pr3+ and Ag/CASO:Pr3+ phosphors were characterized using SEM and EDS. Fig. 1a and d show the SEM images of CASO:Pr3+ and Ag/CASO:Pr3+. The SEM images reveal that the CASO:Pr3+ exhibits an irregular shape. The Ag/CASO:Pr3+ also possesses an irregular morphology. Ag is coated densely on the surface of CASO:Pr3+. The EDS spectrum of CASO:Pr3+ reveals the presence of constituent elements, including Ca, Al, O, Si and Pr (Fig. 1b). The Ca, Al, O, Si, Pr and Ag elements appeared in the Ag/CASO:Pr3+ EDS spectrum (Fig. 1e). The uniform distribution of elements indicates that CASO:Pr3+ and Ag/CASO:Pr3+ exhibit homogeneous chemical properties, and the composites of Ag and CASO:Pr3+ were successfully obtained (Fig. 1c and f). Fig S2 (ESI†) displays the XRD patterns of CASO:Pr3+ and Ag/CASO:Pr3+. All diffraction peaks are in excellent agreement with the standard Ca2Al2SiO7 (PDF # 98-000-0226). It indicates the successful synthesis of the CASO hosts. The Raman spectra of CASO:Pr3+ and Ag/CASO:Pr3+ were recorded at an excitation wavelength of 532 nm (Fig. S3, ESI†). The peaks at the 242 cm−1 and 302 cm−1 bands can be assigned to the stretching vibrations of the Ca–O bond. The peak at 627 cm−1 is the symmetric stretching vibration of the Al–O bond. The band at 797 cm−1 is the antisymmetric stretching vibration of the Al–O bond. The peaks at 911 cm−1 and 978 cm−1 are attributed to the symmetric stretching vibrations of Al–O and Si–O and antisymmetric stretching vibrations of the Si–O bond in the pyrosilicate moiety, respectively. The peak positions of CASO:Pr3+ and Ag/CASO:Pr3+ are nearly the same. The Raman scattering intensity of Ag/CASO:Pr3+ is significantly higher than that of CASO:Pr3+. This phenomenon can be attributed to the SPR effect introduced by Ag loading.42 The results also demonstrate the successful loading of Ag on the CASO:Pr3+ surface.
 |
| | Fig. 1 (a) and (d) SEM images of CASO:Pr3+ and Ag/CASO:Pr3+. (b) and (e) EDS spectra of CASO:Pr3+ and Ag/CASO:Pr3+. (c) and (f) EDS elemental mapping images of CASO:Pr3+ and Ag/CASO:Pr3+. | |
Fig. 2a and b show the TEM images of CASO:Pr3+. The prepared pristine CASO:Pr3+ displays irregular granularity with a relatively smooth surface. It is in good agreement with the results of the SEM image, as shown in Fig. 1a. Fig. 2c and d depict that Ag/CASO:Pr3+ has a granular surface. Ag nanoparticles are uniformly dispersed in the main lattice of CASO:Pr3+. High-resolution TEM (HRTEM) further confirmed the microstructure of Ag/CASO:Pr3+ as well as the connectivity patterns between the components. As shown in Fig. 2e, the lattice stripe with a lattice spacing of 0.37 nm belongs to the CASO crystals. The lattice stripe spacing of 0.23 nm corresponds to the (111) lattice planes of Ag nanoparticles.43 It further indicates the presence of Ag nanoparticles on the surface of the CASO host. The diffraction pattern in Fig. 2f also reveals the presence of Ag nanoparticles. The diffraction peaks (1 1 1), (2 0 0) and (2 2 0) of the Ag nanoparticles are observed in the selected area electron diffraction (SAED) image.44 The EDS elemental mapping of CASO:Pr3+ and Ag/CASO:Pr3+ is illustrated in Fig. 2g and h. It clearly shows that Ca, Al, Si, O, Pr and Ag are homogeneously distributed in the CASO matrix. The results are consistent with those of the SEM and TEM images. The tight binding of Ag and CASO:Pr3+ contributes to the Schottky junction formation, maximizing the inhibition of photogenerated carrier complexation and enhancing photocatalytic performance.
 |
| | Fig. 2 (a) and (b) TEM images of CASO:Pr3+. (c) and (d) TEM images of Ag/CASO:Pr3+. (e) HRTEM image of Ag/CASO:Pr3+. (f) Selected area electron diffraction (SAED) images of Ag. (g) EDS mapping images of CASO:Pr3+. (h) EDS mapping images of Ag/CASO:Pr3+. | |
The full XPS spectra of CASO:Pr3+ and Ag/CASO:Pr3+ indicate the presence of multiple elements on the sample surface. The binding energies at around 347, 74, 102, 532 and 367 eV correspond to Ca 2p, Al 2p, Si 2p, O 1s and Ag 2p in CASO:Pr3+ and Ag/CASO:Pr3+, respectively (Fig. 3a). As previously reported, the binding energy of metallic Ag ranges between 371.1 and 374.1 eV.34 It indicates that Ag particles successfully complexed with the CASO phosphors and Ag+ underwent successful photo-reduction to Ag0 (Fig. 3b). XPS scanning results of CASO:Pr3+ and Ag/CASO:Pr3+ are shown in Fig. S4 (ESI†). The binding energies of Ca, Si, Al and O elements remain the same. Fig. 3c shows the absorption spectra of CASO:Pr3+ and Ag/CASO:Pr3+. The absorption peaks are in the same position, and the absorption peak located near 245 nm originates from the 3H4 → 4f 5d transitions of Pr3+.45,46 The SPR effect introduced by Ag loading increases the overall absorption capacity of CASO:Pr3+. Ag becomes an effective light trap to improve the storage capacity of the photogenerated carriers.47 It implies that more photogenerated carriers can be released for the photocatalytic reaction under its own afterglow-driven process. The composite of Ag and CASO can form a Schottky junction.48 It inhibits the complexation of photogenerated carriers and improves the charge separation efficiency. The inset band gaps are calculated according to the following formula:33,49
A is the absorbance, converted to reflectance
R;
B is a constant;
hν is the photon energy; and
Eg is the optical band gap. The band gaps of CASO:Pr
3+ and Ag/CASO:Pr
3+ are calculated to be 5.15 eV and 5.12 eV. The band gaps after Ag loading did not show a significant change. The results are consistent with previous experimental data.
15 Fig. 3d displays the emission spectra of CASO:Pr
3+ and Ag/CASO:Pr
3+. The Ag loading does not affect the position of the CASO:Pr
3+ and Ag/CASO:Pr
3+ emission peaks. The intensity of the emission peaks increased with the rising Ag concentration. The SPR effect of Ag introduces hot electrons under the 254 nm excitation. It introduces more electrons and holes in the compound and results in stronger emission. The emission bands can be observed at 273 nm and 318 nm. They are assigned to the 4f
15d
1 → 4f
2 transition of Pr
3+.
50,51 The TL spectra of CASO:Pr
3+ and Ag/CASO:Pr
3+ are shown in
Fig. 3e. The TL intensity of the Ag/CASO:Pr
3+ sample is significantly higher than that of CASO:Pr
3+. The Ag loading introduces new energy levels, allowing hot electrons to be excited from the valence band (VB) to the conduction band (CB). The low work function endows Ag can become an effective light trap to improve the storage capacity of photogenerated carriers. The characterization of the internal trap distribution and kinetic processes in CASO:Pr
3+ and Ag/CASO:Pr
3+ can be revealed by analyzing the pattern of the TL intensity as a function of temperature. The concentration of trapped charges
n0 and the trap depth of
E can be estimated using the following equation:
34where
Im is the TL intensity of the glow peaks,
β is the heating rate (4 °C s
−1),
ω is the shape parameter,
δ is the high-temperature half width and
τ is the low-temperature half-width. The asymmetry parameter
μg =
δ/
ω.
kB is the Boltzmann constant (1.38 × 10
−23 J K
−1).
Tm is the temperature. Fig. S5 (ESI
†) illustrates the TL spectra corresponding to the parameters. The parameters of the TL curve and the trap depths of CASO:Pr
3+ and Ag/CASO:Pr
3+ phosphors are listed in Table S1 (ESI
†). It indicates that the Ag/CASO:Pr
3+ can capture more electrons. It is attributed to the low work function of Ag. The Ag can act as electron traps to store electrons.
Fig. 3f exhibits the afterglow decay curves of CASO:Pr
3+ and Ag/CASO:Pr
3+. The afterglow intensity is enhanced in Ag/CASO:Pr
3+. The decay time of the Ag/CASO:Pr
3+ reaches its maximum at the Ag concentration of 0.0005. The decay process includes two stages: rapid decay and slow decay. The decay curves of CASO:Pr
3+ and Ag/CASO:Pr
3+ can be evaluated by fitting a double-exponential equation to assess its decay trend. The equation is as follows:
34,52I(t) = I0 + A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ1) + A2exp(−t/τ2) exp(−t/τ1) + A2exp(−t/τ2) |
I0 is the long afterglow luminescence intensity at a specific time after turning off the excitation source,
t is the time,
τ1 and
τ2 correspond to the rapid and slow decay components, and
A1 and
A2 are the shallow and deep level traps, respectively. The corresponding afterglow decay parameters are listed in Table S2 (ESI
†). The values of
A1/
A2 are 1.72 and 1.77 in CASO:Pr
3+ and Ag/CASO:Pr
3+, respectively. The variability of the amplitude ratio
A1/
A2 indicates a change in the relative contribution of the shallow and deep traps. The Ag/CASO:Pr
3+ stores a large number of photogenerated charge carriers during the light irradiation activation process. After removing the light irradiation, the afterglow is slowly released and drives the Ag/CASO to act as a photocatalyst for photocatalytic degradation reactions. The free electrons in Ag are excited as hot electrons and directly participate in the photocatalytic reaction.
53,54
 |
| | Fig. 3 (a) XPS full spectra of CASO:Pr3+ and Ag/CASO:Pr3+ phosphors. (b) XPS high-resolution spectrum of Ag 3d for pristine Ag/CASO:Pr3+ phosphors. (c) UV-vis absorption spectra of CASO:Pr3+ and Ag/CASO:Pr3+ phosphors, the inset shows the Kubelka–Munk plots with the extrapolation of bandgaps for CASO:Pr3+ and Ag/CASO:Pr3+ phosphors. (d) Emission spectra of CASO:Pr3+ and Ag/CASO:Pr3+ phosphors under 254 nm excitation. (e) TL spectra of the CASO:Pr3+ and Ag/CASO:Pr3+ phosphors in the dark. (f) Long afterglow luminescence decay curves of CASO:Pr3+ and Ag/CASO:Pr3+ monitored at 254 nm. | |
3.2 Photocatalytic degradation
The photocatalytic performance of the Ag/CASO:Pr3+ catalyst was evaluated using RhB, TC and NFX as models. Fig. S6 (ESI†) displays the RhB concentration versus time in the photocatalytic system without catalyst addition and UV irradiation. RhB degradation is negligible without the catalyst addition or UV irradiation. As shown in Fig. 4a, CASO:Pr3+ and Ag/CASO:Pr3+ demonstrate the capacity to degrade RhB after UV light irradiation. The RhB degradation rates of CASO:Pr3+ and Ag/CASO:Pr3+ are 14.3%, 20.4%, 27.5%, 31.9%, 34.7%, 40.4% and 31.2%, respectively. The photocatalytic activity of the CASO:Pr3+ and Ag/CASO:Pr3+ photocatalysts are quantitatively investigated using the pseudo-first-order kinetic model from the following equation:55,56
k is the pseudo-first-order rate constant (min−1), and C0 and C are the initial concentration of the pollutant and the concentration of RhB at the time t, respectively. The pseudo-first-order equation was applied in fitting the apparent rate constant of RhB. The relative results values of k for CASO:Pr3+ and Ag/CASO:Pr3+ are 0.0006 min−1, 0.0035 min−1, 0.0049 min−1, 0.0059 min−1, 0.0065 min−1, 0.0078 min−1 and 0.0058 min−1, respectively (Fig. 4b and c). The Ag/CASO:Pr3+ exhibits the most efficient photocatalytic activity for RhB degradation. The RhB degradation efficiency of the Ag/CASO:Pr3+ is 3.25 times higher than that of the CASO:Pr3+. The enhancement of photocatalytic activity can be attributed to the increased active sites provided by Ag loading and the promotion of photogenerated carrier separation via Ag loading. In addition, the hot electrons generated by Ag loading can be directly involved in the catalytic reaction. This leads to more electrons combining with oxygen to form superoxide radicals and accumulated holes reacting with water to form hydroxyl radicals.57 The results in a higher degradation rate in the Ag/CASO:Pr3+ catalyzed system than in the CASO:Pr3+ system.
