
Discovery and biological evaluation of nitrofuranyl–pyrazolopyrimidine hybrid conjugates as potent antimicrobial agents targeting Staphylococcus aureus and methicillin-resistant S. aureus†
Sapna
Saini‡
be,
G. Lakshma
Reddy‡
ae,
Anjali
Gangwar
be,
Harpreet
Kour
ae,
Gajanan G.
Nadre
ce,
Ramajayan
Pandian
de,
Sunny
Pal
b,
Utpal
Nandi
def,
Rashmi
Sharma
 *be and
Sanghapal D.
Sawant
*ace
*be and
Sanghapal D.
Sawant
*ace
aNatural Products and Medicinal Chemistry Division, CSIR-Indian Institute of Integrative Medicine, Canal Road, Jammu-180001, India. E-mail: sd.sawant@ncl.res.in
bInfectious Diseases Division, CSIR – Indian Institute of Integrative Medicine, Canal Road, Jammu – 180001, India. E-mail: rashmi.sharma.09@iiim.res.in
cOrganic Chemistry Division, CSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pune-411008, India
dPharmacology Division, CSIR – Indian Institute of Integrative Medicine, Canal Road, Jammu – 180001, India
eAcademy of Scientific and Innovative Research, Ghaziabad – 201002, India
fDepartment of Chemical Sciences, Bose Institute, Unified Academic Campus, Kolkata – 700 091, India
First published on 26th December 2024
Abstract
Nitrofuran and pyrazolopyrimidine-based compounds possess a broad antimicrobial spectrum including Gram-positive and Gram-negative bacteria. In the present work, a series of conjugates of these scaffolds was synthesized and evaluated for antimicrobial activity against Staphylococcus aureus and methicillin-resistant S. aureus (MRSA). Many compounds showed MIC values of ≤2 μg ml−1, with compound 35 demonstrating excellent activity (MICs: 0.7 and 0.15 μg ml−1 against S. aureus and MRSA, respectively) and safety up to 50 μg ml−1 in HepG2 cells. Compound 35 also exhibited no hemolytic activity, biofilm eradication, and effectiveness against efflux-pump-overexpressing strains (NorA, TetK, MsrA) without resistance development. It showed synergistic effects with vancomycin (S. aureus) and rifampicin (MRSA). Mechanistic studies revealed that compound 35 exhibits good membrane-targeting abilities, as evidenced by DAPI/PI staining and scanning electron microscopy (SEM). In an intracellular model, it reduced bacterial load efficiently in both S. aureus and MRSA strains. With a strong in vitro profile, compound 35 demonstrated favorable oral pharmacokinetics at 30 mg kg−1 and potent in vivo anti-MRSA activity, highlighting its potential against antibiotic-resistant infections.
Introduction
Antimicrobial resistance (AMR) has emerged as a result of the misuse and overconsumption of antibiotics, providing bacteria with opportunities to evolve and develop various resistance mechanisms.1–4 ESKAPE pathogens, which cause complex infections, have developed resistance to available antibiotics and drugs, either through intrinsic or acquired mechanisms, contributing to significant global health challenges in the 21st century.2,4–7 AMR caused an estimated 1.27 million fatalities around the world and indirectly contributed to approximately 4.95 million deaths in 2019.8 According to the latest report, AMR is projected to cause over 300 million premature deaths by 2050, with an additional 10 million deaths expected annually (Antimicrobial Resistance: Tackling a Crisis for the Future Health and Wealth of Nations, 2014, downloaded from http://amr-review.org/, last accessed on August 30, 2024). Methicillin-resistant Staphylococcus aureus (MRSA) has been identified as a high-priority pathogen needing urgent control.9 MRSA resistance to methicillin was first reported in the UK in the 1960s, and since then, it has rapidly spread across Europe, Asia, the USA, and Africa.10–13 Data from 15 European countries indicate that MRSA accounted for over 10% of bloodstream S. aureus infections (EARS-Net data, 2013), while S. aureus infections overall contributed to 22.1%.14 MRSA is also developing resistance to new antibiotics, including ceftaroline, daptomycin, oxazolidinones, and tetracycline.12,15,16There is an urgent need to find newer molecules and scaffolds to overcome the limitation of these resistances. In this study, we present a series of molecules bearing conjugates of nitrofuran and pyrazolopyrimidine; two distinct and biologically important classes of molecules with significant MICs against S. aureus and MRSA as potent antibacterial agents targeting S. aureus and MRSA.
The nitrofurans are widely studied and typically used as antibiotics or antimicrobials.17 They appear to inhibit a number of microbial enzyme systems but their mechanism is still unclear. Their primary action is bacteriostatic, but at high doses, they are also bactericidal. They appear to inhibit a number of microbial enzyme systems but their mechanism is still unclear. There are many nitrofuran-based molecules that are used as a drug or being clinically investigated. Nitrofurantoin (1) is used to treat urinary tract infection; nifurtimox (2) is used for the treatment of Chagas disease.18 There are several other molecules reported in the nitrofuran class as antibacterial agents like nifuroxazide (3), furazolidone (4), nifuratel, ranbezolid, furylfuramide, nitrofurazone, nifurquinazole, nifurtoinol, nifurzide, etc.19–23 Some of the representative molecules that possess nitrofuran cores are shown in Fig. 1. On the other hand, the pyrazolopyrimidine scaffold has also been very well exploited and many analogs based on this scaffold are reported as anti-bacterial agents such as compounds (5) and (6), as shown in Fig. 1.24–27 The pyrazolopyrimidine scaffold is considered as a bioisostere of the biogenic purine class and this nucleus has a high impact in the field of pharmaceutical sciences with a vast spectrum of biological activities that includes adenosine receptor antagonists,28,29 anti-viral,30,31 anti-cancer and some others32,33 Further, there are a good number of literature reports on their PDE inhibitory potential34,35 including some approved drugs that are known for anti-inflammatory36,37 activities also.
 | ||
| Fig. 1 Structures of some biologically important nitrofuran and pyrazolopyrimidine class of molecules. | ||
Both the nitrofuran and pyrazolopyrimidine scaffolds represent promising candidates as antibacterial agents and it is expected that these molecules could further give some potential candidates in synergy. Keeping in mind the importance of these scaffolds, we started a program based on the literature report of H. A. Burch in 1968, in which 4-amino-6-(5-nitro-2-furyl)-lH-pyrazolo[3,4-d]pyrimidines were presented to show significant activity against S. aureus with a low MIC of 0.1 μg ml−1.25 The concept of bioisosterism was used while designing our present series, where the idea was that the molecules that exhibit similar volume, shape, and/or physicochemical properties can produce broadly similar biological effects.38 Considering this report, we have designed a bioisostere series based on nitrofuranyl–pyrazolopyrimidine scaffold (Fig. 2).
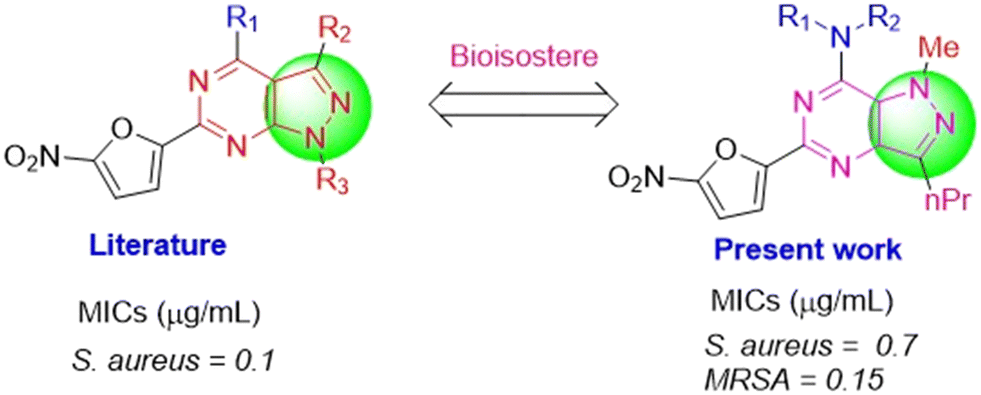 | ||
| Fig. 2 Series of molecules as bioisosteres of 4-amino-6-(5-nitro-2-furyl)-lH-pyrazolo[3,4-d]pyrimidine as earlier reported by H. A. Burch et al. as potent antibacterial agents active against S. aureus. | ||
We synthesized 38 novel compounds exemplified as 7-amino-1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidine analogs and screened against S. aureus and MRSA. This synthesis was based on known methods and one of our recent reports, where a microwave-assisted strategy was established for the preparation of 5-substituted-1H-pyrazolo[4,3-d]pyrimidin-7(6H)-one as mTOR inhibitors; similar procedures were adopted for the preparation of this series for the synthesis of compound 8.39 This was utilized further for the preparation of final compounds using literature-reported methods.40
After evaluating the in vitro antibacterial activity of various derivatives, we identified a potent compound, compound 35, which demonstrated remarkable activity against both S. aureus ATCC 29213 and MRSA ATCC 15187 isolates. This compound was selected for further investigation. We evaluated its safety index by assessing its cytotoxicity and hemolytic activity. Additionally, we investigated time-kill kinetics, the potential for resistance development, combinational effects with standard drugs, and its mechanism of action through mechanistic studies, including scanning electron microscopy and DAPI/PI staining. Ex vivo studies were conducted to evaluate its efficiency in reducing bacterial burden within macrophages infected with bacterial cells. Pharmacokinetic (PK) studies, along with evaluations of in vivo safety and antibacterial efficacy, were performed using a mouse model of systemic MRSA infection. The present study offers valuable insights into developing new antimicrobials based on nitrofuranyl–pyrazolopyrimidine hybrid conjugates.
Results and discussion
Synthesis of nitrofuranyl–pyrazolopyrimidine hybrid conjugates 1–46
A series of compounds based on nitrofuranyl–pyrazolopyrimidines were synthesized by following the procedures reported by our group recently and using known methods. We started our synthesis (Scheme 1) by cyclization of 4-amino-1-methyl-3-propyl-1H-pyrazole-5-carboxamide with 5-nitrofuran-2-carbaldehyde in the presence of MW and K2S2O8 as oxidizing agent in DMSO/H2O; it gave an intermediate compound 8 (1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7(6H)-one). Chlorination of this intermediate compound was carried out using POCl3, which gave intermediate compound 9 (7-chloro-1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidine). Further, the replacement of chlorine with different respective aliphatic amines in DMF and K2CO3 as base and aromatic amines in iPrOH/conc. HCl (1–2 drops) gave the amino derivatives as final compounds (10–46).40 We further prepared different analogs bearing aliphatic or aromatic amines that are listed in Tables 1–3. | ||
Scheme 1 Synthesis of analogs of nitrofuranyl–pyrazolopyrimidines. Reagents and conditions: (a) 5-nitrofuran-2-carbaldehyde, K2S2O8, MW 350 W, 100 °C, DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O, 10 min. (b) POCl3, reflux, 5 h. (c) Aliphatic amines, K2CO3, DMF, room temperature, 4 h. (d) Ar amines, iPrOH, conc. HCl, reflux, 4 h. :H2O, 10 min. (b) POCl3, reflux, 5 h. (c) Aliphatic amines, K2CO3, DMF, room temperature, 4 h. (d) Ar amines, iPrOH, conc. HCl, reflux, 4 h. | ||
| Compound | R | S. aureus ATCC 29213 | MRSA ATCC 15187 | E. coli ATCC 25922 | HepG2 |
|---|---|---|---|---|---|
| MIC (μg ml−1) | IC50 (μg ml−1) | ||||
| Gram-positive strains include S. aureus and its resistant variant methicillin-resistant S. aureus (MRSA), while Gram-negative strains include E. coli. ND represents not determined while ‘−ve’ represents negative results for antibacterial activity. MIC values are calculated in μg ml−1. The outcome is representative of three experiments. | |||||
| 8 |
|
8 | 8 | −ve | >50 |
| 10 |
|
>32 | >32 | −ve | >50 |
| 11 |
|
>32 | >32 | −ve | >50 |
| 12 |
|
>32 | >32 | −ve | >50 |
| 13 |
|
>32 | >32 | −ve | >50 |
| 14 |
|
>32 | >32 | −ve | >50 |
| 15 |
|
>32 | 16 | −ve | >50 |
| 16 |

|
16 | 0.5 | −ve | >50 |
| 17 |
|
>32 | >32 | −ve | >50 |
| 18 |

|
>32 | >32 | −ve | >50 |
| 19 |

|
>32 | >32 | −ve | >50 |
| 20 |

|
>32 | >32 | −ve | >50 |
| 21 |

|
>32 | >32 | −ve | >50 |
| 22 |

|
>32 | >32 | −ve | >50 |
| 23 |

|
>32 | >32 | −ve | >50 |
| Ciprofloxacin | 0.25 | 16 | 0.006 | ND | |
| Compound | R | S. aureus ATCC 29213 | MRSA ATCC 15187 | E. coli ATCC 25922 | HepG2 |
|---|---|---|---|---|---|
| MIC (μg ml−1) | IC50 (μg ml−1) | ||||
| The MIC values are calculated in μg ml−1. ND and ‘−ve’ represent not determined and negative results for antibacterial activity, respectively. The data are representative of three experiments. | |||||
| 24 |

|
32 | 2 | −ve | >50 |
| 25 |
|
4 | 0.5 | −ve | 38.1 |
| 26 |

|
4 | 0.25 | −ve | >50 |
| 27 |
|
4 | 2 | −ve | >50 |
| 28 |

|
>32 | 8 | −ve | >50 |
| 29 |

|
>32 | >32 | −ve | >50 |
| 30 |
|
>32 | >32 | −ve | >50 |
| 31 |

|
>32 | >32 | −ve | >50 |
| 32 |

|
>32 | >32 | −ve | >50 |
| 33 |

|
0.5 | 1 | −ve | >50 |
| 34 |
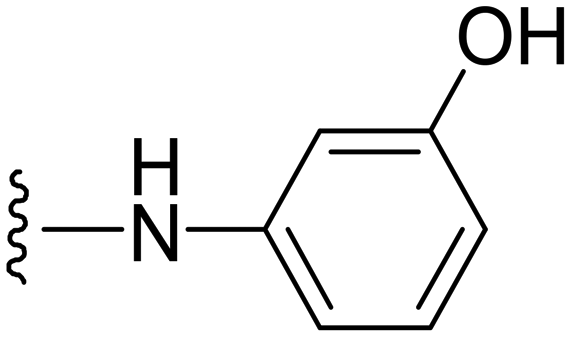
|
4 | 1 | −ve | >50 |
| 35 |
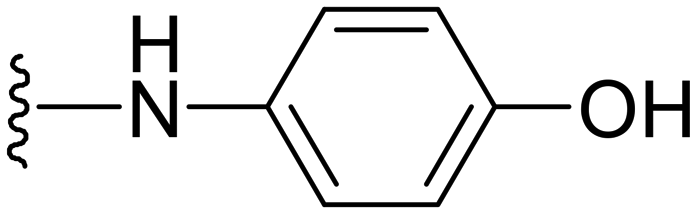
|
0.64 | 0.15 | −ve | >50 |
| 36 |

|
>32 | >32 | −ve | >50 |
| 37 |
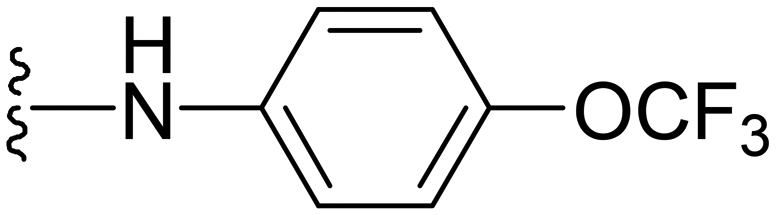
|
>32 | >32 | −ve | >50 |
| Ciprofloxacin | 0.25 | 16 | 0.006 | ND | |
| Compound | R | S. aureus ATCC 29213 | MRSA ATCC 15187 | E. coli ATCC 25922 | HepG2 |
|---|---|---|---|---|---|
| MIC (μg ml−1) | IC50 (μg ml−1) | ||||
| ND represents not determined, while ‘−ve’ shows the negative effect of compounds on antibacterial activity. MIC values are calculated in μg ml−1 units. The data are representative of three experiments. | |||||
| 38 |

