
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diastereoselective 1,3-nitrooxygenation of bicyclo[1.1.0]butanes†
Anirban
Maity
,
Kuruva
Balanna
,
Constantin G.
Daniliuc
 and
Armido
Studer
*
and
Armido
Studer
*
Organisch-Chemisches Institut, Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: studer@uni-muenster.de
First published on 25th March 2025
Abstract
Cyclobutanes are strained carbocycles found in many drugs and natural products. Herein, we report a diastereoselective 1,3-nitrooxygenation of bicyclo[1.1.0]butanes with tert-butylnitrite and TEMPO to access 1,1,3-trisubstituted cyclobutanes. Various bicyclo[1.1.0]butanes effectively participated in the radical reaction yielding the substituted cyclobutane scaffolds with excellent yields and diastereoselectivity. The reaction is catalyst-free, easy to perform, and scalable and can be conducted in open air. Products obtained serve as substrates for the synthesis of 1,1,3,3-tetrasubstituted cyclobutanes with good yields and diastereoselectivity.
Introduction
Cyclobutanes are important structural units found in many naturally occurring and pharmaceutically active bio-molecules (Scheme 1a).1 In this regard, nitrocyclobutanes1e,f are highly valuable intermediates in organic synthesis1g due to their unique dual reactivity, arising from both the strained cyclobutane core and the electron-withdrawing nitro group. The nitro functionality serves as a versatile handle for various transformations, including reductions to amines, Henry reactions, and Nef reactions, which facilitate the formation of ketones and other functional groups. More specifically, highly functionalized and well-decorated cyclobutanes are often challenging to synthesize in a straightforward manner. Major synthetic routes for the synthesis of these strained carbocycles include radical cyclization,2 Wolff-rearrangement,3 oxidative pinacol-rearrangement,4 and the recently reported nitrogen-deletion strategy.5 Photocatalyst mediated intermolecular [2 + 2] cycloaddition6 has been extensively used for the construction of such strained carbocycles (Scheme 1b). However, despite significant advancement in energy transfer mediated photochemical [2 + 2] cycloadditions the outcome of these reactions is often complicated because of regio- and diastereoselectivity issues,6 and careful selection of π-systems is often a prerequisite criterion to achieve the desired selectivity. In recent years an additional approach has emerged, which involves the exploitation of strained bicyclo[1.1.0]butanes (BCBs)7 to access a variety of complex cyclobutane scaffolds (Scheme 1c). Due to the 96% p-character of the central C–C bond of a BCB, such compounds show a diverse range of reactivities varying from nucleophilic, electrophilic and radicalophilic.7 The pioneering work of nucleophilic additions to BCBs was realized by Gaoni et al.8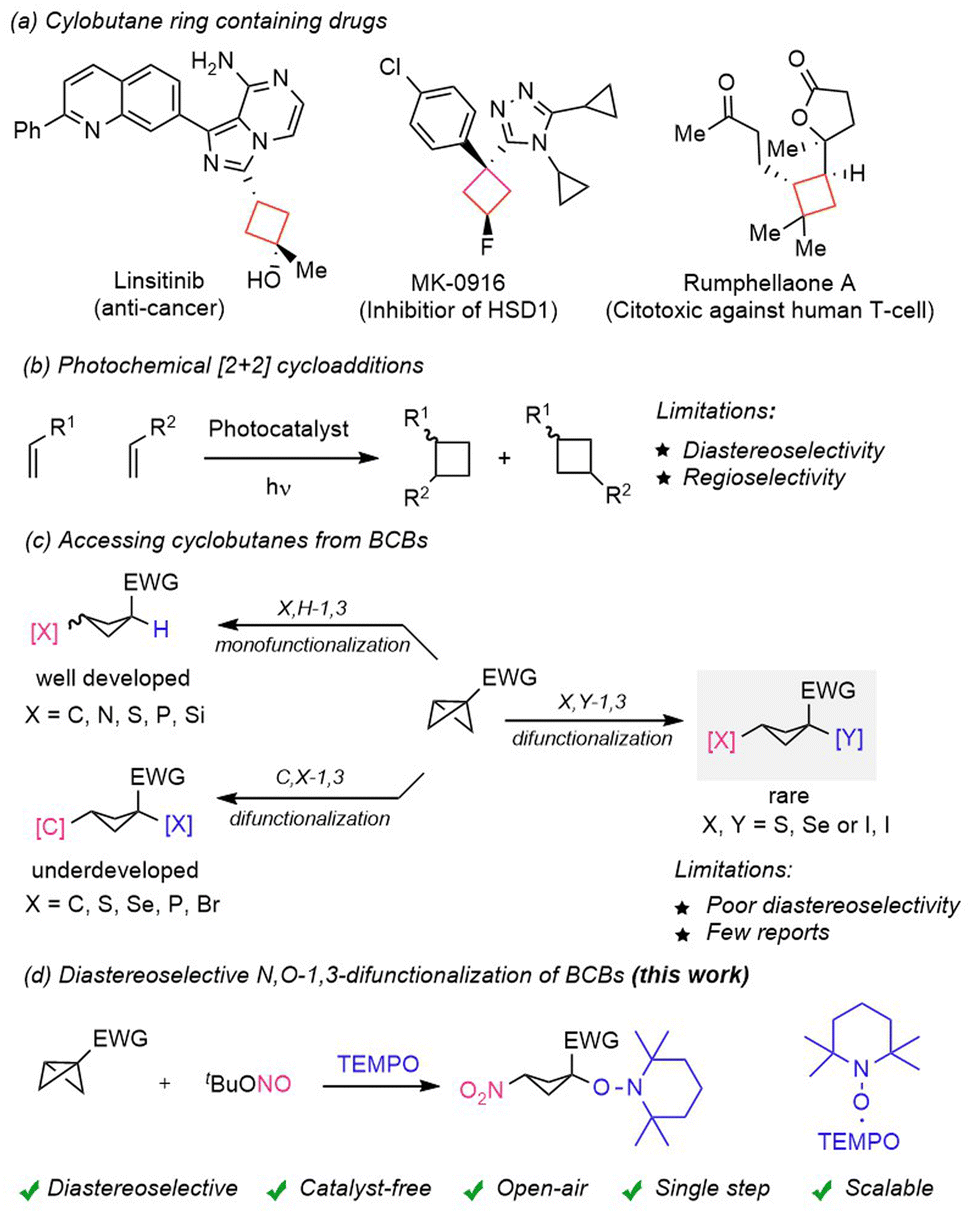 | ||
| Scheme 1 (a) Cyclobutane ring containing drugs. (b) Photochemical [2 + 2] cycloaddition. (c) Accessing cyclobutanes from BCBs. (d) This work: diastereoselective N,O-1,3-difunctionalization of BCBs. | ||
Moreover, the Baran group9 developed amination of BCBs using amines as nucleophiles. Biju et al.10 and Feng, Qu, Yu et al.11 independently developed diastereoselective arylation of disubstituted-BCBs. Along with nucleophilic ring-openings, several radical additions1,12 to BCBs were reported, predominantly for mono-functionalization. Considering difunctionalizing ring-opening of BCBs, except for few cases, the majority of these transformations suffer from low diastereoselectivity. Along these lines, diastereoisomerically enriched 1,1,3-trisubstituted cyclobutanes were successfully prepared by the Fox group13 through organocuprate addition and subsequent trapping using an external electrophile. The Glorius group14 developed a 1,3-carbothiolation of BCBs through an intramolecular group transfer process to secure high syn-selectivity. The Hari group15 presented a 1,3-carboheterodifunctionalization of BCBs for the synthesis of spirocyclobutyl lactones and lactams, albeit with moderate diastereocontrol. Boronate-BCBs were investigated by the Aggarwal group16 who achieved highly diastereoselective 1,3-carbodifunctionalization by employing various electrophiles or electrophilic radicals to induce the ring-opening process. The preinstalled boronate functionality controls both regio- and diastereoselectivity of the products, 1,1,3-trisubstituted cyclobutanes.
In addition to mono and 1,3-difunctionalization, cycloaddition17 and insertion reactions1 of BCBs have offered synthetic chemists direct and straightforward routes to access cyclobutane rings containing complex 3D-enriched scaffolds. However, only carbon–carbon (C–C) and carbon–heteroatom (C–S) 1,3-difunctionalization was presented in a diastereoselective manner to date. The diastereoselective heteroatom–heteroatom (X,Y) 1,3-difunctionalization of bicyclo[1.1.0]butanes is rare.18 In order to achieve a heteroatom–heteroatom, particularly (O–N) 1,3-difunctionalization, we considered using tert-butyl nitrite (tBuONO)19 as a nitrogen atom source in combination with the TEMPO radical20 as an oxygen atom source (Scheme 1d). tBuONO upon thermal homolysis will generate a persistent NO-radical, which in the presence of molecular oxygen will be oxidized to a transient NO2-radical. The NO2-radical should then add along the central C–C bond of the BCB to generate a transient adduct cyclobutyl radical, which can engage in a selective cross-coupling with the persistent TEMPO radical to afford the desired 1,3-difunctionalized cyclobutane product. Interestingly, in this reaction two persistent radicals (NO and TEMPO) are present and the persistent NO-radical is selectively converted to a transient NO2-radical during the course of the reaction, while TEMPO remains unchanged. The suggested difunctionalizing BCB ring-opening reaction presents challenges that are product stability21 and diastereoselectivity control, as radical/radical cross couplings generally occur with very low barriers.