 |
| | Fig. 4 (a) Photocatalytic performance of RhB degradation by CASO:Pr3+ and Ag/CASO:Pr3+ phosphors under irradiation with a 254 nm UV lamp. (b) Corresponding pseudo-first-order kinetics of CASO:Pr3+ and Ag/CASO:Pr3+ phosphors. (c) Degradation rates of CASO:Pr3+ and Ag/CASO:Pr3+ phosphors. | |
The photocatalytic degradation performance of Ag/CASO:Pr3+ still has room to improve. It may be attributed to the competitive effects between photocatalysis and long afterglow on photogenerated carriers. The photogenerated carrier recombination in the afterglow leads to a decrease in the photocatalytic efficiency of Ag/CASO:Pr3+. The key is to maximize the photocatalytic degradation efficiency within the limited afterglow duration. The application of the photocatalytic Fenton reaction offers the potential to further raise the degradation efficiency under UV afterglow conditions. The photo-Fenton degradation system consisting of FeCl3, H2O2, and Ag/CASO:Pr3+ is constructed.24,58,59 The CASO:Pr3+ and Ag/CASO:Pr3+ degradation efficiencies for RhB in the Fenton system are 29.5%, 59.4%, 62.4%, 64.0%, 65.2%, 82.1% and 79.6%, respectively (Fig. 5a). It demonstrates that the degradation efficiency of Ag/CASO:Pr3+ for RhB is significantly enhanced in the photo-Fenton system. Fig. S7 (ESI†) shows the comparison of the photodegradation performance of Ag/CASO:Pr3+ with other existing photocatalysts.11,16,60–66 Ag/CASO:Pr3+ exhibits superior performance over time compared to other nanoprecious metal-loaded long afterglow photocatalysts. Fig. 5b illustrates that the degradation of RhB follows a pseudo-first-order kinetic model.67 The reaction rate coefficients are shown in Fig. 5c. The k values for CASO:Pr3+ and Ag/CASO:Pr3+ are 0.0057 min−1, 0.0132 min−1, 0.0146 min−1, 0.0155 min−1, 0.0149 min−1, 0.0273 min−1 and 0.0256 min−1, respectively. The degradation rates of Ag/CASO:Pr3+ for RhB are 2.3, 2.6, 15.8, 2.7, 2.6, 4.8 and 4.8 times that of CASO:Pr3+, respectively. The hot electrons generated by the Ag loading can promote the reduction of Fe3+ to Fe2+ and accelerate the conversion of Fe3+/Fe2+ in the Fenton system. The Fenton reaction produces ˙OH and ˙O2−. It increases the number of reactive radicals and improves the photocatalytic activity.68 Due to the energy level matching between Ag nanoparticles and CASO:Pr3+, the electrons accumulate on Ag nanoparticles to form a Schottky junction. It inhibits the complexation of photogenerated carriers. Consumption of electrons is also required during the afterglow luminescence, thus avoiding the probability of forming a new complex center on Ag.69 It maximizes the photocatalytic efficiency. Fig. 5d displays the cyclic experiments of photocatalytic RhB degradation of the Ag/CASO:Pr3+-5 phosphor. The photo-Fenton degradation efficiency of Ag/CASO:Pr3+ remained above 80% after 5 cycles. It indicates that the Ag/CASO:Pr3+ catalyst has photostability. The degradation curves and primary kinetic parameters of Ag/CASO:Pr3+ on TC and NFX are shown in Fig. S8 and S9 (ESI†). The direct degradation of TC and NFX occurs with maximum efficiencies of 15% and 20%, respectively. The maximum efficiencies for degradation of TC and NFX in the Fenton environments are 55% and 60%, respectively. As shown in Table S3 (ESI†), the TC and NFX degradation constants k were fitted using the pseudo-first-order equations. It further demonstrates that the introduction of the Fenton environment improves the pollutant reduction efficiency.