|
>32 | 4 | −ve | 38.86 |
| 39 |
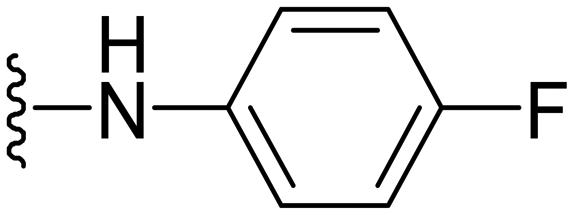
|
8 | 0.25 | −ve | >50 |
| 40 |

|
>32 | >32 | −ve | >50 |
| 41 |

|
>32 | >32 | −ve | >50 |
| 42 |

|
>32 | >32 | −ve | >50 |
| 43 |
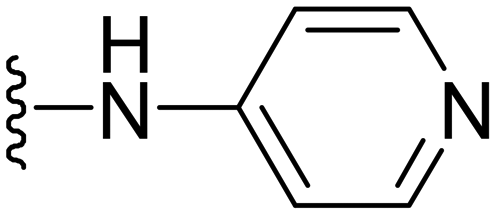
|
0.5 | 4 | 4 | >50 |
| 44 |
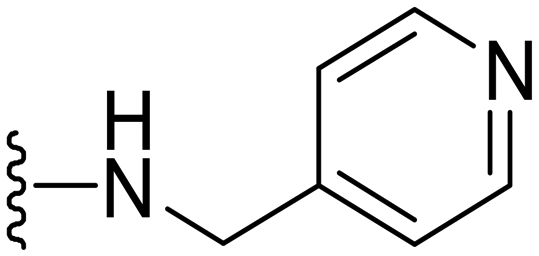
|
1 | 1 | −ve | >50 |
| 45 |
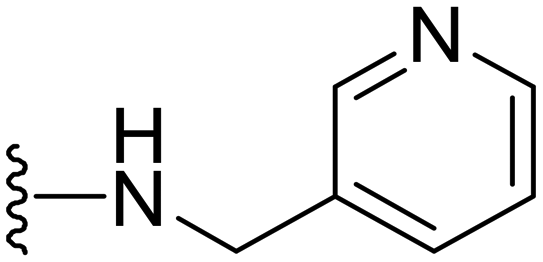
|
8 | 4 | −ve | >50 |
| 46 |
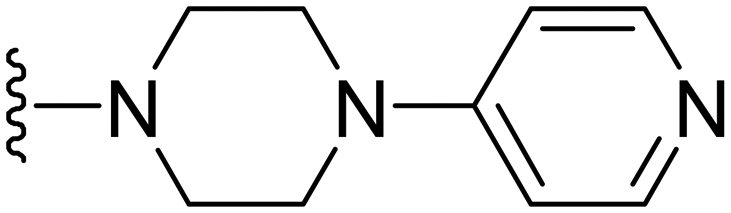
|
1 | 0.25 | −ve | >50 |
| Ciprofloxacin | 0.25 | 16 | 0.006 | ND | |
Biological activity
Relationship of structure and activity by in vitro evaluation (MICs) against S. aureus and MRSA
Different primary or secondary amines at the 7th position of the pyrazolopyrimidine ring of this scaffold were synthesized and screened. In our initial in vitro screening for compounds 10–23, the data revealed that the compound having 4-hydroxy substitution on the piperidinyl ring placed at the 7th position of pyrazolopyrimidine was found to be active against both strains, as shown in Table 1. From these initial results, compound 16 was selected for exploring the role of the hydroxyl group present on this piperidinyl ring by replacing it with aromatic or aliphatic hydroxy substituted compounds. Here, we hypothesized that the activity could be attributed to presence of the hydroxyl group and it can play a vital role in manipulating the activity. Therefore, we drew a strategy for preparing more analogs with varying substitutions including the hydroxyl group on the arylamine rings or directly placing aliphatic amines with the hydroxyl group at different positions and varying chain lengths at the 7th position of the pyrazolopyrimidine ring. In this direction, different aliphatic and aromatic amines were prepared and screened against the selected strains. Many of the compounds have shown significant activity with lower MICs (Table 2). Among the aliphatic and aromatic hydroxyl group-bearing amines, aromatic amines with hydroxyl substitution at different positions were found to be highly active against all three strains as compared to aliphatic hydroxy amines. However, the compound with 4-OH substitution of aniline substrate (compound 35) turned out to be the most active with MIC of 0.7 and 0.15 μg ml−1 against S. aureus and MRSA, respectively, as compared to the 2-OH- or 3-OH-substituted arylamine substrates.Further in our observation, we screened more analogs with different substitutions on arylamines, including the – OMe (compound 36) or –OCF3 (compound 37) substitutions at the para-position of arylamines, to see the effect of protection on active compound 35. However, these compounds turned out to be inactive with MICs >32 μg ml−1. Some compounds with heteroatom bearing cyclic aliphatic rings along with the presence of the hydroxyl group at varying positions were also designed, synthesized and screened (Table 2, entries 29–32), all these molecules were found to be inactive.
Apart from this, various halo substitutions were tried on arylamine rings (Table 3), in which the compounds with 3- and 4-fluoro substitution on the arylamine ring were found active, i.e., compound 38 and 39, respectively. Other compounds from the halo series were not having significant activity. Next, as shown in Table 3, different hetero aryl amines were placed to see the effect on the activity; some of the compounds have shown good activity, and further work has been undertaken to see the structure–activity relationship of these substitutions on this scaffold.
These antibacterial activity results prompted us to further examine the cytotoxicity effect on the safety of the nitrofuranyl–pyrazolopyrimidine compounds on HepG2 cell lines using MTT assay. As a result, no compound was found to have a cytotoxic effect up to 50 μg ml−1 concentrations (Tables 1–3). Thus, the overall observation of the results indicates that compound 35 was the most active.
Consequently, further studies, including in vivo efficacy, physicochemical parameter analysis, and pharmacokinetic profiling of this compound, were planned. Fig. 3 illustrates the dose–response curve for compound 35 against S. aureus and MRSA. The curve shows a clear growth inhibition with increasing concentrations of the compound, with MIC values of 0.7 and 0.15 μg ml−1, respectively. Additionally, the minimum bactericidal concentration (MBC) of compound 35 was found to be equivalent to the MIC.
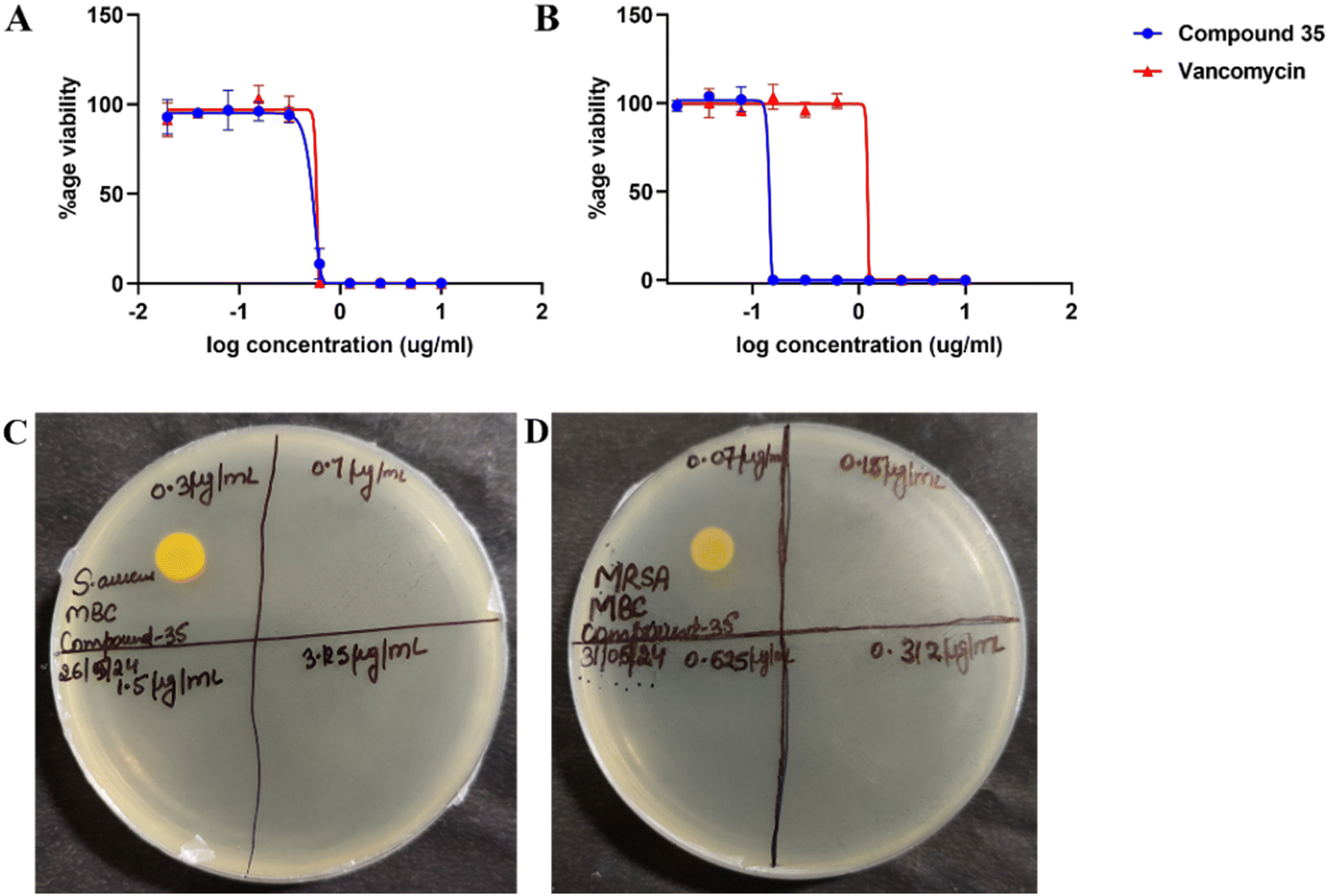 | ||
| Fig. 3 Dose–response curves exhibiting growth inhibition of (A) S. aureus and (B) methicillin-resistant S. aureus (MRSA) in response to an increase in the concentration of compound 35 (blue line) while vancomycin was used as a standard drug control (red line). Compound 35 showed similar viability inhibition to vancomycin in S. aureus and higher inhibition was observed in MRSA. The minimum bactericidal concentration (MBC) of compound 35 is shown against (C) S. aureus and (D) MRSA. The compound shows a bactericidal effect at the minimum inhibitory concentration (MIC). The figure is representative of three experiments. | ||
Based on the excellent antibacterial activity of compound 35, we further evaluated the ability of compound 35 to inhibit S. aureus and MRSA biofilms. Compound 35 was tested at 1/2×, 1×, 2×, 4×, and 6× MIC concentrations, with vancomycin being used as a drug control at the respective concentrations. The compound effectively inhibited biofilm formation, with a notable decrease in biofilm formation as its concentration increased, as shown in Fig. 4. Compound 35 inhibited biofilm formation at all tested concentrations, whereas vancomycin was ineffective at 1/2× MIC concentration. The compound was further assessed for its ability to disrupt preformed biofilms in S. aureus and MRSA where compound 35 was able to show maximum 30% disruption in preformed biofilm in both bacterial strains at 4× and 6× MIC concentration, while the drug control vancomycin exhibited 40–50% disruption at 4× and 6× MIC concentrations (ESI,† Fig. S1). Since the compound effectively prevents biofilm formation but shows limited ability to disrupt existing biofilms, it is better suited for preventing biofilm development than for treating established infections.
 | ||
| Fig. 4 The graph represents the biofilm inhibition at different concentrations of compound 35. The untreated cells/growth control in (A) S. aureus and (B) MRSA demonstrate the ability to form biofilms, while vancomycin, used as the drug control, shows its effect on biofilm inhibition. Compound 35 effectively inhibits biofilm formation at all tested concentrations (1/2×, 1×, 2×, 4×, and 6× MIC), showing similar efficacy to vancomycin (at 1× to 6× MIC). The figure is representative of three experiments. | ||
Antimicrobial resistance, particularly due to overexpression of efflux pumps, poses a significant hurdle in the discovery of antibacterial drugs. As discussed by Poole (2007), targeting efflux pumps can be an effective approach to combat antibiotic resistance.41 In this context, compound 35 was further tested against several strains of S. aureus that overexpress different efflux pumps: NorA (SA-1199B), MsrA (SA-K2191), and TetK (SA-K2192) as well as a ciprofloxacin-sensitive strain (SA-1199) used as a control. MIC values for these strains are provided in Table 4.
| Strain | Phenotype | MIC (μg ml−1) | ||
|---|---|---|---|---|
| Ciprofloxacin | Vancomycin | Compound 35 | ||
| SA-1199 | Ciprofloxacin-sensitive | 0.25 | 0.25 | 0.3 |
| SA-1199B | NorA overexpressing | 4.14 | 0.25 | 0.6 |
| SA-K2191 | MsrA overexpressing | 4.14 | 0.25 | 0.15 |
| SA-K2192 | TetK overexpressing | 0.25 | 0.56 | 0.3 |
Compound 35 showed similar MIC values for both S. aureus 29213 and NorA overexpressing strain, S. aureus 1199B, while strains (SA-1199 and SA-K2192) showed a onefold decrease in MIC value and SA-K2191 showed a twofold decrease in MIC value clearly shown in Fig. 5 representing a dose–response curve of compound 35 against these strains. These results indicate that compound 35 remains effective against strains with overexpressed efflux pumps, highlighting its potential for targeting resistant strains.
 | ||
| Fig. 5 The graph represents the dose–response growth inhibition of different S. aureus efflux pump overexpressing strains, SA-1199–ciprofloxacin sensitive (blue line), SA-1199B-overexpressing NorA (green line), SA-K2191-overexpressing MsrA (red line) and SA-2192-overexpressing TetK (purple line). The wild-type strain of S. aureus 29213 is used for comparison (black line). The data indicate a similar range of inhibition possessed by compound 35 against all the strains used with minor variations. | ||
Cell viability and hemolytic assays
Compound 35 was analyzed further to assess its impact on cell viability and toxicity in other cell lines including RAW264.7 and J774A.1 macrophage cell lines with 24 h of incubation time. The compound was evaluated at different concentrations ranging from 200 to 0.19 μg ml−1. Doxorubicin at a concentration of 10 μM served as the drug control, while 0.1% DMSO, growth, and medium controls were used for comparison. As the compound showed safety up to 200 μg ml−1 in mammalian cell lines, we further assessed its safety over 48 and 72 h periods using the RAW264.7 cell line. Compound 35 was found to be safe at concentrations up to 100 μg ml−1, with IC50 values exceeding 100 μg ml−1 at both 48 and 72 h of compound treatment (ESI,† Fig. S2).Further, the hemolytic activity (HC50) of compound 35 against rabbit erythrocytes was determined to confirm their toxicity and membrane selectivity. This assay helps evaluate the safety profile of compound 35 with respect to erythrocytes. We assessed the effect of compound 35 on erythrocytes at various concentrations, with vancomycin serving as a drug control. Triton-X, a lysing agent, was used as a positive control at 1% concentration, which demonstrated 100% hemolytic activity, while PBS with 0% lysis served as the negative control. The growth control, which contained undisturbed RBCs, exhibited minimal hemolysis. Compound 35 showed no hemolytic activity comparable to the standard drug vancomycin, whereas 1% Triton-X induced substantial hemolysis. This assay further predicted compound 35 as safe against erythrocytes.42Fig. 6 presents the cell viability and percentage hemolysis of the test compounds at different concentrations. Compound 35 shows a safe profile against macrophage cell lines used and erythrocytes up to a concentration of 200 μg ml−1.
 | ||
| Fig. 6 Compound 35 (blue lines) shows 100% growth of macrophage cells (A) RAW264.7 and (B) J774A.1 similar to growth control. Drug control doxorubicin was used at a concentration of 10 μM, which showed a greater reduction in the growth of respective cells. (C) The graph represents the membrane disruption of erythrocytes (% age hemolysis) caused by compound 35 (blue line), vancomycin (red line), and 1% Triton-X (green line), while untreated RBCs (orange line) were used as growth control. Triton-X displayed a strong hemolytic effect, whereas compound 35 showed no hemolytic activity, highlighting its favorable safety profile. | ||
