
Discovery and structure–activity relationship study of novel isoxazole-based small molecules targeting Zika virus infections†
Berehe Solomon
Girmay
 ab,
Sileshi Abera
Ayele
ab,
Syed Azeem
Abbas
ab,
Su San
Jang
a,
Eunhye
Jung
a,
Jin Soo
Shin
a,
Soo Bong
Han
*ab and
Hyejin
Kim
*abc
ab,
Sileshi Abera
Ayele
ab,
Syed Azeem
Abbas
ab,
Su San
Jang
a,
Eunhye
Jung
a,
Jin Soo
Shin
a,
Soo Bong
Han
*ab and
Hyejin
Kim
*abc
aInfectious Diseases Therapeutic Research Center, Therapeutics and Biotechnology Division, Korea Research Institute of Chemical Technology, Daejeon 34114, Republic of Korea. E-mail: sbhan@krict.re.kr; hjinkim@krict.re.kr
bDepartment of Medicinal Chemistry and Pharmacology, University of Science and Technology, Daejeon 34113, Republic of Korea
cSchool of Pharmacy, Sungkyunkwan University, Suwon 16419, Republic of Korea
First published on 22nd July 2024
Abstract
The Zika virus (ZIKV), a significant public health threat, is transmitted by Aedes aegypti mosquitoes and is associated with severe neurological disorders, particularly in newborns. Currently, there are no approved vaccines or specific therapeutics for ZIKV. Our study focuses on the identification and optimization of isoxazole-based small molecules, specifically through the structural modification of KR-26827, to combat ZIKV infections. Among the synthesized derivatives, 7l emerged as the most promising candidate, showing potent antiviral activity against ZIKV strains and an improved safety profile in vitro. This research underlines the potential of 7l for further development as a ZIKV therapeutic agent.
1 Introduction
The Zika virus (ZIKV), an enveloped, positive-sense single-stranded RNA virus classified within the genus Flavivirus and family Flaviviridae, was first identified in the Zika forest of Uganda in 1947.1,2 Subsequently, cases of ZIKV infections in humans have been reported worldwide.3–9 The primary vector of ZIKV is the Aedes aegypti mosquito, similar to that of the other members of the genus Flavivirus.10,11 Additionally, ZIKV transmission can occur through blood transfusion or sexual contact.12,13 Of notable concern is the vertical transmission from a pregnant mother to her fetus, resulting in severe consequences for newborns, including microcephaly, fetal loss, and premature delivery.14,15 Furthermore, ZIKV infections are associated with neurological abnormalities,16 prompting the World Health Organization to declare ZIKV infection a global threat to public health in 2016.17 Despite extensive research endeavors, the absence of an approved vaccine or targeted pharmaceutic intervention remains a significant gap in addressing ZIKV infections.18,19Efforts in medicinal chemistry have been directed towards the development of drugs for ZIKV infections (Fig. 1). For example, compound 17, characterized by a carbazole scaffold, has been observed to inhibit the enzymatic activity of the NS2B/NS3 protease (50% inhibitory concentration [IC50] = 0.52 μM) and impede ZIKV replication in infected HuH-7 cells (50% effect concentration [EC50] = 1.25 μM; 50% cytotoxicity concentration [CC50] = 47.5 μM).20 Similar to compound 17, compound 1 was identified as a protease inhibitor, although it exhibited a diminished inhibitory effect on both protease inhibition and cellular activity (IC50 = 158 μM; EC50 = 13.9 μM).21 Likewise, a pyrazine analog, denoted as compound 47, exhibited enzymatic inhibitory effects on the ZIKV protease at a submicromolar level (IC50 = 0.2 μM) and demonstrated enhanced potency against ZIKV in U87 cells (EC68 = 0.3–0.6 μM).22 Another type of carbazole compound, compound 15, featuring urea substituents at the C3 position of the carbazole core, demonstrated antiviral activity against ZIKV in Vero E6 cells (EC50 = 1.67 μM; CC50 > 12.5 μM).23 Other types of chemical scaffolds have also been highlighted; for example, the andrographolide derivative 17B, exhibited cellular efficacy against ZIKV in Vero cells (EC50 = 4.5 μM; CC50 = 88.7 μM).24 Furthermore, C-SD5, a chloroquinoline with an alkyl sulfonamide substituent, exhibited micromolar cell-based potency against ZIKV-BR in cervical cells (EC50 = 3.2 μM).25 Additionally, a 4-amino-2-(4-benzylpiperazin-1-yl)methyl benzonitrile analog 3p, suppressed ZIKV replication in infected Vero E6 cells (EC50 = 5.1 μM; CC50 > 100 μM).26
 | ||
| Fig. 1 Reported compounds for ZIKV inhibitors. | ||
In 2022, our research group reported significant findings on a series of 1,2,4-oxadiazole derivatives, notably KR-26827 (Fig. 1).27 The optimized compound KR-26827, demonstrated antiviral activity against ZIKV MR766 strains (EC50 = 1.35 μM) and exhibited low cellular toxicity (CC50 > 50 μM). However, despite the demonstrated efficacy of KR-26827, comprehensive investigations into other in vitro safety parameters and biochemical profiles remain pending, encompassing assessments for inhibition of CYP enzymes and human ether a-go-go-related gene (hERG), as well as cytotoxicity. Therefore, in this study, we aimed to identify potent and safe compounds for the treatment of ZIKV infections through the structural modification of KR-26827.
2 Results and discussion
2.1 Synthesis
Several compounds, including 6d, 7a–7r and 8a–8m, were synthesized using a general synthetic procedure, as described in Scheme 1 (see ESI†). The initial step involved the reaction of benzaldehyde derivatives (1a and 1b) and hydroxylamine hydrochloride in ethanol at room temperature, yielding the target product oximes (2a and 2b). Subsequently, the oxime intermediates (2a and 2b) were subjected to a chlorination reaction with N-chlorosuccinimide in dimethylformamide at room temperature, resulting in N-hydroxybenzimidoyl chloride derivatives (3a and 3b). The chlorinated intermediates (3a and 3b) were combined with alkynyl derivatives in t-butanol/tetrahydrofuran in the presence of copper(I) iodide and potassium bicarbonate at room temperature for 24 h, resulting in the formation of isoxazole derivatives 4a–4s and 5a–5m. Finally, the intermediates 4a–4s and 5a–5m were subjected to a reaction with 3-picolylamine in dimethyl sulfoxide in the presence of copper(I) iodide, L-proline, and potassium carbonate, to obtain the products 6d, 7a–7r and 8a–8m.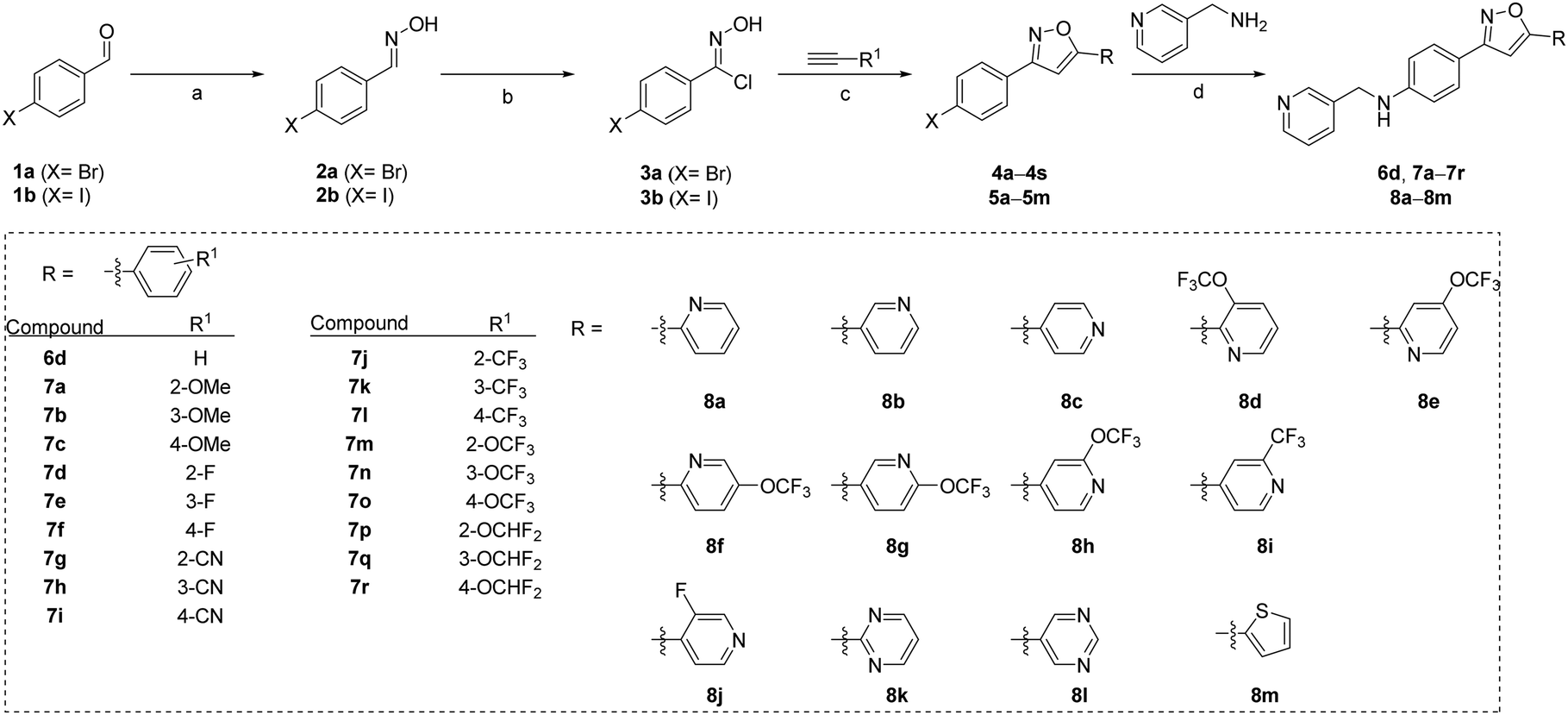 | ||
Scheme 1 Synthesis of compounds 6d, 7a–7r and 8a–8m. Reagents and conditions: (a) hydroxylamine hydrochloride (1.2 eq.), pyridine (2.4 eq.), ethanol (0.5 M), room temperature, 1 h; (b) N-chlorosuccinimide (2.4 eq.), dimethylformamide (0.5 M), room temperature, 4 h; (c) copper(I) iodide (0.07 eq.), t-butanol/tetrahydrofuran (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 0.3 M), room temperature, 10 min, potassium bicarbonate (1.0 eq.), room temperature, 24 h; (d) copper(I) iodide (10 mol%), L-proline (20 mol%), potassium carbonate (3.0 eq.), dimethyl sulfoxide (0.5 M), 80 °C, 14 h. :1, 0.3 M), room temperature, 10 min, potassium bicarbonate (1.0 eq.), room temperature, 24 h; (d) copper(I) iodide (10 mol%), L-proline (20 mol%), potassium carbonate (3.0 eq.), dimethyl sulfoxide (0.5 M), 80 °C, 14 h. | ||
The synthetic route described in Scheme 2 (see ESI†) was used to synthesize the compound 4-(5-(1-methyl-1H-pyrrol-2-yl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (9d). The initial step involved the mixing of 1-(4-bromophenyl)ethan-1-one (9a) and 1-methyl-1H-pyrrole-2-carbaldehyde in ethanol in the presence of a 40% aqueous solution of sodium hydroxide at room temperature to obtain the chalcone derivative (E)-1-(4-bromophenyl)-3-(1-methyl-1H-pyrrol-2-yl)prop-2-en-1-one (9b). Subsequently, the intermediate (9b) was treated with hydroxylamine hydrochloride in ethanol, followed by the addition of sodium acetate in hot acetic acid. The resulting mixture was refluxed, leading to cyclization and formation of 3-(4-bromophenyl)-5-(1-methyl-1H-pyrrol-2-yl)isoxazole (9c). Finally, the intermediate 9c was combined with 2-picolyiamine in dimethyl sulfoxide in the presence of copper(I) iodide, L-proline, and potassium carbonate to generate the required product 9d.