 |
| | Fig. 5 (a) Photo-Fenton degradation performance for RhB degradation by CASO:Pr3+ and Ag/CASO:Pr3+ phosphors. (b) Corresponding pseudo-first-order kinetics of CASO:Pr3+ and Ag/CASO:Pr3+ phosphors in the photo-Fenton system. (c) Degradation rates of CASO:Pr3+ and Ag/CASO:Pr3+ phosphors in the photo-Fenton system. (d) Cyclic degradation kinetic curves of RhB. | |
The reaction formula for the auto-luminescence of Ag/CASO:Pr3+ for the photocatalytic decomposition of RhB is (1)–(3). The Ag/CASO:Pr3+ produces electrons (e−) and holes (h+) under the excitation of UV light (1). Ag/CASO:Pr3+ has a more negative CB potential (−0.71 eV vs. NHE) than the standard potential E0 (O2/˙O2−) (−0.46 eV vs. NHE). It indicates that the photogenerated electrons can react with O2 to produce superoxide radicals (˙O2−) (2).70 The valence band potential of Ag/CASO:Pr3+ (+4.41 eV vs. NHE) is more positive than the standard redox potential E0 (˙OH/H2O) (+2.7 eV vs. NHE). It demonstrates that photogenerated holes can directly oxidize H2O to produce ˙OH (3).71
| | |
Ag/CASO:Pr3+ + hv → h+ + e−
| (1) |
The enhanced degradation efficiency of RhB is summarized in eqn (4) and (5) under the Fenton conditions. Fe3+ combines with e− to produce Fe2+ in the Fenton system. The Fe2+ reaction with H2O2 releases ˙OH and Fe3+ to maintain the cyclic reaction.72,73
| | |
Fe2+ + H2O2 → Fe3+ + ˙OH + OH−
| (5) |
The N2 adsorption–desorption isotherms revealed the specific surface area and pore size distribution of the CASO:Pr3+ and Ag/CASO:Pr3+ phosphors (Fig. S10, ESI†). The BET-specific surface area of Ag/CASO:Pr3+ (2.0092 m2 g−1) is much higher than that of CASO:Pr3+ (0.5999 m2 g−1). The total pore volume of Ag/CASO:Pr3+ (14.6252 nm) is also higher than that of CASO:Pr3+ (12.1879 nm). The larger specific surface area and total pore volume demonstrate that the Ag/CASO:Pr3+ has more active sites for the photocatalytic reaction.74
In order to further verify the above speculations, the ESR spectra (DMPO–˙OH) of CASO:Pr3+ and Ag/CASO:Pr3+ were recorded.75,76 The results are shown in Fig. S11 (ESI†). No ˙OH signal was detected in either CASO:Pr3+ or Ag/CASO:Pr3+ under dark conditions, indicating that the catalytic system does not perform the photocatalytic degradation reaction in the absence of light. The ˙OH signals appeared in both CASO:Pr3+ and Ag/CASO:Pr3+ after 5 min of UV irradiation. The signal intensity in Ag/CASO:Pr3+ is higher than that in CASO:Pr3+. The ˙OH signal from Ag/CASO:Pr3+ remained stronger than that from CASO:Pr3+ even after the irradiation was removed. It suggests that the Ag/CASO:Pr3+ catalytic system contains a higher concentration of ˙OH. The ˙OH signals persisted for 5 min after the irradiation was removed for 5 min. It confirms that the afterglow photocatalyst can continuously generate ˙OH for degrading pollutants even in the absence of light. The above results not only demonstrate that Ag loading enhances the concentration of ˙OH but also confirm that catalytic degradation could continue in the dark for both CASO:Pr3+ and Ag/CASO:Pr3+.
3.3 Afterglow generation and photodegradation mechanisms
The increased photocatalytic efficiency can be attributed to the increased concentration of reactive species in the reacting system. The active species quenching experiments were performed to further investigate the reaction mechanism in the Ag/CASO:Pr3+ Fenton system. Active species, including the ˙O2−, ˙OH, and h+, were scavenged by p-benzoquinone (BQ), isopropanol (IPA) and ethylenediamine tetraacetic acid (EDTA), respectively.70,77 Fig. 6a displays that the RhB degradation efficiency of Ag/CASO:Pr3+ with the addition of the scavengers. The results indicate that the addition of the scavengers reduced the degradation efficiency. Among them, the addition of BQ produced the highest inhibition of RhB degradation, and the addition of EDTA exhibited the slightest inhibitory effect on RhB degradation. Fig. 6b shows that the degradation efficiency of RhB reached 82% within 1 h without the addition of capturing agents. The RhB degradation efficiency changed to 66%, 54% and 32% after the addition of BQ, EDTA and IPA, respectively. These results demonstrate that ˙OH is the key radical responsible for the degradation of RhB. The mechanism of the Ag resonance-enhanced afterglow luminescence of Ag/CASO:Pr3+ is illustrated in Fig. 6c. The ground-state electrons are excited to the 4f 5d level under UV excitation (process 1). The excited electrons are more easily trapped in the deep and shallow level traps of Ag/CASO:Pr3+ due to the formation of an electrostatic field (processes 2 and 4). The Ag/CASO:Pr3+ exhibits long afterglow luminescence after removing the UV light. Some of the trapped electrons thermally escape from the deep and shallow level traps to the conduction band at room temperature and subsequently recombine with the excited states of Pr3+ (processes 3 and 5). The recombination of electrons and holes in Pr3+ leads to the UV afterglow luminescence (process 6). Fig. 6d illustrates the photocatalytic degradation mechanism of Ag/CASO:Pr3+ under dark conditions. Ag can act as an electron trap, capturing electrons from Ag/CASO:Pr3+. The SPR effect of Ag enhances the light absorption ability.15,78 The generation of photogenerated electrons and holes is enhanced on the Ag/CASO:Pr3+ surface. This phenomenon increases the number of electrons and holes involved in the photocatalytic reaction during the catalytic process (eqn (1), (2) and (3)). It increases the photocatalytic degradation capability, improves the number of photogenerated carriers and effectively inhibits their recombination. The introduction of Fe3+ and H2O2 forms the Fenton system. It increases the concentration of reactive species and improves the photocatalytic degradation efficiency (eqn (4) and (5)). Ag/CASO:Pr3+ is able to degrade RhB efficiently in the dark environment under the synergistic effect of Ag and the photo-Fenton system. The surface plasmon excitations of the Ag nanoparticles capture energy from the light field via multiple relaxation pathways as the frequency of the irradiation light matches the vibrational frequency of Ag.79 It significantly enhances the ability to absorb light and improves the photocatalytic reaction efficiency. The high-energy thermal electrons on the Ag surface can be directly transferred to the CB of the photocatalyst and promote the separation and transfer of photogenerated carriers. The CB electrons can capture O2 and reduce it to ˙O2−. The formation of the Schottky junction by Ag and CASO:Pr3+ inhibits the complexation of photogenerated carriers.80 A large number of ˙OH are generated by the cycling process of Fe3+/Fe2+ in the Fenton reaction. The synergistic effect of the enhanced light-absorbing capacity of Ag, hot electrons that facilitate the separation of photogenerated carriers, Schottky junction and increased concentration of active species during the Fenton process resulted in a significant increase in the degradation efficiency of the Ag/CASO:Pr3+ catalyst system.
