
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling the mechanisms of ketene generation and transformation in syngas-to-olefin conversion over ZnCrOx|SAPO-34 catalysts†
Zhuo-Yan Yaoa,
Sicong Ma *b and
Zhi-Pan Liu*ab
*b and
Zhi-Pan Liu*ab
aState Key Laboratory of Porous Materials for Separation and Conversion, Collaborative Innovation Center of Chemistry for Energy Material, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Key Laboratory of Computational Physical Science, Department of Chemistry, Fudan University, Shanghai 200433, China. E-mail: scma@mail.sioc.ac.cn; zpliu@fudan.edu.cn
bState Key Laboratory of Metal Organic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
First published on 10th April 2025
Abstract
Ketene was identified as an intermediate in syngas-to-olefin (STO) conversion catalyzed by metal oxide–zeolite composites, which sparked a hot debate regarding its formation mechanism and catalytic roles. Here, we employed large-scale atomic simulations using global neural network potentials to explore the STO reaction pathways and microkinetic simulations to couple the reaction kinetics in ZnCrOx|SAPO-34 composite sites. Our results demonstrate that the majority of ketene (86.1%) originates from the methanol carbonylation-to-ketene route 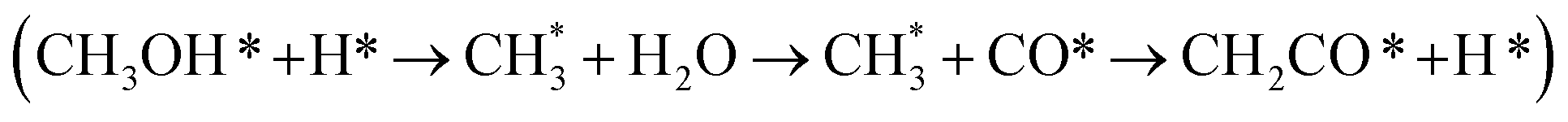 via nearby zeolite acidic sites, where methanol is produced through conventional syngas-to-methanol conversion on the Zn3Cr3O8 (0001) surface, while the minority of ketene (13.9%) arises from a direct CHO*–CO* coupling pathway (CHO* + CO* + H* → CHOCO* + H* → CH2CO + O*) on Zn3Cr3O8. The presence of the ketene pathway significantly alters the catalytic performance in the zeolite, as methanol carbonylation to ketene is kinetically more efficient in competing with conventional methanol-to-olefins (MTO) conversion and thus predominantly drives the product to ethene. Based on our microkinetic simulation, it is the methanol carbonylation activity in the zeolite that dictates the performance of STO catalysts.
via nearby zeolite acidic sites, where methanol is produced through conventional syngas-to-methanol conversion on the Zn3Cr3O8 (0001) surface, while the minority of ketene (13.9%) arises from a direct CHO*–CO* coupling pathway (CHO* + CO* + H* → CHOCO* + H* → CH2CO + O*) on Zn3Cr3O8. The presence of the ketene pathway significantly alters the catalytic performance in the zeolite, as methanol carbonylation to ketene is kinetically more efficient in competing with conventional methanol-to-olefins (MTO) conversion and thus predominantly drives the product to ethene. Based on our microkinetic simulation, it is the methanol carbonylation activity in the zeolite that dictates the performance of STO catalysts.
1. Introduction
Recent years have witnessed significant progress in the one-step syngas-to-olefin (STO) process enabled by OX–ZEO (metal oxide–zeolite) composite catalysts. In this strategy, syngas is first converted into intermediates on metal oxides, which are subsequently transformed into light olefins over zeolites.1–5 In contrast to the combined prior knowledge on syngas-to-methanol (STM) conversion on oxides and methanol-to-olefin (MTO) conversion in zeolites, the STO process exhibits its own unique features, particularly the observation of ketene (H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO) intermediates1 and the distinct product distribution—ethene selectivity reaching up to 80% (ref. 6) compared to ∼50% in typical MTO processes. It was believed that novel reaction pathways must be present in the STO process, which are, however, largely elusive due to the lack of experimental characterization techniques and large-scale theoretical calculation methods to explore all likely pathways. How the ketene intermediate is generated and consumed ranks as the top concern in the catalytic process.
CO) intermediates1 and the distinct product distribution—ethene selectivity reaching up to 80% (ref. 6) compared to ∼50% in typical MTO processes. It was believed that novel reaction pathways must be present in the STO process, which are, however, largely elusive due to the lack of experimental characterization techniques and large-scale theoretical calculation methods to explore all likely pathways. How the ketene intermediate is generated and consumed ranks as the top concern in the catalytic process.
