
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chasing isolable late d-block metal nitrenes and imidyls
C. Gunnar Werncke ab
ab
aChemistry Department, Philipps-University Marburg, Hans-Meerwein-Str. 4, D-35032 Marburg, Germany. E-mail: gunnar.werncke@chemie.uni-marburg.de
bInstitute of Inorganic Chemistry, Faculty of Chemistry, Leipzig University, Johannisallee 29, D-04103, Leipzig, Germany. E-mail: gunnar.werncke@chemie.uni-leipzig.de
First published on 8th April 2025
Abstract
Imido complexes of late transition metals are of fundamental interest as transient species in bond activation catalysis. In this perspective article, we present late imido metal complexes in uncommon and highly reactive states, concentrating on structurally authenticated imido metal complexes with higher spin states and imidyl or even nitrene character. Further, the few examples of analogous metalla nitridyls and metallanitrenes are discussed. This work serves as an overview of the existing array of these compounds and provides insights into their unequivocal identification or ambiguity, offering an entry point for interested molecular organic/inorganic chemists to contribute to this area.
Introduction
Organic nitrenes [RNs] play important roles as transient species in chemical transformations;1,2 however, they are intrinsically unstable owing to their electronic unsaturated state. Hence, nitrenes are historically examined under in situ or matrix conditions.2,3 Spectroscopic evidences of persistent nitrenes in solutions date back to several decades (Fig. 1A).4 For example, in N-(2,2,6,6)-tetramethylpiperidyl nitrene 1a, the nitrene is stabilized via π-donation from the adjacent nitrogen and can thus be described as a 1,1-diazene. In 2012, Dielmann et al. reported the first isolable nitrene (2, Fig. 1A) via irradiation of an encumbered azido phosphine.5 In this singlet phosphinonitrene, the nitrene nitrogen is stabilized via π-donation from the phosphorous atom, resulting in a phosphazene resonance structure, in analogy to those of aminonitrenes. The first isolable triplet organo nitrenes (3) were independently reported with the last 12 months by the groups of Beckmann6 and Tan,7 with each using very bulky hydrindacene ligands (Fig. 1C). These nitrenes exhibited remarkable thermal stability and nitrene-like reactivity at elevated temperatures. | ||
| Fig. 1 Persistent in situ formed amino nitrenes (A) and sole examples of isolable phosphino (B) and aromatic (C) nitrenes (Dipp = 2,6-iPr2-C6H3). | ||
Owing to the high reactivity of nitrenes, control over their selectivity is challenging. In this regard metal-mediated nitrene transfer has been increasingly proven as a viable tool in catalysis.8,9 Since the first example of a catalytic nitrene transfer by Breslow,10,11 the identification and isolation of the imido metal key intermediates have been a focus of inorganic chemists. The aim is to gain profound insights into their properties to provide an experimentally proven understanding of the metal-nitrene interaction. This is crucial to validate any mechanistic proposal in terms of speciation, geometry and/or relevant spin states.
For consistency, we will refer to all complexes bearing a [MNR] unit as metal imidos and use imide, imidyl or nitrene together with the oxidation state of the metal for addressing the specific electronic structures.12 This originates from the isolobal dianionic oxido ligand whereas the alternatively used term nitrenoid is in analogy to (neutral) carbenes. The classic view of imido ligands is that of a dianionic imide [NR]2−. It can engage with vacant, energetically well separated, high-lying d-orbitals of an electropositive (early) transition metal to form either a double or a triple bond by one or two π-interactions, respectively (Fig. 2A–C). When moving to the mid and later transition metals, the increasing population of d-orbitals leads to an unfavorable occupation of antibonding π*-orbitals. This lowers the bond order and reduces the stability of any late 3d metal imido complex akin to the situation for isolobal oxido species, which is also referred to as the oxo wall.13 Discounting for further electronic effects, it creates a zwitterionic situation with highly basic imido nitrogen (Fig. 2C, right), for which only a singular, isolable example of late d-block metals is known (vide infra).14
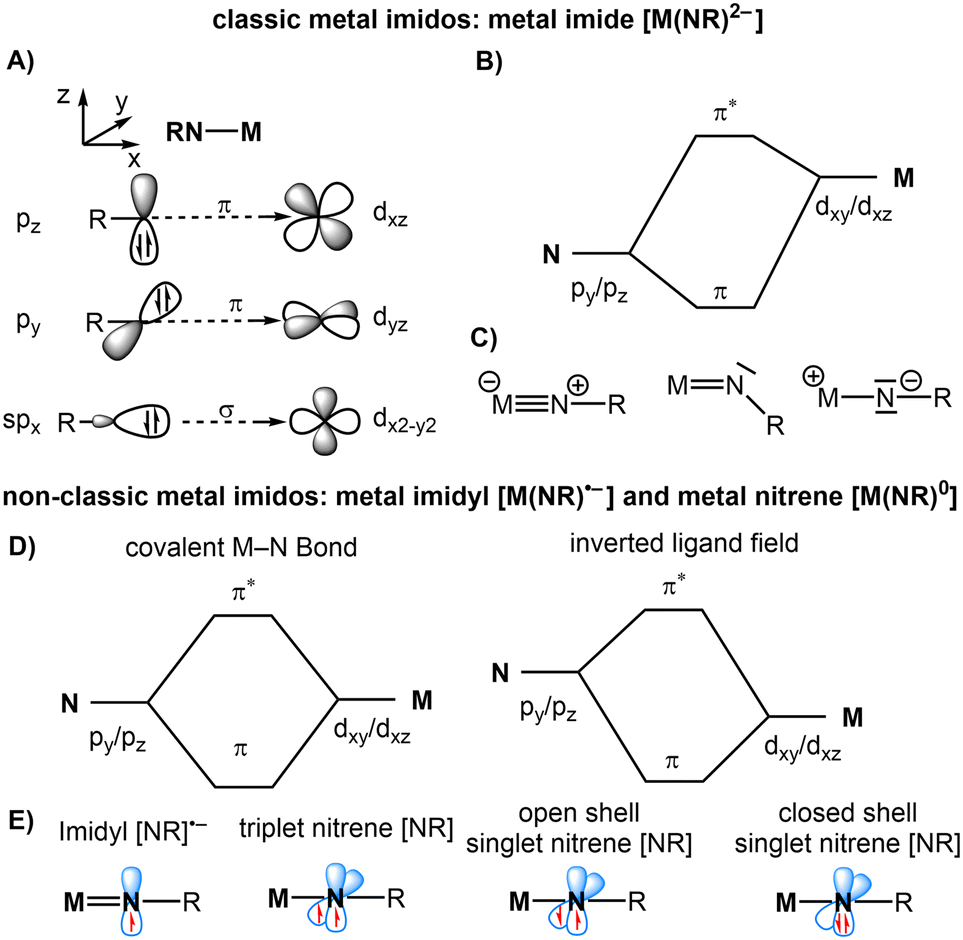 | ||
| Fig. 2 Generalized orbital interaction (A), simplified MO scheme (B) and resonance structures of metal imides (C). Simplified MO scheme for covalent and inverted metal-imido interactions (D). Spin states of metal imidyl and nitrenes (E). | ||
Apart from the mere occupation of their d-orbitals, the late 3d and late 4d/5d-block metals are also subject to lower d-orbital energies. This leads to less polarized and more covalent imido metal bonds or even results in an inverted order of the metal and imido orbitals (Fig. 2D). Accordingly, the respective electronic structures are better described as metal imidyl radical anions [NR]˙− or a metal-bound singlet or triplet nitrene [NR]0 (Fig. 2E). In the presence of an aromatic substituent, the spin density can further delocalise onto the aromatic ring, thus resulting in an iminyl-type situation, with substantial spin/charge localisation at the para-position for the free nitrene, as depicted in Fig. 1C. The situation becomes more complicated for complexes in a higher spin state, which is mostly relevant for late 3d metal imidos. It leads to a further population of the anti-bonding π* orbitals. Additional complications can arise from the participation of low-lying excited spin states, which hamper the precise denomination of the electronic situation of the MNR unit. Its computational analysis requires multireference approaches (e.g. CASSCF) to disentangle the contributions of the different electronic structures and spin states. Thus, the Enemark–Feltham15 notation for the imido metal unit as {MNR}n can be more suitable, as it avoids (wrongly) singling out one of the resonance structures.
With this caveat in mind, we want to give an overview of the isolable or structurally authenticated metal nitrenes, imidyls and imidos in higher spin-states. For a more extensive analysis and overview of late transition metal imidos (including in situ formed species), their reactivity and catalytic applications, the reader is referred to respective reviews.9,12,16 The general synthetic approach to metal imido constitutes the reaction of a suitable metal complex in a low-oxidation state with the nitrene transfer agent. These are mostly organo and tosyl azides, hydroxylamines and lately also dioxazolones.