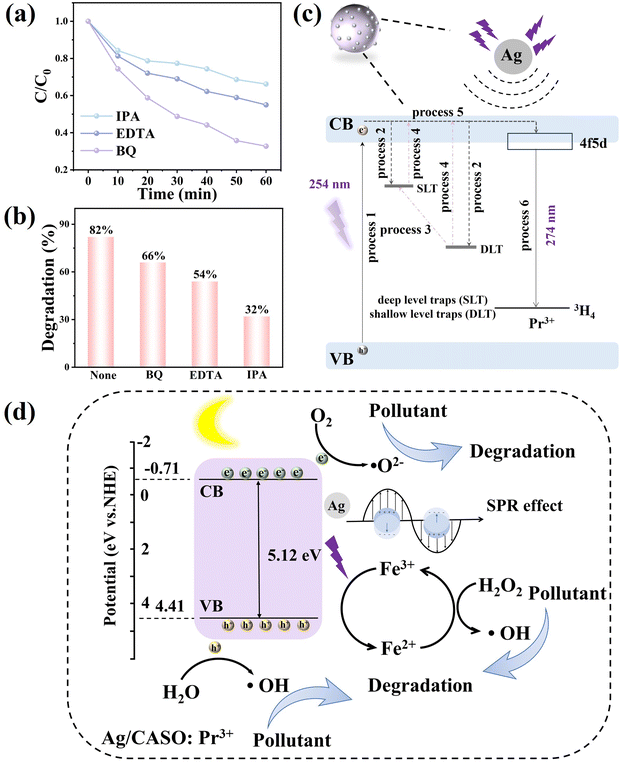 |
| | Fig. 6 (a) Effect of various scavengers (EDTA, IPA, BQ) on the degradation of RhB for Ag/CASO:Pr3+. (b) Degradation efficiency of RhB after adding the scavengers. (c) The mechanism diagram of Ag enhancing the long afterglow performance of Ag/CASO:Pr3+. (d) The possible photocatalytic mechanisms for the degradation of pollutant by Ag/CASO:Pr3+. | |
3.4 Electrochemical properties
Fig. 7a shows the carrier release process over a period of 1 h in the dark. The afterglow photocurrent of CASO:Pr3+ decayed from 3824.36 nA to 39.86 nA. The afterglow photocurrent of Ag/CASO:Pr3+ decayed from 820.65 nA to 65.52 nA. It indicates that CASO:Pr3+ and Ag/CASO:Pr3+ can continuously release charge carriers in the dark, thereby maintaining the progress of the photocatalytic reaction. The steady-state photocurrent response of CASO:Pr3+ and Ag/CASO:Pr3+ is shown in Fig. 7b. The photocurrent intensity of Ag/CASO:Pr3+ (161.44 nA) increased by 9.53 times compared to that of CASO:Pr3+ (16.94 nA). It demonstrates that the recombination of Ag increases the trap density in Ag/CASO:Pr3+. It leads to the detection of more photogenerated carrier signals. Fig. 7c displays the EIS spectra of CASO:Pr3+ and Ag/CASO:Pr3+, and the Ag/CASO:Pr3+ exhibits lower EIS than CASO:Pr3+. It indicates that Ag/CASO:Pr3+ is more conducive to the progress of photocatalytic reactions. The Mott–Schottky plots of CASO:Pr3+ and Ag/CASO:Pr3+ are displayed in Fig. 7d. The positive slope of the CASO:Pr3+ and Ag/CASO:Pr3+ curves confirm that both materials exhibit an n-type semiconductor behavior. The flat-band potential of an n-type semiconductor is approximately its ECB; hence, the ECB of Ag/CASO:Pr3+ is approximately −0.61 V (vs. Ag/AgCl). The flat band potential (Efb) values for n-type semiconductors can be converted to standard H2 electrode (NHE) potentials using the following equation:33,81
| Efb (vs. NHE) = Efb (vs. Ag/AgCl) + EAg/AgCl |
EAg/AgCl is the energy of Ag/AgCl (about 0.197 V). The Efb (vs. Ag/AgCl) of Ag/CASO:Pr3+ is −0.61 V. The Efb (vs. NHE) of Ag/CASO:Pr3+ is calculated to be −0.41 V. The Efb (vs. NHE) is 0.3 V higher than the ECB potential. Therefore, the ECB of Ag/CASO:Pr3+ is −0.71 V. The energy band structure diagram of Ag/CASO:Pr3+ is given in Fig. S12 (ESI†). Ag loading introduces new energy levels and enhances the generation of hot electrons. When Ag/CASO:Pr3+ is excited, electrons transition from the VB to the CB, generating hot electrons on the Ag surface. The defect sites in the catalyst trap a large number of electrons and release them back into the conduction band after the excitation is stopped. It ensures that the reaction continues. Hot electrons have a higher energy level than ordinary electrons. It helps separate and transfer photogenerated electron–hole pairs within Ag/CASO:Pr3+. They can be transferred directly to the CB and participate in the photocatalytic reaction. The efficient separation and transfer of photogenerated carriers within Ag/CASO:Pr3+ significantly enhance the photocatalytic performance.
