
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Yttrium-mediated ring-opening polymerization of functionalizable dihydrocarvide: tunable terpene-based polyesters using grafting from and block copolymerization strategies†
Lea-Sophie
Hornberger
a,
Julian
Fischer
 b,
Alexandra
Friedly
c,
Ingo
Hartenbach
c,
Thomas
Sottmann
b and
Friederike
Adams
*ade
b,
Alexandra
Friedly
c,
Ingo
Hartenbach
c,
Thomas
Sottmann
b and
Friederike
Adams
*ade
aChair of Macromolecular Materials and Fiber Chemistry, Institute of Polymer Chemistry, University of Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany. E-mail: friederike.adams@ipoc.uni-stuttgart.de
bInstitute of Physical Chemistry, University of Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany
cInstitute of Inorganic Chemistry, University of Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany
dSection for Translational Research in Ophthalmology, Center for Ophthalmology, University Eye Hospital Tübingen, Elfriede-Aulhorn-Strasse 7, 72076 Tübingen, Germany
eWacker-Chair of Macromolecular Chemistry, TUM School of Natural Sciences, Department of Chemistry, Technical University of Munich, Lichtenbergstr. 4, 85748 Garching, Germany
First published on 12th May 2025
Abstract
Poly(dihydrocarvide) (PDHC) is synthesized through ring-opening polymerization (ROP) of terpene-based 7-membered lactone dihydrocarvide (DHC) using an amino-alkoxy-bis(phenolate) yttrium amido catalyst and isopropanol (iPrOH) as a chain transfer agent while retaining the pendant-group double bond in the monomer unit. Polymerization under conditions found to be favorable (60 °C, 1 eq. iPrOH) yielding PDHC with tunable molecular weights and low to moderate polydispersities (Đ = 1.2–1.5). Crystalline fractions are introduced into amorphous PDHC by producing block copolymers with 16-membered ω-pentadecalactone (PDL) or 4-membered racemic β-butyrolactone (BBL) via sequential addition following the coordination strength hierarchy (PDL < DHC < BBL). This resulted in semi-crystalline renewable block copolymers P(PDL-b-DHC) and P(DHC-b-PHB) that were further analyzed by PXRD and SAXS measurements. Additionally, PDHC is functionalized via thiol–ene reaction with 2-mercaptoethanol, introducing hydroxyl functionality and opening up a multitude of functionalization possibilities. As one example, atom transfer radical polymerization (ATRP) initiators are attached, and SARA and ARGET ATRP techniques are employed to graft poly(ethyl acrylate) (PEA) as model compound, forming PDHC-g-PEA brush polymers. The TPMANMe2-based ARGET ATRP system demonstrates superior control over molecular weight and polydispersity compared to SARA ATRP, though both methods yield well-defined polymer brushes with molecular weight growth correlating with the initial amount of ethyl acrylate. This approach demonstrates the potential of PDHC for constructing diverse polymer architectures from different types of lactones or vinyl monomers by combining ROP and ATRP.
Introduction
With growing public awareness of climate change, environmental deterioration, and resource depletion, sustainability and green chemistry are gaining significant attention. Due to their extensive applications, polymers are crucial in supporting sustainable development and reducing environmental impact.1 Besides sustainability, material properties play an essential role in determining the capability of a renewable biopolymer to be a promising alternative to petroleum-based polymers.The class of terpenes is amongst the biggest classes of renewable feedstocks with more than 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 naturally occurring aliphatic substrates that derive from the basic unit isoprene.2,3 These terpenes are isolated from various sources such as herbs, trees, flowers, and fruits like orange, ginger, clove, and peppermint. Terpenoids (terpenes with functional groups) derived from mint oil are, e.g. menthol or carvone.4 The cyclic terpene carvone is an α,β-unsaturated ketone naturally occurring in spearmint and caraway oil. It occurs as (S)-(+)- and (R)-(−)-carvone, also known as (+)-carvone, and (−)-carvone. While (+)-carvone is the main component of the caraway (Carum carvi) and dill (Oleum anethi) oils, (−)-carvone is found in spearmint (Mentha spicata) and kuromoji oil.5 Synthetic routes from alternative feedstocks are also available.5–8 Synthetic carvone is mostly derived from limonene, another terpene found in citrus peels, and is a co-product of citrus juice production (30000 tons per year).9 Various methods for converting (+)-limonene to (−)-carvone are reported, including nitrosochlorination,5,10–13 allylic oxidation,5,10,14–17 or epoxidation,5,17–19 and thus significant amounts of carvone can be sourced from renewable waste streams of the juicing industry.
000 naturally occurring aliphatic substrates that derive from the basic unit isoprene.2,3 These terpenes are isolated from various sources such as herbs, trees, flowers, and fruits like orange, ginger, clove, and peppermint. Terpenoids (terpenes with functional groups) derived from mint oil are, e.g. menthol or carvone.4 The cyclic terpene carvone is an α,β-unsaturated ketone naturally occurring in spearmint and caraway oil. It occurs as (S)-(+)- and (R)-(−)-carvone, also known as (+)-carvone, and (−)-carvone. While (+)-carvone is the main component of the caraway (Carum carvi) and dill (Oleum anethi) oils, (−)-carvone is found in spearmint (Mentha spicata) and kuromoji oil.5 Synthetic routes from alternative feedstocks are also available.5–8 Synthetic carvone is mostly derived from limonene, another terpene found in citrus peels, and is a co-product of citrus juice production (30000 tons per year).9 Various methods for converting (+)-limonene to (−)-carvone are reported, including nitrosochlorination,5,10–13 allylic oxidation,5,10,14–17 or epoxidation,5,17–19 and thus significant amounts of carvone can be sourced from renewable waste streams of the juicing industry.
Selective hydrogenation of carvone yields dihydrocarvone. Recently, an additional highly efficient way to directly transform limonene-1,2-epoxide into cis-dihydrocarvone (cis-D), and trans-dihydrocarvone (trans-D) was reported by Gallego-Villada, Murzin, and Serrano.20,21 Further, Baeyer–Villiger rearrangement from dihydrocarvone yields the seven-membered lactone, dihydrocarvide (DHC).22 Classical Baeyer–Villiger oxidation with organic peracids favors side reactions, particularly the epoxidation of the double bond in dihydrocarvone, rather than selective lactone formation.23 Additionally, organic peracids have limited commercial applications due to their shock sensitivity, which makes them hazardous.24 Tolman and Hillmyer reported on a greener Bayer–Villiger oxidation of dihydrocarvone using Oxone®, which is a commercially available potassium triple salt (KHSO5·KHSO4·K2SO4) to yield the lactone DHC, bearing a double bond in the C5 substituent.22 The oxidation from dihydrocarvone to dihydrocarvide can also be conducted enzymatically with Baeyer–Villiger monooxygenases, such as the wild-type of a cyclohexanone monooxygenase from Acinetobacter calcoaceticus.25
To produce polyesters from DHC, the isopropenyl group could cause unwanted side-reactions during a coordination ring-opening polymerization (ROP), such as migratory-insertion polymerization, which can compete with the ROP mechanism leading to uncontrolled polymer growth, lower molecular weights, or broader polydispersity of the polymers. Only a few examples report the controlled ROP of dihydrocarvide.22,26–28 Tolman and Hillmyer conducted such polymerizations at 100 °C using a catalyst/initiating system consisting of diethyl zinc/benzyl alcohol to yield poly(dihydrocarvide) (PDHC) with a molecular weight of up to 10.5 kg mol−1 (Đ = 1.24) while preserving the double-bond in the pendant group of the repeating unit.22,26 The groups of Scrutton and Dove both applied the metal–organic catalyst magnesium 2,6 di-tert-butyl-4-methylphenoxide and benzyl alcohol initiator for homo- or copolymerization of DHC.27,28 To the best of our knowledge, these are the only examples of polymerizing DHC as a homo- or copolymer. Post-polymerization modifications of PDHC and related copolymers have been explored to utilize the reactivity of the double bonds to create robust thermoplastic polyurethanes.22,26,29
Due to their activity, stereoselectivity, and control over the reaction, yttrium complexes are highly promising catalysts for the coordination–insertion ROP of lactones with diverse ring sizes especially for producing block copolymers.30–36 Aminoalkoxybis(phenolate) yttrium complexes with a tert-butyl substituted amino-methoxy bis(phenolate) ligand ([ONOO]tBu) and a bis(dimethylsilyl)amide (bdsa) initiator ([(ONOO)tBuY(bdsa)(THF)]) were first reported by Carpentier et al. for the controlled ROP of lactones such as lactide and racemic β-butyrolactone (BBL).37–40In situ generation of the alkoxide initiator by reaction of the complex with one equivalent of isopropanol or performing an immortal ROP with up to 50 equivalents of isopropanol increased the efficiency for polymerizing heterotactic lactide from a racemic monomer mixture, producing polylactic acid with molecular weights of up to 160 kg mol−1 and narrow molar mass distributions (Đ = 1.06–1.40).40,41 In the presence of alcohol, the [ONOO]tBu ligand stabilizes the oxophilic and electrophilic metal center of the catalyst.42 Immortal ROP with isopropanol and racemic BBL produced syndiotactic enriched polyhydroxybutyrate (PHB).39,43 However, the use of more than five equivalents of iPrOH significantly decreased the activity when using BBL as a monomer.41,42 The monomer scope was extended to include seven-membered rings such as ε-caprolactone37 and (−)-menthide.44 Recently, we reported on the controlled homopolymerization and copolymerization of 16-membered macrolactone ω-pentadecalactone (PDL) with BBL using amino-alkoxy-bis(phenolate) yttrium amido and alkoxide catalysts yielding poly(pentadecalactone) (PPDL) and PPDL-co-PHB with high molecular weights of up to 134.5 kg mol−1 and with moderate polydispersities (Đ = 1.3–2.1).30
In this study, DHC was polymerized using amino-alkoxy-bis(phenolate) yttrium amido ([(ONOO)tBuY(bdsa)(THF)]) and alkoxide ([(ONOO)tBuY(iPrO)(THF)]) catalysts via ROP, yielding PDHC with functionalizable double bonds in each repeating unit, enabling thiol–ene modification. The simplicity and efficiency of thiol–ene reactions make it a commonly used method for polymer modification, either pre-45–47 or post-polymerization.48–53 Hawker et al. designed alkene–functionalized copolymers and demonstrated that photochemical thiol–ene reactions with mercaptans were faster and more tolerant toward diverse backbones and orthogonal functional groups than thermal thiol–ene reactions.48
Although side-chain hydroxyl functionality introduced via thiol–ene reaction with mercaptoethanol offers various applications, we report on a grafting from approach by attaching an atom transfer radical polymerization (ATRP) initiator. ARGET ATRP (activators regenerated by electron transfer atom transfer radical polymerization) is a copper-mediated reversible-deactivation radical polymerization where alkyl halides undergo redox reactions with a transition-metal complex, continuously regenerated by reducing agents.54–58 It operates under mild conditions using low metal concentrations, yielding uniform polymers with controllable architectures.54–58 SARA ATRP (supplemental activator and reducing agent ATRP), another variation of ATRP, employs copper(0) as a reducing agent and additional activator.59 In 2018, Matyjaszewski et al. reported on an ARGET ATRP system utilizing a Cu-based catalyst with the tetradentate ligand tris[(4-dimethylaminopyridyl)methyl]amine (TPMANMe2) and silver as the reducing agent.60 Recently, this ARGET ATRP system has been employed to successfully polymerize a range of active esters and more challenging monomers.61
In this work, an ATRP initiator was introduced onto the PDHC backbone using post-polymerization functionalization, allowing brush polymers to grow directly from the backbone in a grafting from strategy. This approach facilitates the formation of high-density brush polymers.62,63 Besides producing graft copolymers, block copolymers of DHC with BBL or PDL were synthesized by sequential monomer addition, yielding materials that combine the amorphous properties of PDHC with the semi-crystalline characteristics of syndiotactic PHB or PPDL.