3d metal imidyls and nitrenes
Manganese and earlier transition metals
As observed in the introduction, imidyl- or nitrene-type situations for chromium and earlier metals are usually disregarded owing to the energetic mismatch between the involved d and pN orbitals. Only a report by Wieghardt and Lu on a chromium imido complex (4, Fig. 3)17 revealed so far a measurable chromium imidyl character based on computational and X-ray absorption spectroscopic analysis. Here, it is likely connected to the high cationic charge as well as the ambiguous role of the redox non-innocent of the iminopyridine co-ligands. Authenticated, unequivocal manganese nitrenes, imidyls or even high-spin imidos are absent to this day although they have been invoked in several catalytic transformations since the seminal report by Breslow using a manganese porphyrine for amination and aziridination reactions in the 1980s.10,11 Abu-Omar and co-workers obtained a manganese(V) imide by photolysis of tosyl azide in the presence of a manganese(III) corrole (5, Fig. 3),18 yet it proved to be unreactive. Oxidation of an anionic TAML-ligated imido manganese complex facilitated nitrene transfer to a thioether,19 but spectroscopic analysis showed a manganese(VI) imide character (6, Fig. 3), with only marginal unpaired spin density on the imido nitrogen. The participation of the MnIV nitrene or MnV imidyl resonance structures was thus deemed irrelevant to the ground state.19 The closest report in this regard concerned a tosyl imido manganese porphyrine by Powers (7, Fig. 3) obtained from the photolysis of a MnIII chloroamidate precursor (7Cl).20 Low-temperature X-band EPR-spectroscopy revealed a high-spin ground state (S = 5/2). A multireference character of the Mn-NR bond was invoked owing to computationally similar spin state energies. It include the possibility of manganese(III) imidyl and manganese(II) nitrene, but a conclusive determination of the electronic structure was not achieved. | ||
| Fig. 3 Chromium imidyl and manganese imidos in high-oxidation states (Ada = adamantyl, Ts = –S(O)2(4-Me-C6H4), Mes = 2,4,6-Me3-C6H2). | ||
Iron
For manganese, the potential for iron-mediated nitrene transfer was discovered by Breslow.10 The earliest isolable imido iron complex dates back to the early 2000s21 owing to the intrinsic lability of later 3d-metal imido bonds. In 2011 Betley reported for the first time the feasibility of imidyl-type ligands for iron (e.g. in 8Cl, Fig. 4)22,23 and their use in cyclisative amination reactions.24 Employing a low-coordinate setting in conjunction with a monoanionic weak-field dipyrromethene ligand allowed us to obtain an overall high-spin situation. Here, an iron(III) ion is antiferromagnetically coupled to an aromatic imidyl ligand with a very long imido iron bond of 1.768(2) Å (8Cl, Fig. 4).22 The aromatic substituent contributes to the stability of the imidyl ligand via the delocalisation of considerable spin density onto the aromatic ring. A related alkyl imidyl complex could be isolated, with its structural metrics being similar (1.761(6) Å) using X-ray absorption techniques.25 The reduction of the imidyl complexes is in both cases little Fe-centred as perceived from little changes of the 1s → 3d absorption in the iron K-edge pre-edge region and a marginal 57Fe Mössbauer spectroscopic isomer shift difference. In both cases, a shortening of the Fe–N bond to 1.708(4) Å for the aromatic imido (8) and to 1.674(11) Å for an analogous aliphatic imido complex was observed. Computational analyses corroborate a ligand-centred reduction and loss of the N-based spin density of the former imidyl. The reduction was accompanied by a loss of C–H activation rates by two orders of magnitude for both aromatic and aliphatic imidos. It inferred the importance of the N-based spin density that is predisposed to the H atom abstraction process. Reith et al. reported an anionic, trigonal iron(II) imidyl complex in a high-spin state with a similar elongated Fe–N bond of 1.775(2) Å (9−, Fig. 4). X-band EPR spectroscopy was hinted at the imido nitrogen spin density via a large 14N hyperfine coupling constant. Despite its clear imidyl character, 9− is highly nucleophilic and acts as an iron(III) imide under the insertion of CS2 into the imido metal bond, forming an iron(III) dithiocarbonimidate [FeIII(η2-S2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR)] unit. The oxidation of 9− yielded the neutral high-spin compound 9, which shows mixed imidyl, nitrene and imide character, as revealed by ab initio complete active space self-consistent field (CASSCF) calculations.26 The loss of an electron upon oxidation is equally shared by the metal and the imido nitrogen as corroborated by 57Fe Mössbauer spectroscopy and marginally impacts the M–N bond length. For the similar neutral complex 10, computations gave only an imidyl type character.27 This points to the substantial impact of slight changes in the ligand set but also cautions to attribute the electronic structure using only computational methods. Attempts to obtain an aliphatic iron derivative via this route were not possible owing to complications from immediate intramolecular C–H amination.27 This was in part overcome independently by the groups of Albrecht and Werncke via the transformation of defined aliphatic iron(II) organo azide adducts (11R, Fig. 5).28,29 Although 1H NMR spectroscopic evidence pointed to the subsequent in situ formation of imido iron species (12R), it could not accumulate owing to rapid intramolecular C–H bond amination (13R). More insights were gained by the in crystallo irradiation of the isolated azide adduct 11R with blue light (390 nm). For 11tBu, the elimination of 35% N2 prior to loss of crystallinity was achieved, effectuating the shortening of the Fe–N bond length from 2.160(2) Å to 2.066(6) Å.28 This is accompanied by the beginning rearrangement of one of the ancillary silylamide ligands that probably aligns for intramolecular nitrene insertion as seen in the solution. The transferring iron imido species was calculated as rather linear (176°) with a Fe–N bond length of 1.70 Å and predicted as a pentet by CASSCF/NEVPT2 calculations with a highly covalent Fe–N bond. In the paralleling report by Albrecht, near complete irradiative N2 extrusion of 12Ada allowed a better determination of the imido iron bond length to 1.691(9) Å with a bent Fe–N-R unit (146°).29 In this case, CASSCF calculations revealed a preference for the triplet state with an iron(III) ion being antiferromagnetically coupled to an imidyl. This assignment was supported by a comparison of the calculated and experimentally obtained 57Fe Mössbauer parameters from the irradiation of a frozen solution of the azide adduct. Owing to the even weaker bonding capabilities of aromatic azides, a corresponding adduct to iron could not be obtained.
NR)] unit. The oxidation of 9− yielded the neutral high-spin compound 9, which shows mixed imidyl, nitrene and imide character, as revealed by ab initio complete active space self-consistent field (CASSCF) calculations.26 The loss of an electron upon oxidation is equally shared by the metal and the imido nitrogen as corroborated by 57Fe Mössbauer spectroscopy and marginally impacts the M–N bond length. For the similar neutral complex 10, computations gave only an imidyl type character.27 This points to the substantial impact of slight changes in the ligand set but also cautions to attribute the electronic structure using only computational methods. Attempts to obtain an aliphatic iron derivative via this route were not possible owing to complications from immediate intramolecular C–H amination.27 This was in part overcome independently by the groups of Albrecht and Werncke via the transformation of defined aliphatic iron(II) organo azide adducts (11R, Fig. 5).28,29 Although 1H NMR spectroscopic evidence pointed to the subsequent in situ formation of imido iron species (12R), it could not accumulate owing to rapid intramolecular C–H bond amination (13R). More insights were gained by the in crystallo irradiation of the isolated azide adduct 11R with blue light (390 nm). For 11tBu, the elimination of 35% N2 prior to loss of crystallinity was achieved, effectuating the shortening of the Fe–N bond length from 2.160(2) Å to 2.066(6) Å.28 This is accompanied by the beginning rearrangement of one of the ancillary silylamide ligands that probably aligns for intramolecular nitrene insertion as seen in the solution. The transferring iron imido species was calculated as rather linear (176°) with a Fe–N bond length of 1.70 Å and predicted as a pentet by CASSCF/NEVPT2 calculations with a highly covalent Fe–N bond. In the paralleling report by Albrecht, near complete irradiative N2 extrusion of 12Ada allowed a better determination of the imido iron bond length to 1.691(9) Å with a bent Fe–N-R unit (146°).29 In this case, CASSCF calculations revealed a preference for the triplet state with an iron(III) ion being antiferromagnetically coupled to an imidyl. This assignment was supported by a comparison of the calculated and experimentally obtained 57Fe Mössbauer parameters from the irradiation of a frozen solution of the azide adduct. Owing to the even weaker bonding capabilities of aromatic azides, a corresponding adduct to iron could not be obtained.
