
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTi3C2Tx MXene augments osmo-adaptive repression of the inflammatory stress response for improved wound repair†
Sertan Kiziloz‡
a,
Emma J. Ward‡a,
Daniel Hawthornea,
Avick Sinhaab,
Grace Cooksley c,
Dipak Sarkera,
Cyril Cruad,
Andrew Lloyda,
Christopher E. Shucke,
Yury Gogotsif and
Susan Sandeman*a
c,
Dipak Sarkera,
Cyril Cruad,
Andrew Lloyda,
Christopher E. Shucke,
Yury Gogotsif and
Susan Sandeman*a
aCentre for Regenerative Medicine and Devices, School of Applied Sciences, University of Brighton, Brighton BN2 4GJ, UK. E-mail: s.sandeman@brighton.ac.uk
bAdvanced Engineering Centre, University of Brighton, Brighton BN2 4GJ, UK
cDevelopmental Biology and Cancer (DBC) Research and Teaching Department, UCL GOS Institute of Child Health, London WC1N 1EH, UK
dSchool of Engineering and Informatics, University of Sussex, Brighton, BN1 9QJ, UK
eDepartment of Chemistry and Chemical Biology, Rutgers University, Piscataway, NJ 08854, USA
fDepartment of Materials Science and Engineering and A. J. Drexel Nanomaterials Institute, Drexel University, Philadelphia, PA 19104, USA
First published on 11th April 2025
Abstract
Chronic non-healing wounds represent a growing global health challenge that is poorly addressed by current advances in wound care dressings. Hyperosmotic stress linked, for example, to poor glycaemic control, is a known but under-investigated contributor to the chronic wound environment and a known inflammatory stimulus. MXene (Ti3C2Tx) has been considered for smart dressing applications but has not been investigated for use with bioactive agents to directly moderate hyperosmotic stress for improved wound care. In this study, Ti3C2Tx, in combination with osmolyte betaine, was used to investigate hyperosmotic stress-induced effects on wound closure. The effect of these materials was measured using a wound closure scratch assay, and data was used to mathematically model changes in HaCaT human keratocyte migratory rate and velocity. Changes in the upregulation of apoptotic and inflammatory markers were measured, and qualitative changes in phalloidin-labelled actin cytoskeletal structure were observed. A tert-butyl glycine betainate (tBu-GB) polyacrylate microgel loaded Ti3C2Tx dressing was then fabricated and tested for biocompatibility and slow elution of osmolyte over time. Osmotic stress at levels that did not induce cell death reduced the migratory capacity of keratocytes to close the scratch. Migration by osmotically stressed keratocytes was reduced by more than 50% at 24 h and remained at 65% (±5%) at 48 h compared to complete scratch closure at 24 h in the cell only control. This reduction was reversed by a Ti3C2Tx coating, allowing complete scratch closure by 48 h in the osmotically stressed group. Exposure of osmotically stressed cells to betaine increased normalised wound closure in the osmotically stressed keraotycte group at each time point and this was augmented by the presence of a Ti3C2Tx coating. Osmotic stress induced upregulation of inflammatory markers IL-6, IL-1α, IL-1β, CXCL1, and CXCL8 by at least 10-fold. The effect was significantly greater in the presence of bacterial LPS and this was significantly reduced by the presence of Ti3C2Tx alone and in combination with betaine. Sustained and slow release of betaine was demonstrated from a tBu-GB-microgel loaded Ti3C2Tx dressing over 48 h supporting the use of such dressings to improve osmotic stress induced, poor wound closure rates.
1. Introduction
Advances in nanomaterials for wound repair have yet to significantly impact the growing global clinical health challenge of chronic non-healing wounds. In the UK alone, an estimated 3.8 million patients receive treatment for chronic wounds at an estimated cost of £5.6 billion.1 A partial explanation lies in the complexity of non-healing wounds and the impact of co-morbidities, such as diabetes, which can negatively influence the success of targeted treatments.2,3 An underexplored aspect of this challenge is the role of osmotic imbalance in delayed wound repair and potential strategies to address it using bioactive wound dressings.4,5 A promising, relatively new class of transition metal carbides, nitrides, and carbonitrides, called MXenes, have recently been used within a smart dressing application.6 However, MXenes have yet to be considered in combination with other bioactive agents to moderate hyperosmotic stress and stimulate a shift beyond a chronic hyper-inflammatory state to wound resolution.The chronic wound, though complex, is defined by several parameters. It remains stalled in the inflammatory stage beyond 12 weeks rather than progressing through the classic stages of normal wound repair, moving from haemostasis, inflammation and proliferation to remodelling.7–9 The wound bed tends to be characterised by increased reactive oxygen species, inflammatory cytokines, matrix metalloprotease (MMP), lysozyme and other protease activity, fibroblast senescence, increased pH, increased tendency to infection, increased glucose concentration in the case of diabetes, delayed angiogenesis and delayed proliferative re-epithelialisation.10,11 A key challenge is to address the impact of local and systemic factors that maintain prolonged and dysregulated inflammation rather than progressing sequentially through the stages of wound repair to close the wound. Bioactive dressings synthesised with materials that aim to adaptively respond to changes in the wound bed and release active drugs or molecules to enable improved wound repair are a promising approach to improve treatment.12 However, combination materials that allow slow payload release into the wound bed, potentially using biological signals (e.g., MMPs, lysozyme or other proteases, pH, temperature, reactive oxygen species) from the wound bed, have yet to be effectively utilised.
Developments beyond basic gauze dressings provide an array of available routes for chronic wound care. Advanced dressings, acting by physical and chemical means, include films, foams, hydrocolloids, and hydrogels and tend to be selected based on the presence of infection, moisture control, and requirements to remove excess wound exudate where necessary.13,14 However, evidence indicating the benefits of one dressing over another for chronic wound closure is limited.14 Additional therapeutic options have been considered, incorporating both non-cellular and cellular, biologically active components.12,15,16 These include biopolymers and nanomaterials within the dressing which are responsive to changes in the wound bed as well as regenerative factors which can be slowly eluted into the wound bed following triggered release. Examples include the use of growth factors to promote wound closure and neovascularisation,17 antimicrobial peptides (AMPs) to target infection associated delays in wound repair,18,19 collagen/mesenchymal stem cell combinations to modulate macrophage behaviour and stimulate skin flap repair,20,21 and the use of silver nanoparticles as antibacterial and anti-inflammatory dressing components.22,23 However, major challenges exist in the application of these approaches. For example, AMP-loaded systems must maintain the stability of the AMP on variations in the wound environment such as temperature and pH in addition to improved, selective toxicity towards bacterial cells only.19–21 Stem cell therapies face limitations to efficacy based on challenges in preparation and delivery. The intensively studied silver nanoparticles face challenges on repeated and prolonged use due to tissue accumulation and toxicity.22,24 Whilst investigated bioactives show benefits for aspects of wound bed pathophysiology issues such as manufacturing complexity, cost, stability, regulatory requirements, sensitivity to degradation, and potential destruction by the immune system remain.5 In addition, methods by which bioactive agents can be protected from the harsh wound bed environment for controlled and sustained efficacy remain an open challenge.
Ti3C2Tx MXenes are inflammation-repressive nanomaterials,25,26 and have been used as sensor materials for dermal applications and within dressings to improve wound healing.6 Ti3C2Tx is synthesised through selective chemical etching of the Ti3AlC2, MAX phase, precursor and follows the general formula, Mn+1XnTx, where M represents the early transition metal, titanium, X is carbon, and T refers to surface functional groups, typically –O, –OH, or –F. Useful properties in a biomedical context include biocompatibility, flexibility, metallic electrical conductivity, hydrophilicity, and tuneable surface chemistry, making these highly versatile materials.25,27,28 Extensive reviews have highlighted the potential of MXenes in the wound dressing field and the need for refinement for optimisation of current approaches.29 Here, we propose their use in combination with an encapsulated osmolyte to anchor the hydrophilic osmolyte to Ti3C2Tx and the wound dressing for slow and sustained elution over time into the wound bed.
Osmolytes are part of the osmo-adaptive response, which exists physiologically to control tissue osmolarity and maintain an osmotic environment conducive to normal cell function.30 Among the osmolytes endogenous to mammals, glycine betaine (GB) is the most well characterised as the primary osmoprotectant utilised to restore osmotic balance in key tissues including those of the kidneys, heart, liver, and brain.31–36 It is synthesised endogenously in humans via oxidation of choline in addition to exogenous dietary sources.37 Topical application of GB, at concentrations of 2–200 mM, has been shown to increase resistance to osmotic stress in corneal tissue in addition to modulation of epidermal tight junctional barrier function for subsequently improved skin wound repair.38–41 On exposure to a hyperosmotic environment, osmolytes are synthesised and taken up by cells to protect protein and DNA function, stabilise the intracellular environment, and prevent water efflux.42 However, pathological variations in osmotic control result in cell shrinkage through dehydration, cytoskeletal damage, protein and DNA disruption, cell cycle arrest, and apoptosis.42 Such shifts in osmotic control can occur within the wound bed microenvironment, particularly in chronic conditions such as diabetes, where raised glucose levels alter cell capacity for osmo-adaptive control and a normal wound healing response.2,11 The goal of the current study is, therefore, to improve cell resilience and address osmotic imbalance for resolution of the chronic non-healing wound using a microencapsulated osmoprotectant/Ti3C2Tx combination materials approach. It is hypothesised that Ti3C2Tx, in combination with a well-studied osmoprotectant GB, will significantly improve sustained wound repair characteristics, repress hyper-inflammatory stimulus and improve the potential for wound closure.
2. Experimental
2.1. Synthesis and spray-coating of Ti3C2Tx
Ti3C2Tx was synthesised using the minimally intensive layer delamination (MILD) method,43 etching MAX phase (Ti3AlC2; synthesis conditions described in previous literature44) and delaminating to yield single flake Ti3C2Tx. Continuous stirring of 9 M hydrochloric acid (20 mL) (Sigma Aldrich, UK), 1.6 g lithium fluoride (Sigma Aldrich, UK), and 1 g Ti3AlC2 at 35–40 °C was carried out for 24 h. The synthesised sediment was resuspended in 50 mL deionised (DI) water and centrifuged (ALC PK 120R Centrifuge, DJB Labcare Ltd, UK) at 2191g for 5 min. The sediment was resuspended, and the centrifugation/wash step was repeated until pH 6 was achieved. The sediment was stirred with 1 g lithium chloride (Sigma Aldrich, UK), for 4 h to improve delamination yield. The centrifugation/wash step was repeated to achieve pH 6 again, and the supernatant was collected. The supernatant was then centrifuged further at a higher speed of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 10 min to isolate the delaminated Ti3C2Tx flakes. The resultant sediment from the final centrifugation step was resuspended in DI water and stored at −20 °C before use.
000g for 10 min to isolate the delaminated Ti3C2Tx flakes. The resultant sediment from the final centrifugation step was resuspended in DI water and stored at −20 °C before use.
A 12-well tissue culture plate was spray-coated with Ti3C2Tx at a concentration of 5 mg mL−1. The tissue culture plate was mounted on a clamp stand, and the spray gun nozzle was 15 cm away from the plate. The plate was covered with parafilm, only exposing the relevant wells for spray-coating of the appropriate amount per well.
2.2. Characterisation of Ti3C2Tx and Ti3C2Tx coatings
:16, 1:8, 1:4 to 1:2 were prepared. Absorbance was measured by UV-Vis spectrometry (UV 2401-PC, SHIMADZU Ltd, UK) using a wavelength range of 200 to 1000 nm and an optical path length of 1 cm.2.3. Assessing the osmoprotective effects of GB-Ti3C2Tx using an in vitro HaCaT model of wound repair
 | (1) |
In this equation, M represents the maximum potential closure percentage, while the coefficient α corresponds to the reference size from the curve fit, ensuring dimensional consistency. The variable t denotes time in hours, tracking the progression of wound closure. Parameters b and c influence the closure rate, with b affecting the initial exponential increase and c controlling the hyperbolic sine function asinh(t). The modified wound closure model starts with a rapid phase driven by the exponential term, reflecting the fast initial healing after a wound is created. As time progresses, the influence of the exponential term diminishes, and the hyperbolic asinh(t) becomes more prominent, indicating a slowing closure rate as the wound nears full closure. This combination of exponential decay and hyperbolic sine functions provides the model with the flexibility to accurately fit diverse wound closure data. Adjusting the parameters allows the model to capture the non-linear, time-dependent nature of the healing process. The equation was fitted to the experimental data using a MATLAB 2023 cftool inbuilt function.
To calculate cell migration velocity, time-lapse microscopy experiments were conducted using a CytoSMART Lux2 inverted microscope to track cell migration across the scratch in one field of view. Images were captured at 2 h intervals over 48 h, and velocity calculations were performed using a custom MATLAB code developed in-house (ESI Fig. 1†). Edge detection algorithms in MATLAB were used to generate contours and precisely trace the boundary between the migrating cells and the cell-free zone. Accurate identification of these edges was used to quantify cell migration velocity by tracking the movement of the cell fronts over time. An example scratch assay micrograph with a red contour trace to delineate the scratch cell edge margins is shown in ESI Fig. 2.† The central cleared area represents the wounded region within the confluent cell monolayer, with surrounding cells migrating into this space over time. The red lines superimposed on the image mark the detected edges of the migrating cells.
000 events per sample at a medium flow rate. Cells were gated to exclude debris, and compensation settings were applied using the appropriate unstained and single-stained controls to account for spectral overlap between the FITC and PI stains. The inbuilt CytExpert software was used for data analysis.2.4. Synthesis and testing of a Ti3C2Tx-tBu-GB-microgel wound dressing
:1 weight ratio. 1.5 mL of this monomer mixture was added dropwise to 20 mL of a vigorously stirred deionised (DI) water solution containing the initiator, ammonium persulfate (2.0% w/v). 150 μL of tetramethylethylenediamine (TEMED) (Fisher Scientific, UK) were then added to the resulting coarse emulsion to catalyse the generation of radical species from APS, enabling the polymerisation of AA and EGDMA to progress at room temperature. After mixing for 15 min under high shear, poly(acrylic acid-co-EGDMA) was isolated by filtration, washed three times each with acetone, DI water, and methanol and dried.The acrylate-based microgels were then loaded with tBu-GB by a simple soak-swell-rinse-dry method designed to saturate the gel with tBu-GB through electrostatic interaction. Briefly, 0.2 g of the dry gel was stirred in 2.5 mL DI water containing 0.5 g tBu-GB for 5 min to initiate electrostatic pairing of negatively charged acrylate and positively charged betaine ester, i.e. [tBu-GB]+/[PAA-co-EGDMA]−. Excess water and unbound solutes were removed by filtration, and the solids were allowed to air dry. The loading procedure was repeated three times to saturate the gels with tBu-GB. Approximately 10 mg of plain polyacrylate microgels and tBu-GB loaded polyacrylate microgels were dispersed in 1 mL DI water and were analysed for changes in size and polydispersity using Dynamic Light Scattering (DLS) (Mastersizer 3000, Malvern, UK). A suspension prepared in the same way was imaged using light microscopy. tBu-GB loading into the polyacrylate based microgels was confirmed by FTIR.
LC-MS analysis was conducted using a Burker HCT Ultra Electrospray ionisation/ion trap mass spectrometer (Bruker, USA) interfaced with an Agilent 1200 series higher performance liquid chromatograph (HPLC) (Agilent Technologies, USA). The stationary phase was a Kinetex silica 100 × 3.0 mm hydrophilic interaction chromatography (HILIC) column (Phenomenex, USA). An isocratic elution was performed with a mobile phase comprising 65% v/v acetonitrile, 17% v/v water, 17% v/v methanol, 1% v/v acetic acid, and 3.9 mg mL−1 ammonium acetate. All LC-MS solvents and additives were purchased from Fisher Scientific, UK. Extracted ion count (EIC) chromatograms were used for data analysis; the target ion, 140 m/z, was the most abundant for GB (corresponding to the sodium adduct of GB: [117GB·23Na+]). The molecular ion corresponding to tBu-GB, 174 m/z, was used to generate the EIC chromatograms for tBu-GB.
The tBu-GB microgels and Ti3C2Tx-tBu-GB microgel dressings were UV-sterilised. Samples were incubated at 37 °C for 28 h on an orbital shaker (SI50 Orbital Incubator Platform Shaker, Stuart Scientific) in 1 mL media, adjusted to 300, 550 or 650 mOsM, prior to collection of the conditioned media. HaCaT cells were seeded at 5 × 104 cells per well in a 48-well plate and incubated at 37 °C in 5% CO2 for 24 h. 500 μL of the conditioned media for each sample type (cell only, tBu-GB microgel, Ti3C2Tx-tBu-GB microgel dressing) was then transferred to the relevant well and plates were incubated for 24 h at 37 °C in 5% CO2. The MTS assay was then carried out as described in 2.3.4.
2.5. Statistical analysis
Data was analysed using GraphPad Prism version 8.0.1 (GraphPad Software, San Diego, CA). Significance was determined using a two-way analysis of variance (ANOVA) and a Tukey's multiple comparison post hoc test with a significance value set at p < 0.05. All data was expressed as mean ± standard error of the mean (SEM) for 3 experimental repeats unless otherwise stated. First-order curve fitting was performed in MATLAB 2021 using the Curve Fitting Tool, allowing visualization of 95% confidence bounds and extraction of key metrics such as coefficient values, R2 and RMSE.3. Results and discussion
3.1. Characterisation of Ti3C2Tx and Ti3C2Tx coatings
UV-Vis spectroscopy was used to measure the absorbance of dilute colloidal Ti3C2Tx solutions, as shown in Fig. 1(a). A broad peak was detected in the 700–800 nm range with a peak maximum at 766 nm. Following Beer-Lambert's law, a calibration curve was constructed (ESI Fig. 4†), leading to the calculation of an extinction coefficient of 29.78 mL mg−1 cm−1, comparable to values reported in the literature.28 DLS was used to estimate the average hydrodynamic size distribution of the Ti3C2Tx colloidal solutions (Fig. 1(b)). Two peaks were identified: a major peak, which accounted for >97% of the distribution, with an average size of 260.6 nm, and a minor peak representing 3% of the distribution, estimated to be 51.3 nm, with an average polydispersity index of 0.22. The XRD pattern of Ti3AlC2 exhibited sharp, well-defined peaks (Fig. 1(c)). In contrast, on etching away the Al layer to form Ti3C2Tx, the XRD pattern showed a significant shift of the (002) peak due to increased interlayer spacing, as well as a disappearance of the higher ordered peaks. These changes demonstrate the successful topochemical transformation from bulk Ti3AlC2, MAX phase, to the 2D Ti3C2Tx flakes.46 The SEM images of the Ti3C2Tx spray-coated well plate indicated a 1–2 μm thick uniformly distributed surface layer (ESI Fig. 4†).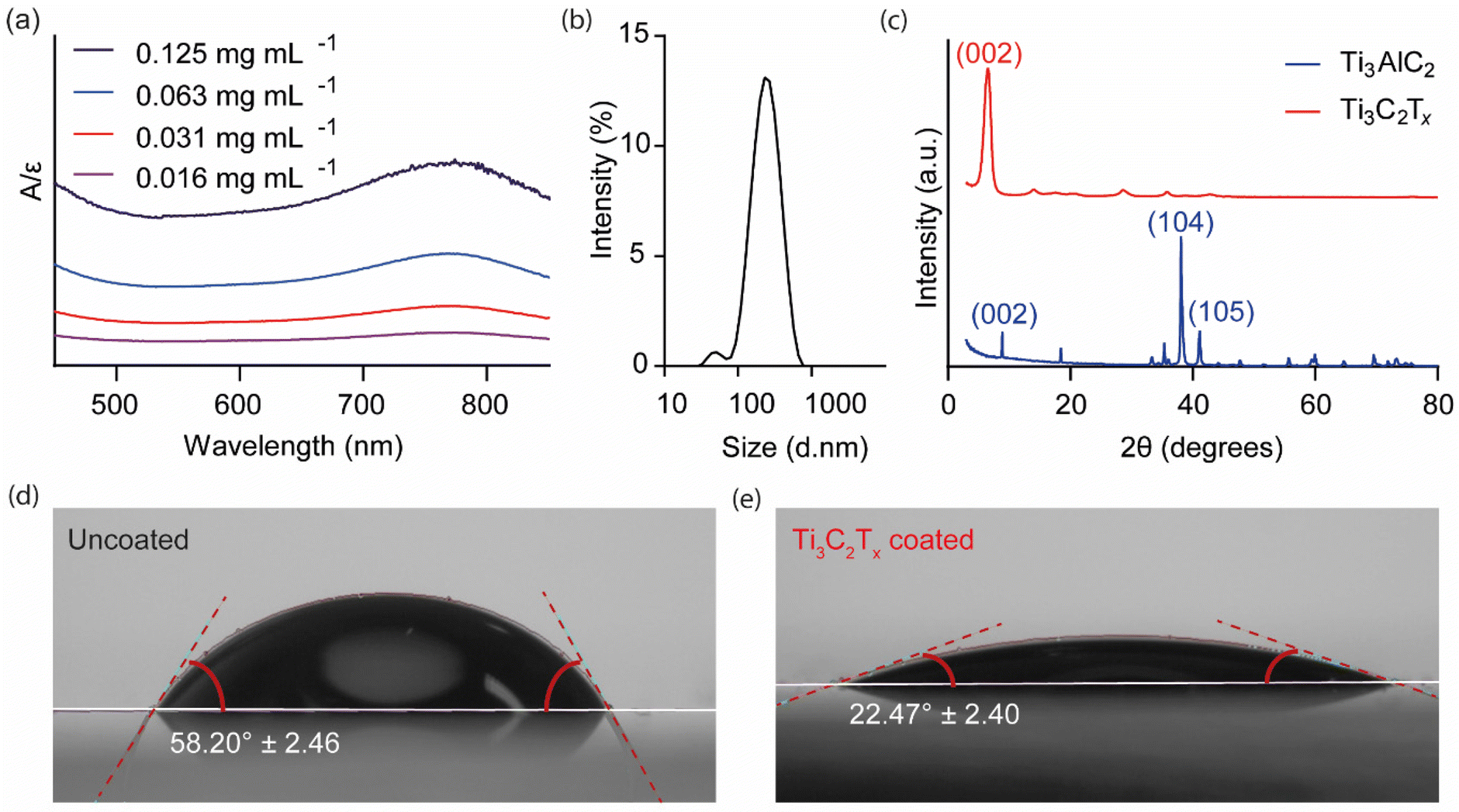 | ||
| Fig. 1 Physical characterisation of Ti3C2Tx. (a) UV-Vis spectroscopy of Ti3C2Tx solutions of varying concentrations normalised to the molar extinction coefficient (ε = 29.78 L g−1 cm−1) calculated from the calibration curve (ESI Fig. 5†). (b) Dynamic light scattering was used to determine the particle size and distribution of the Ti3C2Tx solution. (c) X-ray diffraction comparing the crystal structure of Ti3AlC2 to the synthesised and vacuum-filtered Ti3C2Tx film (Cu Kα radiation source (λ = 1.5406 Å) at 40 kV and 15 nA beam current). (d) The contact angle of the uncoated (58.20 ± 2.46°) and (e) Ti3C2Tx coated (22.47 ± 2.40°) glass slide surface. | ||
The hydrophilicity of Ti3C2Tx as a coating, when spray-coated onto a solid support, was investigated using contact angle analysis (Fig. 1(d and e)). The presence of the coating significantly reduced the contact angle from 58.20° ± 2.46 to 22.47° ± 2.40, indicating enhanced wettability and suitability for a smart wound dressing application. Hydrophilic materials enhance the ability of dressings to absorb and retain moisture and solutes away from the wound bed, including inflammogenic proteins, which may delay the wound healing process,26 facilitating the efficient loading and sustained release of hydrophilic drugs.27 Hydrophobic coatings, on the other hand, are less suitable for wound dressing applications as, for example, they reduce the ability of the substrate material to absorb wound exudate.
3.2. Assessing the osmoprotective effects of GB-Ti3C2Tx using an in vitro HaCaT model of wound repair
 | ||
| Fig. 2 Cell migration scratch assay results indicating a significant reduction in keratocyte migratory capacity under mannitol-induced, hyperosmotic stress conditions and a significant protective effect for cells grown on Ti3C2Tx coatings to more rapidly close the scratch, (a) normalised wound closure (WC) over time, comparing keratocyte scratch closure under iso-osmotic conditions to scratch closure under hyperosmolar conditions in the presence and absence of Ti3C2Tx coatings and GB. Across all cases, the curve fit yielded an R2 value >0.99. (b) Mean migration velocity (MV) of cells over time under normal iso-osmolarity (cell-only), mannitol-induced hyperosmolarity (mannitol), and mannitol-induced hyperosmolarity with GB (mannitol GB) in uncoated and (c) Ti3C2Tx coated conditions, (n = 3, mean ± SD). | ||
The rate coefficient data also confirms the positive impact of Ti3C2Tx on wound closure (ESI Tables 2 and 3†). Coefficient b values, representing the initial migration rate, were consistently higher for the Ti3C2Tx groups compared to the cell-only, cells with mannitol, and cells with mannitol plus GB groups. Coefficient c values, modulating the impact of the hyperbolic sine term in the model and representing the later stages of wound closure, were lower for the Ti3C2Tx group compared to the mannitol and mannitol GB group in late-phase keratocyte closure of the wound. The lower value of coefficient c reflects a slower transition in the middle phase but enables rapid closure at later time points due to less influence from the hyperbolic term. It may be that Ti3C2Tx is less influential in the mid to late stages of migration. Alternatively, the removal of Ti3C2Tx between the cell margins on the application of the scratch may reduce the impact of Ti3C2Tx in mid to late-phase migration.
The positive impact of Ti3C2Tx and GB of keratocyte migration was confirmed by the keratocyte velocity data. Time-lapse microscopy imaging and a custom MATLAB code were used to determine the velocity of the wound edge keratocytes as they migrated to close the scratch (ESI Fig. 2†). Fig. 2(b) and (c) show the mean migration velocity (MV) of cells over time without and with a Ti3C2Tx coating, respectively, and indicate a significant reduction in initial keratocyte MV under mannitol-induced osmotic stress conditions, an increase in keratocyte MV for keratocytes in contact with Ti3C2Tx coatings under iso-osmotic and hyperosmotic conditions. They also show a further increase in keratocyte MV in combined GB and Ti3C2Tx coated conditions. Under iso-osmotic cell-only conditions, keratocyte MV remained relatively high (∼0.018 μm s−1) until wound closure by 24 h. Iso-osmotic Ti3C2Tx coated keratocyte MV was initially rapid (∼0.022 μm s−1), declining at 8 h until wound closure by 24 h. For mannitol-treated, osmotically stressed cells initial MV was much slower (∼0.014 μm s−1), with no improvement or closure of the scratch wound by 48 h. In contrast, keratocytes cultured on Ti3C2Tx coatings under mannitol conditions showed a rapid MV (∼0.018 μm s−1), which slowed at 8 h to close the wound by 36 h. GB mannitol also induced an initial rapid MV (∼0.017 μm s−1) with a gradual decline to close the wound at 48 h. GB mannitol in the presence of a Ti3C2Tx coating induced an initial rapid MV (∼0.021 μm s−1) to close the wound by 36 h and more rapidly than in the GB mannitol alone, indicating the protective effect of Ti3C2Tx and GB combined (ESI Table 4†).
Whilst normal wound repair relies on a more complex series of signalling cues involving multiple cell types, in vitro modelling using a single cell type scratch assay allowed the impact of individual variables on keratocyte behaviour to be more easily defined. Mechanisms by which these variables affect scratch closure can then be considered in more detail. Under normal conditions epidermal keratocyte wound repair is typically dependent on the inherent regenerative abilities of both a proliferative keratocyte fraction and the cut edge keratocytes. These cells respond to signalling cues and change their cytoskeletal network to migrate across a scratch and gradually close the wound.48 Hyperosmotic stress provokes an altered cell repair response, including cell shrinkage, protein and DNA damage, and changes to cytoskeletal structure, all of which will limit migratory capacity. Ti3C2Tx has a unique combination of properties to mitigate the upregulation of damage-associated pathways in keratocytes. These include high electrical conductivity, small solute intercalation properties, hydrophilicity, and a reactive surface chemistry, all of which are likely to contribute to a favourable environment for cellular activities necessary for wound healing. For example, Ti3C2Tx may augment the impact of intracellular transient electrical behaviour arising from changes in molecular machinery, such as microfibrillar rearrangement to more rapidly mediate cell migration and wound closure. The presence of a naturally occurring, water-soluble, osmo-adaptive mediator, such as GB, may also benefit the disrupted wound bed environment. GB resists the effects of osmotic stress by stabilising cell volume, protein structure, enzyme activity, inflammatory upregulation, and other damaging effects of osmotic shock. Pre-incubation of keratocytes with GB stabilised the intracellular environment, allowing retention of migratory response and scratch repair despite external hyperosmotic stress conditions. Given the combined, synergistic effects of Ti3C2Tx and GB on improved wound closure rate, subsequent experiments sought to determine the mechanisms by which they may act for therapeutic benefit to reverse the negative impact of osmotic stress on aspects of the keratocyte wound repair response.
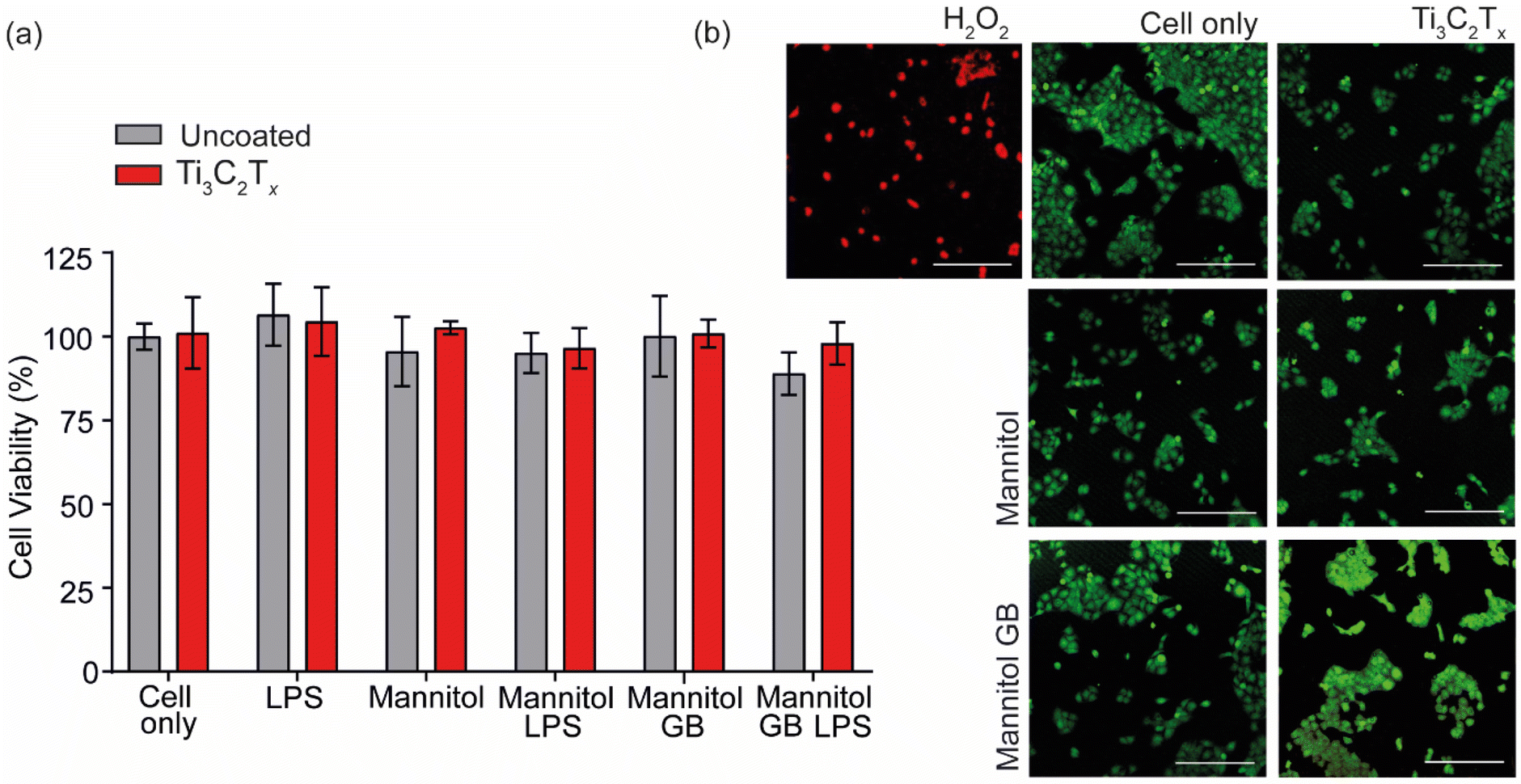 | ||
| Fig. 3 (a) Cell viability assay indicating no significant cytotoxic effects on keratocytes exposed to all conditions (cell-only, LPS, mannitol (550 mOsM), mannitol and LPS, mannitol and GB (10 mM), mannitol, GB and LPS) in the presence and absence of Ti3C2Tx, coatings (n = 3, mean ± SEM, p < 0.05, scale = 250 μm). (b) representative confocal images of live/dead stained keratocytes 24 h after treatment with each condition in comparison to a cytotoxic H2O2 (4 mM) control. | ||
 | ||
| Fig. 4 Repression of HaCaT keratocyte inflammatory response to osmotic stress following Ti3C2Tx GB treatment. (a) ELISA IL-6 production of keratocytes treated with Ti3C2Tx, LPS, mannitol (550 mOsM), GB (10 mM). (b) qRT-PCR IL-6 production of keratocytes exposed to the same treatments detailed above. (c) qRT-PCR IL-1α. (d) qRT-PCR IL-1β. (e) qRT-PCR CXCL1. (f) qRT-PCR CXCL8. Significance was determined using a two-way ANOVA and post-hoc Tukey's multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) (mean ± SEM, n = 3). | ||
The results of the qPCR and ELISA support the results of the keratocyte scratch closure assay, demonstrating that both Ti3C2Tx and GB are potent anti-inflammatory agents, but moreover, that the combination of the two exerts a synergistic effect which has a greater effect than either treatment alone. Indeed, it is well-known that GB stabilises cell volume and protein structure in plant, animal, and bacterial cells exposed to osmotic, cryogenic, or desiccant stress,50–55 and that its presence has striking effects on the downregulation of proinflammatory cytokines in, for example, osmotically stressed ocular and hepatic cells.34,38,56–58 Ti3C2Tx, while emerging only relatively recently compared to GB, is known to possess potent anti-inflammatory properties, with studies reporting its ability to reduce proinflammatory cytokine expression in LPS-stressed cells.25,26 Ti3C2Tx is also known to have antioxidant properties that could reduce oxidative stress at the wound site,59 providing an environment that is more conducive to rapid healing.
 | ||
| Fig. 5 Ti3C2Tx coatings protect against osmotic and H2O2 stress induced HaCaT cell apoptosis. (a) Flow cytometry data indicating viable and apoptotic cell fraction under conditions of iso-osmotic cell only and cell-Ti3C2Tx controls, 550 mOsM cell only, GB, Ti3C2Tx and GB-Ti3C2Tx conditions, 650 mOsM cell only, GB, Ti3C2Tx and GB-Ti3C2Tx conditions with viable cell fractions shown in green and combined early and late-stage apoptotic cell fractions shown in red. (b) Representative dot plots of the flow cytometry data shown in a. (c) Flow cytometry data demonstrating oxidative stress induced apoptosis using 4 mM H2O2 and the protective effect of Ti3C2Tx, comparing conditions of cells with H2O2 GB, H2O2 Ti3C2Tx and H2O2 GB-Ti3C2Tx to an H2O2 cell only control. (d) Representative dot plots of the flow cytometry data shown in c. Significance was determined using a two-way ANOVA and post-hoc Tukey's multiple comparison test (Viable cell fraction, ***p < 0.001, ****p < 0.0001) (Apoptotic cell fraction, ###p < 0.001, ####p < 0.0001) (mean ± SEM; n = 3). | ||
 | ||
| Fig. 6 Confocal micrographs showing actin cytoskeletal structure of HaCaT keratocytes following 4 hour exposure to mannitol and Ti3C2Tx. Actin cytoskeletal structure was stained using TRITC conjugated rhodamine phalloidin (red), and nuclei were counterstained using DAPI (blue). Scale bar = 100 μm. | ||
It was hypothesised that forming a cationic ester of GB with a short-chain alcohol and embedding this within a negatively charged reservoir/platform material for incorporation with Ti3C2Tx into a wound dressing would enhance the former's release lifetime through a combination of two main mechanisms. Firstly, GB release from the ester would be controlled by a relatively slow chemical reaction (hydrolysis) rather than solely by (relatively rapid) diffusion. Secondly, the intact, cationic ester could be retained by electrostatic interaction at anionic pendant groups in a reservoir/platform material with an overall negative charge. A GB-releasing system was therefore designed based on tBu-GB for use in combination with a negatively charged acrylic acid-based microgel, as shown in Fig. 7(a).
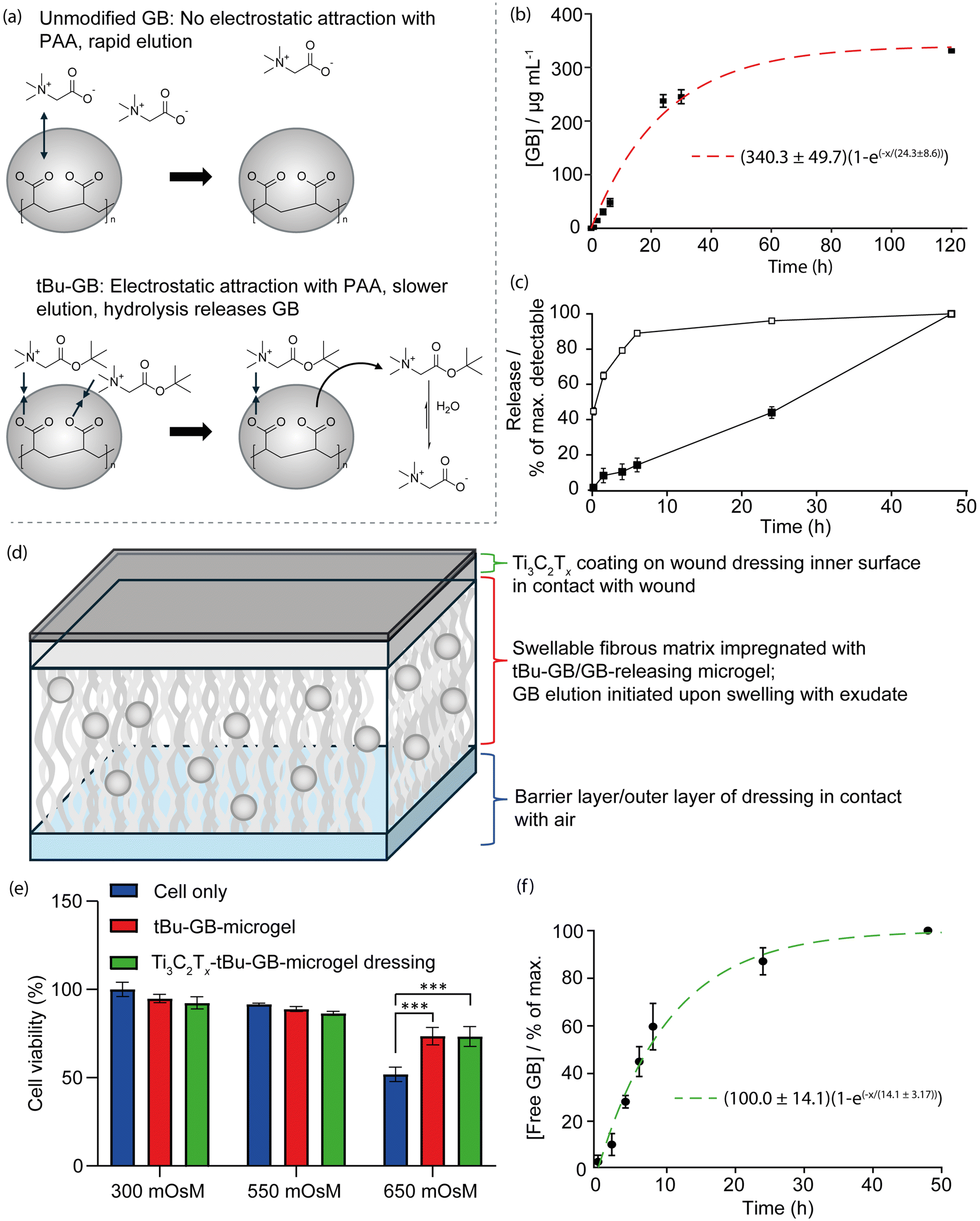 | ||
| Fig. 7 Synthesis and testing of a Ti3C2Tx-tBu-GB-microgel wound dressing for slow elution of GB from a tBu-GB impregnated microgel within the dressing over time. (a) Illustration of the GB release mechanism from the acrylate-based PAA microgels. (b) Best fit curve plot showing first-order release of GB from a 2.5 mg mL−1 solution of tBu-GB at pH 7.2 in 32 °C PBS, as absolute concentration μg mL−1. (c) Release of free GB (black squares) and tBu-GB (white squares) from [tBu-GB]+/[PAA-co-EGDMA]- microgels in PBS at pH 7.2 at 32 °C, as concentration relative to the maximum concentration detected after 48 h. (d) Schematic of the cross-section of the bioactive wound dressing employing both Ti3C2Tx layer and GB elution technologies to enhance the wound healing process. (e) MTS cytotoxicity plot for HaCaT cells incubated with tBu-GB/acrylate microgel-loaded, Ti3C2Tx-coated dressing under isotonic and hyperosmotic conditions (550 and 650 mOsM) (n = 3, mean ± SEM, ***p < 0.001) (f) Best fit curve plot showing first-order elution of GB from the Ti3C2Tx-tBu-GB-microgel wound dressing over time. | ||
A successful synthesis of tBu-GB was confirmed by mass spectrometry (ESI Fig. 6†) and FTIR (ESI Fig. 7†). Hydrolytic conversion of tBu-GB into GB was confirmed by repeated sampling and LC-MS analysis of the same 2.5 mg mL−1 solution of tBu-GB, in PBS, at 32 °C and pH 7.2 over 5 days. After 5 days, the LC-MS peak corresponding to tBu-GB was not evident, while that corresponding to GB dominated the spectrum, confirming the conversion of tBu-GB to GB in aqueous surroundings. The release profile of GB from tBu-GB in this solution is shown in Fig. 7(b) and is approximately linear over the initial 24 h (during which 70% of the total measured GB is evolved), before entering a lag phase between 30 h and 120 h. A total of 330 μg mL−1 GB was released after 5 days. This is ca. 85% of the total releasable GB, recovered after degradation of a separate aliquot of the same 2.5 mg mL−1 stock, which was degraded into Na·GB and tert-butyl alcohol by exposure to highly alkaline conditions after spiking with 0.25 M NaOH. The release curve was fitted to a first order kinetic plot with curve in the form M(1 − e(−x/T)). Here, M = 340.3 ± 49.7 M μg mL−1 represents the asymptotic maximum concentration of GB released, and T = 1.01 ± 0.36 is the time constant representing the rate at which the release approaches saturation.
An AA/EGDMA-based microgel was selected as the base material for the entrapment of the positively charged tBu-GB. Polymerisation of the AA/EGDMA monomer combination is known to produce highly water-swellable, porous, and strongly negatively charged structures which can retain small, positively charged molecules, and even larger molecules such as proteins.62 The polymerisation method was based on the works of Tuncel et al., who studied the development of non-swellable and swellable AA/EGDMA microspheres by altering synthetic parameters,63 and Hoare et al., whose study detailed the impact of ionisable group distribution in microgels in the delivery of ionic drugs.64 Due to the small molecular cross-section of tBu-GB, a lower degree of swelling is more desirable in this case, as it can be expected to impact rate of elution via diffusion inhibition;65 hence, a high cross-linker ratio of 2:1 AA:EGDMA was selected. Nonetheless, the retention and elution of the GB precursor, tBu-GB, is expected to be primarily driven by the electrostatic interaction between negatively charged COO− and positively charged –N+Me3 groups in the respective materials.
The prepared [tBu-GB]+/[PAA-co-EGDMA]− microgels presented as a fine powder once dried, with an average particulate size of 11.9 μm and an asymmetrical, aspherical morphology, forming aggregates and sediments readily in unstirred solution (ESI Fig. 8†). As shown in Fig. 7(c), submerging the [tBu-GB]+/[PAA-co-EGDMA]− microgels in aqueous surroundings immediately initiated the elution of both free GB and tBu-GB. The release of GB was approximately linear over the 48 h study period (2.0% h−1, R2 = 0.995). This is a desirable feature in any drug or therapeutic molecule-releasing system, as it enables accurate dose estimation at any given time following initial administration. Additionally, the persistent release of new GB and tBu-GB, despite the repeated removal and replenishment of aqueous supernatant, indicates that the cross-linked acrylate gels are effective in entrapping tBu-GB and GB. Interestingly, the release of GB from the gels continues for at least 24 h after no new release of tBu-GB is evident. tBu-GB release is 89% complete within 6 h, yet significant GB release continues between 24–48 h. This may indicate that significant quantities of unreleased tBu-GB remain bound to the gel and do not enter the solution, or hydrolysis of tBu-GB occurs predominantly in/at the gels rather than the aqueous surroundings.
The scratch assay data indicated that the presence of Ti3C2Tx and GB at 10 mM in a simulated osmotically challenged non-healing scratch wound increased the scratch closure rate such that the scratch reached 100% closure within 48 h. Thus, from the release data above, the release duration of GB from the tBu-GB-loaded microgels appears suitable for promoting scratch wound closure under osmotically challenged conditions. Over the 48 h study period, 1.75 μg of GB and 31.25 μg tBu-GB were released per mg of microgel material. Assuming 31.25 μg tBu-GB evolves ca. 24.9 μg GB (as indicated previously), the total releasable GB is ca. 26 μg mg−1. Based on the approximately linear 2.0% h−1 release gradient, this is equal to 0.5 μg mg−1 h−1. It is likely that the total loading and release profiles can be adjusted by varying the ratio of AA to EGDMA in the system.66,67
Following demonstration of a successful encapsulation mechanism by which bioactive osmolyte GB could be incorporated into a Ti3C2Tx dressing a Ti3C2Tx-tBu-GB-microgel dressing was fabricated and tested for biocompatibility and efficacy. The wound dressing was prepared by dispersing dry microgels of ca. 10 μm in size in the fibrous absorbent inner layers of the dressing material, designed to absorb and swell with retained wound exudate. The simple and efficient spray-coating method developed in this work, was used to coat the wound-facing surface of the dressing, shown in Fig. 7(d). The therapeutic Ti3C2Tx layer would then remain in contact with the wound bed, and the absorption of exudate by the dressing would result in the swelling of the embedded microgels, slowly releasing GB into the wound bed by the mechanisms described in section 3.3. FTIR spectral analysis of the microgels with and without tBu-GB loading in addition to the cellulose based commercial dressing with and without tBu-GB microgel loading and an outer Ti3C2Tx coating indicated successful incorporation of each component within the final dressing and is described in ESI Fig. 9.†
The fabricated Ti3C2Tx-tBu-GB-microgel dressing was assessed for keratocyte cytotoxicity, protective effects against hyperosmotic stress and slow elution of GB over time. Using a 24 h conditioned media study, it was found that the tBu-GB microgel alone and within the Ti3C2Tx-tBu-GB-microgel dressing, had no effect on cell viability indicating its biocompatibility. This was in agreement with the direct contact cell viability data shown in Fig. 3a for the GB and Ti3C2Tx components (Fig. 7(e)). In addition, under hyperosmotic (650 mOsM) conditions, sufficient to significantly reduce cell viability, the tBu-GB-microgel and Ti3C2Tx-tBu-GB-microgel dressing conditioned media, provided a protective effect, significantly increasing cell viability compared to that of the cell only control. This indicated that slow elution of GB from both the tBu-GB-microgel alone and the Ti3C2Tx-tBu-GB-microgel dressing into the media was sufficient to then protect the HaCaT cells from osmotic stress induced cell death. The result was confirmed by a slow elution study from the Ti3C2Tx-tBu-GB-microgel dressing over time indicating the release of free GB from the dressing over a period of 48 h (Fig. 7(f)). A single-term exponential saturation model, given by M(1 − e(−x/T)), was fitted to the release curve as shown in Fig. 7f, and based on an R2 value of 0.9797, this model effectively captures the release kinetics of GB from the microgel dressing. The parameter M represents the total amount of GB that can be released over time, corresponding to the final plateau value in the plot. The coefficient T defines the characteristic time required to reach approximately 63% of the total release. Beyond this point, the release rate slows down, asymptotically approaching 100%, which represents the final equilibrium amount of GB released. A higher T value suggests a more prolonged release due to slower diffusion, whereas a lower T value indicates a faster elution process.
This work aims to engineer a wound dressing that exerts the synergistic therapeutic and anti-inflammatory properties of GB-Ti3C2Tx to alleviate hyperosmotic stress and promote a transition from persistent hyper-inflammation to wound healing. This bio-interactive dressing approach also provides the potential for a smart dressing design that would enable release by pathologically raised triggers within the chronic wound bed. There are other examples of this approach within the drug delivery literature, exploiting the body's own interactions with polymer and liposome surface biomolecule conjugates to initiate targeted drug release at specific sites.68–70
4. Conclusion
In this study, Ti3C2Tx was investigated for use in a bioactive device designed to mitigate the mechanisms active within the chronic wound bed, which lead to poor wound repair. Ti3C2Tx was combined with a bioactive osmoprotectant to specifically consider the potential of these materials to protect against the damaging effects of hyperosmotic stress. In the context of wound repair, hyperosmotic stress is an underexplored but potentially significant contributor to inflammation and infection-associated delays in the activation of cellular repair pathways. Whilst normal osmo-adaptive mechanisms exist to regulate any osmotic flux within physiological limits, chronic shifts linked to systemic disease can disrupt this balance to provoke damage to the wound bed environment and prevent wound closure. This study showed that the ability of dermal keratocytes to close a scratch wound was prevented under mannitol-induced hyperosmotic stress conditions and that Ti3C2Tx could reverse this effect to allow wound closure. Ti3C2Tx coatings significantly increased the rate and velocity of keratocyte migration to close the scratch wound. This protective effect was increased by including osmoprotectant GB, demonstrating a synergistic effect of the Ti3C2Tx coating with this bioactive molecule. The same effect was demonstrated on measuring inflammatory pathway upregulation by migratory keratocytes. Hyperosmotic stress-induced upregulation of IL-6, IL-1α, IL-1β, CXCL1, and CXCL8, and this effect was primed by pre-incubation of keratocytes with bacterial LPS to further increase inflammatory cytokine and chemokine upregulation. Ti3C2Tx was able to significantly reduce the production of these inflammatory markers towards that of the non-stressed, cell-only control in the presence and absence of bacterial LPS. The effect was augmented by the presence of GB, again suggesting a synergistic role for these materials. Ti3C2Tx demonstrated a protective effect towards apoptotic stimuli and oxidative stressors, in addition to preventing osmotic stress induced cytoskeletal changes. The study also presents a slow elution mechanism by which GB may be released into the wound bed within a bioactive Ti3C2Tx dressing in a tissue engineering approach to repress inflammation and oxidative stress and improve migratory stimulus for chronic wound repair.Author contributions
SK, EJW, DH, AS and SS were responsible for the conceptualisation, implementation, planning, writing and revision of the paper. SK, DH and AS carried out the experimental and mathematical modelling work. SS, DH, AS, DS and CC supervised the work of SK. GC, CC, AL, CS and YG provided conceptual input, critical feedback and comments for draft writing and improvement.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
University of Brighton authors acknowledge the EPSRC grant MyDress (EP/W023164/1). XRD was carried out by Prof. Martin Smith at the University of Brighton.References
- J. F. Guest, G. W. Fuller and P. Vowden, BMJ Open, 2020, 10, e045253 CrossRef PubMed.
- H. Deng, B. Li, Q. Shen, C. Zhang, L. Kuang, R. Chen, S. Wang, Z. Ma and G. Li, J. Diabetes, 2023, 15, 299–312 CrossRef PubMed.
- M. J. Portou, R. Yu, D. Baker, S. Xu, D. Abraham and J. Tsui, Eur. J. Vasc. Endovasc. Surg., 2020, 59, 117–127 CrossRef PubMed.
- Q. Pang, F. Yang, Z. Jiang, K. Wu, R. Hou and Y. Zhu, Mater. Des., 2023, 229, 111917 CrossRef CAS.
- P. Alves, C. Barrias, P. Gomes and M. Martins, Mater. Today Chem., 2021, 22, 100623 CrossRef CAS.
- Y. Hu, F. Wang, H. Ye, J. Jiang, S. Li, B. Dai, J. Li, J. Yang, X. Song and J. Zhang, npj Flexible Electron., 2024, 8, 30 CrossRef CAS.
- E. Bachar-Wikstrom, M. Manchanda, R. Bansal, M. Karlsson, P. Kelly-Pettersson, O. Sköldenberg and J. D. Wikstrom, Int. Wound J., 2021, 18, 49–61 CrossRef PubMed.
- S. Akita, Int. J. Mol. Sci., 2019, 20, 6328 CrossRef PubMed.
- H. N. Wilkinson and M. J. Hardman, Open Biol., 2020, 10, 200223 CrossRef CAS PubMed.
- P. Rousselle, F. Braye and G. Dayan, Adv. Drug Delivery Rev., 2019, 146, 344–365 CrossRef CAS PubMed.
- U. A. Okonkwo and L. A. DiPietro, Int. J. Mol. Sci., 2017, 18, 1419 CrossRef PubMed.
- P. Lu, D. Ruan, M. Huang, M. Tian, K. Zhu, Z. Gan and Z. Xiao, Signal Transduction Targeted Ther., 2024, 9, 166 CrossRef PubMed.
- D. V. Verdolino, H. A. Thomason, A. Fotticchia and S. Cartmell, Emerging Top. Life Sci., 2021, 5, 523–537 Search PubMed.
- R. Dong and B. Guo, Nano Today, 2021, 41, 101290 CrossRef CAS.
- G. Zhao, F. Liu, Z. Liu, K. Zuo, B. Wang, Y. Zhang, X. Han, A. Lian, Y. Wang and M. Liu, Stem Cell Res. Ther., 2020, 11, 1–18 CrossRef PubMed.
- Z. Xu, S. Han, Z. Gu and J. Wu, Adv. Healthcare Mater., 2020, 9, 1901502 CrossRef CAS PubMed.
- A. Hautmann, D. Kedilaya, S. Stojanović, M. Radenković, C. K. Marx, S. Najman, M. Pietzsch, J. F. Mano and T. Groth, Biomater. Adv., 2022, 142, 213166 CrossRef CAS PubMed.
- S. Obuobi, H. K.-L. Tay, N. D. T. Tram, V. Selvarajan, J. S. Khara, Y. Wang and P. L. R. Ee, J. Controlled Release, 2019, 313, 120–130 CrossRef CAS PubMed.
- S. Gera, E. Kankuri and K. Kogermann, Pharmacol. Ther., 2022, 232, 107990 CrossRef CAS PubMed.
- C.-F. Hsieh, C.-H. Chen, H.-H. Kao, D. T. Govindaraju, B. S. Dash and J.-P. Chen, Biomedicines, 2022, 11, 2902 CrossRef PubMed.
- X. Lin, B. Kong, Y. Zhu and Y. Zhao, Adv. Sci., 2022, 9, 2201226 CrossRef CAS PubMed.
- J. Franková, V. Pivodová, H. Vágnerová, J. Juráňová and J. Ulrichová, J. Appl. Biomater. Funct. Mater., 2016, 14, 137–142 Search PubMed.
- C. You, Q. Li, X. Wang, P. Wu, J. K. Ho, R. Jin, L. Zhang, H. Shao and C. Han, Sci. Rep., 2017, 7, 10489 CrossRef PubMed.
- H. Hanif, G. Sanjay, Z. K. Vasilev and J. C. Allison, Wound Pract. Res., 2020, 28, 173–180 Search PubMed.
- G. Cooksley, M. K. Dymond, N. A. Stewart, G. Bucca, A. Hesketh, J. Lacey, Y. Gogotsi and S. Sandeman, 2D Mater., 2022, 10, 014003 CrossRef.
- T. Ozulumba, G. Ingavle, Y. Gogotsi and S. Sandeman, Biomater. Sci., 2021, 9, 1805–1815 RSC.
- A. Zarepour, N. Rafati, A. Khosravi, N. Rabiee, S. Iravani and A. Zarrabi, Nanoscale Adv., 2024, 6, 3513–3532 RSC.
- E. J. Ward, J. Lacey, C. Crua, M. K. Dymond, K. Maleski, K. Hantanasirisakul, Y. Gogotsi and S. Sandeman, Adv. Funct. Mater., 2020, 30, 2000841 CrossRef CAS.
- Y. Li, R. Fu, Z. Duan, C. Zhu and D. Fan, ACS Nano, 2022, 16, 7486–7502 CrossRef CAS PubMed.
- E. K. Hoffmann, I. H. Lambert and S. F. Pedersen, Physiol. Rev., 2009, 89, 193–277 Search PubMed.
- S. A. Craig, Am. J. Clin. Nutr., 2004, 80, 539–549 CrossRef CAS PubMed.
- S. A. Kempson, Y. Zhou and N. C. Danbolt, Front. Physiol., 2014, 5, 159 Search PubMed.
- S. M. Hosseiniyan Khatibi, F. Zununi Vahed, S. Sharifi, M. Ardalan, M. Mohajel Shoja and S. Zununi Vahed, Biochimie, 2019, 158, 156–164 CrossRef CAS PubMed.
- M. K. Arumugam, M. C. Paal, T. M. Donohue Jr, M. Ganesan, N. A. Osna and K. K. Kharbanda, Biology, 2021, 10, 456 CrossRef CAS PubMed.
- M. Bhatt, A. Di Iacovo, T. Romanazzi, C. Roseti and E. Bossi, Basic Clin. Pharmacol. Toxicol., 2023, 133, 485–495 CrossRef CAS PubMed.
- L. S. Knight, Q. Piibe, I. Lambie, C. Perkins and P. H. Yancey, Neurochem. Res., 2017, 42, 3490–3503 CrossRef CAS PubMed.
- C. R. Day and S. A. Kempson, Biochim. Biophys. Acta, 2016, 1860, 1098–1106 CrossRef CAS PubMed.
- X. Hua, Z. Su, R. Deng, J. Lin, D.-Q. Li and S. C. Pflugfelder, Curr. Eye Res., 2015, 40, 657–667 CrossRef CAS PubMed.
- K.-H. Hsu, P. Lazon de la Jara, A. Ariyavidana, J. Watling, B. Holden, Q. Garrett and A. Chauhan, Curr. Eye Res., 2015, 40, 267–273 CrossRef CAS PubMed.
- J. J. López-Cano, M. A. González-Cela-Casamayor, V. Andrés-Guerrero, R. Herrero-Vanrell, J. M. Benítez-Del-Castillo and I. T. Molina-Martínez, Exp. Eye Res., 2021, 211, 108723 CrossRef PubMed.
- C. El-Chami, A. R. Foster, C. Johnson, R. P. Clausen, P. Cornwell, I. S. Haslam, M. C. Steward, R. E. B. Watson, H. S. Young and C. A. O'Neill, Br. J. Dermatol., 2021, 184, 482–494 CrossRef CAS PubMed.
- C. Brocker, D. C. Thompson and V. Vasiliou, Biomol. Concepts, 2012, 3, 345–364 CAS.
- M. Alhabeb, K. Maleski, B. Anasori, P. Lelyukh, L. Clark, S. Sin and Y. Gogotsi, Chem. Mater., 2017, 29, 7633–7644 Search PubMed.
- C. E. Shuck, A. Sarycheva, M. Anayee, A. Levitt, Y. Zhu, S. Uzun, V. Balitskiy, V. Zahorodna, O. Gogotsi and Y. Gogotsi, in MXenes, Jenny Stanford Publishing, 2023, pp. 539–560 Search PubMed.
- M. Tabatabai, D. K. Williams and Z. Bursac, Theor. Biol. Med. Modell., 2005, 2, 1–13 CrossRef PubMed.
- M. Shekhirev, C. E. Shuck, A. Sarycheva and Y. Gogotsi, Prog. Mater. Sci., 2021, 120, 100757 CrossRef CAS.
- W. Chen, X. Zhang, J. Li, Y. Wang, Q. Chen, C. Hou and Q. Garrett, Invest. Ophthalmol. Visual Sci., 2013, 54, 6287–6297 CrossRef CAS PubMed.
- F. Ascione, A. Vasaturo, S. Caserta, V. D'Esposito, P. Formisano and S. Guido, Exp. Cell Res., 2016, 347, 123–132 CrossRef CAS PubMed.
- H. Li, M. Mu, B. Chen, L. Zhou, B. Han and G. Guo, Mater. Res. Lett., 2024, 12, 67–87 CrossRef CAS.
- J. Yang, N. Cai, H. Zhai, J. Zhang, Y. Zhu and L. Zhang, Sci. Rep., 2016, 6, 37458 CrossRef CAS PubMed.
- P. G. Petronini, E. De Angelis, P. Borghetti, A. Borghetti and K. P. Wheeler, Biochem. J., 1992, 282, 69–73 CrossRef CAS PubMed.
- M. D'Souza-Ault, L. T. Smith and G. Smith, Appl. Environ. Microbiol., 1993, 59, 473–478 CrossRef PubMed.
- A. Sakamoto and N. Murata, Plant, Cell Environ., 2002, 25, 163–171 CrossRef CAS PubMed.
- M. Ashraf and M. R. Foolad, Environ. Exp. Bot., 2007, 59, 206–216 CrossRef CAS.
- A. Bonaterra, J. Camps and E. Montesinos, FEMS Microbiol. Lett., 2005, 250, 1–8 CrossRef CAS PubMed.
- G. Zhao, F. He, C. Wu, P. Li, N. Li, J. Deng, G. Zhu, W. Ren and Y. Peng, Front. Immunol., 2018, 9, 1070 CrossRef PubMed.
- M. Veskovic, D. Mladenovic, M. Milenkovic, J. Tosic, S. Borozan, K. Gopcevic, M. Labudovic-Borovic, V. Dragutinovic, D. Vucevic and B. Jorgacevic, Eur. J. Pharmacol., 2019, 848, 39–48 CrossRef CAS PubMed.
- C.-Y. Fan, M.-X. Wang, C.-X. Ge, X. Wang, J.-M. Li and L.-D. Kong, J. Nutr. Biochem., 2014, 25, 353–362 CrossRef CAS PubMed.
- Y. He, H. Sun, Y. Wang, C. Mu and L. Chen, Chem. Eng. J., 2024, 485, 150047 CrossRef CAS.
- H. Geng, Y. Ren, G. Qin, T. Wen, Q. Liu, H. Xu and W. He, RSC Adv., 2022, 12, 11128–11138 RSC.
- L. Hou, F. Gong, B. Liu, X. Yang, L. Chen, G. Li, Y. Gong, C. Liang, N. Yang and X. Shen, Theranostics, 2022, 12, 3834 CrossRef CAS PubMed.
- M. A. Velazco-Medel, L. A. Camacho-Cruz and E. Bucio, Radiat. Phys. Chem., 2020, 171, 108754 CrossRef CAS.
- A. Tuncel, K. Ecevit, K. Kesenci and E. Pişkin, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 45–55 CrossRef CAS.
- T. Hoare and R. Pelton, Langmuir, 2008, 24, 1005–1012 CrossRef CAS PubMed.
- S. W. Kim, Y. H. Bae and T. Okano, Pharm. Res., 1992, 9, 283–290 CrossRef CAS PubMed.
- B. Lu, M. Tarn, N. Pamme and T. Georgiou, J. Mater. Chem. B, 2015, 3, 4524–4529 RSC.
- T.-L. Tsou, S.-T. Tang, Y.-C. Huang, J.-R. Wu, J.-J. Young and H.-J. Wang, J. Mater. Sci.: Mater. Med., 2005, 16, 95–100 CrossRef CAS PubMed.
- M. T. Manzari, Y. Shamay, H. Kiguchi, N. Rosen, M. Scaltriti and D. A. Heller, Nat. Rev. Mater., 2021, 6, 351–370 CrossRef CAS PubMed.
- A. Tewabe, A. Abate, M. Tamrie, A. Seyfu and E. Abdela Siraj, J. Multidiscip. Healthcare, 2021, 1711–1724 CrossRef PubMed.
- Y. Li, C. Zhang, G. Li, G. Deng, H. Zhang, Y. Sun and F. An, Acta Pharm. Sin. B, 2021, 11, 2220–2242 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04622f |
| ‡ Joint first authors. |
| This journal is © The Royal Society of Chemistry 2025 |