 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Using phase-resolved vibrational sum-frequency imaging to probe the impact of head-group functionality on hierarchical domain structure in lipid membranes†
Ben John ,
Sarabjeet Kaur,
Martin Wolf,
Martin Thämer and
Alexander P. Fellows*
,
Sarabjeet Kaur,
Martin Wolf,
Martin Thämer and
Alexander P. Fellows*
Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, Berlin, 14195, Germany. E-mail: fellows@fhi-berlin.mpg.de
First published on 1st May 2025
Abstract
The substantial diversity in phospholipids within a plasma membrane, varying in tail length, degree of saturation, and head-group functionality, generates widespread structural heterogeneity. This exists both laterally across the membrane through the spontaneous formation of condensed domains that differ from their surrounding expanded phase in density, composition, and molecular packing order, as well as between its two leaflets, which normally maintain significant compositional asymmetry. Of particular importance is the exposure of phosphatidylserine (PS) lipids which is a marker for important physiological processes e.g. apoptosis. Despite this, the molecular-level alterations to the phase-structure of the membrane that result from PS exposure remain generally unknown. In this work, we utilise recently developed phase-resolved azimuthal-scanned sum-frequency generation (SFG) microscopy to investigate structural changes that occur heterogeneously across model membranes as a result of PS-lipid exposure. Specifically, by probing mixed monolayers of 1,2-dipalmitoylphosphatidylcholine (DPPC) and deuterated 1-palmitoyl-2-oleoylphosphatidylcholine (dPOPC) in both the C–H and C–D stretching regions as well as equivalent films with DPPC exchanged with DPPS, we analyse the variations in the apparent phase distributions and domain morphologies, and quantitatively extract the density, composition, and relative out-of-plane packing order for both mixtures. We find that, in these mixtures, DPPS shows vast differences in the domain growth and coalescence behaviour compared to DPPC, as well as in the relative compositions and molecular ordering within each phase. This demonstrates the critical role the head-group plays in the heterogeneous phase structure of the membrane and may give insights into their impact on important physiological processes.
Introduction
The phospholipids within cell membranes play a pivotal role in determining their physicochemical properties and physiological function. One of the most pertinent and characteristic features of the membranes is their widespread heterogeneity, which exists both laterally across the membrane as well as between its two leaflets.1–3 The lateral heterogeneity is often expressed by spontaneous separation of the membrane constituents into distinct phases, which can differ in density, composition, and packing structure.4–7 The condensed lipid phase domains (rafts) that form during this phase separation are at the heart of many important cellular processes e.g., by providing specific binding sites for proteins and enzymes,2,8 altering the adhesion properties of the membrane,9 and displaying characteristic signalling markers.8 Overall, the functional behaviour of these domains, and thus of the membrane as a whole, is highly reliant on the specific details of their molecular structure.Beyond the lateral heterogeneity, membranes also possess substantial heterogeneity in the lipid composition between their two leaflets, with the proximal (internal) leaflet being enriched in lipids with phosphatidylethanolamine (PE) and phosphatidylserine (PS) head-groups, whilst the distal (external) leaflet is enriched in phosphatidylcholine (PC) head-group lipids.3,10 This asymmetry is actively maintained by the balance between active ‘flippase’ and ‘scramblase’ transport proteins11 but can easily be disturbed, leading to a mixing of lipids and thus substantial compositional changes between the two leaflets. These changes in the lipid constitution can hence have marked effects on the lateral structure of each leaflet, with potentially significant alterations to any formed domains and thus also to the macroscopic physiological membrane properties.
This loss in leaflet asymmetry is particularly critical for the PS lipids as their presence in the proximal leaflet is linked to specific docking sites for signalling proteins12,13 as well as them being important enzyme cofactors.14 Equally, the loss of internalised PS is concomitant with PS exposure in the distal leaflet, which has been implicated in many disease pathogeneses15 as well as being an early marker for apoptosis.16 Specifically, externalisation of PS lipids has been suggested to impact the susceptibility of the membrane for viral entry13 and neuronal re-/degeneration,17,18 as well as generally altering the adhesive properties of the membrane, leading to an increased likelihood of aggregation.19,20 Nevertheless, despite the importance of this compositional heterogeneity in the membrane, relatively little is known about the molecular-level structural alterations induced by PS exposure, and thus about the mechanistic pathways leading to the wider changes in membrane behaviours that are crucial in these critical physiological phenomena.
One important route to understand these changes to the membrane structure due to the varying concentration of PS lipids is through fundamental studies of the inter-lipid interactions within the liquid-condensed (LC) and liquid-expanded (LE) phases in model systems comprising phospholipid mono- or bi-layers. By understanding the deterministic factors controlling the fluctuations in the hierarchical packing structure in these films, one gets a unique insight into the molecular-level origins of the aforementioned physiological processes. Techniques such as atomic force microscopy (AFM) and Brewster angle microscopy (BAM) have been combined with this bottom-up approach to elucidate the structural characteristics in lipid membranes and have revealed many of the crucial details underlying the observed changes in phase behaviour i.e., differences in the size and morphology of the formed LC domains, as well as their growth and coalescence mechanisms.21–23 Beyond this, both Raman imaging24,25 and fluorescence microscopy (FM)26,27 have been used to study differences in their composition, showing a general enrichment of saturated lipids in the LC phase. Furthermore, through the use of polarised light, BAM28 and FM29–33 have demonstrated that certain lipid mixtures can lead to substantial crystallinity in the LC molecular packing arrangements. While these studies have indeed revealed important details about the molecular-level factors underlying PS lipid exposure, many crucial structural properties within these model membranes nevertheless remain elusive.34
In this context, one particularly important structural property of the lipid films is the relative packing order in the different phases. While the lipids are known to pack with their tail-groups generally oriented in an ‘upright’ fashion,35 the extent to which they are conformationally ordered can be highly variable.36,37 This out-of-plane packing order is a parameter that is intimately connected to other structural properties such as the local packing density and composition (which both modulate the local intermolecular environment), but itself can lead to substantial differences in the membrane behaviour e.g., through changing its thickness or energy barrier to transport processes, as well as altering the exposed structural motifs that are linked to signalling interactions.38–40 Therefore, without this important piece of information, our understanding of the changes in molecular-to-mesoscopic phase structure of the membrane due to PS exposure remains limited.