 | ||
| Fig. 4 Oxidation state-dependent electronic structures of isolable imidyl/nitrene iron systems in higher spin states. | ||
 | ||
| Fig. 5 In crystallo irradiation of iron(II) organoazides, and the subsequent formation of aliphatic imido iron complexes and their intramolecular, nitrene-like reactivity. | ||
The benefit of a higher spin state for H atom abstraction (HAA) was exemplified by Deng for a bis(imido) iron system (Fig. 6A). The purposeful manipulation of the imido substituents in a formal iron(IV) bis(imide) led to the isolation of structurally similar complexes in either the S = 0 (14Me) or S = 1 (14CF3) ground state, with only the latter showing intramolecular HAA reactivity. Different spin states arise from the synergistic structural and steric effects of the N-substituents and their electron-withdrawing properties, effectuating Fe–N bond polarisation and a more acute bending of the imido substituent (140° vs. 160°). This yields an imido nitrogen in 14CF3 with a sufficient imidyl character and an exposed pN orbital that is predisposed to H atom abstraction.
 | ||
| Fig. 6 NHC-based bis(imido) (A, B) and imido iron systems (C). | ||
It is important to note that a high formal oxidation state does not automatically evoke a nitrene or imidyl character, as impressively shown for formal iron(V) and iron(VI) (sic!) bis(imides) (15(+), Fig. 6B). Here, the overall oxidation state change is mostly metal centred, thus underscoring the strongly donating properties of imide ligands, as observed for earlier transition metals.30 Moreover, Smith observed for the anionic high-spin iron(II) imide 16− (Fig. 6C) with a very long Fe–N bond length of 1.779(2) Å only imide character. This was attributed to nitrogen-centred π-interactions, and the unpaired spin density resided mostly on iron-centred orbitals. The imido nitrogen is highly nucleophilic and can be employed in the guanilidation of carbodiimides.31 The oxidation of 16− to 16 is metal-centred to yield an intermediate-spin iron(III) imide.
Cobalt
Cobalt imidyl complexes and imido complexes in higher spin states have been known for a short time. Coincidently, a low-spin cobalt(III) imidyl (17),32 an intermediate-spin cobalt(II) imidyl (18)33 and a high-spin cobalt(II) imidyl (19)34,35 were reported in 2021 (Fig. 7). This shows that the imidyl character is feasible for cobalt for different charges, spin states and complex geometries, all with Co-NR bond lengths of over 1.70 Å. Hereby, the cobalt(II) imidyl 19 complex is informative, as the X-band EPR spectroscopic signature exhibits simultaneous signals for the cobalt(II) ion as well as that of an N-centred organic radical. In the case of a trigonal bipyramidal cationic environment, DeBeer, Munz and Meyer nicely showed the impact of the formal oxidation state on the imido ligand.36 Starting with the monocationic CoIII imide 20+ (Fig. 7) oxidation leads first to a dicationic mixed CoIV imide/CoIII imidyl (202+) and is accompanied by an increase in the Co–N bond length. Subsequent oxidation to tricationic 203+ shortens the bond, and a mixed CoIV imidyl/CoIII nitrene character was invoked on an analytical and computational level. Furthermore, the exchange of the aromatic substituent at the para-position is observed for 203+, a known behaviour of singlet nitrenes.37 C–H amination is mitigated owing to steric hindrance.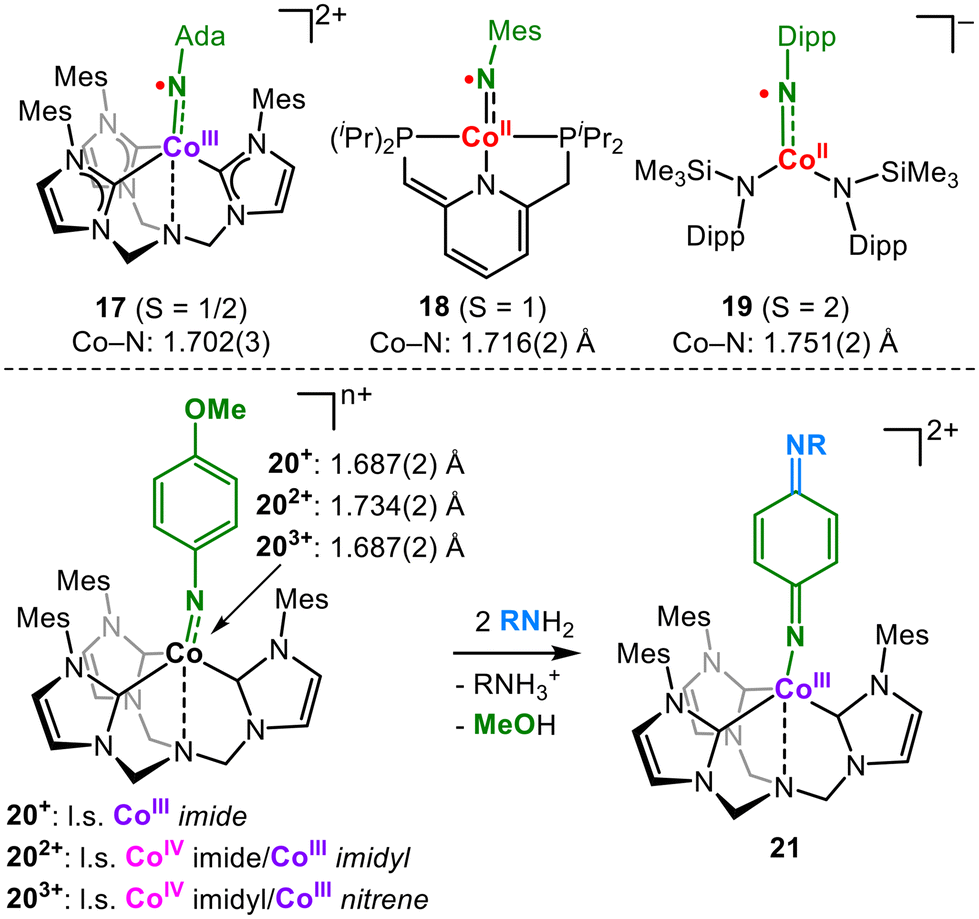 | ||
| Fig. 7 Isolable cobalt imidyl complexes (top) and oxidation state-dependent electronic structure of a cobalt imido system and its nitrene-like reactivity (bottom). | ||
Betley exploited the in crystallo conversion of a catalytically relevant low-coordinate cobalt(II) aliphatic organo azide adduct (22, Fig. 8).38 Thermal treatment of the crystal yields approx. 20% N2 depletion prior to crystal degradation. This could be enhanced to nearly 70% at 100 K using a synchrotron-based high-flux X-ray source. Upon irradiation, a Co–Nα bond contraction from 2.1 Å to 1.79 Å is observed. This latter bond length is longer than the imido cobalt bonds in very similar systems (1.63–1.65 Å). This indicates an incomplete relaxation of the bond situation owing to the constraints imposed by the crystal matrix. In agreement with the supposed elongated Co–NR bond, the computational analysis indicated the electronic structure of a cobalt(III) imidyl (23) rather than a cobalt(IV) imide. Reacting an electron-poor aromatic azide with a cobalt(I) dipyrromethene precursor gives a dimeric compound with a bis(ketimine) ligand (24dimer, Fig. 8). This is the result of the dimerization of a monomeric cobalt(II) ketiminyl radical species (24mono). The process is reversible; thus, complex 24dimer can effectively act as a monomeric CoII imidyl in HAA reactivity.
 | ||
| Fig. 8 In crystallo formation of an aliphatic imidyl cobalt(III) complex (top) and reversible dimerization of an aromatic imidyl cobalt(II) complex (bottom). | ||
Nickel
For nickel, the non-classical electronic structures of late 3d-metal imidos were pointed out first by Warren for a trigonal low-spin imido nickel complex using a nacnac (β-diketiminate) ligand (25, Fig. 9).39,40 Although the antibonding π* SOMOs are nickel centred, the imido nitrogen carries substantial amounts of the overall spin density (57%). This is also reflected in the X-band EPR hyperfine coupling to nitrogen (A = 22 G).39 The imido complex can be reversibly captured using a second nickel fragment yielding a bridging imido ligand, which acts primarily as an imide. For HAA from substrates, dissociation is required. The isostructural aromatic imido complex could not be isolated given that it dimerizes irreversibly by coupling at the para-position of the aromatic ring. Hillhouse reported a two-coordinate high-spin (S = 1) aromatic imido nickel complex (28, Fig. 10A).41 | ||
| Fig. 9 Formation and behaviour of nacnac-based nickel imido/imidyl complexes. | ||
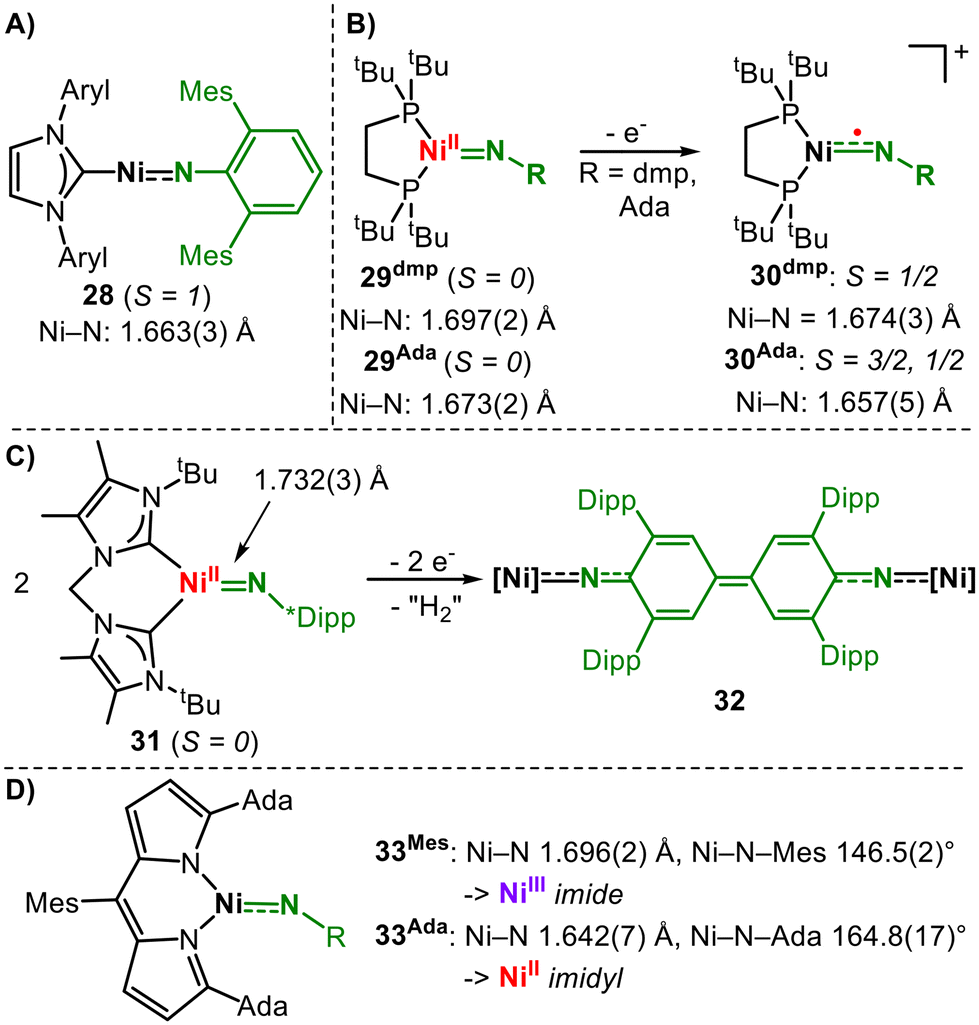 | ||
| Fig. 10 Covalent nickel imido complex (A), oxidation state-dependent behaviour of nickel imido complexes (B and C) (dmp = 2,6-bis(mesityl)phenyl) and substituent-dependent electronic structure of nickel imido complexes (D). | ||
In 28 both imido metal π-interactions are highly covalent (ligand![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :metal = 5:5); thus, the description of a metal bound triplet nitrene can be attributed.12,41 The aromatic substituent contributes to the stability of the nitrene, as generally observed for other high-spin aromatic imido complexes mentioned herein.25,26,34,42,43 For three-coordinate, diphosphine ligated alkyl and aryl imido nickel complexes by Hillhouse (29dmp and 29Ada, Fig. 10B), the situation is that of diamagnetic low-spin compounds with DFT-calculated π/π* centred HOMO and LUMO.44 Upon oxidation, the low-spin-state persists in the case of the aromatic 30dmp, while the aliphatic 30Ada is subject to a high-spin/low-spin equilibrium.45 X-band EPR spectroscopic examinations revealed the contribution of a low-spin NiIII ion (d7). DFT calculations showed that for both cationic complexes, the spin density is shared by nitrogen and nickel. The slightly larger spin density on nitrogen in the case of the alkyl imido 30Ada, in conjunction with less steric encumbrance as well as an energetically accessible high-spin state, renders it highly potent in H atom abstraction reactions.45 The oxidation of a related trigonal bis(NHC) nickel imide (31, Fig. 10C) yielded a more imido-centred oxidation. The resulting elusive cationic imido complex immediately dimerizes via the backbone of the imido substituent with subsequent dehydrogenation and quinonediimine formation (32).46 In a comprehensive study, Betley gained spectroscopically more evidence for the imidyl-type situation for nickel.42 Using a monoanionic dipyrromethene ligand, they isolated low-spin (S = ½) aromatic and aliphatic imido nickel complexes (Fig. 10D). In the case of aliphatic 33Ada, a rhombic X-band EPR signal (77 K) with hyperfine coupling to the imido nitrogen is observed, which is unresolved in the case of an aromatic substituent. This is explained by the larger contributions of the imido Np orbital to the single-occupied molecular orbital (SOMO) of 33Ada, yielding a Ni(II) imidyl character. In contrast, the SOMO of aromatic 33Mes is polarized towards nickel, which is attributed to the delocalisation effect of the aromatic ring. Thus, 33Mes corresponds more to a nickel(III) imide.
:metal = 5:5); thus, the description of a metal bound triplet nitrene can be attributed.12,41 The aromatic substituent contributes to the stability of the nitrene, as generally observed for other high-spin aromatic imido complexes mentioned herein.25,26,34,42,43 For three-coordinate, diphosphine ligated alkyl and aryl imido nickel complexes by Hillhouse (29dmp and 29Ada, Fig. 10B), the situation is that of diamagnetic low-spin compounds with DFT-calculated π/π* centred HOMO and LUMO.44 Upon oxidation, the low-spin-state persists in the case of the aromatic 30dmp, while the aliphatic 30Ada is subject to a high-spin/low-spin equilibrium.45 X-band EPR spectroscopic examinations revealed the contribution of a low-spin NiIII ion (d7). DFT calculations showed that for both cationic complexes, the spin density is shared by nitrogen and nickel. The slightly larger spin density on nitrogen in the case of the alkyl imido 30Ada, in conjunction with less steric encumbrance as well as an energetically accessible high-spin state, renders it highly potent in H atom abstraction reactions.45 The oxidation of a related trigonal bis(NHC) nickel imide (31, Fig. 10C) yielded a more imido-centred oxidation. The resulting elusive cationic imido complex immediately dimerizes via the backbone of the imido substituent with subsequent dehydrogenation and quinonediimine formation (32).46 In a comprehensive study, Betley gained spectroscopically more evidence for the imidyl-type situation for nickel.42 Using a monoanionic dipyrromethene ligand, they isolated low-spin (S = ½) aromatic and aliphatic imido nickel complexes (Fig. 10D). In the case of aliphatic 33Ada, a rhombic X-band EPR signal (77 K) with hyperfine coupling to the imido nitrogen is observed, which is unresolved in the case of an aromatic substituent. This is explained by the larger contributions of the imido Np orbital to the single-occupied molecular orbital (SOMO) of 33Ada, yielding a Ni(II) imidyl character. In contrast, the SOMO of aromatic 33Mes is polarized towards nickel, which is attributed to the delocalisation effect of the aromatic ring. Thus, 33Mes corresponds more to a nickel(III) imide.
