
A dual initiator approach for oxygen tolerant RAFT polymerization†
Nicholas G.
Taylor
,
Marcus H.
Reis
,
Travis P.
Varner
,
Johann L.
Rapp
,
Alexis
Sarabia
and
Frank A.
Leibfarth
 *
*
Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. E-mail: FrankL@email.unc.edu
First published on 8th August 2022
Abstract
Reversible-deactivation radical polymerizations are privileged approaches for the synthesis of functional and hybrid materials. A bottleneck for conducting these processes is the need to maintain oxygen free conditions. Herein we report a broadly applicable approach to “polymerize through” oxygen using the synergistic combination of two radical initiators having different rates of homolysis. The in situ monitoring of the concentrations of oxygen and monomer simultaneously provided insight into the function of the two initiators and enabled the identification of conditions to effectively remove dissolved oxygen and control polymerization under open-to-air conditions. By understanding how the surface area to volume ratio of reaction vessels influence open-to-air polymerizations, well-defined polymers were produced using acrylate, styrenic, and methacrylate monomers, which each represent an expansion of scope for the “polymerizing through” oxygen approach. Demonstration of this method in tubular reactors using continuous flow chemistry provided a more complete structure–reactivity understanding of how reaction headspace influences PTO RAFT polymerizations.
Introduction
The radical polymerization of vinyl monomers represents nearly half of industrially produced polymers.1 Radical polymerization is prized as a method of chain growth polymerization due to its tolerance to a wide variety of functional groups and ability to produce materials with a range of thermomechanical properties.2,3 Such polymerizations are typically run under an inert atmosphere due to the propensity for the propagating radical intermediate to react with triplet oxygen and form peroxy radicals or hydroperoxides.4 This undesired reactivity can inhibit polymerization completely or result in a lowering of the number average molar mass (Mn), and an increase in the dispersity (Đ).5 As such, the need to remove oxygen increases the production cost of material synthesis at an industrial scale.6,7Reversible-deactivation radical polymerizations (RDRPs) are privileged approaches for the synthesis of functional and hybrid materials with a well-defined Mn and low Đ.8,9 RDRP approaches typically exert control over chain-growth kinetics by maintaining a low concentration of propagating radicals in solution, making them particularly sensitive to the presence of dissolved oxygen. This has sparked the development of several innovative methods to make RDRP more oxygen tolerant.5 For example, a variety of different approaches were developed that enabled oxygen tolerant atom transfer radical polymerizations (ATRP).10–14 In contrast, less methods have been explored that enable oxygen tolerant reversible addition fragmentation chain transfer (RAFT) polymerizations. The most widely employed method for oxygen-tolerant RAFT polymerization leverages the reactivity of photocatalysts in their excited state for the conversion of triplet oxygen to singlet oxygen.15 Other strategies to enable oxygen tolerant RAFT polymerizations rely on enzymatic degassing16–24 or photoiniferter systems.25–32 In all these examples, however, specific reaction conditions and/or additives are required to achieve oxygen tolerance. A more general and operationally simple method that enables the synthesis of a wide variety of well-defined polymers under diverse reactions conditions remains an unsolved challenge.
“Polymerizing through” oxygen (PTO) is a nascent approach to achieve oxygen tolerant RAFT polymerizations.33 PTO relies on the generation of a high flux of radicals at the beginning of a polymerization to rapidly convert oxygen into peroxy radicals or hydroperoxides that are inefficient at initiation and have high thermal stability.6 Subsequent radical generation initiates polymerization. Early observations of PTO indicated that oxygen did not significantly retard RAFT polymerizations when using high initiator concentrations.34 Perrier and coworkers intentionally exploited this concept for the preparation of multiblock copolymers by demonstrating that the generation of a high flux of radicals at an elevated temperature in water can simultaneously remove oxygen and provide high-fidelity polymerization of acrylamide monomers (Fig. 1A).35–37 The selection of an azo initiator that decomposes quickly at the reaction temperature (half-life of 77 seconds at 100 °C) and a monomer class with a rapid propagation rate enabled this approach by minimizing inhibition periods typically observed during RAFT polymerization in the presence of oxygen.38 Despite the success of this PTO strategy for high throughput synthesis, this approach has been restricted to the polymerization of acrylamide monomers in water.39,40 Previous work from our lab empirically discovered that oxygen tolerant RAFT polymerizations of acrylic comonomers in organic solvents was possible using two azo initiators with different decomposition temperatures.41 We hypothesized that a two-stage decomposition profile originating from the initiators could provide a more broadly applicable approach to polymerizing through oxygen, where the high concentration of radicals early in the polymerization would deplete dissolved oxygen, and the low and consistent radical generation thereafter would provide initiation of RAFT polymerization.
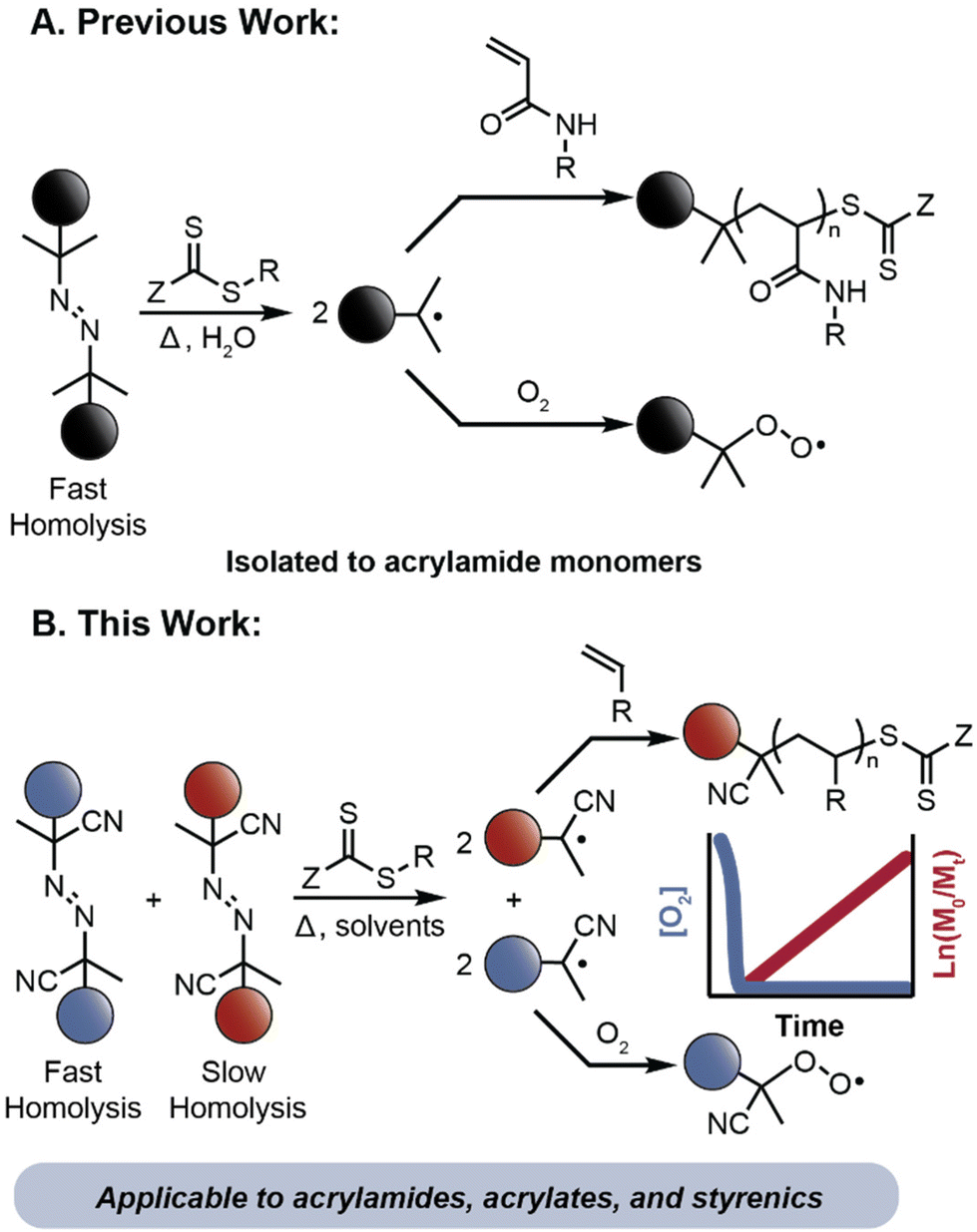 | ||
| Fig. 1 Previous work (A) uses a low decomposition-temperature azo initiator to enable oxygen-tolerant polymerizations of acrylamide monomers in aqueous solution. This work (B) uses two azo initiators with different rates of homolysis to expand the scope of oxygen-tolerant RAFT polymerization to a diverse range of monomers and reaction conditions. | ||
Herein, we describe a dual initiator approach to PTO that enables temporal control over radical flux during a polymerization, where one initiator decomposes rapidly to consume dissolved oxygen and a second initiator generates radicals slowly to initiate controlled RAFT polymerizations (Fig. 1B). The synergistic combination of radical initiators provides controlled polymerization under open-to-air conditions of acrylates, styrenics, and methacrylates, which each represent new monomers classes for use in a PTO RAFT strategy. The ability to simultaneously monitor the kinetics of monomer and oxygen consumption in situ allowed us to identify initiator concentrations that led to controlled polymerizations. Translation of these conditions into a flow reactor demonstrated the feasibility of performing controlled, oxygen tolerant RAFT polymerizations in a rapid, continuous system. This operationally simple approach to PTO RAFT polymerizations will be potentially valuable for both large scale process chemistry and the high throughput synthesis of complex materials on small scale.42–46
Results and discussion
Optimization of initiator ratios for oxygen-tolerant polymerizations
In order to expand the dual initiator approach41 to work for a variety of monomers with different reactivity profiles, a more quantitative understanding of reaction rates governing oxygen depletion and monomer polymerization was required. Azobisisobutyronitrile (AIBN) and 2,2′-azobis(4-methoxy-2.4-dimethyl valeronitrile) (V-70) where chosen as azo initiators with significantly different decomposition profiles, demonstrating 10-hour half-life decomposition temperatures of 65 °C and 30 °C, respectively. These initiators are also both soluble in organic solvents, which we hypothesized would enhance the scope of PTO RAFT beyond reactions conducted in water. To aid in understanding the time evolution of radical concentration in a reaction that includes both AIBN and V-70, a simulation was performed that estimated the radical concentration at 80 °C in a reaction containing either 2 mM V-70, 6 mM AIBN, or a combination of the two (Fig. 2). The initiator concentrations were chosen based on our previous empirical observations, and the homolysis rate constants were estimated assuming Arrhenius behavior. The simulation, which does not consider the effect of radical termination, demonstrated that V-70 homolysis creates a high flux of radicals within the first 5 minutes, whereas AIBN decomposes more slowly. The combination of the two initiators showed an initial high flux of radicals, followed by a steady flux of radicals after approximately 8 minutes. | ||
| Fig. 2 Simulated radical concentrations with respect to time in polymerizations containing 2 mM V-70 (blue dashed line), 6 mM AIBN (black dotted line), and a combination of 6 mM AIBN and 2 mM V-70 (red solid line) at 80 °C. The rate constants for each initiator were estimated using the activation energies and frequency factors supplied by Wako chemical company. These simulations do not include any contributions from radical termination events. | ||
The RAFT polymerization of n-butyl acrylate was used for method development. Acrylic monomers had not previously been optimized for PTO RAFT approaches; therefore, this experiment represents an increase in the scope of this nascent approach for controlled polymerizations. The real time, simultaneous monitoring of both solution oxygen concentration ([O2]) and monomer conversion was particularly enabling. Polymerizations were conducted at 80 °C in a closed triple neck flask equipped with two probes–one enabling the in situ quantification of [O2] and the other helping to determine reaction kinetics using in situ infrared spectroscopy by monitoring the disappearance of the n-butyl acrylate alkene stretch at 1650 cm−1. Dioxane was initially chosen as solvent for its high oxygen solubility compared to other organic solvents.47 We hypothesized that optimizing reaction conditions with dioxane would enable the generalization of the approach to other organic solvents. Initiating the polymerization with 6 mM V-70 as the sole initiator demonstrated oxygen removal after 1.5 minutes and subsequent non-linear kinetics with a plateau in monomer conversion after 15 minutes of polymerization (61% n-butyl acrylate conversion, 96% of V-70 decomposition) (Fig. S1†). In contrast, a separate experiment with 6 mM AIBN as the sole initiator resulted in a 31 minute inhibition period (Fig. S2 and S3†), which underscored the importance of using an initiator that undergoes quick decomposition for reproducible PTO RAFT polymerizations.
As indicated by the simulation, a V-70 concentration of 2 mM rapidly depleted dissolved oxygen in solution experimentally and polymerized n-butyl acrylate to high conversion in a non-degassed reaction vial (Fig. S4†). An inflection point in pseudo first-order behavior was apparent that resulted in a kinetic profile with two distinct linear regimes. The first and steepest regime was observed during the first 10 minutes of polymerization, at which time an estimated 90% of the V-70 and 8% of the AIBN had homolyzed. Polymerization during this initial period, therefore, is largely attributed to polymerization initiated by V-70 after oxygen consumption. A similar but less pronounced two-stage polymerization was observed when decreasing the V-70 concentration to 500 μM (Fig. 3A). Reducing the V-70 concentration further to 300 μM was sufficient for the complete removal of dissolved oxygen after 4.5 minutes (Fig. 3B) and resulted in a polymerization that demonstrated pseudo first-order kinetic behavior. We hypothesize that this concentration of V-70 was sufficient to consume dissolved oxygen, which enabled the radicals generated by the decomposition of AIBN to be predominately responsible for the observed polymerization kinetics. Presumably, radicals formed from AIBN and V-70 homolysis may also contribute to oxygen consumption or monomer initiation, respectively. Decreasing V-70 concentration further (100 μM) led to a significant inhibition period of 21 minutes before observable monomer conversion, likely due to incomplete removal of dissolved oxygen by the V-70 initiator (Fig. 3C).
 | ||
| Fig. 3 Oxygen consumption and monomer conversion of n-butyl acrylate RAFT polymerizations conducted with 6 mM of AIBN and 500 μM V-70 (A), 300 μM V-70 (B), and 100 μM V-70 (C). Conditions: [n-butyl acrylate] = 2 M, [CTA] = 10 mM in dioxane at 80 °C. Radical initiator half-lives estimated from the Arrhenius equation at 80 °C. | ||
Identification of experimental parameters to enable open-to-air polymerizations
With initiator concentrations optimized, the diffusion of oxygen from the headspace into solution was investigated. Extensive work has shown that the diffusion of oxygen into solution is proportional to its interfacial surface area.48–54 For example, oxygen-tolerant RAFT approaches relying on enzymatic degassing have reported the need to match enzymatic activity to the reaction vessel surface area so that the rate of oxygen consumption by the enzyme is faster than oxygen diffusion into solution.17,22 PTO RAFT polymerizations could be viewed analogously, where the generation of new radical species over time could consume oxygen radicals at a faster rate than oxygen diffusion into solution. Accordingly, we hypothesized that optimizing the polymerization surface-area-to-volume ratio (SA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V) would influence the rate of oxygen diffusion and enable controlled polymerizations in vessels open to air.
:V) would influence the rate of oxygen diffusion and enable controlled polymerizations in vessels open to air.
To test this hypothesis, a series of n-butyl acrylate polymerizations were left uncapped under ambient conditions in 2-dram vials with optimized initiator concentrations (6 mM AIBN and 300 μM V-70). This experimental design maintained a constant interfacial surface area while changing the total volume of each polymerization (Fig. 4). The infinite headspace intrinsic to open-to-air polymerizations kept the rate of oxygen diffusion constant during these experiments, while also posing a significant challenge for achieving controlled radical polymerizations. In these experiments, we identified a trend where increasing SA:V (by decreasing solution volume) lowered monomer conversion and polymer molecular weight; the polymerization with the largest SA:V value produced no observable polymer. Conversely, a polymerization with a SA:V value of 0.60 produced low dispersity (<1.30) materials that achieved their target molar mass (Mn). Capping the reaction vessels with an oxygen impermeable cap demonstrated less sensitivity to oxygen, with reaction outcomes that mirrored degassed polymerizations when SA:V < 1.2 (Table S1†). This approach also displayed a high tolerance for stirring, only showing a deviation from expected monomer conversion with stirring speeds that effectively increased interfacial surface area through disrupting the solution-air interface (Table S2†).
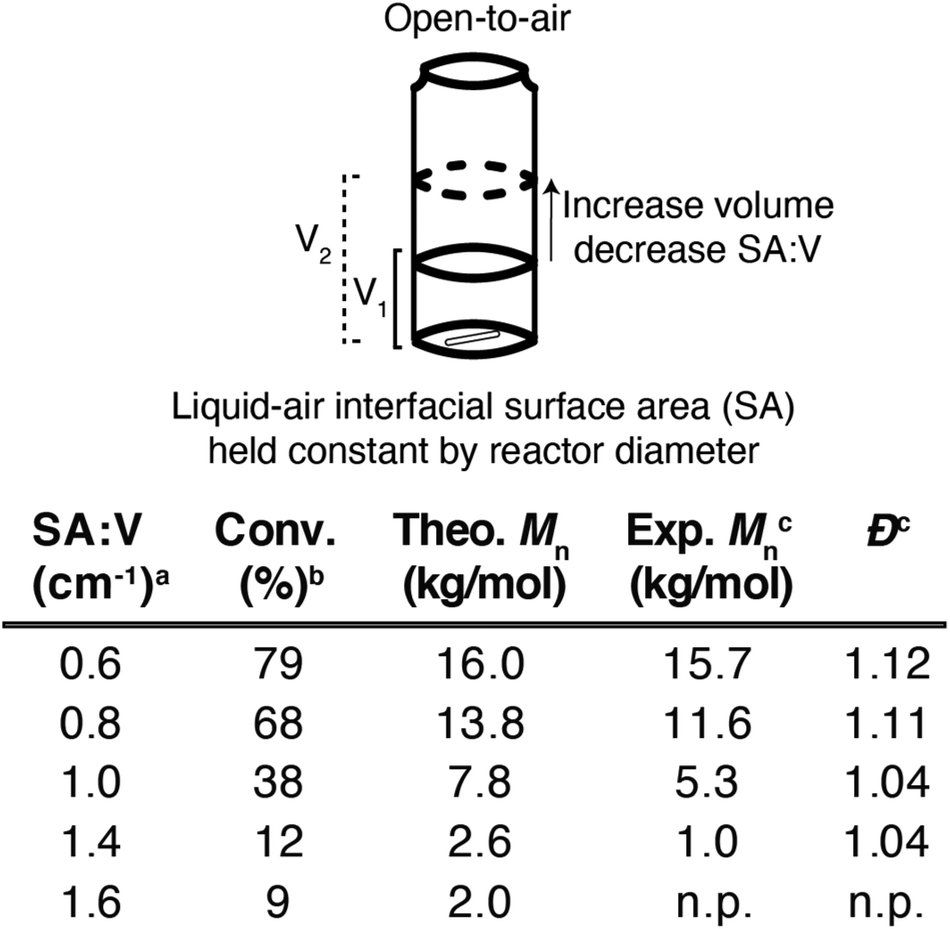 | ||
| Fig. 4 The effect of SA:V on control of thermal RAFT polymerizations. SA:V was systematically altered by changing the volume of the polymerization solution while keeping the surface area constant. Conditions: [n-butyl acrylate] = 2 M, [CTA] = 10 mM, [AIBN] = 6 mM, [V-70] = 300 μM, 80 °C, uncapped 2-dram vial, stirring for 30 minutes. aInterfacial surface area estimated by measuring the base of the vial. bMonomer conversion as determined from 1H NMR using an internal standard. cMolecular weight and dispersity calculated using MALS on a THF GPC. n.p. indicates no observed polymerization. | ||
The importance of the SA:V was further supported by using different sized vials, which intrinsically altered the interfacial surface area of each polymerization. Reactions with a SA:V value of 0.60 produced similar polymers regardless of reactor size (Table S3†), demonstrating the generality of these experimental conditions for oxygen tolerant RAFT polymerizations. Polymers were even synthesized under open-to-air conditions on a larger scale (12 mL, SA:V = 0.60), which demonstrated the feasibility of scaling up open-to-air RAFT polymerizations (Fig. S5†).
The kinetics of open-to-air n-butyl acrylate polymerizations were evaluated at a SA:V value of 0.60. A plot of experimental Mn against monomer conversion with polymerizations targeting a degree of polymerization (DP) of 200 shows a close agreement with target molar masses (Fig. S6 and S7†). The linear increase in molar mass corroborated the initial pseudo first-order kinetic data observed from monomer conversion, further confirming the control over these thermally initiated RAFT polymerizations. Higher molecular weights were achieved by targeting a DP of 400. The molar masses of the resulting polymers deviate from their expected values at high conversion (>60%) and demonstrated dispersities approaching 1.30 (Fig. S8†). An experiment excluding the RAFT CTA demonstrated that the kinetic chain length under these conditions is <100 kg mol−1, which indicates an inherent limitation of achieving high molar mass under these experimental conditions (Table S4†).
Expanding the monomer scope
Expanding the scope of PTO RAFT to acrylates motivated the exploration of this method for other monomers.41 We hypothesized that the use of this dual initiator platform would enable controlled polymerization using different RAFT chain transfer agents in a variety of solvents, thus demonstrating the versatility of this method compared to previous strategies run exclusively in DMSO or water.17–24,55–63 First, polymerizations of tert-butyl acrylate and 2-ethylhexyl acrylate were investigated open-to-air under identical conditions to n-butyl acrylate polymerizations and displayed similar control, producing poly(acrylates) near theoretical Mn with low dispersities (Fig. 5 and Fig. S9†). | ||
| Fig. 5 PTO RAFT polymerizations of different monomer classes under batch conditions. Conditions: [AIBN] = 6 mM, [V-70] = 300 uM, uncapped 2-dram vial, 300 rpm stirring, SA:V value of 0.60. All polymer molecular weights were calculated using multi-angle light scattering. aConversion calculated from 1H NMR relative to an internal standard. b2-(Butylthiocarbonothioylthio) propanoic acid (BTTCP) used as the CTA in a monomer:CTA molar ratio of 200:1 in dioxane at 80 °C and ran for 30 minutes. cCyanomethyl dodecyl trithiocarbonate (CMDT) used as the CTA in a monomer:CTA molar ratio of 100:1 in DMF at 80 °C and ran for 15 minutes. dDibenzyl trithiocarbonate (DBTTC) used as the CTA in a monomer:CTA molar ratio of 300:1 in dichlorobenzene with 6 mM ABCN used in place of AIBN at 100 °C and ran for 2 hours. e2-Cyano-2-propyl benzodithioate (CPDB) used as the CTA in a monomer:CTA molar ratio of 200:1 in toluene and reaction vessel capped at 100 °C and ran for 20 minutes. | ||
The reaction conditions were separately optimized for the other monomer classes (Fig. S10A–E†). For instance, when using dimethylformamide (DMF) as a solvent, this approach was successful in polymerizing the acrylamide monomers N-acryloylmorpholine and N-(isobutoxymethyl)acrylamide with excellent control (Fig. 5 and Fig. S11†). Increasing monomer concentration enabled access to higher molecular weight poly(N-acryloylmorpholine) (DP = 200, Mn = 20.5 kg mol−1, Đ = 1.08) (Fig. S12†).
The effectiveness of a two azo initiator approach was further applied to control the RAFT polymerization of challenging monomers that demonstrate a slower rate of propagation. For the polymerization of styrenics, higher monomer concentrations (3 M), higher temperatures (100 °C), and longer reaction times (2 hours) were required to expedite the synthesis of high molecular weight polymers (Fig. 5 and Fig. S13†). Optimization revealed that switching AIBN to an azo initiator that decomposes at a slower rate, such as 1,1′-azobis(cyclohexanecarbonitrile) (ABCN), was essential for keeping radical flux to a minimum at higher operating temperatures.64 The resulting polystyrene materials closely matched the target molar mass and maintained low dispersities (Đ < 1.2). Copolymerization of styrene with oxygen to form copolymer peroxides has been reported;65–70 however, no peroxide was observed using 1H NMR and 13C NMR analyses under these conditions (Fig. S14 and S15†).66 Styrene polymerizations performed with no exogenous initiators demonstrated the minor role of styrene self-initiation, showing low monomer conversion (9%) and negligible molar mass (Mn = 1500 g mol−1) (Table S5†). Polymerization of tert-butyl styrene using the PTO RAFT strategy demonstrated behavior similar to the analogous degassed polymerization (Fig. S10B†) and considerably improved from the open-to-air polymerizing using a single initiator (Table S7†).
Methacrylate polymerizations were more sensitive to oxygen, and required high temperatures, short reaction times, and capping of the reaction vessel to achieve controlled PTO RAFT polymerization. The polymers produced from this approach achieved molar masses near their targeted value with dispersities below 1.30 (Fig. 5 and Fig. S16†). The dual initiating system is considerably better than either of the two initiators individually, which produced polymer with low molar masses and higher dispersities (Table S5 and S6†). Expanding the exploration to poly(isobornyl methacrylate) also demonstrated molar masses and dispersity values similar to the analogous degassed polymerization (Fig. S10E†).
The chain-end fidelity of different polymer classes made from this oxygen-tolerant approach was explored through chain-extension experiments (Fig. 6). Polymerizations of n-butyl acrylate, N-acryoylmorpholine, and tert-butylstyrene were performed under their optimized, open-to-air reaction to target a DP of approximately 20. The resulting macro-RAFT CTAs of n-butyl acrylate (Mn = 1.7 kg mol−1, Đ = 1.06), N-acryloylmorpholine (2.7 kg mol−1, Đ = 1.11), and tert-butylstyrene (Mn = 1.7 kg mol−1, Đ = 1.07) were then subjected to open-to-air reaction conditions targeting higher DPs. The resulting polymerizations produced higher molecular weight acrylate (DP = 200, Mn = 18.3 kg mol−1, Đ = 1.12), acrylamide (DP = 200, Mn = 31.8 kg mol−1, Đ = 1.16), and styrenic (DP = 300, Mn = 46.3 kg mol−1, Đ = 1.33) materials. In each case, a monomodal GPC chromatogram was observed with complete extension of the macro-RAFT CTA, which indicated a high degree of chain-end fidelity resulting from this dual initiator approach. Furthermore, the apparent low molecular weight tailing following chain extension was consistent with the tailing observed in degassed polymerizations (Fig. S17†).
 | ||
| Fig. 6 GPC chromatograms of polymers made from chain-extension experiments. Macro-RAFT CTAs and chain-extended polymers made from n-butyl acrylate (blue), N-acryloylmorpholine (red), and tert-butylstyrene (black) were synthesized under optimized, open-to-air reaction conditions and purified for analysis. | ||
In a set of control experiments, the two initiators were used individually to assess their performance with a diverse range of monomers in open-to-air polymerizations at a SA:V of 0.60. The use of only V-70 at concentrations of either 1 mM or 6 mM resulted in a decrease in conversion and Mn for all substrates (Table S5†). Notably, styrene polymerizations initiated with V-70 only achieved one-fourth of the molecular weight achieved by the dual initiator approach, despite having very similar monomer conversions. Similarly, methacrylate polymerizations required higher concentrations of V-70 to achieve polymerization, with 6 mM V-70 only achieving 25% conversion and low molecular weight materials. The use of an AIBN concentration of 6 mM as the sole initiator displayed similar kinetics and polymer molecular weights as the dual initiating approach for styrene and N-acryloyl morpholine, but for n-butyl acrylate and cyclohexyl-methacrylate exhibited an approximately 20–30% lower monomer conversion and molar mass (Table S6†).
Translation of the dual initiator approach into continuous flow
Continuous-flow chemistry is a platform technology that has several advantages over batch processes, such as improved heat transfer, mixing, and the potential for scale-up and automation.71 Upon appropriate selection of oxygen-impermeable tubing, polymerizations run in flow provide a SA:V value approaching zero. Therefore, studying PTO RAFT polymerizations in tubular reactors would extend our understanding of how SA:V influences reaction outcome under a broader set of experimental conditions. A series of experiments were performed to directly compare batch n-butyl acrylate polymerizations to analogous polymerizations performed in a continuous flow system (Fig. S18†). A method developed by Moore and coworkers was used to obtain continuous flow polymerization kinetics.72 A polymerization initiated using the dual initiator conditions (6 mM AIBN and 300 μM V-70) under continuous-flow conditions produced well-defined materials (Mn = 12 kg mol−1, Đ = 1.10), with an inhibition period of 2 minutes and 50 seconds (Fig. 7A). This inhibition period was similar to what was observed under batch conditions (SA:V = 0.60). Additionally, the observed pseudo-first order kinetics indicated the successful translation of controlled PTO RAFT polymerizations into continuous flow. A faster rate was observed than the analogous batch reaction, which is likely attributed to the lack of headspace, improved heat transfer and enhanced mixing intrinsic to continuous flow chemistry. Polymerizations initiated with only 6 mM AIBN demonstrated a longer inhibition period of 3 minutes and 30 seconds, producing a material with similar properties (Mn = 14.9 kg mol−1, Đ = 1.08) at a similar rate to the dual initiator approach (Fig. 7B). Lastly, polymerizations initiated with 6 mM V-70 exhibited the shortest inhibition time of 2 minutes but demonstrated a kinetic profile that deviated from first order at moderate conversions (Fig. 7C).
 | ||
| Fig. 7 PTO RAFT polymerizations of n-butyl acrylate run with 6 mM AIBN (A), 6 mM AIBN and 300 μM V-70 (B), and 6 mM V-70 (C). PTO RAFT polymerizations conducted in continuous flow with either 6 mM AIBN (Condition A) or 300 μM V-70 and 6 mM AIBN (Condition B) (D). All polymer molecular weights were calculated using multi-angle light scattering. Conditions: [AIBN] = 6 mM, [V-70] = 300 uM (Condition A) or solely [AIBN] = 6 mM (Condition B). aConversion calculated from 1H NMR relative to an internal standard. b2-(Butylthiocarbonothioylthio)propanoic acid (BTTCP) used as the CTA in a monomer:CTA molar ratio of 200:1 in dioxane at 80 °C and ran for 30 minutes. cCyanomethyl dodecyl trithiocarbonate (CMDT) used as the CTA in a monomer:CTA molar ratio of 100:1 in DMF at 80 °C and ran for 15 minutes. dDibenzyl trithiocarbonate (DBTTC) used as the CTA in a monomer:CTA molar ratio of 300:1 in dichlorobenzene with 6 mM ABCN used in place of AIBN at 100 °C and ran for 2 hours. e2-Cyano-2-propyl benzodithioate (CPDB) used as the CTA in a monomer:CTA molar ratio of 200:1 in toluene at 100 °C and ran for 20 minutes. | ||
The success of the PTO RAFT polymerization of n-butyl acrylate in a continuous flow reactor prompted the exploration of the other monomer classes. Using the same optimized polymerization conditions previously described in Fig. 5, polymerizations of acrylates, acrylamides, styrenics, and methacrylates were conducted in continuous flow. Here, all monomers were polymerized effectively with a single initiator approach (AIBN or ABCN) and the dual initiator approach to produce polymers with similar molar mass and dispersity (Fig. 7D). We attribute the success of the single and dual initiator approaches to the diminished oxygen diffusion present in flow due to a lack of headspace, resulting in shorter inhibition periods for AIBN- or ABCN-initiated polymerizations. In contrast, identical polymerizations run with 6 mM V-70 had slower kinetics with every monomer, despite having a large initial flux of radicals (Table S7†).
The polymerization results in continuous flow illuminated the critical role that reaction head space plays in PTO RAFT polymerizations. We conclude that the dual initiator approach is useful for open-to-air polymerizations with SA:V < 0.60. In contrast, the use of sufficient quantities of a single, high homoylsis temperature initiator is sufficient for continuous flow polymerizations where SA:V is effectively zero, assuming the application can tolerate a slightly longer incubation period. These results in flow reactors provide a more complete understanding of the structure–reactive relationships in PTO RAFT polymerizations.
Conclusions
Kinetic and experimental studies demonstrated that a synergistic combination of two thermally latent radical initiators facilitated the temporal control of radical flux during a polymerization. This dual initiator approach to PTO RAFT enabled the polymerization of a variety of monomers in open-to-air conditions that achieve values of Mn and Đ analogous to polymerization performed under an inert atmosphere. SA:V ratio is an important parameter to consider to achieve controlled RAFT polymerizations conducted under open-to-air conditions, with a SA:V ratio < 0.60 producing well defined polymers. Further, continuous flow polymerizations, where reaction headspace is not a concern, proved to be an operationally simple, scalable, and efficient technology to conduct PTO RAFT polymerization without the need to degas reagent solutions. We envision that the dual initiator strategy for PTO RAFT polymerizations will prove to be a broadly applicable and versatile approach for controlled polymerizations run at small and large scale.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
F. A. L. acknowledges support from the National Institute of General Medical Sciences under award no. R35-GM142666. The UNC Department of Chemistry's NMR Core Laboratory provided expertise and instrumentation that enabled this study with support from National Science Foundation (CHE-1828183 and CHE-0922858).References
- P. Nesvadba, Radical Polymerization in Industry, in Encyclopedia of Radicals in Chemistry, Biology and Materials, John Wiley & Sons, Ltd, Chichester, UK, 2012 Search PubMed.
- R. A. Hutchinson and A. Penlidis, Free-Radical Polymerization: Homogeneous Systems, in Polymer Reaction Engineering, Blackwell Publishing Ltd, Oxford, UK, 2007, pp 118–178 Search PubMed.
- G. Moad and D. Solomon, The Chemistry of Radical Polymerization, Elsevier, 2005 Search PubMed.
- V. A. Bhanu and K. Kishore, Role of Oxygen in Polymerization Reactions, Chem. Rev., 1991, 91(2), 99–117 CrossRef CAS.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Up in the Air: Oxygen Tolerance in Controlled/Living Radical Polymerisation, Chem. Soc. Rev., 2018, 47(12), 4357–4387 RSC.
- S. C. Ligon, B. Husár, H. Wutzel, R. Holman and R. Liska, Strategies to Reduce Oxygen Inhibition in Photoinduced Polymerization, Chem. Rev., 2014, 114(1), 557–589 CrossRef CAS PubMed.
- F. R. Wight, Oxygen Inhibition of Acrylic Photopolymerization, J. Polym. Sci., Polym. Lett. Ed., 1978, 16(3), 121–127 CrossRef CAS.
- C. J. Hawker and K. L. Wooley, The Convergence of Synthetic Organic and Polymer Chemistries, Science, 2005, 309(5738), 1200–1205 CrossRef CAS PubMed.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Reversible-Deactivation Radical Polymerization (Controlled/Living Radical Polymerization): From Discovery to Materials Design and Applications, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- K. Matyjaszewski, H. Dong, W. Jakubowski, J. Pietrasik and A. Kusumo, Grafting from Surfaces for “Everyone”: ARGET ATRP in the Presence of Air, Langmuir, 2007, 23(8), 4528–4531 CrossRef CAS PubMed.
- W. Jakubowski, K. Min and K. Matyjaszewski, Activators Regenerated by Electron Transfer for Atom Transfer Radical Polymerization of Styrene, Macromolecules, 2006, 39(1), 39–45 CrossRef CAS.
- Y. Sun, S. Lathwal, Y. Wang, L. Fu, M. Olszewski, M. Fantin, A. E. Enciso, G. Szczepaniak, S. Das and K. Matyjaszewski, Preparation of Well-Defined Polymers and DNA–Polymer Bioconjugates via Small-Volume EATRP in the Presence of Air, ACS Macro Lett., 2019, 8(5), 603–609 CrossRef CAS PubMed.
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Electrochemically Mediated Atom Transfer Radical Polymerization, Science, 2011, 332(6025), 81–84 CrossRef CAS PubMed.
- G. Szczepaniak, L. Fu, H. Jafari, K. Kapil and K. Matyjaszewski, Making ATRP More Practical: Oxygen Tolerance, Acc. Chem. Res., 2021, 54(7), 1779–1790 CrossRef CAS PubMed.
- J. Phommalysack-Lovan, Y. Chu, C. Boyer and J. Xu, PET-RAFT Polymerisation: Towards Green and Precision Polymer Manufacturing, Chem. Commun., 2018, 54(50), 6591–6606 RSC.
- B. Yuan, T. Huang, X. Wang, Y. Ding, L. Jiang, Y. Zhang and J. Tang, Oxygen–Tolerant RAFT Polymerization Catalyzed by a Recyclable Biomimetic Mineralization Enhanced Biological Cascade System, Macromol. Rapid Commun., 2022, 43(1), 2100559 CrossRef CAS PubMed.
- D. K. Schneiderman, J. M. Ting, A. A. Purchel, R. Miranda, M. V. Tirrell, T. M. Reineke and S. J. Rowan, Open-to-Air RAFT Polymerization in Complex Solvents: From Whisky to Fermentation Broth, ACS Macro Lett., 2018, 7(4), 406–411 CrossRef CAS PubMed.
- Z. Liu, Y. Lv and Z. An, Enzymatic Cascade Catalysis for the Synthesis of Multiblock and Ultrahigh-Molecular-Weight Polymers with Oxygen Tolerance, Angew. Chem., 2017, 129(44), 14040–14044 CrossRef.
- Y. Lv, Z. Liu, A. Zhu and Z. An, Glucose Oxidase Deoxygenation–redox Initiation for RAFT Polymerization in Air, J. Polym. Sci., Part A: Polym. Chem., 2017, 55(1), 164–174 CrossRef CAS.
- B. Zhang, X. Wang, A. Zhu, K. Ma, Y. Lv, X. Wang and Z. An, Enzyme-Initiated Reversible Addition–Fragmentation Chain Transfer Polymerization, Macromolecules, 2015, 48(21), 7792–7802 CrossRef CAS.
- R. Chapman, A. J. Gormley, M. H. Stenzel and M. M. Stevens, Combinatorial Low-Volume Synthesis of Well-Defined Polymers by Enzyme Degassing, Angew. Chem., Int. Ed., 2016, 55(14), 4500–4503 CrossRef CAS PubMed.
- R. Chapman, A. J. Gormley, K.-L. Herpoldt and M. M. Stevens, Highly Controlled Open Vessel RAFT Polymerizations by Enzyme Degassing, Macromolecules, 2014, 47(24), 8541–8547 CrossRef CAS.
- N. Corrigan, L. Zhernakov, M. H. Hashim, J. Xu and C. Boyer, Flow Mediated Metal-Free PET-RAFT Polymerisation for Upscaled and Consistent Polymer Production, React. Chem. Eng., 2019, 4(7), 1216–1228 RSC.
- S. R. Zavada, T. Battsengel and T. F. Scott, Radical-Mediated Enzymatic Polymerizations, Int. J. Mol. Sci., 2016, 17(2), 195 CrossRef PubMed.
- B. Zhao, J. Li, Y. Xiu, X. Pan, Z. Zhang and J. Zhu, Xanthate-Based Photoiniferter RAFT Polymerization toward Oxygen-Tolerant and Rapid Living 3D Printing, Macromolecules, 2022, 55(5), 1620–1628 CrossRef CAS.
- L. Lu, H. Zhang, N. Yang and Y. Cai, Toward Rapid and Well-Controlled Ambient Temperature RAFT Polymerization under UV-Vis Radiation: Effect of Radiation Wave Range, Macromolecules, 2006, 39(11), 3770–3776 CrossRef CAS.
- Q. Fu, K. Xie, T. G. McKenzie and G. G. Qiao, Trithiocarbonates as Intrinsic Photoredox Catalysts and RAFT Agents for Oxygen Tolerant Controlled Radical Polymerization, Polym. Chem., 2017, 8(9), 1519–1526 RSC.
- J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo and T. P. Davis, Reversible Addition–Fragmentation Chain Transfer Polymerization Initiated with Ultraviolet Radiation, Macromolecules, 2002, 35(20), 7620–7627 CrossRef CAS.
- T. G. McKenzie, Q. Fu, E. H. H. Wong, D. E. Dunstan and G. G. Qiao, Visible Light Mediated Controlled Radical Polymerization in the Absence of Exogenous Radical Sources or Catalysts, Macromolecules, 2015, 48(12), 3864–3872 CrossRef CAS.
- J. Wang, M. Rivero, A. Muñoz-Bonilla, J. Sanchez-Marcos, W. Xue, G. Chen, W. Zhang and X. Zhu, Natural RAFT Polymerization: Recyclable-Catalyst-Aided, Opened-to-Air, and Sunlight-Photolyzed RAFT Polymerizations, ACS Macro Lett., 2016, 5(11), 1278–1282 CrossRef CAS PubMed.
- A. Bagheri, K. E. Engel, C. W. A. Bainbridge, J. Xu, C. Boyer and J. Jin, 3D Printing of Polymeric Materials Based on Photo-RAFT Polymerization, Polym. Chem., 2020, 11(3), 641–647 RSC.
- J. R. Lamb, K. P. Qin and J. A. Johnson, Visible-Light-Mediated, Additive-Free, and Open-to-Air Controlled Radical Polymerization of Acrylates and Acrylamides, Polym. Chem., 2019, 10(13), 1585–1590 RSC.
- M. D. Nothling, Q. Fu, A. Reyhani, S. Allison-Logan, K. Jung, J. Zhu, M. Kamigaito, C. Boyer and G. G. Qiao, Progress and Perspectives Beyond Traditional RAFT Polymerization, Adv. Sci., 2020, 7(20), 2001656 CrossRef CAS PubMed.
- F. M. Calitz, M. P. Tonge and R. D. Sanderson, Kinetic and Electron Spin Resonance Analysis of RAFT Polymerization of Styrene, Macromolecules, 2003, 5–8 CrossRef CAS.
- J. Tanaka, P. Gurnani, A. B. Cook, S. Häkkinen, J. Zhang, J. Yang, A. Kerr, D. M. Haddleton, S. Perrier and P. Wilson, Microscale Synthesis of Multiblock Copolymers Using Ultrafast RAFT Polymerisation, Polym. Chem., 2019, 10(10), 1186–1191 RSC.
- A. Kuroki, I. Martinez-Botella, C. H. Hornung, L. Martin, E. G. L. Williams, K. E. S. Locock, M. Hartlieb and S. Perrier, Looped Flow RAFT Polymerization for Multiblock Copolymer Synthesis, Polym. Chem., 2017, 8(21), 3249–3254 RSC.
- G. Gody, R. Barbey, M. Danial and S. Perrier, Ultrafast RAFT Polymerization: Multiblock Copolymers within Minutes, Polym. Chem., 2015, 6(9), 1502–1511 RSC.
- S. Cosson, M. Danial, J. R. Saint-Amans and J. J. Cooper-White, Accelerated Combinatorial High Throughput Star Polymer Synthesis via a Rapid One-Pot Sequential Aqueous RAFT (Rosa-RAFT) Polymerization Scheme, Macromol. Rapid Commun., 2017, 38(8), 1600780 CrossRef PubMed.
- S. Parkinson, S. T. Knox, R. A. Bourne and N. J. Warren, Rapid Production of Block Copolymer Nano-Objects via Continuous-Flow Ultrafast RAFT Dispersion Polymerisation, Polym. Chem., 2020, 11(20), 3465–3474 RSC.
- P. Gurnani, T. Floyd, J. Tanaka, C. Stubbs, D. Lester, C. Sanchez-Cano and S. Perrier, PCR-RAFT: Rapid High Throughput Oxygen Tolerant RAFT Polymer Synthesis in a Biology Laboratory, Polym. Chem., 2020, 11(6), 1230–1236 RSC.
- M. Reis, F. Gusev, N. G. Taylor, S. H. Chung, M. D. Verber, Y. Z. Lee, O. Isayev and F. A. Leibfarth, Machine-Learning-Guided Discovery of 19 F MRI Agents Enabled by Automated Copolymer Synthesis, J. Am. Chem. Soc., 2021, 143(42), 17677–17689 CrossRef CAS PubMed.
- M. Rubens, J. H. Vrijsen, J. Laun and T. Junkers, Precise Polymer Synthesis by Autonomous Self–Optimizing Flow Reactors, Angew. Chem., 2019, 131(10), 3215–3219 CrossRef.
- M. H. Reis, T. P. Varner and F. A. Leibfarth, The Influence of Residence Time Distribution on Continuous-Flow Polymerization, Macromolecules, 2019, 52(9), 3551–3557 CrossRef CAS.
- D. J. Walsh, D. A. Schinski, R. A. Schneider and D. Guironnet, General Route to Design Polymer Molecular Weight Distributions through Flow Chemistry, Nat. Commun., 2020, 11(1), 3094 CrossRef CAS PubMed.
- M. H. Reis, C. L. G. Davidson and F. A. Leibfarth, Continuous-Flow Chemistry for the Determination of Comonomer Reactivity Ratios, Polym. Chem., 2018, 9(13), 1728–1734 RSC.
- M. H. Reis, F. A. Leibfarth and L. M. Pitet, Polymerizations in Continuous Flow: Recent Advances in the Synthesis of Diverse Polymeric Materials, ACS Macro Lett., 2020, 9(1), 123–133 CrossRef CAS PubMed.
- T. Sato, Y. Hamada, M. Sumikawa, S. Araki and H. Yamamoto, Solubility of Oxygen in Organic Solvents and Calculation of the Hansen Solubility Parameters of Oxygen, Ind. Eng. Chem. Res., 2014, 53(49), 19331–19337 CrossRef CAS.
- M. Robischon, Surface-Area-to-Volume Ratios, Fluid Dynamics & Gas Diffusion: Four Frogs & Their Oxygen Flux, Am. Biol. Teach., 2017, 79(1), 64–67 CrossRef.
- A. Kucernak and J. Jiang, Mesoporous Platinum as a Catalyst for Oxygen Electroreduction and Methanol Electrooxidation, Chem. Eng. J., 2003, 93(1), 81–90 CrossRef CAS.
- K. D. Vandegriff and J. S. Olson, Morphological and Physiological Factors Affecting Oxygen Uptake and Release by Red Blood Cells, J. Biol. Chem., 1984, 259(20), 12619–12627 CrossRef CAS PubMed.
- L. Z. Flores-López, H. Espinoza-Gómez and R. Somanathan, Silver Nanoparticles: Electron Transfer, Reactive Oxygen Species, Oxidative Stress, Beneficial and Toxicological Effects. Mini Review, J. Appl. Toxicol., 2019, 39(1), 16–26 CrossRef PubMed.
- R. W. Mair, G. P. Wong, D. Hoffmann, M. D. Hürlimann, S. Patz, L. M. Schwartz and R. L. Walsworth, Probing Porous Media with Gas Diffusion NMR, Phys. Rev. Lett., 1999, 83(16), 3324–3327 CrossRef CAS PubMed.
- S. Wilken, J. Parisi and H. Borchert, Role of Oxygen Adsorption in Nanocrystalline ZnO Interfacial Layers for Polymer–Fullerene Bulk Heterojunction Solar Cells, J. Phys. Chem. C, 2014, 118(34), 19672–19682 CrossRef CAS.
- N. D. Gershon, K. R. Porter and B. L. Trus, The Cytoplasmic Matrix: Its Volume and Surface Area and the Diffusion of Molecules through It, Proc. Natl. Acad. Sci. U. S. A., 1985, 82(15), 5030–5034 CrossRef CAS PubMed.
- S. Shanmugam, J. Xu and C. Boyer, Utilizing the Electron Transfer Mechanism of Chlorophyll a under Light for Controlled Radical Polymerization, Chem. Sci., 2015, 6(2), 1341–1349 RSC.
- S. Shanmugam, J. Xu and C. Boyer, Photoinduced Electron Transfer–Reversible Addition–Fragmentation Chain Transfer (PET-RAFT) Polymerization of Vinyl Acetate and N -Vinylpyrrolidinone: Kinetic and Oxygen Tolerance Study, Macromolecules, 2014, 47(15), 4930–4942 CrossRef CAS.
- S. Shanmugam, J. Xu and C. Boyer, Aqueous RAFT Photopolymerization with Oxygen Tolerance, Macromolecules, 2016, 49(24), 9345–9357 CrossRef CAS.
- L. Shen, Q. Lu, A. Zhu, X. Lv and Z. An, Photocontrolled RAFT Polymerization Mediated by a Supramolecular Catalyst, ACS Macro Lett., 2017, 6(6), 625–631 CrossRef CAS PubMed.
- J. Xu, S. Shanmugam, C. Fu, K.-F. Aguey-Zinsou and C. Boyer, Selective Photoactivation: From a Single Unit Monomer Insertion Reaction to Controlled Polymer Architectures, J. Am. Chem. Soc., 2016, 138(9), 3094–3106 CrossRef CAS PubMed.
- Y. Song, Y. Kim, Y. Noh, V. K. Singh, S. K. Behera, A. Abudulimu, K. Chung, R. Wannemacher, J. Gierschner and L. Lüer, et al., Organic Photocatalyst for Ppm-Level Visible-Light-Driven Reversible Addition–Fragmentation Chain-Transfer (RAFT) Polymerization with Excellent Oxygen Tolerance, Macromolecules, 2019, 52(15), 5538–5545 CrossRef CAS.
- J. Niu, Z. A. Page, N. D. Dolinski, A. Anastasaki, A. T. Hsueh, H. T. Soh and C. J. Hawker, Rapid Visible Light-Mediated Controlled Aqueous Polymerization with In Situ Monitoring, ACS Macro Lett., 2017, 6(10), 1109–1113 CrossRef CAS PubMed.
- J. Xu, K. Jung and C. Boyer, Oxygen Tolerance Study of Photoinduced Electron Transfer–Reversible Addition–Fragmentation Chain Transfer (PET-RAFT) Polymerization Mediated by Ru(Bpy)3Cl2, Macromolecules, 2014, 47(13), 4217–4229 CrossRef CAS.
- S. Shanmugam, J. Xu and C. Boyer, Exploiting Metalloporphyrins for Selective Living Radical Polymerization Tunable over Visible Wavelengths, J. Am. Chem. Soc., 2015, 137(28), 9174–9185 CrossRef CAS PubMed.
- M. Benaglia, J. Chiefari, Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, Universal (Switchable) RAFT Agents, J. Am. Chem. Soc., 2009, 131(20), 6914–6915 CrossRef CAS PubMed.
- T. Mukundan and K. Kishore, Synthesis, Characterization and Reactivity of Polymeric Peroxides, Prog. Polym. Sci., 1990, 15(3), 475–505 CrossRef CAS.
- Z. Zhang, J. Zhu, Z. Cheng and X. Zhu, Reversible Addition–Fragmentation Chain Transfer (RAFT) Polymerization of Styrene in the Presence of Oxygen, Polymer, 2007, 48(15), 4393–4400 CrossRef CAS.
- R. E. Cais and F. A. Bovey, Carbon-13 Nuclear Magnetic Resonance Study of the Microstructure and Molecular Dynamics of Poly(Styrene Peroxide), Macromolecules, 1977, 10(1), 169–178 CrossRef CAS.
- K. Kishore, Spectral and Thermal Data on Polystyrene Peroxide, J. Chem. Eng. Data, 1980, 25(1), 92–94 CrossRef CAS.
- S. Jayanthi and K. Kishore, Oxidative Copolymerization: Microstructure Analysis of the Terpolymer of Styrene, Methyl Methacrylate, and Oxygen, Macromolecules, 1993, 26(8), 1985–1989 CrossRef CAS.
- A. J. L. Villaraza, A. Bumb and M. W. Brechbiel, Macromolecules, Dendrimers, and Nanomaterials in Magnetic Resonance Imaging: The Interplay between Size, Function, and Pharmacokinetics, Chem. Rev., 2010, 110(5), 2921–2959 CrossRef CAS PubMed.
- N. Zaquen, M. Rubens, N. Corrigan, J. Xu, P. B. Zetterlund, C. Boyer and T. Junkers, Polymer Synthesis in Continuous Flow Reactors, Prog. Polym. Sci., 2020, 107, 101256 CrossRef CAS.
- J. S. Moore and K. F. Jensen, “Batch” Kinetics in Flow: Online IR Analysis and Continuous Control, Angew. Chem., Int. Ed., 2014, 53(2), 470–473 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00603k |
| This journal is © The Royal Society of Chemistry 2022 |