DOI:
10.1039/D5SC00102A
(Edge Article)
Chem. Sci., 2025,
16, 5512-5517
In situ uncovering the catalytic cycle of electrochemical and chemical oxygen reduction mediated by an iron porphyrin†
Received
7th January 2025
, Accepted 24th February 2025
First published on 27th February 2025
Abstract
As one of the critical reactions in biotransformation and energy conversion processes, the oxygen reduction reaction (ORR) catalyzed by iron porphyrins has been widely explored by electrochemical, spectroscopic, and theoretical methods. However, experimental identification of all proposed intermediates of iron porphyrins in one catalytic cycle is rather challenging in the mechanistic studies of the ORR driven by electrochemical or chemical methods. Herein, we report the application of electrochemical mass spectrometry (EC-MS) and chemical reaction mass spectrometry (CR-MS) to in situ uncover the catalytic cycle of electrochemical and chemical ORRs mediated by an iron porphyrin molecular catalyst. Five crucial iron–oxygen intermediates detected by both EC-MS and CR-MS help to build the whole catalytic cycle and indicate the details of the 4e−/4H+ pathway to produce H2O in the electrochemical and chemical ORRs. By combining in situ MS methods with electrochemical and spectroscopic methods to characterize the intermediates and study the selectivities, this work provides a mechanistic comparison of the electrochemical and chemical ORRs catalyzed by one model iron porphyrin.
Introduction
The oxygen (O2) reduction reaction (ORR) is a critical step in biotransformation and energy conversion processes, including biological respiration,1,2 fuel cells,3,4 metal–air batteries,5,6etc. Inspired by the heme active sites in natural cytochrome c oxidases,2 iron porphyrins have been widely used and studied as molecular catalysts for the ORR.7–26 This simplified model reaction helps researchers to better understand the mechanisms and structure–activity relationships of the ORR under controlled conditions with different catalyst ligands, solvents, and proton/electron sources. Depending on electron sources, the ORR catalyzed by molecular complexes can be driven by electrodes or chemical reductants such as ferrocene derivatives.21,27 Accordingly, most of the previous in situ characterization studies in this field investigated the kinetics and mechanisms of the ORR by electrochemical, spectroscopic, and microscopy methods assisted by theoretical calculations.19,28–46 Furthermore, the evidence of a series of reactive iron–oxygen intermediates has also been obtained through spectroscopic methods in single-turnover experiments mainly under extremely low temperatures (Table S1 in the ESI†).47–52 However, the identification of all possible key intermediates proposed in one complete cycle under catalytic conditions is important but inadequate in either the electrochemical or chemical ORR catalyzed by iron porphyrins,12,18,28–31,33,43,45 which limits comprehensive comparison and understanding of the mechanistic details of the ORR driven by electrochemical and chemical methods. Therefore, developing and applying novel in situ characterization methods to detect as many intermediates as possible under electrochemical and chemical conditions is urgently necessary to reveal the mechanism of the ORR catalyzed by molecular complexes including iron porphyrins.
Mass spectrometry (MS) is a powerful analytical technique to characterize analytes with high sensitivity and selectivity, which is widely employed in monitoring chemical/electrochemical reactions and in elucidating their mechanisms by detecting intermediates and products.53–58 Dual micropipettes pulled from glass theta capillaries are commonly applied as microreactors, sampling collectors, and spray emitters in MS analysis.59–67 Utilizing hybrid ultramicroelectrodes (UMEs) fabricated from dual micropipettes, the in situ electrochemical mass spectrometry (EC-MS) developed by Shao and Luo et al. has been applied in the mechanistic studies of a series of complicated electrochemical reactions.68–73 The in situ MS method based on dual micropipettes and hybrid UMEs would be an effective tool to identify intermediates and investigate mechanisms in both chemical and electrochemical ORRs.
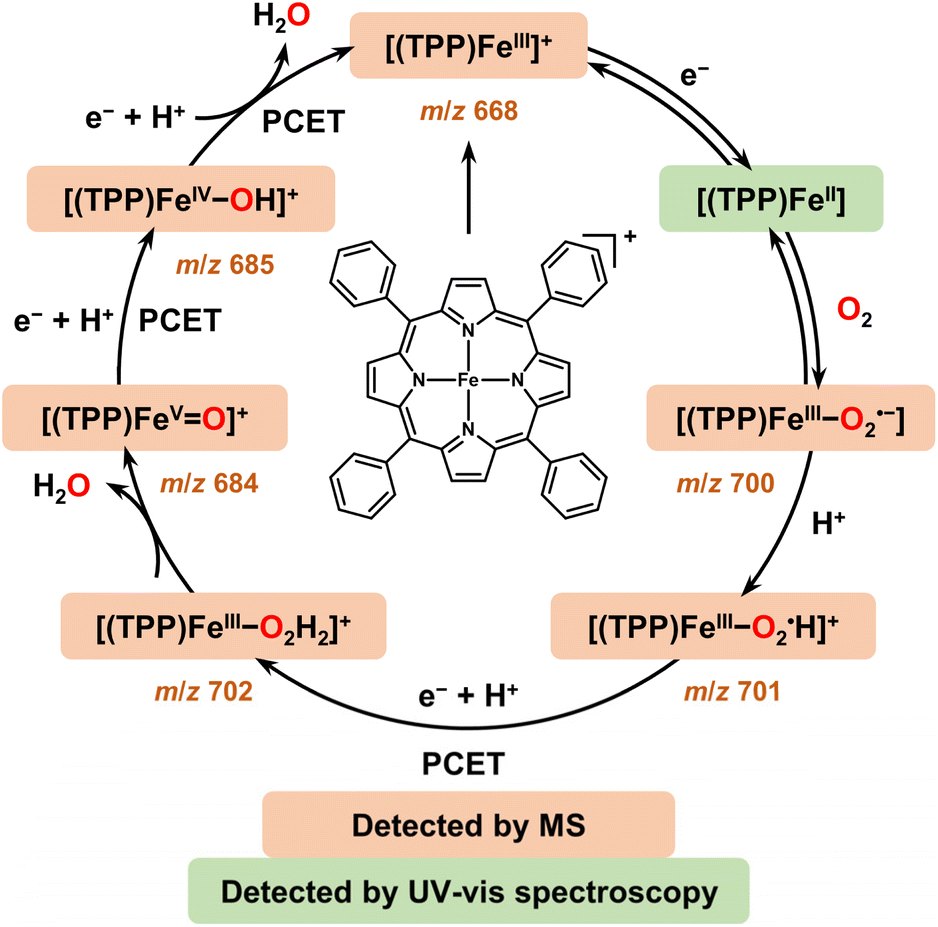
Herein, we apply EC-MS based on hybrid UMEs and chemical reaction mass spectrometry (CR-MS) recently developed by our group74 to in situ reveal the mechanism of the ORR mediated by a molecular catalyst 5,10,15,20-meso-tetraphenylporphyrin iron(III) perchlorate ([(TPP)FeIII]ClO4) (Scheme 1). Five key suggested intermediates involved in the electrochemical/chemical ORR were simultaneously detected for the first time by both in situ MS methods, providing full experimental evidence and mechanistic details to build the whole catalytic cycle of the ORR mediated by [(TPP)FeIII]+.29,31,33 Assisted by electrochemical and spectroscopic methods to determine the selectivity, the same five iron–oxygen intermediates ([(TPP)FeIII–O2˙−],  , [(TPP)FeIII–O2H2]+, [(TPP)FeV
, [(TPP)FeIII–O2H2]+, [(TPP)FeV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O]+, and [(TPP)FeIV–OH]+) detected with the exact chemical compositional information by both EC-MS and CR-MS suggest a good agreement of a 4e−/4H+ mechanism in the electrochemical and chemical ORRs catalyzed by [(TPP)FeIII]+. This work provides a methodology containing two sets of complementary methods to characterize the intermediates and study the mechanisms of electrochemical/chemical redox reactions mediated by molecular catalysts, including the ORR catalyzed by iron porphyrins.
O]+, and [(TPP)FeIV–OH]+) detected with the exact chemical compositional information by both EC-MS and CR-MS suggest a good agreement of a 4e−/4H+ mechanism in the electrochemical and chemical ORRs catalyzed by [(TPP)FeIII]+. This work provides a methodology containing two sets of complementary methods to characterize the intermediates and study the mechanisms of electrochemical/chemical redox reactions mediated by molecular catalysts, including the ORR catalyzed by iron porphyrins.
 |
| | Scheme 1 Schematic illustration of the in situ MS setups to study (a) electrochemical and (b) chemical ORRs mediated by [(TPP)FeIII]+. | |
Results and discussion
Electrochemical ORR
The electrochemical ORR mediated by [(TPP)FeIII]+ was studied by voltammetry and in situ EC-MS. The electrochemical behaviors of [(TPP)FeIII]ClO4 as an ORR molecular catalyst were first investigated on the carbon hybrid UME in N,N-dimethylformamide (DMF) with excess perchloric acid (HClO4) as the proton source in the absence and presence of O2 (Cell 1 in the ESI†). The carbon hybrid UME was fabricated from quartz dual micropipettes according to the reported procedures (Fig. S1†).73,75 In the cyclic voltammetry (CV) results (Fig. 1), black (1 atm N2) and red (1 atm O2) curves show the potential windows of the carbon UME using Cell 1 without [(TPP)FeIII]ClO4. When [(TPP)FeIII]ClO4 was added under 1 atm N2, a new reversible steady-state curve (blue) emerged, which could be ascribed to the reduction of [(TPP)FeIII]+ to [(TPP)FeII] (the half-wave potential E1/2 = −0.55 V vs. Fc+/Fc). When [(TPP)FeIII]ClO4 was added under 1 atm O2, the CV curve (green) showed much higher currents than in the presence of [(TPP)FeIII]+ and N2 (blue), indicating the occurrence of the catalytic ORR. The electrochemical ORR mediated by [(TPP)FeIII]+ on the carbon UME is comparable to those observed on the glassy carbon (GC) electrode in previous reports,29,41 which proves that the redox state of [(TPP)FeIII]+ can be controlled to catalyze the ORR by applying the potential on the carbon UME under steady-state conditions. Furthermore, the selectivity of the electrochemical ORR mediated by [(TPP)FeIII]ClO4 was also studied by rotating ring-disk voltammetry (RRDV) under the same conditions (Cell 2 in the ESI†). The results of RRDV prove that the catalytic ORR (GC disk current) occurred and a trace of H2O2 (Pt ring current) was generated simultaneously when the voltage was less than −0.55 V vs. Fc+/Fc (Fig. S2†). The average number of electrons ncat transferred in the electrochemical ORR equals 3.88e−/O2 from −0.6 V to −0.9 V vs. Fc+/Fc (Table S2†). The selectivity studies by RRDV confirmed that the 4e−/4H+ pathway is dominant in the electrochemical ORR catalyzed by [(TPP)FeIII]ClO4.
 |
| | Fig. 1 CV curves of 0.5 mM [(TPP)FeIII]ClO4 in the absence (blue) and presence (green) of 1 atm O2 on a carbon UME in DMF (Cell 1). Black (with N2) and red (with O2) curves are the potential windows of the carbon UME using Cell 1 without [(TPP)FeIII]ClO4. | |
Based on the above results by electrochemical characterization of the ORR mediated by [(TPP)FeIII]ClO4, in situ EC-MS experiments were performed with Cell 3 (in the ESI†) under 1 atm O2 using the oxygenated EC-MS setup which combines hybrid UME techniques and relay electrospray ionization MS (Scheme 1a and Fig. S3†).68,76 When the voltage of carbon UME was off, only [(TPP)FeIII]+ (m/z 668), [(TPP)FeIII–DMF]+ (m/z 741), and background signals were detected (Fig. S4†). When the voltage was on (at −0.6 V to −0.9 V vs. Fc+/Fc), five new signals, m/z 684, m/z 685, m/z 700, m/z 701, and m/z 702, could be simultaneously detected by MS along with the catalyst signal (Fig. 2 and S5†). The signals of m/z 700, m/z 701, and m/z 702 differing by m/z 1 probably correspond to the intermediate [(TPP)FeIII–O2]+ (ionized from [(TPP)FeIII–O2˙−] by the principle of relay electrospray ionization),76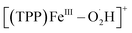 , and [(TPP)FeIII–O2H2]+, respectively. According to the relative abundance, the signal of m/z 701 and the signal of m/z 702 may contain the isotope peak of [(TPP)FeIII–O2]+ and
, and [(TPP)FeIII–O2H2]+, respectively. According to the relative abundance, the signal of m/z 701 and the signal of m/z 702 may contain the isotope peak of [(TPP)FeIII–O2]+ and  , respectively. Likewise, the signal of m/z 685 might also correspond to a mixture including the monoisotopic mass peak of [(TPP)FeIV–OH]+ and the isotope peak of [(TPP)FeVO]+. The iron-oxo species could also be of the form [(TPP˙)FeIVO]+77 and we have uniformly represented it in the formal [(TPP)FeVO]+ form in this work. The crucial intermediates [(TPP)FeVO]+ and [(TPP)FeIV–OH]+ detected by EC-MS indicate that the electrochemical ORR catalyzed by [(TPP)FeIII]+ involves a 4e−/4H+ pathway to produce H2O under the described conditions.
, respectively. Likewise, the signal of m/z 685 might also correspond to a mixture including the monoisotopic mass peak of [(TPP)FeIV–OH]+ and the isotope peak of [(TPP)FeVO]+. The iron-oxo species could also be of the form [(TPP˙)FeIVO]+77 and we have uniformly represented it in the formal [(TPP)FeVO]+ form in this work. The crucial intermediates [(TPP)FeVO]+ and [(TPP)FeIV–OH]+ detected by EC-MS indicate that the electrochemical ORR catalyzed by [(TPP)FeIII]+ involves a 4e−/4H+ pathway to produce H2O under the described conditions.
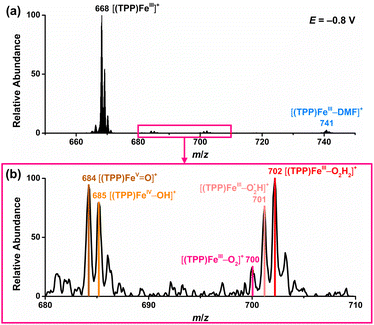 |
| | Fig. 2 Mass spectra of the electrochemical ORR catalyzed by [(TPP)FeIII]+ in DMF when the voltage was at −0.8 V vs. Fc+/Fc. (a) Full-scale mass spectra. (b) Zoomed-in mass spectra of iron–oxygen intermediates. | |
Chemical ORR
For the chemical ORR mediated by [(TPP)FeIII]+, stopped-flow UV-vis spectroscopy was first used to detect the intermediates and study the selectivity. The experiments were performed by mixing a solution of air-saturated DMF containing 0.05 mM [(TPP)FeIII]ClO4 and 5 mM HClO4 with an equal volume of an air-saturated DMF solution of 5 mM decamethylferrocene (Me10Fc) in a stopped-flow instrument.41Fig. 3a shows that the signals of iron porphyrin intermediates gradually changed with the increasing concentration of Me10Fc+ (715 nm), suggesting the occurrence of the catalytic ORR. By comparing the results with known spectra of iron porphyrins,78 the intermediate of FeII can be identified, while the spectra of other intermediates are overlapped and cannot be distinguished. The selectivity of the chemical ORR catalyzed by [(TPP)FeIII]ClO4 was indicated by determining the amount of Me10Fc+ produced in the ORR. The results in Fig. 3b suggest that the ncat consumed in the chemical ORR equals 3.90e−/O2, which is close to that in the electrochemical ORR.
 |
| | Fig. 3 (a) UV-vis spectra observed during the chemical ORR with [(TPP)FeIII]ClO4. (b) Time profiles of the Me10Fc+ formation during the chemical ORR with and without [(TPP)FeIII]ClO4. | |
To further investigate the mechanism of the chemical ORR catalyzed by [(TPP)FeIII]+ in DMF, in situ CR-MS experiments were performed with 10 mM Me10Fc and 1 mM [(TPP)FeIII]ClO4 separately in each barrel of a quartz dual micropipette reactor under 1 atm O2 using the same oxygenated MS setup (Scheme 1b and Fig. S3†). HClO4 (5 mM) and [Bu4N][PF6] (10 mM) were added in both barrels of the dual micropipette. Fig. S6† shows that only [(TPP)FeIII]+ (m/z 668), [(TPP)FeIII–DMF]+ (m/z 741), and background signals were detected when no Me10Fc was added. When the reductant Me10Fc was added, the signals of the five intermediates, [(TPP)FeVO]+ (m/z 684), [(TPP)FeIV–OH]+ (m/z 685), [(TPP)FeIII–O2]+ (m/z 700), 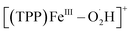 (m/z 701), and [(TPP)FeIII–O2H2]+ (m/z 702) were simultaneously detected by MS (Fig. 4). The detected species of iron–oxygen intermediates in the chemical ORR are the same as those in the electrochemical ORR, which proves the same 4e−/4H+ mechanism in the ORR driven by both the electrode and the chemical reductant. Additionally, it should be pointed out that the peaks of m/z 686 and m/z 703 in both electrochemical and chemical ORRs are attributed to the second strongest isotopic peaks of MS spectra for [(TPP)FeIV–OH]+ and [(TPP)FeIII–O2H2]+, respectively.
(m/z 701), and [(TPP)FeIII–O2H2]+ (m/z 702) were simultaneously detected by MS (Fig. 4). The detected species of iron–oxygen intermediates in the chemical ORR are the same as those in the electrochemical ORR, which proves the same 4e−/4H+ mechanism in the ORR driven by both the electrode and the chemical reductant. Additionally, it should be pointed out that the peaks of m/z 686 and m/z 703 in both electrochemical and chemical ORRs are attributed to the second strongest isotopic peaks of MS spectra for [(TPP)FeIV–OH]+ and [(TPP)FeIII–O2H2]+, respectively.
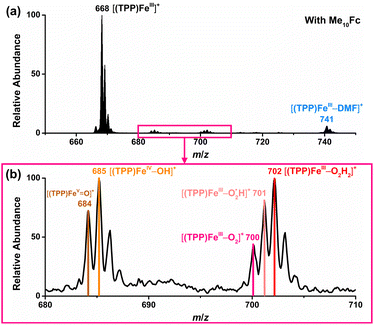 |
| | Fig. 4 Mass spectra of the chemical ORR catalyzed by [(TPP)FeIII]+ with Me10Fc as the reductant in DMF. (a) Full-scale mass spectra. (b) Zoomed-in mass spectra of iron–oxygen intermediates. | |
Isotope-labeling experiments and ORR mechanism
To further confirm the iron–oxygen intermediates, isotope-labeling electrochemical and chemical ORRs were performed and measured by in situ MS, using 18O2 (97 atom% 18O) in place of 16O2 under the same conditions. As shown in Fig. 5, [(TPP)FeV18O]+ (m/z 686), [(TPP)FeIII–18O2]+ (m/z 704), and [(TPP)FeIII–18O2H2]+ (m/z 706) were all detected by MS in both electrochemical and chemical ORRs, while [(TPP)FeIV–18OH]+ (m/z 687) can only be identified in the electrochemical ORR because its signal intensity is much higher than the relative abundance of the second strongest isotopic peak of [(TPP)FeV18O]+. In addition, possible 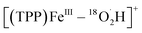 cannot be identified in the isotopic experiments due to the signal intensity of m/z 705 being close to that of the second strongest isotopic peak of [(TPP)FeIII–18O2]+.
cannot be identified in the isotopic experiments due to the signal intensity of m/z 705 being close to that of the second strongest isotopic peak of [(TPP)FeIII–18O2]+.
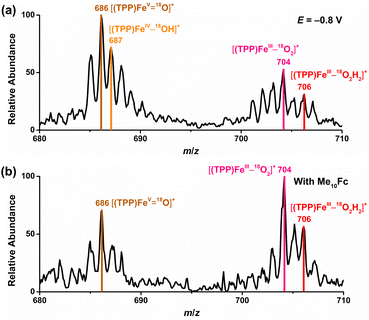 |
| | Fig. 5 Mass spectra of 18O2-labeling (a) electrochemical and (b) chemical ORRs catalyzed by [(TPP)FeIII]+. | |
According to the above in situ EC-MS/CR-MS, RRDV, and UV-vis spectroscopy results, five iron–oxygen intermediates, including [(TPP)FeIII–O2˙−], 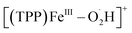 , [(TPP)FeIII–O2H2]+, [(TPP)FeVO]+, and [(TPP)FeIV–OH]+, are experimentally confirmed to be produced in both electrochemical and chemical ORRs catalyzed by [(TPP)FeIII]+. Scheme 2 shows the proposed mechanism of the 4e−/4H+ ORR based on the experimental results in this work and kinetic analysis in previous reports.31,33,77 Note that we used formal oxidation/reduction states to assign these intermediates. In the catalytic cycle of the ORR with [(TPP)FeIII]+ as the catalyst, [(TPP)FeIII]+ is first reduced by the electrode or Me10Fc to give [(TPP)FeII]. After that, [(TPP)FeII] binds an O2 molecule to produce the ferric superoxide [(TPP)FeIII–O2˙−], and then [(TPP)FeIII–O2˙−] is protonated to form
, [(TPP)FeIII–O2H2]+, [(TPP)FeVO]+, and [(TPP)FeIV–OH]+, are experimentally confirmed to be produced in both electrochemical and chemical ORRs catalyzed by [(TPP)FeIII]+. Scheme 2 shows the proposed mechanism of the 4e−/4H+ ORR based on the experimental results in this work and kinetic analysis in previous reports.31,33,77 Note that we used formal oxidation/reduction states to assign these intermediates. In the catalytic cycle of the ORR with [(TPP)FeIII]+ as the catalyst, [(TPP)FeIII]+ is first reduced by the electrode or Me10Fc to give [(TPP)FeII]. After that, [(TPP)FeII] binds an O2 molecule to produce the ferric superoxide [(TPP)FeIII–O2˙−], and then [(TPP)FeIII–O2˙−] is protonated to form 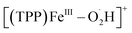 .31,33 The perhydroxyl complex
.31,33 The perhydroxyl complex  is next protonated at the distal oxygen along with 1 eq. electron (proton-coupled electron transfer, PCET) to form [(TPP)FeIII–O2H2]+ and then [(TPP)FeIII–O2H2]+ releases 1 eq. H2O to produce [(TPP)FeVO]+.12,20 Subsequently, [(TPP)FeVO]+ undergoes successive PCET processes to produce [(TPP)FeIV–OH]+ and [(TPP)FeIII]+ along with 1 eq. H2O to finally restart the catalytic cycle of the ORR.77 Among these intermediates, [(TPP)FeIII–O2H2]+ is the key to determining the selectivity of the ORR catalyzed by [(TPP)FeIII]+ and its exact configuration has two possible forms ([(TPP)FeIII–OOH2]+ and [(TPP)FeIII–HOOH]+).12 In the 4e−/4H+ pathway, the water could be produced via the [(TPP)FeIII–OOH2]+ configuration. For the ORR catalyzed by [(TPP)FeIII]+, the main difference between electrochemical and chemical-driven methods is the resource of electrons, one is from the electrode, and the other is from the reductant. The consistent results of the mechanism and selectivity together confirm the agreement of the ORR driven by these two methods.
is next protonated at the distal oxygen along with 1 eq. electron (proton-coupled electron transfer, PCET) to form [(TPP)FeIII–O2H2]+ and then [(TPP)FeIII–O2H2]+ releases 1 eq. H2O to produce [(TPP)FeVO]+.12,20 Subsequently, [(TPP)FeVO]+ undergoes successive PCET processes to produce [(TPP)FeIV–OH]+ and [(TPP)FeIII]+ along with 1 eq. H2O to finally restart the catalytic cycle of the ORR.77 Among these intermediates, [(TPP)FeIII–O2H2]+ is the key to determining the selectivity of the ORR catalyzed by [(TPP)FeIII]+ and its exact configuration has two possible forms ([(TPP)FeIII–OOH2]+ and [(TPP)FeIII–HOOH]+).12 In the 4e−/4H+ pathway, the water could be produced via the [(TPP)FeIII–OOH2]+ configuration. For the ORR catalyzed by [(TPP)FeIII]+, the main difference between electrochemical and chemical-driven methods is the resource of electrons, one is from the electrode, and the other is from the reductant. The consistent results of the mechanism and selectivity together confirm the agreement of the ORR driven by these two methods.
 |
| | Scheme 2 Proposed mechanism of the 4e−/4H+ ORR with [(TPP)FeIII]+ as the catalyst.12,20,31,33,77 | |
Conclusions
In summary, we have investigated the mechanism of the electrochemical/chemical ORR catalyzed by [(TPP)FeIII]ClO4 using in situ EC-MS/CR-MS assisted by electrochemical characterization and stopped-flow UV-vis spectroscopy. The crucial iron–oxygen intermediates detected by in situ MS give detailed experimental evidence to establish the complete catalytic cycle of the ORR with [(TPP)FeIII]+ as a molecular catalyst and indicate a 4e−/4H+ mechanism of this reaction, further supplementing the mechanistic details lacking in previous studies.29,31,33 This work provides a set of systematical in situ analytical methods to study the mechanisms of electrochemical and chemical redox reactions mediated by molecular catalysts. Future work needs to try more kinds of hybrid UMEs and explore the mechanisms of PCET reactions mediated by other molecular catalysts, not limited to the ORR.
Data availability
All experimental data are available in the manuscript and ESI.†
Author contributions
Y. Shao and R. Cao conceived and designed the project. X. Zhang and J. Zhan developed the in situ mass spectrometry techniques. Y. Shao supervised the experiments of electrochemistry and mass spectrometry. R. Cao supervised the catalyst synthesis and the experiments of UV-vis spectroscopy. X. Zhang and J. Zhan carried out the experiments of electrochemistry and mass spectrometry. H. Qin carried out the experiments of catalyst synthesis and UV-vis spectroscopy. J. Deng assisted in the experiments of electrochemistry and mass spectrometry. All authors contributed to the discussion of the results and the preparation of the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22034001, 22304003, 22325202, and 22171176), the Beijing National Laboratory for Molecular Sciences (BNLMS-CXXM-202008), and Research Funds of Shaanxi Normal University. We thank Prof. Yan Li and Dr Jian Sheng at Peking University for their help on RRDV experiments.
References
- G. T. Babcock and M. Wikström, Nature, 1992, 356, 301–309 CrossRef CAS PubMed.
- S. Yoshikawa and A. Shimada, Chem. Rev., 2015, 115, 1936–1989 CrossRef CAS PubMed.
- Y. Li, M.-Y. Chen, B.-A. Lu and J.-N. Zhang, J. Electrochem., 2023, 29, 2215002 Search PubMed.
- S. Li, L. Shi, Y. Guo, J. Wang, D. Liu and S. Zhao, Chem. Sci., 2024, 15, 11188–11228 RSC.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- Y. Su, Z. Zhao, J. Huang, E. Wang and Z. Peng, J. Phys. Chem. C, 2022, 126, 1243–1255 CrossRef CAS.
- M. L. Rigsby, D. J. Wasylenko, M. L. Pegis and J. M. Mayer, J. Am. Chem. Soc., 2015, 137, 4296–4299 CrossRef CAS PubMed.
- Y. Xuan, X. Huang and B. Su, J. Phys. Chem. C, 2015, 119, 11685–11693 CrossRef CAS.
- X. Huang, Y. Xuan, L. Xie and B. Su, ChemElectroChem, 2016, 3, 1781–1786 CrossRef CAS.
- S. Bhunia, A. Rana, P. Roy, D. J. Martin, M. L. Pegis, B. Roy and A. Dey, J. Am. Chem. Soc., 2018, 140, 9444–9457 CrossRef CAS PubMed.
- A. Ghatak, S. Bhakta, S. Bhunia and A. Dey, Chem. Sci., 2019, 10, 9692–9698 RSC.
- A. C. Brezny, S. I. Johnson, S. Raugei and J. M. Mayer, J. Am. Chem. Soc., 2020, 142, 4108–4113 CrossRef CAS PubMed.
- A. Singha, A. Mondal, A. Nayek, S. G. Dey and A. Dey, J. Am. Chem. Soc., 2020, 142, 21810–21828 CrossRef CAS PubMed.
- A. C. Brezny, H. S. Nedzbala and J. M. Mayer, Chem. Commun., 2021, 57, 1202–1205 RSC.
- B. D. Groff and J. M. Mayer, ACS Catal., 2022, 12, 11692–11696 CrossRef CAS.
- S. Bhunia, A. Ghatak, A. Rana and A. Dey, J. Am. Chem. Soc., 2023, 145, 3812–3825 CrossRef CAS PubMed.
- D. Nishiori, J. P. Menzel, N. Armada, E. A. Reyes Cruz, B. L. Nannenga, V. S. Batista and G. F. Moore, J. Am. Chem. Soc., 2024, 146, 11622–11633 Search PubMed.
- A. K. Surendran, A. Y. Pereverzev and J. Roithová, J. Am. Chem. Soc., 2024, 146, 15619–15626 Search PubMed.
- S. Chatterjee, K. Sengupta, B. Mondal, S. Dey and A. Dey, Acc. Chem. Res., 2017, 50, 1744–1753 CrossRef CAS PubMed.
- W. Zhang, W. Lai and R. Cao, Chem. Rev., 2017, 117, 3717–3797 CrossRef CAS PubMed.
- M. L. Pegis, C. F. Wise, D. J. Martin and J. M. Mayer, Chem. Rev., 2018, 118, 2340–2391 Search PubMed.
- H. Lei, X. Li, J. Meng, H. Zheng, W. Zhang and R. Cao, ACS Catal., 2019, 9, 4320–4344 CrossRef CAS.
- C. W. Machan, ACS Catal., 2020, 10, 2640–2655 CrossRef CAS.
- Y. Li, N. Wang, H. Lei, X. Li, H. Zheng, H. Wang, W. Zhang and R. Cao, Coord. Chem. Rev., 2021, 442, 213996 CrossRef CAS.
- S. Bhunia, A. Ghatak and A. Dey, Chem. Rev., 2022, 122, 12370–12426 CrossRef CAS PubMed.
- X. Li, H. Lei, L. Xie, N. Wang, W. Zhang and R. Cao, Acc. Chem. Res., 2022, 55, 878–892 CrossRef CAS PubMed.
- A. W. Nichols, E. N. Cook, Y. J. Gan, P. R. Miedaner, J. M. Dressel, D. A. Dickie, H. S. Shafaat and C. W. Machan, J. Am. Chem. Soc., 2021, 143, 13065–13073 CrossRef CAS PubMed.
- K. Sengupta, S. Chatterjee, S. Samanta and A. Dey, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8431–8436 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Rodríguez, M. L. Pegis and J. M. Mayer, J. Am. Chem. Soc., 2014, 136, 12544–12547 Search PubMed.
- C. Costentin, H. Dridi and J.-M. Savéant, J. Am. Chem. Soc., 2015, 137, 13535–13544 Search PubMed.
- M. L. Pegis, B. A. McKeown, N. Kumar, K. Lang, D. J. Wasylenko, X. P. Zhang, S. Raugei and J. M. Mayer, ACS Cent. Sci., 2016, 2, 850–856 CrossRef CAS PubMed.
- C. Costentin and J.-M. Savéant, J. Am. Chem. Soc., 2018, 140, 16669–16675 CrossRef CAS PubMed.
- M. L. Pegis, D. J. Martin, C. F. Wise, A. C. Brezny, S. I. Johnson, L. E. Johnson, N. Kumar, S. Raugei and J. M. Mayer, J. Am. Chem. Soc., 2019, 141, 8315–8326 Search PubMed.
- Y. Liu, G. Zhou, Z. Zhang, H. Lei, Z. Yao, J. Li, J. Lin and R. Cao, Chem. Sci., 2020, 11, 87–96 RSC.
- A. Facchin, T. Kosmala, A. Gennaro and C. Durante, ChemElectroChem, 2020, 7, 1431–1437 Search PubMed.
- J. Meng, H. Qin, H. Lei, X. Li, J. Fan, W. Zhang, U.-P. Apfel and R. Cao, Angew. Chem., Int. Ed., 2023, 62, e202312255 CrossRef CAS PubMed.
- J. Han, H. Tan, K. Guo, H. Lv, X. Peng, W. Zhang, H. Lin, U.-P. Apfel and R. Cao, Angew. Chem., Int. Ed., 2024, 63, e202409793 Search PubMed.
- T. Liu, H. Qin, Y. Xu, X. Peng, W. Zhang and R. Cao, ACS Catal., 2024, 14, 6644–6649 Search PubMed.
- A. Facchin, D. Forrer, M. Zerbetto, F. Cazzadori, A. Vittadini and C. Durante, ACS Catal., 2024, 14, 14373–14386 CrossRef CAS.
- A. Santra, A. Das, S. Kaur, P. Jain, P. P. Ingole and S. Paria, Chem. Sci., 2024, 15, 4095–4105 RSC.
- H. Qin, J. Kong, X. Peng, Z. Wang, X. Li, H. Lei, W. Zhang and R. Cao, ChemSusChem, 2025, 18, e202401739 CrossRef CAS PubMed.
- C. Costentin, Chem. Rev., 2008, 108, 2145–2179 CrossRef CAS PubMed.
- J.-M. Savéant, Chem. Rev., 2008, 108, 2348–2378 CrossRef PubMed.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Rev., 2010, 110, PR1–PR40 Search PubMed.
- K. Sengupta, S. Chatterjee and A. Dey, ACS Catal., 2016, 6, 6838–6852 CrossRef CAS.
- C. Costentin and J.-M. Savéant, Nat. Rev. Chem., 2017, 1, 0087 CrossRef CAS.
- J.-G. Liu, Y. Shimizu, T. Ohta and Y. Naruta, J. Am. Chem. Soc., 2010, 132, 3672–3673 CrossRef CAS PubMed.
- Z. Halime, H. Kotani, Y. Li, S. Fukuzumi and K. D. Karlin, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13990–13994 CrossRef CAS PubMed.
- M. T. Kieber-Emmons, M. F. Qayyum, Y. Li, Z. Halime, K. O. Hodgson, B. Hedman, K. D. Karlin and E. I. Solomon, Angew. Chem., Int. Ed., 2012, 51, 168–172 CrossRef CAS PubMed.
- H. Kim, P. J. Rogler, S. K. Sharma, A. W. Schaefer, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2020, 142, 3104–3116 CrossRef CAS PubMed.
- S. Samanta, S. Sengupta, S. Biswas, S. Ghosh, S. Barman and A. Dey, J. Am. Chem. Soc., 2023, 145, 26477–26486 CrossRef CAS PubMed.
- E. E. Chufán, S. C. Puiu and K. D. Karlin, Acc. Chem. Res., 2007, 40, 563–572 CrossRef PubMed.
- A. Ray, T. Bristow, C. Whitmore and J. Mosely, Mass Spectrom. Rev., 2018, 37, 565–579 Search PubMed.
- D. Freitas, X. Chen, H. Cheng, A. Davis, B. Fallon and X. Yan, ChemPlusChem, 2021, 86, 434–445 Search PubMed.
- W. Li, J. Sun, Y. Gao, Y. Zhang, J. Ouyang and N. Na, TrAC, Trends Anal. Chem., 2021, 135, 116180 Search PubMed.
- J. Sun, Y. Yin, W. Li, O. Jin and N. Na, Mass Spectrom. Rev., 2022, 41, 70–99 CrossRef CAS PubMed.
- X. Zhang, J. Zhan, Z. Yu, J. Deng, M. Li and Y. Shao, Chin. J. Chem., 2023, 41, 214–224 CrossRef CAS.
- K. Chen, Q. Wan, S. Wei, W. Nie, S. Zhou and S. Chen, Chem.–Eur. J., 2024, 30, e202402215 CrossRef CAS PubMed.
- L. P. Mark, M. C. Gill, M. Mahut and P. J. Derrick, Eur. J. Mass Spectrom., 2012, 18, 439–446 CrossRef CAS PubMed.
- D. N. Mortensen and E. R. Williams, Anal. Chem., 2014, 86, 9315–9321 CrossRef CAS PubMed.
- D. N. Mortensen and E. R. Williams, J. Am. Chem. Soc., 2016, 138, 3453–3460 CrossRef CAS PubMed.
- R. M. Bain, S. Sathyamoorthi and R. N. Zare, Angew. Chem., Int. Ed., 2017, 56, 15083–15087 CrossRef CAS PubMed.
- E. T. Jansson, Y.-H. Lai, J. G. Santiago and R. N. Zare, J. Am. Chem. Soc., 2017, 139, 6851–6854 CrossRef CAS PubMed.
- A. Saha-Shah, J. A. Karty and L. A. Baker, Analyst, 2017, 142, 1512–1518 RSC.
- N. Sahota, D. I. AbuSalim, M. L. Wang, C. J. Brown, Z. Zhang, T. J. El-Baba, S. P. Cook and D. E. Clemmer, Chem. Sci., 2019, 10, 4822–4827 RSC.
- Y. Li, L. Meng, G. Wang, X. Zhou, Z. Ouyang and Z. Nie, Anal. Chem., 2020, 92, 12049–12054 CrossRef CAS PubMed.
- L. A. Baker and G. S. Jagdale, Curr. Opin. Electrochem., 2019, 13, 140–146 CrossRef CAS PubMed.
- R. Qiu, X. Zhang, H. Luo and Y. Shao, Chem. Sci., 2016, 7, 6684–6688 RSC.
- W. Guo, H. Ding, C. Gu, Y. Liu, X. Jiang, B. Su and Y. Shao, J. Am. Chem. Soc., 2018, 140, 15904–15915 CrossRef CAS PubMed.
- C. Gu, X. Nie, J. Jiang, Z. Chen, Y. Dong, X. Zhang, J. Liu, Z. Yu, Z. Zhu, J. Liu, X. Liu and Y. Shao, J. Am. Chem. Soc., 2019, 141, 13212–13221 CrossRef CAS PubMed.
- M. Li, P. He, Z. Yu, S. Zhang, C. Gu, X. Nie, Y. Gu, X. Zhang, Z. Zhu and Y. Shao, Anal. Chem., 2021, 93, 1515–1522 CrossRef CAS PubMed.
- Z. Yu, Y. Shao, L. Ma, C. Liu, C. Gu, J. Liu, P. He, M. Li, Z. Nie, Z. Peng and Y. Shao, Adv. Mater., 2022, 34, 2106618 CrossRef CAS PubMed.
- X. Zhang, Q.-F. Chen, J. Deng, X. Xu, J. Zhan, H.-Y. Du, Z. Yu, M. Li, M.-T. Zhang and Y. Shao, J. Am. Chem. Soc., 2022, 144, 17748–17752 CrossRef CAS PubMed.
- Q.-F. Chen, X. Zhang, J. Shi, J. Zhan, F. Xie, H.-T. Zhang, J. Deng, J. Liu, M. Li, Y. Shao and M.-T. Zhang, CCS Chem., 2025, 7, 893–904 CrossRef.
- Y. Takahashi, A. I. Shevchuk, P. Novak, Y. Zhang, N. Ebejer, J. V. Macpherson, P. R. Unwin, A. J. Pollard, D. Roy, C. A. Clifford, H. Shiku, T. Matsue, D. Klenerman and Y. E. Korchev, Angew. Chem., Int. Ed., 2011, 50, 9638–9642 CrossRef CAS PubMed.
- A. Li, A. Hollerbach, Q. Luo and R. G. Cooks, Angew. Chem., Int. Ed., 2015, 54, 6893–6895 CrossRef CAS PubMed.
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS PubMed.
- P. Mondal, I. Ishigami, S.-R. Yeh and G. B. Wijeratne, Angew. Chem., Int. Ed., 2022, 61, e202211521 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Rui
Cao
a,
Rui
Cao
 , [(TPP)FeIII–O2H2]+, [(TPP)FeV
, [(TPP)FeIII–O2H2]+, [(TPP)FeV

 , and [(TPP)FeIII–O2H2]+, respectively. According to the relative abundance, the signal of m/z 701 and the signal of m/z 702 may contain the isotope peak of [(TPP)FeIII–O2]+ and
, and [(TPP)FeIII–O2H2]+, respectively. According to the relative abundance, the signal of m/z 701 and the signal of m/z 702 may contain the isotope peak of [(TPP)FeIII–O2]+ and  , respectively. Likewise, the signal of m/z 685 might also correspond to a mixture including the monoisotopic mass peak of [(TPP)FeIV–OH]+ and the isotope peak of [(TPP)FeV
, respectively. Likewise, the signal of m/z 685 might also correspond to a mixture including the monoisotopic mass peak of [(TPP)FeIV–OH]+ and the isotope peak of [(TPP)FeV

 (m/z 701), and [(TPP)FeIII–O2H2]+ (m/z 702) were simultaneously detected by MS (Fig. 4). The detected species of iron–oxygen intermediates in the chemical ORR are the same as those in the electrochemical ORR, which proves the same 4e−/4H+ mechanism in the ORR driven by both the electrode and the chemical reductant. Additionally, it should be pointed out that the peaks of m/z 686 and m/z 703 in both electrochemical and chemical ORRs are attributed to the second strongest isotopic peaks of MS spectra for [(TPP)FeIV–OH]+ and [(TPP)FeIII–O2H2]+, respectively.
(m/z 701), and [(TPP)FeIII–O2H2]+ (m/z 702) were simultaneously detected by MS (Fig. 4). The detected species of iron–oxygen intermediates in the chemical ORR are the same as those in the electrochemical ORR, which proves the same 4e−/4H+ mechanism in the ORR driven by both the electrode and the chemical reductant. Additionally, it should be pointed out that the peaks of m/z 686 and m/z 703 in both electrochemical and chemical ORRs are attributed to the second strongest isotopic peaks of MS spectra for [(TPP)FeIV–OH]+ and [(TPP)FeIII–O2H2]+, respectively.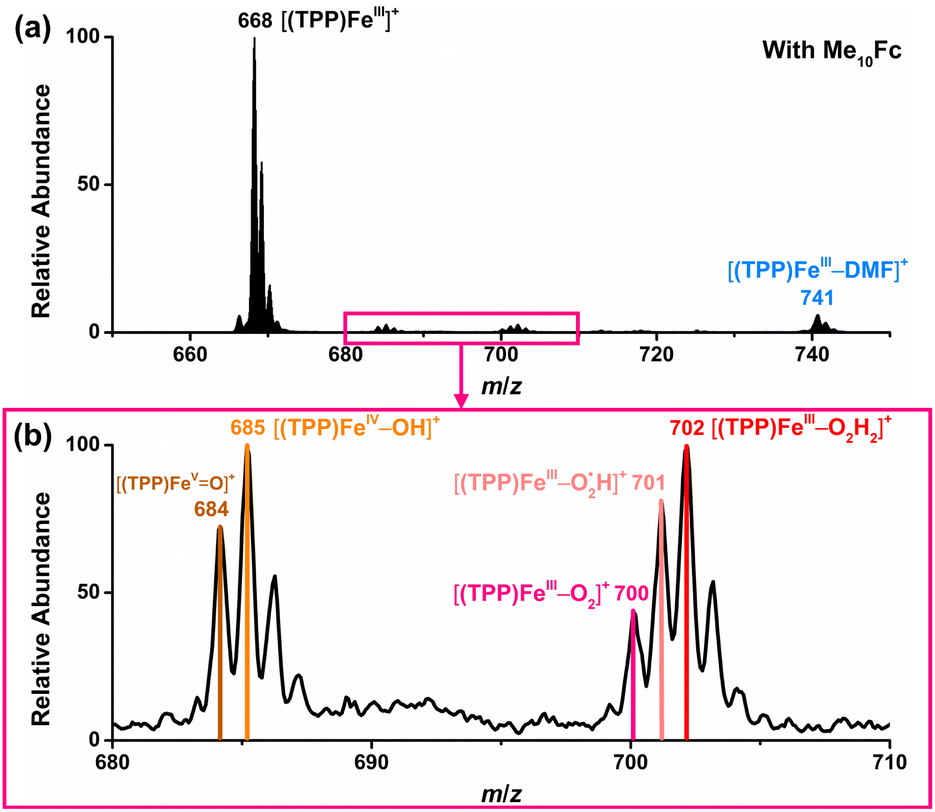
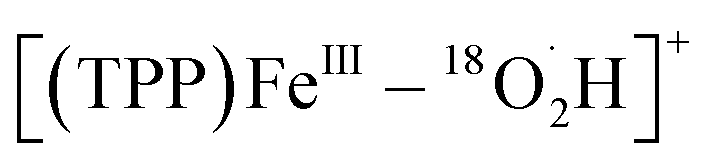 cannot be identified in the isotopic experiments due to the signal intensity of m/z 705 being close to that of the second strongest isotopic peak of [(TPP)FeIII–18O2]+.
cannot be identified in the isotopic experiments due to the signal intensity of m/z 705 being close to that of the second strongest isotopic peak of [(TPP)FeIII–18O2]+.

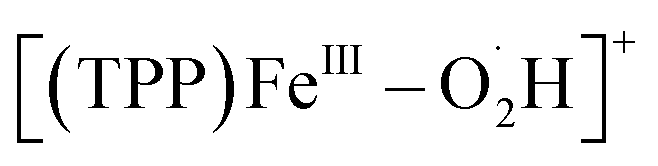 , [(TPP)FeIII–O2H2]+, [(TPP)FeV
, [(TPP)FeIII–O2H2]+, [(TPP)FeV .31,33 The perhydroxyl complex
.31,33 The perhydroxyl complex 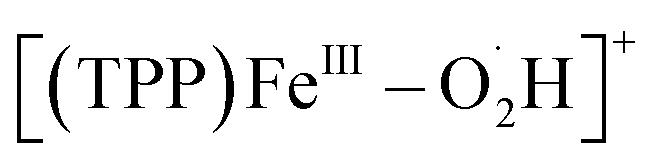 is next protonated at the distal oxygen along with 1 eq. electron (proton-coupled electron transfer, PCET) to form [(TPP)FeIII–O2H2]+ and then [(TPP)FeIII–O2H2]+ releases 1 eq. H2O to produce [(TPP)FeV
is next protonated at the distal oxygen along with 1 eq. electron (proton-coupled electron transfer, PCET) to form [(TPP)FeIII–O2H2]+ and then [(TPP)FeIII–O2H2]+ releases 1 eq. H2O to produce [(TPP)FeV