 |
| | Fig. 7 (a) Afterglow photocurrent of CASO:Pr3+ and Ag/CASO:Pr3+. (b) Steady-state photocurrent response of CASO:Pr3+ and Ag/CASO:Pr3+. (c) Nyquist diagram of CASO:Pr3+ and Ag/CASO:Pr3+. (d) Mott–Schottky plots of CASO:Pr3+ and Ag/CASO:Pr3+. | |
4. Conclusion
A CASO:Pr3+ long afterglow phosphor was successfully prepared via a high-temperature solid-state method. Ag loading onto CASO:Pr3+ was achieved using the photoreduction deposition method. Both CASO:Pr3+ and Ag/CASO:Pr3+ enabled the photocatalytic degradation of RhB, TC and NFX under their own long-afterglow-driven processes. Ag/CASO:Pr3+ exhibits superior performance to CASO:Pr3+ in terms of the afterglow intensity and photocatalytic degradation performance. It is attributed to the SPR effect from Ag loading, increasing the light absorption capability. The afterglow duration was extended and the afterglow intensity was improved by the storage and release of hot electrons. The Schottky junction between Ag and CASO:Pr3+ inhibits the recombination of photogenerated electrons and holes and increases the active sites in photocatalytic reactions. The Fenton reaction cycle was sustained by a variety of diverse active species, including ˙OH, h+ and ˙O2−. The synergistic effect of these factors promotes the photocatalytic performance boost of Ag/CASO:Pr3+. Ag/CASO:Pr3+ can achieve a 55% removal rate of TC and a 60% removal rate of NFX under its own afterglow-driven process within 1 h after 10 min of UV light irradiation. This work opens a novel perspective for developing all-weather photocatalytic technology without continuous light irradiation.
Author contributions
Xiaoxuan Fan: software, data Curation and writing – original draft. Yonggui Zheng: validation and visualization. Zimin Yao: visualization. Pingping Liang: methodology. Xuemei Lu: methodology and resources. Jianbo Xu: visualization. Baijie Guan: visualization. Tianya Tan: visualization. Shuo Cao: visualization and project administration. Yang Zhao: visualization. Kexin Wang: methodology, formal analysis, resources, supervision and writing, review and editing. Feifei Yin: resources and supervision. Jiwei Wang: conceptualization, project administration and funding acquisition.
Data availability
All relevant data are within the manuscript and its additional files.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 12374181 and 52403324), the Fundamental Research Funds for Public Universities in Liaoning (LJ212410140035, LJ212410140037 and LJ212410140048), the Shenyang Science and Technology Bureau (22-315-6-06), the General Project of the Department of Education of Liaoning Province (JYTMS20230777), Science and Technology Talents Project of Shenyang – U35 Outstanding Young Talent (Grant No. RC230348), the Youth Research Fund in Liaoning University (LDYBJC2401), Fund of Liaoning Provincial for Excellent Young Scholars (Grant No. 2024JH3/10200045) and the Liaoning Province Science and Technology Plan Joint Program (Natural Science Foundation General Project, 2024-MSLH-188).
References
- B. Al-Ghafri, T. Bora, P. Sathe, S. Dobrestov and M. Al-Abri, Appl. Catal., B, 2018, 233, 136–142 CrossRef CAS.
- W. Xiang, J. Yuan, Y. Wu, H. Luo, C. Xiao, N. Zhong, M. Zhao, D. Zhong and Y. He, Chin. Chem. Lett., 2022, 33, 3632–3640 Search PubMed.
- J. Liu, Q. Zhang, X. Tian, Y. Hong, Y. Nie, N. Su, G. Jin, Z. Zhai and C. Fu, Chem. Eng. J., 2021, 404, 127146 CrossRef CAS.
- S. Silvestri, A. R. Fajardo and B. A. Iglesias, Environ. Chem. Lett., 2021, 20, 731–771 Search PubMed.
- S. Dong, Y. Gong, Z. Zeng, S. Chen, J. Ye, Z. Wang and D. D. Dionysiou, Water Res., 2023, 242, 120297 Search PubMed.
- D. Chen, Y. Cheng, N. Zhou, P. Chen, Y. Wang, K. Li, S. Huo, P. Cheng, P. Peng, R. Zhang, L. Wang, H. Liu, Y. Liu and R. Ruan, J. Cleaner Prod., 2020, 268, 121725 CrossRef CAS.
- N. A. Mohd Razali, W. N. Wan Salleh, F. Aziz, L. W. Jye, N. Yusof and A. F. Ismail, J. Cleaner Prod., 2021, 309, 127438 CrossRef CAS.
- Y. Zheng, Y. Liu, X. Guo, Z. Chen, W. Zhang, Y. Wang, X. Tang, Y. Zhang and Y. Zhao, J. Mater. Sci. Technol., 2020, 41, 117–126 CrossRef CAS.
- R. Tai, S. Gao, Y. Tang, X. Ma, P. Ding, R. Wu, P. Li, X. Song, S. Chen and Q. Wang, Small, 2024, 20, 2310785 Search PubMed.
- Y. Xiao, B. Luo, B. Cheng, Q. Huang, Y. Ye, L. Fang, L. Zhou and S. Lei, Dalton Trans., 2018, 47, 7941–7948 Search PubMed.
- V. Havasi, B. Vödrédi and Á. Kukovecz, Catal. Today, 2017, 284, 107–113 CrossRef CAS.
- S.-S. Li, M. Liu, L. Wen, Z. Xu, Y.-H. Cheng and M.-L. Chen, Environ. Sci. Pollut. Res., 2022, 30, 322–336 CrossRef PubMed.
- W. Zhang, Y. Niu, H. Xia, Z. Wu, L. Song, X. Li and L. Cui, Sep. Purif. Technol., 2025, 359, 130752 Search PubMed.
- X. Yang, B. Tang, X. Cao, Y. Ding and M. Huang, J. Photochem. Photobiol., A, 2021, 411, 113302 Search PubMed.
- O. Hai, M. Pei, E. Yang, Q. Ren, X. Wu, J. Zhu, Y. Zhao and L. Du, J. Alloys Compd., 2021, 866, 158752 CrossRef CAS.
- H. Li, S. Yin, Y. Wang, T. Sekino, S. W. Lee and T. Sato, J. Mater. Chem. A, 2013, 1, 1123–1126 RSC.
- C. R. García, L. A. Diaz-Torres, P. Salas, M. Guzman and C. Angeles-Chavez, Mater. Sci.
Semicond. Process., 2015, 37, 105–111 CrossRef.