Copper
Copper-bound nitrenes hold prominence for aziridination and C–H bond amination. However, for a long time, they eluded their isolation, with identification being mostly restricted to in situ spectroscopic studies.47 The first copper nitrene complex stemmed from the coordination of the isolable phosphinonitrene 2 to a copper(I) ion yielding nitrene copper complexes with terminal or bridging coordination modes of the nitrene (34 and 35, Fig. 11).48 The observed Cu–N bonds are approx. 1.8 Å, which is rather long compared to those previously calculated for copper nitrenes (approx. Cu–N of 1.75 Å),49 or those for related complexes of the lighter Fe to Ni. This is a testament to the unique electronic structure of the free phosphinonitrene that limits its comparability to catalytically relevant species. A genuine isolable copper nitrene was reported by Betley from the reaction of an encumbered copper(I) pyrromethene complex with aromatic azides (36mono, Fig. 11). The isolable products exhibit a near-linear [CuNR] unit with Cu–N bonds ranging from 1.745(2) to 1.759(2) Å. | ||
| Fig. 11 Phosphinonitrene (top) and organonitrene copper (bottom) complexes. | ||
X-ray absorption spectroscopy and quantum chemical calculations revealed a triplet nitrene adduct of copper(I) and the absence of Cu–N multiple-bond character.50 Accordingly, copper(II) imidyl or copper(III) imide contributions were found to be minimal. If the para-position is not blocked, dimerization can occur (36di). Interestingly, in this system, the nitrene formation via N2 elimination becomes rate-limiting compared to the C–H bond activation step for most of the imido complexes shown herein. This can be rationalized by the difference in the reduction potential of copper(I) with regard to iron(I) or cobalt(I). This is exemplified by the stability of an analogous alkylazide copper(I) adduct that is resistant to N2 elimination.51 Warren gave further insights into imido copper complexes52 via imido bridged dicopper species that exhibit a Cu–N distance of around 1.8 Å (38, Fig. 12). They dissociate in solution to give the active monomeric imido copper unit (37), which is responsible for nitrene transfer. The aliphatic imidos were predicted by CASSCF calculations as a copper-bound open-shell singlet nitrene (DFT methods preferring a triplet ground state), with excitation within a covalent πCuN/π*CuN manifold. For aromatic substituents, the monomeric imido copper species 39mono is predicted with either a triplet or an open-shell singlet ground state. In both cases, substantial spin density in the aromatic para position is found, which rationalizes the (reversible) dimerization found in the solid state (39di, Fig. 12).53
 | ||
| Fig. 12 Masked imido copper nitrenes (aryl = Mes or 2,6-Cl2-C6H3). | ||
Late 4d/5d nitrene and imidyl complexes
Apart from ruthenium and osmium, for which several imido complexes have been reported, structurally characterized late 4d/5d metal imidos have become increasingly scarce. Although Ru9,54 and to a lesser extent Os55 play an important role in catalytic nitrene transfer catalysis, isolable ruthenium and osmium imidos with predisposed, detectable nitrene characters are so far absent. The possibility of stabilizing an imidyl ligand for both metals was reported by Peters (40M, Fig. 13A). The spin density resides mostly on nitrogen as perceived by EPR spectroscopy (e.g.: 40Ru: giso = 2.02, AN = 98 MHz, and ARu = 48 MHz).56 Schneider and co-workers presented square planar iridium imidos in three different oxidation states (41n, Fig. 13B).57 Although dicationic 412+ presents as a diamagnetic iridium(V) imide, monocationic derivative 41+ is best described as an IrIII imidyl with an Ir–N bond length of 1.805(2) Å.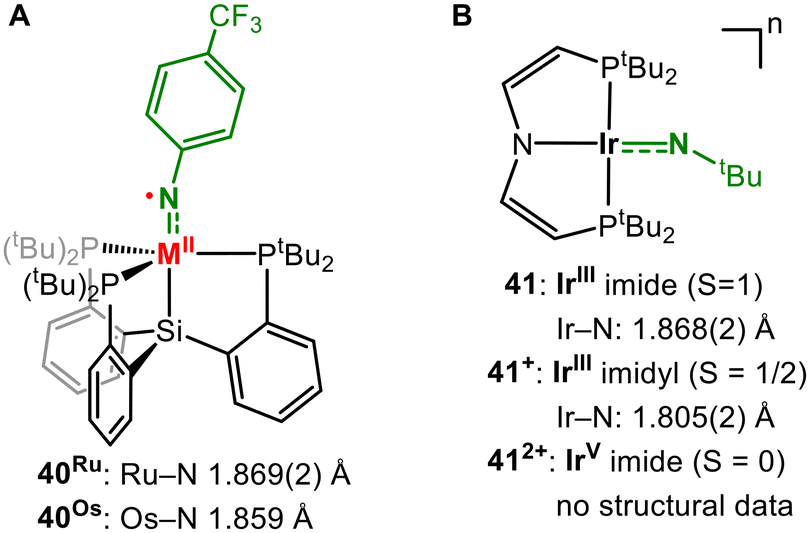 | ||
| Fig. 13 Ruthenium, osmium (A) and iridium imidyl (B) complexes. | ||
Further reduction is N-centred to yield iridium(III) imide 41 and effectuates an Ir-bond elongation owing to the further population of an antibonding π* orbital. Chang irradiated a mononuclear rhodium pyridyl dioxazolone adduct (41, Fig. 14A). To embed into the coordination sphere of the metal, the otherwise poorly binding dioxazolone ligand was tethered by applying a pyridine linker. The in crystallo irradiation of the complex yielded 33% CO2 elimination and the formation of a formal rhodium acylnitrene (43).58 In the due process, the relevant Rh–N bond contracted noticably from 2.153(2) Å to 2.02(2) Å. 43 was described by CASSCF computations as a rhodium(III) singlet acylnitrene, with minor contributions from a singlet Rh(V) acylimide resonance structure. A rhodium(III) triplet nitrene was ruled out based on the absence of radical-like reactivity. The co-crystallization of acetone allowed a subsequent in crystallo reaction with the formed acyl nitrene, providing snapshots of a complete nitrene transfer process to an external substrate.
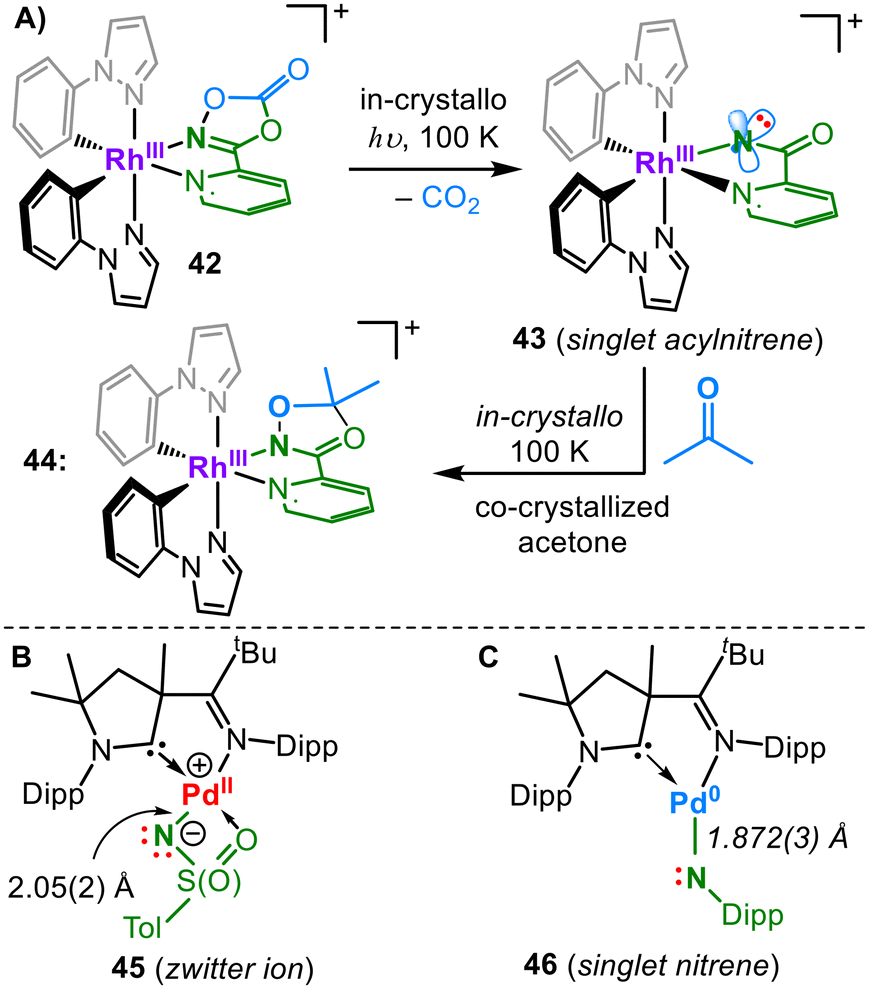 | ||
| Fig. 14 Rhodium nitrene (A) and palladium imide (B) and nitrene (C). | ||
Munz reported two palladium complexes that nicely show the differences in imido metal bonding depending on the choice of the imido substituent. The reaction of tosyl azide with a Pd(0) precursor yields the imido complex 45 (Fig. 14B).14 The metal-imido bond is weak owing to the complete population of the π/π* orbitals, which are also highly asymmetric owing to the chelating nature of the tosyl imido unit. The Pd–N bond order approaches zero, which is however compensated by the admixture of the 5s orbital as well as the additional chelating O-donor. This gives an exceptionally long Pd–N bond of 2.043(19) Å, thereby resulting in a zwitterionic situation with a highly nucleophilic imido nitrogen. In the case of a monodentate aromatic azide, the π-interactions within the corresponding complex 46 are less distorted.59
Computational analysis of 46, aided by UV/Vis and NIR spectroscopic examinations, yielded a substantial palladium(0) singlet nitrene character, with a comparably stronger Pd/N interaction as represented by a Pd–N bond length of 1.872(3) Å. The corresponding aliphatic imido palladium complexes were too reactive to be isolated. Distinct from triplet nitrenes and radical imidyl ligands, 46 is strongly ambiphilic, e.g. it swiftly activates C–F and C–H bonds at or below room temperature. Imidyls or nitrenes of silver and gold are absent, apart from a bis(phosphinonitrene) adduct of silver(I) ([Ag(NPR2)2]+), akin to copper complex 34 (Fig. 11).
