
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStable cationic nanobelts synthesized by chemical oxidation of methylene-bridged [6]cycloparaphenylene†
Nobushige Kai‡
a,
Hideya Kono‡a,
Timo Stünkela,
Daiki Imotoa,
Riccardo Zanasi *b,
Guglielmo Monacob,
Francesco F. Summab,
Lawrence T. Scottc,
Akiko Yagi*ad and
Kenichiro Itami*de
*b,
Guglielmo Monacob,
Francesco F. Summab,
Lawrence T. Scottc,
Akiko Yagi*ad and
Kenichiro Itami*de
aDepartment of Chemistry, Graduate School of Science, Nagoya University, Chikusa, Nagoya 464-8602, Japan. E-mail: yagi.akiko@itbm.nagoya-u.ac.jp
bDipartimento di Chimica e Biologia “A. Zambelli”, Università, Degli Studi di Salerno, Fisciano 84084, SA, Italy. E-mail: rzanasi@unisa.it
cDepartment of Chemistry, University of Nevada, Reno, Nevada 89557-0216, USA
dInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Nagoya 464-8602, Japan
eMolecule Creation Laboratory, RIKEN Cluster for Pioneering Research, Wako, Saitama 351-0198, Japan. E-mail: kenichiro.itami@riken.ac.jp
First published on 23rd April 2025
Abstract
Nanobelts are cyclic arenes that only consist of annulated structures. Recently, various types of nanobelts have been synthesized and their unique properties have been unveiled. However, cationic nanobelts without heteroatoms have been rarely synthesized, and their properties are of significant interest from both fundamental and application perspectives. Herein, we report the synthesis of radical cationic and dicationic hydrocarbon nanobelts by chemical oxidation of methylene-bridged [6]cycloparaphenylene ([6]MCPP). These cationic species are remarkably stable in air, which made it possible to measure and uncover their structural and electronic properties. Notably, the [6]MCPP dicationic salt has sharp absorption and fluorescence bands at longer wavelengths than those of neutral [6]MCPP, close to the near-infrared region. From both experimental and theoretical investigation, the existence of a strong diatropic belt current in [6]MCPP dication was indicated. In addition, a longer lifetime was observed for the hexamethyl[6]MCPP dicationic salt than for the [6]MCPP dicationic salt in solution.
Cationic molecules have long been studied as reaction intermediates in a range of cation-triggered organic transformations and also have been utilized in numerous materials such as dyes, liquid crystals, conducting polymers, and photocatalysts.1 Among them, cationic arenes, in which π-electrons have been removed from aromatic rings, have been recognized as important cationic molecules because they can act as intermediates of charge carriers in p-type organic semiconductors.2 To utilize intrinsically moisture-sensitive cationic arenes as stable species, electron-donating heteroatoms such as nitrogen and oxygen atoms are often introduced into arene scaffolds. In contrast, cationic arenes without heteroatoms, namely cationic aromatic hydrocarbons, are relatively rare.3 While the introduction of multiple bulky substituents such as mesityl groups to gain air-stability has been a common strategy in the field, this sometimes is not enough to stabilize cations of π-conjugated arenes. Delocalization of positive charge is another powerful strategy for stabilizing cationic species of π-extended arenes. In the past decade, the groups of Jasti and Yamago have extensively studied the cationic species of cycloparaphenylenes (CPPs) and other charged CPP derivatives, revealing their unique size-dependent properties.4 Considering the recent progress of the chemistry of π-extended arenes,5 their cationic species are also worth investigating.
Nanobelts and aromatic belts are a class of synthetically challenging and recently emerging cyclic π-extended arenes (Fig. 1a).6,7 Since the first synthesis of (6,6)carbon nanobelts ((6,6)CNB) in 2017,6 different types of nanobelts have been synthesized, and their properties have been investigated. Despite the recent advances in nanobelts, cationic nanobelts are limited to certain dicationic/tetracationic species of nonalternant nanobelts doped with nitrogen atoms and bowl-shaped nanobelts.8 There are no reports on the synthesis of stable cationic species of nanobelts, probably because of their assumed instability; thus, their synthesis is in strong demand.
 | ||
| Fig. 1 (a) The structures of neutral aromatic hydrocarbon nanobelts. Ar = p-tBuC6H4. (b) The structures of [6]MCPP radical cation SbCl6− salt 1 and [6]MCPP dication (SbF6−)2 salt 2. | ||
Herein, we report the synthesis of stable cationic hydrocarbon nanobelts. Methylene-bridged [6]cycloparaphenylene ([6]MCPP) was selected as our target nanobelt for obtaining a cationic nanobelt.9 [6]MCPP is a nanobelt synthesized by our group in 2020, in which all the benzene rings of [6]CPP are bridged by methylene units. The synthesis of [6]MCPP requires only two steps from pillar[6]arene,10 which has enabled the large-scale synthesis and commercialization of [6]MCPP.11 Unique properties of [6]MCPP, such as a small energy gap (2.66 eV) and paratropic belt current, have led the progress of MCPP chemistry, as demonstrated by the syntheses of larger MCPPs ([8]MCPP and [10]MCPP),12 a naphthalene-based MCPP,13 and functionalized MCPPs.14 Owing to the high highest occupied molecular orbital (HOMO) energy (−4.40 eV), electron-donating methylene units, and rigid cyclic π-conjugated system of [6]MCPP, we anticipated that cationic species of [6]MCPP can be easily and stably produced under air. The [6]MCPP radical cation SbCl6− salt 1 and [6]MCPP dication (SbF6−)2 salt 2 were synthesized and found to be very stable as solutions in CH2Cl2 and even as solids in air. Nuclear magnetic resonance (NMR) analysis, ultraviolet (UV)–visible (vis)–near-infrared region (NIR) absorption/fluorescence measurements, X-ray crystal structure analysis, and electron spin resonance (ESR) measurement of 1 and 2 revealed several unique properties of the cationic nanobelts.
Chemical oxidation of [6]MCPP with 3 equivalents of NOSbF6 in CH2Cl2 solution, at 23 °C under an argon atmosphere, changed the red color of the reaction solution, derived from [6]MCPP, to a yellow green color. From 1H NMR, 13C NMR, and electrospray ionization (ESI) mass spectrometric analyses, as well as UV–vis–NIR absorption measurements, the full conversion of [6]MCPP was observed and 2 was obtained in 92% isolated yield (Scheme 1, bottom). When using 1 equivalent of NOSbF6, the formation of 1 was indicated by the ESR and absorption spectra of the reaction mixture. Eventually, under these conditions, 1 was isolated in 73% yield (see the Section 2.1 in ESI† for details). By treating [6]MCPP with Et3OSbCl6, a slightly weaker oxidant than NOSbF6 (Eox; 0.87 V and 0.91 V (vs. Fc/Fc+), respectively), 1 was also selectively formed as judged by ESR analysis and absorption measurements and isolated in 99% yield (Scheme 1, top). In the ESR spectrum of the CH2Cl2 solution of 1 measured under argon, the g value of 1 is 2.0037 (329.059 mT) and the hyperfine structure was observed, which are reasonable based on the results reported for [n]CPP radical cations (see Fig. S14 in ESI†). The half-life time of 1 and 2 are determined as about 143 hours and 26 hours, respectively, from the decay of the maximum absorption peak intensities in CH2Cl2 solution under air (see Fig. S1 and S2 in ESI†). The determined values of half-life time should be longer than those of CPP cations, which have been characterized as air-sensitive; however, no lifetime data have been reported.4 The comproportionation reaction by mixing 2 and neutral [6]MCPP was conducted in CH2Cl2 at room temperature. This oxidant-free method of generating the radical cation was previously reported in the case of CPP by the Yamago group.4d As a result, 1 was obtained quantitatively, also supporting high stability of 1 (see the Section 2.4 in ESI†).
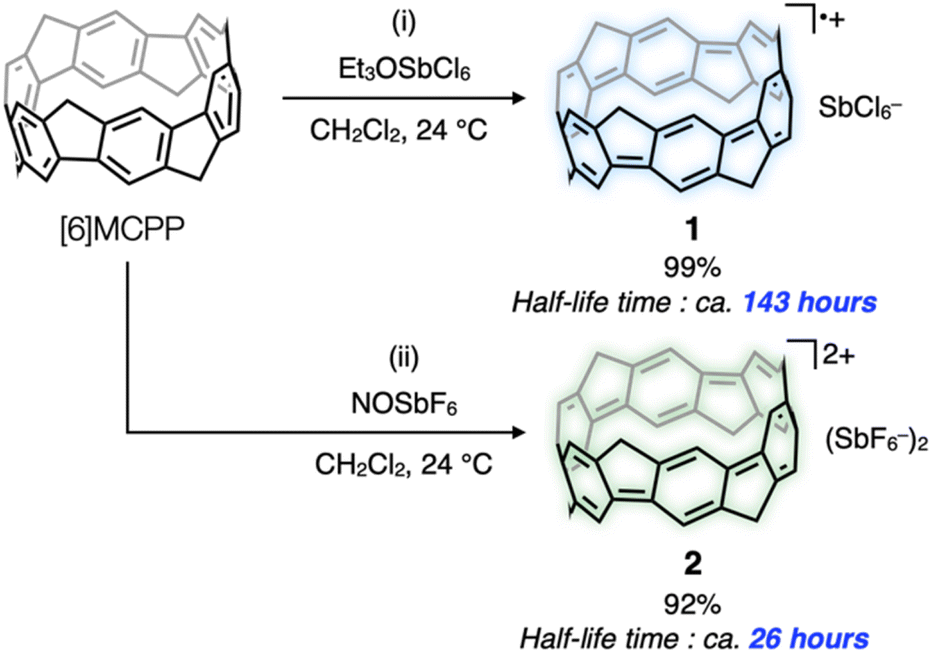 | ||
| Scheme 1 Synthesis of 1 and 2. Reaction conditions: (i) [6]MCPP (9.6 μmol, 5.1 mg, 1.0 equiv.), Et3OSbCl6 (14 μmol, 6.2 mg, 1.5 equiv.), CH2Cl2 (2.0 mL), 30 min. (ii) [6]MCPP (7.6 μmol, 4.0 mg, 1.0 equiv.), NOSbF6 (23 μmol, 6.0 mg, 2.2 equiv.), CH2Cl2 (3.0 mL), 2 hours. Half-life time was determined in CH2Cl2 solution at 23 °C under air. | ||
A single crystal of 2 was successfully obtained by slow evaporation of a CH2Cl2 solution at −30 °C under an argon atmosphere for 6 days. X-ray single-crystal structural analysis revealed the crystal structure of 2 (Fig. 2a) and its packing structure, where the [6]MCPP2+ scaffold is aligned in columns with two sandwiched SbF6− anions and CH2Cl2 molecules (Fig. 2b).15 The diameter of the [6]MCPP2+ scaffold is estimated as 7.675 Å, which is 0.083 Å shorter than that of neutral [6]MCPP (Table 1). Considering the carbon–carbon bonds, the difference between 2 and [6]MCPP strongly indicates that the [6]MCPP2+ scaffold has a quinoidal character. The harmonic oscillator model of aromaticity (HOMA) values for the 6-membered rings are estimated using the crystal structures of [6]MCPP and 2, and density functional theory (DFT) calculations at the B3LYP/6-31G(d) level of theory (see the Section 5 in ESI† for details). The average HOMA value is 0.702 for [6]MCPP2+ scaffold in contrast to 0.955 for [6]MCPP, supporting the decreased benzenoid character of 2. This difference was also observed in the case of cationic CPPs, which explains the effective delocalization of the positive charge across the entire [6]MCPP2+ scaffold.
 | ||
| Fig. 2 X-ray crystal structure of 2. (a) Oak Ridge Thermal Ellipsoid Plot (ORTEP) drawing of 2 with 50% thermal probability. (b) Packing structure viewed along the a-axis. | ||
|
|||
|---|---|---|---|
| [6]MCPP | [6]MCPP2+ | Δ([6]MCPP2+–[6]MCPP) | |
| Diameter [Å] | 7.758 | 7.675 | −0.083 |
| C1–C2 [Å] | 1.518 | 1.514 | −0.004 |
| C2–C3 [Å] | 1.384 | 1.368 | −0.016 |
| C2–C4 [Å] | 1.408 | 1.436 | 0.028 |
| C3–C4 [Å] | 1.395 | 1.415 | 0.020 |
| C4–C4 [Å] | 1.478 | 1.428 | −0.050 |

The UV–vis–NIR absorption and fluorescence spectra of 1 and 2, in CH2Cl2 solutions, are recorded and compared with those of [6]MCPP (Fig. 3a). For 1, four absorption peaks are observed at 415, 890, 1037, and 1054 nm. The absorption band in the NIR region clearly indicates the radical cationic character of 1, which undergoes transitions with a singly occupied molecular orbital (SOMO). Using DFT calculations, the frontier molecular orbitals of 1 and 2 were depicted and the features similar to those seen in most of radical cations and cations were found. The SOMO of 1 is distributed exactly the same way as the HOMO of [6]MCPP, and the other frontier molecular orbitals of 1 are shifted by one (see Fig. 3b and S10 in ESI†). Similarly, the LUMO of 2 has the same orbital distribution as that of the HOMO of [6]MCPP, whereas the other orbitals are shifted by one. As estimated by time-dependent DFT (TDDFT) calculation at the UB3LYP/6-31G(d) level of theory, the absorption bands of 1, with peaks at approximately 415 nm, 890 nm, 1037 nm and 1054 nm can be attributed to HOMO/HOMO−1 to LUMO, and SOMO → LUMO, HOMO/HOMO−1 → SOMO transitions, respectively, though the absorption at 890 nm is very weak, presumably because SOMO → LUMO transition is forbidden (f = 0). Fluorescence of the CH2Cl2 solution of 1 was measured but not observed.
 | ||
| Fig. 3 (a) UV–vis–NIR absorption (solid line) and fluorescence (dashed line) spectra of CH2Cl2 solutions of [6]MCPP, 1, and 2. Fluorescence spectrum of 2 was recorded upon excitation at 450 nm. (b) Frontier molecular orbitals of [6]MCPP and transitions estimated by TDDFT calculation. | ||
The absorption bands of the CH2Cl2 solution of 2 are observed with peaks at 453 and 745 nm. The positions of the peaks correspond well with the HOMO/HOMO−1 → LUMO+1 (415 nm) and HOMO/HOMO−1 → LUMO (701 nm) transitions calculated using TDDFT. Notably, the absorption band with a peak around 745 nm is very sharp with a full width at half maximum (FWHM) of 14.5 nm. The fluorescence spectrum of 2 was observed, whereas neutral [6]MCPP exhibited negligible fluorescence due to forbidden transitions. The FWHM of the fluorescence band of 2 is also small (20 nm), comparable to that of quantum dots or dyes with heteroatoms.16 No vibronic structure was observed in the fluorescence band, probably because the structure of 2 is too rigid to see obvious vibronic structures. The fluorescence quantum yield (ΦF) of 2 is determined to be 0.23. Considering that the Stokes shift is only 5 nm and self-absorption is not negligible, the value of ΦF is significant. Compared to [6]CPP2+ (SbF6−)2 CH2Cl2 solution, 2 has over 10 times larger ΦF (0.23 vs. 0.018).4f The oscillator strengths (f) for fluorescence of 2 and [6]CPP2+ (SbF6−)2 were estimated to be 0.22 and 0.21, respectively, from TDDFT calculation based on the optimized structures of S1 states for [6]MCPP2+ and [6]CPP2+ (see the Section 5 in ESI† for details). Because ΦF is defined as kf/(kf + knr + kISC), where kf is fluorescence rate, knr is nonradiative deactivation rate, kISC is intersystem crossing rate, and kISC is negligible for [6]MCPP2+ and [6]CPP2+, the difference in ΦF may be due to knr. Nonradiative deactivation, particularly thermal deactivation, is expected to be small in the [6]MCPP2+ scaffold compared to [6]CPP2+ by virtue of the rigid structure resulting from the methylene bridges. These results clearly show that methylene-bridging modulates the spectroscopic properties of the parent [6]CPP dication structure, enabling fluorescence, which is a remarkable structural effect of methylene-bridging firstly revealed by this work.
In our previous work, the paratropic belt current of MCPP, which changes depending on the ring size, was first unveiled.12 The chemical shifts of the methylene hydrogen atoms in 1H NMR spectra indicate the existence of a paratropic belt current that decreases with increasing ring size of [n]MCPP (n = 6, 8, and 10), which is also supported by a theoretical study of the magnetically induced current density. To investigate the belt-current effect in the [6]MCPP2+ scaffold, the 1H NMR spectrum of 2 in CD2Cl2 was recorded (Fig. 4). As observed for neutral [6]MCPP, 2 also exhibited three types of signals: a singlet signal at 4.18 ppm, two doublet signals at 3.61 ppm and −0.34 ppm. This suggests a D3d symmetry for 2 in CH2Cl2 solution, indicating a delocalized positive charge on the entire [6]MCPP2+ scaffold. The observed chemical shifts of the signals were further supported by simulations using gauge-including atomic orbitals (GIAO) calculations at the B3LYP/6-311+G(2d,p) level (see Section 5 in ESI† for details). The singlet signal of 4.18 ppm is assigned to the hydrogen atoms on benzene rings (highlighted as green color in Fig. 4). The doublet signal of 3.61 ppm is assigned to the methylene hydrogen atoms facing outside of nanobelt (Hout, highlighted as red color) and the doublet signal of −0.34 ppm is from those facing inside of nanobelt (Hin, highlighted as blue color).
 | ||
| Fig. 4 1H NMR spectra of (a) [6]MCPP and (b) 2 at 600 MHz. TMS: tetramethylsilane. | ||
A paratropic/diatropic belt current refers to a delocalized electronic current induced in cyclic π-conjugated molecules, such as nanobelts, when exposed to a magnetic field aligned with their symmetry axis. This current flows in a direction that generates a paramagnetic/diamagnetic (paratropic/diatropic) response, often associated with antiaromaticity/aromaticity.12,17 As described in our previous reports concerning the neutral MCPP, 1H NMR chemical shifts of hydrogen atoms, especially ones directed to inner side of the belt, provide evidence for the existence of the paratropic belt current in neutral MCPPs, further supported by a theoretical study of the magnetically induced current density. The extraordinary upfield shift of the 1H NMR signals of 2 can only be attributed to a drastic change in the current regime induced by the magnetic field of the spectrometer. As we will show in the following, the paratropic belt current that characterizes the magnetic response of [6]MCPP disappears in the dication, giving way to an intense diatropic belt current that causes a large anisotropic effect on the 1H NMR signals. The deshielding effect is particularly pronounced for Hin, whose δ moves up-field by 4.41 ppm. The aromatic hydrogen atoms undergo a similar shift equal to 3.67 ppm, while Hout moves only 0.62 ppm for their external disposition.
Magnetically-induced electron current density and aryl–aryl bond current strength of 2 have been calculated assuming the combination B97-2/6-311+G(2d,p) of density functional and basis set, using the SYSMOIC program package as in previous studies, adopting the GSGT method for having origin-independent results.17a The wave functions for the calculation have been obtained using the G16 program package,17b including, in this case of a charged molecule, the solvent (CH2Cl2) effect by means of the CPCM method.17c
We find that the dominant contribution to the delocalized electron current density is given by the HOMO. This contribution is shown in Fig. 5, in which intensities lower than 0.04 a.u. were cut to clearly show the shape of the current and how its flow bifurcates and comes together around the six-membered rings of the nanobelts. To facilitate the comparison with [6]MCPP, we placed side by side in the bottom of Fig. 6 the current density calculated for the neutral (left) and charged (right) species adopting the same filter, this time equal to 0.015 a.u., to cut the low magnitude current. The inset on the left is therefore identical to that used for depiction of the paratropic belt current in [6]MCPP.
 | ||
| Fig. 5 HOMO contribution to the electron current density induced in 2 by a magnetic field parallel to the main symmetry axis (blue arrow). The current lower than 0.04 a.u. are not shown. Maximum current is 0.34 a.u. Red square delimits the integration region for the calculation of aryl–aryl bond current strength. | ||
 | ||
| Fig. 6 The summary of belt current in neutral [6]MCPP (left) and [6]MCPP dication 2 (right). The belt current strength is calculated at B97-2/6-311+G(2d,p) level of theory adopting the CSGT method. | ||
In comparison with [6]MCPP studied in ref. 12, first, we observe the opposite direction of circulation of the current, i.e., diatropic instead of paratropic; second, the magnitude of the current is much larger. To obtain a quantitative estimate, bond current strengths17d have been calculated, integrating the current crossing a 5 × 5 a.u. square perpendicular to one aryl–aryl bond in its middle (shown in red in both Fig. 5 and 6) for the three instances: only HOMO, all-but-HOMO, all-MOs. Values are reported in Table 2 as a percentage of the benzene ring current strength (IBEN) calculated using the same method, i.e., I/|IBEN| × 100, with IBEN = 12 nA T−1. Customarily, the sign of the IBEN is taken as negative, so that paratropic/diatropic current strengths are identified by a positive/negative sign.17e
| Molecule | HOMO | All-but-HOMO | All-MOs |
|---|---|---|---|
| 2 | −241 | −1 | −242 |
| [6]MCPP | +138 | −63 | +75 |
As it can be seen, the delocalized diatropic belt current of 2 is almost entirely due to the HOMO. Two contributions were instead found for [6]MCPP, one dominant paratropic and another less intense diatropic. All that can be very easily understood by considering the symmetries of the frontier molecular orbitals shown in Fig. 3.
According to the few electrons model by Steiner and Fowler,17f,g an occupied-to-unoccupied virtual transition gives a paratropic contribution if the product of symmetries contains a match to a rotation, and a diatropic contribution if it contains a match to a translation. For [6]MCPP the HOMO → LUMO transition (a2g × a1g = a2g) gives the already well-known paratropic belt current whose strength is 138% of |IBEN|, while the HOMO−1 → LUMO transition (eu × a1g = eu) makes a contribution that accounts almost entirely for the diatropic contribution of −63% of |IBEN|. The smaller HOMO–LUMO gap is consistent with the bigger paratropic contribution. Now, as already observed above, the HOMO of [6]MCPP becomes the LUMO of 2. Thus, two transitions originate from the HOMO of the dication (eu × a2g = eu and eu × a1g = eu) which give the large diatropic belt current shown in Fig. 5, whose strength is estimated to be −242% of |IBEN|.
The summary of the belt current of neutral [6]MCPP and 2 is shown in Fig. 6. Two-electron oxidation turns a paratropic belt current of MCPP skeleton into a diatropic belt current, which is significantly increased to reflect experimentally observed huge upfield 1H NMR chemical shift of inner methylene hydrogen atoms of 2. Owing to the large strength of the diatropic belt current in the dication, the current predicted based on the “in-plane aromaticity” concept4c appears to be qualitatively correct. The belt current has a sizable effect also on the carbon NMR signals (see Tables S5–S9 in ESI† for a brief account). In addition, nucleus-independent chemical shift (NICS) was also calculated, which also indicates the increased aromaticity of 2 (see Table S4 in ESI†).
The interesting properties of 1 and 2 described above are unveiled owing to their high stability under air as both solution and solid. In order to discuss the extraordinarily high stability of 2, the chemical oxidation of hexamethyl[6]MCPP, which is a known molecule reported in 2021,14a has been tried in a same manner as the synthesis of 2 (Scheme 2). The dicationic species of hexamethyl[6]MCPP 3 has been obtained successfully to investigate its half-life time in CH2Cl2 solution under air. As a result, it was found that 3 has a much longer half-life time than 2, which is estimated to be about 2640 hours (110 days). Although discussion for high stability was difficult because neither 13C NMR analysis nor X-ray diffraction analysis was successful, kinetic protection, electron-donating effect and hyperconjugation effect of methyl groups likely improve the stability of [6]MCPP dication.
 | ||
| Scheme 2 Chemical oxidation of hexamethyl[6]MCPP. Reaction conditions: hexamethyl[6]MCPP (7.6 μmol, 4.6 mg, 1.0 equiv.), NOSbF6 (23 μmol, 6.0 mg, 3.0 equiv.), CH2Cl2 (3.0 mL). Half-life time was elucidated in CH2Cl2 solution at 23 °C under air. | ||
Conclusions
In conclusion, we have synthesized [6]MCPP radical cationic salt 1 and [6]MCPP dicationic salt 2 from [6]MCPP. The appropriate choice of chemical oxidants has led the successful synthesis of the [6]MCPP cationic species. The half-life times of the cationic species are longer than those of CPP cationic species, possibly owing to the methylene-bridged structures which fix the quinoidal CPP structure to a belt form. As important properties of [6]MCPP cations, we found that the crystal structure of 2 has an obvious quinoidal character and a smaller diameter in contrast to neutral [6]MCPP. The UV–vis–NIR absorption and fluorescence spectra of 2 have small FWHM in long wavelength close to NIR, which indicates the possibility of 2 to be used as a unique photo material. The drastic change in the fluorescence quantum yields of 2 (ΦF = 0.23) and neutral [6]MCPP (ΦF ∼0) means that turning the fluorescence on and off is possible by using oxidation of [6]MCPP. Notably, experimental and theoretical 1H NMR studies of 2 uncovered its interesting magnetic property: a diatropic belt current. The belt current of 2 is much stronger than that of [6]MCPP, which is reasonable from allowed transition in frontier molecular orbitals of 2. The diatropic belt current should be present in already reported [n]CPP dications; however, evidence for it is difficult to obtain without the characteristic methylene hydrogen atoms in 2, the 1H NMR chemical shifts of which are strongly affected by the diatropic belt current. In order to create highly stable MCPP dications, hexamethyl[6]MCPP has been oxidized similarly. The thus-obtained dication 3 was extraordinarily stable to strongly indicate the hyperconjugation effect, electron-donating effect and kinetic protection by the methyl groups in 3. The compounds synthesized in this study are remarkably stable radical and cationic nanobelts. Their striking properties would give new insights into the chemistry of π-conjugated molecules and materials science.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
A. Y. and K. I. conceived the concept and directed the project. N. K. and H. K. synthesized compounds 1, 2, 3 and performed all experimental measurements and DFT calculations. T. S. contributed to synthesize compound 3. D. I. performed and analyzed X-ray crystallography. R. Z., G. M. and F. F. S. performed the DFT calculations for NMR studies. R. Z., L. T. S., A. Y., and K. I. wrote the manuscript with feedback from other authors. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Kin-ichi Oyama and Ms. Rio Yamada (Nagoya University) for their support in ESI-MS measurement and analysis. We also thank Ms Yui Ito, Associate Prof. Masahito Murai and Prof. Shigehiro Yamaguchi (Nagoya University) for their support in NIR absorption and FL measurements. The authors also thank Dr Hiroki Shudo (Nagoya University) for fruitful discussion. We also thank the referees of our first manuscript for their valuable suggestions. H. K. and D. I. thank the Nagoya University Graduate Program of Transformative Chem-Bio Research (WISE Program) supported by MEXT. T. S. thanks and the Deutsche Forschungsgemeischaft (DFG, German Research Foundation – GRK2678-437785492). Computations were performed using the resources of the Research Center for Computational Science, Okazaki, Japan (23-IMS-C087 and 24-IMS-C123). This work is supported by JSPS Promotion of Joint International Research (grant number 22K21346 to A. Y.), JSPS KAKENHI (grant number 19H05463 to K. I.) and Sumitomo Foundation (to A. Y.).Notes and references
- (a) K. S. Peters, Chem. Rev., 2007, 107, 859–873 CrossRef CAS PubMed; (b) J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS PubMed; (c) J. Bosson, J. Gouin and J. Lacour, Chem. Soc. Rev., 2014, 43, 2824–2840 RSC; (d) K. Goossens, K. Lava, C. W. Bielawski and K. Binnemans, Chem. Rev., 2016, 116, 4643–4807 CrossRef CAS PubMed; (e) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- (a) T. Nishinaga Organic Redox Systems, John Wiley & Sons, Inc, Hoboken, NJ, 2015 Search PubMed; (b) P. W. Antoni and M. M. Hansmann, J. Am. Chem. Soc., 2018, 140, 14823–14835 CrossRef CAS PubMed; (c) T. Harimoto and Y. Ishigaki, ChemPlusChem, 2022, 87, e202200013 CrossRef CAS PubMed.
- (a) A. Matsuura, T. Nishinaga and K. Komatsu, J. Am. Chem. Soc., 2000, 122, 10007–10016 CrossRef CAS; (b) K. Komatsu, S. Aonuma, Y. Jinbu, R. Tsuji, C. Hirosawa and K. Takeuchi, J. Org. Chem., 1991, 56, 195–203 CrossRef CAS; (c) M. Banerjee, V. S. Vyas, S. V. Lindeman and R. Rathore, Chem. Commun., 2008, 1889–1891 RSC; (d) M. Banerjee, R. Shukla and R. Rathore, J. Am. Chem. Soc., 2009, 131, 1780–1786 CrossRef CAS PubMed; (e) T. Nishiuchi, S. Aibara and T. Kubo, Angew. Chem., Int. Ed., 2018, 57, 16516–16519 CrossRef CAS PubMed; (f) C. Zhu, K. Shoyama and F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 21505–21509 CrossRef CAS PubMed; (g) Y. Ishigaki, T. Harimoto, K. Sugawara and T. Suzuki, J. Am. Chem. Soc., 2021, 143, 3306–3311 CrossRef CAS PubMed; (h) H. Miyoshi, R. Sugiura, R. Kishi, S. N. Spisak, Z. Wei, A. Muranaka, M. Uchiyama, N. Kobayashi, S. Chatterjee, Y. Ie, I. Hisaki, M. A. Petrukhina, T. Nishinaga, M. Nakano and Y. Tobe, Angew. Chem., Int. Ed., 2022, 61, e202115316 CrossRef CAS PubMed; (i) Z. Li, X. Hou, Y. Han, W. Fan, Y. Ni, Q. Zhou, J. Zhu, S. Wu, K. Huang and J. Wu, Angew. Chem., Int. Ed., 2022, 61, e202210697 CrossRef CAS PubMed; (j) T. Yoshihara, H. Shudo, A. Yagi and K. Itami, J. Am. Chem. Soc., 2023, 145, 11754–11763 CrossRef CAS PubMed.
- (a) M. R. Golder, B. M. Wong and R. Jasti, Chem. Sci., 2013, 4, 4285–4291 RSC; (b) E. Kayahara, T. Kouyama, T. Kato, H. Takaya, N. Yasuda and S. Yamago, Angew. Chem., Int. Ed., 2013, 52, 13722–13726 CrossRef CAS PubMed; (c) N. Toriumi, A. Muranaka, E. Kayahara, S. Yamago and M. Uchiyama, J. Am. Chem. Soc., 2015, 137, 82–85 CrossRef CAS PubMed; (d) E. Kayahara, T. Kouyama, T. Kato and S. Yamago, J. Am. Chem. Soc., 2016, 138, 338–344 CrossRef CAS PubMed; (e) M. P. Alvarez, M. C. Ruiz Delgado, M. Taravillo, V. G. Baonza, J. T. López Navarrete, P. Evans, R. Jasti, S. Yamago, M. Kertesz and J. Casado, Chem. Sci., 2016, 7, 3494–3499 RSC; (f) Y. Masumoto, N. Toriumi, A. Muranaka, E. Kayahara, S. Yamago and M. Uchiyama, J. Phys. Chem. A, 2018, 122, 5162–5167 CrossRef CAS PubMed; (g) E. R. Darzi, E. S. Hirst, C. D. Weber, L. N. Zakharov, M. C. Lonergan and R. Jasti, ACS Cent. Sci., 2015, 1, 335–342 CrossRef CAS PubMed; (h) J. M. Van Raden, E. R. Darzi, L. N. Zakharov and R. Jasti, Org. Biomol. Chem., 2016, 14, 5721–5727 RSC; (i) F. Schwer, S. Zank, M. Freiberger, R. Kaur, S. Frühwald, C. C. Robertson, A. Görling, T. Drewello, D. M. Guldi and M. von Delius, Org. Mater., 2022, 4, 7–17 CrossRef CAS.
- (a) Y. Segawa, H. Ito and K. Itami, Nat. Rev. Mater., 2016, 1, 15002 CrossRef CAS; (b) Y. Segawa, D. R. Levine and K. Itami, Acc. Chem. Res., 2019, 52, 2760–2767 CrossRef CAS PubMed; (c) N. Panwar, A. M. Soehartono, K. K. Chan, S. Zeng, G. Xu, J. Qu, P. Coquet, K. Yong and X. Chen, Chem. Rev., 2019, 119, 9559–9656 CrossRef CAS PubMed; (d) A. Tielens, Annu. Rev. Astron. Astrophys., 2008, 46, 289–337 CrossRef CAS; (e) R. G. Harvey Polycyclic Aromatic Hydrocarbons, Wiley-VCH, New York, 1997 Search PubMed; (f) A. Narita, X. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC; (g) K. Yoon and G. Dong, Mater. Chem. Front., 2020, 4, 29–45 RSC.
- G. Povie, Y. Segawa, T. Nishihara, Y. Miyauchi and K. Itami, Science, 2017, 356, 172–175 CrossRef CAS PubMed.
- (a) S. E. Lewis, Chem. Soc. Rev., 2015, 44, 2221–2304 RSC; (b) Y. Segawa, A. Yagi, K. Matsui and K. Itami, Angew. Chem., Int. Ed., 2016, 55, 5136–5158 CrossRef CAS PubMed; (c) Q. Guo, Y. Qiu, M. Wang and J. Fraser Stoddart, Nat. Chem., 2021, 13, 402–419 CrossRef CAS PubMed; (d) D. Imoto, A. Yagi and K. Itami, Precis. Chem., 2023, 1, 516–523 CrossRef CAS.
- (a) X. Lu, T. Y. Gopalakrishna, Y. Han, Y. Ni, Y. Zou and J. Wu, J. Am. Chem. Soc., 2019, 141, 5934–5941 CrossRef CAS PubMed; (b) H. Sato, R. Suizu, T. Kato, A. Yagi, Y. Segawa, K. Awaga and K. Itami, Chem. Sci., 2022, 13, 9947–9951 RSC.
- Y. Li, Y. Segawa, A. Yagi and K. Itami, J. Am. Chem. Soc., 2020, 142, 12850–12856 CrossRef CAS PubMed.
- (a) T. Ogoshi, S. Kanai, S. Fujinami, T. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed; (b) T. Ogoshi, T. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed.
- https://www.tcichemicals.com/assets/brochure-pdfs/Brochure_FF122_E.pdf.
- H. Kono, Y. Li, R. Zanasi, G. Monaco, F. F. Summa, L. T. Scott, A. Yagi and K. Itami, J. Am. Chem. Soc., 2023, 145, 8939–8946 CrossRef CAS PubMed.
- N. Kai, H. Kono, A. Yagi and K. Itami, Synlett, 2023, 34, 1433–1436 CrossRef CAS.
- (a) X. Du, D. Zhang, Y. Guo, J. Li, Y. Han and C. Chen, Angew. Chem., Int. Ed., 2021, 60, 13021–13028 CrossRef CAS PubMed; (b) F. Zhang, X. Du, D. Zhang, Y. Wang, H. Lu and C. Chen, Angew. Chem., Int. Ed., 2021, 60, 15291–15295 CrossRef CAS PubMed.
- CCDC number of 2: 2359239.
- (a) J. M. Pietryga, Y. Park, J. Lim, A. F. Fidler, W. K. Bae, S. Brovelli and V. I. Klimov, Chem. Rev., 2016, 116, 10513–10622 CrossRef CAS PubMed; (b) W. Zhao and E. M. Carreira, Angew. Chem., Int. Ed., 2005, 44, 1677–1679 CrossRef CAS PubMed.
- (a) G. Monaco, F. F. Summa and R. Zanasi, J. Chem. Inf. Model., 2021, 61, 270–283 CrossRef CAS PubMed; (b) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed; (c) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed; (d) J. Jusélius, D. Sundholm and J. Gauss, J. Chem. Phys., 2004, 121, 3952–3963 CrossRef PubMed; (e) G. Monaco and R. Zanasi, J. Phys. Chem. A, 2014, 118, 1673–1683 CrossRef CAS PubMed; (f) E. Steiner and P. W. Fowler, J. Phys. Chem. A, 2001, 105, 9553–9562 CrossRef CAS; (g) E. Steiner and P. W. Fowler, Chem. Commun., 2001, 21, 2220–2221 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2359239. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01305d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |