
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Eu3Ta2ON5: an n = 2 Ruddlesden–Popper ferromagnetic oxynitride with long-range ordered anion vacancies
Judith Oró-Solé†
a,
Jhonatan Ricardo Guarín†a,
Carlos Frontera *a,
Jaume Gàzqueza,
Bernat Mundetb,
Josep Fontcuberta*a and
Amparo Fuertes*a
*a,
Jaume Gàzqueza,
Bernat Mundetb,
Josep Fontcuberta*a and
Amparo Fuertes*a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus UAB, 08193 Bellaterra, Spain. E-mail: amparo.fuertes@icmab.es
bInstitut Català de Nanociència i Nanotecnologia (ICN2), Campus UAB-08193 Bellaterra, Barcelona, Spain
First published on 10th June 2025
Abstract
The new compound Eu3Ta2ON5 with an n = 2 Ruddlesden–Popper structure has been prepared by solid state reaction between EuN, Eu2O3 and Ta3N5 at 1200 °C under N2. It crystallizes in the I4/mmm space group with unit cell parameters a = 3.98240(1) Å and c = 20.42020(6) Å and has anion vacancies in central A-site layers of the perovskite blocks, where tantalum is in a square pyramidal coordination polyhedron with composition [TaO0.5N4.5] and europium is 8-coordinated. The perovskite blocks intergrow with [EuO0.5N0.5] NaCl-type layers of the Ruddlesden–Popper structure. Electron Energy Loss spectrum imaging shows a mixed Eu2+/Eu3+ valence with preferred occupation of Eu3+ in the 8-coordinated sites. Magnetic susceptibility measurements are consistent with the formal composition Eu22+Eu3+Ta5+2ON5 and indicate ferromagnetic order of Eu2+ ions below TC = 20 K. Interestingly, despite the oxynitride containing non-magnetic Eu3+ ions (7F0), the Curie temperature is found to be much larger than in related Eu2TiO4 and Eu3Ti2O7 Ruddlesden–Popper compounds (≈9–10 K), which is attributed to the covalency-reinforced Eu2+–N/O–Eu2+ superexchange interactions.
Introduction
Simple perovskite oxynitrides ABO3−xNx -where A is an alkaline earth metal or rare earth metal and B is a transition metal- have been widely investigated because of their electronic and photocatalytic properties.1 There are numerous examples of compounds with different combinations of cations and anion stoichiometries, and some paradigmatic materials are non-toxic pigments La1−xCaxTaO2−xN1+x,2 BaTaO2N and SrTaO2N with high dielectric permittivity,3 EuNbO2N that shows colossal magnetoresistance at low temperature,4 and LaTiO2N that is a photocatalyst for water splitting.5 Compounds with more complex structures are less common, and include magnetic double or triple perovskites such as La2MnTaO5N,6 Ba2MnWO4.42N1.58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 or Eu3Ta3O3.66N5.34,8 and polar hexagonal perovskite BaWON2.9 Perovskite related Ruddlesden–Popper oxynitrides AX(ABX3)n (X = O,N), formed by intergrowth of n perovskite blocks with a rock salt AX layer, have been reported as a few examples restricted to aluminum, tantalum and niobium at the B sites. Existing n = 1 members for alkaline earth cations at A sites are Sr2TaO3N, Ba2TaO3N10 and Sr2NbO3N.11 Lanthanide derivatives were initially reported by Marchand for aluminum, R2AlO3N (R = La, Sm, Nd),12 and more recently the tantalum compounds R2Ta(O,N)4 (R = La, Nd, Sm, Eu) have been discovered.13 To the best of our knowledge the only reported n = 2 compounds are Sr3Nb2O5N211 and Eu3Ta2O3N4.14 The strontium niobium compound was prepared using a solid state reaction at room pressure under NH3 at 1050 °C, and crystallizes in the aristotype space group for Ruddlesden–Popper phases, I4/mmm. The europium tantalum phase was obtained in the form of single crystals by ammonothermal synthesis at 797 °C and 170 MPa. It was described as a europium mixed valence compound of detailed composition Eu3+2Eu2+Ta2O3N4, with a distorted symmetry P42/mnm and cell parameters a = 5.7278(1), c = 19.8149(5) Å.
7 or Eu3Ta3O3.66N5.34,8 and polar hexagonal perovskite BaWON2.9 Perovskite related Ruddlesden–Popper oxynitrides AX(ABX3)n (X = O,N), formed by intergrowth of n perovskite blocks with a rock salt AX layer, have been reported as a few examples restricted to aluminum, tantalum and niobium at the B sites. Existing n = 1 members for alkaline earth cations at A sites are Sr2TaO3N, Ba2TaO3N10 and Sr2NbO3N.11 Lanthanide derivatives were initially reported by Marchand for aluminum, R2AlO3N (R = La, Sm, Nd),12 and more recently the tantalum compounds R2Ta(O,N)4 (R = La, Nd, Sm, Eu) have been discovered.13 To the best of our knowledge the only reported n = 2 compounds are Sr3Nb2O5N211 and Eu3Ta2O3N4.14 The strontium niobium compound was prepared using a solid state reaction at room pressure under NH3 at 1050 °C, and crystallizes in the aristotype space group for Ruddlesden–Popper phases, I4/mmm. The europium tantalum phase was obtained in the form of single crystals by ammonothermal synthesis at 797 °C and 170 MPa. It was described as a europium mixed valence compound of detailed composition Eu3+2Eu2+Ta2O3N4, with a distorted symmetry P42/mnm and cell parameters a = 5.7278(1), c = 19.8149(5) Å.
Reported perovskite oxynitrides of europium show ferromagnetic order of Eu2+ S = 7/2 spins at low temperatures. EuNbO2N, EuTaO2N4 and EuWO1−xN215 show Curie temperatures up to 12 K, and Ruddlesden–Popper n = 1 Eu2TaO2.8N1.2 orders below 8 K.13 Additionally the Nb and W perovskites show colossal magnetoresistances at low temperatures, originating from the coupling between the carrier spins of the transition metal and the localized Eu spins.
Here we report the new n = 2 Ruddlesden–Popper oxynitride Eu2+2Eu3+Ta2ON5 that has been prepared by a solid-state reaction between EuN, Eu2O3 and Ta3N5 at room pressure, under N2 at 1200 °C, and shows a different crystal structure and richer Eu2+ contents than early reported Eu3+2Eu2+Ta2O3N4.14 The differences in anion composition and crystal chemistry between these two compounds are due to the drastic changes in the synthesis conditions, as Eu3+2Eu2+Ta2O3N4 was obtained at much lower temperature and under high pressure conditions, starting with Ta, Eu, NaN3 and NaOH.14 Rietveld refinement of synchrotron X-ray diffraction data and integrated differential phase contrast imaging indicate that tantalum is in square pyramidal coordination analogously to the nitrides R3B2N6 (R = La, Ce, Pr; B = Nb, Ta),16 resulting from long-range ordered anion vacancies in the central europium layer of the perovskite blocks. The new compound represents the first example of a stoichiometric anion deficient oxynitride perovskite. Magnetization measurements show that Eu2+ ions, that are concentrated at the NaCl-type layers, are ferromagnetically ordered below 20 K. The Curie temperature is significantly larger than reported for Eu2+ perovskite-related oxides, where TC has been found to be lower than 10 K. It will be argued that the ordering of magnetic Eu2+and non-magnetic Eu3+ ions and the reinforced Eu–N/O–Eu superexchange interactions account for this remarkable observation.
Experimental
Synthetic procedures and chemical characterization
Black Eu3Ta2ON5 samples of 100 mg were prepared by solid state reaction between Eu2O3 (Sigma-Aldrich 99.9%), EuN (Materion, 99.9%) and Ta3N5 at 1200 °C under N2 gas (Air Liquide, 99.9999%) using one single treatment of 3 hours at this temperature. Ta3N5 was prepared by reaction of Ta2O5 (Sigma-Aldrich 99.99%) with NH3 (Carburos Metálicos 99.9%) at 850 °C during several cycles of 15 hours with intermediate regrinding, using a flow rate of 600 cm3 min−1. Eu2O3 was treated at 900 °C under vacuum of 10−3 torr during 12 hours for dehydration. The reactants were weighed, mixed and pelletized in a glove box under recirculating Ar. The pellets were placed in molybdenum crucibles covered by zirconium foil. Another crucible with zirconium was placed near the sample in the reaction tube to scavenge H2O and O2 impurities from the gas. The reaction tube was evacuated to 10−3 torr and purged three times with N2 before heating at 300 °C h−1 up to the maximum temperature. The obtained samples were stable in air.Nitrogen analyses were performed in a Thermo Fisher Scientific instrument heating the samples up to 1060 °C under O2, using MgO, WO3 and Sn as additives and atropine as reference standard.
Structural characterization
Laboratory X-ray powder diffraction patterns were obtained on capillary samples of diameter 0.3 mm using a Bruker D8 Advance A25 diffractometer in a Debye Scherrer configuration with Mo Kα1 radiation (λ = 0.7093 Å). High resolution synchrotron X-ray powder diffraction (SXRPD) data were collected at room temperature on the same capillaries in the angular range 2.0° ≤ 2θ ≤ 60° at the MSPD beamline of the ALBA Synchrotron (Cerdanyola del Vallès, Spain),17 using multi analyser detector and 30 keV energy that resulted in a wavelength of 0.41376 Å determined by refining SRM640d NIST Si standard. Rietveld analysis was performed using the program Fullprof.18 Background refinement was performed by linear interpolation and data were corrected from absorption.Electron diffraction patterns were taken in a JEOL 1210 transmission electron microscope operating at 120 kV using a double tilt side entry specimen holder with tilting angles ±60°/±30°. The samples were deposited in powder form on a copper grid coated by a Lacey carbon film with a continuous layer of ultrathin carbon film.
Atomic resolution images were obtained at the Joint Electron Microscopy Center at ALBA (Cerdanyola del Vallès, Spain) on a double-corrected Thermo Fisher Spectra 300 (S)TEM microscope operated at 200 kV. High-angle annular dark field (HAADF) images were acquired using a semi-convergence angle of 19.5 mrad. Integrated differential phase contrast scanning transmission electron microscopy (iDPC-STEM) method has also been used for imaging of all elements using the segmented Panther detector from Thermo Fisher. This imaging mode detects the deflection of the beam produced by the atomic electrostatic fields thus allowing to image simultaneously heavy and light atoms.19 Electron Energy Loss Spectroscopy (EELS) spectra were collected in a continuum spectrometer equipped with a fast direct electron detection camera K3 from Gatan, using a collection angle of around 40 mrad, a probe current of around 90p A and a dwell time of 50 ms. A principal component analysis filter was used after acquisition to minimize the random noise of the EELS spectrum images. To improve the energy resolution, the beam was monochromated using an excitation of 0.6, a spot size of 14 and a 1μm diameter C1 condenser aperture. This leads to a zero-loss peak full width at half maximum of around 0.4 eV. EDX spectra were acquired using a four quadrant Super-X windowless silicon drift detector system and beam currents of ≈100–250 pA. The crystal composition was quantified using the Thermo Fisher Scientific Velox software, applying the Cliff–Lorimer approach with Brown–Powell ionization cross-section models.
Magnetic measurements
Magnetic susceptibility measurements were performed on a Quantum Design SQUID magnetometer at temperatures between 2 and 300 K and magnetic fields of 25 Oe and 10 kOe. Magnetization-field loops were measured between −70 kOe and +70 kOe at temperatures below 50 K.Results and discussion
Synthesis and structural study
The synthesis of the new anion deficient n = 2 Ruddlesden–Popper oxynitride takes place through the formal reaction:| 2 Eu2O3 + 5 EuN + 2 Ta3N5 → 3 Eu3Ta2ON5 + 3/2 O2, |
:EuN:Ta3N5 = 0.45:8.1:2 with N/O = 13.4 produced a sample free of impurities according to synchrotron X-ray powder diffraction data (Fig. 1).
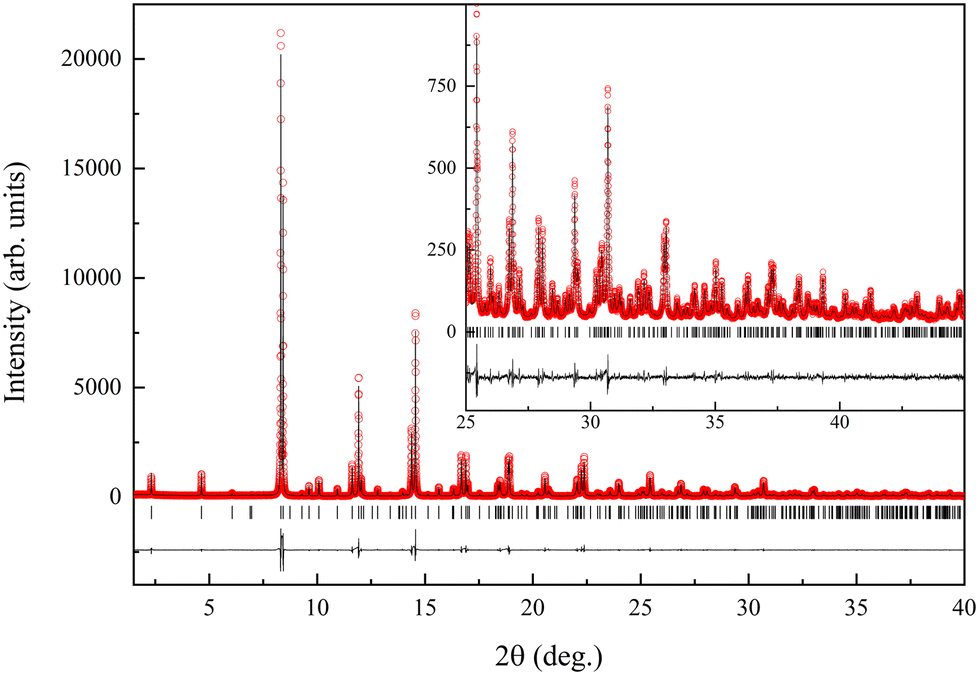 | ||
| Fig. 1 Rietveld fit to synchrotron X-ray powder diffraction data (λ = 0.41376 Å) of Eu3Ta2ON5, performed in the I4/mmm space group with a = 3.98240(1), c = 20.42020(6) Å. The inset shows the high 2θ region enlarged. | ||
Syntheses starting with N/O ratios below this value led biphasic samples with different proportions of the n = 1 member Eu2TaO2.37N0.63. This phase is prepared at the same temperature but has a lower N/O stoichiometric ratio compared to Eu3Ta2ON5,13 hence the increase of N/O favours the formation of the n = 2 member. In the biphasic samples, Eu2TaO2.37N0.63 was detected either in the X-ray diffraction patterns or as small intergrowth domains in the scanning transmission electron microscopy (STEM) images.
Electron diffraction patterns of Eu3Ta2ON5 (Fig. 2) were indexed in a cell with a ≂ b ≂ 3.97 and c ≂ 20.54 Å in the I4/mmm aristotype space group, with reflection conditions: hkl, h + k + l = 2n; hk0, h + k = 2n; 0kl, k + l = 2n; hhl, l = 2n; 00l, l = 2n; h00, h = 2n. For some crystals, very weak additional reflections were observed in one of the 〈110〉 planes, that could be indexed in a √2a0 × √2a0 × c0 superstructure with the orthorhombic space group Amaa (no. 66), where a0 and c0 are the parameters of the I4/mmm cell.
 | ||
| Fig. 2 Electron diffraction patterns of Eu3Ta2ON5 along [010], [001] and [110] axes. Orange arrows indicate superstructure reflections observed in some crystals, that can be indexed in a √2a0 × √2a0 × c0 unit cell. | ||
High-resolution HAADF-STEM imaging in combination with EDX and iDPC allowed mapping of the position of all atomic sublattices in real space. Fig. 3a shows a high-resolution HAADF-STEM image of a Eu3Ta2ON5 crystal with the characteristic n = 2 Ruddlesden–Popper structure. EDX maps (Fig. 3b) showed no Eu/Ta intermixing, giving a ratio Eu/Ta of 2.8(2)/2; the nitrogen content of 7.56% determined by combustion analysis indicated 4.88 atoms per formula, close to the formal stoichiometry Eu3Ta2ON5 where the oxygen content has been adjusted considering 6 anions. EDX spectra also indicated the presence of oxygen and nitrogen in the sample, but their quantitative analysis was not possible because of the large error observed for these light elements.