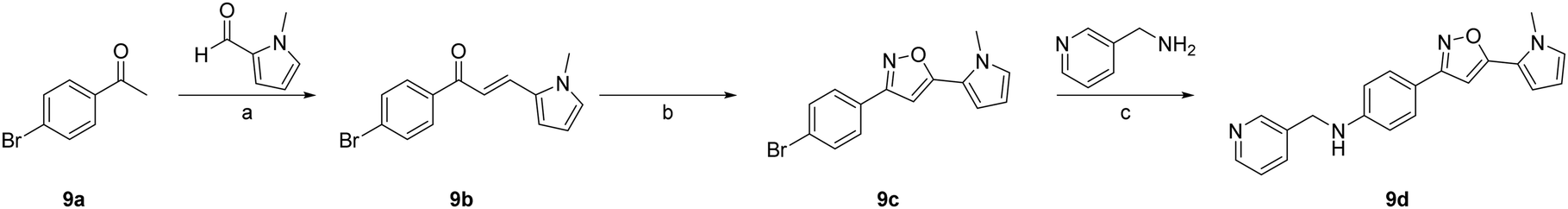 | ||
| Scheme 2 Synthesis of compound 9d. Reagents and conditions: (a) aqueous sodium hydroxide (40%), ethanol (0.3 M), 0 °C to room temperature, 12 h; (b) hydroxylamine hydrochloride (1.5 eq.), sodium acetate (1.5 eq.), acetic acid (0.5 M), ethanol (0.01 M), reflux for 8 h; (c) copper(I) iodide (10 mol%), L-proline (20 mol%), potassium carbonate (3.0 eq.), dimethyl sulfoxide (0.5 M), 80 °C, 14 h. | ||
The synthetic route illustrated in Scheme 3 (see ESI†) was used to synthesize the compound 4-(5-morpholinoisoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (10f). The sequence commenced with the synthesis of 3-(4-nitrophenyl)isoxazol-5(4H)-one (10b), achieved by reacting ethyl 3-(4-nitrophenyl)-3-oxopropanoate (10a) with hydroxylamine hydrochloride in ethanol in the presence of potassium carbonate at 78 °C for 4 h. Subsequently, the intermediate (10b) was subjected to a chlorination reaction using phosphorus oxychloride and triethylamine at 0 °C to 100 °C for 16 h to obtain the desired product 5-chloro-3-(4-nitrophenyl)isoxazole (10c). Next, the intermediate 10c was combined with morpholine in dimethylformamide in the presence of potassium carbonate at 80 °C for 12 h, resulting in the formation of 4-(3-(4-nitrophenyl)isoxazole-5-yl)morpholine (10d). The conversion of the nitro derivative (10d) to an amine was conducted using tetrahydroxydiboron and 4,4′-bipyridine in dimethylformamide at room temperature for 5 min to obtain the required product 4-(5-morpholinoisoxazol-3-yl)aniline (10e). Finally, the intermediate 10e was subjected to a reductive amination reaction with pyridine-2-carbaldehyde and trifluoroacetic acid in dry tetrahydrofuran using sodium cyanoborohydride from 0 °C to room temperature for 12 h to obtain the desired product 10f.
 | ||
| Scheme 3 Synthesis of compound 10f. Reagents and conditions: (a) hydroxylamine hydrochloride (1.5 eq.), potassium carbonate (1.5 eq.), ethanol (0.5 M), room temperature to 78 °C, 4 h; (b) phosphorus oxychloride (7.0 eq.), triethylamine (1.0 eq.), 0 °C to 100 °C, 16 h; (c) potassium carbonate (2.5 eq.), dimethylformamide (0.5 M), 80 °C, 12 h; (d) tetrahydroxydiboron (3.0 eq.), 4,4′-bipyridine (0.05 eq.), dimethylformamide (0.5 M), 5 min; (e) sodium cyanoborohydride (3.0 eq.), trifluoroacetic acid (1.5 eq.), tetrahydrofuran (0.2 M), 0 °C to room temperature, 12 h. | ||
2.2 In vitro safety profile of KR-26827
According to the in vitro safety profile for KR-26827 (Table 2), KR-26827 displayed inhibitory effects on five cytochrome P450 (CYP) isozymes (IC50 < 17 μM), indicating a potential risk of adverse drug–drug interactions. Moreover, significant inhibition of hERG was observed, raising concerns related to cardiac toxicity. Evaluation of the cytotoxicity of five distinct mammalian cell lines (VERO, HFL-1, L929, NIH 3T3, and CHO-K1) revealed IC50 values ranging from 33.0 to 43.7 μM. These findings indicate safety concerns associated with KR-26827, underscoring the imperative for further structural modifications to improve its safety profile.2.3 Structure–activity relationship (SAR) of KR-26827
Commencing from the preceding compound, KR-26827, structural modifications were conducted on the A- and B-rings (Fig. 2). In this study, all compounds in Tables 1, 3, and 4 (6a–6f, 7a–7r, 8a–8m, 9d, and 10f), except for KR-26827, were newly synthesized and evaluated for their antiviral efficacy. Regarding the A-ring modification, various 5-membered heteroaromatic fragments were employed as substitutes for the 1,2,4-oxadiazole moiety (Table 1). Analysis of a structural isomer of 1,2,4-oxadiazole with a different position of the oxygen atom revealed that the compound 6a demonstrated lower potency (entry 2; EC50 = 5.3 μM) than KR-26827. A 1,3,4-oxadiazole-containing compound, 6b, demonstrated no activity against ZIKV (entry 3; EC50 > 100 μM). After the removal of one of the nitrogen atoms, resulting in 6c, the activity was restored despite its cellular toxicity (entry 4). Compared to the oxazole moiety in 6c, the isoxazole demonstrated better cellular activity and toxicity. 6d exhibited a similar EC50 level without apparent cytotoxicity, prompting further investigation into its properties. In contrast to the oxazole derivatives, imidazole did not display activity against ZIKV-infected cells (6e and 6f; entries 6 and 7).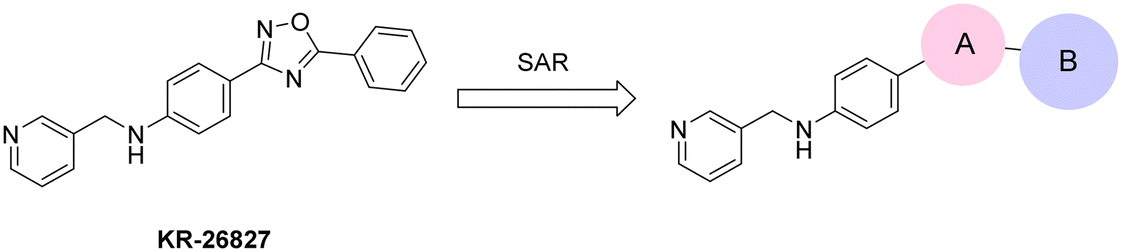 | ||
| Fig. 2 Structural modification of 1,2,4-oxadiazole derivative. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Compound | A | ClogP | EC50a (μM) | CC50 (μM) | SIb |
| a The antiviral activity (EC50 value) was assessed using the ZIKV MR766 strain. b SI: selectivity index. c nd: not determined. | ||||||
| 1 | KR-26827 |

|
3.4 | 1.35 | >50 | >37.0 |
| 2 | 6a |
|
3.4 | 5.3 | 61.4 | 11.6 |
| 3 | 6b |
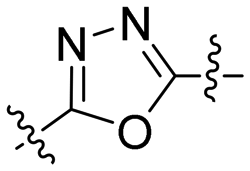
|
2.53 | >100 | >100 | ndc |
| 4 | 6c |
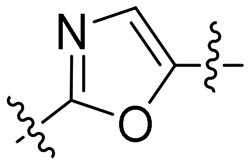
|
3.66 | 5.1 | 12.9 | 2.5 |
| 5 | 6d |
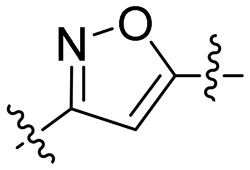
|
3.95 | 2.8 | >100 | >35.9 |
| 6 | 6e |
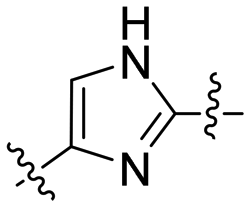
|
3.81 | 36.8 | 94.1 | 2.6 |
| 7 | 6f |

|
3.83 | >100 | >100 | ndc |

2.4 In vitro safety and biochemical profile of 6d
Table 2 summarizes the results of the in vitro safety and biochemical profile of 6d. The IC50 value for CYP inhibition by 6d indicated improved CYP inhibitory effects compared to the previous compound, KR-26827. However, concerns remain, particularly regarding its effect on 2C9 isozyme (IC50 = 2.6 μM). The ligand-binding assay for hERG also demonstrated a reduced value of percentage inhibition, resulting in a two-fold improvement over KR-26827. Cytotoxic effects were examined in various mammalian cell lines, revealing no detectable cytotoxic effects. Additionally, tests for liver microsomal stability and plasma protein binding were conducted, culminating in the final summary of the biochemical profiles of 6d.| In vitro assay | KR-26827 | 6d |
|---|---|---|
| a CYP inhibition in human liver microsomes using cocktail substrate assay. b hERG ligand binding assay % inhibition at 10 μM concentration. c The IC50 in various mammalian cell lines; (VERO: African green monkey kidney cell line, HFL-1: human embryonic lung cell line; L929: NCTC clone 929 mouse fibroblast cell line; NIH 3T3: mouse embryonic fibroblast cell line, CHO-K1: Chinese hamster ovary cell line). d Liver microsomal phase I stability (%) remaining after 30 min. e Plasma protein binding rate (%) remaining after 4 h incubation at 37 °C. f na: not applicable. Assay conditions are described in ESI.† | ||
| CYP inhibition (IC50, μM) | ||
| 1A2 | 8.3 | 21.5 |
| 2C9 | 2.0 | 2.6 |
| 2C19 | 14.3 | 25.8 |
| 2D6 | 16.9 | >100 |
| 3A4 | 12.1 | 23.4 |
| Cardiotoxicity (% inhibition, 10 μM) | ||
| hERG | 43.5 | 19.2 |
| Cytotoxicity assay (IC50, μM) | ||
| VERO | 40.0 | >100 |
| HFL-1 | 43.7 | >100 |
| L929 | 39.2 | >100 |
| NIH 3T3 | 33.0 | >100 |
| CHO-K1 | 36.5 | >100 |
| Liver microsomal phase I stability (%) | ||
| Mouse | naf | 74.5 |
| Rat | naf | 41.6 |
| Human | naf | 93.0 |
| Plasma protein binding (%) | ||
| Mouse | naf | 99.92 |
| Human | naf | 98.16 |
2.5 SAR study of 6d