A technique that is sensitive to the orientational order is vibrational sum-frequency generation (SFG) spectroscopy. Like Raman, by probing the vibrational resonances of the different lipids, SFG can provide molecular recognition and is thus sensitive to the film composition and lipid density. As a second-order optical technique, however, the unique symmetry selection rules of SFG mean it is dependent on absolute orientations (instead of just molecular alignment), and thus the output signals are highly sensitive to the orientational distribution i.e., the amount of orientational order.41–47 These properties have made SFG spectroscopy a powerful tool in the structural elucidation of model lipid membranes,48–57 but it was only recently that microscopic imaging of these systems has been demonstrated.58–62 Through these latest technical advances, it was shown that SFG microscopy can reveal previously unattainable structural features in the lipid packing structure.59,60
In this work, we employ this state-of-the-art vibrational SFG microscopy to study phase-separated phospholipid monolayers with different compositions to gain an unprecedented insight into the impact of PS exposure on the membrane structure. Specifically, by using our recently developed phase-resolved azimuthal-scanned SFG microscope,60 we elucidate the differences in domain size and morphology, lipid composition, and out-of-plane packing order between mixed PS/PC monolayers with ones containing only PC lipids. The obtained results give a new perspective on the structural changes that occur when saturated PC lipids are replaced with those possessing a negatively charged PS head-group, and thus also provide insight on the molecular-level mechanisms resulting in the wider membrane alterations associated with PS lipid exposure.
Results and discussion
To elucidate the structural impact of PS lipid exposure, phase-resolved hyperspectral SFG images were recorded in the PPP polarisation combination (SFG, visible, infrared) of mixed monolayers of either DPPC or DPPS with POPC. In order to separate the spectral signatures of the two constituent lipids within each film, deuterated POPC (dPOPC) was used, and the hyperspectral images recorded in both the C–H (∼2800–3000 cm−1) and C–D (∼2050–2250 cm−1) stretching regions. Furthermore, we isolate the out-of-plane structural information of the lipids from the overall PPP response that contains both in-plane (x-direction) and out-of-plane (z-direction) contributions by recording SFG images with azimuthal scanning of the sample. Full theoretical details of this procedure can be found elsewhere.60 In short, the azimuthal scanning modulates the in-plane contributions whilst leaving the out-of-plane contributions unchanged. Therefore, taking the 0-frequency response of the rotational Fourier transform of this four-dimensional dataset yields hyperspectral images solely containing the out-of-plane signals. A brief schematic of this structural characterisation procedure is presented in Fig. 1a. The resulting out-of-plane hyperspectral information for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of DPPS with dPOPC is then shown in Fig. 1b and c. Specifically, Fig. 1b presents the imaginary parts (absorptive line-shapes) of the average spectra across the two frequency ranges, also showing their resonant assignments based on the literature44,63 and a comparison between pure dPOPC and d31-DPPC SFG spectra shown in the ESI.† Fig. 1c then shows images of the out-of-plane SFG response at selected frequencies through both spectral regions.
:1 mixture of DPPS with dPOPC is then shown in Fig. 1b and c. Specifically, Fig. 1b presents the imaginary parts (absorptive line-shapes) of the average spectra across the two frequency ranges, also showing their resonant assignments based on the literature44,63 and a comparison between pure dPOPC and d31-DPPC SFG spectra shown in the ESI.† Fig. 1c then shows images of the out-of-plane SFG response at selected frequencies through both spectral regions.
 | ||
| Fig. 1 Azimuthal-scanned phase-resolved SFG microscopy of a mixed lipid monolayer of 1:1 DPPS:dPOPC. (a) Schematic of the SFG microscope. (b) Imaginary parts of the out-of-plane spectra averaged over the entire image, in both the C–D (black) and C–H (red) frequency ranges, with labels showing the assignment of the resonant features. (c) Images at selected frequencies through the two frequency ranges. A single condensed lipid domain is encircled in each image for reference. All SFG spectra and images are from the PPP polarisation combination and are given relative to the response from a z-cut quartz reference. | ||
An initial comparison of the spatially averaged spectra shown in Fig. 1b highlights two important points about the structure of the membrane. Firstly, the C–H spectrum, corresponding to the DPPS lipid, contains predominantly CH3 resonances, with only minimal contributions from the (more numerous) CH2 groups. This strongly contrasts to the C–D spectrum of dPOPC which shows substantial CD2 contributions amongst relatively modest CD3 resonances. The presence of CH2/CD2 signals in such systems is typically indicative of decreased packing order given that an ‘all-trans’ tail conformation will present perfectly cancelling methylene signals (as there is an equal number of oppositely oriented groups, and implying an effective local centro-symmetry).44 Only deviations from this well-ordered conformation e.g., the introduction of gauche ‘defects’, can therefore give rise to a net CH2/CD2 response. Even considering the partial tail-group oxidation of dPOPC which can reduce the effective CD3 density by up to a factor of 2 (both the saturated and unsaturated chains are still present after oxidation, but only the former maintains its terminal CD3),64 the observed methyl-to-methylene ratios strongly indicate that the DPPS lipid is more ordered than dPOPC. Such an observation aligns with previous SFG spectroscopy measurements of similar lipid systems52,64–66 as well as with expectation as the fully saturated DPPS lipid can pack more closely and thus form highly restricted packing motifs that favour the all-trans ‘pointing up’ conformation with its smaller molecular footprint. On the other hand, the unsaturated dPOPC lipid (as well as its oxidised forms) cannot pack as efficiently64 and thus has tail-groups with more spatial freedom, favouring the formation of the higher entropy conformations containing gauche defects.
The second important insight from the comparison of the C–H and C–D spectra in Fig. 1b is that, despite originating from a 1:1 mixing ratio of the two lipids and despite the effects of oxidation of dPOPC, the amplitude of the average C–D spectrum is notably larger than that of the average C–H spectrum. This is even more surprising given that aliphatic C–H resonances typically have larger amplitudes than their corresponding C–D resonances (by a factor of ∼1.5, see ESI†). Nevertheless, as mentioned earlier, SFG signals are not simply related to the local molecular density, but also to the orientational order. Therefore, this observation could also originate from a reduced order in the DPPS (cf. dPOPC). In the discussion above, however, we determined that the DPPS showed signs of greater packing order given its relative lack of CH2 signals. This apparent contradiction suggests that many of the DPPS molecules do not contribute to the observed spectrum. This could be due to them having largely in-plane contributions from their tail-groups (thus no out-of-plane signals), or equally that they are so disordered that their signals entirely cancel (including CH2 signals from conformational defects). Both of these are characteristic of relatively low packing densities with greater spatial freedom. The combination of some DPPS lipids that contribute highly ordered SFG signals with others that appear to be highly disordered hence suggests a distinct two-phase structure of the monolayer.