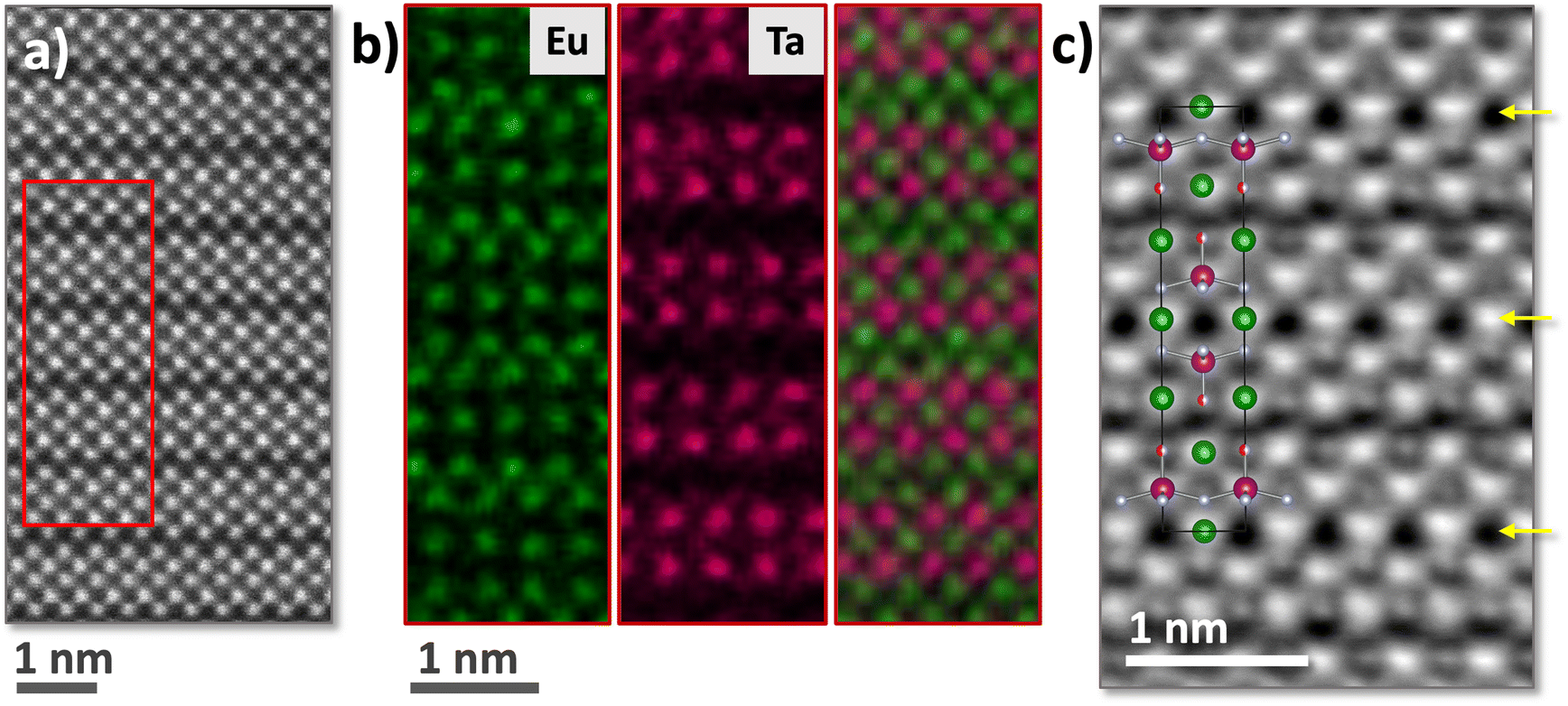 | ||
| Fig. 3 (a) HAADF-STEM image of a Eu3Ta2ON5 crystal along the [010] zone axis. The image shows an n = 2 Ruddlesden–Popper structure, with two perovskite blocks intergrowing with a rock salt-type layer. The inset shows the corresponding area for the elemental EDX maps. (b) From left to right, atomic resolution EDX maps of Eu (in green), Ta (in magenta) and a combined colour map. (c) iDPC image with the Eu3Ta2ON5 structure overlay (Eu: green, Ta: magenta, O: red and N: light gray). Yellow arrows point to the planes in which the anion vacancies are located. | ||
In order to probe the O and N sublattices, iDPC-STEM imaging mode was used, which is a direct phase imaging process that relies on atomic number. As the atomic number increases, the scattering intensity and signal brightness also rise. This imaging principle results in a linear relationship between the intensity in iDPC images and the atomic number.19 In contrast, high-angle annular dark field (HAADF) images exhibit an approximate proportionality between intensity and the square of the atomic number. Consequently, iDPC-STEM offers greater information about lighter elements, which is highly valuable for investigating the O and N sublattices of Eu3Ta2ON5. Notice that the iDPC contrast of the image of a crystallite oriented along [010] allows for the identification of all sublattices in Eu3Ta2ON5. Remarkably, the planes signalled with an arrow show a much dimmer contrast due to an anion deficiency, consistent with a n = 2 Ruddlesden–Popper structure with 6 anions per formula and vacancies located between the two [TaX2] planes of the perovskite block, resulting in square pyramidal coordination of tantalum atoms.
Rietveld refinement of synchrotron X-ray powder diffraction data (Fig. 1) were performed in the I4/mmm space group starting with the structural model of La3Ta2N6 that has two crystallographically independent anions at 8g and 4e sites, corresponding respectively to equatorial and axial positions of the TaX5 pyramids.16 The refined cell parameters were a = 3.98240(1), c = 20.42024(6) Å, with agreement factors of Rp = 6.90%, Rwp = 8.95%, χ2 = 6.35 and RBragg = 1.97% (Table 1 and Fig. 4). Refinements were also performed in the Amaa (no. 66) space group using a unit cell with parameters √2a0 × √2a0 × c0 and three positions for the anions, but the observed Bragg R factor was significantly larger than for the I4/mmm model (5.06% vs. 1.97%).
 | ||
| Fig. 4 Structural model in the I4/mmm space group and cation coordination polyhedra of Eu3Ta2ON5 showing bond distances. | ||
| Atom | Wyckoff site | x | y | z | Bb (Å2) | Occ. factor |
|---|---|---|---|---|---|---|
| Eu1 | 2b | 0 | 0 | 0.5 | 0.44 | 1 |
| Eu2 | 4e | 0 | 0 | 0.31422(2) | 0.65 | 1 |
| Ta | 4e | 0 | 0 | 0.09922(3) | 0.47 | 1 |
| N1 | 8g | 0 | 0.5 | 0.0738(3) | 1.04(9) | 1 |
| O2/N2 | 4e | 0 | 0 | 0.1910(3) | 1.04 | 0.5/0.5 |
| Bond length(Å) | ||
|---|---|---|
| a O/N occupation factors were fixed considering the predicted distribution using Pauling's second crystal rule. Refined cell parameters were a = 3.98240(1), c = 20.42024(6) Å. Agreement factors: Rp = 6.90%, Rwp = 8.95%, χ2 = 6.35, RBragg = 1.97%.b Beq in Å2 is calculated for Eu and Ta atoms from refined anisotropic temperature factors (×104): Eu1 β11 = β22 = 64(3) β33 = 3.1(2); Eu2 β11 = β22 = 120(4) β33 = 2.5(2); Ta β11 = β22 = 59(2) β33 = 3.9(1). The temperature factors were common for the two anion sites. | ||
| Ta–N1 | 2.0595(6) × 4 | Ta–O2,N2 1.865(6) |
| Eu1–N1 | 2.493(4) × 8 | |
| Eu2–N1 | 3.036(5) × 4 | Eu2–O2,N2 2.525(6) |
| Eu2–O2,N2 | 2.8177(2) × 4 | |
The europium atoms in Eu3Ta2ON5 show two well differentiated polyhedra with coordination numbers 8 and 9 for Eu1 at 2b and Eu2 at 4e sites respectively, and corresponding average bond distances of 2.493(4) and 2.871 Å. Considering the difference between both distances and the ionic radii of Eu3+ for CN = VIII (1.01 Å) and Eu2+ for CN = IX (1.30 Å),20 the new oxynitride can be formally formulated as Eu2+2Eu3+Ta5+2ON5, a charge-ordered compound where Eu3+ occupies the 8-coordinated sites and Eu2+ ions are in the 9-coordinated sites.
The distribution of O and N in the 8g and 4e sites could not be determined from X-ray diffraction because this technique does not provide enough contrast between the two anions. Neutron powder diffraction is the optimal technique to investigate the anion ordering in oxynitrides, but for Eu3Ta2ON5 the large absorption cross section of europium would make difficult to obtain accurate information from these data. Pauling's second crystal rule (PSCR) can be used to predict the anion distribution in mixed anion compounds,21,22 from the calculation of the bond strength sums (b) with the equation:

According to PSCR, the electrical charge of each anion (q) tends to compensate the strength of the electrostatic valence bonds from the cations (b). Considering the ordering of Eu3+ at 2b sites and Eu2+ at 4e sites, the calculated bond strength sums are 2.85 and 1.94 for the anions at 8g and 4e positions respectively, predicting the preferred occupancy of N atoms at the equatorial positions of the [TaX5] pyramids. Accordingly, in the refinement we used fixed 100% occupancy of nitrogen on 8g sites and 50/50 N/O at 4e sites.
The observed equatorial Ta–N bond distance (2.0595(6) Å) is similar to those reported for La3Ta2N6 and Ce3Ta2N6 (2.053(3) and 2.0465(19) Å respectively).16 In contrast, the Ta–O,N apical distance (1.865(6) Å) is slightly shorter than in the two nitrides (1.96(3) and 1.951(19) Å for La3Ta2N6 and Ce3Ta2N6 respectively). This difference is consistent with the mixed O/N occupancy at axial sites, according with the larger ionic radius of N3− compared to O2− (1.46 vs. 1.38 Å, both for CN = IV).20
EELS measurements provided detailed atomic scale information of the oxidation state of both Eu2 and Eu1 sites by analysing the energy onset of the Eu M4,5 edge. Fig. 5(a–c) shows a HAADF-STEM image of a Eu3Ta2ON5 crystal and two EEL spectra from the crystallographic sites Eu2 and Eu1. The fine structure of the two Eu M4,5 spectra is clearly different, although the source is not as straight forward. For Eu2+ species the onset is lower in energy than that of the Eu3+ species.23 If both Eu2+ and Eu3+ are present, EEL spectra typically display two peaks with the stronger one belonging to the dominant oxidation state. The thickness of the sample may also play an important role, as it widens the electron beam, it lowers the spatial resolution and increases the contribution of nearest unit cells,24 which may be the reason of a presence of a Eu2+ signal in the spectra acquired from the Eu1 site, or of Eu3+ in the spectra acquired for the Eu2 site. Nonetheless, these EELS measurements support the preferred occupation of Eu2+ and Eu3+ ions in Eu2 and Eu1 sites, respectively, of Eu3Ta2ON5.
 | ||
| Fig. 5 (a) HAADF-STEM image of the Eu3Ta2ON5 crystal along the [010] zone axis with the Eu3Ta2ON5 structure overlay (Eu: green, Ta: magenta, O: red and N: light gray). (b) and (c) show the averaged EEL M4,5 spectra obtained from Eu2 and Eu1 sites of the Eu3Ta2ON5 structure, marked in blue and red in (a), respectively. | ||
Magnetic properties
The temperature-dependent magnetic susceptibility χ(T) (black symbols) and χ−1(T) (red symbols) of Eu3Ta2ON5, recorded at H = 5k Oe, are shown in Fig. 6. It can be appreciated that χ−1(T) displays an appreciable bending, suggesting an additional temperature-dependent contribution other than the Curie–Weiss behavior. As indicated by the solid lines thorough the data, the susceptibility per mole can be well fitted by the contributions from nEu2+ moles of Eu2+ (S = 7/2, J = 7/2), modelled as a Curie–Weiss law, and a small temperature-dependent Van Vleck contribution arising from nEu3+ moles of Eu3+, in addition to a background contribution (χ0). Notice that although Eu3+ ground state is nonmagnetic (7F0, S = 3, L = 3, J = 0), thermal excitation to higher lying states (for instance the first one (7F1, S = 3, L = 3, J = 1) is only at about 46 meV25) shall produce an additional contribution to the magnetic susceptibility (χEu3+), that will add to the Eu2+ contribution, and to the diamagnetic contribution. Accordingly, the magnetic susceptibility per Eu ion can be expressed as:8,13| χ(T) = nEu2+ χEu2+(T) + nEu3+ χEu3+(T) + χ0, |

with:

and gJ = 3/2, except g0 that equals 2 + L = 2 + S = 5.26,27

 | ||
| Fig. 6 Magnetic susceptibility χ(T) (left axis) and the reciprocal χ−1(T), (right axis) recorded at 5 kOe. Solid lines are the results of the fit described in the text. | ||
Fitting to experimental inverse susceptibility was done by varying nEu2+, nEu3+, θ and χ0, while fixing λ/kB = 531.5 K (equivalent to an energy splitting, λ = 46 meV, between the non-magnetic ground state of Eu3+ and its first excited state),13,26 as well as the effective paramagnetic moment of Eu2+. From the fit, we obtain nEu2+ = 1.98, nEu3+ = 1.02, θ = 20.2 K and χ0 = 4.6 × 10−3 emu mol−1. These results are quite consistent with chemical analysis and crystal structure results, that suggest 2 Eu2+ and 1 Eu3+ per formula. The extrapolated Curie–Weiss temperature is θ > 0 (≈20 K), indicating the prevalence of ferromagnetic interactions.
In agreement with the susceptibility data of Fig. 6, the M(H) loops (Fig. 7) develop a ferromagnetic-like shape at T ≤ 20 K. At 2 K the saturation magnetization is about 14μB per f.u., which is fully consistent with the presence of two ferromagnetically ordered Eu2+ ions per f.u in this oxynitride. The ferromagnetic ordering occurring at T ≈ 20K is evidenced in the zero-field-cooling and field-cooling susceptibility data recorded at low field (inset in Fig. 7), that show a perceptible hysteresis.
 | ||
| Fig. 7 Magnetization loops recorded at several temperatures (indicated) up to ±5 T. Inset: zero field-cooled/field-cooled magnetization vs. temperature measured under 25 Oe applied magnetic field. | ||
The most remarkable feature of these results is that the Curie temperature (≈20 K) is significantly larger than those reported for similar systems, such as Ruddlesden–Popper n = 1 Eu2TiO4 and n = 2 Eu3Ti2O7,25,28 where TC is around 9–10 K. To rationalize the observed dramatic enhancement of TC in Eu3Ta2ON5, it is worth to compare these results with isostructural Eu3Ti2O7, that for this purpose is described as a sequence of EuTiO3 (perovskite-type) and Eu2TiO4 (K2NiF4-type, rock salt) blocks.25 We recall that, as argued above, the non-magnetic Eu3+ ions in Eu3Ta2ON5 occupy Eu1 sites equivalent to those in EuTiO3 structure and the magnetic Eu2+ ions occupy Eu2 sites as in Eu2TiO4. Therefore, the magnetic interactions in Eu3Ta2ON5 are analogous to those for Eu2TiO4.
Fig. 8 illustrates the most relevant magnetic nearest-neighbor (nn) (J11 and J12) and next-nearest-neighbor (nnn) magnetic interactions (J21) in Eu3TaON5. Notice that we omit magnetic interactions involving Eu3+ as this ion is nonmagnetic. Here, we follow the nomenclature of Chien et al.25 who employed a mean field approach to describe the TC of Eu3Ti2O7 in terms of the nn and nnn magnetic interactions (eqn (19) in ref. 25). The model can be easily adapted to the present case by keeping only interactions involving Eu2+ ions; namely J11, J12 and J21. It follows that:

 | ||
| Fig. 8 Sketch of the structure and the most relevant nn (J11 and J12) and nnn (J21) magnetic interactions in Eu3Ta2ON5. Ta, N and O atoms are represented by magenta, light gray and red spheres respectively; Eu3+ and Eu2+ are shown as light and dark green spheres respectively. The two structural blocks for Eu are labelled as P, (perovskite) and RS (rock salt, K2NiF4-type blocks). For comparison with ref. 25, the structure has been shifted by (0.5,0.5,0) with respect to Fig. 4. | ||
We note that the 4f7 electrons of Eu2+ have well localized wave functions with tiny overlapping with neighboring Eu2+ ions. It follows that the direct 4f–4f interaction is negligible. Instead, as proposed long ago by Goodenough29 and Kasuya,30 intra-atomic 4f–5d intermixing offers a mechanism to propagate magnetic interactions. The 4f–5d intermixing is dictated by the relative position of the 4f and 5d orbitals, the latter being determined by the crystal field.25,29,30 As the coordination polyhedra of Eu2+ in Eu3Ta2ON5 and Eu2TiO4 have rather similar bond lengths, it can be suspected the 4f–5d mixing in these compounds to be similar. As the nn distances in both compounds differ only by some 1.5%, it follows that the nn Eu–Eu interactions (J11, J12) should be also similar, justifying the assumption made above.