Our next endeavor involved the structural modification of 6d through the exploration of substituted B-ring structures (Table 3). In conjunction with EC50 and CC50 values for antiviral and cytotoxic effects, respectively, CYP inhibition was examined to evaluate the potential safety of compounds. Phenyl groups with different substituents at ortho-, meta-, and para-methoxy positions were introduced. Methoxy-substituted aromatic compounds exhibited reduced antiviral effects (entries 2–4; 7a–7c), among which meta-substitution demonstrated better EC50 values, albeit with a lower CC50 value (entry 3; 7b). For aryl fluoride-containing compounds, the introduction of fluoride at the ortho position improved antiviral activity; however, the CC50 value yielded a lower selective index (SI) compared to that of 6d (entry 1 vs. entries 5–7; 6dvs.7d–7f). Regarding meta-fluoro phenyl substituted compound 7e, the SI value increased without showing the cytotoxic effect, although the CYP inhibition of five isozymes raised concerns regarding drug–drug interactions (entry 6).|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Compound | R | ClogP |
Antiviral assay result | CYP inhibitiond (IC50) | ||||||
| EC50a (μM) | C 50 (μM) | SIb | 1A2 | 2C9 | 2C19 | 2D6 | 3A4 | ||||
| a The antiviral activity (EC50 value) was assessed using the ZIKV MR766 strain. b SI: selectivity index. c nd: not determined. d The IC50 of CYP inhibition in human liver microsomes using cocktail substrate assay. | |||||||||||
| 1 | 6d | H | 3.95 | 2.9 | >100 | >35.9 | 21.5 | 2.6 | 25.8 | >100 | 23.4 |
| 2 | 7a | 2-OMe | 3.42 | 13.2 | >100 | >7.6 | 25.4 | 3.2 | 15.0 | 27.1 | 7.6 |
| 3 | 7b | 3-OMe | 3.98 | 7.3 | 27.1 | 3.7 | 10.9 | 1.2 | 5.0 | 15.0 | 3.8 |
| 4 | 7c | 4-OMe | 3.98 | 20.3 | >100 | >4.9 | >100 | 0.6 | 43.2 | >100 | 4.2 |
| 5 | 7d | 2-F | 4.12 | 2.3 | 33.3 | 14.8 | 14.5 | 3.8 | 20.8 | 23.4 | 18.5 |
| 6 | 7e | 3-F | 4.12 | 3.3 | >100 | >30.7 | 12.5 | 2.8 | 12.2 | 25.4 | 6.0 |
| 7 | 7f | 4-F | 4.12 | >100 | >100 | ndc | >100 | 5.7 | 52.4 | 56.4 | 32 |
| 8 | 7g | 2-CN | 3.46 | 73.1 | >100 | >1.4 | 21.2 | 4.0 | 32.9 | 118.6 | 55.8 |
| 9 | 7h | 3-CN | 3.46 | 21.5 | >100 | >4.7 | 153.1 | 4.9 | 21.6 | 172.6 | 1.2 |
| 10 | 7i | 4-CN | 3.46 | 81.4 | >100 | >1.2 | >99 | >99 | >99 | >99 | 52.3 |
| 11 | 7j | 2-CF3 | 4.89 | 1.5 | >100 | >65.4 | 10.6 | 1.8 | 17.2 | 28.4 | 1.8 |
| 12 | 7k | 3-CF3 | 4.89 | 5.9 | >100 | >16.9 | 61.5 | 7.9 | 31.7 | 85.4 | 11.4 |
| 13 | 7l | 4-CF3 | 4.89 | 1.1 | >100 | >90.9 | >99 | >99 | 81.6 | >99 | 15.9 |
| 14 | 7m | 2-OCF3 | 4.53 | 44.5 | >100 | >2.3 | 10.6 | 0.9 | 6.2 | 3.5 | 6.2 |
| 15 | 7n | 3-OCF3 | 5.09 | 18.6 | >100 | >5.4 | 48.1 | 9.4 | 16.8 | 30.9 | 15.1 |
| 16 | 7o | 4-OCF3 | 5.09 | 2.4 | >100 | >41.8 | >99 | 47.9 | 88.6 | >99 | 7.1 |
| 17 | 7p | 2-OCHF2 | 3.86 | 44.7 | >100 | >2.2 | 3.3 | 1.2 | 3.1 | 13.8 | 1.9 |
| 18 | 7q | 3-OCHF2 | 4.42 | 1.8 | >100 | >55.2 | 13.9 | 0.9 | 6.8 | 17.9 | 1.7 |
| 19 | 7r | 4-OCHF2 | 4.42 | >100 | >100 | ndc | >99 | 69.7 | 82.4 | >99 | 11.4 |
The introduction of cyano groups into the phenyl moiety resulted in compounds with low antiviral potency (entries 8–10; 7g–7i). Subsequently, trifluoromethyl phenyl compounds were prepared and tested, with all three types of derivatives exhibiting comparable activity for ZIKV inhibition in cellular assays (entries 11–13; 7j–7l). Among this series, 7l demonstrated the highest potency with no detectable cytotoxicity (entry 13). Moreover, the presence of compound 7l did not raise any safety concerns related to CYP inhibition (entry 13). Compounds with fluoromethoxy phenyl substituents, such as trifluoromethoxy and difluoromethoxy phenyl compounds, consistently exhibited no cytotoxicity (entries 14–19; 7m–7r). Among these compounds, those with para-trifluoromethoxy and meta-difluoromethoxy substituents showed improved potency and SI values compared to 6d; however, CYP inhibition still remained a concern (entries 16 and 18; 7o and 7q).
In addition, the activity and toxicity of compounds incorporating heterocyclic substituents were assessed (Table 4). The synthesis of 2-, 3-, and 4-pyridyl compounds was performed based on the positional isomerism of nitrogen (entries 1–3; 8a–8c). Notably, 3-pyridyl compound demonstrated the better antiviral activity and SI value than its 2-pyridyl counterpart. Among these, 4-pyridyl compound exhibited the most favorable activity, with its EC50 value of 0.79 μM and SI value of 52.3. However, all three compounds exhibited low inhibitory concentrations (IC50) against CYP isozymes.
|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Compound | B | ClogP |
Antiviral assay result | CYP inhibitiond (IC50) | ||||||
| EC50a (μM) | CC50 (μM) | SIb | 1A2 | 2C9 | 2C19 | 2D6 | 3A4 | ||||
| a The antiviral activity (EC50 value) was assessed using the ZIKV MR766 strain. b SI: selectivity index. c nd: not determined. d The IC50 of CYP inhibition in human liver microsomes using cocktail substrate assay. | |||||||||||
| 1 | 8a |

|
2.84 | 15.2 | 60.7 | 4.0 | 47.1 | 2.4 | 12.4 | 27.5 | 9.0 |
| 2 | 8b |
|
2.63 | 8.2 | 55.1 | 6.7 | 13.5 | 1.7 | 18.3 | 19.2 | 3.7 |
| 3 | 8c |

|
2.63 | 0.79 | 41.3 | 52.3 | 55.2 | 8.4 | 39.6 | 15.6 | 4.2 |
| 4 | 8d |

|
3.80 | 2.1 | >100 | >47.8 | 16.5 | 1.7 | 9.5 | 20.5 | 4.4 |
| 5 | 8e |

|
4.37 | 3.5 | >100 | >28.5 | >99 | 3.2 | 48.4 | 41 | 3.3 |
| 6 | 8f |
|
4.36 | 1.2 | >100 | >83.3 | >99 | 14.2 | 50.4 | >99 | 11.6 |
| 7 | 8g |
|
4.37 | 1.4 | >100 | >71.4 | 57.1 | 3.4 | 45.3 | >99 | 16.9 |
| 8 | 8h |
|
3.55 | 84.4 | >100 | >1.2 | 44.6 | >99 | 86.6 | 73.7 | >99 |
| 9 | 8i |
|
3.63 | >100 | >100 | ndc | 90.9 | 3.3 | 40.8 | 130 | 11.6 |
| 10 | 8j |
|
2.82 | 3.0 | >100 | >33.9 | 10.5 | 2.2 | 12.1 | 10.6 | 2.6 |
| 11 | 8k |
|
1.88 | 46.2 | >100 | >2.2 | 28.4 | 2.1 | 12.6 | 11.3 | 15.8 |
| 12 | 8l |

|
1.67 | >100 | >100 | ndc | 10.6 | 0.9 | 6.2 | 3.5 | 6.2 |
| 13 | 8m |

|
3.86 | 16.4 | >100 | >6.1 | 36.4 | 22.3 | 53.8 | 80.2 | 54.5 |
| 14 | 9d |

|
3.30 | 5.0 | 76 | 15.1 | 36.4 | 2.1 | 30.9 | 85.5 | 14.1 |
| 15 | 10f |

|
1.84 | 10.1 | >100 | >9.9 | 38.9 | 3.4 | 29.5 | 11.8 | 8.2 |

Compounds with substituted pyridyl moieties were also synthesized (entries 4–6). Compounds 8d–8f, featuring a trifluoromethoxy substituent in the 2-pyridyl group, displayed notable antiviral efficacy. Particularly, 8f demonstrated the highest SI value, with an EC50 1.2 μM and no observed toxicity (entry 6). However, these compounds displayed low IC50 values against 2C9 and 3A4 CYP isozymes, suggesting the necessity for further structural optimization.
Similarly, a 3-pyridyl compound with a 4-trifluoromethoxy group, 8g, exhibited cellular efficacy comparable to that of 8f but displayed pronounced CYP inhibition (entry 7). In contrast, the introduction of 3-trifluoromethoxy and 3-trifluoromethyl moiety into 8c, exemplified by 8h and 8i, resulted in a significant decrease in antiviral activity (entries 8 and 9). While 8h displayed lowered CYP inhibition, 8i posed challenges in CYP inhibition. Additionally, the antiviral efficacy of the 2-fluoro-4-pyridyl group rebounded; however, all five CYP isozymes tested were highly inhibited (entry 3 vs. 10; 8cvs.8j).
The effects of the number of nitrogen atoms in the B-ring were investigated. Compounds 8k and 8l, featuring 2-pyrimidyl and 5-pyrimidyl moieties, respectively, exhibited no activity in inhibiting ZIKV and showed severe CYP inhibition (entries 11 and 12). The 5-membered heteroaromatic compounds 8m and 9d exhibited reduced antiviral efficacy (entries 13 and 14), compared to 7l. Finally, as an example of aliphatic heterocyclic compounds, the efficacy of the morpholine compound 10f was examined (entry 15). The results indicated no improvement in antiviral efficacy or CYP inhibition.