Results and discussion
Readily prepared (see the ESI†) bicyclo[1.1.0]butan-1-yl(naphthalen-2-yl)methanone (1) was chosen as the model substrate. We were pleased to observe that the reaction of 1 with tBuONO (0.20 mmol, 2.0 equiv.) and TEMPO (0.15 mmol, 1.5 equiv.) in DMSO at 70 °C for 18 hours afforded the desired 1,3-nitrooxygenated product 2 in 38% NMR-yield, albeit with a moderate 2.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereoselectivity (Table 1, entry 1). Then, different solvents were tested under otherwise identical conditions. In CHCl3, yield of the product 2 increased to 82% but diastereoselectivity further decreased (Table 1, entry 2). Other solvents such as DCE (76%), toluene (48%), 1,4-dioxane (64%), and CH3CN (74%) afforded lower yields and lower diastereoselectivities (Table 1, entries 3–6). The lower efficiency of the transformation in toluene and 1,4-dioxane can be rationalized by considering the competitive hydrogen atom transfer (HAT) reaction with the reactive NO2-radical from the solvent.
:1 diastereoselectivity (Table 1, entry 1). Then, different solvents were tested under otherwise identical conditions. In CHCl3, yield of the product 2 increased to 82% but diastereoselectivity further decreased (Table 1, entry 2). Other solvents such as DCE (76%), toluene (48%), 1,4-dioxane (64%), and CH3CN (74%) afforded lower yields and lower diastereoselectivities (Table 1, entries 3–6). The lower efficiency of the transformation in toluene and 1,4-dioxane can be rationalized by considering the competitive hydrogen atom transfer (HAT) reaction with the reactive NO2-radical from the solvent.
|
|
||||
|---|---|---|---|---|
| # | Solvent | Temperature | dr | Yielda,b (%) |
| a Reaction conditions: 1 (0.1 mmol), tBuONO (0.20 mmol), TEMPO (0.15 mmol), solvent (0.1 M), air, 18 h. b Determined by 1H NMR using 1,3,5-trimethoxy benzene as an internal standard. c After removal of solvent the crude was re-dissolved in CHCl3 (0.1 M) in the presence of the given base (20 mol%) and stirred for 18 h at rt. d Isolated yield and diastereomeric ratio after silica gel column chromatography of the reaction mixture. | ||||
| 1 | DMSO | 70 °C | 2.2:1 |
38 |
| 2 | CHCl 3 | 70 °C |
1.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
82 |
| 3 | DCE | 70 °C | 1.4:1 |
76 |
| 4 | Toluene | 70 °C | 1.2:1 |
48 |
| 5 | 1,4-Dioxane | 70 °C | 1.1:1 |
64 |
| 6 | CH3CN | 70 °C | 1.0:1 |
74 |
| 7 | CHCl3 | 25 °C | 1.7:1 |
63 |
| 8 | CHCl3 | 100 °C | 3.9:1 |
81 |
| 9c | CHCl3 (DMAP) | 70 °C | 4.4:1 |
82 |
| 10c | CHCl3 (DBU) | 70 °C | 5.8:1 |
61 |
| 11c | CHCl3 (Cs2CO3) | 70 °C | 5.3:1 |
81 |
| 12 | CHCl 3 | 70 °C |
>20:1
|
74d |
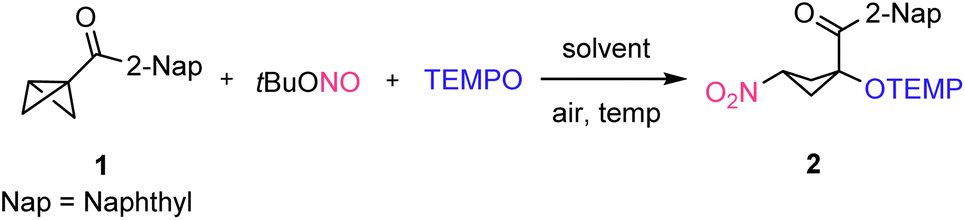
The optimization campaign was continued by varying reaction temperature using CHCl3 as the solvent. At room temperature a significantly lower yield (63%) resulted, possibly due to the slow decomposition of tBuONO (Table 1, entry 7). Upon increasing temperature from 70 °C to 100 °C the yield of the reaction remained unchanged (81%); however, an improved diastereoselectivity was noted. This surprising stereochemical outcome indicated that isomerization likely occurred after initial BCB-nitrooxygenation, probably through deprotonation/re-protonation of the acidic proton next to the nitro group. Despite the fact that reversible alkoxyamine C–O-bond homolysis in 2 could not be excluded as the isomerization process at that temperature, we tested whether the poor diastereomeric ratio can be improved by applying a deprotonation/re-protonation strategy through simple addition of a base. To this end, several bases