Combinational studies
A combination study was performed to check the compound's activity on the MIC of other antibiotics using checkerboard analysis. Test compound 35 was studied with 3 different antibiotics, rifampicin, vancomycin, and ciprofloxacin, which have different mechanisms of action targeting the β-subunit of RNA polymerase,43 cell wall44 and DNA gyrase45 respectively. The data were determined using FICI which predicts the effect of the compound on a standard drug, mentioned in the table present in Fig. 7. Data suggested that in combination with rifampicin, compound 35 shows a synergistic effect on MRSA and an additive effect on S. aureus. In contrast, combination with vancomycin shows synergy of compound 35 in S. aureus and additive effect on MRSA. Compound 35 does not show any positive or negative impact (indifference) in combination with ciprofloxacin in the case of MRSA but shows an additive effect on S. aureus. There had not been any antagonistic effect from any of the combinations. The data for each combination of compound 35 with antibiotic are shown in Fig. 7. The heatmap plot of compound 35 shows the degree of inhibition in Fig. 8 displaying the darkest point (maximum growth of bacteria), while the lightest point represents maximum growth inhibition).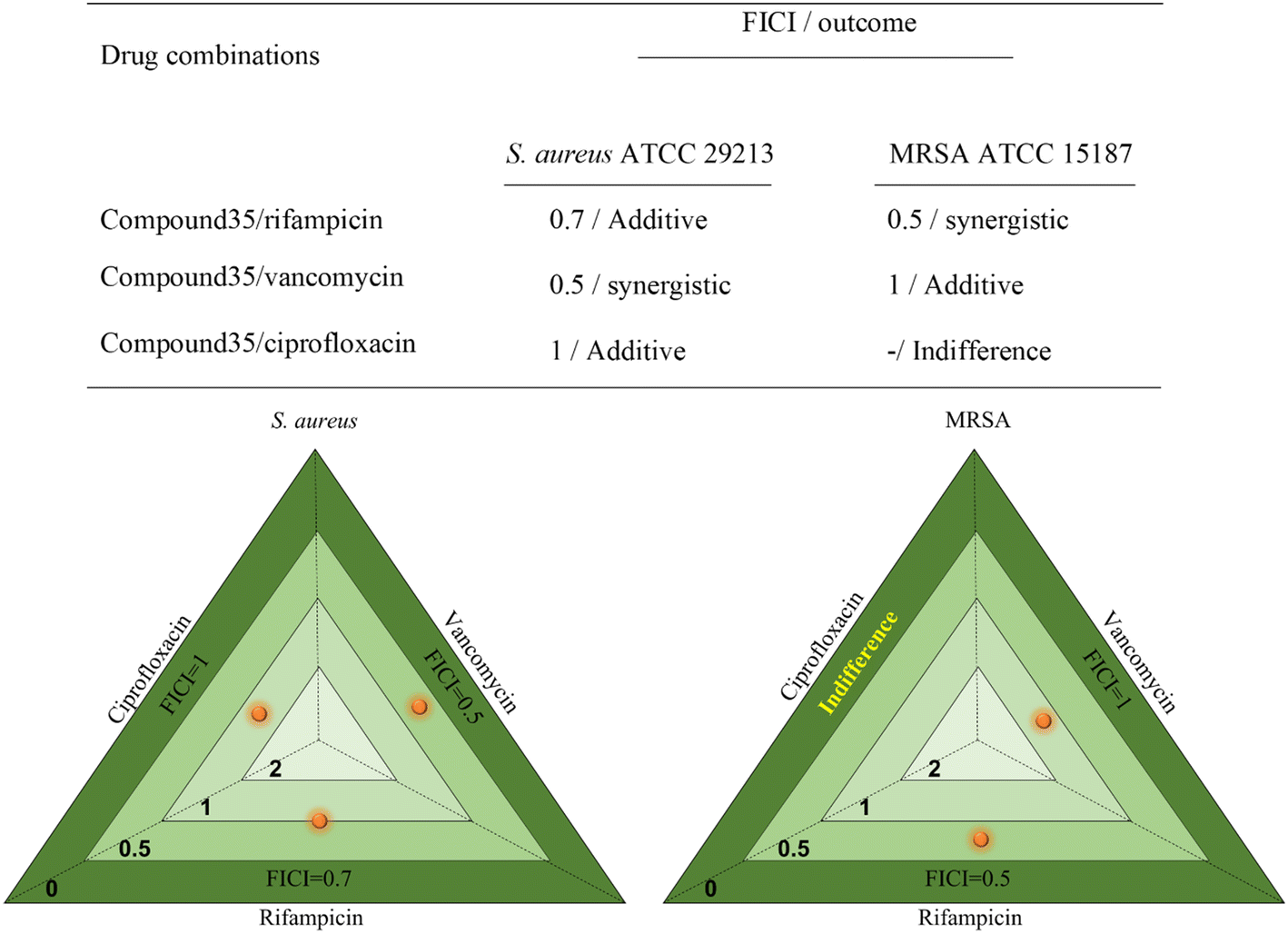 | ||
| Fig. 7 The table displays the calculated FIC indices and their outcome against S. aureus and MRSA with a graphical image showing outcomes in one place for S. aureus and MRSA. | ||
 | ||
| Fig. 8 The given heatmaps display the combinational effect of compound 35 with (A1) rifampicin, (A2) vancomycin and (A3) ciprofloxacin, analyzed through checkerboard analysis against S. aureus, while in MRSA compound 35 shows a combinational effect with (B1) rifampicin, (B2) vancomycin and (B3) ciprofloxacin. The degree of inhibition is shown as a heat plot where the lightest point represents maximum inhibition while the darkest point represents maximum growth of S. aureus and MRSA. The red dot indicates FICI and the outcome for the respective antibiotic. | ||
Time-kill kinetics and resistance development studies
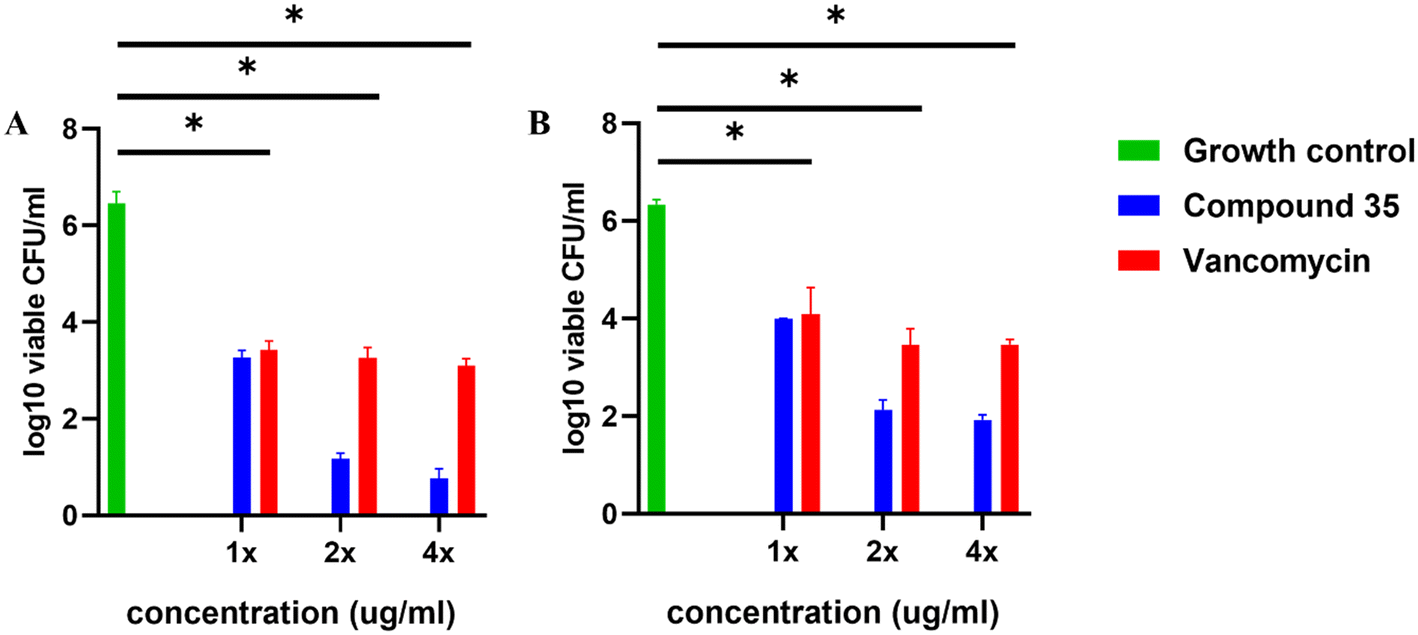
Given the excellent in vitro antibacterial activity of compound 35, its time-kill kinetics were investigated to evaluate its bactericidal properties against both S. aureus ATCC 29213 and MRSA. We tested three different concentrations of compound 35, 1× MIC, 2× MIC, and 4× MIC, alongside two concentrations of vancomycin, 1× MIC and 2× MIC. Untreated bacterial cells served as the growth control. Bactericidal activity is defined as a 99.9% reduction in the original inoculum (>3log10 CFU ml−1) while bacteriostatic activity is indicated by a reduction of less than 99.9% (<3log10 CFU ml−1).46 The results were analyzed by plotting log10 colony-forming units per milliliter (CFU ml−1) versus time (in hours). The killing curves presented in Fig. 9(A and B) illustrate the reduction in total CFU ml−1 in the compound-treated bacterial samples. Compound 35 exhibited a concentration-dependent effect against S. aureus where the compound at 1× MIC is acting as a bacteriostatic (reducing or maintaining <3log10 CFU of initial inoculum). In contrast, at 2× and 4× MIC, it displayed bactericidal activity (reducing 99.9% of the initial CFU ml−1) and demonstrated killing in the initial hours (within 2 h) compared to vancomycin.
 | ||
| Fig. 9 The time-kill curves show the effect of compound 35 in the reduction of log10 CFU ml−1 against (A) S. aureus and (B) MRSA. Untreated bacterial cells (blue lines) were used to determine the initial inoculum without any treatment. Vancomycin at 1× MIC (red line) and 2× MIC (green line) served as drug controls for comparison. Compound 35 at different concentrations, 1× MIC (purple line), 2× MIC (yellow line) and 4× MIC (pink line), were used. The black dashed line represents the bactericidal threshold (3 log10 CFU ml−1), indicating a 99.9% reduction in log10 CFU ml−1 below this level, while bactericidal activity is observed above this threshold. At 1× MIC, the compound exhibits bacteriostatic behaviour. The graph representing analysis of resistance development for compound 35 (blue line), vancomycin (red line), and the fluoroquinolone norfloxacin (black line) against (C) S. aureus and (D) MRSA is plotted. Bacteria were serially passaged following exposure to a sublethal concentration of drug over 20 days and MIC at each passage was determined. The graph clearly indicates the inability of compound 35 to develop resistance similar to vancomycin control while sublethal concentration of norfloxacin makes (C) S. aureus resistant by increasing MIC to 200-fold at 20 days and (D) MRSA by increasing MIC to 3200-fold. | ||
For MRSA, compound 35 showed both concentration- and time-dependent killing. At 1× MIC, it was bacteriostatic. At 2× MIC, it exhibited initial killing but exhibited some regrowth after 8–10 h, possibly due to the compound's labile nature. At 4× MIC, compound 35 was bactericidal and showed killing at the initial duration (within 2 h), earlier than vancomycin.
In addition, the synergistic effect of compound 35 with vancomycin in S. aureus and with rifampicin in MRSA has been verified using time-kill curves.47 The combination of compound 35 with vancomycin at synergistic concentrations demonstrated bacterial growth inhibition but was less effective, achieving a >10log reduction in CFU for up to 8 h. However, regrowth was observed after 24 h, likely due to the bacteriostatic nature of the combination. The combination of compound 35 and rifampicin at synergistic concentrations exhibits a moderately improved effect compared to rifampicin alone at the FIC concentration, which is already effective, as shown in Fig. S3.†
Assessing the tendency of bacteria to develop resistance is a crucial factor in determining the effectiveness of newly developed antimicrobials. Consequently, we investigated the resistance propensity of compound 35 against both S. aureus and MRSA by exposing the bacteria to sublethal concentrations of the compound over 20 days. Vancomycin and norfloxacin were employed as control agents in this assay. Fig. 9(C and D) depict that compound 35, similar to vancomycin, has a minimal tendency to induce resistance in both S. aureus and MRSA strains over 16 days. In contrast, fluoroquinolone norfloxacin (positive control) significantly increased the original MIC by 200-fold for S. aureus and 3200-fold for MRSA by day 20.48,49 The lack of resistance development observed with compound 35 may be attributed to its rapid bactericidal action and membrane-targeting mechanism.50
Antimicrobial mechanism studies
 | ||
| Fig. 10 Scanning electron micrographs of S. aureus exposed to compound 35 for 2 h, including images at 5 μm and 1 μm scales, with the 1 μm scale image providing a magnified view of a portion of the 5 μm scale image. A1 and A2 depict the morphology of untreated cells, which appear in clusters. The treated samples with compound 35 at 1× MIC (B1 and B2), 2× MIC (C1 and C2), and 4× MIC (D1 and D2) exhibit concentration-dependent morphological disruption. The drug controls vancomycin 1× MIC (E1 and E2), 2× MIC (F1 and F2) and daptomycin 1× MIC (G1 and G2), 2× MIC (H1 and H2) were used, which demonstrated significant disruption at both concentrations. | ||
 | ||
| Fig. 11 Scanning electron micrographs of S. aureus exposed to compound 35 for 6 h, including two images of 5 μm and 1 μm scale where the image with 1 μm scale is the magnified view of the selected portion of the 5 μm scale image. A1 and A2 represent the morphological appearance of untreated cells which are grown in bunches; the treated samples with compound 35 at 1× MIC (B1 and B2), 2× MIC (C1 and C2) and 4× MIC (D1 and D2) show morphological disruption in a concentration-dependent manner. The vancomycin drug control showed great disruption in both concentrations, 1× MIC (E1 and E2) and 2× MIC (F1 and F2). | ||
Subsequently, we assessed the integrity of bacterial membranes using fluorescence microscopy and percentage permeability analysis. The fluorescence LIVE/DEAD bacterial viability assay employs two dyes: propidium iodide (PI) and 4′,6-diamidino-2-phenylindole (DAPI). DAPI, a blue fluorescent nucleic acid stain, can penetrate cell membranes regardless of their integrity, staining both live and damaged cells. In contrast, PI, a red fluorescent nucleic acid stain, passes only through damaged cell membranes. To further explore the antimicrobial mechanism of compound 35, S. aureus and MRSA were treated with or without the compound and stained with DAPI and PI dyes. As shown in Fig. 12, blue fluorescence was visible in both the growth control and compound 35-treated groups, while strong red fluorescence appeared only in the compound-treated group. The growth control exhibited blue fluorescence with minimal traces of red, indicating an intact bacterial membrane. In contrast, both the drug control and compound-treated groups showed red and blue fluorescence in their respective channels, signifying their effectiveness in disrupting the bacterial membrane. MRSA also shows similar results as shown in Fig. S6 of the ESI.† These findings demonstrate that compound 35 compromises the bacterial cell membrane integrity in both S. aureus ATCC 29213 and MRSA ATCC 15187.
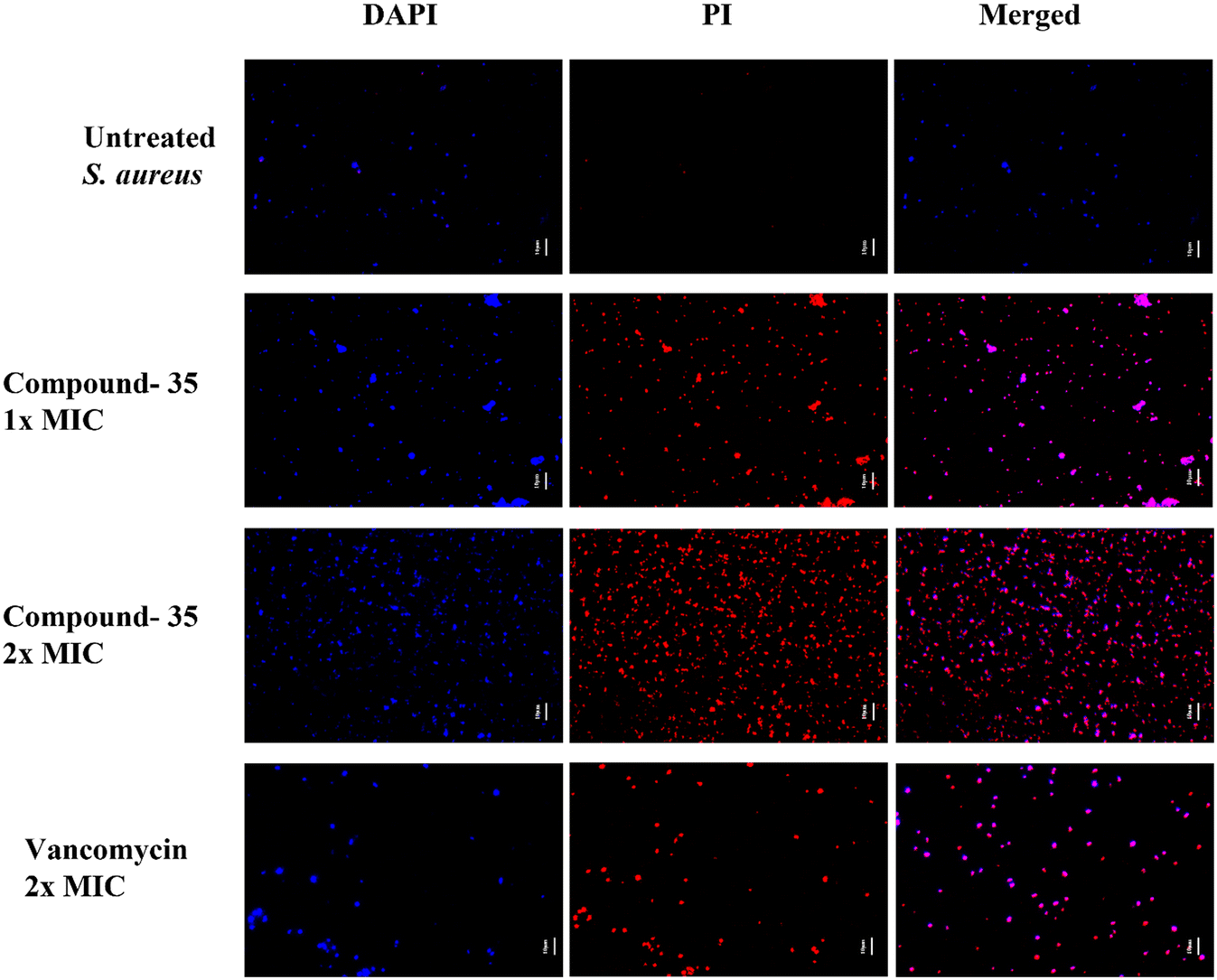 | ||
| Fig. 12 Fluorescence micrographs of S. aureus ATCC 29213 stained with DAPI and PI after treatment with compound 35 at 1× and 2× MIC. Vancomycin, at 2× MIC, served as a positive control. In the blank control (bacteria without drug treatment), DAPI staining was visible, with no PI staining, indicating intact bacterial membranes. In contrast, treatment with compound 35 resulted in both intense blue (DAPI) and red (PI) fluorescence, indicating a disruption of the bacterial cell wall integrity. Scale bar: 10 μm. | ||
The efficiency of compound 35 to permeate the bacterial membrane was further evaluated using a propidium iodide (PI) permeabilization assay. Various concentrations of the compound (1×, 2×, 4×, 8× MIC) were tested, and permeability in both S. aureus and MRSA was analyzed over time. In both strains, permeability steadily increased, reaching a maximum of 60% in S. aureus and 45% in MRSA at 80 min compared to drug control vancomycin (4× MIC and 8× MIC) which is giving maximum permeability of 22% in S. aureus and 18% in MRSA (Fig. 13). The effect of increasing compound concentration on bacterial permeability was minimal, as shown in Fig. 13.
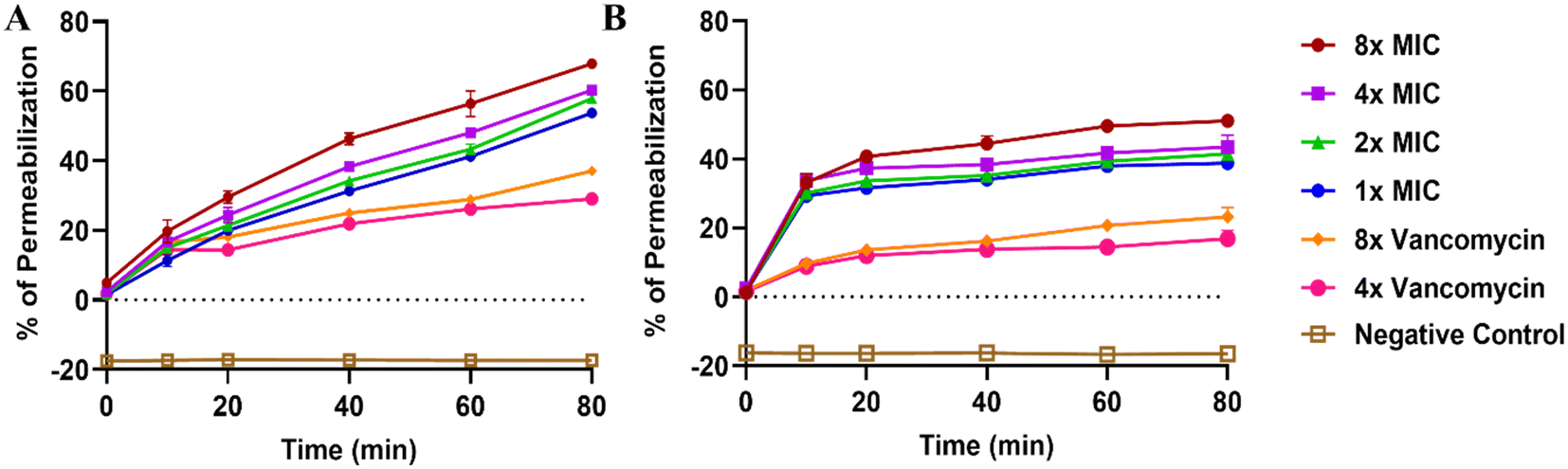 | ||