2.6 In vitro safety and biochemical profile of 7l
In the course of our studies, 6d was initially selected due to its promising safety profile and improved CYP inhibitory effects compared to the previous compound, KR-26827. However, despite its advancements, 6d exhibited significant concerns, particularly with the 2C9 isozyme, necessitating further structural modifications. Consequently, 7l emerged through these modifications, demonstrating an enhanced safety profile and potent antiviral efficacy, thereby justifying its selection for further in vitro safety and biochemical profiling.By optimizing the structural–activity relationship and drawing insights from previous research, our primary objective was to ameliorate the potential drug toxicity resulting from the compounds, such as 6d, through CYP inhibition assessment. Concurrently, we aimed to identify compounds with enhanced therapeutic efficacy. Ultimately, 7l emerged as the promising compound, demonstrating potent antiviral efficacy without inducing cytotoxic effects. Notably, the IC50 values for CYP inhibition revealed negligible inhibitory activity across four of the five tested isozymes (1A2, 2C9, 2C19, and 2D6), even at elevated concentrations, with an IC50 value of 15.9 μM for 3A4, indicating its comparatively elevated safety profile among analogs (Table 3).
Subsequently, further experiments were conducted using 7l to obtain additional in vitro safety and biochemical profiles (Table 5). The hERG ligand-binding assay confirmed low hERG inhibition even at a high concentration of 10 μM, indicating a low potential for cardiotoxicity. Moreover, toxicity evaluations across diverse mammalian cell lines corroborated the acceptable safety profile of this compound. Assessment of hepatic metabolism via liver microsomal phase l stability experiments also confirmed its favorable metabolic profile. Finally, we conducted measurements to determine the affinity of the compound for binding to plasma proteins to evaluate its significance.
| In vitro assay | 7l |
|---|---|
| a hERG ligand binding assay %inhibition at 10 μM concentration. b The IC50 in various mammalian cell lines; (VERO: African green monkey kidney cell line, HFL-1: human embryonic lung cell line; L929: NCTC clone 929 mouse fibroblast cell line; NIH 3T3: mouse embryonic fibroblast cell line, CHO-K1: Chinese hamster ovary cell line). c Liver microsomal phase I stability (%) remaining after 30 min. d Plasma protein binding rate (%) remaining after 4 h incubation at 37 °C. Assay conditions are described in ESI.† | |
| Cardiotoxicity (% inhibition, 10 μM) | |
| hERG | 5.37 |
| Cytotoxicity assay (IC50, μM) | |
| VERO | >100 |
| HFL-1 | >100 |
| L929 | >100 |
| NIH3T3 | >100 |
| CHO-K1 | >100 |
| Liver microsomal phase I stability (%) | |
| Mouse | 79.6 |
| Rat | 51.7 |
| Human | 71.5 |
| Plasma protein binding (%) | |
| Mouse | 99.7 |
| Human | 99.53 |
3 Conclusions
Our study represents a significant step forward in the development of treatments against Zika virus (ZIKV) infections. Through the optimization of KR-26827, we identified 6d, which showed improved CYP inhibitory effects and a promising safety profile. However, further structural modifications led to the identification of 7l, which demonstrated enhanced safety and potent antiviral efficacy.Using systematic medicinal chemistry approaches, we focused on structural modifications to improve the antiviral activities and safety profiles of various compounds. 7l emerged as the most promising candidate, showing potent antiviral activity against ZIKV strains and an improved safety profile in vitro. However, high plasma protein binding was observed for 7l, indicating the need for future optimization to reduce this binding.
Future studies will focus on elucidating the mechanism of action of 7l and conducting additional preclinical evaluations to confirm its safety and efficacy. These steps are essential for advancing 7l as a potential therapeutic agent. We are committed to furthering this research and will share our findings to address the urgent need for effective ZIKV therapeutics.
4 Experimental section
4.1 Biology
4.2 Chemistry
Synthesis of 4-(5-phenylisoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (6d). The title compound was prepared from 4a (0.17 g, 0.56 mmol) and pyridine-3-ylmethanamine (0.18 g, 1.68 mmol) in the manner described above general procedure to obtain desired product (91.0 mg, 49%). 1H NMR (300 MHz, DMSO-d6): δ 8.63 (s, 1H), 8.48 (s, 1H), 7.89 (dd, J = 8.1, 1.8 Hz, 2H), 7.77 (d, J = 7.8 Hz, 1H), 7.63 (d, J = 8.7 Hz, 2H), 7.59–7.50 (m, 3H), 7.41 (s, 1H), 7.39–7.35 (m, 1H), 6.83–6.79 (m, 1H), 6.73 (d, J = 8.7 Hz, 2H), 4.39 (d, J = 6.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6): δ 169.30, 163.09, 150.47, 149.40, 148.57, 135.51, 130.72, 129.71, 128.14, 127.60, 125.94, 116.44, 112.84, 98.45, 44.17. HRMS (FAB+) m/z calcd. for C21H18N3O+ [M + H]+: 328.1444 found 328.1448.
Synthesis of 4-(5-(2-methoxyphenyl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (7a). The title compound was prepared from 4b (0.07 g, 0.21 mmol) and pyridine-3-ylmethanamine (0.07 g, 0.63 mmol) in the manner described above general procedure to obtain desired product (5.40 mg, 2%, two step yield). 1H NMR (300 MHz, DMSO-d6): δ 8.61 (s, 1H), 8.47–8.46 (m, 1H), 7.86 (dd, J = 7.8, 1.8 Hz, 1H), 7.79–7.75 (m, 1H), 7.66–7.64 (m, 2H), 7.53–7.47 (m, 1H), 7.39–7.35 (m, 1H), 7.25–7.19 (m, 2H), 7.14–7.09 (m, 1H), 6.79 (t, J = 6.3 Hz, 1H), 6.73–6.70 (m, 2H), 4.39 (d, J = 5.7 Hz, 2H), 3.98 (s, 3H). 13C NMR (101 MHz, DMSO-d6): δ 165.54, 162.91, 156.42, 150.36, 149.40, 148.57, 135.67, 135.53, 132.08, 128.17, 127.41, 123.97, 121.24, 116.66, 116.12, 112.83, 112.59, 101.56, 56.22, 44.18. HRMS (FAB+) m/z calcd. for C22H20N3O2+ [M + H]+: 358.1550 found 358.1563.
Synthesis of 4-(5-(3-methoxyphenyl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (7b). The title compound was prepared from 4c (0.07 g, 0.21 mmol) and pyridine-3-ylmethanamine (0.07 g, 0.63 mmol) in the manner described above general procedure to obtain the desired product (7.0 mg, 9%). 1H NMR (300 MHz, CD3OD): δ 8.58 (s, 1H), 8.43 (s, 1H), 7.85 (d, J = 7.8 Hz, 1H), 7.63 (d, J = 9.0 Hz, 2H), 7.42–7.37 (m, 4H), 7.06–7.00 (m, 2H), 6.70 (d, J = 8.7 Hz, 2H), 4.44 (s, 2H), 3.86 (s, 3H). 13C NMR (101 MHz, DMSO-d6): δ 169.17, 163.09, 160.17, 150.47, 149.38, 148.56, 135.64, 135.54, 130.94, 128.80, 128.13, 123.99, 118.22, 116.60, 116.42, 112.84, 111.06, 98.79, 55.82, 44.15. HRMS (FAB+) m/z calcd. for C22H20N3O2+ [M + H]+: 358.1550 found 358.1554.
Synthesis of 4-(5-(4-methoxyphenyl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (7c). The title compound was prepared from 4d (0.09 g, 0.27 mmol) and pyridine-3-ylmethanamine (0.09 g, 0.81 mmol) in the manner described above general procedure to obtain the desired product (26.0 mg, 27%). 1H NMR (300 MHz, DMSO-d6): δ 8.62 (d, J = 2.4 Hz, 1H), 8.46 (dd, J = 4.5, 1.5 Hz, 1H), 7.83–7.76 (m, 3H), 7.61 (d, J = 8.4 Hz, 2H), 7.39–7.34 (m, 1H), 7.24 (s, 1H), 7.10 (d, J = 9 Hz, 2H), 6.79 (t, J = 6 Hz, 1H), 6.72 (d, J = 8.7 Hz, 2H), 4.39 (d, J = 6.0 Hz, 2H), 3.83 (s, 3H). 13C NMR (101 MHz, DMSO-d6): δ 169.34, 162.99, 161.17, 150.39, 149.39, 148.56, 135.65, 135.53, 128.09, 127.62, 123.96, 120.32, 116.62, 115.11, 112.81, 96.93, 55.84, 44.16. HRMS (FAB+) m/z calcd. for C22H20N3O2+ [M + H]+: 358.1550 found 358.1565.
Synthesis of 4-(5-(2-fluorophenyl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (7d). The title compound was prepared from 4e (0.20 g, 0.63 mmol) and pyridine-3-ylmethanamine (0.20 g, 1.89 mmol) in the manner described above general procedure to obtain the desired product (30.0 mg, 14%). 1H NMR (500 MHz, CD3OD): δ 8.59 (s, 1H), 8.44 (d, J = 5 Hz, 1H), 7.99–7.96 (m, 1H), 7.90–7.88 (m, 1H), 7.67 (d, J = 8.5 Hz, 2H), 7.55–7.52 (m, 1H), 7.44–7.42 (m, 1H), 7.39–7.31 (m, 2H), 7.08 (d, J = 3.5 Hz, 1H), 6.74 (d, J = 8.5 Hz, 2H), 4.49 (s, 2H). 13C NMR (101 MHz, DMSO-d6): δ 163.57 (d, J = 2.6 Hz), 163.16, 150.84 (d, J = 253.0 Hz), 150.54, 149.37, 148.56, 135.63, 135.52, 132.83 (d, J = 8.6 Hz), 128.29, 128.15 (d, J = 2.0 Hz), 125.77 (d, J = 3.4 Hz), 123.98, 117.10 (d, J = 21.2 Hz), 116.12, 115.64 (d, J = 12.22 Hz) 112.84, 101.71 (d, J = 8.9 Hz), 44.14. HRMS (FAB+) m/z calcd. for C21H17FN3O+ [M + H]+: 346.1350 found 346.1355.
Synthesis of 4-(5-(3-fluorophenyl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (7e). The title compound was prepared from 4f (0.05 g, 0.16 mmol) and pyridine-3-ylmethanamine (0.05 g, 0.48 mmol, 3.0 eq.) in the manner described above general procedure to obtain the desired product (20.0 mg, 37%). 1H NMR (500 MHz, DMSO-d6): δ 8.65 (s, 1H), 8.49 (s, 1H), 7.79–7.72 (m, 3H), 7.63–7.59 (m, 3H), 7.51 (s, 1H), 7.39–7.35 (m, 2H), 6.85–6.82 (m, 1H), 6.74 (d, J = 8.0 Hz, 2H), 4.40 (d, J = 6.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6): δ 168.00 (d, J = 2.9 Hz), 163.19, 162.89 (d, J = 245.4 Hz), 150.56, 149.41, 148.58, 135.49, 132.00 (d, J = 8.6 Hz), 129.61 (d, J = 8.6 Hz), 128.13, 122.08 (d, J = 2.8 Hz), 117.55 (d, J = 21.0 Hz), 116.18, 112.90, 112.85, 112.67, 99.57, 44.15. HRMS (FAB+) m/z calcd. for C21H17FN3O+ [M + H]+: 346.1350 found 346.1360.