Dirhodium catalysed C–H amination was developed as a broadly applicable, reliable synthetic methodology in recent years,9,60 yet the nitrene transferring species eluded their characterisation for a long time. In this regard in crystallo photo chemistry has recently emerged as a powerful strategy; Powers could irradiate a bis(organoazide) adduct of a paddlewheel dirhodium precursor (47, Fig. 15) and observed N2 liberation in the solid state.61 Conversion of up to 70% could be achieved before loss of crystallinity with the partial detection of liberated N2 in the crystal lattice. Upon N2 extrusion, the Rh–N bond of the forming imido ligand contracts from 2.335(3) Å in 47 to 2.12(1) Å in 48, accompanied by an expansion of the Rh–N–C angle from 129.1° to 147.2°. The bond lengths and angles of the remaining complex fragments were subject to only marginal changes. The metrics of the Rh–N-Ada unit allowed to delineate a triplet state by DFT calculations and rule out the singlet spin state alternative. Similarly, an aromatic azide adduct (49) could be converted in crystallo by 90% (50, Fig. 15).62 It revealed a contraction of Rh–N from 2.244(3) Å to 2.055(4) Å. It was corroborated by EXAFS examination that gave indistinguishable Rh–N/O interactions at 2.03(1) Å. Concurrently, the N–C bond contracted from 1.441(5) Å to 1.335(7) Å and the Rh–N–C bond angle expanded from 125.0(2)° to 140.6(4)°.
 | ||
| Fig. 15 In crystallo formation of dirhodium nitrenes. | ||
Metallanitrenes
Similar to the electronic ambivalence of imido metal complexes, for terminal nitrido complexes (LnM![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N3−), the divalent nitridyl (LnMN2−˙) or even metallanitrene (LnM–N−) description can be considered for mid or late transition metals. A profound proof of an N-centred nitridyl was found in the case of a square planar iridium system (52n, Fig. 16).63 The reduction of a regular, cationic IrV nitride (52+) with a short Ir–N bond of 1.677(4) Å leads to the population of a π* orbital that is covalently shared by Ir and N and effectuates the (computed) elongation of the Ir–N bond to 1.714 Å in the resulting neutral 52. The X-band EPR spectrum of 52 shows a rhombic signal with large g-anisotropy without resolved (super)hyperfine couplings to nitrogen. However, 14N hyperfine interactions were observed using X-band electron-nuclear double resonance (ENDOR) spectroscopy. The obtained neutral compound can best be described as an IrIII nitridyl, with the spin density nearly equally residing on Ir and N, which is in resonance with an IrIV nitride.
N3−), the divalent nitridyl (LnMN2−˙) or even metallanitrene (LnM–N−) description can be considered for mid or late transition metals. A profound proof of an N-centred nitridyl was found in the case of a square planar iridium system (52n, Fig. 16).63 The reduction of a regular, cationic IrV nitride (52+) with a short Ir–N bond of 1.677(4) Å leads to the population of a π* orbital that is covalently shared by Ir and N and effectuates the (computed) elongation of the Ir–N bond to 1.714 Å in the resulting neutral 52. The X-band EPR spectrum of 52 shows a rhombic signal with large g-anisotropy without resolved (super)hyperfine couplings to nitrogen. However, 14N hyperfine interactions were observed using X-band electron-nuclear double resonance (ENDOR) spectroscopy. The obtained neutral compound can best be described as an IrIII nitridyl, with the spin density nearly equally residing on Ir and N, which is in resonance with an IrIV nitride.
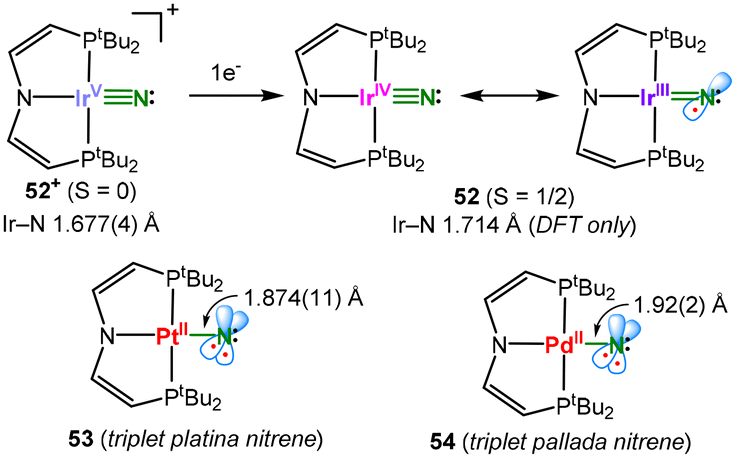 | ||
| Fig. 16 Iridium nitridyl (top) and metallanitrene (bottom) complexes. | ||
This was pushed further by the bona-fide platina nitrene 53 (Fig. 16).64 It was obtained as a transient species via irradiation of a PtII azide precursor in solution, yet irradiation in a crystalline state allowed for its structural characterisation. The observed Pt–N distance of 1.874(11) Å is at the lower end of the reported Pt–N bond lengths. Magnetic, spectroscopic and computational studies gave a triplet state with two N-centred unpaired electrons (91% spin density on N). Moreover, 53 exhibits a covalent single Pt–N bond character in the absence of significant π-bonding. Thus, it is akin to an organic triplet nitrene with the diamagnetic Pt complex fragment replacing the organic substituent. The platinum nitrene shows an ambiphilic character and thus can facilitate reactions with nucleophiles (e.g. phosphines) and electrophiles (boranes) under N-transfer and bond insertion, respectively. Similarly, the Pd triplet nitrene 54 was reported,65 which shows even more nitrogen-centred spin density (96%). Despite the subvalent nature of the nitrogen ligand, it undergoes facile C–H insertion with aldehydes via a nucleophilic rather than an electrophilic or radical attack.
Conclusion and outlook
The pursuit of the isolation and/or structural characterisation of highly reactive nitrene transferring species, especially in the context of C–H amination reactions, has seen substantial advances in recent years. In particular, complexes carrying either considerable unpaired spin density on the N atom or even a nitrene character are of interest given that they are predestined for initial H atom abstraction. Herein, three synthetic approaches have been successfully established: (a) direct isolation via the reaction of metal precursors with the nitrene transfer agent. These are intrinsically often of moderate reactivity to allow for their isolation in the first place. (b) Reduction/oxidation of suitable imido metal precursors, which enables the installation of the imido function in a more stable form. Therefore, it overcomes the possible problems concerning the activation of the nitrene transfer agent in the “right” oxidation state of the metal. The additional benefits of this approach are insights into the oxidation state-dependent electronic balance between the metal and the imido nitrogen in an otherwise unchanged complex geometry. (C) In crystallo irradiation provides a more recent and highly potent access to metal nitrenes. This is particularly of interest for catalytically relevant and highly reactive species. Here, a fundamental challenge is to obtain a suitable complex for irradiation studies given that the respective nitrene transfer agents are often poor ligands. The recently used approach of tethering the respective functionalities to stronger donor functions is thus a prospective of significant advancement in this regard. Further, the recently reported “true” free organo nitrene 3 likely provides a platform for the coordination of small metal fragments, e.g. metal carbonyls. It presumably yields imido complexes in analogy to classic Fischer carbene chemistry. The structural and analytical characterisation (especially using EPR spectroscopy) of all such complexes continue to provide an increasing body of evidence to correctly describe the electronic structure of the metal-bound imido unit using quantum chemical methods. Here, structural metrics are a valuable first indicator for identifying the imidyl-/nitrene-type species. For example, an M–N bond length of around 1.75 Å for late 3d-metal imidos is a strong indicator thereof. However, in some cases, an elongated M–N bond is merely a result of a reduced bond order with nucleophilic imide nitrogen akin to a zwitterion. To complicate the situation, some 3d-metal complexes can switch between different resonance structures, which enable them to respond to the specific requirements of a substrate. A stable metal fragment in a low-spin state, either via a high-oxidation state or using 4d/5d late transition metals, can also provide a suitable platform in which the imido nitrogen is electrophilic, either directly or via ligand-centred oxidation. This is convincingly illustrated in a square planar environment with a d8 configuration or an octahedral geometry with a d6 configuration owing to the pronounced ligand-field stabilisation. The delicate interplay between the metal's identity, oxidation/spin state, coordination geometry, charge and donor strength of the ancillary co-ligands, among themselves and in conjunction with the variety of nitrene fragments (e.g. aromatic, aliphatic tosyl or acylnitrenes), with each exhibiting substantially different electronics and coordination modes (end-on with bent or linear substituent orientation and the possibility of bidentate coordination as for tosylnitrenes) makes this field still not well understood. The current advancements on a structural and isolable level are, however, prospective of rapid advances in understanding the imido metal bond. Ultimately, these are fundamental to be able to judiciously navigate between the possible spin states and electronic structures to alleviate constructive bond activation and transfer reactions. This may help disclose new reactivity pathways and will likely transcend to other metal/main-group multiple bond functionalities.Author contributions
C. G. W. conceptualised and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. G. W. acknowledges the Deutsche Forschungsgemeinschaft (grants WE 5627/7-1 and WE 5627/8-1).References
- (a) G. Dequirez, V. Pons and P. Dauban, Angew. Chem., Int. Ed., 2012, 51, 7384–7395 CrossRef CAS PubMed; (b) G. Smolinsky, J. Am. Chem. Soc., 1960, 82, 4717–4719 CrossRef CAS; (c) L. Horner and A. Christmann, Angew. Chem., Int. Ed. Engl., 1963, 2, 599–608 CrossRef; (d) P. A. S. Smith and B. B. Brown, J. Am. Chem. Soc., 1951, 73, 2435–2437 CrossRef CAS; (e) F. Tiemann, Ber. Dtsch. Chem. Ges., 1891, 24, 4162–4167 CrossRef.