Similarly, as the Eu–Eu nnn distances in both structures are also similar (within 1.8%), we conclude that the reinforced ferromagnetic Eu–Eu interaction results from the fact that the Eu–(N/O)–Eu hybridization is much enhanced. This can be explained by the higher hybridization (lower electronegativity of 2p(N) orbitals compared to 2p(O)) with 4f–5d orbitals of Eu. Enhanced ferromagnetic interactions induced by the large covalency of Mn–N bonds have also been observed in Ba2MnWO4.42N1.58 oxynitride.7
In short, the selective occupation of Eu3+ and Eu2+ in the two A sites of Eu3Ta2ON5 precludes magnetic dilution, that would be present if both cations would occupy the same site, and the reinforced hybridization of Eu–(N/O)–Eu bonds strengthens the ferromagnetic superexchange. On the other hand, this selective occupation implies the absence of bridging magnetic ions in the perovskite block (see Fig. 8). It follows that ferromagnetic order takes place basically within the Eu2+-containing bilayers of the rock-salt block. Therefore, a natural question arises: if the Eu2+ bilayers are magnetically decoupled, why do they appear similarly magnetically aligned in the magnetic measurements shown above? Tiny magnetic interactions between them may exist, either via direct dipolar coupling or via the presence of a small fraction of Eu2+ at the perovskite positions. However, none of those mechanisms, nor the combination of them, seem to be compatible with the observed large TC (≈20 K). Alternatively, the internal field associated to the Eu2+ ordering, may induce mixing of J = 1 and J = 0 in the ground state of Eu3+ (7FJ) mimicking the role of an external field which is responsible for the temperature-independent paramagnetism of Eu3+, with the consequent induction of a magnetic moment in an otherwise non-magnetic ion. The induced moment in Eu3+ may be responsible for the interlayer ferromagnetic coupling, as observed in Eu1−xYxMnO3.31 Therefore, the possibility arises that the two RS blocks of Eu2+ in the Eu3Ta2ON5 structure spontaneously order separately but are further aligned due to the magnetic applied field. Elucidating this possibility would only be possible by characterizing the magnetic order at zero field (e.g. by muon spectroscopy).
Conclusions
The new n = 2 Ruddlesden–Popper oxynitride Eu22+Eu3+Ta2ON5 has been prepared by a solid state reaction between EuN, Eu2O3 and Ta3N5 during 3 hours under N2 at 1200 °C. It crystallizes in the I4/mmm space group, with cell parameters a = 3.98240(1) and c = 20.42024(6) Å. Contrast inverted STEM iDPC images show long range ordered anion vacancies in the Eu layers between the TaX2 planes of the perovskite blocks, which decrease the coordination number of the transition metal from 6 to 5 anions, analogously to reported rare earth nitrides R3B2N6 (R = La, Ce, Pr; B = Nb, Ta). The anion vacancies generate two different environments for europium, with 8-coordinated Eu3+ located between the TaX2 planes and 9-coordinated Eu2+ in rock-salt layers. The magnetization data of Eu22+Eu3+Ta2ON5 shows ferromagnetic order of Eu2+ S = 7/2 spins below TC = 20 K. The selective occupation of Eu3+ and Eu2+ in the two sites gives rise to Eu3+ magnetically inert perovskite blocks that alternate with rock-salt blocks of ferromagnetic Eu2+ bilayers, where the presence of nitrogen at the anion sites leads to stronger Eu–(N/O)–Eu superexchange interactions and a record ordering temperature. The nitride introduction in the n = 2 Ruddlesden–Popper structure allows electronic and chemical orders and subsequent tailoring of magnetic interactions. New magnetic properties may emerge by further modification of the N/O ratio that certainly will modify the balance between the two europium oxidation states.Author contributions
J. O. S. and J. R. G.: investigation, formal analysis. B. M.: investigation. C. F. and J. G: investigation, formal analysis, methodology, writing. A. F. and J. F: conceptualization, supervision, writing.Data availability
The data used in this publication are available from the author on reasonable request. The synchrotron X-ray powder diffraction data are available through the following link https://doi.org/10.20350/digitalCSIC/17261.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants PID2023-146263NB-I00, PID2023-152225NB-I00, Severo Ochoa MATRANS42 CEX2023-001263-S funded by MCIN/AEI/10.13039/501100011033 (Ministerio de Ciencia e Innovación/Agencia Estatal de Investigación) and FEDER, EU, TED2021-129857B-I00 and PDC2023-145824-I00 funded by MCIN/AEI/10.13039/501100011033 and European Union Next Generation EU/PRTR, and grants 2021SGR00439 and 2021SGR00445 funded by the Generalitat de Catalunya. We thank ALBA synchrotron (experiment number AV-2024028275) for the provision of beam time; we also thank Dr François Fauth (ALBA) for assistance during data collection and Dr Bernat Bozzo (ICMAB-CSIC) for performing the magnetic measurements. JRG acknowledges AEI predoctoral fellowship PRE2018-085204. We are thankful for the assistance of ICMAB Scientific and Technological services of X-ray Diffraction, Low Temperature and Magnetism and Electron Microscopy. We acknowledge the Joint Electron Microscopy Center at ALBA (JEMCA) and funding from Grant IU16-014206 (METCAM-FIB) to ICN2 from the European Union through the European Regional Development Fund (ERDF), with the support of the Ministry of Research and Universities, Generalitat de Catalunya.References
- A. Fuertes, APL Mat., 2020, 8, 020903 CrossRef CAS.