| Fig. 13 PI membrane permeabilization studies in S. aureus and MRSA. The graph illustrates the ability of PI to permeate the cell membrane in (A) S. aureus and (B) MRSA. The data indicate that the percentage of membrane permeabilization increases over time and occurs in a concentration-dependent manner, with different MIC concentrations represented by the following: 8× MIC (brown line), 4× MIC (purple line), 2× MIC (green line), and 1× MIC (blue line) compared to 8× MIC (orange line) and 4× MIC (pink line) of vancomycin (drug control). The brown line represents the blank control where no permeabilization of PI is determined. | ||
Overall, our findings demonstrate that compound 35 effectively kills bacteria by disrupting the cell membrane. However, we cannot exclude the possibility that other mechanisms may also contribute to bacterial killing.
 | ||
| Fig. 14 (A) S. aureus-infected and (B) MRSA-infected RAW macrophages were treated with the compounds, following which intracellular bacterial load was assessed by CFU ml−1 enumeration. The graph represents the log CFU ml−1 at various concentrations of the compound-35 (blue bar). Vancomycin was used as drug control in the assay (red bars). Significance was calculated by the one-way ANOVA test using GraphPad prism software. The samples were compared for significance with the infection control using one-way ANOVA, where * denotes p < 0.005. | ||
| Parameter | Unit | IV @ 1 mg kg−1 | PO @ 30 mg kg−1 |
|---|---|---|---|
| t 1/2,β | h | 0.50 | 0.35 |
| T max | h | — | 0.50 |
| C max | ng ml−1 | — | 2010 |
| C 0 | ng ml−1 | 552 | — |
| AUC0−t | ng h ml−1 | 90.8 | 3146.1 |
| AUC0−∞ | ng h ml−1 | 94.8 | 3149.8 |
| CL | ml min−1 kg−1 | 176 | 159 |
| V d | L kg−1 | 1.69 | 4.75 |
After a 24 h incubation period, all mice were sacrificed, and their lungs and kidneys were isolated to analyze the bacterial load reduction. The organs were thoroughly homogenized and plated on Mueller–Hinton agar (MHA) supplemented with 20 μg ml−1 ciprofloxacin to detect the bacterial load (CFU ml−1). The plates were incubated for 24 h, and bacterial loads in each group were compared. Fig. 15 illustrates the reduction in bacterial load (CFU ml−1) in kidneys and lungs across different groups: untreated, treated with compound 35, and vancomycin as the drug control. Statistical analysis was conducted using an unpaired t-test in GraphPad Prism, with a p-value <0.05 considered significant.
 | ||
| Fig. 15 In vivo antibacterial efficacy of compound 35. (A) Schematic representation of the experimental protocol for the MRSA-induced peritonitis model in mice. Treatment groups included saline, compound 35 at doses of 50 and 100 mg kg−1, and vancomycin at 50 mg kg−1. Bacterial burden in the kidneys (B) and lungs (C) is shown in the corresponding graphs, depicting the reduction of CFU ml−1 of MRSA. The graphs compare bacterial counts in treated samples (50 mg kg−1 and 100 mg kg−1 compound 35) against untreated controls in both organs. Vancomycin (50 mg kg−1) was included as a reference drug control. The t-test clearly predicts the significant difference between different treated samples and untreated controls, where **** indicates p < 0.0001, *** indicates p < 0.001 and ** indicates p < 0.01. | ||
In the kidneys, the group treated with 50 mg kg−1 exhibited a significantly greater difference (P < 0.0001) compared to the untreated group than the group treated with 100 mg kg−1 (P < 0.0004) and the vancomycin control group (P < 0.0023). In contrast, in the lungs, the groups treated with 50 mg kg−1 and 100 mg kg−1 compound 35 showed lesser significant differences compared to the untreated group, with P-values of 0.0068 and 0.0085, respectively, compared to the vancomycin control group (P < 0.0009).
Conclusions
In conclusion, we have discovered a series of conjugates of nitrofuranyl–pyrazolopyrimidines as antibacterial agents specifically active against S. aureus and also against the methicillin resistant strain, MRSA. In a series of 38 molecules, compound 35 showed excellent antimicrobial activity with MICs of 0.7 and 0.15 μg ml−1 against S. aureus and MRSA, respectively. Compound 35 demonstrated a robust safety profile, including safety in mammalian cell lines, no hemolytic activity, and effectively inhibiting bacterial biofilm formation with little effect in eradicating preformed biofilm. It showed strong potency against bacterial strains with overexpressed efflux pumps (NorA, TetK, MsrA) and did not induce resistance in S. aureus or MRSA. Additionally, it exhibited synergistic effects with vancomycin in S. aureus and rifampicin in MRSA, while mechanistic studies revealed its membrane-targeting abilities. Compound 35 significantly reduced bacterial load in intracellular models and displayed a favorable pharmacokinetic profile in vivo. These findings highlight its promise as a potential candidate for treating MRSA infections.Experimental section
Chemistry
Synthesis of 4-(1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-yl)morpholine (10). In a typical procedure, the following steps were followed for the preparation of compound 10.
Step 1: 1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7(6H)-one (8)
In a typical procedure, a solution of 4-amino-1-methyl-3-propyl-1H-pyrazole-5-carboxamide (500 mg, 2.74 mmol) and 5-nitrofuran-2-carbaldehyde (395 mg 2.8 mmol) in DMSO:H2O (1:1) with K2S2O8 added (2.225 g 8.74 mmol) was taken in a sealed reaction tube and the reaction mixture was irradiated under microwave conditions for 3 min with a power of 350 W at 100 °C. After completion, the reaction mass was diluted with EtOAc (20 ml) and water (30 ml) was added. The organic layer was separated and extracted with EtOAc (2 × 10 ml). The combined organic layer was then washed with brine solution, concentrated under vacuum and the residue was purified by column chromatography, affording a yellow solid, 1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7(6H)-on, i.e., compound 8 (707 mg) in 85% yield.
Yellow solid; 1H NMR (400 MHz, DMSO) δ 8.30 (s, 1H), 7.86 (s, 1H), 4.15 (s, 3H), 2.81 (t, J = 7.6 Hz, 2H), 1.84–1.68 (m, 2H), 0.96 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 152.00, 147.45, 145.70, 139.85, 136.82, 124.70, 115.06, 114.06, 68.9, 37.92, 27.02, 21.50, 13.80; HRMS (ESI) calcd for C13H14N5O4 [M + H]+: 304.1045, found 304.1040.
Step 2: 7-chloro-1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidine (9)
Compound 8 1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7(6H)-one (700 mg, 2.31 mmol) was dissolved in POCl3 (4 ml), DIPEA (402 μl 2.31 mmol) was added, and the mixture was refluxed for 5 h. After the reaction was completed, the POCl3 was removed under vacuum. Then, workup was carried out using CHCl3 (20 ml) and ice water (15 ml). The organic layer was separated and the water layer re-extracted with CHCl3 (2 × 15 ml). The combined organic layers were washed with brine solution, dried over anhydrous sodium sulfate, and concentrated. The residue was purified by column chromatography. Compound 9 was obtained as a light yellow solid (592 mg, yield 80%).
Yellow solid; 1H NMR (400 MHz, DMSO) δ 8.32 (s, 1H), 7.88 (s, 1H), 4.16 (s, 3H), 2.82 (t, J = 7.4 Hz, 2H), 1.84–1.68 (m, 2H), 0.96 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 153.75, 152.07, 147.43, 145.67, 139.96, 136.88, 124.72, 115.02, 114.06, 37.92, 27.02, 21.52, 13.79; mass ESI [M + H]+: 321.9.
Step 3: (a) procedure for aliphatic amines
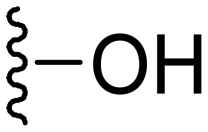
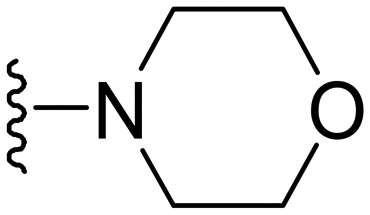
Synthesis of 4-(1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-yl)morpholine ( 10 )
In a typical procedure, morpholine (20 mg, 0.23 mmol, 1 eq.) was dissolved in dry DMF (2 ml) and to it was added K2CO3 (96 mg, 0.70 mmol 3q); then, 7-chloro-1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidine (75 mg. 0.23 mmol, 1 eq.) was added to the mixture. The reaction mixture was stirred for 3–4 h at 25 °C. After completion of the reaction, 20 ml EtOAc and 30 ml water were added and the organic layer was separated. The water layer was re-extracted with EtOAc (2 × 20 ml) and the combined organic layers were washed with brine solution, dried over anhydrous sodium sulfate, and concentrated. The residue was purified by column chromatography. Compound 10 was obtained as a yellow solid (75 mg, yield 86%).
Yellow solid; m.p.: 206–208 °C; 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 3.7 Hz, 1H), 7.32 (d, J = 3.7 Hz, 1H), 4.14 (s, 3H), 3.94 (t, J = 4.4 Hz, 4H), 3.62 (t, J = 4.4 Hz, 4H), 3.02 (t, J = 7.6 Hz, 2H), 1.94–1.84 (m, 2H), 1.05 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 154.14, 153.40, 151.70, 146.98, 145.93, 143.54, 123.63, 114.74, 114.09, 65.57, 49.39, 39.20, 27.17, 21.35, 13.85. HRMS (ESI) calcd for C17H21N6O4 [M + H]+: 373.1624, found 373.1614.
(b) Procedure for aromatic amines
4-((1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-yl)amino)phenol ( 35 )
In a typical procedure, 7-chloro-1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidine (75 mg, 0.23 mmol) and 4-aminophenol (38 mg, 0.35 mmol) were dissolved in iPrOH (5 ml). To this solution was added 37% conc. HCl (1–2 drops). The mixture was heated at reflux for 3–6 h. Then the reaction was cooled and evaporated at reduced pressure. The residue was diluted with CHCl3 (20 ml), neutralized by Et3N and washed with water (20 ml), and the organic layer was separated. The water layer was re-extracted with CHCl3 (2 × 10 ml) and the combined organic layer was washed with brine solution, dried over anhydrous sodium sulfate, and concentrated. The residue was purified by column chromatography; compound 35 was obtained as a yellow solid (75 mg, yield 76%).
Yellow solid; m.p.: 251–253 °C; 1H NMR (400 MHz, acetone d6) δ 7.66 (d, J = 9.2 Hz, 2H), 7.6 (d, J = 4 Hz, 1H), 7.25 (d, J = 4 Hz, 1H), 6.93 (d, J = 9.2 Hz, 2H), 4.4 (s, 3H), 2.93 (t, J = 7.6 Hz, 2H), 1.91–1.82 (m, 2H), 1.01 (t, J = 7.2 Hz, 3H); 13C (126 MHz, acetone-d6), δ 156.06, 155.36, 152.94, 149.03, 148.73, 146.19, 143.76, 131.27, 125.41, 122.11, 115.91, 114.47, 114.06, 39.76, 29.36, 22.35, 14.26; HRMS (ESI) calcd for C19H19N6O4 [M + H]+: 395.1468, found 395.1463.
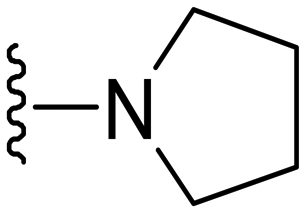
1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-7-(pyrrolidin-1-yl)-1H-pyrazolo[4,3-d]pyrimidine ( 11 )
Orange-colored solid; m.p.: 156–158 °C; 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 4 Hz, 1H), 7.29 (d, J = 4 Hz, 1H), 4.20 (s, 3H), 3.87 (t, J = 6.4 Hz, 4H), 3.0 (t, J = 7.6 Hz, 2H), 2.05 (t, J = 6.4 Hz, 4 Hz), 1.93–1.83 (m, 2H), 1.05 (t, J = 7.6 Hz, 3H); 13C NMR (126 MHz, DMSO) δ 154.81, 151.46, 149.92, 146.72, 144.64, 142.80, 122.08, 114.76, 113.54, 50.15, 42.07, 27.10, 25.07, 21.42, 13.88; HRMS (ESI) calcd for C17H21N6O3 [M + H]+: 357.1675, found 357.1667.
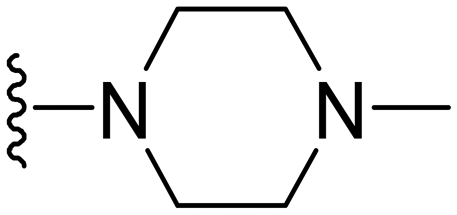
1-Methyl-7-(4-methylpiperazin-1-yl)-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidine ( 12 )