- N. P. Gritsan and M. S. Platz, Chem. Rev., 2006, 106, 3844–3867 CrossRef CAS PubMed.
- (a) H. H. Wenk and W. Sander, Angew. Chem., Int. Ed., 2002, 41, 2742–2745 CrossRef CAS; (b) R. Feng, Y. Lu, G. Deng, J. Xu, Z. Wu, H. Li, Q. Liu, N. Kadowaki, M. Abe and X. Zeng, J. Am. Chem. Soc., 2018, 140, 10–13 CrossRef CAS PubMed; (c) W. T. Borden, N. P. Gritsan, C. M. Hadad, W. L. Karney, C. R. Kemnitz and M. S. Platz, Acc. Chem. Res., 2000, 33, 765–771 CrossRef CAS PubMed.
- (a) P. B. Dervan, M. E. Squillacote, P. M. Lahti, A. P. Sylwester and J. D. Roberts, J. Am. Chem. Soc., 1981, 103, 1120–1122 CrossRef CAS; (b) G. B. Robertson, P. A. Tucker and P. O. Whimp, Inorg. Chem., 1980, 19, 2307–2315 CrossRef CAS; (c) W. D. Hinsberg and P. B. Dervan, J. Am. Chem. Soc., 1978, 100, 1608–1610 CrossRef CAS; (d) P. G. Schultz and P. B. Dervan, J. Am. Chem. Soc., 1980, 102, 878–880 CrossRef CAS.
- F. Dielmann, O. Back, M. Henry-Ellinger, P. Jerabek, G. Frenking and G. Bertrand, Science, 2012, 337, 1526–1528 CrossRef CAS PubMed.
- M. Janssen, T. Frederichs, M. Olaru, E. Lork, E. Hupf and J. Beckmann, Science, 2024, 385, 318–321 CrossRef CAS PubMed.
- D. Wang, W. Chen, H. Chen, Y. Chen, S. Ye and G. Tan, Nat. Chem., 2025, 17, 38–43 CrossRef CAS PubMed.
- (a) Y. Luo, X. Zhang and Y. Xia, Chin. Chem. Lett., 2024, 35, 108778 CrossRef CAS; (b) T. Shimbayashi, K. Sasakura, A. Eguchi, K. Okamoto and K. Ohe, Chem. – Eur. J., 2019, 25, 3156–3180 CrossRef CAS PubMed; (c) B. Du, C.-M. Chan, C.-M. Au and W.-Y. Yu, Acc. Chem. Res., 2022, 55, 2123–2137 CrossRef CAS PubMed; (d) F. Collet, R. H. Dodd and P. Dauban, Chem. Commun., 2009, 5061–5074 RSC.
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed.
- R. Breslow and S. H. Gellman, J. Chem. Soc., Chem. Commun., 1982, 1400–1401 RSC.
- R. Breslow and S. H. Gellman, J. Am. Chem. Soc., 1983, 105, 6728–6729 CrossRef CAS.
- A. Grünwald, S. S. Anjana and D. Munz, Eur. J. Inorg. Chem., 2021, 2021, 4147–4166 Search PubMed.
- H. B. Gray and J. R. Winkler, Acc. Chem. Res., 2018, 51, 1850–1857 CAS.
- A. Grünwald, N. Orth, A. Scheurer, F. W. Heinemann, A. Pöthig and D. Munz, Angew. Chem., Int. Ed., 2018, 57, 16228–16232 CrossRef PubMed.
- J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef CAS.
- (a) K. Ray, F. Heims and F. F. Pfaff, Eur. J. Inorg. Chem., 2013, 2013, 3784–3807 CAS; (b) P. F. Kuijpers, J. I. van der Vlugt, S. Schneider and B. de Bruin, Chem. – Eur. J., 2017, 23, 13819–13829 CrossRef CAS PubMed.
- C. C. Lu, S. DeBeer George, T. Weyhermüller, E. Bill, E. Bothe and K. Wieghardt, Angew. Chem., Int. Ed., 2008, 47, 6384–6387 CAS.
- R. A. Eikey, S. I. Khan and M. M. Abu-Omar, Angew. Chem., Int. Ed., 2002, 41, 3591–3595 Search PubMed.
- H. Shi, J. Xie, W. W. Y. Lam, W.-L. Man, C.-K. Mak, S.-M. Yiu, H. K. Lee and T.-C. Lau, Chem. – Eur. J., 2019, 25, 12895–12899 Search PubMed.
- G. P. van Trieste, K. A. Reid, M. H. Hicks, A. Das, M. T. Figgins, N. Bhuvanesh, A. Ozarowski, J. Telser and D. C. Powers, Angew. Chem., Int. Ed., 2021, 60, 26647–26655 Search PubMed.
- J. S. Duncan, T. M. Nazif, A. K. Verma and S. C. Lee, Inorg. Chem., 2003, 42, 1211–1224 CrossRef CAS PubMed.
- E. R. King, E. T. Hennessy and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 4917–4923 Search PubMed.
- (a) D. A. Iovan and T. A. Betley, J. Am. Chem. Soc., 2016, 138, 1983–1993 CrossRef CAS PubMed; (b) M. J. T. Wilding, D. A. Iovan and T. A. Betley, J. Am. Chem. Soc., 2017, 139, 12043–12049 CrossRef CAS PubMed.
- E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591–595 CrossRef CAS PubMed.
- M. J. T. Wilding, D. A. Iovan, A. T. Wrobel, J. T. Lukens, S. N. MacMillan, K. M. Lancaster and T. A. Betley, J. Am. Chem. Soc., 2017, 139, 14757–14766 Search PubMed.
- S. Reith, S. Demeshko, B. Battistella, A. Reckziegel, C. Schneider, A. Stoy, C. Lichtenberg, F. Meyer, D. Munz and C. G. Werncke, Chem. Sci., 2022, 13, 7907–7913 RSC.
- P.-C. Yang, K.-P. Yu, C.-T. Hsieh, J. Zou, C.-T. Fang, H.-K. Liu, C.-W. Pao, L. Deng, M.-J. Cheng and C.-Y. Lin, Chem. Sci., 2022, 13, 9637–9643 Search PubMed.
- A. Gonzalez, S. Demeshko, F. Meyer and C. G. Werncke, Chem. Commun., 2023, 59, 11532–11535 RSC.
- W. Stroek, M. Keilwerth, L. A. Malaspina, S. Grabowsky, K. Meyer and M. Albrecht, Chem. – Eur. J., 2024, 30, e202303410 CrossRef CAS PubMed.
- J. L. Martinez, S. A. Lutz, H. Yang, J. Xie, J. Telser, B. M. Hoffman, V. Carta, M. Pink, Y. Losovyj and J. M. Smith, Science, 2020, 370, 356–359 Search PubMed.
- Y. Gao, V. Carta, M. Pink and J. M. Smith, J. Am. Chem. Soc., 2021, 143, 5324–5329 Search PubMed.
- W. Mao, D. Fehn, F. W. Heinemann, A. Scheurer, D. Munz and K. Meyer, Angew. Chem., Int. Ed., 2021, 60, 16480–16486 Search PubMed.
- Y. Park, S. P. Semproni, H. Zhong and P. J. Chirik, Angew. Chem., Int. Ed., 2021, 60, 14376–14380 Search PubMed.