Far greater insight into the phase structure of the model membrane can clearly be obtained by looking beyond the spatially averaged spectra and into the hyperspectral images in Fig. 1c. Unlike the average spectra, these showcase the significant and expected lateral heterogeneity present in the film. Through the C–H stretching frequencies, the images at selected resonant features show roughly circular surface regions with increased amplitude. This indicates that they possess increased molecular densities of DPPS and/or greater packing order than their surroundings. By contrast, the same surface regions appear to show a diminished amplitude at the C–D stretching frequencies, suggesting either a decrease in the local dPOPC density or a reduced packing order of this lipid. These observations are consistent with the expectations for condensed phase (LC) domains, which are typically enriched in saturated lipids like DPPS at the expense of unsaturated lipids like POPC, which predominantly occupy the surrounding expanded (LE) phase.67,68 Overall, this confirms the presence of a clear two-phase, LC–LE equilibrium structure in the membrane.
Domain size and morphology
The hyperspectral images in Fig. 1c clearly contain a vast wealth of structural information that can be used to assess the impact of PS lipid exposure. In the next step, we directly compare the phase-separated structure of the membranes by contrasting images of the integrated C–H magnitudes taken from similar SFG microscopy measurements to those presented in Fig. 1. These comparisons are made between the DPPS:dPOPC and DPPC:dPOPC mixtures, thus essentially replacing the DPPS with DPPC, at both the 1:1 mixture that is shown for the DPPS mixture in Fig. 1, as well as the higher, 4:1 ratio of the saturated lipids. The resulting images are presented in Fig. 2, showcasing the different sizes and morphologies of the formed LC domains. For the two 1:1 mixtures, while both form roughly circular domains, those formed with DPPS are considerably smaller, with ∼5 μm diameters compared to ∼10–15 μm for those formed with DPPC. For the 4:1 mixtures, while the increased proportion of DPPC still presents roughly circular LC domains (albeit smaller than those in the corresponding 1:1 mixture), the DPPS sample essentially shows a continuous, interconnected condensed phase network.
 | ||
| Fig. 2 SFG magnitude images for lipid mixtures of both DPPS and DPPC with dPOPC in 1:1 and 4:1 ratios, calculated by integrating across the C–H stretching region. | ||
These vastly contrasting phase structures within the two lipid mixtures (with and without PS) demonstrates that important differences exist in the intermolecular interactions between the lipids in each system. These different interactions are emphasised by the presence of the smaller isolated domains in the 1:1 mixture with DPPS (cf. with DPPC). This is because the domain size is linked to thermodynamic properties such as line tension which, in turn, relates to the lateral lipid interactions in the film.69,70 Therefore, the exposure of PS lipids appears to vastly perturb the domain growth mechanism and thus also the resulting condensed structural motifs present in the membrane. Equally important are the differences between the two 4:1 mixtures, with the DPPS sample showing that the condensed domains have merged into a continuous LC network unlike those containing DPPC that maintain their separation. This suggests that the domains formed with PS lipids have complementary packing structures which present a much smaller energy barrier to coalescence.69 In contrast, the apparent large coalescence energy barrier for the DPPC domains may well be an underlying factor determining the smaller domain sizes in the 4:1 mixture. This results from the combination of unfavourable coalescence and larger LC area coverage favouring smaller domains for a more optimal 2D packing. Overall, these observations accentuate the substantial structural impact that PS lipids can have on the phase-separated membrane structure.
Composition and packing order
As a further step in the structural comparison between model membranes with and without PS lipids, we look deeper into the structure of the LC and LE phases within the monolayers by probing their density, composition, and relative packing orders. Fig. 3 shows equivalent SFG magnitude images to those in Fig. 2, but for both the C–H and C–D spectral regions of the two 1:1 mixture samples. Additionally, the imaginary parts of the spectra averaged over the LC (red) and LE (black) phases are shown below each image. For each sample, both the magnitude images and spectra in Fig. 3a and c show that the LC phase (red trace) has greater C–H signal than the surrounding LE phase (black trace), and vice versa for the C–D signals in Fig. 3b and d. Although the SFG amplitudes are not solely a measure of the specific molecular density, as mentioned earlier, this is consistent with the expected composition of the two phases i.e. enrichment of saturated lipids in the LC phase. Similarly, for both samples, the C–H spectra (Fig. 3a and c) across both phases show more order than the C–D spectra (Fig. 3b and d, even accounting for the oxidation of dPOPC mentioned previously). As above, this is characterised by the ratio of methyl to methylene resonances, which is emphasised here by the green (methylene, CH2/CD2) and orange (methyl, CH3/CD3) bars shown within each of the spectral plots representing the amplitudes of their symmetric stretches in the LC phase spectra. Once again, this aligns with the expected packing structure of the saturated vs. unsaturated lipids.
 | ||
| Fig. 3 SFG magnitude images for both DPPS:dPOPC ((a) and (b)) and DPPC:dPOPC ((c) and (d)) 1:1 lipid mixtures in the C–H ((a) and (c)) and C–D ((b) and (d)) stretching regions. A single LC domain in each system is encircled for reference. Also shown are the imaginary parts of the PPP spectra averaged over the LC (red) and LE (black) phases. The spectra are given relative to the response from a z-cut quartz reference. The amplitudes of the methyl (CH3/CD3, orange) and methylene (CH2/CD2, green) symmetric stretches within the LC spectra are indicated by bars at the bottom of each spectral plot. | ||
Beyond these similarities, however, the two samples also show notable differences. Firstly, comparing the C–H spectra, the DPPS in the LC phase (Fig. 3a, red trace) appears to be generally more ordered than the LC phase DPPC (Fig. 3c, red trace), with the latter presenting larger relative CH2 signals (see orange/green bars). For the LE phase, however, while the DPPC (Fig. 3c, black trace) shows a relatively weak spectrum with significant CH2 signals (thus indicating more disorder than the corresponding LC phase DPPC), the DPPS sample (Fig. 3a, black trace) essentially shows no C–H amplitude. This could indicate a general lack of DPPS density in the LE phase, or equally that the DPPS is strongly disordered there. The above comparison of the average spectra for this sample (Fig. 1b) indicated that there must be a significant proportion of DPPS molecules not contributing to the SFG signals. This would hence suggest that the latter is true i.e., the LE phase does contain DPPS, but it is largely disordered.
Comparing the C–D spectra shows generally larger amplitudes in the DPPS sample (Fig. 3b, cf. with DPPC in Fig. 3d). Again, this indicates either a higher density or greater packing order of dPOPC. Interestingly, the C–D spectra for the LC and LE phases shows a greater contrast in the DPPC sample (Fig. 3d) than for the mixture with DPPS (Fig. 3b). Specifically, the LC spectrum with DPPC (Fig. 3d, red trace) is much weaker than that for the LE phase (Fig. 3d, black trace), and has a notably different ratio of the CD3 and CD2 symmetric stretches, indicating greater order in the former. By contrast, while the LC spectrum for the DPPS sample (Fig. 3b, red trace) is also weaker and has a reduced CD3 to CD2 ratio (cf. LE), the differences are more modest and the spectra generally appear closer in overall amplitude and line-shape. This indicates that the structure of the dPOPC lipids within the two phases are likely more similar for the DPPS sample than with DPPC.
While the above analysis of the spectra from the LC and LE phases in the two samples does give some snippets of structural insight into the differences between the two membranes, it is nevertheless not straightforward to combine these pieces of information into a comprehensive overview of the different membrane structures. This is predominantly due to the qualitative nature of this comparison. For better insight, therefore, the measured signals should be quantitatively related to the structural details. To do this, the observed SFG signals need to be separated into their two main contributions: density and out-of-plane order.
Overall, SFG signals can be described as in eqn (1), where ρ is the molecular density, β is the molecular hyperpolarisability, and R represents a rotation matrix as part of the Euler transformation converting the molecular coordinates (a, b, c) to the lab frame (x, y, z).
 | (1) |
![[small theta, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c9.gif) ,
, ![[small phi, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d6.gif) , and
, and ![[small psi, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d9.gif) ).
).|
SSFG ∝ ρ·O·τ(, , )·β
| (2) |
For the comparison of the different lipid mixtures presented here, eqn (2) can be further simplified. Firstly, as we are solely isolating the out-of-plane contribution, the in-plane Euler angle, , can be neglected. Secondly, as the SFG signals are generally dominated by the lipid tail-groups,52,65 they are modulated by the same hyperpolarisabilities, meaning β can be absorbed into the proportionality constant. Finally, as the lipids all present a general ‘upright’ orientation of their tail-groups, the variation in their two remaining average Euler angles is likely not particularly large. In fact, comparing the ratio of the symmetric and antisymmetric methyl stretches (which is highly dependent on both of these parameters46,65) across the spectra for both phases in both lipid mixtures shows very little variation (see ESI†). This means, to a reasonable approximation, the τ parameter can also be absorbed into the proportionality constant, leaving any variations in signals being dictated only by changes in density or the out-of-plane orientational order coefficient. This is shown in eqn (3), which forms the basis of the analysis of the SFG signals in this work.
| SSFGOP ∝ ρ·OOP | (3) |
With this description, and assuming these structural parameters are constant within the LC and LE phases (which appears reasonable given the relative homogeneity of their observed SFG amplitudes), there are still 10 unknown structural parameters that describe each membrane. These are the densities and order coefficients of each lipid within each phase, as well as the area coverages of the two phases (ALC and ALE). The microscopy measurements shown in Fig. 3 provide 7 observables for each membrane, namely the area coverages of each phase (thus directly reducing the number of unknown structural parameters to 8), the SFG amplitudes in both spectral regions for the LC and LE phases (defined here as the amplitude separation between the symmetric and antisymmetric methyl stretches), and the average mixing ratio of the two lipids. This latter observable gives us eqn (4) (given the 1:1 ratios), relating the densities of each lipid in phase to each other through the area coverages.
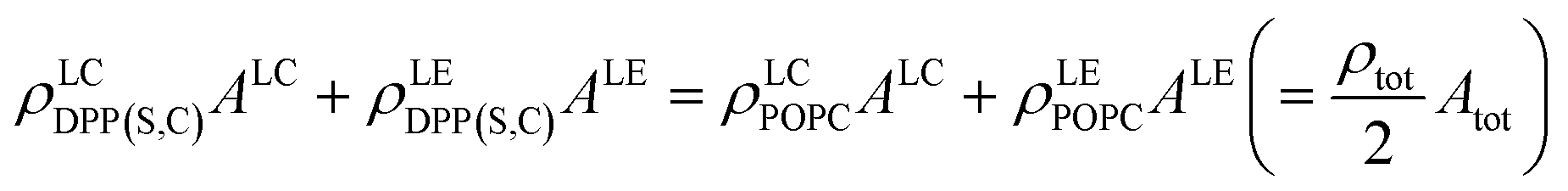 | (4) |
As the SFG amplitudes are only given by proportionalities (see eqn (3)), however, the number of unknowns is then increased to 9 (the remaining 8 structural parameters plus the proportionality constant). Clearly, the aforementioned observables obtained by SFG microscopy alone are insufficient for the full quantitative elucidation of the different structural parameters. Nevertheless, this can be overcome by combining these measurements with information from the pressure–area (Π−A) isotherms of the different monolayers shown in Fig. 4a, as well as reference spectra from pure DPPC and dPOPC monolayers which are shown in Fig. 4b. The isotherms can be used to determine the total (average) molecular density, ρtot (see eqn (4)), providing one additional observable. Equally, the average SFG signals from the two pure monolayers, when divided by their measured average densities (also obtained from their isotherm data), yield spectra that only depend on their orientational order coefficient, as shown in eqn (5) and (6) (based on eqn (3)).
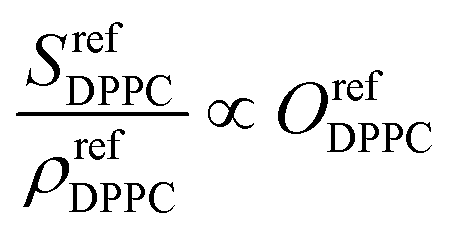 | (5) |
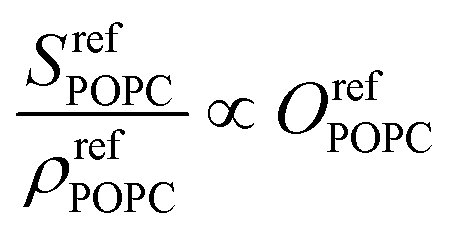 | (6) |
 | ||
| Fig. 4 Analysis of the molecular structure in the 1:1 DPPS:dPOPC and DPPC:dPOPC membranes. (a) Pressure–area isotherms for the three pure lipids as well as the two 1:1 mixtures. (b) SFG spectra in the PPP polarisation combination of a pure DPPC and dPOPC monolayer, referenced to quartz and divided by their average molecular density to provide compositional and structural reference spectra. (c) Calculated compositions of the LC and LE phases for the two lipid mixtures, given in absolute molecular densities. (d) Relative out-of-plane order coefficients in the LC and LE phases for the two lipids within each 1:1 mixture. The saturated lipid (DPPC or DPPS) within the LC phase is taken to have a coefficient of unity. | ||
Therefore, by assuming that these condensed pure monolayers (reference measurements) have equal orientational coefficients to the LC domains in the mixed lipid samples, as in eqn (7) and (8), we obtain two further observables from the sizes of their SFG signals, without any further unknowns. This assumption is justified by the similarity between the spectral line-shapes of the pure monolayer references and the LC phases across both samples (see ESI†). Furthermore, the saturated lipids in the LC phase in the mixed systems and the lipids in the pure condensed monolayers both generally have well-packed structures. As they should therefore present narrow orientational distributions, it is reasonable to take their orientational order coefficients to be unity, as in eqn (7).
| OrefDPPC = OLCDPP(S,C) = 1 | (7) |
| OrefPOPC = OLCPOPC | (8) |
With these reasonable assumptions, the two density-normalised reference spectra shown in Fig. 4b can hence act as absolute density calibrations for the observed C–H and C–D signals. Of course, the validity of this C–D calibration reference also requires the relative oxidation of the dPOPC molecules to be equivalent in the reference sample and for the two lipid mixtures. Based on previous studies from the literature,64 it is likely that the dPOPC is essentially fully oxidised under the ambient conditions during our sample preparation, which are the same for across all samples. This assumption is further validated by the fact that they also present highly overlapping average spectra (see ESI†), indicating relatively similar surface structures.
With this combined dataset, we can hence fully deconvolute the SFG signals and obtain the compositional densities and out-of-plane orientational order coefficients. Full details of this deconvolution are given in the ESI,† with the obtained results presented in Fig. 4c, which reports on the density, and Fig. 4d, which shows the out-of-plane orientational order coefficients. Firstly, the absolute molecular densities of each lipid in the two phases presented in Fig. 4c confirm the previous suggestion that the LC phase is enriched in DPPC/DPPS (shown in red) and depleted in dPOPC (shown in blue) for both sample systems. Secondly, for both samples, the LE phases show a lesser total (combined) density than the LC phases, and a greater relative proportion of dPOPC, also aligning with expectation. An overall comparison between the densities of both samples then shows that the DPPS mixture generally leads to less enrichment of the saturated lipids into the LC phase i.e., there is less de-mixing of the two lipids. This hence indicates that the ratio of homo- to hetero-molecular interaction energies is also diminished, leading to more hetero-molecular mixing. This is not entirely surprising given that the PS head-group is doubly negatively charged and thus provides a strong electrostatic repulsion, unlike the zwitterionic PC head-group.
Another important comparison is the total (combined) density of each phase between the samples with the two different head-groups. Both phases of the DPPS sample have a higher total density than those with DPPC. This observation is likely the origin of the higher CH3 to CH2 ratio observed for DPPS cf. DPPC discussed above. The observed increase in density for the DPPS sample is also reflected in the isotherm data in Fig. 4a. This shows that a greater density is required to achieve the same surface pressure (which was the defining parameter used to form the samples) and means that at the same molecular density, the PS-containing membrane has a higher surface energy density, which is generally an indicator for a less ordered structure. This clearly demonstrates that PS exposure can have a pronounced effect on compositional heterogeneity within the proximal membrane leaflet as well as its overall thermodynamic properties.
The results on orientational ordering are highlighted in Fig. 4d, showing the difference in out-of-plane order coefficients in the two films obtained from these measurements. As mentioned above, the values for DPPC/DPPS in the respective LC domains are taken to be equal to that in the condensed DPPC reference measurement, and set to unity based on them having well-packed structures (eqn (7)). Equally, the values for dPOPC in the LC phase are also taken to be equal for both samples as well as the reference (eqn (8)), but are not set to unity. This value is instead determined by comparing the two reference measurements (DPPC and dPOPC), as discussed in ESI,† yielding a reduced value of ∼0.45. This value assumes no loss in effective CD3 density due to oxidation and thus represents the lower limit of its relative out-of-plane order. That means for the likely case of almost complete oxidation, we obtain a value of ∼0.9, which suggests a surprisingly high order for dPOPC (although still being smaller than for the saturated lipids, as expected). However, as the extent to which the dPOPC has oxidised cannot be fully quantified from our measurements, we simply consider the minimum order parameter and present the corresponding values in the mixed lipid samples (which are determined relative to this) having been scaled by this reduced factor.
With these fixed order parameters for the LC phase, we can now compare the deviations observed for the LE phase. For the DPPC sample, the orientational order of both lipids is found to be somewhat lower in the LE phase compared to the LC phase, again fitting with expectation. This, however, strongly contrasts with the observations for the DPPS sample. Here, the DPPS order in the LE phase is shown to be substantially reduced to ∼10% of the relative order in the LC phase. This observation aligns with our previous discussions relating to the seemingly low amplitudes of the C–H average spectra and the general lack of DPPS signals from the LE phase. On the other hand, the dPOPC in the LE phase shows a very similar order coefficient to the LC phase. This also lines up with the above observation of similar C–D spectra for the two phases in this sample. Overall, these substantial differences demonstrate that the simple exchange of PC lipids with their PS equivalents can also lead to pronounced changes in the molecular packing order, particularly for DPPS within the LE phase.
Conclusions and outlook
Based on the individual findings presented and discussed above, a broader picture of the properties of the PS lipid in model membranes and its substantial impact on their structure and behaviour can be derived. Just as with solely PC lipids, the LC domains in the PS/PC mixture show the expected enrichment of saturated lipids, with a higher density and greater orientational order than the surrounding LE phase. However, the PS/PC mixture generally forms a higher energy monolayer and shows distinctly less compositional de-mixing between the two phases, pointing towards a lesser distinction between homo- and hetero-molecular interactions in these films. This shows that important thermodynamic properties in these model membranes are altered due to the presence of the PS lipids. Furthermore, the PS lipids clearly tend to show large disorder unless they are conformationally restricted in the LC phase, in direct contrast to PC lipids. On the other hand, it seems that there is a high barrier for forming these LC domains containing the PS lipids. These two observations may well be linked, since significant energy must be input to overcome both the entropic cost of ordering the largely disordered PS lipids in the LE phase, as well as the significant electrostatic repulsion of their negatively charged head-groups. Interestingly, the LC domains with DPPS, once they are formed, also show no significant barrier to coalescence at higher relative concentrations, in stark contrast to those with DPPC. For DPPC, it even seems that the high barrier to coalescence leads to the formation of smaller domains so as to optimise their 2D packing and avoid contact. This indicates that the boundary structures of the LC domains with DPPS are easily coupled without a significant disruption to either structure unlike those with DPPC.While these observations lead to important insights into the molecular-to-mesoscopic structural changes that accompany the externalisation of PS lipids, many questions remain unanswered. It would thus be interesting to extend these studies by varying more sample parameters such as, mixing ratios, surface pressures, and other relevant head-groups, to shed more light on the thermodynamic and mechanistic origins of these structural effects. As demonstrated in this work, with our microscope, we nevertheless have a tool which is capable of obtaining advanced structural characterisation of these systems. Importantly, the accuracy of the quantitative structural parameters obtained here, namely densities and orientational order coefficients, are also highly supported by the complementary analysis of methyl-to-methylene ratios in the different mean spectra that provide independent confirmation. Therefore, this unprecedented insight into the molecular-level details within these systems can be extended to directly address many of these open questions. Furthermore, the elucidative potential of this technique opens possibilities for the structural investigation of a broader range of sample systems.
Experimental methods
Sample preparation
The lipids 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC, >99%), 1,2-dipalmitoyl-sn-glycero-3-phosphoserine (DPPS, >99%), and d82-1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (dPOPC, >99%) were procured from Avanti Polar Lipids (Alabaster, AL, USA). Stock solutions of DPPC and dPOPC were prepared in chloroform (99.0–99.4% purity, VWR International GmbH, Darmstadt, Germany) at a concentration of 1 mg mL−1. The DPPS stock solution was prepared in a 2:1 (v/v) mixture of chloroform and methanol (≥99.8% purity, VWR International GmbH, Darmstadt, Germany) also at a concentration of 1 mg mL−1, as the addition of methanol aids in solubilising the polar and charged serine head-group of DPPS.
Mixed lipid monolayers were prepared by combining lipids in mass ratios of 4:1 and 1:1 (DPPC:dPOPC and DPPS:dPOPC, respectively) and depositing them dropwise onto the water surface of a PTFE Langmuir–Blodgett trough (MicroTrough G1, Kibron, Helsinki, Finland) containing ultrapure water (Milli-Q, 18.2 MΩ cm, <3 ppb total organic carbon). Prior to lipid deposition, the Langmuir–Blodgett trough was cleaned using chloroform and ultrapure water, followed by repeated aspiration of the water surface until the surface pressure variation was within 0.1 mN m−1 under full compression. The platinum Wilhelmy plate pressure sensor was flamed and rinsed with chloroform and ultrapure water to remove residual contamination. After deposition, the films were left undisturbed for approximately 5 minutes to allow for the evaporation of chloroform (or the chloroform–methanol mixture for DPPS). The monolayers were then compressed to a surface pressure of 20 mN m−1 at a barrier speed of 10 mm min−1 and equilibrated for 2 hours to ensure stabilisation and oxidation of the POPC lipid by ambient ozone exposure.64,71
To immobilise the monolayers for microscopy measurements, a Langmuir–Blodgett transfer method was employed to suppress both intrinsic dynamic motion and convection-driven flows, such as Bénard–Marangoni convection caused by localised subphase heating.72 Monolayers were transferred at a rate of 2 mm min−1 using a LayerX dipper (Kibron, Helsinki, Finland) onto ultra-flat fused silica substrates (Korth Kristalle, Altenholz, Germany, 5 mm thickness, 25.4 mm diameter, <2 nm surface roughness). The substrates were cleaned with chloroform and ultrapure water, followed by UV-ozone treatment (UV/Ozone ProCleaner Plus, BioForce Nanosciences, Virginia Beach, VA, USA) for at least 30 minutes prior to use.
The surface pressure–area isotherms of pure DPPC, DPPS, and POPC, as well as the 1:1 mass mixtures of DPPC:dPOPC and DPPS:dPOPC, were recorded using the same Langmuir–Blodgett trough. The trough was cleaned following the procedure described above and the lipid stock solutions were deposited dropwise onto the water surface using an Eppendorf pipette, ensuring sufficient coverage. After deposition, the films were left undisturbed for a few minutes to ensure complete solvent evaporation. Isotherms were acquired using the compression isotherm module integrated into the Langmuir–Blodgett trough system, with the barriers compressed at a constant speed of 10 mm min−1.
SFG microscope
The heterodyned phase-resolved widefield vibrational sum-frequency generation (vSFG) microscope used in this work is based on our previously developed system, details of which can be found in our prior publications.59,60,73 In brief, the microscope operates fully in the time-domain, utilising a home-built interferometer driven by the broadband IR and visible outputs from a Ti:sapphire laser system.74 These outputs, combined with a local oscillator (LO) generated from z-cut quartz, are aligned in a collinear geometry and focused onto the sample through a custom-designed reflective objective (0.78 NA, Pike Technologies, Madison, WI, USA). The incident beams (IR, visible, and LO) are frequency-filtered, temporally aligned, and focused onto the sample surface at an incidence angle of 36° using a 38 cm focal length off-axis parabolic mirror. The reflected vSFG signal is isolated and recorded on an electrically cooled CCD camera (ProEM-HS:1024BX3, Teledyne Princeton Instruments, Trenton, NJ, USA) using paired-pixel balanced imaging to reduce noise.73 A PPP polarisation combination is employed throughout, probing both in-plane and out-of-plane molecular vibrations.The hyperspectral SFG images were acquired as interferometric images recorded over time delays between the IR and other input beams (visible and LO) from −300 fs to 3000 fs in 2 fs steps. Fast Fourier transforms (FFT) of the time-domain data provided spectral images, which were normalised in phase and amplitude using a reference measurement of z-cut quartz (from −300 to 300 fs). Post-processing included removing dark counts, subtracting a linear non-resonant baseline, and averaging over 4–12 interferometric scans depending on the dataset.
For rotational analysis, vSFG images were obtained at rotational increments of 60° across the full azimuthal range. The processed images were back-rotated to align corresponding pixels, generating a 4D dataset correlating surface location, spectral frequency, and azimuthal angle. A complex Fourier transform was then applied directly to the azimuthal angle data to extract azimuthal frequencies. Two sets of measurements were performed to probe specific vibrational regions: one with the IR frequency centred at 2900 cm−1 to target CH stretching modes and another centred at 2100 cm−1 to target CD stretching modes.
Background correction
To accurately isolate the resonant vibrational signals, the SFG magnitude images were corrected for background contributions. Background correction was performed by subtracting the mean signal intensity from spectral regions outside the frequency ranges of interest, specifically the CD stretching region (∼2100 cm−1) and the CH stretching region (∼2900 cm−1). These out-of-interest regions primarily contain non-resonant contributions and noise, which do not correspond to the targeted vibrational modes. This correction step ensured that the processed images reflected only the resonant signals associated with the molecular vibrations under investigation.Data availability
Due to the size, relative complexity, and multi-level processing of the raw data presented in this work, it has not been made available on a data repository, but any and all parts will be made available upon reasonable request to the corresponding author.Conflicts of interest
There are no conflicts of interest.Acknowledgements
We thank M. Krenz along with the technicians group at the Fritz Haber Institute for providing invaluable technical support along with the Bluhm group for use of their Langmuir–Blodgett trough in these measurements. Open Access funding provided by the Max Planck Society.References
- T. Harayama and H. Riezman, Nat. Rev. Mol. Cell Biol., 2018, 19, 281–296 CrossRef PubMed.
- E. Sezgin, I. Levental, S. Mayor and C. Eggeling, Nat. Rev. Mol. Cell Biol., 2017, 18, 361–374 CrossRef PubMed.
- W. Stillwell, An Introduction to Biological Membranes: from Bilayers to Rafts, Academic Press, London, UK, 2013 Search PubMed.
- D. Lingwood and K. Simons, Science, 2010, 327, 46–50 CrossRef PubMed.
- P. J. Quinn and D. Chapman, CRC Crit. Rev. Biochem., 1980, 8, 1–117 CrossRef.
- M. Carquin, L. D’Auria, H. Pollet, E. R. Bongarzone and D. Tyteca, Prog. Lipid Res., 2016, 62, 1–24 CrossRef PubMed.
- J. J. Kinnun, D. Bolmatov, M. O. Lavrentovich and J. Katsaras, Chem. Phys. Lipids, 2020, 232, 104976 CrossRef PubMed.
- D. A. Brown and E. London, Annu. Rev. Cell Dev. Biol., 1998, 14, 111–136 CrossRef CAS PubMed.
- S. Munro, Cell, 2003, 115, 377–388 CrossRef CAS.
- Y. Ma, K. Poole, J. Goyette and K. Gaus, Front. Immunol., 2017, 8, 01513 CrossRef.
- M. Doktorova, J. L. Symons and I. Levental, Nat. Chem. Biol., 2020, 16, 1321–1330 CrossRef CAS PubMed.
- T. Yeung, G. E. Gilbert, J. Shi, J. Silvius, A. Kapus and S. Grinstein, Science, 2008, 319, 210–213 CrossRef CAS.
- E. M. Bevers and P. L. Williamson, Physiol. Rev., 2016, 96, 605–645 CrossRef CAS PubMed.
- J. E. Vance and R. Steenbergen, Prog. Lipid Res., 2005, 44, 207–234 CrossRef CAS.
- D. C. Calianese and R. B. Birge, Cell Commun. Signal., 2020, 18, 41 CrossRef PubMed.
- B. Fadeel and S. Orrenius, J. Intern. Med., 2005, 258, 479–517 CrossRef.
- H.-W. Shin and H. Takatsu, Crit. Rev. Biochem. Mol. Biol., 2020, 55, 166–178 CrossRef CAS.
- H. Faris, M. Almasieh and L. A. Levin, Cell Death Discovery, 2021, 7, 247 CrossRef.
- R. F. A. Zwaal, P. Comfurius and E. M. Bevers, Cell. Mol. Life Sci., 2005, 62, 971–988 CrossRef PubMed.
- A. P. Fellows, M. T. L. Casford, P. B. Davies, J. S. Gibson, J. N. Brewin and D. C. Rees, Colloids Surf., B, 2021, 197, 111383 CrossRef PubMed.
- D. Vollhardt, Curr. Opin. Colloid Interface Sci., 2014, 19, 183–197 CrossRef.
- R. I. S. Romão, Q. Ferreira, J. Morgado, J. M. G. Martinho and A. M. P. S. Gonçalves da Silva, Langmuir, 2010, 26, 17165–17177 CrossRef PubMed.
- J. Sanchez and A. Badia, Chem. Phys. Lipids, 2008, 152, 24–37 CrossRef.
- S. H. J. Donaldson and H. B. de Aguiar, J. Phys. Chem. Lett., 2018, 9, 1528–1533 CrossRef.
- J. Ando, M. Kinoshita, J. Cui, H. Yamakoshi, K. Dodo, K. Fujita, M. Murata and M. Sodeoka, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4558–4563 CrossRef PubMed.
- R. F. M. de Almeida, L. M. S. Loura and M. Prieto, Chem. Phys. Lipids, 2009, 157, 61–77 CrossRef.
- I. C. Shieh and J. A. Zasadzinski, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E826–E835 CrossRef.
- P. Krüger and M. Lösche, Phys. Rev. E:Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 7031–7043 CrossRef.
- V. T. Moy, D. J. Keller, H. E. Gaub and H. H. McConnell, J. Phys. Chem., 1986, 90, 3198–3202 CrossRef.
- V. T. Moy, D. J. Keller and H. M. McConnell, J. Phys. Chem., 1988, 92, 5233–5238 CrossRef.
- U. Bernchou, J. Brewer, H. S. Midtiby, J. H. Ipsen, L. A. Bagatolli and A. C. Simonsen, J. Am. Chem. Soc., 2009, 131, 14130–14131 CrossRef PubMed.
- J. Dreier, J. R. Brewer and A. C. Simonsen, J. Phys. Chem. Lett., 2013, 4, 2789–2793 CrossRef.
- J. Dreier, J. Brewer and A. C. Simonsen, Langmuir, 2014, 30, 10678–10685 CrossRef PubMed.
- A. J. Sodt, M. L. Sandar, K. Gawrisch, R. W. Pastor and E. Lyman, J. Am. Chem. Soc., 2014, 136, 725–732 CrossRef.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Garland Science, New York, 2002 Search PubMed.
- J. L. Ranck, L. Mateu, D. M. Sadler, A. Tardieu, T. Gulik-Krzywicki and V. Luzzati, J. Mol. Biol., 1974, 85, 249–277 CrossRef.
- K. Kjaer, J. Als-Nielsen, C. A. Helm, L. A. Laxhuber and H. Möhwald, Phys. Rev. Lett., 1987, 58, 2224–2227 CrossRef CAS PubMed.
- R. Lipowsky, Nature, 1991, 349, 475–481 CrossRef CAS.
- J. F. Nagle and S. Tristram-Nagle, Biochim. Biophys. Acta, 2000, 1469, 159–195 CrossRef CAS PubMed.
- J. Frallicciardi, J. Melcr, P. Siginou, S. J. Marrink and B. Poolman, Nat. Commun., 2022, 13, 1605 CrossRef CAS.
- Y. R. Shen, Fundamentals of Sum-Frequency Spectroscopy, Cambridge University Press, Cambridge, 2016 Search PubMed.
- A. Morita, Theory of Sum Frequency Generation Spectroscopy, Springer, Singapore, 2018 Search PubMed.
- R. W. Boyd, Nonlinear Optics, Elsevier Inc., London, UK, 4th edn, 2020 Search PubMed.
- A. G. Lambert, P. B. Davies and D. J. Neivandt, Appl. Spectrosc. Rev., 2005, 40, 103–145 CrossRef CAS.
- A. J. Moad and G. J. Simpson, J. Phys. Chem. B, 2004, 108, 3548–3562 CrossRef CAS.
- H. F. Wang, W. Gan, R. Lu, Y. Rao and B. H. Wu, Int. Rev. Phys. Chem., 2005, 24, 191–256 Search PubMed.
- H.-F. Wang, L. Velarde, W. Gan and L. Fu, Annu. Rev. Phys. Chem., 2015, 66, 189–216 CrossRef CAS PubMed.
- G. L. Richmond, Chem. Rev., 2002, 102, 2693–2724 CrossRef CAS PubMed.
- S. Roke, ChemPhysChem, 2009, 10, 1380–1388 CrossRef CAS.
- J. Zhang, W. Yang, J. Tan and S. Ye, Phys. Chem. Chem. Phys., 2018, 20, 5657–5665 RSC.
- J. Liu and J. C. Conboy, J. Am. Chem. Soc., 2004, 126, 8376–8377 CrossRef CAS PubMed.
- G. Ma and H. C. Allen, Langmuir, 2006, 22, 5341–5349 CrossRef CAS.
- J. F. D. Liljeblad, V. Bulone, E. Tyrode, M. W. Rutland and C. M. Johnson, Biophys. J., 2010, 98, L50–L52 CrossRef CAS.
- A. P. Fellows, M. T. L. Casford and P. B. Davies, AIP Adv., 2021, 11, 045119 CrossRef CAS.
- S. A. Goussous, A. P. Fellows, M. T. L. Casford and P. B. Davies, Biochim. Biophys. Acta, Biomembr., 2019, 1861, 1568–1578 CrossRef CAS PubMed.
- W. Liu, Z. Wang, L. Fu, R. M. Leblanc and E. C. Y. Yan, Langmuir, 2013, 29, 15022–15031 CrossRef CAS PubMed.
- X. Chen, J. Wang, C. B. Kristalyn and Z. Chen, Biophys. J., 2007, 93, 866–875 CrossRef CAS.
- F. Yang and S. Baldelli, J. Phys. Chem. B, 2022, 126, 5637–5645 CrossRef CAS.
- A. P. Fellows, B. John, M. Wolf and M. Thämer, Nat. Commun., 2024, 15, 3161 CrossRef CAS PubMed.
- A. P. Fellows, B. John, M. Wolf and M. Thämer, J. Phys. Chem. Lett., 2024, 15, 10849–10857 CrossRef CAS PubMed.
- S. M. Ortiz and S. Baldelli, Surf. Sci., 2025, 751, 122627 CrossRef CAS.
- H. Wang and W. Xiong, Annu. Rev. Phys. Chem., 2021, 72, 279–306 CrossRef CAS.
- E. Tyrode and J. Hedberg, J. Phys. Chem. C, 2012, 116, 1080–1091 CrossRef CAS.
- L. Qiao, A. Ge, Y. Lang and S. Ye, J. Phys. Chem. B, 2015, 119, 14188–14199 CrossRef CAS.
- R. J. Feng, X. Li, Z. Zhang, Z. Lu and Y. Guo, J. Chem. Phys., 2016, 145, 244707 CrossRef.
- K. I. Inoue, C. Takada, L. Wang, A. Morita and S. Ye, J. Phys. Chem. B, 2020, 124, 5246–5250 CrossRef CAS PubMed.
- P. J. Quinn and C. Wolf, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 33–46 CrossRef CAS.
- G. Ma and H. C. Allen, Langmuir, 2007, 23, 589–597 CrossRef CAS.
- M. Flörsheimer and H. Möhwald, Chem. Phys. Lipids, 1989, 49, 231–241 CrossRef.
- C. W. McConlogue and T. K. Vanderlick, Langmuir, 1997, 13, 7158–7164 CrossRef.
- A. P. Fellows, M. T. L. Casford and P. B. Davies, Biophys. J., 2023, 122, 2007–2022 CrossRef CAS.
- A. P. Fellows, M. T. L. Casford and P. B. Davies, J. Chem. Phys., 2022, 156, 164701 CrossRef CAS.
- T. Khan, B. John, R. Niemann, A. Paarmann, M. Wolf and M. Thämer, Opt. Express, 2023, 31, 28792–28804 CrossRef PubMed.
- M. Thämer, R. K. Campen and M. Wolf, Phys. Chem. Chem. Phys., 2018, 20, 25875–25882 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4fd00187g |
| This journal is © The Royal Society of Chemistry 2025 |