Results and discussion
Homopolymerization of dihydrocarvide
Initially, (+)-dihydrocarvone (D) was purchased as a mixture of isomers, and the GC (gas chromatography) chromatogram showed two peaks, which could be attributed to the presence of trans and cis isomers of D. Integration of the peak areas revealed a ratio of 17:83 (Fig. S3 and S4†). D was oxidized via a Bayer–Villiger reaction using Oxone to obtain crude dihydrocarvide (DHC). Conversion of D to DHC was confirmed via1H NMR (nuclear magnetic resonance) and 13C NMR spectroscopy (Fig. S1 and S2† for D, Fig. S5 and S6† for DHC). An 80:20 ratio of doublets was observed for the splitting of the side-chain methyl group adjacent to the cyclic ketone in the crude dihydrocarvone product in the 1H NMR spectrum (Fig. S1†), due to the presence of the isomers. The signal with the smaller integral diminished following the purification of DHC by column chromatography (Fig. S5†). DHC isomers were successfully separated due to their distinct Rf values.22 The trans isomer (4R,7R)-4-isopropenyl-7-methyloxepan-2-one ((4R,7R)-dihydrocarvide), from here on referred to as DHC, eluted first and was collected. Elemental analysis and GC-MS (gas chromatography-mass spectrometry) confirmed the purity of DHC and the absence of epoxide byproducts (Fig. S7 and S8†).
Polymerization of DHC was conducted via coordination–insertion ring-opening mechanism with the amino-alkoxy-bis(phenolate) yttrium amido [(ONOO)tBuY(bdsa)(THF)] and alkoxide [(ONOO)tBuY(iPrO)(THF)] catalysts to synthesize poly(dihydrocarvide) (PDHC) with varying molecular weights at different reaction temperatures (room temperature and 60 °C, Scheme 1). The immortal coordination–insertion ring-opening polymerization of DHC is depicted in Scheme S1.† Results for DHC polymerization are shown in Table 1. The preservation of the isopropenyl group in the PDHC polymer pendant groups was proven by 1H NMR spectroscopy (Fig. 1a). Relative molar masses of PDHC determined by size-exclusion chromatography (SEC) were used to evaluate relative trends in molar mass and polydispersity. End-group analysis via1H NMR spectroscopy was not possible, as no distinct end-group signals could be identified due to weak or overlapping end-group signals (Fig. S9†).
 | ||
| Scheme 1 Synthesis of PDHC via ring-opening polymerization of DHC, performed with a [(ONOO)tBuY(bdsa)(THF)] amido catalyst with or without isopropanol for the in situ preparation of the [(ONOO)tBuY(iPrO)(THF)] alkoxide catalyst or as a chain transfer agent. | ||
 | ||
| Fig. 1 (a) 1H NMR spectrum of trans isomer (4R,7R)-dihydrocarvide (top) and PDHC (bottom; Table 1, entry 4); (b) kinetic measurements of DHC polymerization at 60 °C. Conversion, determined by 1H NMR measurements, was plotted against polymerization time and the growth of number-average molar mass with time for a polymerization at 60 °C; (c) dependency of relative number-average molar mass (Mn,SEC) and polydispersity (Đ), measured via size-exclusion chromatography (SEC) in chloroform, on the conversion for a polymerization at 60 °C using [(ONOO)tBuY(iPrO)(THF)] as a catalyst. | ||
| Entry | [DHC]:[Y]:[iPrOH]a |
T [°C] | Conv.b [%] | M n,calc [kg mol−1] | M n,SEC [kg mol−1] | Đ | T g [°C] |
|---|---|---|---|---|---|---|---|
| a Reactions performed for 120 min with 24.9 μmol [(ONOO)tBuY(bdsa)(THF)] in 0.5 mL toluene with a respective amount of isopropanol. b Determined via1H NMR spectroscopy integrating the –C–H– groups of DHC and PDHC (Fig. S12†). c M n,calc calculated by Mn,calc = Mmonomer × ([M]/[Y]) × conversion, with [iPrOH]: Mn,calc = (Mmonomer × ([M]/[Y]) × conversion) × (1/([iPrOH])). d Number-average molecular weight measured via SEC in chloroform at 40 °C relative to polystyrene standards. e Measured via DSC with a heating rate of 10 K min−1 (second heat cycle), n.d. = not detected. f Reaction performed for 1250 min. g Neat reaction without solvent. h Polymerization in 0.5 mL THF. | |||||||
| 1 | 25:1:0 |
RT | >99 | 4.2 | 23.5 | 1.5 | n.d. |
| 2f | 50:1:0 |
RT | >99 | 8.3 | 34.5 | 1.5 | −10 |
| 3 | 100:1:0 |
RT | >99 | 16.7 | 46.5 | 1.5 | −12 |
| 4 | 25:1:0 |
60 | >99 | 4.2 | 14.3 | 1.3 | n.d. |
| 5g | 50:1:0 |
60 | 95 | 8.1 | 21.3 | 1.5 | −8 |
| 6h | 50:1:0 |
60 | >99 | 8.3 | 32.5 | 1.4 | n.d. |
| 7 | 50:1:0 |
60 | >99 | 8.3 | 26.2 | 1.4 | −8 |
| 8 | 100:1:0 |
60 | >99 | 16.7 | 34.5 | 1.5 | −17 |
| 9 | 50:1:1 |
60 | >99 | 8.3 | 8.0 | 1.2 | n.d. |
| 10 | 50:1:3 |
60 | >99 | 2.8 | 4.7 | 1.2 | n.d. |
| 11 | 50:1:5 |
60 | >99 | 1.7 | 3.5 | 1.2 | n.d. |
The end groups of PDHC were investigated by mass spectrometry. Oligomerization experiments of DHC were analyzed by electrospray ionization coupled with mass spectrometry (ESI-MS) and positive ion matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) measurements to give information on the end groups and repeating units. ESI-MS analysis of oligo-DHC synthesized with the [(ONOO)tBuY(bdsa)(THF)] catalyst showed signals for m/z = [n × MDHC + Na]+, as well as m/z = [n × MDHC + K]+ for repeating units (n) n = 4–11, with Na+ and K+ operating as charge carriers (Fig. S14†). Bisdimethylsilyamide end-groups could not be detected as already obtained in previous publications due to ESI-MS fragmentation mechanisms.30,44,64
Further, isopropanol was used for the in situ generation of the alkoxide catalyst [(ONOO)tBuY(iPrO)(THF)]. Oligomerization of DHC using this catalyst at room temperature (Fig. S15†) and at the elevated temperature of 60 °C (Fig. S16†) was analyzed by MALDI-TOF measurements. For reaction at room temperature, signals for m/z = [n × MDHC + MiPrOH + Na]+ were detected for n = 4, 5, 6. Reaction at 60 °C delivered m/z = [n × MDHC + MiPrOH + Na]+ for a slightly broader range of repeating units, n = 4–8. Both results show chain initiation by the isopropoxide unit and an absence of any side reaction with the pendant double bond.
Polymerization experiments with [(ONOO)tBuY(bdsa)(THF)] were conducted at ambient temperature and 60 °C, targeting degrees of polymerization (DP) from 25 to 100 (Table 1, entries 1–3, 4, 7 and 8). For all polymerization reactions, complete monomer conversion was achieved. The isolated polymers exhibited moderate polydispersities (Đ) at ambient temperature, with values around 1.5. At 60 °C, polydispersities were generally lower, ranging from 1.3 for DP = 25 and 50 to 1.5 for DP = 100. Correspondingly, molecular weights increased with targeted DP, from 23.5 to 46.5 kg mol−1 at ambient temperature and from 14.3 to 34.5 kg mol−1 at 60 °C. The lower molecular weights and polydispersities observed at 60 °C suggest enhanced control over polymerization under elevated temperatures.