- A. Reckziegel, M. Kour, B. Battistella, S. Mebs, K. Beuthert, R. Berger and C. G. Werncke, Angew. Chem., Int. Ed., 2021, 60, 15376–15380 Search PubMed.
- R. Weller, L. Ruppach, A. Shlyaykher, F. Tambornino and C. G. Werncke, Dalton Trans., 2021, 50, 10947–10963 Search PubMed.
- (a) W. Mao, D. Fehn, F. W. Heinemann, A. Scheurer, M. van Gastel, S. A. V. Jannuzzi, S. DeBeer, D. Munz and K. Meyer, Angew. Chem., Int. Ed., 2022, 61, e202206848 Search PubMed; (b) W. Mao, Z. Zhang, D. Fehn, S. A. V. Jannuzzi, F. W. Heinemann, A. Scheurer, M. van Gastel, S. DeBeer, D. Munz and K. Meyer, J. Am. Chem. Soc., 2023, 145, 13650–13662 CrossRef CAS PubMed.
- K. Feng and Y. Li, J. Org. Chem., 1996, 61, 398–400 CrossRef CAS.
- Y. Baek, A. Das, S.-L. Zheng, J. H. Reibenspies, D. C. Powers and T. A. Betley, J. Am. Chem. Soc., 2020, 142, 11232–11243 Search PubMed.
- E. Kogut, H. L. Wiencko, L. Zhang, D. E. Cordeau and T. H. Warren, J. Am. Chem. Soc., 2005, 127, 11248–11249 Search PubMed.
- G. Bai and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 1856–1859 CrossRef CAS PubMed.
- C. A. Laskowski, A. J. M. Miller, G. L. Hillhouse and T. R. Cundari, J. Am. Chem. Soc., 2011, 133, 771–773 CrossRef CAS PubMed.
- Y. Dong, J. T. Lukens, R. M. Clarke, S.-L. Zheng, K. M. Lancaster and T. A. Betley, Chem. Sci., 2020, 11, 1260–1268 Search PubMed.
- Y. Dong, C. J. Lund, G. J. Porter, R. M. Clarke, S.-L. Zheng, T. R. Cundari and T. A. Betley, J. Am. Chem. Soc., 2021, 143, 817–829 CAS.
- (a) D. J. Mindiola and G. L. Hillhouse, J. Am. Chem. Soc., 2001, 123, 4623–4624 CAS; (b) R. Waterman and G. L. Hillhouse, J. Am. Chem. Soc., 2008, 130, 12628–12629 CrossRef CAS PubMed.
- V. M. Iluc, A. J. M. Miller, J. S. Anderson, M. J. Monreal, M. P. Mehn and G. L. Hillhouse, J. Am. Chem. Soc., 2011, 133, 13055–13063 Search PubMed.
- C. A. Laskowski, G. R. Morello, C. T. Saouma, T. R. Cundari and G. L. Hillhouse, Chem. Sci., 2013, 4, 170–174 Search PubMed.
- (a) S.-L. Abram, I. Monte-Pérez, F. F. Pfaff, E. R. Farquhar and K. Ray, Chem. Commun., 2014, 50, 9852–9854 Search PubMed; (b) S. Kundu, E. Miceli, E. Farquhar, F. F. Pfaff, U. Kuhlmann, P. Hildebrandt, B. Braun, C. Greco and K. Ray, J. Am. Chem. Soc., 2012, 134, 14710–14713 Search PubMed; (c) T. Corona, L. Ribas, M. Rovira, E. R. Farquhar, X. Ribas, K. Ray and A. Company, Angew. Chem., Int. Ed., 2016, 55, 14005–14008 CAS; (d) P. Brandt, M. J. Södergren, P. G. Andersson and P.-O. Norrby, J. Am. Chem. Soc., 2000, 122, 8013–8020 CAS; (e) Z. Li, R. W. Quan and E. N. Jacobsen, J. Am. Chem. Soc., 1995, 117, 5889–5890 Search PubMed.
- F. Dielmann, D. M. Andrada, G. Frenking and G. Bertrand, J. Am. Chem. Soc., 2014, 136, 3800–3802 Search PubMed.
- T. R. Cundari, A. Dinescu and A. B. Kazi, Inorg. Chem., 2008, 47, 10067–10072 Search PubMed.
- K. M. Carsch, I. M. DiMucci, D. A. Iovan, A. Li, S.-L. Zheng, C. J. Titus, S. J. Lee, K. D. Irwin, D. Nordlund, K. M. Lancaster and T. A. Betley, Science, 2019, 365, 1138–1143 Search PubMed.
- K. M. Carsch, S. C. North, I. M. DiMucci, A. Iliescu, P. Vojáčková, T. Khazanov, S.-L. Zheng, T. R. Cundari, K. M. Lancaster and T. A. Betley, Chem. Sci., 2023, 14, 10847–10860 Search PubMed.
- (a) Y. M. Badiei, A. Krishnaswamy, M. M. Melzer and T. H. Warren, J. Am. Chem. Soc., 2006, 128, 15056–15057 CAS; (b) M. J. B. Aguila, Y. M. Badiei and T. H. Warren, J. Am. Chem. Soc., 2013, 135, 9399–9406 CAS; (c) Y. M. Badiei, A. Dinescu, X. Dai, R. M. Palomino, F. W. Heinemann, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2008, 47, 9961–9964 CAS.
- A. G. Bakhoda, Q. Jiang, J. A. Bertke, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2017, 56, 6426–6430 CAS.
- (a) D. Intrieri, D. M. Carminati and E. Gallo, J. Porphyrins Phthalocyanines, 2016, 20, 190–203 CAS; (b) C.-X. Ye and E. Meggers, Acc. Chem. Res., 2023, 56, 1128–1141 CAS; (c) Y. Liu, K.-P. Shing, V. K.-Y. Lo and C.-M. Che, ACS Catal., 2023, 13, 1103–1124 Search PubMed.
- G. Wang, Z. Zhou, X. Shen, S. Ivlev and E. Meggers, Chem. Commun., 2020, 56, 7714–7717 RSC.
- (a) A. Takaoka, L. C. H. Gerber and J. C. Peters, Angew. Chem., Int. Ed., 2010, 49, 4088–4091 CrossRef CAS PubMed; (b) A. Takaoka, M.-E. Moret and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 6695–6706 Search PubMed.
- M. Kinauer, M. Diefenbach, H. Bamberger, S. Demeshko, E. J. Reijerse, C. Volkmann, C. Würtele, J. van Slageren, B. de Bruin, M. C. Holthausen and S. Schneider, Chem. Sci., 2018, 9, 4325–4332 RSC.
- H. Jung, J. Kweon, J.-M. Suh, M. H. Lim, D. Kim and S. Chang, Science, 2023, 381, 525–532 CrossRef CAS PubMed.
- A. Grünwald, B. Goswami, K. Breitwieser, B. Morgenstern, M. Gimferrer, F. W. Heinemann, D. M. Momper, C. W. M. Kay and D. Munz, J. Am. Chem. Soc., 2022, 144, 8897–8901 CrossRef PubMed.
- (a) M. P. Paudyal, A. M. Adebesin, S. R. Burt, D. H. Ess, Z. Ma, L. Kürti and J. R. Falck, Science, 2016, 353, 1144–1147 Search PubMed; (b) J.-L. Ma, X.-M. Zhou, J.-L. Chen, J.-X. Shi, H.-C. Cheng, P.-H. Guo and H.-B. Ji, Org. Biomol. Chem., 2022, 20, 7554–7576 RSC.
- A. Das, Y.-S. Chen, J. H. Reibenspies and D. C. Powers, J. Am. Chem. Soc., 2019, 141, 16232–16236 CrossRef CAS PubMed.
- A. Das, C.-H. Wang, G. P. van Trieste, C.-J. Sun, Y.-S. Chen, J. H. Reibenspies and D. C. Powers, J. Am. Chem. Soc., 2020, 142, 19862–19867 CrossRef CAS PubMed.
- M. G. Scheibel, B. Askevold, F. W. Heinemann, E. J. Reijerse, B. de Bruin and S. Schneider, Nat. Chem., 2012, 4, 552–558 CrossRef CAS PubMed.
- J. Sun, J. Abbenseth, H. Verplancke, M. Diefenbach, B. de Bruin, D. Hunger, C. Würtele, J. van Slageren, M. C. Holthausen and S. Schneider, Nat. Chem., 2020, 12, 1054–1059 CrossRef CAS PubMed.
- T. Schmidt-Räntsch, H. Verplancke, J. N. Lienert, S. Demeshko, M. Otte, G. P. van Trieste, K. A. Reid, J. H. Reibenspies, D. C. Powers, M. C. Holthausen and S. Schneider, Angew. Chem., Int. Ed., 2022, 61, e202115626 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |