
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoinduced copper-catalysed enantioselective amination of allylic and propargylic C–H bonds†
Ling Dai,
Ying-Ying Chen,
Jing-Jun Wang,
Jun-Jia Chen and
Qi-Lin Zhou *
*
State Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Frontiers Science Center for New Organic Matter, Nankai University, Tianjin 300071, China. E-mail: qlzhou@nankai.edu.cn
First published on 17th April 2025
Abstract
The hydrogen atom transfer (HAT)-mediated strategy has emerged as a straightforward and powerful approach for oxidative C–H bond functionalization. However, despite remarkable progress in this field, enantioselective allylic and propargylic C–H aminations remain a challenge. In this study, we developed highly enantioselective allylic and propargylic C–H aminations by combining a visible light-activated HAT process with copper catalysis. Using this method, a wide range of alkenes and alkynes can be converted into high-value chiral allylic and propargylic amines with high enantio-, regio-, and E/Z-selectivity. These enantio-enriched amines serve as versatile building blocks in organic synthesis and hold significant potential for applications in the synthesis of pharmaceuticals, natural products and other bioactive molecules.
Introduction
Chiral amines, particularly allylic amines, are ubiquitous structure motifs in pharmaceuticals, natural products, and other biologically active molecules (Scheme 1A).1–3 Enantioselective C(sp3)–H amination provides a straightforward approach for synthesizing chiral amines due to the abundant availability of starting materials and the elimination of substrate pre-activation.4–6 Transition metal-catalysed C–H activation via π-allyl metal intermediates is an efficient method for the synthesis of chiral allylic amines;7–10 however, successful examples of this method are limited to intramolecular reactions.11,12 Metal-nitrenoid insertion into the C–H bond has been reported for achieving enantioselective allylic C–H amination,13–18 but this catalytic system suffers from issues such as alkene isomerization, competing aziridination, and the requirement for pre-functionalized nitrene precursors (Scheme 1B).19,20 Therefore, it is desirable to develop efficient and practical methods for enantioselective allylic C–H amination.21 | ||
| Scheme 1 Catalytic asymmetric allylic C–H amination reactions. | ||
Recently, the radical-mediated hydrogen-atom transfer (HAT) process has emerged as an efficient strategy for enantioselective C–H bond functionalization.22–24 In 2022, Gevorgyan and co-workers presented an open-shell approach that enables palladium-catalysed enantioselective allylic C–H amination with nucleophilic aliphatic amines (Scheme 1C).25 Nevertheless, the substrate scope remained relatively narrow, with only a few substrates achieving optimal enantioselectivity control. We have previously reported a copper-catalysed enantioselective benzylic C–H amination using peroxide as the HAT reagent and amides as amination reagents.26 However, this cationic copper catalyst exhibited very low yield in the enantioselective allylic C–H amination of alkenes. Through a systematic investigation of various reaction parameters, we found that the reaction proceeds under visible light irradiation with carbamate as the amination reagent. Herein, we describe the photoinduced copper-catalysed enantioselective allylic C–H amination of alkenes, which produces chiral primary allylamines with high enantio-, regio-, chemo-, and E/Z-selectivity. Moreover, this strategy can also be successfully applied to enantioselective propargylic C–H amination (Scheme 1D).
Results & discussion
The copper-catalysed amination of (E)-but-1-ene-1-phenyl (1a) with methyl carbamate (2a) was carried out to find efficient catalysts, oxidants, amine sources, and optimal reaction conditions (Table 1, see Tables S1–S6† for details). A comparison of ligands showed that the Evans bisoxazoline ligand L9 with bulky phenyl groups was the optimal ligand, producing amination product 3a with high yield and enantioselectivity. The study of catalyst precursors revealed that the combination of CuCl and NaBArF provided the best yield and enantioselectivity (entries 1–4, see Table S2† for details). Among the tested oxidants, di-tert-butyl peroxide (DTBP) offered the most satisfactory results, generating tert-butoxy radicals under visible light irradiation and subsequently alkyl radicals from alkenes via the HAT process27,28 (entries 5, 6 vs. 1, see Table S3† for details). In addition to C6F6, in which the reaction has the highest yield and enantioselectivity, C6H5Cl and dichloroethane (DCE) were also viable solvents, albeit with lower yield and enantioselectivity (entries 7, 8 vs. 1). Control experiments demonstrated that each of the following components—copper, sodium tetrakis[3,5-bis(trifluoromethyl)phenyl] borate (NaBArF), ligand L9, DTBP, and light—is indispensable for the reaction (entries 9–13). The reaction was also successfully carried out with one equivalent of alkene, although the yield and enantioselectivity were slightly lower (entry 14).