In addition to reactions conducted in toluene, polymerizations were also executed as neat reactions (Table 1, entry 5), omitting the use of supplementary solvents. The conversion of DHC reached 95%, resulting in an Mn,SEC of 21.3 kg mol−1, and a polydispersity of 1.5. This resulted in a lower molecular weight but a similar polydispersity compared to reactions conducted in toluene at both room temperature and 60 °C for a targeted DP of 50 (Table 1, entries 2 and 7). However, the use of neat reactions was discontinued due to the persistent residues of catalyst in the dried PDHC (Fig. S11†). Using tetrahydrofuran (THF) as the solvent for polymerization at a temperature of 60 °C (Table 1, entry 6) resulted in the synthesis of PDHC with nearly complete conversion of DHC. The Mn,SEC was slightly elevated (32.5 kg mol−1) accompanied by a slightly higher polydispersity index of 1.4, compared to the reaction conducted in toluene suggesting that toluene might be the preferred solvent for DHC polymerization. However, it is noteworthy that polymerization in THF remains a viable and effective alternative.
Isopropanol served either as an initiator in the [(ONOO)tBuY(iPrO)(THF)] complex or as a chain transfer agent in the polymerization of DHC using [(ONOO)tBuY(bdsa)(THF)]. The generation of the [(ONOO)tBuY(iPrO)(THF)] catalyst was achieved in situ by stirring one equivalent of iPrOH with one equivalent of the dissolved [(ONOO)tBuY(bdsa)(THF)] catalyst before polymerization. Polymerizations involving the CTA (if more than one equivalent of isopropanol was used) were conducted at 60 °C with varying amounts of iPrOH (1, 3, and 5 equivalents) and [(ONOO)tBuY(bdsa)(THF)] (Table 1). All reactions achieved nearly complete conversion of DHC and displayed low polydispersities (Đ = 1.2). Chain length control was demonstrated by adjusting the amount of CTA, which corresponded to the quantity of chain-initiating species. The Mn,SEC decreased as the CTA equivalent increased, from 8.0 kg mol−1 (1 equivalent) to 3.5 kg mol−1 (5 equivalents). Furthermore, contrasting the amide (bdsa) and alkoxide (iPrOH) initiators (Table 1, entry 7 vs. entry 9) reveals that iPrOH led to superior results, yielding a lower molecular weight, due to a higher initiator efficiency, and reduced polydispersity. The improved performance of iPrOH can be attributed to its higher nucleophilicity. In contrast, polymerizations initiated with bdsa exhibited broader molecular weight distributions, indicating possible side reactions. Additionally, the absence of a detectable bdsa-derived end-group in ESI-MS suggests its high reactivity, which may contribute to undesired side reactions and reduced control over the polymerization. Overall, employing 1 equivalent of iPrOH as a CTA at 60 °C yielded the most favorable polymerization conditions, establishing it as the optimal choice for the studied system.
Differential scanning calorimetry (DSC) measurements, conducted between −40 and 180 °C, revealed the absence of crystalline regions (Fig. S17†). Amorphous PDHC displayed a single glass transition temperature (Tg) in the range of −8 to −17 °C (Table 1).
Kinetic measurements were systematically carried out to elucidate the influence of temperature on the catalytic activity of both the amido and the alkoxide catalyst. Samples were taken regularly from the polymerization reaction to determine conversion, molecular weight and polydispersity. At room temperature, [(ONOO)tBuY(iPrO)(THF)] induced a more rapid progression towards the plateau region, reaching 97% conversion in 70 minutes, compared to 110 minutes with [(ONOO)tBuY(bdsa)(THF)] (Fig. S18†). The turn-over frequency (TOF) was 80 h−1 for the amide initiator and 195 h−1 for the alkoxide initiator. Since both systems share the same catalytic site (metal + ligand system), the lower TOF and higher polydispersity of [(ONOO)tBuY(bdsa)(THF)] suggest increased side reactions and a lower initiator efficiency caused by the bdsa initiator. For both catalysts, molecular weight increased linearly with conversion (Fig. S20 and S21†). The alkoxide-initiated ROP maintained a stable polydispersity index (Đ < 1.2) up to full conversion during polymerization at room temperature, indicating fewer side reactions. At 60 °C, reaction kinetics were significantly accelerated. [(ONOO)tBuY(iPrO)(THF)] achieved the plateau region with nearly complete conversion in 16 minutes, while [(ONOO)tBuY(bdsa)(THF)] required 30 minutes (Fig. 1b and Fig. S19†). TOFs increased to 315 h−1 (bdsa) and 585 h−1 (isopropoxide). The amide catalyst exhibited a gradual ascent in polydispersity to 1.45 (Fig. S22†), whereas the alkoxide system maintained a polydispersity below 1.3, though it increased at high conversions over 90% (Fig. 1c and Fig. S23†), suggesting that side reactions become more prominent at elevated temperatures and high conversions. The deviation from the expected linear trend in molecular weight increase further indicates possible transesterification reactions.
Block copolymerizations
Amorphous polymers such as PDHC offer flexibility and impact resistance due to their lack of long-range order.65 Semi-crystalline polymers, such as poly(pentadecalactone) (PPDL) and syndiotactic poly(hydroxybutyrate) (PHB), provide mechanical strength and thermal stability through their crystalline regions.66 PPDL is an aliphatic polyester with thermal and material properties comparable to low- or high-density polyethylene, depending on its molecular weight.67Syndiotactic PHB exhibits promising characteristics as a substitute for fossil-based thermoplastics, such as poly(propylene).44,68 Combining amorphous polymers with semi-crystalline polymers to form block copolymers is a strategic approach to tailor material properties and develop thermoplastic elastomers. Incorporating PDHC as an amorphous polymer into block copolymers with PHB or PPDL, while retaining the double bond within the copolymer structure, enables twofold control over material properties: (1) through the inherent characteristics of the block components and (2) via post-polymerization modifications.Block copolymers from PDL and DHC, as well as DHC and BBL were synthesized via sequential addition with [(ONOO)tBuY(bdsa)(THF)] or [(ONOO)tBuY(iPrO)(THF)]. The monomer addition sequence was determined by the coordination strength of the monomer to the catalyst, starting with the monomer with the weakest coordination (PDL < DHC < BBL). Using 13C NMR spectroscopy, coordination strength was assessed by reacting [(ONOO)tBuY(bdsa)(THF)] with 10 equivalents of PDL and DHC, revealing that only DHC was polymerized in this mixture (Fig. S30†). Consequently, PDL was selected as the first block in polymerizations yielding PPDL-b-PDHC. Prior studies on the terpene-based 7-membered (−)-menthide showed that BBL exhibits a higher coordination strength to the catalyst than (−)-menthide.44 By analogy, BBL was assumed to coordinate stronger than DHC. For all block copolymerizations, a temperature of 60 °C was chosen, as it provided high monomer conversions while maintaining excellent control over molecular weight and dispersity, both in the present results for DHC and in previous studies involving PDL.30 For the block copolymerization synthesis, the first monomer was polymerized as the first block, before the second monomer was added to build the second block. The resulting copolymers were characterized using NMR spectroscopy (1H, DOSY, 13C), SEC, and DSC (Tables 2 and 3). Additionally, selected samples underwent in-depth crystallographic analysis by powder X-ray diffraction (PXRD) and small-angle X-ray scattering (SAXS) to further analyze the crystalline and amorphous regions in these block copolymers (Fig. 3 and 4).
| Entry | [Y]:[iPrOH]:[PDL]:[DHC]a |
TimePDL [h] | Conv.PDLb [%] | M n, PPDL, SEC [kg mol−1] | Đ PPDL | TimeDHC [h] | Conv.DHCd [%] | M n, AB, SEC [kg mol−1] | Đ AB | M PPDL/MPDHCe | T g,1 [°C] | T m [°C] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Reactions performed with 24.9 μmol [(ONOO)tBuY(bdsa)(THF)] in 0.5 mL toluene at 60 °C with a respective amount of isopropanol. b Determined via1H NMR spectroscopy integrating the –O–CH2– signal of PDL and PPDL as shown in Fig. S31.† c Measured via SEC in chloroform at 40 °C relative to polystyrene standards. d Determined via1H NMR spectroscopy integrating the –C–H– groups of DHC and as illustrated in Fig. S32.† e Calculated via1H NMR spectroscopy as depicted in Fig. S29.† f Measured via DSC with a heating rate of 10 K min−1 (second heating cycle), n.d. = not detected. Tg,2 could not be detected. | ||||||||||||
| 1 | 1:0:25:24 |
14.7 | 81 | 29.2 | 1.4 | 2.0 | 92 | 53.4 | 1.5 | 64/36 | n.d. | 88 |
| 2 | 1:1:25:42 |
15.5 | 92 | 9.3 | 1.3 | 3.2 | 93 | 12.3 | 1.4 | 55/45 | −18 | 84 |
| 3 | 1:1:50:49 |
16.8 | 94 | 12.6 | 1.6 | 2.0 | 96 | 21.3 | 1.6 | 50/50 | n.d. | 87 |
| Entry | [Y]:[iPrOH]:[DHC]:[BBL]a |
TimeDHC [h] | Conv.DHCb [%] | M n, PDHC, SEC [kg mol−1] | Đ PDHC | TimeBBL [h] | ConvBBLd [%] | P r [%] | M n, AB, SEC [kg mol−1] | Đ AB | M PDHC/MPHBf | T g,1 [°C] | T g,2 [°C] | T m [°C] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Reactions performed with 24.9 μmol [(ONOO)tBuY(bdsa)(THF)] in 2 mL toluene at 60 °C with a respective amount of isopropanol. b Determined via1H NMR spectroscopy integrating the –C–H– groups of DHC and PDHC as illustrated in Fig. S43.† c Measured via SEC in chloroform at 40 °C relative to polystyrene standards. d Determined via1H NMR spectroscopy by integrating the –O–CH– signal of BBL and PHB as shown in Fig. S44.† e Probability of racemic linkages between monomer units, determined via the carbonyl region of 13C NMR spectroscopy as shown in Fig. S45.† f Calculated via1H NMR spectroscopy as depicted in Fig. S42.† g Measured via DSC with a heating rate of 10 K min−1 (second heating cycle), n.d. = not detected. | ||||||||||||||
| 1 | 1:0:49:99 |
2.0 | >99 | 32.0 | 1.6 | 16.0 | 71 | 73 | 39.8 | 1.6 | 33/67 | −6 | 4 | 112 |
| 2 | 1:1:25:104 |
2.1 | 94 | 5.0 | 1.3 | 17.3 | 97 | 73 | 9.4 | 1.3 | 16/84 | −4 | n.d. | 103 |
| 3 | 1:1:50:42 |
2.1 | 96 | 10.6 | 1.2 | 17.2 | 99 | 76 | 13.0 | 1.2 | 52/48 | −10 | 5 | 106 |
| 4 | 1:1:50:114 |
2.1 | >99 | 7.6 | 1.2 | 16.0 | 94 | 73 | 15.2 | 1.2 | 27/73 | −7 | 0 | 103 |
PPDL-b-PDHC copolymers
For PPDL-b-PDHC block copolymers, PDL polymerization was carried out first due to its lower coordination strength towards the [(ONOO)tBuY(bdsa)(THF)] catalyst.PPDL-b-PDHC polymers were synthesized both with [(ONOO)tBuY(bdsa)(THF)] (Table 2, entries 2 and 3) and with [(ONOO)tBuY(OiPr)(THF)] (Table 2, entry 1). The ratio of PPDL to PDHC in the copolymers was calculated using 1H NMR spectroscopy, based on the distinct proton signals of the two homopolymers (Fig. S29†). The DOSY NMR spectrum of PPDL-b-PDHC produced with [(ONOO)tBuY(bdsa)(THF)] (Table 2, entry 1) indicated the presence of two species (Fig. S33†) corresponding to both the block copolymer PPDL-b-PDHC and PDHC homopolymer. This suggests the presence of inactive [(ONOO)tBuY(bdsa)(THF)] catalyst species during the polymerization, which solely polymerizes the second block after its addition, or the presence of side reactions with the bdsa end group further initiating new chains. However, active chains from polymerization of PDL stayed active during chain extension since no PPDL homopolymer was formed, indicating a living polymerization mechanism. In contrast, synthesis with [(ONOO)tBuY(OiPr)(THF)] catalyst, exhibited a single set of signals in the DOSY NMR spectrum (Fig. S34†), confirming the successful linkage of the two blocks without formation of any homopolymers and the presence of all characteristic signals from both homopolymers in the NMR spectrum further confirmed successful block copolymerization (Fig. 2a). SEC measurements demonstrated an increase in the number-average molecular weight from the first to the second block (Fig. S36 and S37†). Thermal analysis via DSC revealed that PDHC is completely amorphous with a single glass transition temperature (Tg) (Fig. S17†), while PPDL homopolymers are semi-crystalline, displaying both a Tg and a melting temperature (Tm) of −27 °C and 100 °C, respectively.67 However, due to the high crystallinity of PPDL, its Tg is often difficult to detect.30 In the PPDL-b-PDHC copolymers, the Tg of PDHC (−18 °C) was only observable in samples with a higher PDHC content (Table 2, entry 2). The Tm of PPDL was consistently retained across all samples, ranging from 84 °C to 88 °C (Fig. S38 and S39†). A minor endothermic event was observed prior to the main melting transition of PPDL, indicating the presence of less stable crystalline regions. This effect could be related to the incorporation of amorphous PDHC segments, which may disrupt the formation of highly ordered crystalline domains in PPDL. Additionally, phase separation effects within the copolymer might influence the crystalline structure, potentially limiting the formation of large, well-defined crystallites.69,70
 | ||
| Fig. 2 1H NMR spectra (CDCl3, 400 MHz) of block copolymers: (a) PPDL-b-PDHC (PPDL/PDHC = 50/50; Table 2, entry 3) and (b) PDHC-b-PHB (PDHC/PHB = 27/73; Table 2, entry 7). | ||
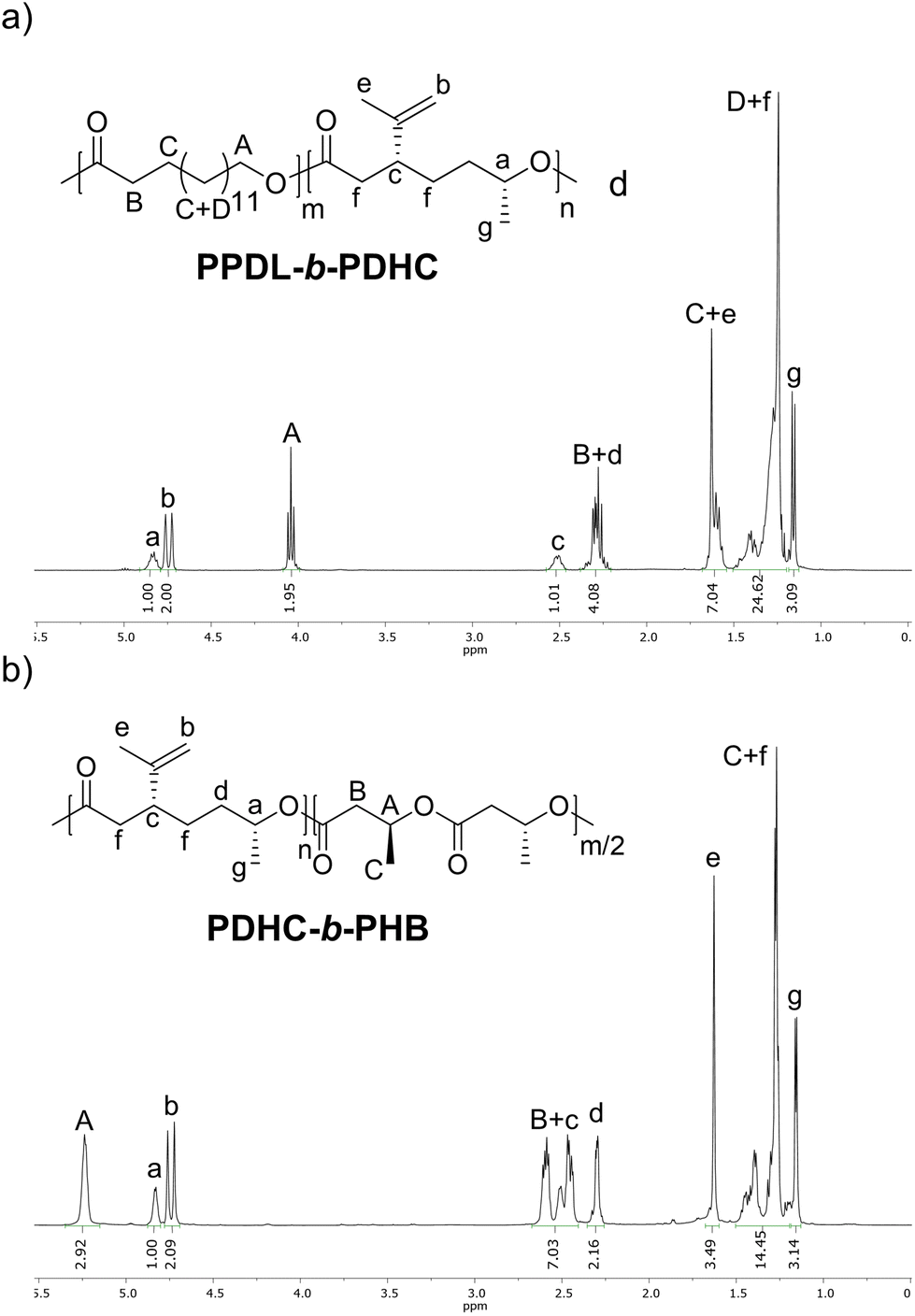
For further investigating the crystallinity of PPDL-b-PDHC, PXRD analysis was performed. The baseline-corrected data are shown in Fig. S40,† while Fig. 3a presents the normalized PXRD pattern of PPDL-b-PDHC with a 50/50 composition, normalized to the reflection at 21.3° (green). The PPDL homopolymer exhibits two strong reflections at 21.3° and 23.8°, corresponding to crystalline regions, with a minor amorphous background between 12° and 27°.30,71 In PPDL-b-PDHC block copolymers, PPDL reflections at 21.3 and 23.8° are retained, albeit slightly broadened, while the halo, corresponding to amorphous regions is more pronounced. These results confirm the preservation of crystalline PPDL domains, even with the addition of more amorphous PDHC portions.
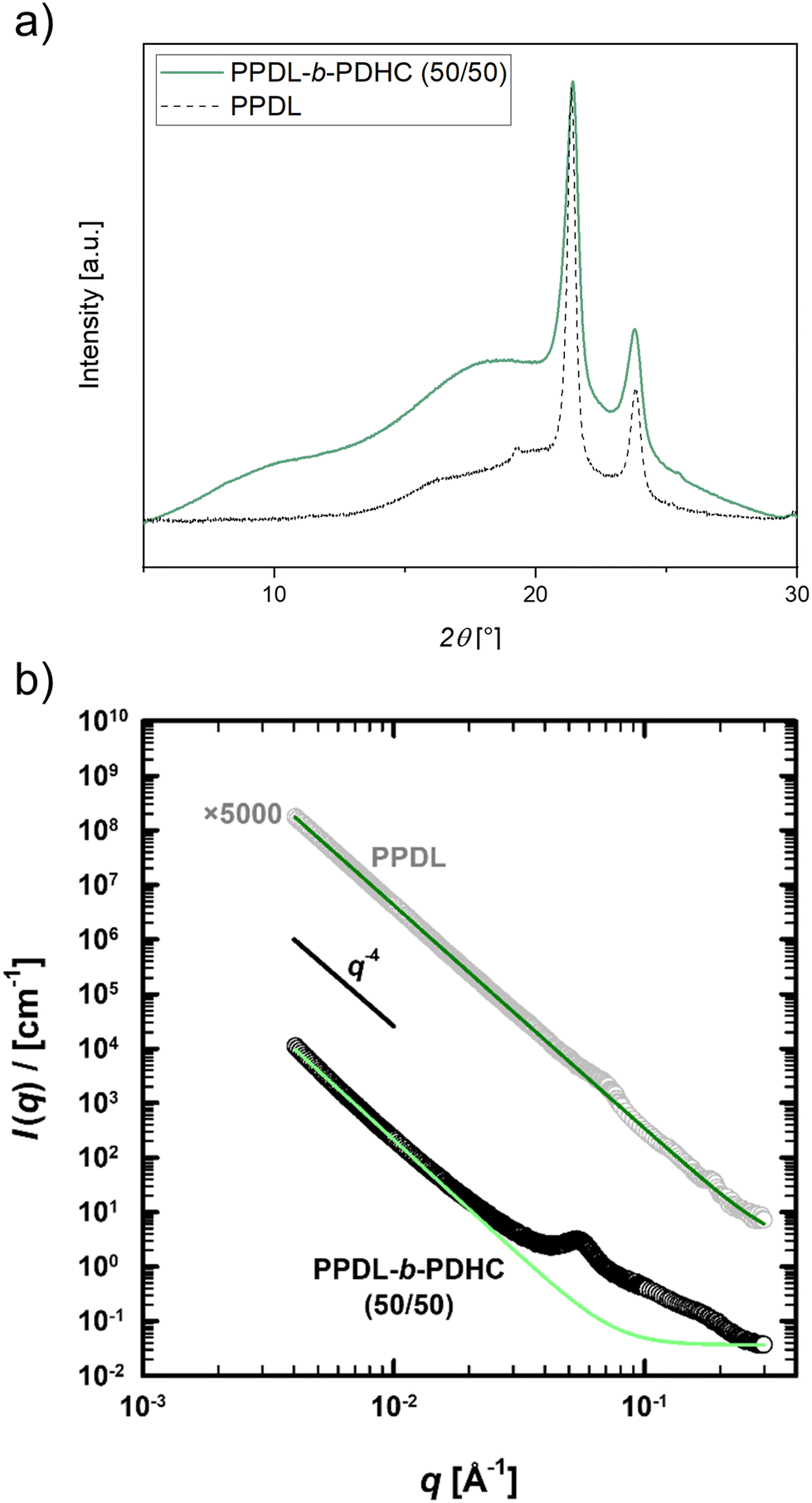 | ||
| Fig. 3 (a) Powder X-ray diffractograms of PPDL-b-PDHC (50/50) (Table 2, entry 3) and PPDL. For PPDL, as reported in a previous publication.30 (b) Background-corrected SAXS curves (circles) fitted according to eqn (S2)† (green lines) for PPDL homopolymer (light grey circles) synthesized with [(ONOO)tBuY(iPrO)(THF)] (Mn,SEC = 15800 g mol−1; Đ = 1.6) and PPDL-b-PDHC (black circles; 50/50; Table 2, entry 3). For clarity, the homopolymer plot is vertically offset by a factor of 5000. | ||
As more information on the size of these domains might be gained from scattering data at smaller angles, SAXS analysis was performed for both the PPDL homopolymer as well as the PPDL-b-PDHC block copolymer with a block composition of 50/50 (Table 2, entry 3). The corresponding scattering curves are shown in Fig. 3b as a function of the momentum transfer q = 4π sin(θ)/λ (scattering angle 2θ, wavelength λ = 1.54 Å). For small q-values, both curves show the characteristic q−4-decay of a two-phase system with a sharp interface,72 which results from the microscopic granular structure of these polymers and was fitted according to eqn (S2)† (green lines). The scattering curve of the block copolymer (black circles) further shows an additional scattering contribution in the q-range from 0.02 to 0.3 Å−1 including a distinct scattering peak at qmax = 0.053 Å−1, which can be attributed to amorphous or crystalline domains. To a much lesser extent, a scattering shoulder can also be recognized in the scattering curve of the PPDL homopolymer (light grey circles) at qmax = 0.065 Å−1. Besides the rise in intensity, the analysis of the position of the two maxima (eqn (S3), Fig. S41†) with regard to the corresponding domain size d ∼ 2π/qmax shows the growth of these domains from 97 Å for the homopolymer to 118 Å with the addition of PDHC, indicating that the respective domains may be amorphous. Overall, the SAXS results thus verify the semi-crystalline nature of both the PPDL homopolymer and the PPDL-b-PDHC block copolymer.
PDHC-b-PHB copolymers
For synthesizing PDHC-b-PHB copolymers, DHC was incorporated as the first block due to its lower coordination strength compared to the smaller four-membered β-butyrolactone. Similar to the PPDL-b-PDHC copolymers, these block copolymers were produced using [(ONOO)tBuY(bdsa)(THF)] (Table 3, entry 1) and [(ONOO)tBuY(iPrO)(THF)] (Table 3, entries 2–4) via sequential addition (Fig. 2b, Fig. S42†). The DOSY NMR spectrum obtained with the amide catalyst revealed two distinct species corresponding to the homopolymers (Fig. S46†), suggesting insufficient linkage as also observed in the SEC measurement (Fig. S49†), suggesting side reactions and an absence of a living-type polymerization. In contrast, block copolymers were produced using the alkoxide catalyst as proven by DOSY NMR spectroscopy and SEC (Fig. S47 and S50†). This conclusion was further supported when comparing the DOSY spectrum of the block copolymer to that of a blend of PDHC and PHB homopolymers with the same composition (Fig. S48†), where two distinct homopolymer species were visible. SEC traces showed no shoulders, indicating a well-defined molar mass distribution and a shift towards higher number-average molecular weights upon the addition of the second block (Fig. S50†). 13C NMR spectroscopy was used for the determination of tacticity by calculating the probability of racemic linkages between BBL monomer units (Fig. S45†), since aminoalkoxybis(phenolate) rare-earth metal complexes are known for their stereoselective BBL polymerization.39,42,43,68,73The tacticity analysis revealed a syndiotactic stereostructure of PHB, with Pr values between 0.73 and 0.76. The block copolymerization process did not alter the tacticity in comparison the respective PHB homopolymers, consistent with previous findings in PPDL-b-PHB copolymerization.30
Thermal analysis using DSC revealed two glass transition temperatures and a melting temperature, consistent with the amorphous nature of PDHC and the semi-crystalline nature of PHB. The DSC curves showed microphase separation in both samples, with Tg values of −10 to −4 °C and 0 to 5 °C, and Tm values between 103 and 112 °C (Table 3, entries 1–4) (Fig. S51 and S52†). The presence of an exothermic event prior to melting suggests cold crystallization or a reorganization process within the crystalline domains.
The crystallinity of PDHC-b-PHB was examined using PXRD measurements, with the baseline-corrected diffraction patterns normalized to the 22.8° reflection for copolymers with varying block compositions (16/84, 27/73, 52/48) (Fig. 4a). The homopolymer PHB exhibits characteristic reflections at 11.5°, 16.2°, 19.7°, 22.8°, 25.2° and 28.4°, which are attributed to its crystalline regions, beside the diffuse amorphous background.30 The PXRD patterns of PDHC-b-PHB reveal these crystalline reflections, indicating that the PHB block maintains its semi-crystalline structure within the block copolymer. However, as the PDHC content increases, these reflections become broadened, and the intensity of the amorphous background increases, suggesting that the presence of PDHC disrupts the crystallinity of the PHB phase to some extent. These results confirm the partial preservation of the crystalline structure of PHB, even with the integration of the amorphous PDHC phase.
 | ||
| Fig. 4 (a) Powder X-ray diffractogram of PDHC-b-PHB (16/84) (Table 3, entry 2); PDHC-b-PHB (27/73) (Table 3, entry 4); PDHC-b-PHB (52/48) (Table 3, entry 3) and PHB. For PHB, as reported in a previous publication.30 (b) Background-corrected SAXS curves (circles) fitted according to eqn (S15)† (blue lines) for: PHB homopolymer (light grey circles) synthesized with [(ONOO)tBuY(iPrO)(THF)] (Mn,SEC = 5800 g mol−1; Đ = 1.2), PDHC-b-PHB (dark grey circles; 16/84; Table 3 entry 2) and PDHC-b-PHB (black circles; 52/48; Table 3, entry 3). For clarity, plots and curves are vertically offset by the given factor. | ||
Domain structures of the PHB homopolymer and the PDHC-b-PHB block copolymers were investigated via SAXS (Fig. S53†). Fig. 4b shows the corresponding I(q)-scattering curves of the homopolymer (light grey circles) as well as of the block copolymers with block compositions of 16/84 (dark grey circles; Table 3, entry 2) and 52/48 (black circles; Table 3, entry 3). Due to their microscopic granular structure, the PHB-based (co)polymers also display a scattering intensity which follows I(q) ∝ q−4 for small q values. Likewise, their semi-crystalline nature leads to additional scattering contributions in the range of q = 0.06 to 0.4 Å−1 (PHB), and q = 0.005 to 0.4 Å−1 for PDHC-b-PHB, corresponding to amorphous or crystalline domains. Similar to other PHB-based (co)polymers,44 the scattering contribution of these domains can be described according to eqn (S15)† (blue lines) using a structural model combining the form factor of randomly oriented spheroids74 with an effective structure factor75 based on the hard-sphere model.76–78 For the PHB homopolymer, the analysis of the form factor provides a domain size of 10 Å along (polar) and 55 Å perpendicular (equatorial) to the rotational axis of the spheroid. By adding PDHC, the domains grow in both of these direction with increasing PDHC content, from 10 over 58 to 78 Å in the polar and from 55 over 204 to 272 Å in the equatorial direction. With polar-to-equatorial aspect ratios of 0.18 (PHB), 0.28 (16/84) and 0.29 (52/48), all form factors however indicate disc-like domain structures. In addition to the growth of the domains, the structure factors further show the increase of the effective volume fraction of these domains with increasing amount of the amorphous PDHC, from 18.9% in the PHB homopolymer via 29.5% in the PDHC-b-PHB block copolymer with a block composition of 16/84 to 35.2% with a block composition of 52/48. As for the PPDL-b-PDHC, the SAXS analysis of the PHB-based (co)polymers thus emphasize the semi-crystalline character of these polymers, while also suggesting that the additional scattering contribution originates from amorphous domains.
Post-polymerization functionalization by a “grafting from” approach
Grafting from polymer brushes offer a precise method for growing polymer chains directly from a reactive polymer backbone (Fig. 5). In this work, the combination of ROP to form the PDHC backbone, with its pendant double bonds, and ATRP with activator regeneration (particularly, SARA and ARGET ATRP) was utilized. | ||
| Fig. 5 Top: Scheme of the synthesis of the PDHC–2ME–Br macroinitiator. Bottom: 1H NMR spectrum of PDHC–2-ME–Br (CDCl3, 400 MHz). | ||
Macroinitiators were synthesized with varying molecular weights in which PDHC was either polymerized, precipitated, and redissolved for further thiol–ene reaction or, in the case of low-molecular weight PDHC (Table 1, entry 11; 3.5 kg mol−1), thiol–ene reactions were directly performed with the reaction mixture from polymerization. Double bonds were transformed via a thiol–ene reaction with 2-mercaptoethanol resulting in PDHC–2ME–OH, introducing hydroxyl groups. This reaction proceeded under UV irradiation overnight using 2,2-dimethoxy-2-phenylacetophenone as a photoinitiator. The absence of the doublet signal corresponding to the side-chain double bonds in the 1H NMR spectrum confirmed successful and complete modification of the double bonds (Fig. S54†).
Additionally, the triplet associated with the thiol proton of 2-mercaptoethanol, corresponding to unreacted species, was no longer detectable, while a new signal for the hydroxyl proton emerged, which was a broad singlet at 3.0 ppm. Multiplet signals at 3.7 and 2.7 ppm were attributed to the methylene groups in the attached 2-mercaptoethanol units, as confirmed by COSY NMR spectroscopy (Fig. S55†). DOSY NMR spectroscopy provided further evidence of successful modification, as only a single diffusion coefficient was detected (Fig. S56†). Elemental analysis supported the modification, with measured values matching the theoretical composition. The successful attachment of the ATRP initiator to the PDHC–2ME–OH backbone was achieved using the acid halide-functionalized 2-bromoisobutyryl bromide under full consumption of the hydroxyl group resulting in PDHC–2ME–Br (Fig. 5 and Fig. S58†). A distinct singlet at 1.9 ppm, assigned to the two methyl groups of the ATRP initiator, appeared, further supporting the successful attachment. For the detailed assignment of proton signals, COSY NMR spectroscopy was employed (Fig. S59†). Additionally, DOSY NMR spectroscopy and SEC analysis verified the presence of one single species (Fig. S60, S62 and S63†). Using elemental analysis, the measured carbon and hydrogen content closely matched the calculated values.
PDHC–2ME–Br was utilized as a macroinitiator for ATRP reactions, specifically SARA and ARGET ATRP, to polymerize ethyl acrylate to synthesize PDHC-g-PEA brush polymers. In SARA ATRP, copper wire served as a source of copper(0) species, acting as a supplemental activator and reducing agent, combined with a copper(II) bromide/tris(2-dimethylaminoethyl)amine (CuBr2/Me6TREN) catalyst system. Ethyl acrylate was added in two different amounts reaching target DPs of 10 (Table 4, entry 1) and 15 (Table 4, entry 2), relative to the double bonds present on the PDHC backbone. Higher target DPs were excluded to prevent solubility issues. The NMR spectra of the SARA ATRP grafting from reactions confirmed a successful polymerization: 1H NMR spectroscopy indicated a PDHC:PEA ratio matching the targeted DPs and conversions, facilitating a precise grafting approach (Fig. S66†). DOSY NMR spectra confirmed the covalent linkage between PDHC and PEA, ruling out the presence of unbound polymer chains (Fig. 6a, Fig. S68†). SEC analysis showed molecular weight increase with increasing target DPs, from 7.8 kg mol−1 for the PDHC–2ME–Br macroinitiator to 28.7 kg mol−1 for DP = 15 (Table 3, entry 2), while maintaining low polydispersities (Fig. S69†).
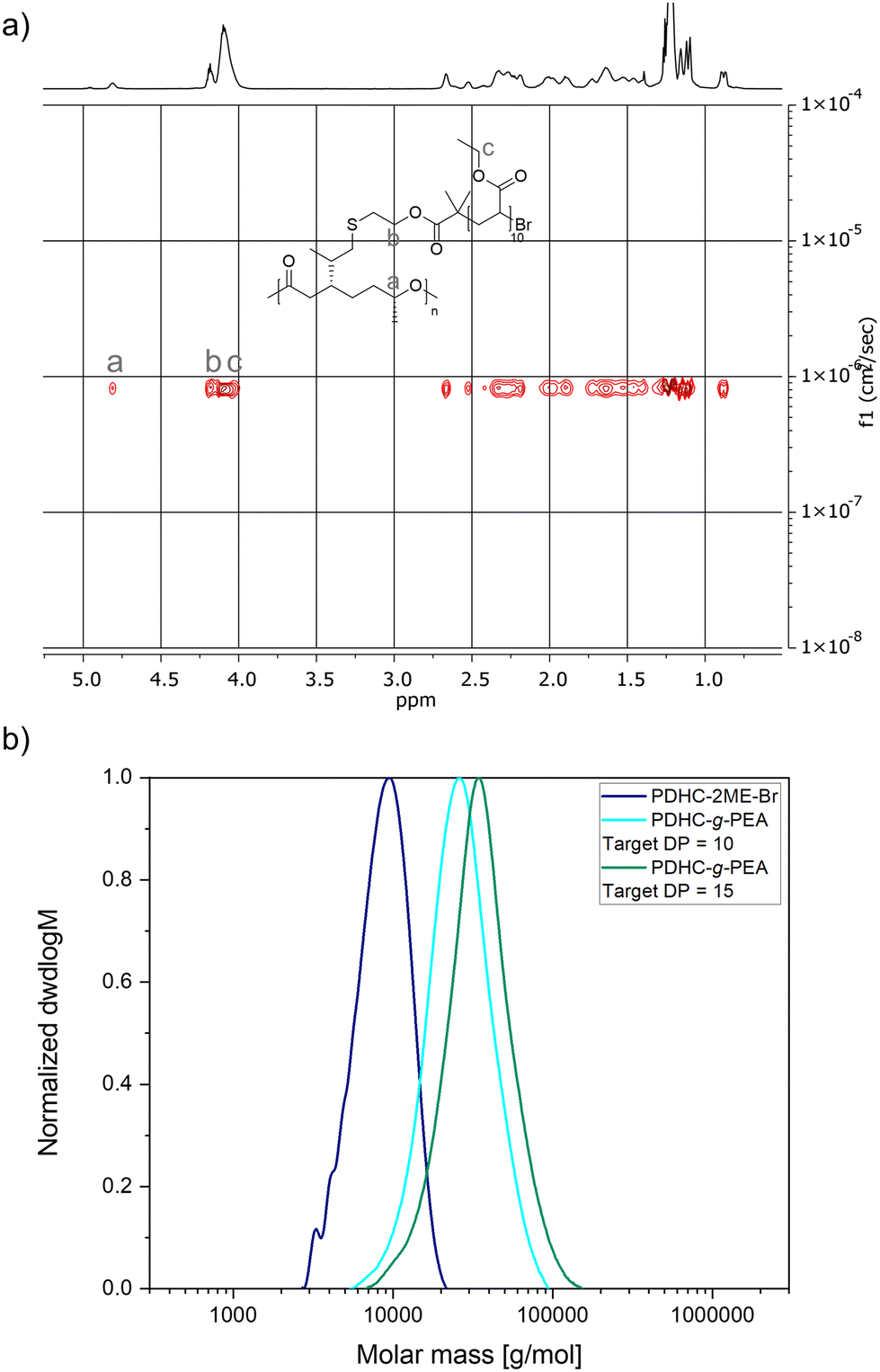 | ||
| Fig. 6 (a) DOSY NMR spectrum of PDHC-g-PEA grafted via SARA ATRP with 10 equivalents of ethyl acrylate (CDCl3, 400 MHz, number of scans: 16, resolution factor: 1, repetitions: 1, points in diffusion dimension: 128). (b) SEC traces of PDHC–2ME–Br and PDHC-g-PEA with targeted DP = 10 and target DP = 15, synthesized via ARGET ATRP. | ||
| Entrya | ATRP method | Target DP | Time [min] | T [°C] | Conv.EAd [%] | M n,SEC [kg mol−1] | Đ |
|---|---|---|---|---|---|---|---|
| a ATRP reactions were performed with PDHC–2ME–Br with Mn,SEC = 7.8 kg mol−1 (Đ = 1.2) in N,N-dimethylformamide. b Feed ratio (SARA ATRP): ethyl acrylate/double bond per repeating unit in PDHC–2ME–Br/Me6TREN/CuBr2 = target DP/1/0.18/0.05. c Feed ratio (ARGET ATRP): ethyl acrylate/double bond per repeating unit in PDHC–2ME–Br/TPMANMe2/CuBr2 = target DP/1/0.18/0.18. d Determined via1H NMR spectroscopy, using the integration of the –CH2– signals from PEA and comparing them to the monomer vinyl signals (Fig. S67†). e Measured via SEC in chloroform at 40 °C relative to polystyrene standards. | |||||||
| 1 | SARA | 10b | 247 | rt | 80 | 18.6 | 1.3 |
| 2 | SARA | 15b | 247 | rt | 96 | 28.7 | 1.4 |
| 3 | ARGET | 10c | 179 | 50 | 84 | 23.4 | 1.2 |
| 4 | ARGET | 15c | 180 | 50 | 90 | 30.5 | 1.2 |
Given the recent advances in ATRP, polymerization was also performed in an ARGET ATRP approach with tris[(4-dimethylaminopyridyl)methyl]amine (TPMANMe2) as a highly active ligand and combined with elemental silver as a potent reducing agent (Table 4, entries 3 and 4).61 As with SARA ATRP, polymerization proceeded efficiently, yielding defined graft polymers. SEC analysis revealed molecular weight increases consistent with the target DPs (Fig. 6b, Fig. S72†), while 1H and DOSY NMR confirmed successful grafting (Fig. S70 and S71†).
A key distinction was the slightly lower polydispersities (Đ = 1.2) observed in ARGET ATRP, suggesting improved control over the polymerization. However, both methods effectively produced well-defined polymer-brushes with molecular weights correlating well with the amount of monomer applied.
Thermogravimetric analysis (TGA) revealed that PDHC and its derivates exhibited distinct thermal stabilities (Fig. S73†). PDHC and PDHC–2ME–OH degraded around 300 °C, whereas the modified PDHC–2ME–Br showed lower thermal stability, with earlier degradation at approximately 200 °C. Likely, the introduction of bromo isobutyryl reduces the thermal stability.
In contrast, PDHC-g-PEA exhibited enhanced thermal stability, similar to that of poly(ethyl acrylate) (PEA), with degradation occurring between 300 °C and 450 °C, likely due to the stabilizing influence of the PEA side chains. DSC analysis of PDHC–2ME–Br and PDHC-g-PEA showed that all samples were completely amorphous (Fig. S74 and S75†).
Conclusions
This work presents a comprehensive approach to the polymerization and functionalization of dihydrocarvide, resulting in the synthesis of poly(dihydrocarvide) and its subsequent modification. Through the use of a controlled ring-opening polymerization using amino-alkoxy-bis(phenolate) yttrium catalysts, PDHC was successfully synthesized via ROP, preserving all double bonds of DHC within the PDHC polymer. With the [(ONOO)tBuY(bdsa)(THF)] catalyst at ambient temperature and 60 °C, target DPs from 25 to 100, and molecular weights of up to 34.5 kg mol−1 were achieved, with moderate polydispersities (Đ < 1.5). Reaction conditions identified as favourable were achieved by a polymerization temperature of 60 °C and using the [(ONOO)tBuY(iPrO)(THF)] catalyst to improve control over chain length, molecular weight, and molecular weight distribution while maintaining complete retention of the double bonds.The introduction of semi-crystalline blocks via sequential ROP, including PPDL and syndiotactic PHB, enhanced the structural properties of amorphous PDHC, adding crystallinity to the resulting PPDL-b-PDHC and PDHC-b-PHB block copolymers. DOSY NMR confirmed the successful block copolymer formation, while PXRD analysis demonstrated the preservation of crystalline PPDL and PHB domains. However, the broadening of PXRD reflections suggests that amorphous PDHC disrupted the crystallinity of the other block. SAXS analysis further verified the semi-crystalline morphology, revealing amorphous and crystalline domains, with additional scattering contributions arising from the amorphous regions upon increasing the PDHC content.
Additionally, PDHC was functionalized for graft polymerization in a post-polymerization functionalization approach by introducing hydroxyl groups, enabling the attachment of ATRP initiators. This approach allowed subsequent grafting of ethyl acrylate via SARA and ARGET ATRP with different target DPs. Both methods yielded defined PDHC-g-PEA brush polymers with molecular weights correlating well with the amount of EA used with ARGET ATRP indicating a slightly better polymerization control.
Overall, this study shows the versatility of PDHC in various polymer architectures, ranging from block copolymers to graft polymers, further demonstrating the potential of combining ROP and ATRP techniques. The results of well-structured polymer architectures emphasize the utility of controlled polymerization strategies in tailoring polymer properties for advanced materials. The approach of synthesizing PDHC via a living-type polymerization enables twofold control over material properties: (1) through the inherent characteristics of the block components and (2) via post-polymerization modifications.
Author contributions
Lea-Sophie Hornberger: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – initial draft; Julian Fischer: investigation, data curation, formal analysis, validation, writing – review & editing; Alexandra Friedly: investigation, data curation, formal analysis, validation; Ingo Hartenbach: resources, validation; Thomas Sottmann: resources, validation; Friederike Adams: conceptualization, funding acquisition, methodology, project administration, resources, supervision, validation, visualization, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.† Further data will be made available on request.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
L.-S. H. and F. A. are thankful for funding by the Federal Ministry of Education and Research (BMBF) and the Baden-Württemberg Ministry of Science as part of the Excellence Strategy of the German Federal and State Governments. The authors thank Dr. Dongren Wang (University of Stuttgart) for help with the MALDI-TOF measurements, and Philipp Weingarten (Technical University of Munich) for help with the ESI-MS measurements and the synthesis of TPMANMe2. The authors thank the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; award number 504929269) and the Carl-Zeiss-Foundation for funding of the Xeuss diffractometer.References
- R. Heijungs, G. Huppes and J. B. Guinée, Polym. Degrad. Stab., 2010, 95, 422–428 CrossRef CAS.
- L. Ruzicka, Experientia, 1953, 9, 357–367 CrossRef CAS PubMed.
- E. M. Davis and R. Croteau, in Biosynthesis: Aromatic Polyketides, Isoprenoids, Alkaloids, ed. F. J. Leeper and J. C. Vederas, Springer Berlin Heidelberg, Berlin, Heidelberg, 2000, pp. 53–95 Search PubMed.
- R. G. Berger, Flavours and fragrances: chemistry, bioprocessing and sustainability, Springer Science & Business Media, 2007 Search PubMed.
- B. Singaram and J. Verghese, Perfum. Flavor., 1977, 2, 47–51 CAS.
- T. Suga, Bull. Chem. Soc. Jpn., 1958, 31, 569–577 CrossRef CAS.
- T. Shono, I. Nishiguchi, T. Yokoyama and M. Nitta, Chem. Lett., 1975, 4, 433–436 CrossRef.
- G. P. Hareau, M. Koiwa, S. Hikichi and F. Sato, J. Am. Chem. Soc., 1999, 121, 3640–3650 CrossRef CAS.
- W. Schwab, C. Fuchs and F.-C. Huang, Eur. J. Lipid Sci. Technol., 2013, 115, 3–8 CrossRef CAS.
- A. Thomas and Y. Bessiere, Nat. Prod. Rep., 1989, 6, 291–309 RSC.
- W. A. Tilden and W. Shenstone, J. Chem. Soc., 1877, 31, 554–561 RSC.
- E. E. Royals and S. E. Horne Jr, J. Am. Chem. Soc., 1951, 73, 5856–5857 CrossRef CAS.
- C. Bordenca, R. K. Allison and P. H. Dirstine, Ind. Eng. Chem., 1951, 43, 1196–1198 CrossRef CAS.
- A. D. Silva, M. L. Patitucci, H. R. Bizzo, E. D'Elia and O. A. C. Antunes, Catal. Commun., 2002, 3, 435–440 CrossRef CAS.
- Y. Li, Y. Yang, D. Chen, Z. Luo, W. Wang, Y. Ao, L. Zhang, Z. Yan and J. Wang, Catalysts, 2019, 9, 374 CrossRef CAS.
- J. Li, Z. Li, G. Zi, Z. Yao, Z. Luo, Y. Wang, D. Xue, B. Wang and J. Wang, Catal. Commun., 2015, 59, 233–237 CrossRef CAS.
- K. B. Wiberg and S. D. Nielsen, J. Org. Chem., 1964, 29, 3353–3361 CrossRef CAS.
- S. M. Linder and F. P. Greenspan, J. Org. Chem., 1957, 22, 949–951 CrossRef CAS.
- J. Młodzik, A. Wróblewska, E. Makuch, R. J. Wróbel and B. Michalkiewicz, Catal. Today, 2016, 268, 111–120 CrossRef.