Synthesis of 4-(5-(4-fluorophenyl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (7f). The title compound was prepared from 4g (0.20 g, 0.63 mmol) and pyridine-3-ylmethanamine (0.20 g, 1.89 mmol) in the manner described above general procedure to obtain the desired product (30.0 mg, 14%). 1H NMR (500 MHz, CD3OD): δ 8.60 (s, 1H), 8.45 (s, 1H), 7.94–7.88 (m, 3H), 7.65 (d, J = 8.0 Hz, 2H), 7.44–7.42 (m, 1H), 7.28 (t, J = 8.5 Hz, 2H), 7.09 (s, 1H), 6.73 (d, J = 8.5 Hz, 2H), 4.49 (s, 2H). 13C NMR (101 MHz, DMSO-d6): δ 168.38, 163.50 (d, J = 249.0 Hz), 163.15, 150.49, 149.38, 148.56, 135.64, 135.53, 128.41 (d, J = 8.9 Hz), 128.13, 124.30 (d, J = 3.3 Hz), 123.99, 116.84 (d, J = 22.4 Hz), 116.33, 112.83, 98.39, 44.12, 39.96. HRMS (FAB+) m/z calcd. for C21H17FN3O+ [M + H]+: 346.1350 found 346.1357.
Synthesis of 2-(3-(4-((pyridin-3-ylmethyl)amino)phenyl)isoxazol-5-yl)benzonitrile (7g). The title compound was prepared from 4h (0.20 g, 0.62 mmol) and pyridine-3-ylmethanamine (0.20 g, 1.86 mmol) in the manner described above general procedure to obtain the desired product (50.0 mg, 23%). 1H NMR (500 MHz, CDCl3): δ 8.70 (s, 1H), 8.60 (s, 1H), 8.14 (d, J = 8.0 Hz, 1H), 7.84 (d, J = 7.5 Hz, 1H), 7.79–7.73 (m, 4H), 7.57 (t, J = 7.5 Hz, 1H), 7.42 (s, 1H), 7.33 (s, 1H), 6.73 (d, J = 8.5 Hz, 2H), 4.47 (s, 2H), 4.43–4.41 (m, 1H). 13C NMR (126 MHz, DMSO-d6): δ 166.10, 163.13, 150.69, 149.40, 148.57, 135.51, 135.21, 134.40, 131.27, 129.58, 128.82, 128.27, 124.06, 118.18, 115.78, 112.89, 108.83, 101.85, 44.15. HRMS (FAB+) m/z calcd. for C22H17N4O+ [M + H]+: 353.1397 found 353.1400.
Synthesis of 3-(3-(4-((pyridin-3-ylmethyl)amino)phenyl)isoxazol-5-yl)benzonitrile (7h). The title compound was prepared from 4i (0.15 g, 0.46 mmol) and pyridine-3-ylmethanamine (0.15 g, 1.38 mmol) in the manner described above general procedure to obtain the desired product (20.0 mg, 13%). 1H NMR (400 MHz, CDCl3): δ 8.68 (s, 1H), 8.58 (d, J = 4.8 Hz, 1H), 8.12 (s, 1H), 8.08 (d, J = 8 Hz, 1H), 7.75–7.70 (m, 4H), 7.64 (t, J = 8 Hz, 1H), 7.34–7.30 (m, 1H), 6.85 (s, 1H), 6.73 (d, J = 8.8 Hz, 2H), 4.48–4.47 (m, 2H), 4.43 (d, J = 6.0 Hz, 1H). 13C NMR (101 MHz, DMSO-d6): δ 167.27, 163.27, 150.62, 149.39, 148.58, 135.60, 135.53, 134.04, 131.07, 130.26, 129.62, 128.62, 128.14, 123.98, 118.63, 116.03, 112.96, 112.88, 100.09, 44.11. HRMS (FAB+) m/z calcd. for C22H17N4O+ [M + H]+: 353.1397 found 353.1397.
Synthesis of 4-(3-(4-((pyridin-3-ylmethyl)amino)phenyl)isoxazol-5-yl)benzonitrile (7i). The title compound was prepared from 4j (0.20 g, 0.62 mmol) and pyridine-3-ylmethanamine (0.20 g, 1.86 mmol) in the manner described above general procedure to obtain the desired product (25.0 mg, 12%). 1H NMR (500 MHz, CDCl3): δ 8.70 (s, 1H), 8.60 (s, 1H), 8.14 (d, J = 8.0 Hz, 1H), 7.84 (d, J = 7.5 Hz, 1H), 7.79–7.73 (m, 4H), 7.57 (d, J = 7.5 Hz, 1H), 7.42 (s, 1H), 7.33 (s, 1H), 6.73 (d, J = 8.5 Hz, 2H), 4.48–4.46 (m, 2H), 4.43–4.41 (m, 1H). 13C NMR (126 MHz, DMSO-d6): δ 167.45, 163.34, 150.64, 149.40, 148.58, 135.59, 135.53, 133.74, 131.35, 128.18, 126.60, 123.99, 118.87, 115.94, 112.85, 100.92, 44.10. HRMS (FAB+) m/z calcd. for C22H17N4O+ [M + H]+: 353.1397 found 353.1399.
Synthesis of 4-(5-(2-(trifluoromethyl)phenyl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (7j). The title compound was prepared from 4k (0.15 g, 0.41 mmol) and pyridine-3-ylmethanamine (0.13 g, 1.23 mmol) in the manner described above general procedure to obtain the desired product (30.0 mg, 19%). 1H NMR (500 MHz, CDCl3): δ 8.66 (d, J = 2.5 Hz, 1H), 8.56 (dd, J = 4.5, 1.5 Hz, 1H), 7.83 (d, J = 9 Hz, 2H), 7.72–7.66 (m, 4H), 7.60 (t, J = 8 Hz, 1H), 7.30–7.28 (m, 1H), 6.78 (s, 1H), 6.73–6.70 (m, 2H), 4.54 (t, J = 6 Hz, 1H), 4.45–4.44 (m, 2H). 13C NMR (126 MHz, CDCl3): δ 166.86, 162.68, 149.12, 148.90, 135.05, 134.37, 132.02, 130.94, 130.04, 128.41, 128.18, 127.94 (q, J = 31.4 Hz), 126.84, 126.68 (q, J = 5.8 Hz), 126.58, 123.64, 123.57 (q, J = 274.1 Hz), 118.22, 112.92, 102.05 (q, J = 3.5 Hz), 45.43. HRMS m/z (FAB+) m/z calcd. for C22H17F3N3O+ [M + H]+: 396.1318 found 396.1324.
Synthesis of N-(pyridin-3-ylmethyl)-4-(5-(3-(trifluoromethyl)phenyl)isoxazol-3-yl)aniline (7k). The title compound was prepared from 4l (0.15 g, 0.41 mmol) and pyridine-3-ylmethanamine (0.13 g, 1.23 mmol) in the manner described above general procedure to obtain the desired product (20.0 mg, 12%). 1H NMR (400 MHz, DMSO-d6): δ 8.62 (s, 1H), 8.48–8.46 (m, 1H), 8.21 (s, 1H), 8.18 (d, J = 8.0 Hz, 1H), 7.88 (d, J = 7.6 Hz, 1H), 7.83–7.73 (m, 2H), 7.66 (s, 1H), 7.63 (d, J = 8.8 Hz, 2H), 7.37 (dd, J = 8.0, 4.8 Hz, 1H), 6.83 (t, J = 6.4 Hz, 1H), 6.74 (d, J = 8.8 Hz, 2H), 4.40 (d, J = 6.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6): δ 167.65, 163.29, 150.60, 149.40, 148.58, 135.61, 135.53, 131.05, 130.53 (q, J = 32.9 Hz), 129.70, 128.50, 128.16, 127.13 (d, J = 3.9), 124.32 (q, J = 273.81 Hz), 123.97, 122.52 (q, J = 3.4 Hz), 116.12, 112.86, 99.96, 44.12. HRMS (FAB+) m/z calcd. for C22H17F3N3O+ [M + H]+: 396.1318 found 396.1326.
Synthesis of N-(pyridin-3-ylmethyl)-4-(5-(4-(trifluoromethyl)phenyl)isoxazol-3-yl)aniline (7l). The title compound was prepared from 4m (0.20 g, 0.48 mmol) and pyridine-3-ylmethanamine (0.16 g, 1.44 mmol) in the manner described above general procedure to obtain the desired product (30.0 mg, 16%). 1H NMR (400 MHz, CDCl3): δ 8.62 (s, 1H), 8.47 (d, J = 4.0 Hz, 1H), 8.09 (d, J = 8.0 Hz, 2H), 7.93 (d, J = 8.0 Hz, 2H), 7.78–7.76 (m, 1H), 7.64 (d, J = 8.4 Hz, 2H), 7.61 (s, 1H), 7.37 (dd, J = 7.6, 4.4 Hz, 1H), 6.84 (t, J = 6.0 Hz, 1H), 6.74 (d, J = 8.4 Hz, 2H), 4.40 (d, J = 6.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6): δ 167.70, 163.31, 150.61, 149.40, 148.58, 135.59, 135.52, 131.13, 130.46 (q, J = 32.3 Hz), 128.19, 126.72, 126.67, 124.39 (q, J = 273.5 Hz), 123.96, 116.08, 112.85, 100.35, 44.13. HRMS (FAB+) m/z calcd. for C22H17F3N3O+ [M + H]+: 396.1318 found 396.1326.
Synthesis of 4-(5-(4-(difluoromethoxy)phenyl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (7m). The title compound was prepared from 4n (0.20 g, 0.52 mmol) and pyridine-3-ylmethanamine (0.17 g, 1.56 mmol) in the manner described above general procedure to obtain the desired product (25.0 mg, 12%). 1H NMR (400 MHz, CDCl3): δ 8.68 (s, 1H), 8.58 (s, 1H), 8.09 (d, J = 7.6 Hz, 1H), 7.72 (d, J = 8.4 Hz, 3H), 7.52–7.41 (m, 3H), 7.33–7.29 (m, 1H), 6.96 (s, 1H), 6.73 (d, J = 8.4 Hz, 2H), 4.47–4.42 (m, 3H). 13C NMR (101 MHz, CDCl3): δ 164.02, 163.08, 149.14, 149.10, 148.94, 145.48, 135.02, 134.37, 131.04, 128.60, 128.18, 127.17, 123.66, 120.97, 120.68, 120.52 (q, J = 260 MHz), 118.37, 112.92, 101.63, 45.48. HRMS (FAB+) m/z calcd. for C22H17F3N3O2+ [M + H]+: 412.1267 found 412.1274.
Synthesis of 4-(5-(3-(trifluoromethoxy)phenyl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (7n). The title compound was prepared from 4o (0.20 g, 0.52 mmol) and pyridine-3-ylmethanamine (0.17 g, 1.56 mmol) in the manner described above general procedure to obtain the desired product (70.0 mg, 33%). 1H NMR (400 MHz, CDCl3): δ 8.68 (s, 1H), 8.58 (d, J = 4.8 Hz, 1H), 7.79–7.69 (m, 5H), 7.54 (t, J = 8.0 Hz, 1H), 7.33–7.29 (m, 2H), 6.80 (s, 1H), 6.73 (d, J = 8.4 Hz, 2H), 4.47–4.46 (m, 2H), 4.41–4.40 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 167.59, 163.26, 150.58, 149.39, 149.33, 148.56, 135.60, 135.52, 131.96, 129.59, 128.16, 124.92, 123.96, 123.01, 120.52 (q, J = 257.5 Hz), 118.46, 116.14, 112.84, 99.84, 44.13. HRMS (FAB+) m/z calcd. for C22H17F3N3O2+ [M + H]+: 412.1267 found 412.1271.
Synthesis of 4-(5-(4-(trifluoromethoxy)phenyl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (7o). The title compound was prepared from 4p (0.20 g, 0.52 mmol) and pyridine-3-ylmethanamine (0.17 g, 1.56 mmol) in the manner described above general procedure to obtain the desired product (50.0 mg, 23%). 1H NMR (500 MHz, DMSO-d6): δ 8.65 (s, 1H), 8.49 (s, 1H), 8.01 (d, J = 9.0 Hz, 2H), 7.77 (d, J = 7.5 Hz, 1H), 7.64 (d, J = 8.5 Hz, 2H), 7.55 (d, J = 8.0 Hz, 2H), 7.47 (s, 1H), 7.38–7.36 (m, 1H), 6.84 (t, J = 6.0 Hz, 1H), 6.74 (d, J = 8.5 Hz, 2H), 4.40 (d, J = 6.0 Hz, 2H). 13C NMR (126 MHz, DMSO-d6): δ 167.93, 163.22, 150.55, 149.77, 149.40, 148.56, 135.50, 128.16, 128.07, 126.77, 124.04, 122.20, 120.47 9 (q, 257.7 Hz), 116.23, 112.83, 99.22, 44.16. HRMS (FAB+) m/z calcd. for C22H17F3N3O2+ [M + H]+: 412.1267 found 412.1271.
Synthesis of 4-(5-(2-(difluoromethoxy)phenyl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (7p). The title compound was prepared from 4q (0.24 g, 0.66 mmol) and pyridine-3-ylmethanamine (0.21 g, 1.98 mmol) in the manner described above general procedure to obtain the desired product (50.0 mg, 19%). 1H NMR (300 MHz, CDCl3): δ 8.68 (s, 1H), 8.58 (s, 1H), 8.10–8.07 (m, 1H), 7.73 (d, J = 8.8 Hz, 3H), 7.49–7.45 (m, 1H), 7.39–7.36 (m, 1H), 7.33–7.30 (m, 1H), 7.28–7.24 (m, 1H), 7.04 (s, 1H), 6.82–6.45 (m, 3H), 4.46 (s, 2H), 4.40 (s, 1H). 13C NMR (126 MHz, DMSO-d6): δ 164.70, 163.03, 150.53, 149.39, 148.56, 147.80, 135.51, 133.87, 132.21, 128.65, 128.18, 126.26, 123.99, 119.69, 119.28, 116.91 (t, J = 260.3 MHz), 116.24, 112.89, 101.93, 44.18. HRMS (FAB+) m/z calcd. for C22H18F2N3O2+ [M + H]+: 394.1362 found 394.1371.
Synthesis of 4-(5-(3-(difluoromethoxy)phenyl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (7q). The title compound was prepared from 4r (0.10 g, 0.27 mmol) and pyridine-3-ylmethanamine (0.09 g, 0.81 mmol) in the manner described above general procedure to obtain the desired product (24.0 mg, 22%). 1H NMR (500 MHz, CD3OD): δ 8.58 (s, 1H), 8.43 (s, 1H), 7.87 (d, J = 8.5 Hz, 1H), 7.74 (d, J = 8.0 Hz, 1H), 7.65 (d, J = 6.0 Hz, 3H), 7.58–7.52 (m, 1H), 7.42 (t, J = 7.5 Hz, 1H), 7.26 (d, J = 8.0 Hz, 1H), 7.17 (s, 1H), 6.95 (t, J = 74 Hz, 1H), 6.71 (d, J = 8.5 Hz, 2H), 4.47 (s, 2H). 13C NMR (126 MHz, DMSO-d6): δ 168.12, 163.20, 151.97 (t, J = 3.3 Hz), 150.55, 150.49, 149.40, 148.57, 135.61, 135.53, 131.64, 129.28, 128.15, 123.97, 122.66, 120.83, 116.77 (t, J = 258.9 Hz), 116.19, 115.99, 112.84, 99.47, 44.12. HRMS (FAB+) m/z calcd. for C22H18F2N3O2+ [M + H]+: 394.1362 found 394.1363.
Synthesis of 4-(5-(4-(difluoromethoxy)phenyl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (7r). The title compound was prepared from 4s (0.30 g, 0.82 mmol) and pyridine-3-ylmethanamine (0.27 g, 2.46 mmol) in the manner described above general procedure to obtain the desired product (25.0 mg, 8%, two step yield). 1H NMR (500 MHz, DMSO-d6): δ 8.63 (s, 1H), 8.48 (s, 1H), 7.94 (d, J = 7.5 Hz, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.62 (d, J = 8.0 Hz, 2H), 7.53–7.24 (m, 5H), 6.83 (t, J = 6.5 Hz, 1H), 6.73 (d, J = 8.5 Hz, 2H), 4.40 (d, J = 6.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6): δ 168.41, 163.15, 152.64 (t, J = 3.3 Hz), 150.49, 149.36, 148.55, 135.70, 135.54, 128.14, 127.93, 124.52, 124.04, 119.61, 116.58 (t, J = 259.3 Hz), 116.33, 112.84, 98.44, 44.14. HRMS (FAB+) m/z calcd. for C22H18F2N3O2+ [M + H]+: 394.1362 found 394.1361.
Synthesis of 4-(5-(pyridin-2-yl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (8a). The title compound was prepared from compounds 5a (98.0 mg, 0.33 mmol) and pyridine-3-ylmethanamine (0.11 g, 0.99 mmol) in the manner described above general procedure to obtain the desired product (18.0 mg, 17%). 1H NMR (300 MHz, CD3OD): δ 8.68 (dt, J = 5.1, 1.5 Hz, 1H), 8.59 (s, 1H), 8.44 (s, 1H), 8.00–7.99 (m, 2H), 7.88 (dt, J = 7.8, 1.8 Hz, 1H), 7.69–7.64 (m, 2H), 7.53–7.46 (m, 1H), 7.42 (dd, J = 7.8, 5.1 Hz, 1H), 7.30 (s, 1H), 6.76–6.71 (m, 2H), 4.48 (s, 2H). 13C NMR (101 MHz, DMSO-d6): δ 169.02, 163.19, 150.60, 150.54, 149.38, 148.57, 146.35, 138.14, 135.63, 135.51, 128.28, 125.37, 123.97, 121.28, 116.18, 112.84, 100.62, 44.13. HRMS (FAB+) m/z calcd. for C20H17N4O+ [M + H]+: 329.1397 found 329.1407.
Synthesis of 4-(5-(pyridin-3-yl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (8b). The title compound was prepared from 5b (0.05 g, 0.17 mmol) and pyridine-3-ylmethanamine (0.05 g, 0.51 mmol) in the manner described above general procedure to obtain the desired product (20.0 mg, 37%). 1H NMR (300 MHz, CD3OD): δ 9.07 (d, J = 1.2 Hz, 1H), 8.64 (dd, J = 5.1, 1.5 Hz, 1H), 8.59 (s, 1H), 8.44 (d, J = 4.8 Hz, 1H), 8.33–8.29 (m, 1H), 7.88 (dt, J = 7.8, 1.8 Hz, 1H), 7.69–7.64 (m, 2H), 7.60 (ddd, J = 8.1, 5.1, 0.9 Hz, 1H), 7.43 (dd, J = 7.8, 4.8 Hz, 1H), 7.30 (s, 1H), 6.76–6.71 (m, 2H), 4.48 (s, 2H). 13C NMR (101 MHz, DMSO-d6): δ 166.86, 163.16, 151.37, 150.59, 149.37, 148.56, 146.95, 135.61, 135.54, 133.28, 128.18, 124.70, 123.97, 123.77, 116.08, 112.86, 99.64, 44.12. HRMS (FAB+) m/z calcd. for C20H17N4O+ [M + H]+: 329.1397 found 329.1406.
Synthesis of N-(pyridin-3-ylmethyl)-4-(5-(pyridin-4-yl)isoxazol-3-yl)aniline (8c). The title compound was prepared from 5c (0.05 g, 0.15 mmol) and pyridine-3-ylmethanamine (0.05 g, 0.45 mmol) in the manner described above general procedure to obtain the desired product (10.0 mg, 19%). 1H NMR (300 MHz, CD3OD): δ 8.73–8.70 (m, 2H), 8.59 (s, 1H), 8.44 (d, J = 5.1 Hz, 1H), 7.90–7.88 (m, 3H), 7.69–7.66 (m, 2H), 7.45–7.41 (m, 2H), 6.76–6.72 (m, 2H), 4.49 (s, 2H). 13C NMR (101 MHz, DMSO-d6): δ 166.93, 163.35, 151.24, 150.67, 149.38, 148.58, 135.59, 135.54, 134.09, 128.21, 123.98, 119.79, 115.88, 112.87, 101.43, 44.11. HRMS (FAB+) m/z calcd. for C20H17N4O+ [M + H]+: 329.1397 found 329.1405.
Synthesis of N-(pyridin-3-ylmethyl)-4-(5-(3-(trifluoromethoxy)pyridin-2-yl)isoxazol-3-yl)aniline (8d). The title compound was prepared from 5d (0.05 g, 0.13 mmol) and pyridine-3-ylmethanamine (0.04 g, 0.39 mmol) in the manner described above general procedure to obtain the desired product (12.0 mg, 23%). 1H NMR (300 MHz, CDCl3): δ 8.66 (dd, J = 4.8, 1.5 Hz, 1H), 8.60 (s, 1H), 8.50 (s, 1H), 7.75–7.65 (m, 4H), 7.42 (dd, J = 8.4, 4.5 Hz, 1H), 7.23 (dd, J = 7.8, 4.8 Hz, 1H), 7.14 (s, 1H), 6.67 (d, J = 8.7 Hz, 2H), 4.76 (t, J = 5.7 Hz, 1H), 4.40 (d, J = 5.4 Hz, 2H). 13C NMR (101 MHz, CDCl3): δ 164.86, 162.73, 149.34, 149.00, 148.71, 148.05, 142.91, 142.89 (d, J = 2.0 Hz), 139.72, 135.02, 134.49, 132.09, 129.24, 128.15, 125.16, 123.62, 120.49 (q, J = 261.9 Hz), 117.72, 117.68, 112.86, 103.58, 45.28. HRMS (FAB+) m/z calcd. for C21H16F3N4O2+ [M + H]+: 413.1220 found 413.1219.
Synthesis of N-(pyridin-3-ylmethyl)-4-(5-(4-(trifluoromethoxy)pyridin-2-yl)isoxazol-3-yl)aniline (8e). The title compound was prepared from 5e (0.05 g, 0.13 mmol) and pyridin-3-ylmethanamine (0.04 g, 0.39 mmol) in the manner described above general procedure to obtain the desired product (7.5 mg, 14%). 1H NMR (300 MHz, DMSO-d6): δ 8.79 (dd, J = 4.5, 1.2 Hz, 1H), 8.62 (d, J = 2.4 Hz, 1H), 8.47 (dd, J = 4.8, 1.8 Hz, 1H), 8.15–8.11 (m, 1H), 7.79–7.67 (m, 4H), 7.47 (s, 1H), 7.37 (dd, J = 7.8, 4.5 Hz, 1H), 6.86 (t, J = 6.0 Hz, 1H), 6.75–6.70 (m, 2H), 4.40 (d, J = 6.0 Hz, 2H). 13C NMR (101 MHz, CDCl3): δ 168.16, 163.19, 158.52, 151.91, 149.17, 149.15, 148.99, 148.93, 135.02, 134.27, 128.23, 123.65, 118.12, 114.79, 113.56, 112.93, 110.33, 100.73, 45.48. HRMS (FAB+) m/z calcd. for C21H16F3N4O2+ [M + H]+: 413.1220 found 413.1307.
Synthesis of N-(pyridin-3-ylmethyl)-4-(5-(5-(trifluoromethoxy)pyridin-2-yl)isoxazol-3-yl)aniline (8f). The title compound was prepared from 5f (0.05 g, 0.13 mmol) and pyridin-3-ylmethanamine (0.04 g, 0.39 mmol) in the manner described above general procedure to obtain the desired product (28.0 mg, 53%). 1H NMR (300 MHz, DMSO-d6): δ 8.83 (s, 1H), 8.62 (d, J = 2.4 Hz, 1H), 8.46 (dd, J = 4.8, 1.5 Hz, 1H), 8.12 (d, J = 1.5 Hz, 2H), 7.77 (dt, J = 7.8, 2.1 Hz, 1H), 7.70–7.67 (m, 2H), 7.55 (s, 1H), 7.37 (dd, J = 7.8, 4.8 Hz, 1H), 6.85 (t, J = 6.0 Hz, 1H), 6.74–6.71 (m, 2H), 4.40 (d, J = 6.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6): δ 167.71, 163.33, 150.62, 149.38, 148.57, 145.96 (d, J = 2.0 Hz), 145.31, 143.75, 135.61, 135.50, 130.72, 128.30, 123.97, 122.77, 120.43 (q, J = 259.5), 115.94, 112.83, 101.42, 44.11. HRMS (FAB+) m/z calcd. for C21H16F3N4O2+ [M + H]+: 413.1220 found 413.1224.
Synthesis of N-(pyridin-3-ylmethyl)-4-(5-(6-(trifluoromethoxy)pyridin-3-yl)isoxazol-3-yl)aniline (8g). The title compound was prepared from compounds 5g (0.05 g, 0.13 mmol) and pyridin-3-ylmethanamine (0.04 g, 0.39 mmol) in the manner described above general procedure to obtain the desired product (8.0 mg, 15%). 1H NMR (300 MHz, DMSO-d6): δ 8.59–8.55 (m, 2H), 8.47–8.46 (m, 1H), 7.88–7.72 (m, 8H), 7.38–7.34 (m, 2H), 6.70 (d, J = 9.0 Hz, 1H), 4.60 (d, J = 6.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6): δ 169.65, 161.91, 159.67, 149.38, 148.50, 146.46, 135.89, 135.58, 134.36, 132.69, 132.63, 128.98, 128.44, 124.04, 123.96, 112.36, 109.11, 96.16, 42.20. HRMS (FAB+) m/z calcd. for C21H16F3N4O2+ [M + H]+: 413.1220 found 413.1232.
Synthesis of N-(pyridin-3-ylmethyl)-4-(5-(2-(trifluoromethoxy)pyridin-4-yl)isoxazol-3-yl)aniline (8h). The title compound was prepared from 5h (0.05 g, 0.13 mmol) and pyridin-3-ylmethanamine (0.04 g, 0.39 mmol) in the manner described above general procedure to obtain the desired product (17.0 mg, 32%). 1H NMR (300 MHz, DMSO-d6): δ 8.62 (s, 1H), 8.56 (d, J = 5.1 Hz, 1H), 8.47 (d, J = 4.8 Hz, 1H), 7.87 (dd, J = 5.4, 1.5 Hz, 1H), 7.82 (s, 1H), 7.79–7.74 (m, 2H), 7.62 (d, J = 8.4 Hz, 2H), 7.37 (dd, J = 8.1, 4.8 Hz, 1H), 6.89 (t, J = 6.3 Hz, 1H), 6.74 (d, J = 8.4 Hz, 2H), 4.40 (d, J = 6.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6): δ 165.63, 163.43, 157.25 (d, J = 1.5 Hz), 150.77, 149.73, 149.39, 148.59, 138.93, 135.52, 128.20, 123.98, 120.18 (q, J = 261.3 Hz), 118.95, 115.62, 112.89, 109.22, 102.75, 44.09. HRMS (FAB+) m/z calcd. for C21H16F3N4O2+ [M + H]+: 413.1220 found 413.1223.
Synthesis of N-(pyridin-3-ylmethyl)-4-(5-(2-(trifluoromethyl)pyridin-4-yl)isoxazol-3-yl)aniline (8i). The title compound was prepared from 5i (0.15 g, 0.41 mmol) and pyridine-3-ylmethanamine (0.13 g, 1.23 mmol) in the manner described above general procedure to obtain the desired product (15.0 mg, 9%). 1H NMR (400 MHz, CDCl3): δ 8.90 (d, J = 5.2 Hz, 1H), 8.68 (s, 1H), 8.59 (d, J = 4.8 Hz, 1H), 8.07 (s, 1H), 7.91 (d, J = 4.4 Hz, 1H), 7.74–7.71 (m, 3H), 7.32 (dd, J = 7.6, 4.8 Hz, 1H), 7.05 (s, 1H), 6.75–6.72 (m, 2H), 4.48–4.45 (m, 3H). 13C NMR (101 MHz, CDCl3): δ 165.73, 163.23, 151.06, 149.62, 149.37, 149.27, 149.00, 135.99, 135.03, 128.24, 122.62, 121.99, 119.90, 117.51, 116.48 (q, J = 2.8 Hz), 112.97, 100.79, 45.47. HRMS (FAB+) m/z calcd. for C21H16F3N4O+ [M + H]+: 397.1271 found 397.1275.
Synthesis of 4-(5-(3-fluoropyridin-4-yl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (8j). The title compound was prepared from 8j (0.05 g, 0.16 mmol) and pyridin-3-ylmethanamine (0.05 g, 0.48 mmol) in the manner described above general procedure to obtain the desired product (7.5 mg, 14%). 1H NMR (300 MHz, CDCl3): δ 8.66 (d, J = 2.1 Hz, 2H), 8.59 (dd, J = 5.1, 1.2 Hz, 2H), 7.90 (dd, J = 6.0, 4.8 Hz, 1H), 7.76–7.71 (m, 3H), 7.32 (dd, J = 7.8, 4.8 Hz, 1H), 7.13 (d, J = 3.3 Hz, 1H), 6.73 (d, J = 8.7 Hz, 2H), 4.46 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 163.32, 160.96 (d, J = 2.9 Hz), 154.86 (d, J = 263.4 Hz), 149.28, 149.16, 149.03, 146.44 (d, J = 5.3 Hz), 139.24 (d, J = 23.9 Hz), 135.02, 134.23, 128.27, 123.69, 122.39 (d, J = 10.0 Hz), 120.42, 117.76, 112.95, 104.25 (d, J = 10.4 Hz), 45.48. HRMS (FAB+) m/z calcd. for C20H16FN4O+ [M + H]+: 347.1303 found 347.1313.
Synthesis of N-(pyridin-3-ylmethyl)-4-(5-(pyrimidin-2-yl)isoxazol-3-yl)aniline (8k). The title compound was prepared from 8k (0.15 g, 0.50 mmol) and pyridine-3-ylmethanamine (0.16 g, 1.50 mmol) in the manner described above general procedure to obtain the desired product (20.0 mg, 12%). 1H NMR (400 MHz, CDCl3): δ 8.88 (d, J = 5.2 Hz, 2H), 8.64 (s, 1H), 8.54 (d, J = 4.8 Hz, 1H), 7.73–7.69 (m, 3H), 7.34–7.32 (m, 2H), 7.30–7.27 (m, 1H), 6.70 (d, J = 8.8 Hz, 2H), 4.54–4.52 (m, 1H), 4.44–4.43 (m, 2H). 13C NMR (126 MHz, CDCl3): δ 167.37, 163.07, 157.66, 156.08, 149.20, 149.05, 148.85, 135.08, 134.38, 128.23, 123.66, 120.75, 117.98, 112.92, 112.90, 103.67, 45.40. HRMS (FAB+) m/z calcd. for C19H16N5O+ [M + H]+: 330.1349 found 330.1346.
Synthesis of N-(pyridin-3-ylmethyl)-4-(5-(pyrimidin-5-yl)isoxazol-3-yl)aniline (8l). The title compound was prepared from 5l (0.15 g, 0.50 mmol) and pyridine-3-ylmethanamine (0.16 g, 1.50 mmol) in the manner described above general procedure to obtain the desired product (20.0 mg, 12%). 1H NMR (400 MHz, CDCl3): δ 9.30 (s, 1H), 9.18 (s, 2H), 8.67 (s, 1H), 8.58 (dd, J = 4.8, 1.6 Hz, 1H), 7.74–7.69 (m, 3H), 7.33–7.30 (m, 1H), 6.94 (s, 1H), 6.75–6.71 (m, 2H), 4.47 (s, 3H). 13C NMR (101 MHz, DMSO-d6): δ 164.27, 163.18, 159.50, 154.11, 150.71, 149.39, 148.58, 135.53, 128.21, 123.98, 122.31, 115.77, 112.89, 100.90, 44.12. HRMS (FAB+) m/z calcd. for C19H16N5O+ [M + H]+: 330.1349 found 330.1345.
Synthesis of N-(pyridin-3-ylmethyl)-4-(5-(thiophen-2-yl)isoxazol-3-yl)aniline (8m). The title compound was prepared from 5m (0.10 g, 0.33 mmol) and pyridine-3-ylmethanamine (0.11 g, 0.99 mmol) in the manner described above general procedure to obtain the desired product (6.5 mg, 6%). 1H NMR (300 MHz, DMSO-d6): δ 7.84–7.77 (m, 3H), 7.68 (d, J = 3.6 Hz, 1H), 7.61 (d, J = 8.1 Hz, 3H), 7.27–7.23 (m, 3H), 6.84–6.80 (m, 1H), 6.71 (d, J = 8.1 Hz, 2H), 4.40 (d, J = 6.0 Hz, 2H). 13C NMR (126 MHz, DMSO-d6): δ 164.56 (d, J = 7.8 Hz), 163.11 (d, J = 7.9 Hz), 150.55 (d, J = 8.2 Hz), 149.46, 148.60, 135.42, 129.66 (d, J = 8.1 Hz), 129.14, 129.06 (d, J = 7.3 Hz), 128.22 (d, J = 7.8 Hz), 127.96 (d, J = 7.4 Hz), 116.13 (d, J = 8.2 Hz), 112.83 (d, J = 7.7 Hz), 97.88 (d, J = 8.2 Hz), 44.20 (d, J = 7.2 Hz). HRMS (FAB+) m/z calcd. for C19H16N3OS+ [M + H]+: 334.1009 found 334.1009.
Synthesis of 4-(5-(1-methyl-1H-pyrrol-2-yl)isoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (9d). The title compound was prepared from 9c (0.16 g, 0.53 mmol) and pyridine-3-ylmethanamine (0.17 g, 1.59 mmol) in the manner described above general procedure to obtain the desired product (15.0 mg, 9%). 1H NMR (500 MHz, DMSO-d6): δ 8.61 (s, 1H), 8.47 (s, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.59 (d, J = 6.5 Hz, 2H), 7.39–7.36 (m, 1H), 6.97–6.94 (m, 3H), 6.73 (d, J = 6.0 Hz, 2H), 6.65–6.64 (m, 1H), 6.14–6.13 (m, 1H), 4.40 (d, J = 6.0 Hz, 2H), 3.89 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 169.14, 156.98, 149.12, 149.02, 148.97, 135.04, 134.17, 127.41, 126.40, 123.66, 122.71, 117.34, 112.80, 112.10, 108.18, 96.10, 45.44, 37.18. HRMS (FAB+) m/z calcd. for C20H19N4O+ [M + H]+: 331.1553 found 331.1568.
Synthesis of 4-(5-morpholinoisoxazol-3-yl)-N-(pyridin-3-ylmethyl)aniline (10f). The intermediate 10e (0.12 g, 0.49 mmol) was dissolved in dry tetrahydrofuran (2.5 mL, 0.20 M) and then trifluoroacetic acid (0.06 mL, 0.74 mmol) was added at room temperature followed by nicotinaldehyde (0.08 g, 0.74 mmol). Sodium cyanoborohydride (0.09 g, 1.47 mmol) was gradually added to the reaction mixture at 0 °C. The reaction mixture was stirred at 0 °C for 10 min, then stirred at room temperature for 12 h. Afterward, the reaction mixture was diluted with water and extracted twice with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by Prep-HPLC to afford the desired product (10.0 mg, 6%). 1H NMR (500 MHz, DMSO-d6): δ 8.60 (s, 1H), 8.46–8.45 (m, 1H), 7.76 (d, J = 8.0 Hz, 1H), 7.47–7.44 (m, 2H), 7.36 (t, J = 6.5 Hz, 1H), 6.73–6.70 (m, 1H), 6.65 (d, J = 7.0 Hz, 2H), 5.69 (s, 1H), 4.36 (d, J = 6.0 Hz, 2H), 3.72–3.70 (m, 4H), 3.28–3.26 (m, 4H). 13C NMR (101 MHz, DMSO-d6): δ 171.14, 163.21, 150.12, 149.38, 148.54, 135.70, 135.51, 127.75, 123.94, 117.45, 112.64, 77.02, 65.70, 46.89, 44.17. HRMS (FAB+) m/z calcd. for C19H21N4O2+ [M + H]+: 337.1659 found 337.1660.
Notes
The synthesis procedure, in vitro safety and biochemical assay, spectral data of target compounds (1H and 13C NMR) are mentioned in the ESI.†Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This study was supported by the Korea Research Institute of Chemical Technology (grant no. KK2432-10).References
- A. Wilder-Smith, E. E. Ooi, O. Horstick and B. Wills, Lancet, 2019, 393, 350–363, DOI:10.1016/S0140-6736(18)32560-1.
- L. Arzuza-Ortega, A. Polo, G. Perez-Tatis, H. Lopez-Garcia, E. Parra, L. C. Pardo-Herrera, A. M. Rico-Turca, W. Villamil-Gomez and A. J. Rodriguez-Morales, Emerging Infect. Dis., 2016, 22, 925–927, DOI:10.3201/eid2205.151934.
- G. W. A. Dick, S. F. Kitchen and A. J. Haddow, Trans. R. Soc. Trop. Med. Hyg., 1952, 46, 509–520, DOI:10.1016/0035-9203(52)90042-4.
- F. N. Macnamara, Trans. R. Soc. Trop. Med. Hyg., 1954, 48, 139–145, DOI:10.1016/0035-9203(54)90006-1.
- G. Grard, M. Caron, I. M. Mombo, D. Nkoghe, S. M. Ondo, D. Jiolle, D. Fontenille, C. Paupy and E. M. Leroy, PLoS Neglected Trop. Dis., 2014, 8, e2681, DOI:10.1371/journal.pntd.0002681.
- M. R. Duffy, T. H. Chen, W. T. Hancock, A. M. Powers, J. L. Kool, R. S. Lanciotti, M. Pretrick, M. Marfel, S. Holzbauer, C. Dubray, L. Guillaumot, A. Griggs, M. Bel, A. J. L. Laven, O. K. A. Panella, B. J. Biggerstaff, M. Fischer and E. B. Hayes, N. Engl. J. Med., 2009, 360, 2536–2543, DOI:10.1056/NEJMoa0805715.
- V. M. Cao-Lormeau, C. Roche, A. Teissier, E. Robin, A. L. Berry, H. P. Mallet, A. A. Sall and D. Musso, Emerging Infect. Dis., 2013, 20, 1085–1086, DOI:10.3201/eid2006.140138.
- S. Reid, K. Rimmer and K. Thakur, Neurol. Clin., 2018, 36, 767–787, DOI:10.1016/j.ncl.2018.06.003.
- B. J. Main, J. Nicholson, O. C. Winokur, C. Steiner, K. K. Riemersma, J. Stuart, R. Takeshita, M. Krasnec, C. M. Barker and L. L. Coffey, PLoS Neglected Trop. Dis., 2018, 12, e0006524, DOI:10.1371/journal.pntd.0006524.
- E. S. Paixao, F. Barreto, M. da Gloria Teixeira, N. C. M. da Conceicao and L. C. Rodrigues, Am. J. Public Health, 2016, 106, 606–612, DOI:10.2105/AJPH.2016.303112.
- D. Gatherer and A. Kohl, J. Gen. Virol., 2016, 97, 269–273, DOI:10.1099/jgv.0.000381.
- M. M. Magnus, D. L. A. Esposito, V. A. D. Costa, P. S. Melo, C. Costa-Lima, B. Fonseca and M. Addas-Carvalho, Cell Ther., 2018, 40(3), 250–254, DOI:10.1016/j.htct.2018.01.011.
- B. D. Foy, K. C. Kobylinski, C. J. L. Foy, B. J. Blitvich, A. T. da Rosa and A. D. Haddow, Infect. Dis., 2011, 17(5), 880–882, DOI:10.3201/eid1705.101939.
- R. S. Azevedo, M. T. Araujo, A. J. Martins Filho, C. S. Oliveira, B. T. Nunes, A. C. Cruz, A. G. Nascimento, R. C. Medeiros, C. A. Caldas, F. C. Araujo, J. A. Quaresma, B. C. Vasconcelos, M. G. Queiroz, E. S. da Rosa, D. F. Henriques, E. V. Silva, J. O. Chiang, L. C. Martins, D. B. Medeiros, J. A. Lima, M. R. Nunes, J. F. Cardoso, S. P. Silva, P. Y. Shi, R. B. Tesh, S. G. Rodrigues and P. F. Vasconcelos, J. Clin. Virol., 2016, 85, 56–64, DOI:10.1016/j.jcv.2016.10.024.
- A. R. Plourde and E. M. Bloch, Emerging Infect. Dis., 2016, 22, 1185–1192, DOI:10.3201/eid2207.151990.
- D. Baud, D. J. Gubler, B. Schaub, M. C. Lanteri and D. Musso, Lancet, 2017, 390, 2099–2109, DOI:10.1016/S0140-6736(17)31450-2.
- A. Gulland, BMJ, 2016, 352, i657, DOI:10.1136/bmj.i657.
- B. Wahid, A. Ali, S. Rafique and M. Idrees, Eur. J. Intern. Med., 2017, 44, 12e18, DOI:10.1016/j.ejim.2017.08.001.
- J. M. Richner and M. S. Diamond, Curr. Opin. Immunol., 2018, 53, 130e136, DOI:10.1016/j.coi.2018.04.024.
- G. Rassias, V. Zogali, C. M. D. Swarbrick, K. W. K. Chan, S. A. Chan, C. P. Gwee, S. Wang, E. Kaplanai, A. Canko, D. Kiousis, J. Lescar, D. Luo, M. T. Matsoukas and S. G. Vasudevan, Eur. J. Med. Chem., 2019, 180, 536–545, DOI:10.1016/j.ejmech.2019.07.007.
- A. Coluccia, M. Puxeddu, M. Nalli, C. K. Wei, Y. H. Wu, E. Mastrangelo, T. Elamin, D. Tarantino, J. J. Bugert, B. Schreiner, J. Nolte, F. Schwarze, G. Regina, J. C. Lee and R. Silvestri, ACS Med. Chem. Lett., 2020, 11, 1869–1874, DOI:10.1021/acsmedchemlett.9b00405.
- S. Nie, Y. Yao, F. Wu, X. Wu, J. Zhao, Y. Hua, J. Wu, T. Huo, Y.-L. Lin, A. R. Kneubehl, M. B. Vogt, J. Ferreon, R. Rico-Hesse and Y. Song, J. Med. Chem., 2021, 64, 2777–2800, DOI:10.1021/acs.jmedchem.0c02070.
- S. Spizzichino, G. Mattedi, K. Lauder, C. Valle, W. Aouadi, B. Canard, E. Decroly, S. J. F. Kaptein, J. Neyts, C. Graham, Z. Sule, D. J. Barlow, R. Silvestri and D. Castagnolo, ChemMedChem, 2020, 15, 385–390, DOI:10.1002/cmdc.201900533.
- F. Li, E. M. Lee, X. Sun, D. Wang, H. Tang and G. C. Zhou, Eur. J. Med. Chem., 2020, 187, 111925, DOI:10.1016/j.ejmech.2019.111925.
- A. A. A. De Souza, L. R. Torres, L. R. P. L. Capobianco, V. S. de Paula, C. M. Cascabulho, K. Salomao, M. d. G. Bonecini-Almeida, M. d. L. G. Ferreira, N. Boechat, L. C. d. S. Pinheiro and E. M. de Souza, Viruses, 2020, 13, 36, DOI:10.3390/v13010036.
- Y. Wang, R. Zhou, Y. Quan, S. Chen, X. Shi, Y. Li and S. Cen, Bioorg. Med. Chem. Lett., 2020, 30, 126906, DOI:10.1016/j.bmcl.2019.126906.
- S. Nam, H. G. Nam, E. H. Oh, E. Jung, Y. H. Lee, E. J. Jeong, Y. D. Ou, B. Zhou, S. Ahn, J. S. Shin, S. B. Han and Y. Y. Go, Arch. Pharmacal Res., 2022, 45, 280–293, DOI:10.1007/s12272-022-01380-8.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00240g |
| This journal is © The Royal Society of Chemistry 2024 |