Ketene was first observed as an intermediate on the oxide part (ZnCrOx) of oxide–zeolite composites by Bao and coworkers,1 the pioneering group of OX–ZEO catalysts, using synchrotron-based vacuum ultraviolet photoionization mass spectrometry with the unique ionization signal of ketene at m/z = 42. Consequently, they proposed that ketene forms on the ZnCrOx surface and subsequently migrates into the zeolite for further transformation. Later, ketene was detected within the zeolite framework. Rasmussen et al.7 observed ketene formation in MOR zeolite when deuterated water (D2O) was introduced into a co-feeding system of dimethyl ether (DME) and CO, leading to the formation of CH2DCOOD. Furthermore, infrared spectroscopy and 13C solid-state nuclear magnetic resonance spectroscopy have also provided evidence for ketene adsorption within the zeolites through distinct CCO vibrational shifts and identified surface acetyl as the ketene hydrogenated derivative.8–13
On the other hand, other non-ketene intermediates, mainly methanol and DME, traditionally regarded as intermediates in STM and MTO catalysis, were also detected in the OX–ZEO catalytic system. At low temperatures (<600 K), the ZnO–ZrO2|SSZ-13 composite catalyst predominantly generates methanol and DME, with formate species (HCOO*) detected by in situ IR.14 Elevated reaction temperatures promote the conversion of these intermediates into C2–4 olefins, accompanied by a significant decline in methanol/DME selectivity. Chemical trapping-mass spectrometry further confirmed that the methoxy (CH3O*) species on the ZnO–ZrO2|Zn-modified SAPO-34 catalyst migrates to acidic sites in the zeolite, participating in carbon-chain growth. This observation validates methanol as a critical carbon source kinetically.15
To elucidate the puzzling observations from experiments, theoretical calculations have been utilized to resolve the reaction network of the OX–ZEO catalytic system. Our previous work16 investigated the most stable surfaces with varying Zn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cr ratios of the ZnCrOx system using stochastic surface walking (SSW) and global neural network (G-NN). We identified the metastable Zn3Cr3O8 as the dominant active phase for methanol formation, while ZnCr2O4 was responsible for methane (CH4) formation. However, ketene formation was not considered due to its relatively high Gibbs energy barriers on oxide catalysts alone. Recently, Lai et al.17 explored the possible elementary steps for methanol, methane and ketene formation on the highly reduced ZnCr2O4 (110) surface through DFT calculations and microkinetic simulations. They confirmed that CH4 is the primary product, while minor amounts of ketene and methanol are also produced. When incorporating a hypothetical zeolite conversion reaction with a Gibbs free energy barrier of ≤1.65 eV, ketene selectivity increases significantly, favoring light olefin formation over CH4. Nevertheless, this simplification of the complex reaction network occurring in the zeolite as a hypothetical reaction represents a huge approximation, limiting deep understanding of the quantitative contributions of methanol and ketene routes and also contradicting the experimental facts on negligible CH4 selectivity. In fact, according to a series of studies by Wang et al.18 on MTO within the SAPO-34 zeolite, ketene formation is also kinetically feasible via the methanol carbonylation pathway
:Cr ratios of the ZnCrOx system using stochastic surface walking (SSW) and global neural network (G-NN). We identified the metastable Zn3Cr3O8 as the dominant active phase for methanol formation, while ZnCr2O4 was responsible for methane (CH4) formation. However, ketene formation was not considered due to its relatively high Gibbs energy barriers on oxide catalysts alone. Recently, Lai et al.17 explored the possible elementary steps for methanol, methane and ketene formation on the highly reduced ZnCr2O4 (110) surface through DFT calculations and microkinetic simulations. They confirmed that CH4 is the primary product, while minor amounts of ketene and methanol are also produced. When incorporating a hypothetical zeolite conversion reaction with a Gibbs free energy barrier of ≤1.65 eV, ketene selectivity increases significantly, favoring light olefin formation over CH4. Nevertheless, this simplification of the complex reaction network occurring in the zeolite as a hypothetical reaction represents a huge approximation, limiting deep understanding of the quantitative contributions of methanol and ketene routes and also contradicting the experimental facts on negligible CH4 selectivity. In fact, according to a series of studies by Wang et al.18 on MTO within the SAPO-34 zeolite, ketene formation is also kinetically feasible via the methanol carbonylation pathway 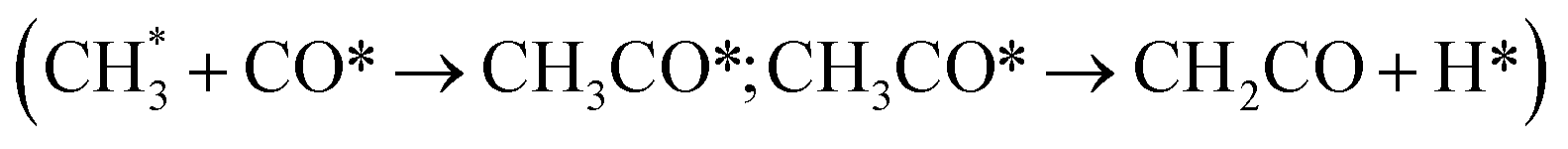 , which suggests that the sole origin of ketene from the oxide part is questionable in the OX–ZEO system. The great complexity of the reaction network highlights the necessity for a systematic investigation to quantitatively determine the contributions of methanol and ketene pathways from both ZnCrOx oxide and SAPO-34 zeolite.