- L. A. Gallego-Villada, J. Cueto, M. D. M. Alonso-Doncel, P. Mäki-Arvela, E. A. Alarcón, D. P. Serrano and D. Y. Murzin, Green Chem., 2024, 26, 10512–10528 RSC.
- L. A. Gallego-Villada, W. Y. Perez-Sena, J. E. Sánchez-Velandia, J. Cueto, M. del Mar Alonso-Doncel, J. Wärmå, P. Mäki-Arvela, E. A. Alarcón, D. P. Serrano and D. Y. Murzin, Chem. Eng. J., 2024, 498, 155377 CrossRef CAS.
- J. R. Lowe, M. T. Martello, W. B. Tolman and M. A. Hillmyer, Polym. Chem., 2011, 2, 702–708 RSC.
- G. R. Krow, Org. React., 2004, 43, 251–798 Search PubMed.
- G. J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Chem. Rev., 2004, 104, 4105–4124 CrossRef PubMed.
- K. Balke, S. Schmidt, M. Genz and U. T. Bornscheuer, ACS Chem. Biol., 2016, 11, 38–43 CrossRef CAS PubMed.
- S. C. Knight, C. P. Schaller, W. B. Tolman and M. A. Hillmyer, RSC Adv., 2013, 3, 20399–20404 RSC.
- H. L. Messiha, S. T. Ahmed, V. Karuppiah, R. Suardiaz, G. A. Ascue Avalos, N. Fey, S. Yeates, H. S. Toogood, A. J. Mulholland and N. S. Scrutton, Biochemistry, 2018, 57, 1997–2008 CrossRef CAS PubMed.
- J. A. Wilson, S. A. Hopkins, P. M. Wright and A. P. Dove, Biomacromolecules, 2015, 16, 3191–3200 CrossRef CAS PubMed.
- J. R. Lowe, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2009, 10, 2003–2008 CrossRef CAS PubMed.
- L.-S. Hornberger, P. Weingarten, P. L. Lange, T. Schleid and F. Adams, Eur. Polym. J., 2023, 199, 112449 CrossRef CAS.
- M. Kränzlein, T. M. Pehl, F. Adams and B. Rieger, Macromolecules, 2021, 54, 10860–10869 CrossRef.
- L. Al-Shok, D. M. Haddleton and F. Adams, in Advances in Polymer Science, Springer Berlin Heidelberg, Berlin, Heidelberg, 2022 Search PubMed.
- D. M. Lyubov, A. O. Tolpygin and A. A. Trifonov, Coord. Chem. Rev., 2019, 392, 83–145 CrossRef CAS.
- J. F. Carpentier, Organometallics, 2015, 34, 4175–4189 CrossRef CAS.
- X. Dong and J. R. Robinson, Chem. Sci., 2020, 11, 8184–8195 RSC.
- F. Adams, Macromol. Rapid Commun., 2024, 45, 2400122 CrossRef CAS PubMed.
- C.-X. Cai, L. Toupet, C. W. Lehmann and J.-F. Carpentier, J. Organomet. Chem., 2003, 683, 131–136 CrossRef CAS.
- C. X. Cai, A. Amgoune, C. W. Lehmann and J. F. Carpentier, Chem. Commun., 2004, 330–331, 10.1039/b314030j.
- A. Amgoune, C. M. Thomas, S. Ilinca, T. Roisnel and J. F. Carpentier, Angew. Chem., Int. Ed., 2006, 45, 2782–2784 CrossRef CAS PubMed.
- A. Amgoune, C. M. Thomas, T. Roisnel and J. F. Carpentier, Chemistry, 2005, 12, 169–179 CrossRef PubMed.
- A. Amgoune, C. M. Thomas and J. F. Carpentier, Macromol. Rapid Commun., 2007, 28, 693–697 CrossRef CAS.
- N. Ajellal, J.-F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363–8376 RSC.
- N. Ajellal, M. Bouyahyi, A. Amgoune, C. M. Thomas, A. Bondon, I. Pillin, Y. Grohens and J.-F. Carpentier, Macromolecules, 2009, 42, 987–993 CrossRef CAS.
- F. Adams, T. M. Pehl, M. Kränzlein, S. A. Kernbichl, J.-J. Kang, C. M. Papadakis and B. Rieger, Polym. Chem., 2020, 11, 4426–4437 RSC.
- M. A. Cortez and S. M. Grayson, Macromolecules, 2010, 43, 4081–4090 CrossRef CAS.
- S. Khan, Z. Wang, R. Wang and L. Zhang, Mater. Sci. Eng., C, 2016, 67, 554–560 CrossRef CAS PubMed.
- M. Claudino, M. Jonsson and M. Johansson, RSC Adv., 2013, 3(27), 11021–11034 RSC.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- M. Hong, S.-R. Liu, B.-X. Li and Y.-S. Li, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2499–2506 CrossRef CAS.
- J. Babinot, E. Renard, B. Le Droumaguet, J. M. Guigner, S. Mura, J. Nicolas, P. Couvreur and V. Langlois, Macromol. Rapid Commun., 2013, 34, 362–368 CrossRef CAS PubMed.
- F. Ercole, A. E. Rodda, L. Meagher, J. S. Forsythe and A. P. Dove, Polym. Chem., 2014, 5, 2809–2815 RSC.
- Z. Li, Y. Qian, Y. Lai, F. S. Du and Z. C. Li, Biomacromolecules, 2022, 23(12), 5213–5224 CrossRef CAS PubMed.
- L. Al-Shok, J. S. Town, D. Coursari, P. Wilson and D. M. Haddleton, Polym. Chem., 2023, 14, 2734–2741 RSC.
- K. Matyjaszewski, H. Dong, W. Jakubowski, J. Pietrasik and A. Kusumo, Langmuir, 2007, 23, 4528–4531 CrossRef CAS PubMed.
- W. Jakubowski, K. Min and K. Matyjaszewski, Macromolecules, 2006, 39, 39–45 CrossRef CAS.
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Huang, W. A. Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15309–15314 CrossRef CAS PubMed.
- V. A. Williams and K. Matyjaszewski, Macromolecules, 2015, 48, 6457–6464 CrossRef CAS.
- V. A. Williams, T. G. Ribelli, P. Chmielarz, S. Park and K. Matyjaszewski, J. Am. Chem. Soc., 2015, 137, 1428–1431 CrossRef CAS PubMed.
- D. Konkolewicz, Y. Wang, M. Zhong, P. Krys, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2013, 46, 8749–8772 CrossRef CAS.
- T. G. Ribelli, M. Fantin, J.-C. Daran, K. F. Augustine, R. Poli and K. Matyjaszewski, J. Am. Chem. Soc., 2018, 140, 1525–1534 CrossRef CAS PubMed.
- P. Weingarten and F. Adams, ACS Macro Lett., 2024, 13, 1318–1324 CrossRef CAS PubMed.
- S. S. Sheiko, F. C. Sun, A. Randall, D. Shirvanyants, M. Rubinstein, H. I. Lee and K. Matyjaszewski, Nature, 2006, 440, 191–194 CrossRef CAS PubMed.
- S. S. Sheiko, S. A. Prokhorova, K. L. Beers, K. Matyjaszewski, I. I. Potemkin, A. R. Khokhlov and M. Möller, Macromolecules, 2001, 34, 8354–8360 CrossRef CAS.
- P. T. Altenbuchner, A. Kronast, S. Kissling, S. I. Vagin, E. Herdtweck, A. Pothig, P. Deglmann, R. Loos and B. Rieger, Chem. – Eur. J., 2015, 21, 13609–13617 CrossRef CAS PubMed.
- P. Sysel, in Encyclopedia of Membranes, ed. E. Drioli and L. Giorno, Springer Berlin Heidelberg, Berlin, Heidelberg, 2016, pp. 66–68 Search PubMed.
- R. Reichardt and B. Rieger, in Synthetic Biodegradable Polymers, ed. B. Rieger, A. Künkel, G. W. Coates, R. Reichardt, E. Dinjus and T. A. Zevaco, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 49–90 Search PubMed.
- J. Cai, C. Liu, M. Cai, J. Zhu, F. Zuo, B. S. Hsiao and R. A. Gross, Polymer, 2010, 51, 1088–1099 CrossRef CAS.
- N. Ajellal, C. M. Thomas and J.-F. Carpentier, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3177–3189 CrossRef CAS.
- Y. Feng, X. Jin and J. N. Hay, Polym. J., 1998, 30, 215–221 CrossRef CAS.
- Y. Hu, J. Zhang, H. Sato, I. Noda and Y. Ozaki, Polymer, 2007, 48, 4777–4785 CrossRef CAS.
- M. Gazzano, V. Malta, M. L. Focarete, M. Scandola and R. A. Gross, J. Polym. Sci., Part B:Polym. Phys., 2003, 41, 1009–1013 CrossRef CAS.
- G. Porod, Kolloid-Z., 1951, 124, 83–114 CrossRef CAS.
- P. T. Altenbuchner, A. Kronast, S. Kissling, S. I. Vagin, E. Herdtweck, A. Pothig, P. Deglmann, R. Loos and B. Rieger, Chemistry, 2015, 21, 13609–13617 CrossRef CAS PubMed.
- A. Guinier, Ann. Phys., 1939, 11, 161–237 Search PubMed.
- M. Kotlarchyk and S. H. Chen, J. Chem. Phys., 1983, 79, 2461–2469 CrossRef CAS.
- J. K. Percus and G. J. Yevick, Phys. Rev., 1958, 110, 1 CrossRef CAS.
- M. Wertheim, Phys. Rev. Lett., 1963, 10, 321 CrossRef.
- D. J. Kinning and E. L. Thomas, Macromolecules, 1984, 17, 1712–1718 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00322a |
| This journal is © The Royal Society of Chemistry 2025 |