Yellow solid; m.p.: 176–178 °C; 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 4 Hz 1H), 7.32 (d, J = 4 Hz, 1H), 4.12 (s, 3H), 3.65 (t, J = 4.2 Hz, 4H), 3.0 (t, J = 7.6 Hz, 2H), 2.65 (t, J = 4.2 Hz, 4H), 2.39 (s, 3H), 1.9 (m, 2H), 1.05 (t, J = 7.2 Hz 3H); 13C NMR (126 MHz, CDCl3) δ 154.78, 153.83, 152.12, 148.19, 147.84, 144.71, 124.51, 113.37, 113.12, 54.39, 49.33, 46.14, 38.89, 27.79, 22.05, 14.10; HRMS (ESI) calcd for C18H24N7O3 [M + H]+: 386.1941, found 386.1927.
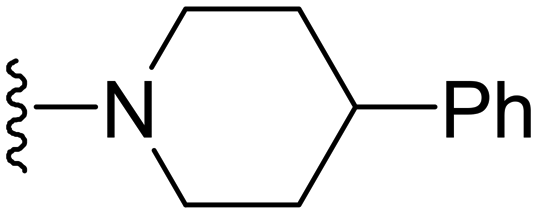
1-Methyl-5-(5-nitrofuran-2-yl)-7-(4-phenylpiperidin-1-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidine ( 13 )
Yellow solid; m.p.: 187–189 °C; 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 3.7 Hz, 1H), 7.37–7.33 (m, 3H), 7.29–7.23 (m, 3H), 4.20–4.17 (m, 5H), 3.26–3.19 (m, 2H), 3.02 (t, J = 7.6 Hz 2H), 2.90–2.82 (m, 1H), 2.12–2.07 (m, 2H), 2.03–1.96 (m, 2H), 1.95–1.86 (m, 2H), 1.06 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 154.90, 154.40, 148.07, 147.90, 145.09, 145.01, 144.57, 128.67, 126.76, 126.67, 124.66, 113.43, 113.09, 50.35, 42.44, 38.86, 32.84, 27.82, 22.10, 14.13; HRMS (ESI) calcd for C24H27N6O3 [M + H]+: 447.2145, found 447.2140.
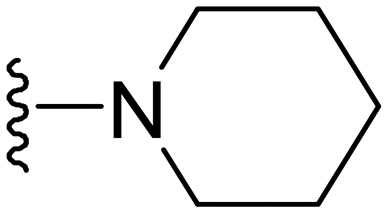
1-Methyl-5-(5-nitrofuran-2-yl)-7-(piperidin-1-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidine ( 14 )
Yellow solid; m.p.: 160–162 °C; 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 4 Hz, 1H), 7.30 (d, J = 4 Hz, 1H), 4.33 (s, 3H), 3.56 (t, J = 4.4 Hz, 4H), 3.0. (t, J = 7.6 Hz, 2H),1.94–1.75 (m, 8H), 1.05 (t, J = 7.2 Hz, 3H); 13C NMR (126 MHz, DMSO) δ 154.30, 153.81, 151.61, 147.03, 145.81, 143.24, 123.70, 114.77, 113.90, 49.90, 39.05, 27.18, 24.92, 23.75, 21.36, 13.87. HRMS (ESI) calcd for C18H23N6O3 [M + H]+: 371.1832, found 371.1831.
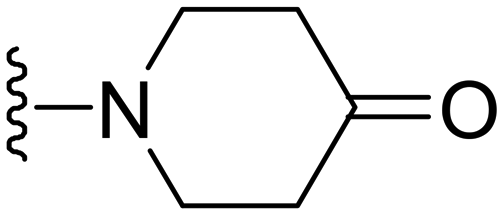
1-(1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-yl)piperidin-4-one ( 15 )
Yellow solid; m.p.: 214–216 °C; 1H NMR (400 MHz, CDCl3) δ 7.4 (d, J = 3.6 Hz, 1H), 7.3 (d, J = 3.6 Hz, 1H), 4.16 (s, 3H), 3.93 (t, J = 6.1 Hz, 4H), 3.0 (t, J = 7.6 Hz, 2H), 2.69 (t, J = 6.1 Hz, 4H), 1.92–1.83 (m, 2H), 1.03 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 207.05, 154.15, 153.03, 151.70, 146.94, 145.87, 143.64, 123.64, 114.77, 114.12, 47.82, 40.19, 39.34, 27.17, 21.37, 13.85; HRMS (ESI) calcd for C18H21N6O4 [M + H]+: 385.1624, found 385.1610.
1-(1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-yl)piperidin-4-ol ( 16 )
Yellow solid; m.p.: 202–204 °C; 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 3.7 Hz, 1H), 7.31 (d, J = 3.7 Hz, 1H), 4.13 (s, 3H), 4.06 (s, 1H), 4.08–4.01 (m, 1H), 3.95–3.89 (m, 2H), 3.39–3.33 (m, 2H), 3.01 (t, J = 7.6 Hz, 2H), 2.19–2.07 (m, 2H), 1.94–1.86 (m, 2H), 1.85–1.75 (m, 2H), 1.05 (t, J = 7.3 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 154.81, 154.14, 152.11, 148.05, 147.83, 144.57, 124.58, 113.42, 113.09, 67.13, 46.92, 38.74, 33.71, 27.79, 22.08, 14.11; HRMS (ESI) calcd for C18H23N6O4 [M + H]+: 387.1781, found 387.1769.
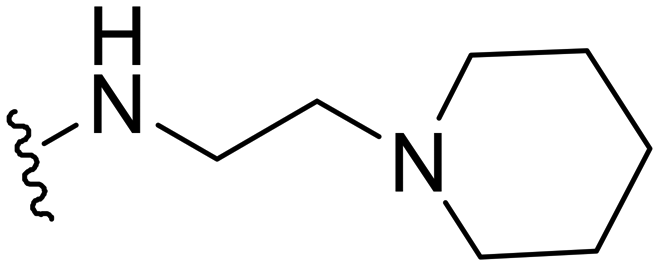
1-Methyl-5-(5-nitrofuran-2-yl)-N-(2-(piperidin-1-yl)ethyl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-amine ( 17 )
Yellow solid; m.p.: 210–213 °C 1H NMR (400 MHz, CDCl3) δ 7.41 (d, J = 3.8 Hz, 1H), 7.30 (d, J = 3.8 Hz, 1H), 6.70 (s, 1H), 4.28 (s, 3H), 3.73 (m, 2H), 2.97 (t, J = 7.6 Hz, 2H), 2.69 (t, J = 5.9 Hz, 2H), 2.51 (br, 4H), 1.94–1.81 (m, 2H), 1.66–1.56 (m, 4H), 1.55–1.46 (m, 2H), 1.04 (t, J = 7.4 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 155.24, 151.98, 150.19, 148.51, 146.46, 142.32, 121.27, 113.45, 112.98, 47.23, 39.19, 37.77, 31.12, 27.68, 26.36, 25.85, 22.13, 14.08; HRMS (ESI) calcd for C20H28N7O3 [M + H]+: 414.2254, found 414.2250.
1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-N-(tetrahydro-2H-pyran-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-7-amine ( 18 )
Orange color solid; m.p.: 238–240 °C; 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 3.7 Hz, 1H), 7.26 (d, J = 3.7 Hz,1H), 5.06 (d, J = 7.0 Hz, 1H), 4.60–4.48 (m, 1H), 4.27 (s, 3H), 4.11–4.04 (m, 2H), 3.65 (m, 2H), 2.97 (t, J = 7.6 Hz, 2H), 2.22 (m, 2H), 1.92–1.81 (m, 2H), 1.64 (m, 2H), 1.03 (t, J = 7.4 Hz, 3H); 13C NMR (126 MHz, DMSO) δ 154.72, 151.54, 148.93, 147.54, 144.48, 141.33, 120.95, 114.77, 113.80, 66.26, 47.30, 39.17, 31.73, 27.11, 21.51, 13.82; HRMS (ESI) calcd for C18H23N6O4 [M + H]+: 387.1781, found 387.1772.
1-Methyl-N-(2-morpholinoethyl)-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-amine ( 19 )
Yellow solid; m.p.: 185–186 °C; 1H NMR (400 MHz, CDCl3) δ 7.41 (d, J = 3.7 Hz, 1H), 7.30 (d, J = 3.7 Hz, 1H), 6.40 (s, 1H), 4.29 (s, 3H), 3.82–3.72 (m, 6H), 2.97 (t, J = 7.6 Hz, 2H), 2.77 (t, J = 5.9 Hz, 2H), 2.63–2.57 (m, 4H), 1.92–1.82 (m, 2H), 1.04 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 154.85, 151.46, 149.62, 147.72, 144.36, 141.08, 120.94, 114.75, 113.39, 66.25, 56.55, 53.26, 39.07, 37.37, 27.12, 21.46, 13.81; HRMS (ESI) calcd for C19H26N7O4 [M + H]+: 416.2046, found 416.2041.
1-Methyl-N-(3-morpholinopropyl)-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-amine ( 20 )
Yellow solid; m.p.: 202–204 °C; 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 3.6 Hz, 1H), 7.29 (d, J = 3.6 Hz, 1H), 6.48 (br s 1H), 4.31 (s, 3H), 3.82 (m, 2H), 3.73 (t, J = 4.4 Hz, 4H), 2.97 (t, J = 7.6 Hz 2H), 2.61 (t, J = 6.0 Hz, 2H), 2.54 (t, J = 4.4 Hz, 4H), 1.99–1.93 (m, 2H), 1.92–1.82 (m, 2H), 1.04 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 154.93, 151.49, 149.68, 147.73, 144.38, 141.03, 121.02, 114.75, 113.47, 66.10, 56.15, 53.33, 39.18, 39.15, 27.11, 25.16, 21.48, 13.80; HRMS (ESI) calcd for C20H28N7O4 [M + H]+: 430.2203, found 430.2200.
N-Benzyl-1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-amine ( 21 )
Yellow solid; m.p.: 190–192 °C; 1H NMR (400 MHz, CDCl3) δ 7.48 (d, J = 7.2 Hz, 2H), 7.42 (d, J = 4 Hz, 1H), 7.39–7.33 (m, 3H), 7.29 (d, J = 4 Hz, 1H), 5.53 (t, J = 5.2 Hz, 1H), 4.92 (d, J = 5.2 Hz, 2H), 4.24 (s, 3H), 2.97 (t, J = 7.6 Hz, 2H), 1.91–1.82 (m, 2H) 1.05 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 154.73, 151.48, 149.31, 147.55, 144.45, 141.24, 139.41, 128.15, 127.70, 126.72, 120.92, 114.68, 113.52, 43.87, 39.24, 27.11, 21.47, 13.80; HRMS (ESI) calcd for C20H21N6O3 [M + H]+: 393.1675, found 393.1669.
N-(4-Methoxybenzyl)-1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-amine ( 22 )
Yellow solid; m.p.: 160–162 °C; 1H NMR (400 MHz, CDCl3) δ 7.41 (m, 3H), 7.31 (d, J = 3.7 Hz, 1H), 6.92 (d, J = 8.4 Hz, 2H), 5.47 (t, J = 4.8 Hz, 1H),4.84 (d, J = 4.8 Hz, 2H), 4.23 (s, 3H), 3.81 (s, 3H), 2.96 (t, J = 7.6 Hz, 2H), 1.91–1.81 (m, 2H), 1.03 (t, J = 7.3 Hz, 3H); 13C NMR (126 MHz, DMSO) δ 158.14, 154.78, 151.47, 149.19, 147.54, 144.38, 141.17, 131.31, 129.16, 120.88, 114.72, 113.50, 54.93, 43.26, 39.27, 27.11, 21.49, 13.81; HRMS (ESI) calcd for C21H23N6O4 [M + H]+: 423.1781, found 423.1771.
N-(3-Fluoro-4-methoxybenzyl)-1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-amine ( 23 )
Yellow solid; m.p.: 198–199 °C; 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 3.7 Hz, 1H), 7.29 (d, J = 3.7 Hz, 1H), 7.24 (s, 1H), 7.19 (dd, J = 11.8, 2.0 Hz, 1H), 6.99–6.94 (m, 1H), 5.52 (t, J = 5.4 Hz, 1H), 4.83 (d, J = 5.4 Hz, 2H), 4.25 (s, 3H), 3.89 (s, 3H), 2.96 (t, J = 7.6 Hz, 2H), 1.91–1.82 (m, 2H), 1.02 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 154.70, 152.41, 151.49, 149.99, 149.14, 147.47, 145.95, 144.39, 141.21, 132.41, 124.06, 120.89, 115.55, 114.67, 113.46, 55.84, 43.00, 39.23, 27.10, 21.48, 13.77; HRMS (ESI) calcd for C21H22FN6O4 [M + H]+: 441.1687, found 441.1678.
6-((1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-yl)amino)hexan-1-ol ( 24 )
Yellow solid; m.p.: 142–144 °C; 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 3.7 Hz, 1H), 7.30 (d, J = 3.7 Hz, 1H), 5.26 (t, J = 5.1 Hz, 1H), 4.27 (s, 3H), 3.74 (m, 2H), 3.68 (t, J = 6.4 Hz, 2H), 2.97 (t, J = 7.6 Hz 2H), 1.92–1.84 (m, 2H), 1.82–1.75 (m, 2H), 1.65–1.59 (m, 2H), 1.52–1.47 (m, 4H), 1.03 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 154.95, 151.48, 149.61, 147.72, 144.38, 141.05, 120.97, 114.71, 113.45, 60.65, 40.41, 39.14, 32.45, 28.46, 27.12, 26.38, 25.26, 21.47, 13.81; HRMS (ESI) calcd for C19H27N6O4 [M + H]+: 403.2094, found 403.2084.
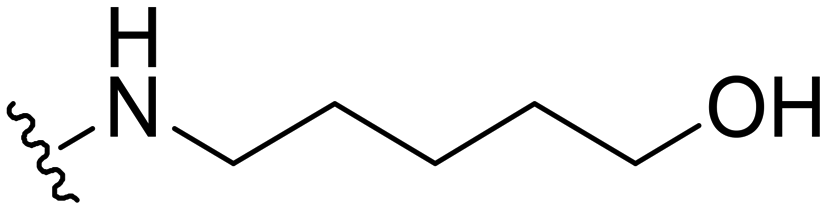
5-((1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-yl)amino)pentan-1-ol ( 25 )
Yellow solid; m.p.: 169–171 °C; 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 3.7 Hz, 1H), 7.30 (d, J = 3.7 Hz, 1H), 5.32 (t, J = 5.3 Hz, 1H), 4.27 (s, 3H), 3.80–3.69 (m, 4H), 2.97 (t, J = 7.6 Hz 2H), 1.92–1.78 (m, 4H), 1.74–1.67 (m, 2H), 1.61–1.53 (m, 2H), 1.03 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 154.94, 151.47, 149.58, 147.70, 144.37, 141.02, 120.95, 114.72, 113.45, 60.63, 40.46, 39.13, 32.17, 28.28, 27.12, 23.01, 21.48, 13.82; HRMS (ESI) calcd for C18H25N6O4 [M + H]+: 389.1937, found 389.1925.
4-((1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-yl)amino)butan-1-ol ( 26 )
Yellow solid; m.p.: 154.5–156.5 °C; 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 3.7 Hz, 1H), 7.30 (d, J = 3.8 Hz, 1H), 5.89 (t, J = 5.0 Hz, 1H), 4.25 (s, 3H), 3.83–3.78 (m, 4H), 2.96 (t, J = 7.6 Hz, 2H) 1.94–1.75 (m, 7H), 1.03 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 154.93, 151.49, 149.60, 147.72, 144.38, 141.03, 120.96, 114.75, 113.57, 60.52, 40.39, 39.13, 29.97, 27.12, 25.19, 21.49, 13.82; HRMS (ESI) calcd for C17H23N6O4 [M + H]+: 375.1781, found 375.1771.
3-((1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-yl)amino)propan-1-ol ( 27 )
Yellow solid; m.p.: 181–183 °C; 1H NMR (400 MHz, CDCl3) δ 7.42 (s, 2H), 6.54 (s, 1H), 4.26 (s, 3H), 3.97–3.89 (m, 4H), 2.96 (t, J = 7.6 Hz, 2H), 2.01 (m, 2H), 1.90–1.80 (m, 2H), 1.02 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 154.93, 151.48, 149.60, 147.71, 144.38, 140.98, 120.97, 114.72, 113.60, 59.10, 39.05, 38.40, 31.48, 27.11, 21.46, 13.80; HRMS (ESI) calcd for C16H21N6O4 [M + H]+: 361.1624, found 361.1620.
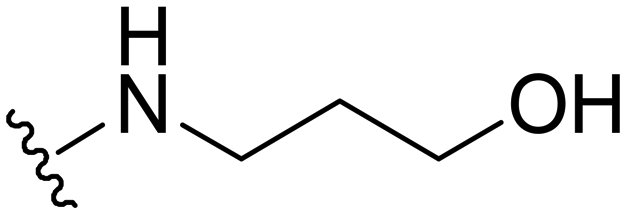
2-((1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-yl)amino)ethanol ( 28 )
Orange-colored solid; m.p.: 204–206 °C; 1H NMR (400 MHz, DMSO) δ 7.81 (d, J = 3.8 Hz, 1H), 7.43 (s, 1H), 7.39 (d, J = 3.8 Hz, 1H), 4.86 (t, J = 5.6 Hz, 1H), 4.22 (s, 3H), 3.69 (s, 2H), 2.84 (t, J = 7.5 Hz, 2H), 1.83–1.72 (m, 2H), 0.95 (t, J = 7.4 Hz, 3H); 13C NMR (126 MHz, DMSO) δ 154.86, 151.52, 149.72, 147.62, 144.33, 141.03, 120.99, 114.78, 113.68, 58.96, 43.12, 39.18, 27.11, 21.52, 13.83; HRMS (ESI) calcd for C15H19N6O4 [M + H]+: 347.1468, found 347.1457.
2-(1-(1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-yl)piperidin-4-yl)ethanol ( 29 )
Yellow solid; m.p.: 195–197 °C; 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 3.7 Hz, 1H), 7.31 (d, J = 3.7 Hz, 1H), 4.12–4.06 (m, 5H), 3.78 (t, J = 6.3 Hz, 2H), 3.13–3.06 (m, 2H), 3.0 (t, J = 7.6 Hz, 2H), 1.99–1.79 (m, 5H), 1.65–1.59 (m, 2H), 1.52–1.45 (m, 2H), 1.05 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 154.34, 153.68, 151.60, 147.04, 145.85, 143.29, 123.74, 114.71, 113.86, 58.19, 49.29, 39.05, 38.99, 31.96, 31.37, 27.18, 21.32, 13.84; HRMS (ESI) calcd for C20H27N6O4 [M + H]+: 415.2094, found 415.2083.
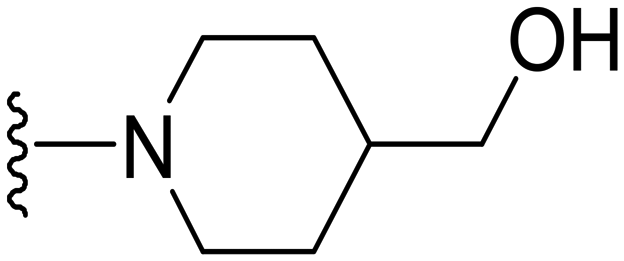
(1-(1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-yl)piperidin-4-yl)methanol ( 30 )
Yellow solid; m.p.: 225–227 °C; 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 3.6 Hz, 1H), 7.31 (d, J = 3.6 Hz, 1H), 4.11 (m, 5H), 3.62 (d, J = 5.9 Hz, 2H), 3.16–3.05 (m, 2H), 3.01 (t, J = 7.6 Hz, 2H), 2.02–1.80 (m, 5H), 1.58–1.47 (m, 3H), 1.05 (t, J = 7.3 Hz, 3H); 13C NMR (126 MHz, DMSO) δ 154.31, 153.72, 151.65, 147.06, 145.83, 143.27, 123.76, 114.80, 113.96, 65.55, 49.05, 39.10, 38.12, 28.11, 27.19, 21.39, 13.88; HRMS (ESI) calcd for C19H25N6O4 [M + H]+: 401.1937, found 401.1923.
3-(4-(1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-yl)piperazin-1-yl)phenol ( 31 )
Orange color solid; m.p.: 225–227 °C; 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 3.7 Hz, 1H), 7.33 (d, J = 3.7 Hz, 1H), 7.17–7.13 (m, 1H), 6.58 (dd, J = 8.2, 1.8 Hz, 1H), 6.48 (d, J = 1.8 Hz, 1H), 6.39 (dd, J = 8.2, 1.8 Hz, 1H), 4.17 (s, 3H), 3.76 (t, J = 4.4 Hz, 4H), 3.42 (t, J = 4.4 Hz, 4H), 3.03 (t, J = 7.6 Hz, 2H), 1.94–1.85 (m, 2H), 1.06 (t, J = 7.4 Hz, 3H); 13C NMR (126 MHz, DMSO) δ 158.12, 154.15, 153.40, 152.18, 151.67, 146.96, 145.91, 143.46, 129.62, 123.63, 114.76, 114.07, 106.85, 106.64, 102.78, 48.80, 47.85, 39.26, 27.19, 21.36, 13.87; HRMS (ESI) calcd for C23H26N7O4 [M + H]+: 464.2046, found 464.2033.
2-(4-(1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-yl)piperazin-1-yl)phenol ( 32 )
Yellow solid; m.p.: 221–223 °C; 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 3.7 Hz, 1H), 7.34 (d, J = 3.7 Hz, 1H), 7.21 (d, J = 7.8 Hz, 1H), 7.17–7.11 (m, 1H), 7.00 (d, J = 8.0 Hz, 1H), 6.96–6.87 (m, 2H), 4.19 (s, 3H), 3.80 (t, J = 4.4 Hz, 4H), 3.14 (t, J = 4.4 Hz, 4H) 3.03 (t, J = 7.6 Hz, 2H) 1.95–1.85 (m, 2H), 1.06 (t, J = 7.3 Hz, 3H); 13C NMR (126 MHz, DMSO) δ 154.21, 153.45, 151.67, 150.15, 147.00, 145.91, 143.43, 139.39, 123.65, 123.25, 119.40, 118.87, 115.64, 114.77, 114.10, 49.64, 49.17, 39.26, 27.20, 21.38, 13.88; HRMS (ESI) calcd for C23H26N7O4 [M + H]+: 464.2046, found 464.2036.
2-((1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-yl)amino)phenol ( 33 )
Light orange color solid; m.p.: 212–214 °C; 1H NMR (500 MHz, DMSO) δ 8.33 (d, J = 7.9 Hz, 1H), 7.78 (d, J = 3.8 Hz, 1H), 7.22 (d, J = 3.8 Hz, 1H), 7.19–6.81 (m, 3H), 4.36 (s, 3H), 2.88 (t, J = 7.5 Hz, 2H), 1.85–1.79 (m, 2H), 0.99 (t, J = 7.3 Hz, 3H); 13C NMR (126 MHz, DMSO) δ 154.35, 151.56, 148.31, 147.29, 147.27, 144.44, 141.74, 126.39, 124.61, 122.41, 121.10, 119.15, 114.83, 114.72, 113.64, 39.88, 27.10, 21.49, 13.87; mass ESI [M + H]+: 395.1.
3-((1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-yl)amino)phenol ( 34 )
Orange-colored solid; m.p.: 227–229 °C; 1H NMR (400 MHz, acetone d6) δ 7.64 (d, J = 3.8 Hz,1H) 7.50–7.48 (m, 1H), 7.39–7.34 (m, 2H), 7.246–7.22 (m, 1H), 6.97 (dd, J = 2.02, 8.08 Hz, 1H), 4.45 (s, 3H) 2.94 (t, J = 7.6 Hz, 2H), 1.93–1.83 (m, 2H), 1.02 (t, J = 7.3 Hz, 3H); 13C NMR (126 MHz, DMSO) δ 157.48, 154.30, 151.62, 147.39, 147.13, 144.75, 142.36, 139.61, 129.14, 121.33, 114.76, 113.90, 112.87, 111.04, 109.07, 39.31, 27.09, 21.48, 13.84; HRMS (ESI) calcd for C19H19N6O4 [M + H]+: 395.1468, found 395.1455.
4-((1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-yl)amino)phenol ( 35 )
Yellow solid; m.p.: 251–253 °C; 1H NMR (400 MHz, acetone d6) δ 7.66 (d, J = 9.2 Hz, 2H), 7.6 (d, J = 4 Hz, 1H), 7.25 (d, J = 4 Hz, 1H), 6.93 (d, J = 9.2 Hz, 2H), 4.4 (s, 3H), 2.93 (t, J = 7.6 Hz, 2H), 1.91–1.82 (m, 2H), 1.01 (t, J = 7.2 Hz, 3H); 13C (126 MHz, acetone) δ 156.06, 155.36,152.94, 149.03, 148.73, 146.19, 143.76, 131.27, 125.41, 122.11, 115.91, 114.47, 114.06, 39.76, 29.36, 22.35, 14.26; HRMS (ESI) calcd for C19H19N6O4 [M + H]+: 395.1468, found 395.1463.
N-(4-Methoxyphenyl)-1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-amine ( 36 )
Orange-colored solid; m.p.: 152–154 °C; 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 9.0 Hz, 2H), 7.39 (d, J = 3.8 Hz, 1H), 7.22 (d, J = 3.7 Hz, 1H), 6.99 (d, J = 9.0 Hz, 2H), 6.92 (s, 1H), 4.31 (s, 3H), 3.86 (s, 3H), 2.94 (t, J = 7.6 Hz, 2H), 1.94–1.82 (m, 2H), 1.05 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 156.95, 154.68, 151.98, 148.05, 147.55, 146.73, 143.37, 130.63, 123.33, 121.02, 114.30, 113.34, 113.14, 55.59, 39.36, 27.64, 22.08, 14.09; HRMS (ESI) calcd for C20H21N6O4 [M + H]+: 409.1624, found 409.1613.
1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-N-(4-(trifluoromethoxy)phenyl)-1H-pyrazolo[4,3-d]pyrimidin-7-amine ( 37 )
Yellow solid; m.p.: 198–200 °C; 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 8.6 Hz, 2H), 7.42 (d, J = 4 Hz, 1H), 7.34 (d, J = 8.6 Hz, 2H), 7.26 (d, J = 4 Hz, 1H), 7.03 (s, 1H), 4.37 (s, 3H), 3.02 (t, J = 7.6 Hz, 2H), 1.96–1.87 (m, 2H), 1.05 (t, J = 7.2 Hz, 3H); 13C NMR (126 MHz, DMSO) δ 154.06, 151.57, 147.10, 146.90, 144.81, 144.01, 142.54, 137.81, 123.60, 121.16, 121.11, 119.15, 114.66, 113.77, 39.33, 27.09, 21.42, 13.82; HRMS (ESI) calcd for C20H18F3N6O4 [M + H]+: 463.1342, found 463.1332.
N-(3-Fluorophenyl)-1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-amine ( 38 )
Yellow solid; m.p.: 179–181 °C; 1H NMR (400 MHz, CDCl3) δ 7.85–7.82 (m, 1H), 7.41 (d, J = 4 Hz, 1H), 7.39–7.34 (m, 2H), 7.27 (d, J = 4 Hz, 1H), 7.06 (s, 1H), 6.93–6.88 (m, 1H), 4.36 (s, 3H), 2.99 (t, J = 7.5 Hz, 2H), 1.93–1.84 (m, 2H), 1.04 (t, J = 7.3 Hz, 3H); 13C NMR (126 MHz, acetone) δ 164.60, 162.68, 155.55, 148.41, 148.21, 146.36, 144.32, 141.50, 130.89, 122.15, 118.33, 114.54, 114.13, 111.18, 109.74, 39.78, 28.25, 22.68, 14.27; HRMS (ESI) calcd for C19H18FN6O3 [M + H]+: 397.1424, found 397.1417.
N-(4-Fluorophenyl)-1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-amine ( 39 )
Yellow solid; m.p.: 201–203 °C; 1H NMR (400 MHz, CDCl3) δ 7.72 (m, 2H), 7.41 (d, J = 3.7 Hz, 1H), 7.23 (d, J = 3.7 Hz, 1H), 7.18 (d, J = 8.5 Hz, 2H), 6.98 (s, 1H), 4.36 (s, 3H), 3.00 (t, J = 7.7 Hz, 2H), 1.95–1.84 (m, 2H), 1.05 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, acetone) δ 161.20, 155.73, 148.61, 146.29, 143.99, 135.79, 125.31, 125.20, 122.03, 115.96, 115.78, 114.49, 114.17, 39.79, 28.27, 22.67, 14.29; HRMS (ESI) calcd for C19H18FN6O3 [M + H]+: 397.1424, found 397.1415.
N-(4-Chlorophenyl)-1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-amine ( 40 )
Orange-colored solid; m.p.: 209–211 °C; 1H NMR (400 MHz, CDCl3) δ 7.74 (d, J = 8.9 Hz, 2H), 7.42 (m, 3H), 7.24 (d, J = 3.8 Hz, 1H), 6.98 (s, 1H), 4.36 (s, 3H), 2.99 (t, J = 7.6 Hz, 2H), 1.93–1.84 (m, 2H), 1.04 (t, J = 7.2 Hz, 4H); 13C NMR (101 MHz, CDCl3) δ 154.41, 152.07, 147.89, 146.95, 146.84, 143.89, 136.41, 129.80, 129.20, 122.36, 120.90, 113.19, 113.05, 39.30, 27.63, 21.98, 14.03. HRMS (ESI) calcd for C19H18ClN6O3 [M + H]+: 413.1129, found 413.1119.
N-(4-Bromophenyl)-1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-7-amine ( 41 )
Orange-colored solid; m.p.: 212–214 °C; 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 8.8 Hz, 2H), 7.58 (d, J = 8.8 Hz, 2H), 7.42 (d, J = 3.7 Hz, 1H), 7.25 (mer with CDCl3, 1H), 6.97 (s, 1H), 4.37 (s, 3H), 3.00 (t, J = 7.6 Hz, 2H), 1.94–1.84 (m, 2H), 1.05 (t, J = 7.3 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 154.73, 152.40, 148.20, 147.27, 147.11, 144.19, 137.26, 132.50, 122.99, 121.25, 117.69, 113.65, 113.49, 39.71, 28.00, 22.39, 14.44; HRMS (ESI) calcd for C19H18BrN6O3 [M + H]+: 457.0624, found 457.0610.
1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-N-(4-(trifluoromethyl)phenyl)-1H-pyrazolo[4,3-d]pyrimidin-7-amine ( 42 )
Yellow solid; m.p.: 216–218 °C; 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 3.7 Hz, 1H), 7.31 (d, J = 3.7 Hz, 1H), 7.16 (s, 1H), 4.42 (s, 3H), 3.02 (t, J = 7.6 Hz, 2H), 1.95–188 (m, 2H), 1.07 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 153.92, 151.61, 146.80, 144.94, 142.89, 142.50, 125.63, 123.57, 123.26, 123.09, 121.53, 121.30, 114.64, 113.91, 39.31, 27.08, 21.38, 13.80; HRMS (ESI) calcd for C20H18F3N6O3 [M + H]+: 447.1392, found 447.1382.
1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-N-(pyridin-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-7-amine ( 43 )
4-Aminopyridine (50 mg, 0.53 mmol 1 eq.) was dissolved in dry DMF (2 ml) and to it was added K2CO3 (220 mg 1.59 mmol 3q); then, 7-chloro-1-methyl-5-(5-nitrofuran-2-yl)-3-propyl-1H-pyrazolo[4,3-d] pyrimidine (170 mg, 0.53 mmol, 1 eq.) was added to the mixture. The reaction mixture was stirred for 3–4 h at 80 °C. After completion, 20 ml EtOAc and 30 ml water were added and the organic layer was separated. The water layer was re-extracted with EtOAc (2 × 20 ml) and the combined organic layers were washed with brine solution, dried over anhydrous sodium sulfate, and concentrated. The residue was purified by column chromatography. Compound 43 was obtained as an orange-colored solid.
Orange-colored solid; m.p.: 265–267 °C; 1H NMR (400 MHz, CDCl3) δ 8.65 (d, J = 4.3 Hz, 2H), 7.83 (d, J = 4.3 Hz, 2H), 7.46 (d, J = 3.8 Hz, 1H), 7.35 (d, J = 3.8 Hz, 1H), 4.41 (s, 3H), 3.02 (t, J = 7.6 Hz, 2H), 1.95–1.85 (m, 2H), 1.05 (t, J = 7.3 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 153.91, 151.67, 149.46, 146.80, 144.99, 143.17, 121.75, 115.06, 114.68, 113.89, 39.28, 27.08, 21.39, 13.81; HRMS (ESI) calcd for C18H18N7O3 [M + H]+: 380.1471, found 380.1461.
1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-N-(pyridin-4-ylmethyl)-1H-pyrazolo[4,3-d] pyrimi-din-7-amine ( 44 )
Yellow solid; m.p.: 218–220 °C; 1H NMR (400 MHz, DMSO) δ 8.51 (d, J = 5.0 Hz, 2H), 8.24 (t, J = 5.7 Hz, 1H), 7.74 (d, J = 3.9 Hz, 1H), 7.55 (d, J = 5.0 Hz, 2H), 7.19 (d, J = 3.9 Hz, 1H), 4.82 (d, J = 5.6 Hz, 1H), 4.29 (s, 3H), 2.84 (t, J = 7.5 Hz, 1H), 1.84–1.70 (m, 2H), 0.95 (t, J = 7.4 Hz, 3H).13C NMR (126 MHz, DMSO) δ 154.49, 151.50, 149.31, 149.16, 148.89, 147.38, 144.44, 141.28, 122.72, 120.90, 114.69, 113.60, 43.16, 39.23, 27.10, 21.53, 13.83; HRMS (ESI) calcd for C19H20N7O3 [M + H]+: 394.1628, found 394.1620.
1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-N-(pyridin-3-ylmethyl)-1H-pyrazolo[4,3-d]pyrimidin-7-amine ( 45 )
Yellow solid; m.p. 243–245 °C; 1H NMR (400 MHz, DMSO) δ 8.77 (s, 1H), 8.43 (d, J = 3.7 Hz, 1H), 8.19 (t, J = 5.7 Hz, 1H), 7.96 (d, J = 7.8 Hz, 1H), 7.77 (d, J = 3.8 Hz, 1H), 7.33 (dd, J = 7.7, 4.8 Hz, 1H), 7.29 (d, J = 3.8 Hz, 1H), 4.80 (d, J = 5.6 Hz, 2H), 4.26 (s, 3H), 2.83 (t, J = 7.5 Hz, 2H), 1.82–1.70 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 154.61, 151.49, 149.33, 149.12, 147.98, 147.42, 144.42, 141.23, 135.63, 134.90, 123.33, 120.89, 114.69, 113.53, 41.69, 39.24, 27.10, 21.48, 13.80; HRMS (ESI) calcd for C19H20N7O3 [M + H]+: 394.1628, found 394.1626.
1-Methyl-5-(5-nitrofuran-2-yl)-3-propyl-7-(4-(pyridin-4-yl)piperazin-1-yl)-1H-pyrazolo[4,3-d]pyrimidine ( 46 )
Yellow solid; m.p.: 249–251 °C; 1H NMR (400 MHz, CDCl3) δ 8.33 (d, J = 4.2 Hz, 2H), 7.44 (d, J = 3.8 Hz, 1H), 7.34 (d, J = 3.8 Hz, 1H), 6.80 (d, J = 5.8 Hz, 2H), 4.19 (s, 3H), 3.83–3.74 (m, 4H), 3.68–3.60 (m, 4H), 3.02 (t, J = 7.6 Hz, 2H), 1.96–1.84 (m, 2H), 1.06 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 154.84, 154.50, 153.74, 152.13, 150.49, 148.26, 147.77, 144.98, 124.38, 113.41, 113.21, 108.76, 49.02, 45.59, 38.89, 27.79, 22.02, 14.12; HRMS (ESI) calcd for C22H25N8O3 [M + H]+: 449.2050, found 449.2039.
Biological assays
Bacterial strains and culture conditions. The bacterial strains S. aureus ATCC 29213, methicillin-resistant S. aureus (MRSA) ATCC 15187 and E. coli ATCC 25922 were obtained from the American Type Culture Collection (ATCC). The bacterial strains SA-1199, SA-1199B, SA-K2191, and SA-K2192 used for efflux pump overexpression studies were procured from the institutional microbial repository. Cation-adjusted Mueller–Hinton broth (MHB; Becton-Dickinson, Cockeysville, MD) was used for minimum inhibitory concentration (MIC) determination. Mueller–Hinton agar (MHA; Becton-Dickinson) was used for minimum bactericidal concentration (MBC) and time-kill studies. Alamar blue (HiMedia) was used for the detection of MIC value. Propidium iodide (HiMedia) and DAPI (Invitrogen) dyes were used to visualize bacterial cells through fluorescence microscopy.
The HepG2, RAW 264.7 and J774A.7 cells were acquired from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco, Life Technologies, NY) supplemented with 10% fetal bovine serum (FBS) at 37 °C and 5% CO2. MTT dye (HiMedia) was used to detect cell viability. The antibiotics ciprofloxacin hydrochloride monohydrate (HiMedia), vancomycin (HiMedia), and rifampicin (HiMedia) were used in different assays.
Antibacterial assay
The minimum bactericidal concentration is the lowest concentration at which complete killing of bacterial cells is observed. The MBC was determined using 10 μl of the suspensions from the wells of the MIC plate where no growth was visible on the MHA plate. The agar plates were incubated for 24 h and the growth of bacterial colony was visualized in the respective concentrations.
Biofilm inhibition. Compound 35 was evaluated for its efficacy to control a major factor of resistance development, biofilm formation. The assay was performed by using different concentrations of compound 35 and vancomycin, 1/2×, 1×, 2×, 4× and 6× MIC. Untreated bacterial cells served as the growth control, and media control was used for comparison. Initially, 100 μL of bacterial suspension with 1.5 × 106 CFU ml−1 (prepared by diluting 100 times the turbid solution of McFarland standard 0.5) was inoculated into a 96-well flat-bottom plate containing TSB medium (tryptic soy broth) + 2% glucose and incubated for 2.5–3 h. Following this, the compound and vancomycin were added at selected concentrations and the mixture was incubated for 27 h at 37 °C with 5% CO2.54 Biofilm formation was observed in the growth control, and the media were decanted from each well. The remaining sample was washed twice with PBS and air-dried. Then, 100 μL of tetrahydrofuran (THF) was added to the dried wells, and absorbance was measured at a wavelength of 600 nm. Absorbance readings were plotted against concentration using GraphPad Prism software to evaluate biofilm inhibition.55
Disruption of preformed biofilm. Compound 35 was tested for its tendency to cause disruption in preformed biofilm in both the bacterial strains, S. aureus and MRSA. The compound and drug control vancomycin were used in the assay at determined concentrations, 1/2×, 1×, 2×, 4× and 6× MIC. The untreated cells were used to determine the biofilm formation ability of bacteria. 100 μL of bacterial suspension with 1.5 × 106 CFU ml−1 was inoculated into a 96-well flat-bottom plate and incubated for 16 h followed by addition of compound and vancomycin at their respective concentrations. The plate was incubated further for 24 h at 37 °C with 5% CO2. The media was removed from well carefully and the biofilm was washed twice with PBS and air-dried. 100 μL of tetrahydrofuran (THF) solvent was added and absorbance was measured at a wavelength of 600 nm. Absorbance versus concentration was plotted using GraphPad Prism software to evaluate the disruption in preformed biofilm.55
MIC against overexpressing strains. The impact of the compound in a multidrug-resistant strain of S. aureus was observed by evaluating the MIC of compound 35 against ciprofloxacin-sensitive (SA-1199), NorA-overexpressing (SA-1199B), MsrA-overexpressing (SA-K2191), and TetK-overexpressing (SA-K2192) strains. The NorA, MsrA and TetK proteins from S. aureus are responsible for extruding the antibiotics and overexpression of these efflux pump proteins develop resistance.56 Among these, the NorA efflux pump plays a major role in resistance development with S. aureus.57 To determine the MIC value, twofold serial dilutions of compound 35 and standard drug control vancomycin were prepared in Muller–Hinton broth (MHB) in a 96-well flat-bottom microtitre plate with final concentrations ranging from 10 to 0.19 μg ml−1 and 10 to 0.19 μM, respectively. The cell suspension of a density of 0.5 McFarland (1.5 × 108 CFU ml−1) was used as inoculum after diluting 1
:100 and 0.1 ml of this inoculum was added to the plate, followed by incubation at 37 °C; the plate was observed for fluorescence measurement after the addition of Alamar blue. The MIC of wild-type strain S. aureus 29213 and respective resistant strains were compared to predict the result.
:100 in MHB; 0.1 ml of this inoculum was added to the plate. The plate was incubated at 37 °C for 24 h, followed by the addition of Alamar blue (0.04%) and the calculation of the FIC index.
The fraction inhibitory concentration index (FICI) is used to categorize the values for the interaction of two antibiotics. It is calculated by using the following formulas:
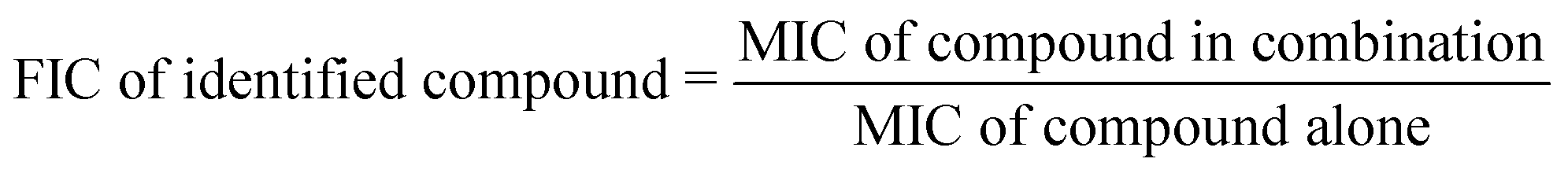
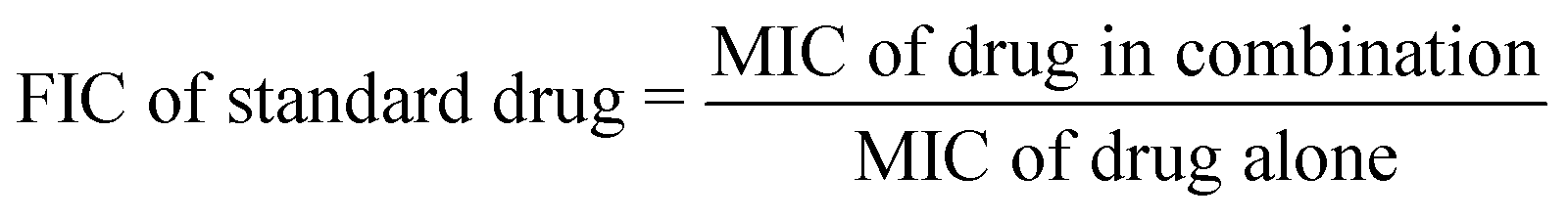
| FICI = FIC of identified compound + FIC of standard drug |
Cytotoxicity assay
The cytotoxic effect of the compounds was initially evaluated on HepG2 cells and then the toxicity effect was determined for the most active compound 35 using RAW 264.7 and J774A.1 macrophage/monocyte cells through 3-(4,5-dimethylthiazol2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in a 96-well-plate format. The MTT assay is a colorimetric assay used to measure the viability and cytotoxicity of cells. The cells were plated at a density of 1 × 104 cells per well in a 96-well flat-bottom plate in DMEM containing 10% FBS and incubated for 24 h (37 °C; 5% CO2). The cell monolayer was exposed to varying concentrations of compounds ranging from 200 to 0.19 μg ml−1 and incubated for 24 h (37 °C; 5% CO2).48 The drug doxorubicin at a concentration of 10 μM and growth and medium controls were used to compare the results. After incubation, 2.5 mg ml−1 MTT dye was added and cells were further incubated for 3 h (37 °C; 5% CO2). The water-insoluble purple and white formazan produced by mitochondrial dehydrogenases was solubilized by 100 μL DMSO and incubated for 15–20 min. Following incubation, the cells were analyzed through a microplate reader in terms of optical density (OD) at 570 nm.61Compound 35 was tested further using RAW 264.7 and J774A.1 macrophage/monocyte cells with changed incubation treatment. The cells were plated at a density of 1 × 104 cells per well in a 96-well flat-bottom plate in DMEM containing 10% FBS and incubated for 48 h and 72 h (37 °C; 5% CO2). The treated cells were then analyzed using OD at 570 nm.
Hemolysis assay
To evaluate the ability of compound 35 to cause membrane disruption of erythrocytes or cytotoxicity, a hemolysis assay was performed. Withdrawn blood from the hepatic vein of a rabbit containing erythrocytes was stabilized by EDTA. The total volume of blood (2 ml) was added to phosphate-buffered saline (PBS) of pH −7.4 and centrifuged at 8000 rpm for 5 min to isolate red blood cells (RBCs). The pellet of RBCs was further washed five times with PBS and diluted in 20 ml of PBS to make a 10% RBC suspension. Varied concentrations of the test compounds, compound 35 and vancomycin (100, 80, 60, 40, 20, 10 μg ml−1), were prepared in a total volume of 400 μl.42 0.1 ml of diluted RBC suspension was added to the prepared test samples and incubated for 4 h. PBS (negative control) and 1% Triton-X (positive control) were used to calculate the percentage of hemolysis. Following incubation, the samples were centrifuged at 8000 rpm for 5 min, and undisturbed supernatant was collected to analyze the absorbance at the wavelength of 577 nm using a microplate reader. The experiment was performed in triplicate. The percentage of hemolysis was calculated using the formula,62
 log10 CFU ml−1) will be classified as bactericidal, while less reduction (<3log10 CFU ml−1) will be referred to as bacteriostatic.46
log10 CFU ml−1) will be classified as bactericidal, while less reduction (<3log10 CFU ml−1) will be referred to as bacteriostatic.46
Antimicrobial mechanism studies
:100 and incubated for 3–4 h till OD reached 0.5. The bacterial culture of the desired concentration was treated with different concentrations of compound 35 and vancomycin, followed by 2.5 h of incubation at 37 °C. The treated and untreated cultures were centrifuged at 5000 rpm for 10 min and washed twice with 1× PBS. The fluorescent dyes PI (60 μM) and DAPI (1 μg ml−1) were added to 1 ml bacterial suspension in the dark and incubated for 30 min to show its full efficiency. The samples were centrifuged at 5000 rpm at 37 °C and washed thrice with 1× PBS to remove extracellular dye. The pellet was resuspended in 500 μl PBS and put in a 24-well plate and allowed to dry properly.65 The samples were then visualized through a Nikon Ts2FL fluorescence microscope using a scale of 10 μm.
:1 and incubated for 2 h. Two hours post-infection, these cells were washed with 100 μg ml−1 gentamycin to remove extracellular bacterial cells and incubated for a further 2 h. This was followed by PBS washing and the addition of compound 35 at different MIC concentrations, 1×, 2× and 4×, along with similar concentrations of vancomycin. The untreated RAW 264.7 cells were used as growth control to observe the reduction of bacterial load. The colonies were counted in the respective dilutions and the colony-forming unit (CFU ml−1) was calculated which was plotted on a graph using GraphPad Prism software. The significant reduction in different groups was determined using one-way ANOVA in GraphPad Prism.69
Pharmacokinetic (PK) studies
Oral and IV route PK studies were performed using male Balb/c mice (n = 5 mice per time point) at a dose of 2.5 and 30 and 1 mg kg−1, respectively. For the oral route, compound 35 was triturated in 2% gum acacia (w/v) and 10 ml distilled water was added to form a 2.5 or 30 mg per 10 ml suspension. In the case of IV route, 5% DMSO, 5% Solutol:absolute alcohol (1:1, v/v), and 90% normal saline were used for the formulation. Samples derived from plasma at different time points were then analyzed by LC-MS/MS to generate the required pharmacokinetic parameters.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
R. S. acknowledges the CSIR-New Delhi (CSPS24/RDSF/IIIM/IHP24/07), DST-SERB (SRG/2022/001567), and CCRAS-New Delhi (GAP-3144) for financial support. The authors express their sincere gratitude to Dr. Vinay Sharma from the Indian Institute of Technology, Jammu, for his invaluable support in the imaging experimentation.References
- F. C. Tenover, Am. J. Infect. Control, 2006, 34(5), S3–S10 CrossRef PubMed.
- F. Prestinaci, P. Pezzotti and A. Pantosti, Pathog. Global Health, 2015, 109(7), 309–318 CrossRef PubMed.
- M. A. Abushaheen, Muzaheed, A. J. Fatani, M. Alosaimi, W. Mansy, M. George, S. Acharya, S. Rathod, D. D. Divakar, C. Jhugroo, S. Vellappally, A. A. Khan, J. Shaik and P. Jhugroo, Dis. Mon., 2020, 66(6), 100971 CrossRef PubMed.
- L. Morrison and T. R. Zembower, Gastrointest. Endosc. Clin. N. Am., 2020, 30(4), 619–635 CrossRef PubMed.
- Sg Kumar, C. Adithan, B. Harish, G. Roy, A. Malini and S. Sujatha, J. Nat. Sci., Biol. Med., 2013, 4(2), 286 CrossRef PubMed.
- J. M. Munita and C. A. Arias, Microbiol. Spectrum, 2016, 4(2), 4.2.15 CrossRef PubMed.
- X. Chen, W. Lou, J. Liu, B. Ding, W. Fan and J. Hong, Biosci. Rep., 2019, 39(10), BSR20192354 CrossRef CAS PubMed.
- Global Antimicrobial Resistance and Use Surveillance System (GLASS) Report 2022, World Health Organization, Geneva, 2022 Search PubMed.
- WHO Bacterial Priority Pathogens List 2024 Bacterial Pathogens of Public Health Importance, to Guide Research, Development, and Strategies to Prevent and Control Antimicrobial Resistance, World Health Organization, Geneva, 2024 Search PubMed.
- S. Santajit and N. Indrawattana, BioMed Res. Int., 2016, 2016, 1–8 CrossRef PubMed.
- A. A. Bitrus, O. M. Peter, M. A. Abbas and M. D. Goni, Vet. Sci. Res. Rev., 2018, 4(2), 43–54 Search PubMed.
- M. Vestergaard, D. Frees and H. Ingmer, Microbiol. Spectrum, 2019, 7(2), 7.2.18 CrossRef PubMed.
- Y. Guo, G. Song, M. Sun, J. Wang and Y. Wang, Front. Cell. Infect. Microbiol., 2020, 10, 107 CrossRef PubMed.
- Antimicrobial Resistance in the EU/EEA (EARS-Net) - Annual Epidemiological Report for 2022, 2022 Search PubMed.
- M. Monaco, F. Pimentel De Araujo, M. Cruciani, E. M. Coccia, A. Pantosti, F. Bagnoli, R. Rappuoli and G. Grandi, Current Topics in Microbiology and Immunology, Springer International Publishing, Cham, 2016, vol. 409, pp. 21–56 Search PubMed.
- R. R. Watkins, M. Holubar and M. Z. David, Antimicrob. Agents Chemother., 2019, 63(12), e01216-19 CrossRef PubMed.
- A. Nayyar, S. R. Patel and R. Jain, Front. Med. Chem., 2009, 4, 506–540 Search PubMed.
- R. R. Bailey, P. E. Gower, A. P. Roberts and H. E. De Wardener, Lancet, 1971, 298(7734), 1112–1114 CrossRef PubMed.
- A. Masunari and L. C. Tavares, Bioorg. Med. Chem., 2007, 15(12), 4229–4236 CrossRef CAS PubMed.
- A. M. Segura, O. Gutiérrez, W. Otero, A. Angel, R. M. Genta and D. Y. Graham, Aliment. Pharmacol. Ther., 1997, 11(3), 529–532 CrossRef CAS PubMed.
- A. P. Tynan, F. R. Macis and J. N. W. McQuaid, Br. J. Urol., 1969, 41, 271–279 CrossRef CAS PubMed.
- B. Das, S. Rudra, A. Yadav, A. Ray, A. V. S. R. Rao, A. S. S. V. Srinivas, A. Soni, S. Saini, S. Shukla, M. Pandya, P. Bhateja, S. Malhotra, T. Mathur, S. K. Arora, A. Rattan and A. Mehta, Bioorg. Med. Chem. Lett., 2005, 15(19), 4261–4267 CrossRef CAS PubMed.
- A. Delsarte, M. Faway, J. M. Frère, J. Coyette, C. M. Calberg-Bacq and E. Heinen, Antimicrob. Agents Chemother., 1981, 19(3), 477–486 CrossRef CAS PubMed.
- A. Ali, G. E. Taylor, K. Ellsworth, G. Harris, R. Painter, L. L. Silver and K. Young, J. Med. Chem., 2003, 46(10), 1824–1830 CrossRef CAS PubMed.
- H. A. Burch, J. Med. Chem., 1968, 11(1), 79–83 CrossRef CAS PubMed.
- M. Bakavoli, G. Bagherzadeh, M. Vaseghifar, A. Shiri, M. Pordel, M. Mashreghi, P. Pordeli and M. Araghi, Eur. J. Med. Chem., 2010, 45(2), 647–650 CrossRef CAS PubMed.
- D. Geffken, R. Soliman, F. S. G. Soliman, M. M. Abdel-Khalek and D. A. E. Issa, Med. Chem. Res., 2011, 20(4), 408–420 CrossRef CAS.
- N. P. Peet, N. L. Lentz, S. Sunder, A. M. L. Ogden and M. Merrell, J. Med. Chem., 1992,(35), 3263–3269 CrossRef CAS PubMed.
- L. Squarcialupi, V. Colotta, D. Catarzi, F. Varano, G. Filacchioni, K. Varani, C. Corciulo, F. Vincenzi, P. A. Borea, C. Ghelardini, L. Di Cesare Mannelli, A. Ciancetta and S. Moro, J. Med. Chem., 2013, 56(6), 2256–2269 CrossRef CAS PubMed.
- P. G. Baraldi, J. D. Simoni, V. Periotto and R. Manservigi, J. Med. Chem., 1984,(27), 986–990 CrossRef CAS PubMed.
- N. K. Saxena, L. A. Coleman, J. C. Drach and L. B. Townsend, J. Med. Chem., 1990,(7), 1980–1983 CrossRef CAS PubMed.
- J. C. Verheijen, D. J. Richard, K. Curran, J. Kaplan, M. Lefever, P. Nowak, D. J. Malwitz, N. Brooijmans, L. Toral-Barza, W.-G. Zhang, J. Lucas, I. Hollander, S. Ayral-Kaloustian, T. S. Mansour, K. Yu and A. Zask, J. Med. Chem., 2009, 52(24), 8010–8024 CrossRef CAS PubMed.
- R. Jorda, L. Havlíček, I. W. McNae, M. D. Walkinshaw, J. Voller, A. Šturc, J. Navrátilová, M. Kuzma, M. Mistrík, J. Bártek, M. Strnad and V. Kryštof, J. Med. Chem., 2011, 54(8), 2980–2993 CrossRef CAS PubMed.
- N. K. Terrett, A. S. Bell, D. Brown and P. M. Ellis, Bioorg. Med. Chem. Lett., 1996, 6, 1819–1824 CrossRef.
- H. A. Flores Toque, F. B. M. Priviero, C. E. Teixeira, E. Perissutti, F. Fiorino, B. Severino, F. Frecentese, R. Lorenzetti, J. S. Baracat, V. Santagada, G. Caliendo, E. Antunes and G. De Nucci, J. Med. Chem., 2008, 51(9), 2807–2815 CrossRef CAS PubMed.
- A. Karoui, F. Allouche, M. Deghrigue, A. Agrebi, A. Bouraoui and F. Chabchoub, Med. Chem. Res., 2014, 23(3), 1591–1598 CrossRef CAS PubMed.
- C. Almansa, A. F. De Arriba, F. L. Cavalcanti, L. A. Gómez, A. Miralles, M. Merlos, J. García-Rafanell and J. Forn, J. Med. Chem., 2001, 44(3), 350–361 CrossRef CAS PubMed.
- G. A. Patani and E. J. LaVoie, Chem. Rev., 1996, 96(8), 3147–3176 CrossRef CAS PubMed.
- G. L. Reddy, S. K. Guru, M. Srinivas, A. S. Pathania, P. Mahajan, A. Nargotra, S. Bhushan, R. A. Vishwakarma and S. D. Sawant, Eur. J. Med. Chem., 2014, 80, 201–208 CrossRef CAS PubMed.
- A. Gangjee, Y. Zhao, S. Raghavan, C. C. Rohena, S. L. Mooberry and E. Hamel, J. Med. Chem., 2013, 56(17), 6829–6844 CrossRef CAS PubMed.
- K. Poole, Ann. Med., 2007, 39(3), 162–176 CrossRef CAS PubMed.
- T. Ito, W. Hashimoto, N. Ohoka, T. Misawa, T. Inoue, R. Kawano and Y. Demizu, ACS Biomater. Sci. Eng., 2023, 9(8), 4654–4661 CrossRef CAS PubMed.
- H. Mosaei and N. Zenkin, EcoSal Plus, 2020, ESP-0017-2019 Search PubMed.
- C. Watanakunakorn, J. Antimicrob. Chemother., 1984, 14(suppl D), 7–18 CrossRef CAS PubMed.
- C. C. Sanders, Rev. Infect. Dis., 1988, 10(3), 516–527 CrossRef CAS PubMed.
- M. H. Sharaf, G. M. El-Sherbiny, S. A. Moghannem, M. Abdelmonem, I. A. Elsehemy, A. M. Metwaly and M. H. Kalaba, Sci. Rep., 2021, 11(1), 4240 CrossRef CAS PubMed.
- Y. Yu, H.-L. Huang, X.-Q. Ye, D.-T. Cai, J.-T. Fang, J. Sun, X.-P. Liao and Y.-H. Liu, Front. Microbiol., 2020, 11, 1919 CrossRef PubMed.
- K. M. S. Herrera, G. F. M. Lopes, M. E. Oliveira, J. F. Sousa, W. G. Lima, F. K. Silva, J. C. M. Brito, A. J. P. S. Gomes, G. H. R. Viana, A. C. Soares and J. M. S. Ferreira, Microbiol. Res., 2022, 261, 127073 CrossRef CAS PubMed.
- W. Guo, Z. Yang, K. Wang, W. Li, Y. Zhao, Y. Yang, W. Chang, Z. Gong, Z. Liu, Y. Chen and Q. Li, J. Med. Chem., 2024, 67(3), 2129–2151 CrossRef CAS PubMed.
- W. Cheng, T. Xu, L. Cui, Z. Xue, J. Liu, R. Yang, S. Qin and Y. Guo, J. Med. Chem., 2023, 66(1), 962–975 CrossRef CAS PubMed.
- X. Chen, W. Lou, J. Liu, B. Ding, W. Fan and J. Hong, Biosci. Rep., 2019, 39(10), BSR20192354 CrossRef CAS PubMed.
- G. Liu, P. Qin, X. Cheng, L. Wu, W. Zhao and W. Gao, Front. Microbiol., 2024, 15, 1389242 CrossRef PubMed.
- P. Parvekar, J. Palaskar, S. Metgud, R. Maria and S. Dutta, Biomater. Invest. Dent., 2020, 7(1), 105–109 CAS.
- S. Cascioferro, B. Maggio, D. Raffa, M. V. Raimondi, M. G. Cusimano, D. Schillaci, B. Manachini, A. Leonchiks and G. Daidone, Med. Chem. Res., 2016, 25(5), 870–878 CrossRef CAS.
- K. Syal, Curr. Microbiol., 2017, 74(10), 1194–1199 CrossRef CAS PubMed.
- P. G. Pinheiro, G. M. P. Santiago, F. E. F. Da Silva, A. C. J. De Araújo, C. R. T. De Oliveira, P. R. Freitas, J. E. Rocha, J. B. D. A. Neto, M. M. C. Da Silva, S. R. Tintino, A. Siyadatpanah, R. Norouzi, S. Dashti, P. Wilairatana, H. D. M. Coutinho and J. G. M. Da Costa, Biotechnol. Rep., 2022, 34, e00717 CrossRef CAS PubMed.
- N. Chandal, R. Tambat, R. Kalia, G. Kumar, N. Mahey, S. Jachak and H. Nandanwar, Microbiol. Spectrum, 2023, 11(5), e04876-22 CrossRef PubMed.
- Y.-C. Lee, P.-Y. Chen, J.-T. Wang and S.-C. Chang, BMC Pharmacol. Toxicol., 2019, 20(1), 25 CrossRef PubMed.
- F. C. Odds, J. Antimicrob. Chemother., 2003, 52(1), 1 CrossRef CAS PubMed.
- S. Perveen, A. Negi, S. Saini, A. Gangwar and R. Sharma, ACS Infect. Dis., 2024, 10(2), 513–526 CrossRef CAS PubMed.
- V. S. De Sena Pereira, C. B. Silva De Oliveira, F. Fumagalli, F. Da Silva Emery, N. B. Da Silva and V. F. De Andrade-Neto, Toxicol. Rep., 2016, 3, 756–762 CrossRef CAS PubMed.
- S. Liu, P. She, Z. Li, Y. Li, Y. Yang, L. Li, L. Zhou and Y. Wu, AMB Express, 2022, 12(1), 150 CrossRef CAS PubMed.
- Z. Chen, H. Duan, X. Tong, P. Hsu, L. Han, S. L. Morris-Natschke, S. Yang, W. Liu and K.-H. Lee, J. Nat. Prod., 2018, 81(3), 465–474 CrossRef CAS PubMed.
- R. Song, B. Yu, D. Friedrich, J. Li, H. Shen, H. Krautscheid, S. D. Huang and M.-H. Kim, Commun. Biol., 2020, 3(1), 529 CrossRef CAS PubMed.
- T. Xu, X. Yan, A. Kang, L. Yang, X. Li, Y. Tian, R. Yang, S. Qin and Y. Guo, J. Med. Chem., 2024, 67(11), 9302–9317 CrossRef CAS PubMed.
- R. Yang, E. Hou, W. Cheng, X. Yan, T. Zhang, S. Li, H. Yao, J. Liu and Y. Guo, J. Med. Chem., 2022, 65(24), 16879–16892 CrossRef CAS PubMed.
- M. Rosenberg, N. F. Azevedo and A. Ivask, Sci. Rep., 2019, 9(1), 6483 CrossRef PubMed.
- L. B. Oyama, H. Olleik, A. C. N. Teixeira, M. M. Guidini, J. A. Pickup, B. Y. P. Hui, N. Vidal, A. R. Cookson, H. Vallin, T. Wilkinson, D. M. S. Bazzolli, J. Richards, M. Wootton, R. Mikut, K. Hilpert, M. Maresca, J. Perrier, M. Hess, H. C. Mantovani, N. Fernandez-Fuentes, C. J. Creevey and S. A. Huws, npj Biofilms Microbiomes, 2022, 8(1), 58 CrossRef CAS PubMed.
- I. Eid, M. M. Elsebaei, H. Mohammad, M. Hagras, C. E. Peters, Y. A. Hegazy, B. Cooper, J. Pogliano, K. Pogliano, H. S. Abulkhair, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2017, 139, 665–673 CrossRef CAS PubMed.
- M. Su, L. Qiu, Y. Deng, C. H. Ruiz, J. D. Rudolf, L.-B. Dong, X. Feng, M. D. Cameron, B. Shen, Y. Duan and Y. Huang, Mol. Pharmaceutics, 2019, 16(7), 3065–3071 CrossRef CAS PubMed.
- N. Arshad, A. Mehreen, I. Liaqat, M. Arshad and H. Afrasiab, BMC Complementary Altern. Med., 2017, 17(1), 498 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00826j |
| ‡ Equal contribution as first author to this work. |
| This journal is © The Royal Society of Chemistry 2025 |