- M. Jansen and H. P. Letschert, Nature, 2000, 404, 980 CrossRef CAS PubMed.
- Y. Kim, P. M. Woodward, K. Z. Baba-Kishi and C. W. Tai, Chem. Mater., 2004, 16, 1267 CrossRef CAS.
- A. B. Jorge, J. Oró-Solé, A. M. Bea, N. Mufti, T. T. M. Palstra, J. A. Rodgers, J. P. Attfield and A. Fuertes, J. Am. Chem. Soc., 2008, 130, 12572 CrossRef CAS PubMed.
- A. Kasahara, K. Nukumizu, G. Hitoki, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Phys. Chem. A, 2002, 106, 6750 CrossRef CAS.
- K. Ishida, C. Tassel, D. Watabe, H. Takatsu, C. M. Brown, G. J. Nilsen and H. Kageyama, Inorg. Chem., 2021, 60, 8252 CrossRef CAS PubMed.
- J. Oró-Solé, C. Frontera, J. R. Guarín, J. Gàzquez, B. Mundet, C. Ritter, J. Fontcuberta and A. Fuertes, Chem. Mater., 2024, 36, 10267 CrossRef PubMed.
- J. R. Guarín, C. Frontera, J. Oró-Solé, J. Gàzquez, C. Ritter, J. Fontcuberta and A. Fuertes, Inorg. Chem., 2023, 62, 17362 CrossRef PubMed.
- J. Oró-Solé, I. Fina, C. Frontera, J. Gàzquez, C. Ritter, M. Cunquero, P. Loza-Alvarez, S. Conejeros, P. Alemany, E. Canadell, J. Fontcuberta and A. Fuertes, Angew. Chem., Int. Ed., 2020, 59, 18395 CrossRef PubMed.
- F. Pors, R. Marchand and Y. Laurent, Ann. Chim., 1991, 16, 547 CAS.
- G. Tobías, J. Oró-Solé, D. Beltrán-Porter and A. Fuertes, Inorg. Chem., 2001, 40, 6867 CrossRef PubMed.
- R. Marchand, C. R. Acad. Sci., Ser. C, 1976, 282, 329 CAS.
- J. R. Guarín, C. Frontera, J. Oró-Solé, B. Colombel, C. Ritter, F. Fauth, J. Fontcuberta and A. Fuertes, Chem. Mater., 2024, 36, 5160 CrossRef PubMed.
- N. Cordes, M. Nentwig, L. Eisenburger, O. Oeckler and W. Schnick, Eur. J. Inorg. Chem., 2019, 2304 CrossRef CAS.
- M. Yang, J. Oró-Solé, A. Kusmartseva, A. Fuertes and J. P. Attfield, J. Am. Chem. Soc., 2010, 132, 4822 CrossRef CAS PubMed.
- L. Cario, Z. A. Gál, T. P. Braun, F. J. Di Salvo, B. Blaschkowski and H.-J. Meyer, J. Solid State Chem., 2001, 162, 90 CrossRef CAS.
- F. Fauth, I. Peral, C. Popescu and M. Knapp, Powder Diffr., 2013, 28, S360 CrossRef CAS.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55 CrossRef.
- I. Lazić, E. G. T. Bosch and S. Lazar, Ultramicroscopy, 2016, 160, 265 CrossRef PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 99, 751 CrossRef.
- L. Pauling, J. Am. Chem. Soc., 1929, 51, 1010 CrossRef CAS.
- A. Fuertes, Inorg. Chem., 2006, 45, 9640 CrossRef CAS PubMed.
- M. Mizumaki, Y. Saitoh, A. Agui, K. Yoshii, A. Fujimori and S. Nakamura, J. Synchrotron Radiat., 2001, 8, 440 CrossRef CAS PubMed.
- R. F. Egerton, Electron Energy-Loss Spectroscopy in the Electron Microscope, Springer, New York, NY, 2011 Search PubMed.
- C.-L. Chien, S. DeBenedetti, F. De and S. Barros, Phys. Rev. B, 1974, 10, 3913 CrossRef CAS.
- M. Adruh, E. Bakalbassis, O. Kahn, J. C. Trobe and P. Porcher, Inorg. Chem., 1993, 32, 1616 CrossRef.
- O. Khan, Molecular Magnetism, VHC Pubs, New York, 1993, pp. 46–48 Search PubMed.
- J. E. Greedan and G. J. McCarthy, Mater. Res. Bull., 1972, 7, 531 CrossRef CAS.
- J. B. Goodenough, Magnetism and Chemical Bond, Interscience, New York, 1963 Search PubMed.
- T. Kasuya, IBM J. Res. Dev., 1970, 14, 214 CAS.
- A. Skaugen, E. Schierle, G. van der Laan, D. K. Shukla, H. C. Walker, E. Weschke and J. Strempfer, Phys. Rev. B:Condens. Matter Mater. Phys., 2015, 91, 180409(R) CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |