
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNi doping in CsPbCl3 nanocrystals: the key to enhanced photoluminescence†
Soumya
Panja
 a,
Prasenjit
Mandal
a,
Subhashri
Mannar
b,
Arpan
Das
c,
Shobhana
Narasimhan
c and
Ranjani
Viswanatha
*ab
a,
Prasenjit
Mandal
a,
Subhashri
Mannar
b,
Arpan
Das
c,
Shobhana
Narasimhan
c and
Ranjani
Viswanatha
*ab
aNew Chemistry Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bangalore 560064, India. E-mail: rv@jncasr.ac.in
bInternational Centre for Material Science, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bangalore 560064, India
cTheoretical Sciences Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bangalore 560064, India
First published on 21st April 2025
Abstract
This study presents a generic method to selectively enhance radiative pathways over non-radiative states by leveraging vibrational coupling between the host lattice and mid-gap states of doped transition metal ions. While previously demonstrated with Mn, this work successfully incorporates Ni2+ ions into CsPbCl3 perovskite nanocrystals (NCs), showcasing the method's versatility and tunability for radiative decay rates. Structural analyses confirm Ni2+ integration, while temperature-dependent photoluminescence studies reveal that Ni-induced shallow trap states enable vibrational coupling, facilitating charge carrier back-transfer to excitonic states. At 2% doping, this mechanism optimally enhances radiative recombination, achieving room-temperature vibrationally assisted delayed fluorescence (VADF). Förster resonance energy transfer (FRET) experiments further validate the improved radiative efficiency. This work establishes transition metal doping as a transformative and selective strategy for tuning optical properties, paving the way for advancements in energy-efficient technologies such as light-emitting diodes, lasers, and photovoltaics.
Introduction
Inorganic lead halide perovskites have attracted considerable attention in electronics and photonics due to their outstanding optoelectronic properties, including high absorption coefficients, tunable bandgaps, extended carrier diffusion lengths, and narrow full width at half maximum (FWHM).1–8 Among these, their photoluminescence quantum yield (PLQY) is a critical factor for applications in light-emitting diodes (LEDs),9,10 lasers,11 and photovoltaic devices.12 However, despite their relatively high PLQY, the efficiency of perovskite-based devices remains limited, primarily due to the intrinsic instability of perovskite halides and significant non-radiative recombination pathways. To address these challenges, recent efforts have concentrated on improving both stability and PLQY through strategies such as materials engineering, surface passivation, and advanced doping techniques. Surface passivation, in particular, has proven highly effective in mitigating surface defects and shielding perovskites from degradation. By employing organic or inorganic passivating layers, researchers have successfully minimized non-radiative recombination at the surface, thereby enhancing PLQY.13,14 These advances underscore the importance of surface engineering in overcoming the inherent limitations of perovskite materials, although industrial-level scalability and uniform surface passivation are still challenging.Building on these advancements, doping quantum dots with transition metal ions has emerged as a well-studied and effective approach for further enhancing PLQY. This technique involves the deliberate incorporation of impurity atoms into the crystal lattice, which not only improves stability but also optimizes optoelectronic properties by reducing defects.13–15 For instance, doping with monovalent, divalent, and trivalent ions have been shown to passivate ionic and electronic defects within the perovskite lattice, leading to a substantial increase in PLQY.16–18 Moreover, strategic dopant selection can enhance phase stability and environmental resilience under operational conditions. Numerous studies have demonstrated the successful incorporation of transition metal and rare earth ions, such as Mn2+, Fe2+, Cu2+, Zn2+, Eu3+, Er3+, and Yb3+etc. into lead halide perovskite matrices.19–25 These doped systems exhibit enhanced band-edge photoluminescence, improved stability, reduced defect state density, and large-scale production, underscoring the versatility and efficacy of doping as a complementary strategy to surface passivation.
Despite advancements in surface passivation and doping strategies, achieving high external quantum efficiency (EQE) in LED devices based on inorganic perovskite halides remains a significant challenge. While these techniques primarily focus on reducing non-radiative recombination and mitigating material degradation, their impact is largely limited to extending device longevity rather than significantly enhancing brightness. Achieving substantial improvements in EQE and color purity necessitates a shift toward optimizing radiative processes, which are directly responsible for efficient photon generation and emission.
One promising strategy for further improving radiative recombination efficiency is engineering materials to couple electronic states with the optical density of states. Microcavity structures, for instance, have demonstrated the ability to enhance spontaneous emission by tailoring the surrounding photonic environment to optimize light-matter interactions.26,27 A more recent and innovative approach involves the use of transition metal dopants, such as manganese (Mn), to further improve emission efficiency.28 Mn dopants, known for their characteristic long-lived forbidden transitions, serve as electron and hole reservoirs. These dopants facilitate vibrational coupling between their mid-gap states and the radiative states of the host lattice, a mechanism referred to as vibrationally assisted delayed fluorescence (VADF). VADF enables a controlled release of photoexcited carriers into radiative pathways, significantly boosting radiative recombination efficiency. This process not only enhances photoluminescence efficiency but also provides a stable and reliable route for photon emission, even during prolonged operational periods.
Briefly, VADF operates by enabling dopant states to capture photoexcited carriers from the host perovskite's valence or conduction band. These carriers are then gradually released into radiative pathways through vibrational assistance, effectively reducing trapping at defect states and increasing the probability of radiative recombination.29 By modulating vibrational modes within the perovskite lattice, VADF establishes a stable and efficient route for photon emission. Crucially, VADF relies on two key factors: long-lived dopant states and strong vibrational coupling between the host lattice and the dopant. Long-lived states arise from phenomena such as phonon bottlenecking30 where excitations are retained for extended periods due to slow de-excitation processes. Strong temperature dependent vibrational coupling may arise due to presence of dopant levels closer to the host excitonic state, that are strongly hybridized in spite of molecular-like flat dopant levels. Transition metals ions such as Mn, Cu, and Ni31,32 exhibit these characteristics, with Mn-doped lead halide perovskite nanocrystals (NCs) being the most extensively studied system demonstrating VADF.28 This ability to leverage dopant-mediated radiative processes underpins the potential of VADF to address key challenges in achieving high EQE, stable perovskite-based energy harvesting optoelectronic devices.
Although previous studies have reported improved PLQY with Ni doping,33 particularly in mixed halide perovskites,34 these improvements were often attributed to increased local structural ordering or potential halide surface passivation arising from the use of excess halide salts during synthesis. VADF has not been explored in transition metal dopants other than Mn. In this work, we focus on unraveling the role of Ni-induced dopant states and their interaction with the perovskite host, specifically investigating the contribution of VADF to PLQY enhancement in Ni-doped perovskite NCs. In particular, we used density functional theory (DFT) calculations to reveal the presence and interaction of the dopant states within the bandgap of the host perovskite lattice. Unlike previous assumptions of improved structural order, our study demonstrates that the PLQY enhancement primarily originates from VADF. Ni2+ incorporation was confirmed using extended X-ray absorption fine structure (EXAFS) spectroscopy and the visual distribution of Ni was confirmed via high-angle annular dark field – scanning transmission electron microscopy with energy dispersive X-ray spectroscopy (HAADF-STEM EDX), while host-dopant interactions were probed through electron spin resonance (ESR) and magnetic circular dichroism (MCD). Temperature dependent gated PL spectroscopy and their FWHM analysis provided compelling evidence that the enhanced PLQY results from vibrational coupling between Ni2+-induced states and the host lattice, facilitating efficient VADF. This study not only highlights the potential of Ni-doped perovskite NCs to boost excitonic emission quantum yield but also demonstrates practical applicability through successful Förster resonance energy transfer (FRET) to a dye, paving the way for energy-efficient optoelectronic materials.
Results and discussion
The role of Ni doping in enhancing PLQY through VADF was explored using a series of Ni-doped CsPbCl3 NCs alongside undoped counterparts. Here we specifically choose CsPbCl3, because among different halides (Cl, Br, and I) based inorganic perovskites, CsPbCl3 experiences the most significant non-radiative energy losses which hinders their industrial applications. The NCs, including undoped CsPbCl3 (P1) and Ni-doped variants (NixP1; x = 0.5, 1, 2, 3), were prepared using modified cation and anion injection methods, as reported in prior literature.1,35 Detailed synthesis protocols are provided in the ESI.† The dopant concentrations were quantified using inductively coupled plasma optical emission spectroscopy (ICP-OES) with values listed in Table S1 in the ESI.†X-ray diffraction (XRD) patterns, shown in Fig. 1(a), confirmed that both undoped and Ni-doped NCs formed cubic (Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) in phase, consistent with bulk CsPbCl3. However, while the crystal structure remained intact, consistent with the Vegard's law, the XRD peaks shifted slightly towards higher angles with increasing Ni percentages shown in Fig. S1 (ESI).† However, due to the peak broadening effects inherent in NCs, attributing this small shift (eg: ∼0.14° in 2θ for the (110) peak) to lattice contraction due to doping of smaller Ni2+ (ionic radius 0.69 Å) in place of larger Pb2+ (ionic radius 1.19 Å), is challenging and falls within the experimental error. This finding underscores the need for complementary characterization techniques, such as EXAFS and DFT, to elucidate the dopant's local environment and confirm its role in modulating the optical and electronic properties of the NCs.
m) in phase, consistent with bulk CsPbCl3. However, while the crystal structure remained intact, consistent with the Vegard's law, the XRD peaks shifted slightly towards higher angles with increasing Ni percentages shown in Fig. S1 (ESI).† However, due to the peak broadening effects inherent in NCs, attributing this small shift (eg: ∼0.14° in 2θ for the (110) peak) to lattice contraction due to doping of smaller Ni2+ (ionic radius 0.69 Å) in place of larger Pb2+ (ionic radius 1.19 Å), is challenging and falls within the experimental error. This finding underscores the need for complementary characterization techniques, such as EXAFS and DFT, to elucidate the dopant's local environment and confirm its role in modulating the optical and electronic properties of the NCs.
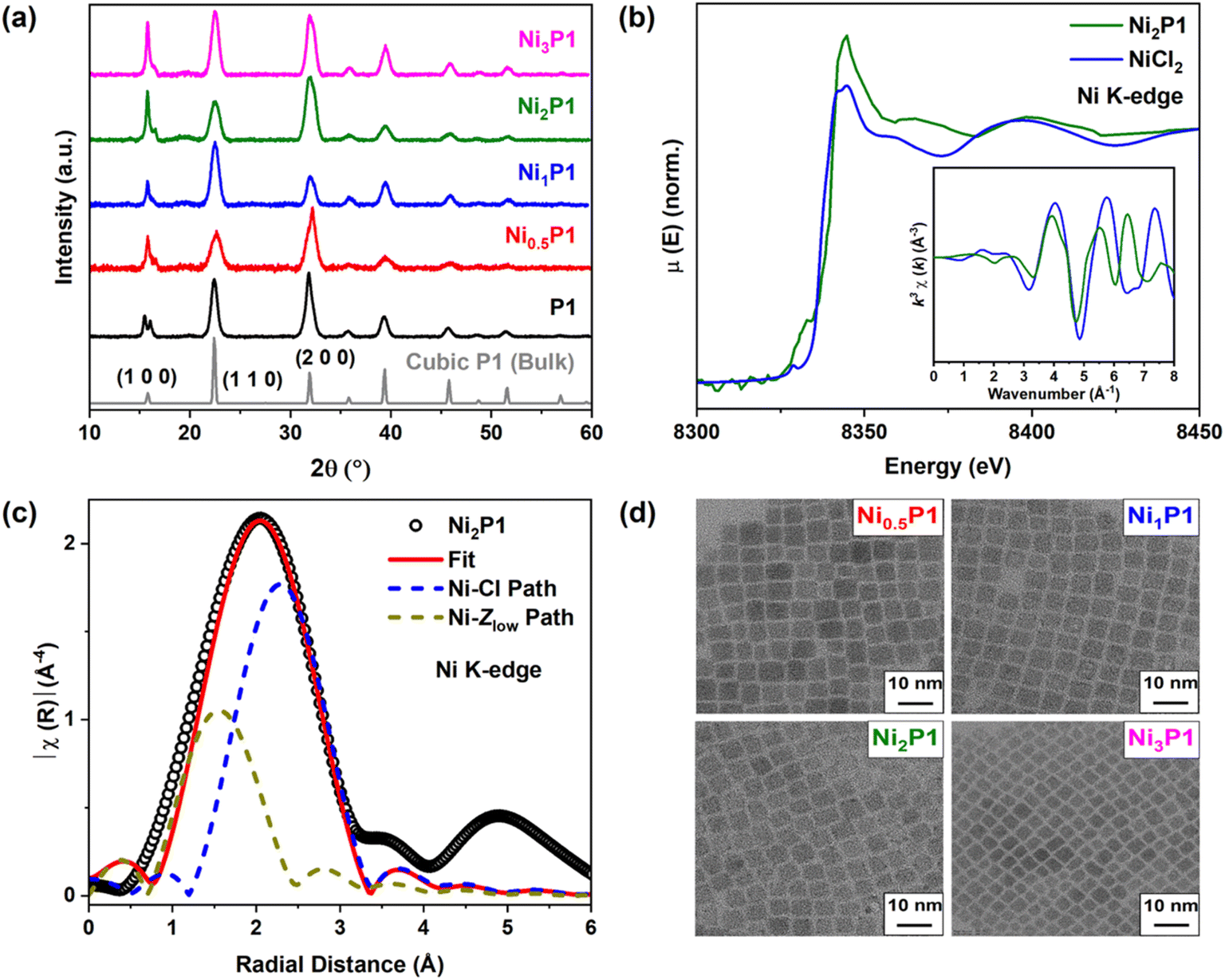 | ||
| Fig. 1 Structural characterization: (a) XRD pattern of undoped (P1) and various Ni-doped CsPbCl3 (NixP1; x = 0.5, 1, 2, 3) NCs with bulk cubic CsPbCl3 data as reference (reference data obtained from inorganic crystal structure database) indicating the cubic (Pmm) phase preserved upon doping. (b) Ni K-edge XANES spectra as a function of incident photoelectron energy along with inset of k3-weighted k-space spectra of Ni2P1 and NiCl2 precursor, suggesting the oxidation state of Ni is +2. (c) k3-weighted magnitude of Fourier-transformed Ni K-edge EXAFS spectra of Ni2P1 (black hollow circle) and best fit (red solid line) along with the possible theoretical paths (dashed lines) for fitting. Dominant Ni–Cl interactions confirm the successful doping of Ni2+. (d) High-resolution TEM images of all Ni-doped CsPbCl3 NCs showing monodisperse cubic NCs. | ||
To corroborate the incorporation of Ni2+ into the CsPbCl3 NCs, we conducted EXAFS analysis on both undoped and Ni-doped NCs. The data were fitted using theoretical paths available in Artemis software. Fig. S2(a) and (b) (ESI)† display the Pb LIII-edge and the k3-weighted magnitude of the Fourier-transformed EXAFS spectra respectively. A dominant peak corresponding to the Pb–Cl interaction was observed at ∼2.29 Å in undoped NCs, which shifted slightly to ∼2.26 Å in Ni-doped NCs. The oxidation state of Ni in the doped NCs was confirmed to be +2 using Ni K-edge X-ray absorption near-edge structure (XANES) spectra, as shown in Fig. 1(b).
From the fitting parameters (Table S2, ESI†), a subtle increase in the Pb2+ coordination number (CN) from 5.4 (undoped) to 6.0 (Ni-doped) was observed, while the Debye–Waller factor (σ2 = 0.016 Å2), representing the mean square displacement between the absorber and the backscattered atom, showed negligible differences. Moreover, the Pb–Cl bond lengths remained nearly unchanged between undoped (2.85 ± 0.01 Å) and Ni-doped (2.84 ± 0.01 Å) NCs, indicating that Ni doping does not disrupt the local structural order of the [PbCl6] octahedra. Although the XANES of the doped sample resembled the precursor, the k3-weighted wavenumber (k) space EXAFS spectra of doped sample, shown in the inset to Fig. 1(b), differed significantly from the bulk NiCl2 precursor, ruling out the presence of unreacted NiCl2 in the NCs.34 Further analysis of the Fourier-transformed spectrum (Fig. 1(c)) for a representative sample, 2% Ni-doped NCs (Ni2P1), revealed a broad peak, which was fitted using a lattice substitution model.36 This broad peak arises from a dominant singly scattered Ni–Cl interaction and a minor Ni–Zlow (Zlow = low atomic number, e.g. O or N) interaction, likely due to surface Ni atoms interacting with the ligands. Best-fit parameters (Tables 1 and S2, ESI†) indicate a Ni–Cl CN of 5.8 and a reduced bond length of 2.67 Å, confirming the successful substitution of Pb2+ by Ni2+ within the NCs lattice.
| Sample | Paths | CN | Bond length (Å) |
|---|---|---|---|
| Ni2P1 | Ni–Cl | 5.8 | 2.67 ± 0.01 |
| Ni–Zlow | 1 | 2.12 ± 0.02 |
To further validate Ni doping, room temperature X-band ESR was performed (Fig. S3, ESI†). The ESR spectra of Ni-doped samples (Ni1P1 and Ni2P1) exhibited a single peak, indicative of unpaired electrons in Ni2+ ions. At higher Ni concentrations (>2%), slight broadening of the ESR spectra was observed, suggesting dipolar exchange interactions among dopant ions.37,38 This structural analysis further supports the incorporation of Ni2+ into the host lattice.
To verify that the synthesized NCs operate within the quantum confinement regime, high-resolution TEM imaging was performed on Ni-doped NCs with varying Ni concentrations as shown in Fig. 1(d). Images of undoped NCs are provided in Fig. S4 (ESI)† for comparison. Size distribution analysis, based on measurements of approximately 300 particles, confirmed monodisperse cubic NCs (Fig. S5, ESI†). Despite maintaining a constant injection temperature during synthesis, Ni incorporation resulted in a decrease in the average particle size from 11.3 (undoped) to 8.4 nm (3% Ni-doped), as summarized in Table S3 (ESI).† This observation aligns with previous studies,33,34 which have reported similar effects of dopant incorporation on particle size. Variation in particle size arose due to different nucleation and growth mechanisms of undoped and doped NCs.39 However, since the average particle size remained around 10 nm and the cubic morphology was unaffected by Ni doping, the NCs were confirmed to lie in the weak quantum confinement regime. Further HAADF-STEM EDX images shown in Fig. S6 (ESI)† indicate uniform distribution of Ni throughout the NCs. The Ni content, as determined from EDX mapping, is consistent with the percentage obtained from ICP-OES elemental analysis. These findings provide a robust basis for further structural and photophysical analyses of Ni-doped NCs.
Building on the confirmation of Ni2+ incorporation into the perovskite lattice, the optical properties of both doped and undoped NCs were investigated using ultraviolet-visible (UV-vis) absorption, MCD, and PL spectroscopy at room temperature. The absorption and PL spectra for both types of samples are presented in Fig. 2(a). The bandgap variation with increasing Ni concentration (x), shown in the inset of Fig. 2(a), exhibits a slight bowing effect – a characteristic feature of doped NCs.40 Size variations within the Bohr diameter of CsPbCl3 (∼10 nm)41,42 could account for the slight blue shift in the bandgap, complicating the precise quantification of the red shift attributed to Ni ion incorporation in the NCs.
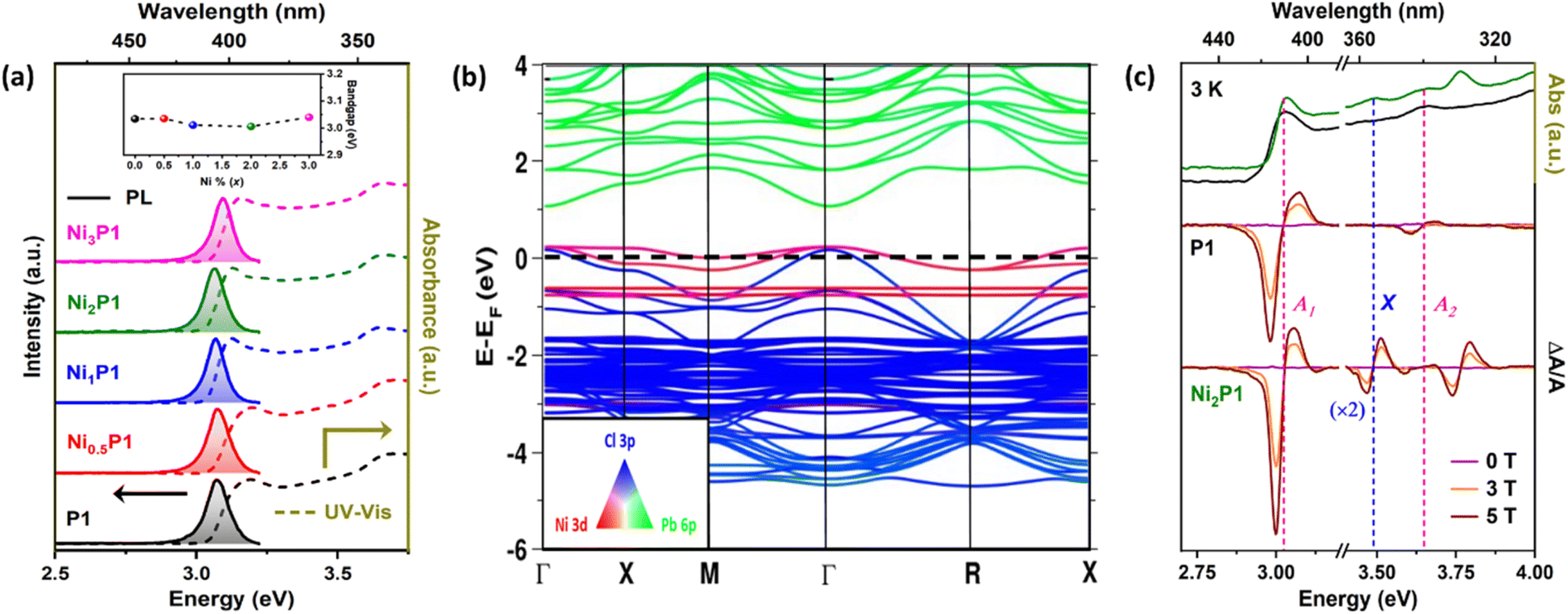 | ||
| Fig. 2 Optical and electronic properties: (a) room temperature absorption and emission spectra of undoped (P1) and various Ni-doped CsPbCl3 (NixP1; x = 0, 0.5, 1, 2, 3) NCs with the inset of bandgap variation with different Ni-percentage (dashed line guides to the eye) showing a bowing effect. (b) Orbital projected band structure of Ni-doped CsPbCl3 with considering spin–orbit coupling (SOC). Red, green, and blue colors represent Ni 3d, Pb 6p, and Cl 3p atomic orbitals respectively, while intermediate colors indicate the extent of mixing or hybridization of these orbital contributions. Band structure confirms significant hybridization of Ni 3d and Cl 3p orbitals near the VBM at Γ point. (c) MCD spectra of P1 and Ni2P1 with their absorption at temperature 3 K with varying magnetic field (0, 3, and 5 T). The presence of extra differential peak (X) confirms the Ni2+-induced exchange interaction. | ||
The PL spectra (Fig. 2(a)) reveal the presence of only band-edge emission with a ∼50 meV Stokes shift from the absorption edge and no additional peaks associated with the Ni mid-gap states, in contrast to the mid-gap emission typically observed for Mn or lanthanide ions.43–46 Interestingly, the PLQY increased with higher Ni doping percentage, as shown in Fig. S7 (ESI).† However, since halide defects in CsPbCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 47 can be passivated by excess chloride salts14,48 introduced during synthesis in the cation injection method, it was crucial to ascertain whether the PLQY enhancement was due to Ni doping or chloride passivation. To address this, we synthesized samples using the sole Cl precursor benzoyl chloride in the anion injection method,35 ensuring that identical amounts of benzoyl chloride were used for both doped and undoped NCs. This approach standardized the chloride ion levels across all samples (details in the experimental section of ESI†) to rule out the possibility of Cl surface passivation. PL spectra of Ni-doped NCs prepared using either cation or anion injection techniques at the same Ni concentrations are shown in Fig. S8 (ESI).† The PLQY enhancement was observed in Ni-doped samples (Inset of Fig. S7, ESI†) regardless of the chloride ion concentration, ruling out surface passivation as the source of the improved band-edge emission. These findings confirm that the increase in PLQY is directly attributed to the incorporation of Ni2+ dopant ions, highlighting that Ni doping effectively enhances the optical properties of the CsPbCl3 perovskite NCs. Further, it should be noted that dopant ion incorporation efficiency in the anion injection method is higher compared to the cation injection method since there are no pre-existing Pb–Cl bond in the precursors. Although the detailed mechanism reported in the literature35 and beyond the scope of this manuscript, we used this method to prepare samples with higher Ni doping concentration.
47 can be passivated by excess chloride salts14,48 introduced during synthesis in the cation injection method, it was crucial to ascertain whether the PLQY enhancement was due to Ni doping or chloride passivation. To address this, we synthesized samples using the sole Cl precursor benzoyl chloride in the anion injection method,35 ensuring that identical amounts of benzoyl chloride were used for both doped and undoped NCs. This approach standardized the chloride ion levels across all samples (details in the experimental section of ESI†) to rule out the possibility of Cl surface passivation. PL spectra of Ni-doped NCs prepared using either cation or anion injection techniques at the same Ni concentrations are shown in Fig. S8 (ESI).† The PLQY enhancement was observed in Ni-doped samples (Inset of Fig. S7, ESI†) regardless of the chloride ion concentration, ruling out surface passivation as the source of the improved band-edge emission. These findings confirm that the increase in PLQY is directly attributed to the incorporation of Ni2+ dopant ions, highlighting that Ni doping effectively enhances the optical properties of the CsPbCl3 perovskite NCs. Further, it should be noted that dopant ion incorporation efficiency in the anion injection method is higher compared to the cation injection method since there are no pre-existing Pb–Cl bond in the precursors. Although the detailed mechanism reported in the literature35 and beyond the scope of this manuscript, we used this method to prepare samples with higher Ni doping concentration.
To investigate the electronic interaction of Ni2+ energy levels with the host CsPbCl3 NCs leading to this increase, we performed ab initio density functional theory (DFT) calculations. A cubic unit cell of CsPbCl3 was modeled with one Pb replaced by a Ni ion in the 2 × 2 × 2 supercell, representing a 12.5% Ni-doped system. Given the heavy Pb2+ ion, spin–orbit coupling (SOC) was incorporated using fully relativistic pseudopotentials for all the atomic types. As is typical with standard DFT, the calculated bandgap (∼1.91 eV) was smaller than the experimental value (∼3.01 eV).49 The band structure, projected onto three atomic orbitals Cl 3p (blue), Pb 6p (green), and Ni 3d (red), is shown in Fig. 2(b). The intermediate colors indicate the extent of mixing or hybridization of these three orbital contributions. Two closely spaced flat Ni 3d bands appear at ∼0.8 eV below the valence band maximum (VBM), along with dopant-induced Ni 3d states near the valence band. These findings are consistent with experimental reports for similar systems.34,50 Interestingly, significant hybridization of Ni 3d and Cl 3p orbitals was observed near the VBM at Γ point, as evident from the mixing of red and blue colors. This hybridization highlights a strong host-dopant interaction, further supporting the role of Ni2+ in influencing the electronic structure of CsPbCl3 NCs.
To investigate the sp–d exchange interaction between the dopant and the host and to pinpoint the relative position of the dopant ions, we measured MCD of undoped and Ni-doped NCs at 3 K with varying magnetic fields of 0, 3, and 5T, as shown in Fig. 2(c). The applied magnetic field induces the Zeeman effect at the band-edge, observed in both undoped and Ni-doped samples, giving rise to a non-zero differential spectrum. MCD, a differential technique, reveals higher-order transitions otherwise buried in the absorption spectrum.51 These differential peaks in the MCD spectra correspond to specific transitions in the absorption spectrum under a magnetic field. We identified the band-edge transition at ∼3.03 eV as A1 (pink dashed line) and the second excited state transition A2 (pink dashed line) in Fig. 2(c). Interestingly, we observe an additional transition at ∼3.5 eV, labeled X (blue dashed line) that appears exclusively in Ni-doped NCs. This extra peak (X) confirms that Ni2+ ions are successfully incorporated into the lattice and actively interact with the host perovskite through exchange interaction.
These results not only confirm successful Ni doping but also underscore the subtle yet impactful influence of Ni2+ on the optical and electronic properties of the NCs. This provides a strong foundation for further exploration of dopant-induced phenomena such as VADF, as discussed earlier. To investigate the origin of the increased QY in excitonic emission, we analyzed the decay kinetics using a micro-second flash lamp, as shown in Fig. 3(a). The undoped NCs exhibited a decay profile closely aligned with the instrument response function (IRF), indicating that the band-edge excitons decay fully within a few nanoseconds (ns).52
 | ||
| Fig. 3 Proof of VADF: (a) lifetime decay curve of undoped (P1) and 2% (Ni2P1) Ni-doped CsPbCl3 NCs with instrument response function (IRF) and fitting curve, indicating the lifetime of doped sample ∼μs and undoped ∼ns. (b) Temperature dependent gated PL spectra of Ni2P1 with 50 μs gating time after subtracting the contribution from undoped (at 295 K the intensity is multiplied by 2 for clarity), showing a significant delayed emission even at room temperature (295 K). (c) Change in integrated intensity as a function of temperature for all Ni-doped CsPbCl3 (NixP1; x = 0.5, 1, 2, 3) with inset of onset VADF temperature (T′) vs. Ni percentage (x) curve with 50 μs gating time (for Ni2P1 solid green line is a guide to the eye), suggesting the optimum doping percentage (∼2%) of Ni2+ can tune VADF up to room temperature. (d) Temperature dependent FWHM (Γ) of gated PL (with 50 μs gating time) band-edge emission peak for Ni-doped NCs (NixP1; x = 1, 2, 3) with the best fitting curve. | ||
In contrast, Ni-doped NCs demonstrated a prolonged tail in their band-edge emission, with Ni2P1 exhibiting a lifetime extending into the microsecond (μs) range. It consists of a fast decay component corresponding to band-edge recombination, and a slow decay component that may be attributed to delayed fluorescence. This behavior is reminiscent of Mn-doped CsPbBr3 NCs,28,53 though the time constant for Ni-doped NCs (∼μs) is shorter than the millisecond-scale lifetimes observed for Mn-doped counterparts. Lifetime decay curve fitting of Ni2P1 NCs shown in Fig. 3(a) indicates while 88% of the sample exhibits fast decay comparable to the IRF, approximately 12% of the NCs exhibit an average lifetime of 45.4 ± 1.6 μs. This value closely matches with the lifetime of Ni-related emission (order of few μs) in II–VI semiconductors.54 To better understand this long-lived emission, we performed temperature dependent gated PL measurement with a 50 μs gating time.55 In this setup, the sample was excited at t = 0 s and emission spectra were collected starting from t = 50 μs onward using the excitation energy of 3.4 eV. This approach effectively discards contributions from direct radiative recombination, which occurs within a few nanoseconds, and isolates long-lived emission components indicative of processes like delayed fluorescence.
Due to the presence of long tail in IRF, we observed minimal intensity in gated PL spectra for undoped NCs as shown in Fig. S9a (ESI)† and has been used as a baseline and subtracted from all samples. Fig. 3(b) presents the gated PL spectra for Ni2P1 after subtracting the contribution from the undoped NCs to eliminate distortions from the IRF's asymmetric tail. Remarkably, significant band-edge emission persisted for several microseconds after the excitation was switched off, even at room temperature (295 K) in Ni-doped NCs. Further evidence supporting this long-lived emission behavior is provided by temperature dependent gated PL measurements, shown in Fig. S9 (ESI).† This observation suggests that Ni doping induces delayed emission pathways, which are likely to contribute to enhanced QY.
To quantify the enhancement in gated PL at different Ni concentrations, we plotted the integrated intensity of band-edge emission for all doped NCs, as shown in Fig. 3(c). The inset displays the temperature (T′) at which gated emission vanishes as a function of Ni concentration. From the figure, a clear trend emerges, namely, as the temperature decreases, the PLQY intensity increases, with the band-edge integrated intensity reaching its peak around 2% Ni doping. Interestingly, the onset temperature for VADF shifts from 190 K at 0.5% Ni to 295 K at 2% Ni doping, highlighting the role of Ni concentration in stabilizing delayed fluorescence at room temperature. However, beyond 2% doping, the benefits diminish, as evidenced by the reduced integrated intensity for Ni3P1 (3% Ni doping), likely due to dopant–dopant interactions that suppress the efficiency of the VADF mechanism.56,57 At higher doping concentration, the probability of Ni2+–Ni2+ interactions rises, leading to enhanced non-radiative recombination pathways, giving less PLQY as observed in Fig. 3(c) and S7 (ESI).†
The possibility of surface passivation as the cause of increased PLQY was ruled out by altering synthesis methods, and EXAFS analysis confirms that Ni doping does not enhance structural order in the NCs. Further power-dependent time-resolved PL (TRPL) decay curve of undoped and Ni-doped NCs shows no power dependency (Fig. S10, ESI†), eliminating the possibility of Ni-induced defect passivation. Instead, the observed delayed fluorescence is attributed to the VADF phenomenon, wherein vibrational coupling between the host perovskite's valence or conduction band and the dopant-induced long-lived states continuously supplies charge carriers to the excitonic system. Unlike Mn-doped emissions, which exhibit significantly longer lifetimes (∼milliseconds), Ni2+ states in II–VI semiconductors have shorter lifetimes (∼μs),54,58 aligning with our observations of microsecond-scale delayed emission in Ni-doped perovskite NCs. Notably, this delayed excitonic emission persisted even at room temperature (295 K) in all Ni-doped mixed halide CsPb(ClyBr1−y)3 perovskites, as shown in Fig. S11 (ESI).† This confirms that delayed fluorescence is an intrinsic property of Ni2+ doping and is independent of the host bandgap. The temperature dependence mirrors that of Mn-doped systems, further supporting the conclusion that the enhanced PLQY arises from VADF.
Further evidence of vibrational assistance to radiative pathways is observed in the FWHM of both steady state and gated excitonic emissions as a function of temperature. As shown in Fig. 3(d), the FWHM decreases with lower temperatures, a trend well documented in the literature.5 Notably, in Ni-doped NCs, the FWHM of gated emission is narrower than that of steady state emission, in contrast to undoped NCs, where both the emission peaks exhibit similar FWHM trends (Fig. S12, ESI†). This distinction arises because steady state emission encompasses contributions from multiple vibrational states, whereas gated emission in doped NCs is primarily influenced by state-specific vibrational coupling, leading to a narrower FWHM as shown in Fig. 3(d). By comparison, in undoped NCs, although gated emission is detectable, its intensity originates from the steady state process, and the FWHM remains unchanged. To further quantify the VADF phenomenon, the temperature-dependent FWHM curve (Fig. 3(d) and S13, ESI†) was analyzed using the linear exciton–phonon coupling model (eqn (1)).5,59
 | (1) |
| Sample | Ni % | Γ 0 (meV) | γ lo (meV) |
|---|---|---|---|
| Ni1P1 | 1 | 46.02 ± 1.32 | 140.45 ± 14.46 |
| Ni2P1 | 2 | 29.76 ± 0.73 | 132.80 ± 11.42 |
| Ni3P1 | 3 | 23.39 ± 4.61 | 268.59 ± 25.15 |
From an electronic structure perspective, the presence of less dispersive Ni 3d bands near the Γ point, just above the Fermi level (Fig. 2(b)), results in a very high effective mass of holes, creating shallow hole trap states. These Ni 3d bands near the VBM trap holes from VB of the host. At lower temperatures, these trapped holes are transferred back to the excitonic system through vibrational assistance, enabling radiative recombination. This process, which is dominant at lower temperatures, selectively channels carriers into radiative pathways, thereby enhancing their contribution to PL.28 Consequently, the radiative recombination of excitons is delayed, resulting in VADF with lifetimes on the order of a few micro-seconds, matching the lifetime of Ni-induced states in Ni-doped CsPbCl3 NCs. This coupling of electronic and vibrational dynamics underpins the enhanced optical properties observed in the doped system. It is important to note here that we are not talking of energy transfer and hence does not necessitate resonance coupling between the dopant's d–d transition and host's radiative transition. In contrast, for a charge carrier transition, we only require the presence of one dopant state close to the conduction or valence band.
To further confirm the presence of VADF and use it for high-performance energy harvesting applications, we performed FRET experiments by titrating doped and undoped NCs with coumarin-30 (C-30), a dye with significant excitonic emission overlap (Fig. 4(a)). FRET efficiency, determined by the radiative transfer from NCs (donor) to the dye (acceptor), serves as a quantitative measure of the radiative transfer efficiency.60,61 Recent studies57 have shown that VADF facilitates an internal mechanism prioritizing radiative channels, resulting in enhanced FRET efficiency. In line with this understanding, we demonstrated how Ni doping in CsPbCl3 NCs enhances charge carrier back-transfer to the host material. This process increases the population of charge carriers within the host lattice, thereby boosting FRET efficiency by favoring radiative transitions over non-radiative ones. This observation further substantiates the role of VADF in promoting radiative pathways in Ni-doped NCs, consistent with the findings from exciton–phonon coupling analysis.
 | ||
| Fig. 4 Effects of VADF: (a) absorption and emission spectra of undoped CsPbCl3 (P1) along with absorption curve of coumarin-30 (C-30) confirming significant excitonic emission overlap between the donor NCs and acceptor dye. (b) Steady state PL spectra of 2% Ni-doped CsPbCl3 and C-30 physical mixture (Ni2P1 + C-30) at 77 K showing quenching of donor emission with a simultaneous increment in acceptor's emission. (c) Lifetime decay curve of (Ni2P1 + C-30) physical mixture, suggesting that with increasing dye concentration lifetime of the band-edge emission decreases. (d) Change of integrated intensity of C-30 emission as a function of C-30 concentration for physical mixture of (P1 + C-30) and (Ni2P1 + C-30) at 77 K (solid circle and solid line, green solid line is a guide to the eye) and at 295 K (open circle and dashed line). It suggests VADF is significantly enhancing the FRET efficiency at 77 K. | ||
Fig. 4(b) displays the steady-state PL spectra of Ni2P1 titrated with varying concentrations of C-30 at 77 K. Additional spectra for undoped NCs at 77 K and 295 K, as well as for Ni-doped NCs at 295 K, are provided in Fig. S14 (ESI).† In all cases, quenching of donor emission accompanied by a concurrent rise in acceptor's emission signifies efficient FRET. The lifetime of the band-edge emission for the physical mixture of undoped (Fig. S15, ESI†) and Ni-doped (Fig. 4(c)) NCs decreases with increasing dye concentration. The FRET efficiency, plotted in Fig. 4(d), for different dye concentrations at 77 K and 295 K, highlights a distinct trend. At 295 K, where VADF is negligible, FRET efficiency remains unaffected by doping. However, at 77 K, where VADF plays a significant role, Ni-doped NCs exhibit a pronounced increase in FRET efficiency around 80% compared to 45% in undoped NCs, confirming that VADF is an intrinsic phenomenon linked to transition metal doping like Ni and Mn. By enabling improved radiative transfer, VADF offers a pathway to selectively enhance energy conversion efficiency through careful dopant selection. These findings not only deepen our understanding of dopant-induced phenomena but also demonstrate the potential of transition metal-doped perovskite NCs for high-performance energy harvesting applications.
Conclusions
In summary, we demonstrated the successful incorporation of Ni2+ into CsPbCl3 perovskite NCs, enhancing PLQY via the VADF phenomenon. Structural integrity was confirmed through EXAFS, XANES, and DFT analysis, while optical studies revealed strong exciton–phonon coupling and efficient radiative energy transfer, particularly at lower temperatures. Power-dependent TRPL measurements, comprehensive EXAFS analysis along with the presence of sp–d exchange interaction as obtained from MCD measurements confirmed the increased PLQY is not due to Ni-induced defects or Cl-induced surface passivation. Vibrational coupling between Ni 3d levels and host VB helped to selectively channel charge carriers into radiative pathways, resulting in increased PLQY. FRET experiments further highlighted improved radiative energy transfer efficiency at lower temperatures.This study highlights Ni2+ doping as a promising strategy to enhance the optical performance of perovskite NCs, enabling applications in light-emitting devices, photovoltaics, lasers, single-photon sources, and photocatalysis. The intrinsic VADF mechanism offers a new paradigm for designing NCs with tailored radiative properties. Future research can optimize energy transfer, extend emission lifetimes, and improve efficiencies while exploring mixed-halide perovskites, other dopants, and hybrid systems to develop multifunctional materials for advanced optoelectronic and energy-harvesting technologies.
Data availability
The detailed experimental data that support our findings are available in the ESI.† It includes experimental methods, characterization and spectroscopic techniques, computational details, elemental analysis table, XRD spectra, EXAFS spectra, fitting parameter table of EXFAS, ESR spectra, TEM images, particle size analysis histograms, and tables, HAADF-STEM EDX elemental mapping, steady state and gated PL emission spectra at different temperatures, power-dependent TRPL decay, FWHM fitting curves along with fitting parameters table, temperature dependent steady state PL spectra and lifetime decay curve of physical mixture of dye with undoped and Ni-doped NCs.Author contributions
S. P. and R. V. designed the project. S. P. synthesized all the samples and carried out all the measurements and analyzed the data. P. M. and S. M. helped in the measurements and data analysis. A. D. did DFT calculation under the supervision of S. N. S. P. wrote the original draft of the manuscript. R. V. edited and reviewed the draft and finalized it. All the authors contributed to preparing the manuscript and approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We would like to thank JNCASR for its experimental facilities, Department of Science and Technology (DST) for funding. S. P., P. M., S. M., and A. D. would like to thank DST-INSPIRE (DST/INSPIRE Fellowship/2020/IF 200284), CSIR, SERB, and JNCASR respectively for research fellowships. A. D. and S. N. would like to thank ParamYukti supercomputer under National Supercomputing Mission (NSM), JNCASR for providing excellent computational facilities. R. V. would like to thank the SERB-POWER fellowship for partial funding of this work. The authors would like to acknowledge SAMat research facilities at JNCASR for temperature dependent PL, TEM, and ESR measurements. EXAFS measurements were carried out at the light source PETRA-III of DESY, a member of the Helmholtz Association (HGF), Germany. The authors would like to thank Dr Aleksandr Kalinko and Dr Maria Naumova at beamline P64. Financial support by the Department of Science and Technology (Government of India) provided within the framework of the India@DESY collaboration is gratefully acknowledged.References
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- S. M. H. Qaid, M. S. Al Sobaie, M. A. Majeed Khan, I. M. Bedja, F. H. Alharbi, M. K. Nazeeruddin and A. S. Aldwayyan, Mater. Lett., 2016, 164, 498–501 CrossRef CAS.
- I. Levchuk, A. Osvet, X. Tang, M. Brandl, J. D. Perea, F. Hoegl, G. J. Matt, R. Hock, M. Batentschuk and C. J. Brabec, Nano Lett., 2017, 17, 2765–2770 CrossRef CAS PubMed.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435–2445 Search PubMed.
- O. H. C. Cheng, T. Qiao, M. Sheldon and D. H. Son, Nanoscale, 2020, 12, 13113–13118 Search PubMed.
- C. M. Sutter-Fella, Y. Li, M. Amani, J. W. Ager, F. M. Toma, E. Yablonovitch, I. D. Sharp and A. Javey, Nano Lett., 2016, 16, 800–806 CrossRef CAS PubMed.
- C. Bi, S. Wang, W. Wen, J. Yuan, G. Cao and J. Tian, J. Phys. Chem. C, 2018, 122, 5151–5160 CrossRef CAS.
- S. Mannar, P. Mandal, A. Roy and R. Viswanatha, J. Phys. Chem. Lett., 2022, 13, 6290–6297 CrossRef CAS PubMed.
- K. Lin, J. Xing, L. N. Quan, F. P. G. de Arquer, X. Gong, J. Lu, L. Xie, W. Zhao, D. Zhang, C. Yan, W. Li, X. Liu, Y. Lu, J. Kirman, E. H. Sargent, Q. Xiong and Z. Wei, Nature, 2018, 562, 245–248 CrossRef CAS PubMed.
- S. Zou, Y. Liu, J. Li, C. Liu, R. Feng, F. Jiang, Y. Li, J. Song, H. Zeng, M. Hong and X. Chen, J. Am. Chem. Soc., 2017, 139, 11443–11450 CrossRef CAS PubMed.
- Z. Li, J. Moon, A. Gharajeh, R. Haroldson, R. Hawkins, W. Hu, A. Zakhidov and Q. Gu, ACS Nano, 2018, 12, 10968–10976 CrossRef CAS PubMed.
- N. G. Park, Mater. Today, 2015, 18, 65–72 Search PubMed.
- M. Gong, R. Sakidja, R. Goul, D. Ewing, M. Casper, A. Stramel, A. Elliot and J. Z. Wu, ACS Nano, 2019, 13, 1772–1783 CrossRef CAS PubMed.
- R. K. Behera, S. Das Adhikari, S. K. Dutta, A. Dutta and N. Pradhan, J. Phys. Chem. Lett., 2018, 9, 6884–6891 CrossRef CAS PubMed.
- D. Chen, G. Fang, X. Chen, L. Lei, J. Zhong, Q. Mao, S. Zhou and J. Li, J. Mater. Chem. C, 2018, 6, 8990–8998 Search PubMed.
- Y. Guo, J. Su, L. Wang, Z. Lin, Y. Hao and J. Chang, J. Phys. Chem. Lett., 2021, 12, 3393–3400 CrossRef CAS PubMed.
- K. M. Hossain, M. Z. Hasan and M. L. Ali, AIP Adv., 2021, 11, 015052 CrossRef CAS.
- Y. Chen, Y. Liu and M. Hong, Nanoscale, 2020, 12, 12228–12248 RSC.
- K. R. Pradeep and R. Viswanatha, Mater. Res. Bull., 2021, 141, 111374 Search PubMed.
- S. Chakraborty, P. Mandal and R. Viswanatha, Chem.–Asian J., 2022, 17, e202200478 CrossRef CAS.
- R. Wu, Z. Bai, J. Jiang, H. Yao and S. Qin, RSC Adv., 2021, 11, 8430–8436 RSC.
- Q. Li, Y. Liu, P. Chen, J. Hou, Y. Sun, G. Zhao, N. Zhang, J. Zou, J. Xu, Y. Fang and N. Dai, J. Phys. Chem. C, 2018, 122, 29044–29050 CrossRef CAS.
- J. P. Ma, Y. M. Chen, L. M. Zhang, S. Q. Guo, J. D. Liu, H. Li, B. J. Ye, Z. Y. Li, Y. Zhou, B. B. Zhang, O. M. Bakr, J. Y. Zhang and H. T. Sun, J. Mater. Chem. C, 2019, 7, 3037–3048 RSC.
- M. Zeng, F. Locardi, D. Mara, Z. Hens, R. Van Deun and F. Artizzu, Nanoscale, 2021, 13, 8118–8125 RSC.
- P. Mandal, S. Panja and R. Viswanatha, Chem.–Asian J., 2025, 20, e202401161 CrossRef CAS PubMed.
- J. Wang, R. Cao, P. Da, Y. Wang, T. Hu, L. Wu, J. Lu, X. Shen, F. Xu, G. Zheng and Z. Chen, Appl. Phys. Lett., 2016, 108, 022103 CrossRef.
- M. V Artemyev, U. Woggon, R. Wannemacher, H. Jaschinski and W. Langbein, Nano Lett., 2001, 1, 309–314 CrossRef.
- K. R. Pradeep, D. Acharya, P. Jain, K. Gahlot, A. Yadav, A. Camellini, M. Zavelani-Rossi, G. Cerullo, C. Narayana, S. Narasimhan and R. Viswanatha, ACS Energy Lett., 2020, 5, 353–359 CrossRef.
- K. R. Pradeep, A. Elumalai and R. Viswanatha, J. Phys. Chem. C, 2022, 126, 9813–9819 CrossRef CAS.
- J. Yang, X. Wen, H. Xia, R. Sheng, Q. Ma, J. Kim, P. Tapping, T. Harada, T. W. Kee, F. Huang, Y.-B. Cheng, M. Green, A. Ho-Baillie, S. Huang, S. Shrestha, R. Patterson and G. Conibeer, Nat. Commun., 2017, 8, 14120 Search PubMed.
- A. Saha, K. Gahlot and R. Viswanatha, ChemNanoMat, 2022, 8, e202100457 CrossRef CAS.
- P. Mondal, S. Chakraborty, G. K. Grandhi and R. Viswanatha, J. Phys. Chem. Lett., 2020, 11, 5367–5372 CrossRef CAS.
- H. Kim, S. Bae, T. H. Lee, H. Lee, H. Kang, S. Park, H. W. Jang and S. Y. Kim, Adv. Funct. Mater., 2021, 31, 1–9 Search PubMed.
- Z. J. Yong, S. Q. Guo, J. P. Ma, J. Y. Zhang, Z. Y. Li, Y. M. Chen, B. B. Zhang, Y. Zhou, J. Shu, J. L. Gu, L. R. Zheng, O. M. Bakr and H. T. Sun, J. Am. Chem. Soc., 2018, 140, 9942–9951 CrossRef CAS PubMed.
- M. Imran, V. Caligiuri, M. Wang, L. Goldoni, M. Prato, R. Krahne, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2018, 140, 2656–2664 CrossRef CAS PubMed.
- S. Chakraborty, M. Makkar and R. Viswanatha, J. Phys. Chem. C, 2023, 127, 18518–18525 CrossRef CAS.
- A. De, N. Mondal and A. Samanta, Nanoscale, 2017, 9, 16722–16727 RSC.
- J. Meng, Z. Lan, M. Abdellah, B. Yang, S. Mossin, M. Liang, M. Naumova, Q. Shi, S. L. Gutierrez Alvarez, Y. Liu, W. Lin, I. E. Castelli, S. E. Canton, T. Pullerits and K. Zheng, J. Phys. Chem. Lett., 2020, 11, 3705–3711 CrossRef CAS.
- P. Mandal, A. Roy, S. Mannar and R. Viswanatha, Nanoscale Adv., 2020, 2, 5305–5311 RSC.
- Y. Nam, M. Kim, S. Y. Kim, J. Jung, G. R. Kumar, J. H. Lee, S. Lee, C. H. Park and Y. W. Heo, Adv. Opt. Mater., 2023, 11, 2300682 CrossRef CAS.
- K. Xu, J. F. Vliem and A. Meijerink, J. Phys. Chem. C, 2019, 123, 979–984 CrossRef CAS PubMed.
- J. Yi, X. Ge, E. Liu, T. Cai, C. Zhao, S. Wen, H. Sanabria, O. Chen, A. M. Rao and J. Gao, Nanoscale Adv., 2020, 2, 4390–4394 RSC.
- S. Das Adhikari, A. K. Guria and N. Pradhan, J. Phys. Chem. Lett., 2019, 10, 2250–2257 CrossRef CAS PubMed.
- P. V Radovanovic, C. J. Barrelet, S. Gradečak, F. Qian and C. M. Lieber, Nano Lett., 2005, 5, 1407–1411 Search PubMed.
- G. Pan, X. Bai, D. Yang, X. Chen, P. Jing, S. Qu, L. Zhang, D. Zhou, J. Zhu, W. Xu, B. Dong and H. Song, Nano Lett., 2017, 17, 8005–8011 CrossRef CAS.
- L. Zhou, T. Liu, J. Zheng, K. Yu, F. Yang, N. Wang, Y. Zuo, Z. Liu, C. Xue, C. Li, B. Cheng and Q. Wang, J. Phys. Chem. C, 2018, 122, 26825–26834 CrossRef CAS.
- Y. Zhang, X. Cheng, D. Tu, Z. Gong, R. Li, Y. Yang, W. Zheng, J. Xu, S. Deng and X. Chen, Angew. Chem., Int. Ed., 2021, 60, 9693–9698 Search PubMed.
- G. H. Ahmed, J. K. El-Demellawi, J. Yin, J. Pan, D. B. Velusamy, M. N. Hedhili, E. Alarousu, O. M. Bakr, H. N. Alshareef and O. F. Mohammed, ACS Energy Lett., 2018, 3, 2301–2307 CrossRef CAS.
- M. A. Becker, R. Vaxenburg, G. Nedelcu, P. C. Sercel, A. Shabaev, M. J. Mehl, J. G. Michopoulos, S. G. Lambrakos, N. Bernstein, J. L. Lyons, T. Stöferle, R. F. Mahrt, M. V Kovalenko, D. J. Norris, G. Rainò and A. L. Efros, Nature, 2018, 553, 189–193 CrossRef CAS PubMed.
- M. N. Islam, M. A. Hadi and J. Podder, AIP Adv., 2019, 9, 1–9 Search PubMed.
- X. Gao, X. Zhang, X. Yang, L. Zhao, B. Han, H. R. Alanagh and Z. Tang, Nanoscale, 2019, 11, 19380–19386 RSC.
- K. Xu, J. F. Vliem and A. Meijerink, J. Phys. Chem. C, 2018, 123, 979–984 CrossRef PubMed.
- S. K. Mehetor, H. Ghosh, B. Hudait, N. S. Karan, A. Paul, S. Baitalik and N. Pradhan, ACS Energy Lett., 2019, 4, 2353–2359 CrossRef CAS.
- S. Jana, B. B. Srivastava, S. Jana, R. Bose and N. Pradhan, J. Phys. Chem. Lett., 2012, 3, 2535–2540 CrossRef CAS PubMed.
- R. Beaulac, P. I. Archer, J. van Rijssel, A. Meijerink and D. R. Gamelin, Nano Lett., 2008, 8, 2949–2953 CrossRef CAS PubMed.
- A. Nag, S. Chakraborty and D. D. Sarma, J. Am. Chem. Soc., 2008, 130, 10605–10611 CrossRef CAS PubMed.
- S. Das, S. Aich, M. Aswathy and R. Viswanatha, ACS Energy Lett., 2024, 9, 5461–5463 CrossRef CAS.
- S. Jana, G. Manna, B. B. Srivastava and N. Pradhan, Small, 2013, 9, 3753–3758 CrossRef CAS PubMed.
- D. P. Strandell and P. Kambhampati, J. Phys. Chem. C, 2021, 125, 27504–27508 CrossRef CAS.
- J. T. DuBose and P. V. Kamat, J. Am. Chem. Soc., 2021, 143, 19214–19223 CrossRef CAS PubMed.
- J. T. DuBose and P. V. Kamat, J. Am. Chem. Soc., 2023, 145, 4601–4612 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00564g |
| This journal is © The Royal Society of Chemistry 2025 |