were tested as catalysts for the isomerization of 2. After radical nitrooxygenation, the solvent was removed and the crude nitro-cyclobutane 2 was then re-dissolved in CHCl3 in the presence of DMAP (20 mol%) and stirred for 18 h at room temperature. The diastereoselectivity improved without compromising the yield (Table 1, entry 9, 82%, dr 4.4:1). With DBU (20 mol%) as the base, the diastereomeric ratio further improved, albeit at the expense of the NMR yield (Table 1, entry 10, 61%, dr 5.8:1). In the presence of Cs2CO3 (20 mol%), product 2 was obtained with a diastereomeric ratio of 5.3:1 without scarifying the yield (Table 1, entry 11). Pleasingly, upon purification of the crude product 2 through silica gel column chromatography without any prior base treatment, we found complete isomerization and 2 (syn-isomer) was isolated in 74% yield with an excellent diastereomeric ratio (dr > 20:1, Table 1, entry 12). The relative configuration was unambiguously assigned by X-ray analysis and other compounds prepared in this study were assigned in analogy. Obviously, the additional isomerization step using an external base is not required, as isomerization to the thermodynamic syn-product efficiently occurs during silica gel chromatography. With these optimized conditions in hand, we then investigated the scope of the reaction.
First, the aryl group of the keto BCB was varied. 1-Naphthyl and 6-methoxy-substituted 2-naphthyl-keto-BCBs afforded the desired products 3 and 4 in 73% and 86% yield with high diastereoselectivity (dr 20:1 and 12:1) (Scheme 2). The unsubstituted phenyl keto-BCB provided the desired cyclobutane 5 in 72% yield and high stereoselectivity (dr 13:1). Aryl keto-BCBs carrying an electron donating or withdrawing group at the aryl ring were eligible substrates. Thus, BCBs carrying an aryl group or an alkyl group at the 4-position of the aryl substituent such as 4-phenyl, 4-methyl, or 4-tert-butyl, afforded the desired products 6–8 with good to excellent yields (75–89%) and good to excellent diastereoselectivity (dr 8:1 to > 20:1). The reason for the varying selectivity as a function of the para-substituent is not fully understood. Along the same lines, electron-rich 4-MeOC6H4 and electron-poor 4-CF3C6H4-substituted BCBs afforded the desired products 9 and 10 in 76% and 83% yield, with good diastereoselectivity (dr 8:1 and 10:1). Renewed chromatography of 9 led to a slightly improved diastereoselectivity (11:1 versus 8:1). 4-Halo-substituted aryl keto-BCBs delivered the targeted 1,3-nitrooxygenated products 11–13 in good to excellent yields (80–94%) and good diastereoselectivity (dr 8:1 to 16:1). Installing a substituent at the meta position of the aryl ring such as in 3,5-dimethyl-C6H3, 3,5-dimethoxy-C6H3, and 3-fluoro-C6H4-substituted keto-BCBs afforded the desired products 14–16 in good to excellent yields (74–92%) and good to excellent diastereoselectivity (dr 8:1 to dr > 20:1). ortho-Methyl substituted phenyl keto-BCB delivered the 1,3-nitrooxygenated product 17 in 76% yield with high diastereoselectivity (dr 17:1). A 2,4-disubstituted phenyl keto-BCB worked well to afford 18 in 80% yield with good diastereoselectivity (dr 10:1). Heterocyclic rings such as thiophene and furan are tolerated and the corresponding 1,3-nitrooxygenated products 19 and 20 were isolated in 79% and 88% yields with high stereoselectivity (dr 16:1 and 11:1). However, a 1,3-disubstituted-BCB did not afford the desired 1,3-nitrooxygenated product 21, possibly because of steric reasons: the 3-phenyl substituent in the BCB hinders NO2-radical attack and ring opening does not occur.
 | ||
| Scheme 2 Reaction conditions: bicyclo[1.1.0]butane (0.2 mmol, 1.0 equiv.), tBuONO (0.4 mmol, 2.0 equiv.), TEMPO (0.3 mmol, 1.5 equiv.), CHCl3 (2.0 mL). Yields and the diastereomeric ratio are reported after isolation via column chromatography. aA second column chromatography was performed. | ||
Moreover, phenylsulfonyl-activated BCBs also engage in the radical cascade reaction, as shown by the successful synthesis of sulfone 23 that was isolated in 70% yield with excellent diastereoselectivity (dr > 20:1). Notably, this methodology is not restricted to aromatic keto-BCBs. It is also effective for the ring-opening di-functionalization of ester-BCBs, amide-BCBs, and alkyl keto-BCBs, yielding trisubstituted cyclobutane derivatives with good to excellent yields and low to moderate diastereoselectivity (22, 87%, dr 7:1; 24, 81%, dr 1:1; 25, 74%, dr 5:1; 26, 57%, dr 5:1).
The synthetic value of the nitrocyclobutane products was documented by conducting several follow-up reactions (Scheme 3). Michael reaction of 2 with methyl acrylate and acrylonitrile afforded the 1,1,3,3-tetrasubstituted cyclobutanes 27 and 28 in 99% and 76% yields with good to excellent diastereoselectivity (dr 7:1 and dr > 20:1). Palladium catalysed allylation of 2 with allyl alcohol was achieved to afford 29 in 68% yield with moderate diastereoselectivity (dr 5.2:1). The relative configuration of the separable isomers was assigned by NOE experiments (see the ESI†). Selectivity of the other products derived from 2 was assigned in analogy. para-Quinomethide as an electrophilic coupling partner delivered cyclobutane 30 in 78% yield, albeit with poor diastereoselectivity. Thermal homolysis of the weak C–O bond of alkoxyamine 2 in the presence of γ-terpene as a hydrogen atom transfer (HAT)22 reagent afforded the 1,3-disubstituted cyclobutane 31 in 44% yield (88%, brsm) and moderate diastereoselectivity. Hydrazination of nitrocyclobutane 2 under basic conditions gave cyclobutene 32 in 48% isolated yield. Hence, the targeted nitrocyclobutane obtained after initial hydrazination eliminates HNO2 under the applied conditions.
 | ||
| Scheme 3 Follow-up chemistry. aReaction conditions: 2 (0.15 mmol, 1.0 equiv.), methyl vinyl ketone (0.18 mmol, 1.2 equiv.), tetramethyl guanidine (0.075 mmol, 0.5 equiv.), THF (0.9 mL). bReaction conditions: 2 (0.15 mmol, 1.0 equiv.), acrylonitrile (0.18 mmol, 1.2 equiv.), tetramethyl guanidine (0.075 mmol, 0.5 equiv.), THF (0.9 mL). cReaction conditions: 2 (0.2 mmol, 1.0 equiv.), allyl alcohol (0.6 mmol, 3.0 equiv.), Pd(PPh3)4 (0.02 mmol, 0.1 equiv.), DMSO (1.2 mL). dReaction conditions: 2 (0.1 mmol, 1.0 equiv.), 2,6-di-tert-butyl-4-(2-hydroxybenzylidene)cyclohexa-2,5-dien-1-one (0.1 mmol, 1 equiv.), Cs2CO3 (0.11 mmol, 1.1 equiv.), Bi(OTf)3 (0.02 mmol, 0.2 equiv.) DCE (1.0 mL). eReaction conditions: 2 (0.1 mmol, 1.0 equiv.), γ-terpene (0.3 mmol, 3.0 equiv.), tert-butanol (5.0 mL). fReaction conditions: 2 (0.10 mmol, 1.0 equiv.), di-tert-butyl (E)-diazene-1,2-dicarboxylate (0.15 mmol, 1.5 equiv.), Cs2CO3 (0.11 mmol, 1.1 equiv.), DCE (1.0 mL). | ||
In order to study the effect of O2, the reaction of 1 was repeated under an argon atmosphere and only traces of product 2 were detected (Scheme 4a). Based on this observation and literature reports19 we propose a plausible mechanism that is depicted in Scheme 4b. Thermal homolysis of the N–O bond in tBuONO leads to the formation of the tert-butoxyl radical along with the persistent nitroso (NO) radical, which in the presence of molecular oxygen is directly oxidized to the NO2-radical. The NO2-radical then engages in a homolytic substitution at carbon cleaving the central C–C bond of the bicyclo[1.1.0]butane to generate the adduct radical 33. This adduct radical is then trapped by the persistent TEMPO radical23 to deliver the isolated product. Of note, the TEMPO-trapping does not occur with a high stereoselectivity and isomerization through a deprotonation/re-protonation sequence forming a thermodynamically more stable isomer during silica gel chromatography and ensures the very high diastereoselectivity observed in many of our transformations.