- Y. Ding, Y. Ye, C. Wang, L. Pei, Q. Mao, M. Liu, R. Zheng, A. Bokhari, N. Han and J. Zhong, J. Cleaner Prod., 2024, 450, 142041 CrossRef CAS.
- A. A. Alothman, N. Ahmad, M. D. Albaqami, Z. A. Alothman, K. N. Alqahtani, M. Rizwan Khan and V. V, Ceram. Int., 2024, 50, 15867–15878 CrossRef CAS.
- Z. Zhang, Z. Bai, S. Yu, X. Meng and S. Xiao, Chem. Eng. Sci., 2024, 288, 119789 CrossRef CAS.
- J. Lin, Y. Deng, X. Yu, J. Yang, W. Zhu, S. Xie and B. Yang, J. Alloys Compd., 2024, 1002, 174863 CrossRef CAS.
- Y. Xiangwei, M. Graells, S. Miralles-Cuevas, A. Cabrera-Reina and M. Pérez-Moya, J. Environ. Chem. Eng., 2021, 9, 105235 CrossRef CAS.
- I. Amildon Ricardo, C. E. S. Paniagua, V. A. B. Paiva, B. R. Gonçalves, R. M. F. Sousa, A. E. H. Machado and A. G. Trovó, Chem. Eng. J., 2018, 339, 531–538 Search PubMed.
- B. Wei, C. Wang, Y. He, G. Ran and Q. Song, Compos. Commun., 2021, 24, 100652 CrossRef.
- J. Hu, J. Li, J. Cui, W. An, L. Liu, Y. Liang and W. Cui, J. Hazard. Mater., 2020, 384, 121399 CrossRef CAS PubMed.
- X. Wang, J. Li, X. Li, Y. Du, X. Li, J. Qian, Q. Wang, Y. Cai and L. Zhang, Appl. Surf. Sci., 2024, 678, 161092 CrossRef CAS.
- X. He, S. Wen, X. Zhang and Y. Yang, Sep. Purif. Technol., 2025, 357, 130181 Search PubMed.
- K. Sun, H. Yuan, Y. Yan, H. Qin, L. Sun, L. Tan, F. Guo, X. Du and W. Shi, J. Water Process Eng., 2024, 58, 104803 Search PubMed.
- C. Chen, S. Wang, F. Han, X. Zhou and B. Li, Sep. Purif. Technol., 2025, 353, 128380 Search PubMed.
- Y. Zhang, Y. Liu, C. Wang, L. Yang, N. Wu and R. Chen, Chem. Eng. Sci., 2024, 289, 119874 CrossRef CAS.
- Q. Wu, H. Yang, L. Kang, Z. Gao and F. Ren, Appl. Catal., B, 2020, 263, 118282 CrossRef CAS.
- W. Zhao, Z. Wei, C. Li and M. Ding, Ceram. Int., 2024, 50, 24063–24069 CrossRef CAS.
- J. Tang, Y. Liu, X. Zhang, Y. Tian, T. Jin, D. Fang and J. Wang, J. Power Sources, 2023, 580, 233433 CrossRef CAS.
- O. Hai, M. Pei, Q. Ren, X. Wu, E. Yang, D. Xu and J. Zhu, Dalton Trans., 2022, 51, 2287–2295 Search PubMed.
- J. Ni, D. Liu, W. Wang, A. Wang, J. Jia, J. Tian and Z. Xing, Chem. Eng. J., 2021, 419, 129969 Search PubMed.
- A. Antuzevics, G. Krieke, G. Doke, P. Rodionovs, D. Nilova, J. Cirulis, A. Fedotovs and U. Rogulis, J. Phys. Chem. C, 2024, 128, 21846–21854 CrossRef CAS.
- X. Fan, L. Xu, W. Liu, F. Yin, J. Xu, S. Cao, Y. Ding, X. Hong, T. Tan, K. Wang and J. Wang, Ceram. Int., 2024, 50, 32583–32590 CrossRef CAS.
- D. Nilova, A. Antuzevics, G. Krieke, G. Doke, I. Pudza and A. Kuzmin, J. Lumin., 2023, 263, 120105 CrossRef CAS.
- X. Zhang, H. Zou, C. Xu, Z. An, R. Dong, K. Zheng, Y. Sheng and Y. Song, Opt. Mater., 2019, 89, 512–520 Search PubMed.
- T. Richhariya, N. Brahme, D. P. Bisen, T. Badapanda, K. Tiwari and E. Chandrawanshi, Mater. Chem. Phys., 2022, 287, 126237 Search PubMed.
- K. Park, H. Kim and D. A. Hakeem, Dyes Pigm., 2017, 136, 70–77 CrossRef CAS.
- Z. Song, K. Song, B. Liu, P. Zheng, H. Barzegar Bafrooei, W. Su, H. Lin, F. Shi, D. Wang and I. M. Reaney, Int. J. Appl. Ceram. Technol., 2019, 17, 771–777 Search PubMed.
- S. Zhang, P. Wang, Z. Xu, M. Gong, J. Cao, J. Shen, X. Fang, X. Xu, J. Xu and X. Wang, J. Catal., 2025, 442, 115893 Search PubMed.
- M. Mokhtar Mohamed, G. Osman and K. S. Khairou, J. Environ. Chem. Eng., 2015, 3, 1847–1859 CrossRef CAS.
- N.-N. Zhang, X.-X. Jiang, Y.-N. Wang, X.-R. Pan, Y.-Y. Zhang, B. Liu, Y.-G. Yang and X.-P. Wang, J. Alloys Compd., 2023, 932, 167626 CrossRef CAS.
- L. Li, P. Yang, W. Xia, Y. Wang, F. Ling, Z. Cao, S. Jiang, G. Xiang, X. Zhou and Y. Wang, Ceram. Int., 2021, 47, 769–775 CrossRef CAS.
- S. Wei, Q. Heng, Y. Wu, W. Chen, X. Li and W. Shangguan, Appl. Surf. Sci., 2021, 563, 150273 CrossRef CAS.