|
|||
|---|---|---|---|
| Entry | Variation from the “standard conditions” | Yielda (%) | eeb (%) |
| a Yields were determined by 1H NMR with dimethyl terephthalate as an internal standard.b Enantiomeric excess (ee) values were determined by chiral HPLC.c Isolated yield. NaBArF, sodium tetrakis [3,5-bis(trifluoromethyl)phenyl] borate. DTBP, di-tert-butyl peroxide. NFSI, N-fluorobenzenesulfonimide. DCP, dicumyl peroxide. | |||
| 1 | None | 81 (76)c | 91 |
| 2 | Cu(OTf)2 instead of CuCl/NaBArF | 16 | 8 |
| 3 | Cu(CH3CN)4BF4 instead of CuCl/NaBArF | 31 | 45 |
| 4 | Cu(CH3CN)4PF6 instead of CuCl/NaBArF | 38 | 59 |
| 5 | NFSI instead of DTBP | 7 | rac |
| 6 | DCP instead of DTBP | 43 | 74 |
| 7 | PhCI instead of C6F6 | 58 | 77 |
| 8 | DCE instead of C6F6 | 62 | 86 |
| 9 | Without CuCl | — | — |
| 10 | Without NaBArF | — | — |
| 11 | Without L9 | — | — |
| 12 | Without DTBP | — | — |
| 13 | Without hv | — | — |
| 14 | 1 equiv. of 1a | 52 | 87 |

Under optimal reaction conditions, we explored the substrate scope of alkenes and carbamates (Scheme 2). Alkene substrates 1, whether as pure E-isomers or E/Z mixtures, reacted with carbamate 2a to yield thermodynamically stable E-products 3 (E/Z > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The excellent E/Z-selectivity enhances the practicability of the reaction, as many alkenes are usually prepared as E/Z-mixtures. Alkenes with para-substituents on the benzene ring, regardless of whether they are electron-donating or electron-withdrawing, showed moderate to good yield and high enantioselectivity (≥90% ee) (1a–1c, 1e–1h, and 1j–1l). Exceptions included 1d (with a para-F) and 1i (with a para-MsO), which exhibited lower enantioselectivity (80% ee). Alkenes with meta-substituents on the benzene ring (1m–1p) afforded the desired amination products with 85–90% ee. However, those with ortho-halogen substituents (1q and 1r) displayed lower enantioselectivity (82% ee and 80% ee, respectively). Similarly, substrates with 2-naphthyl (1s, 80% ee), benzofuran-5-yl (1t, 80% ee) and benzothiophen-5-yl (1u, 85% ee) also showed lower enantioselectivity. Notably, the reaction demonstrated good functional group compatibility with chlorine (1x), acetoxy (1y) and ester (1z) substituents on the alkyl chains of alkene substrates. Other carbamates, such as BocNH2 (2aa), CbzNH2 (2ab), TeocNH2 (2ac) and FmocNH2 (2ad), whose products are more easily deprotected, were also effective amine sources, affording good yields and high enantioselectivity (90–91% ee). The use of carbamates as amine sources provides convenience for the synthesis of allylic amines without a protecting group, as the alkyoxycarbonyl groups in the products can be removed by common methods.29
:1). The excellent E/Z-selectivity enhances the practicability of the reaction, as many alkenes are usually prepared as E/Z-mixtures. Alkenes with para-substituents on the benzene ring, regardless of whether they are electron-donating or electron-withdrawing, showed moderate to good yield and high enantioselectivity (≥90% ee) (1a–1c, 1e–1h, and 1j–1l). Exceptions included 1d (with a para-F) and 1i (with a para-MsO), which exhibited lower enantioselectivity (80% ee). Alkenes with meta-substituents on the benzene ring (1m–1p) afforded the desired amination products with 85–90% ee. However, those with ortho-halogen substituents (1q and 1r) displayed lower enantioselectivity (82% ee and 80% ee, respectively). Similarly, substrates with 2-naphthyl (1s, 80% ee), benzofuran-5-yl (1t, 80% ee) and benzothiophen-5-yl (1u, 85% ee) also showed lower enantioselectivity. Notably, the reaction demonstrated good functional group compatibility with chlorine (1x), acetoxy (1y) and ester (1z) substituents on the alkyl chains of alkene substrates. Other carbamates, such as BocNH2 (2aa), CbzNH2 (2ab), TeocNH2 (2ac) and FmocNH2 (2ad), whose products are more easily deprotected, were also effective amine sources, affording good yields and high enantioselectivity (90–91% ee). The use of carbamates as amine sources provides convenience for the synthesis of allylic amines without a protecting group, as the alkyoxycarbonyl groups in the products can be removed by common methods.29
 | ||
| Scheme 2 Substrate scope of alkenes and carbamates. Isolated yield. Ee values were determined by chiral HPLC, and E/Z ratios were determined by 1H NMR. aThe absolute configuration of compound 3a was determined to be S by comparing the optical rotation of its deprotected amine with that reported in the literature. bThe ratio of site-isomers was determined by 1H NMR. BocNH2, tert-butyl carbamate. CbzNH2, benzyl carbamate. TeocNH2, 2-(trimethylsilyl)ethyl carbamate. FmocNH2, (9H-fluoren-9-yl)methyl carbamate. | ||
Encouraged by the success in the allylic C–H amination, we next studied the enantioselective propargylic C–H amination. We were delighted to find that the standard conditions for the allylic C–H amination also worked for the propargylic C–H amination reaction (Scheme 3). A variety of alkyl aryl alkynes with diverse substituents reacted with carbamate 2a to yield the propargylic C–H amination products (5b–5o) in good yields (56–84%) and with high enantioselectivities (81–92% ee). The amination selectively occurred at the propargylic site when the substrate contained both propargylic and benzylic C–H bonds (5e and 5f). Alkyne substrates featuring naphthyl (5p and 5q, 90% ee), 3-thienyl (5r, 91% ee), benzofuryl (5s, 86% ee), and silyl (5t, 93% ee) also demonstrated high enantioselectivity in the reaction. The effect of substrate's alkyl chain and its functional groups on the enantioselectivity was negligible, indicating the reaction's broad tolerance for functional groups (5u–5ac). Additionally, alkynes derived from oxaprozin (4ad), naproxen (4ae), and isoxepac (4af) reacted smoothly with 2a to yield the corresponding propargylic amines with good enantioselectivity. The absolute configuration of 5n was determined to be S by X-ray single crystal diffraction analysis.
 | ||
| Scheme 3 Substrate scope of alkynes. Isolated yield, ee values were determined by chiral HPLC. aThe ratio of site-isomers was determined by 1H NMR. | ||
To demonstrate the synthetic utility of the method, we investigated the transformations of the amination products (Scheme 4). The deprotection of amination products 3a and 5ag yielded chiral primary amines 630 and 9, without any loss of enantiopurity. Product 3a was successfully converted into saturated aliphatic amine 7 and epoxide 8 by treatment with Pd–C/H2 and m-CPBA, respectively. Similarly, the hydrogenation of product 5a provided Z-allylamine 10. Removal of the silyl group from product 5t afforded terminal alkyne 11, which is a versatile intermediate suitable for Sonogashira coupling or Click reactions. Lysophosphatidic acid receptor 13, a compound used in the treatment of fibrotic diseases,31 and anticancer agent 1632 were readily prepared using asymmetric amination as the key step, further showcasing the method's potential for pharmaceutical synthesis.
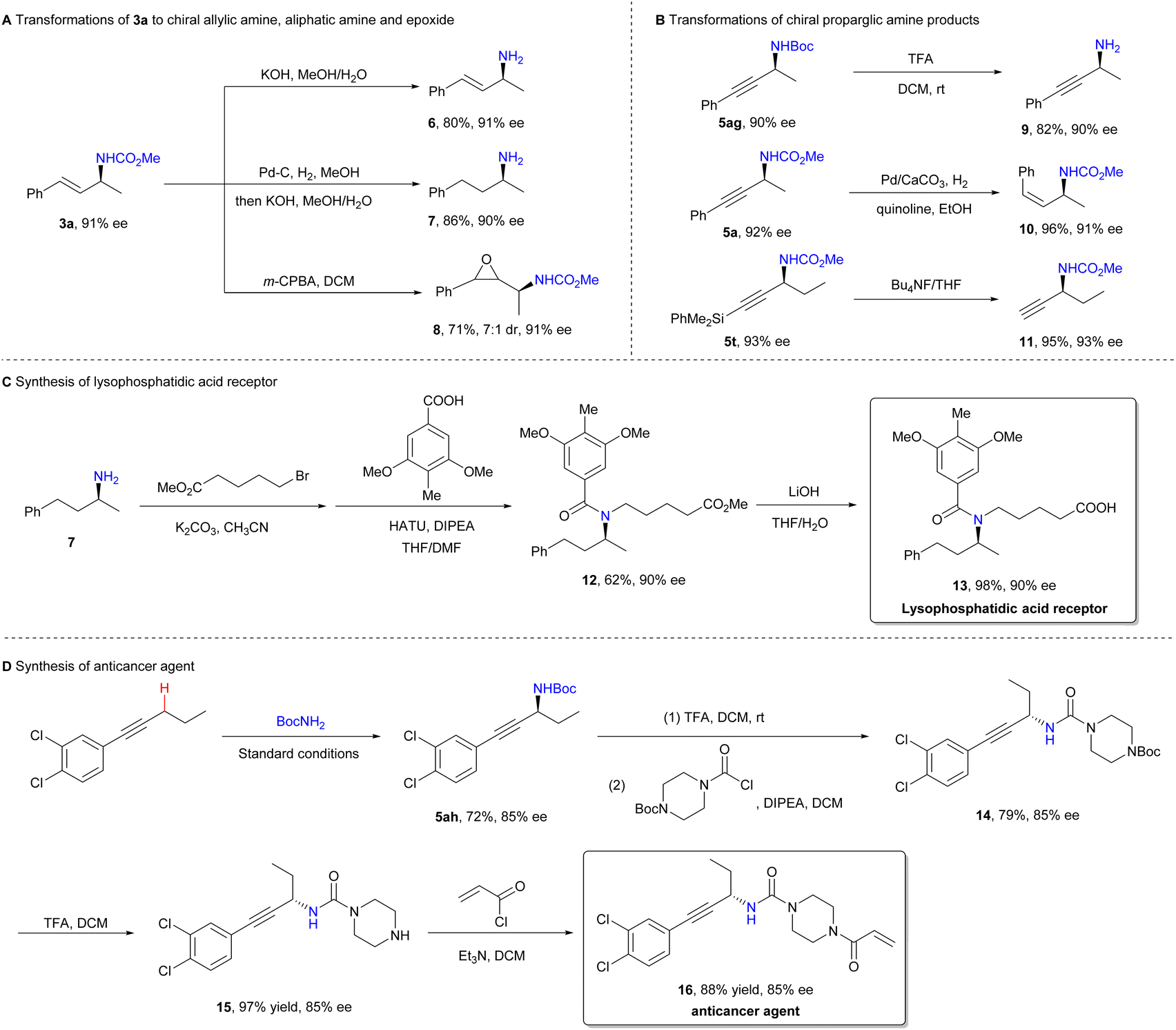 | ||
| Scheme 4 Synthetic applications. | ||
To gain insight into the reaction mechanism, we conducted several experiments to identify the key intermediates of the reaction (Scheme 5). Under the standard conditions, the addition of the radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) completely inhibited the reaction (Scheme 5A), indicating the involvement of a free radical process. Deuterium kinetic isotope effect (KIE) experiments yielded KIE values of 3.0 and 2.4, suggesting that the hydrogen atom abstraction from the C(sp3)–H bond by a tert-butoxy radical might likely be the rate-limiting step (Scheme 5B). When substrate 1a and product 3a were exposed to visible light irradiation, no carbon–carbon double bond isomerization was observed (see the ESI† for details), and the E/Z ratio of the alkene substrate 1a remained unchanged during the reaction (Scheme 5C). These results supported that the E-selection in the allylic C–H amination reaction might be generated through a stereospecific amination of the allyl-copper intermediate, rather than through a photoinduced double bond isomerization process. Using electron paramagnetic resonance (EPR) with the spin-trapping agent 5,5-dimethyl-1-pyrrolidine N-oxide (DMPO), we detected tert-butoxy radicals and found that these radicals are primarily generated by visible light irradiation (Scheme 5D).33 A copper(II) signal was observed in the EPR measurements of the reaction mixture, indicating that copper(II) species, such as CuII–OtBu or CuII–NHCO2Me intermediates, may be involved in the reaction34,35 (Scheme 5E).
 | ||
| Scheme 5 Mechanism studies. | ||
Based on our experimental results and previous reports,22–24 we propose a mechanism for the enantioselective allylic C–H amination, as shown in Scheme 6. Upon visible light irradiation, DTBP decomposes to generate tert-butoxy radicals, which react with the cationic copper(I) complex A to form the Cu(II)–OtBu intermediate B. Ligand exchange of B with carbamate 2a yields the Cu(II)–NHCO2Me intermediate C. Intermediate C interacts with the allyl radical, which is generated by the abstraction of the allylic hydrogen from 1a by the tert-butoxy radical, to form the copper(III) intermediate D. Subsequently, D undergoes reductive elimination, producing product 3a and regenerating the copper(I) complex A.
 | ||
| Scheme 6 Plausible mechanism of allylic C–H amination. | ||
Conclusions
In summary, we have developed a photoinduced copper-catalysed enantioselective C–H amination of alkenes and alkynes. This reaction enables the efficient synthesis of diverse chiral allylic amines and propargylic amines with high enantioselectivity, regioselectivity, and E/Z-selectivity. Mechanistic studies of the reaction reveal that visible-light irradiation decomposes DTBP to generate tert-butoxy radicals, which play a key role in the HAT process and in maintaining the Cu(I)–Cu(II)–Cu(III) catalytic cycle. The protocol shows great potential for applications in the synthesis of bioactive compounds. Future efforts will focus on applying this strategy to other enantioselective C(sp3)–H functionalizations.Data availability
The data supporting this article have been included in the ESI.† Crystallographic data for compound 5n have been deposited at the Cambridge Crystallographic Data Centre, reference numbers CCDC: 2378637.Author contributions
Q.-L. Z. conceived the study; L. D. and Q.-L. Z. designed the experiments and analyzed the data; L. D., Y.-Y. C., J.-J. W. and J.-J. C. prepared substrates and ligands. L. D. performed the reactions and mechanistic study. L. D. and Q.-L. Z. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Key R&D Program of China (2022YFA1504302), the National Natural Science Foundation of China (22188101 and 91956000), the Fundamental Research Funds for the Central Universities, and the Haihe Laboratory of Sustainable Chemical Transformations for financial support.Notes and references
- A. Stütz, Angew. Chem., Int. Ed., 1987, 26, 320–328 CrossRef.
- G. Petranyi, N. S. Ryder and A. Stütz, Science, 1984, 224, 1239–1241 CrossRef CAS PubMed.
- M. Johannsen and K. A. Jorgensen, Chem. Rev., 1998, 98, 1689–1708 CrossRef CAS PubMed.
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed.
- T. A. Ramirez, B. Zhao and Y. Shi, Chem. Soc. Rev., 2012, 41, 931–942 RSC.
- F. Collet, C. Lescot and P. Dauban, Chem. Soc. Rev., 2011, 40, 1926–1936 RSC.
- S. A. Reed and M. C. White, J. Am. Chem. Soc., 2008, 130, 3316–3318 CrossRef CAS PubMed.
- G. Liu, G. Yin and L. Wu, Angew. Chem., Int. Ed., 2008, 47, 4733–4736 CrossRef CAS PubMed.
- H. Lei and T. Rovis, Nat. Chem., 2020, 12, 725–731 CrossRef CAS PubMed.
- Y. Jin, Y. Jing, C. Li, M. Li, W. Wu, Z. Ke and H. Jiang, Nat. Chem., 2022, 14, 1118–1125 CrossRef CAS PubMed.
- P.-S. Wang, M.-L. Shen, T.-C. Wang, H.-C. Lin and L.-Z. Gong, Angew. Chem., Int. Ed., 2017, 56, 16032–16036 CrossRef CAS PubMed.
- Y. Bunno, Y. Tsukimawashi, M. Kojima, T. Yoshino and S. Matsunaga, ACS Catal., 2021, 11, 2663–2668 CrossRef CAS.
- Y. Nishioka, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2013, 52, 1739–1742 CrossRef CAS PubMed.
- P. Xu, J. Xie, D.-S. Wang and X. P. Zhang, Nat. Chem., 2023, 15, 498–507 CrossRef CAS PubMed.
- L.-M. Jin, P. Xu, J. Xie and X. P. Zhang, J. Am. Chem. Soc., 2020, 142, 20828–20836 CrossRef CAS PubMed.
- C. M. B. Farr, A. M. Kazerouni, B. Park, C. D. Poff, J. Won, K. R. Sharp, M.-H. Baik and S. B. Blakey, J. Am. Chem. Soc., 2020, 142, 13996–14004 CrossRef CAS PubMed.
- C. Liang, F. Collet, F. Robert-Peillard, P. Müller, R. H. Dodd and P. Dauban, J. Am. Chem. Soc., 2008, 130, 343–350 CrossRef CAS PubMed.
- H. Ahmed, B. Ghosh, S. Breitenlechner, M. Feßner, C. Merten and T. Bach, Angew. Chem., Int. Ed., 2024, 63, e202407003 CrossRef CAS PubMed.
- J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911–922 CrossRef CAS PubMed.
- H. Lei and T. Rovis, J. Am. Chem. Soc., 2019, 141, 2268–2273 CrossRef CAS PubMed.
- Z.-J. Jia, S. Gao and F. H. Arnold, J. Am. Chem. Soc., 2020, 142, 10279–10283 CrossRef CAS PubMed.
- Z. Zhang, P. Chen and G. Liu, Chem. Soc. Rev., 2022, 51, 1640–1658 RSC.
- D. L. Golden, S.-E. Suh and S. S. Stahl, Nat. Rev. Chem., 2022, 6, 405–427 CrossRef CAS PubMed.
- C. Zhang, Z.-L. Li, Q.-S. Gu and X.-Y. Liu, Nat. Commun., 2021, 12, 475–483 CrossRef CAS PubMed.
- K. P. S. Cheung, J. Fang, K. Mukherjee, A. Mihranyan and V. Gevorgyan, Science, 2022, 378, 1207–1213 CrossRef PubMed.
- L. Dai, Y.-Y. Chen, L.-J. Xiao and Q.-L. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202304427 CrossRef CAS PubMed.
- Y.-W. Zheng, R. Narobe, K. Donabauer, S. Yakubov and B. König, ACS Catal., 2020, 10, 8582–8589 CrossRef CAS.
- D. L. Golden, C. Zhang, S.-J. Chen, A. Vasilopoulos, I. A. Guzei and S. S. Stahl, J. Am. Chem. Soc., 2023, 145, 9434–9440 CrossRef CAS PubMed.
- Y.-F. Zhang, X.-Y. Dong, J.-T. Cheng, N.-Y. Yang, L.-L. Wang, F.-L. Wang, C. Luan, J. Liu, Z.-L. Li, Q.-S. Gu and X.-Y. Liu, J. Am. Chem. Soc., 2021, 143, 15413–15419 CrossRef CAS PubMed.
- T. Tsunoda, O. Sasaki, O. Takeuchi and S. Itô, Tetrahedron, 1991, 47, 3925–3934 CrossRef CAS.
- L. A. Black, W. H. Bunnelle, D. Chen, B. Clapham, D. A. Degoey, X. Deng, L. Fu, L. A. Hazelwood, L. Kong, Q. Lang, C.-H. Lee, M. Li, G. L. Lundgaard, M. V. Patel, R. Tao, L. Zhang, Q. Zang, Q. Zheng and W. Zhu, US Pat., 2017177004A1, 2017 Search PubMed.
- D. Chikkanna, V. V. Khairnar, M. Ramachandra and K. Satyam, IN Pat., 2019142126A1, 2010 Search PubMed.
- J. Hartung, K. Daniel, T. Gottwald, A. Gros and N. Schneiders, Org. Biomol. Chem., 2006, 4, 2313–2322 RSC.
- M. M. Melzer, S. Mossin, X. Dai, A. M. Bartell, P. Kapoor, K. Meyer and T. H. Warren, Angew. Chem., Int. Ed., 2010, 49, 904–907 CrossRef CAS PubMed.
- S. Wiese, Y. M. Badiei, R. T. Gephart, S. Mossin, M. S. Varonka, M. M. Melzer, K. Meyer, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2010, 49, 8850–8855 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2378637. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00872g |
| This journal is © The Royal Society of Chemistry 2025 |