 | ||
| Scheme 4 (a) Control experiment. (b) Plausible reaction mechanism. | ||
Conclusions
In conclusion we have developed a highly diastereoselective 1,3-nitrooxygenation of bicyclo[1.1.0]butanes for the preparation of 1,1,3-trisubstituted cyclobutane scaffolds. It is shown that the radical ring-opening TEMPO-trapping occurs with low or moderate stereoselectivity. Nevertheless, due to the simple isomerization through deprotonation/re-protonation during silica gel chromatography, the thermodynamic syn-isomer can be obtained with good to excellent diastereoselectivity. The reported cascade worked for a wide variety of substrates, delivering the targeted nitrocyclobutane products in good to very good yields. Follow-up chemistry demonstrated that the ring-opening products formed through the radical cascade can be used as substrates for the synthesis of 1,1,3,3-tetrasubstituted cyclobutane scaffolds with good diastereocontrol.Data availability
Experimental procedures and analytical data (NMR, HRMS, IR, melting points) that support the findings of this study are available in the ESI.†Author contributions
A. M. and K. B. conducted all the experiments and characterized all novel compounds. C. G. D. measured and solved the X-ray crystal structure. A. M., K. B. and A. S. designed the experiments and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Münster for supporting this work. Dr Klaus Bergander (University of Münster) is acknowledged for running the NOE experiments.Notes and references
- (a) M. R. van der Kolk, M. A. C. H. Janssen, F. P. J. T. Rutjes and D. Blanco-Ania, ChemMedChem, 2022, 17, e202200020 CrossRef CAS PubMed; (b) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed; (c) A. V. Chernykh, D. S. Radchenko, A. V. Chernykh, I. S. Kondratov, N. A. Tolmachova, O. P. Datsenko, M. A. Kurkunov, S. X. Zozulya, Y. P. Kheylik, K. Bartels, C. G. Daniliuc and G. Haufe, Eur. J. Org Chem., 2015, 6466–6471 CrossRef CAS; (d) B. Ranieri, C. Obradors, M. Mato and A. M. Echavarren, Org. Lett., 2016, 18, 1614–1617 CrossRef CAS PubMed; (e) T. G. Archibald, L. C. Garver, K. Baum and M. C. Cohen, J. Org. Chem., 1989, 54, 2869–2873 CrossRef CAS; (f) G. A. OM, P. Ramaiah and G. K. Surya Prakash, J. Org. Chem., 1993, 58, 763–765 CrossRef; (g) M. A. C. H. Janssen, R. Rappard, M. J. van Well, D. B. Ania and F. P. J. T. Rutjes, Eur. J. Org Chem., 2024, 27, e202301309 CrossRef CAS.
- N. Zhao, S. Yin, S. Xie, H. Yan, P. Ren, G. Chen, F. Chen and J. Xu, Angew. Chem., Int. Ed., 2018, 57, 3386–3390 CrossRef CAS PubMed.
- L. M. Chapman, J. C. Beck, L. Wu and S. E. Reisman, J. Am. Chem. Soc., 2016, 138, 9803–9806 CrossRef CAS PubMed.
- P. S. Baran, T. J. Maimone and J. M. Richter, Nature, 2007, 446, 404–408 CrossRef CAS PubMed.
- C. Hui, L. Brieger, C. Strohmann and A. P. Antonchick, J. Am. Chem. Soc., 2021, 143, 18864–18870 CAS.
- (a) D. I. Schuster, G. Lem and N. A. Kaprinidis, Chem. Rev., 1993, 93, 3–22 CrossRef CAS; (b) S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CAS; (c) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 Search PubMed; (d) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 CAS.
- (a) M. Golfmann and J. C. L. Walker, Commun. Chem., 2023, 6, 9 CAS; (b) Q.-Q. Hu, J. Chen, Y. Yang, H. Yang and L. Zhou, Tetrahedron Chem., 2024, 9, 100070 CAS; (c) J. L. Tyler and V. K. Aggarwal, Chem.–Eur. J., 2023, 29, e202300008 CAS; (d) C. B. Kelly, J. A. Milligan, L. J. Tilley and T. M. Sodano, Chem. Sci., 2022, 13, 11721–11737 RSC.