- H. Liu, X. Liu, W. Yang, M. Shen, S. Geng, C. Yu, B. Shen and Y. Yu, J. Mater. Chem. A, 2019, 7, 2022–2026 Search PubMed.
- A. Balakrishna, V. Kumar, A. Kumar and O. M. Ntwaeaborwa, J. Alloys Compd., 2016, 686, 533–539 CrossRef CAS.
- X. Lv, X. Shan, Y. Zhang and Y. Liang, J. Mater. Chem. C, 2023, 11, 4492–4499 RSC.
- X. Wang, Y. Chen, F. Liu and Z. Pan, Nat. Commun., 2020, 11, 2040 CrossRef CAS PubMed.
- L. Xiao, J. Zhou, G. Liu and L. Wang, J. Alloys Compd., 2017, 712, 24–29 Search PubMed.
- T. Deka, A. Das, S. John and R. G. Nair, Int. J. Hydrogen Energy, 2024, 72, 1169–1183 CrossRef CAS.
- Z. Dehghani and S. R. Hormozi Jangi, Fuel, 2025, 385, 134139 CrossRef CAS.
- D. S. Vavilapalli, L. Qin, J. Palisaitis and J. Rosen, npj Clean Water, 2024, 7, 41 Search PubMed.
- J. Qian, Y. Xue, Y. Ao, P. Wang and C. Wang, Chin. J. Catal., 2018, 39, 682–692 CrossRef CAS.
- M. F. Elshahawy, N. A. Ahmed, Y. H. Gad and A. E.-H. Ali, Int. J. Biol. Macromol., 2024, 262, 129946 Search PubMed.
- W. Zhao, Z. Wei, C. Li and M. Ding, Mater. Res. Bull., 2024, 169, 112508 CrossRef CAS.
- J. Li, Y. Ding, K. Chen, Z. Li, H. Yang, S. Yue, Y. Tang and Q. Wang, J. Alloys Compd., 2022, 903, 174863 Search PubMed.
- C. Zhou, P. Zhan, J. Zhao, X. Tang, W. Liu, M. Jin and X. Wang, Ceram. Int., 2020, 46, 27884–27891 CrossRef CAS.
- F. Li, Z. Li, Y. Cai, M. Zhang, Y. Shen and W. Wang, Mater. Lett., 2017, 208, 111–114 Search PubMed.
- X. Liu, X. Chen, Y. Li, B. Wu, X. Luo, S. Ouyang, S. Luo, A. A. Al Kheraif and J. Lin, J. Mater. Chem. A, 2019, 7, 19173–19186 RSC.
- H. Yin, X. Chen, R. Hou, H. Zhu, S. Li, Y. Huo and H. Li, ACS Appl. Mater. Interfaces, 2015, 7, 20076–20082 CrossRef CAS PubMed.
- H. Wu, Z.-M. Wang, K. Koike, N. Negishi and Y. Jin, Catal. Sci. Technol., 2017, 7, 3736–3746 RSC.
- D. Zhang, H. Liu, C. Su, H. Li and Y. Geng, Sep. Purif. Technol., 2019, 218, 1–7 Search PubMed.
- H. Wu, W. Peng, Z.-M. Wang and K. Koike, RSC Adv., 2016, 6, 37995–38003 Search PubMed.
- X. Yang, Y. Wang, X. Xu, Y. Qu, X. Ding and H. Chen, Chin. J. Catal., 2017, 38, 260–269 CrossRef CAS.
- B. Yang, J. Li, Y. Li, M. Zhang, J. Zhu, T. Zhou and J. Deng, Sep. Purif. Technol., 2022, 299, 121741 CrossRef CAS.
- J. Jin, C. Liu, C. Dai, C. Zeng, Y. Jia and X. Liu, Environ. Res., 2024, 251, 118265 CrossRef PubMed.
- K. G. Motora, C.-M. Wu and S.-T. Lin, J. Water Process Eng., 2023, 52, 103586 CrossRef.
- J. Xin, Y. Liu, L. Niu, F. Zhang, X. Li, C. Shao, X. Li and Y. Liu, Chem. Eng. J., 2022, 445, 136616 CrossRef CAS.
- R. Wang, Y. Chu and H. Zhang, J. Environ. Chem. Eng., 2024, 12, 112479 CrossRef CAS.
- Q. Wu, M. S. Siddique, Y. Yang, M. Wu, L. Kang and H. Yang, J. Cleaner Prod., 2022, 374, 134033 Search PubMed.
- M. R. Espino-Estévez, C. Fernández-Rodríguez, O. M. González-Díaz, J. Araña, J. P. Espinós, J. A. Ortega-Méndez and J. M. Doña-Rodríguez, Chem. Eng. J., 2016, 298, 82–95 Search PubMed.
- P. Wang, L. Wang, R. Xiao, S. Qiu, J. Cao, Y. Fu and Z. Wang, Chem. Eng. J., 2025, 505, 159351 Search PubMed.
- W. Mu, L. Wang, J. Xu and C. Chang, J. Environ. Manage., 2025, 373, 123597 Search PubMed.
- Y. Xu, F. Ge, M. Xie, S. Huang, J. Qian, H. Wang, M. He, H. Xu and H. Li, Catal. Sci. Technol., 2019, 9, 2563–2570 Search PubMed.
- O. Hai, E. Yang, D. Li, W. Bai, Q. Ren and X. Wu, J. Lumin., 2020, 217, 116785 Search PubMed.
- H. Luo, J. Wang, J. Chen and X. Li, J. Environ. Chem. Eng., 2024, 12, 114299 Search PubMed.
- J. Li, Y. Duan, L. Wang and J. Ma, Environ. Res., 2024, 248, 118265 Search PubMed.
- Q. Yang, Y. Wang, Q. Tian, X. Li, A. Pan, M. Zhao, Y. Zhu, T. Wu and G. Fang, J. Mater. Chem. A, 2024, 12, 7207–7214 RSC.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Feifei Yin*a and
Jiwei Wang*a
*a,
Feifei Yin*a and
Jiwei Wang*a