- (a) Y. Gaoni, Tetrahedron Lett., 1982, 23, 5219–5220 CrossRef CAS; (b) Y. Gaoni, Tetrahedron Lett., 1982, 23, 5215–5218 CrossRef CAS; (c) Y. Gaoni, A. Tomazic and E. Potgieter, J. Org. Chem., 1985, 50, 2943–2947 CrossRef CAS; (d) Y. Gaoni and A. Tomazic, J. Org. Chem., 1985, 50, 2948–2957 CrossRef CAS; (e) Y. Gaoni, Tetrahedron Lett., 1988, 29, 1591–1594 CrossRef CAS; (f) Y. Gaoni, Tetrahedron, 1989, 45, 2819–2840 CrossRef CAS.
- (a) R. Gianatassio, J. M. Lopchuk, J. Wang, C.-M. Pan, L. R. Malins, L. Prieto, T. A. Brandt, M. R. Collins, G. M. Gallego, N. W. Sach, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, Science, 2016, 351, 241–246 CrossRef CAS PubMed; (b) J. M. Lopchuk, K. Fjelbye, Y. Kawamata, L. R. Malins, C.-M. Pan, R. Gianatassio, J. Wang, L. Prieto, J. Bradow, T. A. Brandt, M. R. Collins, J. Elleraas, J. Ewanicki, W. Farrell, O. O. Fadeyi, G. M. Gallego, J. J. Mousseau, R. Oliver, N. W. Sach, J. K. Smith, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 3209–3226 CrossRef CAS PubMed.
- (a) A. Guin, S. Bhattacharjee, M. S. Harariya and A. T. Biju, Chem. Sci., 2023, 14, 6585–6591 RSC; (b) A. Guin, S. Deswal, M. S. Harariya and A. T. Biju, Chem. Sci., 2024, 15, 12473–12479 RSC.
- L. Tang, Q.-N. Huang, F. Wu, Y. Xiao, J.-L. Zhou, T.-T. Xu, W.-B. Wu, S. Qu and J.-J. Feng, Chem. Sci., 2023, 14, 9696–9703 RSC.
- (a) J. Liu, Y. Qian, H. Zhao, Y. Liu, Z. Qin, Z. Zhang and L. Rong, J. Org. Chem., 2024, 89, 15914–15923 CrossRef CAS PubMed; (b) G. Chen, D. Tian, X. Wang and H.-J. Zhang, ACS Catal., 2024, 14, 14928–14936 CrossRef CAS; (c) P.-F. Xia, J. Zhou, J. Yuan, R. Zeng, Y. Liu, K.-W. Tang and J.-H. Fan, Org. Lett., 2024, 26, 6486–6490 CAS; (d) J.-H. Fan, J. Yuan, P.-F. Xia, J. Zhou, L.-J. Zhong, P.-F. Huang, Y. Liu, K.-W. Tang and J.-H. Li, Org. Lett., 2024, 26, 2073–2078 CrossRef CAS PubMed; (e) Z. Zhang and V. Gevorgyan, J. Am. Chem. Soc., 2022, 144, 20875–20883 CAS; (f) H. Pickford, V. Ripenko, R. McNamee, S. Holovchuk, A. Thompson, R. Smith, P. Mykhailiuk and E. Anderson, Angew. Chem., Int. Ed., 2023, 62, e202213508 CAS.
- R. Panish, S. R. Chintala, D. T. Boruta, Y. Fang, M. T. Taylor and J. M. Fox, J. Am. Chem. Soc., 2013, 135, 9283–9286 CAS.
- H. Wang, J. E. Erchinger, M. Lenz, S. Dutta, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2023, 145, 23771–23780 CAS.
- (a) K. Das, A. Pedada, T. Singha and D. P. Hari, Chem. Sci., 2024, 15, 3182–3191 CAS; (b) T. Singha, N. A. Bapat, S. K. Mishra and D. P. Hari, Org. Lett., 2024, 26, 6396–6401 CAS; (c) T. Singha and D. P. Hari, Synlett, 2025, 36, 445–451 CAS.
- (a) A. Fawcett, T. Biberger and V. K. Aggarwal, Nat. Chem., 2019, 11, 117–122 CAS; (b) S. H. Bennett, A. Fawcett, E. H. Denton, T. Biberger, V. Fasano, N. Winter and V. K. Aggarwal, J. Am. Chem. Soc., 2020, 142, 16766–16775 CAS; (c) M. Silvi and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 9511–9515 CAS.
- (a) R. Guo, Y.-C. Chang, L. Herter, C. Salome, S. E. Braley, T. C. Fessard and M. K. Brown, J. Am. Chem. Soc., 2022, 144, 7988–7994 CrossRef CAS PubMed; (b) R. Kleinmans, T. Pinkert, S. Dutta, T. O. Paulisch, H. Keum, C. G. Daniliuc and F. Glorius, Nature, 2022, 605, 477–482 CrossRef CAS PubMed; (c) S. Agasti, F. Beltran, E. Pye, N. Kaltsoyannis, G. E. M. Crisenza and D. J. Procter, Nat. Chem., 2023, 15, 535–541 CrossRef CAS PubMed; (d) L. Tang, Y. Xiao, F. Wu, J. Zhou, T. Xu and J. Feng, Angew. Chem., Int. Ed., 2023, 62, e202310066 CrossRef CAS PubMed; (e) T. Yu, J. Yang, Z. Wang, Z. Ding, M. Xu, J. Wen, L. Xu and P. Li, J. Am. Chem. Soc., 2023, 145, 4304–4310 CrossRef CAS PubMed; (f) Y. Zheng, W. Huang, R. K. Dhungana, A. Granados, S. Keess, M. Makvandi and G. A. Molander, J. Am. Chem. Soc., 2022, 144, 23685–23690 CrossRef CAS PubMed; (g) T. V. T. Nguyen, A. Bossonnet, M. D. Wodrich and J. Waser, J. Am. Chem. Soc., 2023, 145, 25411–25421 CrossRef CAS PubMed; (h) N. Radhoff, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2023, 62, e202304771 CrossRef CAS PubMed; (i) D. Ni, S. Hu, X. Tan, Y. Yu, Z. Li and L. Deng, Angew. Chem., Int. Ed., 2023, 62, e202308606 CrossRef CAS PubMed; (j) H. Yan, Y. Liu, X. Feng and L. Shi, Org. Lett., 2023, 25, 8116–8120 Search PubMed; (k) K. J. Woelk, K. Dhake, N. D. Schley and D. C. Leitch, Chem. Commun., 2023, 59, 13847–13850 RSC; (l) D. Liu, X. Guo, S. Zhou, L. Guo and X. Zhang, J. Org. Chem., 2024, 89, 1241–1248 CrossRef CAS PubMed; (m) Y. Liu, Z. Wu, J.-R. Shan, H. Yan, E.-J. Hao and L. Shi, Nat. Commun., 2024, 15, 4374 CrossRef CAS PubMed; (n) M. Borgini, Q.-N. Huang, P.-P. Chen, S. J. Geib, K. N. Houk and P. Wipf, J. Am. Chem. Soc., 2024, 146, 14927–14934 CrossRef CAS PubMed; (o) A. Dasgupta, S. Bhattacharjee, Z. Tong, A. Guin, R. E. McNamee, K. E. Christensen, A. T. Biju and E. A. Anderson, J. Am. Chem. Soc., 2024, 146, 1196–1203 CrossRef CAS PubMed; (p) S. Dutta, C. G. Daniliuc, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2024, 146, 27204–27212 CrossRef CAS PubMed.
- During the preparation of this manuscript, a paper on the diastereoselective 1,3-oxyhalogenation of BCBs appeared, see: Y. Kraemer, A. M. Stephens, J. A. Buldt, A. Young, C. P. McLoughlin, J. C. Fettinger, L. M. Holder and C. R. Pitts, Chem. Commun., 2025, 61, 3159–3162 RSC.
- (a) A. Dahiya, A. K. Sahoo, T. Alam and B. K. Patel, Chem.–Asian J., 2019, 14, 4454–4492 CrossRef CAS PubMed; (b) P. Li and X. Jia, Synthesis, 2018, 50, 711–722 CrossRef CAS.
- (a) D. Leifert and A. Studer, Chem. Rev., 2023, 123, 10302–10380 CrossRef CAS PubMed; (b) L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034–5068 CrossRef CAS PubMed.
- S. Maity, T. Naveen, U. Sharma and D. Maiti, Org. Lett., 2013, 15, 3384–3387 CrossRef CAS PubMed.
- (a) L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed; (b) L. Capaldo and D. Ravelli, Eur. J. Org Chem., 2017, 2056–2071 CAS.
- (a) H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CAS; (b) A. Studer, Chem. Soc. Rev., 2004, 33, 267–273 CAS; (c) A. Studer and T. Schulte, Chem. Rec., 2005, 5, 27–35 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2411255. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08753d |
| This journal is © The Royal Society of Chemistry 2025 |