, which suggests that the sole origin of ketene from the oxide part is questionable in the OX–ZEO system. The great complexity of the reaction network highlights the necessity for a systematic investigation to quantitatively determine the contributions of methanol and ketene pathways from both ZnCrOx oxide and SAPO-34 zeolite.
Here, with the advancement of large-scale atomic simulations based on global neural network potential, we systematically investigated the STO reaction pathways on ZnCrOx|SAPO-34 composite catalysts, including the formation of ketene on ZnCrOx via CO hydrogenation and on SAPO-34 via methanol carbonylation, as well as the subsequent conversion of ketene to ethene. By integrating with microkinetic simulations, we demonstrated that ketene formation follows a dual-site mechanism, wherein the majority of ketene (86.1%) is generated via the methanol carbonylation-to-ketene route within the zeolite, while a minor fraction (13.9%) arises from a direct CHO*–CO* coupling pathway on the ZnCrOx surfaces. The catalytic consequence of this dual-site mechanism is thus revealed, rationalizing the experimental product distribution.
2. Methods
2.1 SSW-NN simulation
Our approach for resolving reactions on ZnCrOx and SAPO-34 zeolite is based on the SSW global optimization with G-NN, known as the SSW-NN method, as implemented in the LASP code.19 The G-NN potential is constructed through iterative self-learning from the plane wave DFT global PES dataset during SSW-NN exploration. The SSW-NN simulation for PES exploration consists of three main steps: generating the global PES dataset using DFT calculations on selected structures obtained from SSW global PES exploration, fitting the G-NN potential, and conducting SSW global PES exploration with the G-NN potential. These steps are iteratively repeated until the G-NN potential becomes transferable and sufficiently robust to accurately describe the global PES. The procedure is summarized as follows.The global dataset is initially constructed through iterative self-learning of the global PES. The initial data of the global dataset come from the DFT-based SSW simulation with subsequent data progressively accumulated through G-NN-based SSW PES exploration. In order to cover all likely compositions of Si–Al–P–C–H–O systems, SSW simulations have been conducted across diverse structural configurations (including bulk, layer and cluster), compositions and atom numbers per unit cell. Overall, these SSW simulations generate more than 107 structures on the PES. The final global dataset, computed from high accuracy DFT calculations, contains 47920 structures.
Subsequently, the G-NN potential is constructed using the method as introduced in our previous work.16,20 To pursue a high accuracy for the PES, we implemented a many-body-function corrected global NN architecture (G-MBNN) and employed 636 power-type structure descriptors for each element to distinguish structures in the global dataset. The neural network architecture consists of three hidden layers, structured as a 636-80-80-80-6 network, containing approximately 386836 network parameters in total. The final layer with 6 nodes is enveloped into a series of many-body functions, which sum to yield the total energy. More details about the G-MBNN architecture can be found in our previous work.21
Min–max scaling is utilized to normalize the training data sets. Hyperbolic tangent activation functions are used for the hidden layers, while a linear transformation is applied to the output layer of all networks. The limited-memory Broyden–Fletcher–Goldfarb–Shanno (L-BFGS) method is used to minimize the loss function to match DFT energy, force, and stress. The final energy and force criteria of the root mean square errors for the Si–Al–P–C–H–O G-NN potential are around 4.1 meV per atom and 0.123 eV Å−1 respectively. All the low-energy structure candidates from G-NN potential calculations are finally verified by plane wave DFT calculations, and thus the energetic data reported in this work, unless specifically mentioned, are obtained from DFT.
2.2 DFT calculations
All DFT calculations were performed using the plane wave VASP code,22 where the electron–ion interaction was represented by the projector augmented wave pseudopotential.23,24 The exchange–correlation functional utilized on the ZnCrOx surface was the spin-polarized GGA–PBE25 with the local Hubbard term U correction (U = 3.3 eV) for Cr element26 given that ZnCrOx structures generally exhibit a strong correction for Cr atoms, while the Bayesian error estimation functional (BEEF) was utilized for the zeolite surface.27 The kinetic energy cutoff was set to 450 eV. The first Brillouin zone k-point sampling used a 3 × 3 × 1 gamma-centered mesh grid for the ZnCrOx surface and a 1 × 1 × 1 gamma centered mesh grid for the SAPO-34 zeolite bulk and surface. The energy and force convergence criteria for electron density and structure optimization were set to 1 × 10−6 eV and 0.02 eV Å−1, respectively. All transition states (TSs) in the Gibbs profile were located using the double-ended surface walking (DESW) method,28 and the stationary structures were optimized through the SSW method.29 All those local minima and transition states obtained from SSW-NN sampling were fully re-optimized using DFT to obtain the final results.Harmonic frequency calculations were conducted using partial Hessian vibrational analysis (PHVA), incorporating all C and H species on both ZnCrOx and SAPO-34 surfaces, as well as the H and O atoms of the acidic site on the zeolite inner surface. Zero-point energies (ZPEs), enthalpies, entropies, and Gibbs free energies were subsequently derived from harmonic frequencies. The DFT calculations, unless otherwise specified, were performed under typical reaction conditions, with the temperature and CO/H2 pressure fixed at 673 K and 2.5 MPa, respectively, and a H2:CO ratio of 2.5:1.1
2.3 Microkinetic simulation
The microkinetic analysis was performed using the CATKINAS package developed by Chen et al.30 The reaction conditions were maintained as reported by Jiao et al.1 in 2016, namely P(CO) = 7.14 bar and P(H2) = 17.86 bar, the total flow rate = 5143 mL g−1 h−1, and the temperature = 673 K.3. Results and discussion
It is known that the Zn:Cr ratio in the oxide component of the ZnCrOx|SAPO-34 system significantly affects the catalytic activity and selectivity. Specifically, the ZnCrOx|SAPO-34 catalyst with a Zn:Cr ratio of 1:1 achieves the highest 85% C2–C4 light olefin selectivity with a CO conversion rate of 58% at 673 K, while a Zn:Cr ratio of 1:2 leads to a sharp decline in CO conversion to 27% and light olefin selectivity to 70%.31 In this context, we first systematically investigated the impact of different Zn:Cr ratios on the ketene-mediated pathways. Apart from the stoichiometric ZnCr2O4 spinel phase with a Zn:Cr ratio of 1:2, the most stable phase32 formed after high-temperature calcination, our previous work16 revealed that ZnCrOx with a Zn:Cr ratio of 1:1 has a metastable Zn3Cr3O8 phase, characterized by its unique [ZnO6] structural motif, as confirmed by experimental observations.33–39 Under reaction conditions, the most stable ZnCr2O4 (111) and Zn3Cr3O8 (0001) surfaces become reduced, which opens the STM pathways.16 In this work, we explored the reaction profiles of ketene formation on both reduced Zn3Cr3O8 (0001) and ZnCr2O4 (111) surfaces, where the surface status is based on the phase diagram determined previously. In short, as illustrated in Fig. S1,† the reduced Zn3Cr3O8 (0001) surface exposes a two-coordinated Zn2c atomic site and two planar [CrO4]pla sites under a typical reductive atmosphere, while the reduced ZnCr2O4 (111) surface exposes two Zn2c atomic sites and two five-coordinated pyramidal [CrO5]pyr sites near the OV.16
3.1 Ketene formation on ZnCrOx
By exploring various reaction pathways, we determined that ketene formation mainly follows the CO → CHO → CHOCO → CH2CO mechanism on the reduced Zn3Cr3O8 (0001) and ZnCr2O4 (111) surfaces, with the CHO species serving as the key intermediate, as illustrated in Fig. 1a. Alternative reaction pathways, local minima, and transition state structures of ketene formation are presented in Fig. S2–S4.† For comparison, two dominant pathways reported in our previous work,16 namely methanol and methane synthesis on these two surfaces from syngas, are also shown in Fig. S2 and S3.†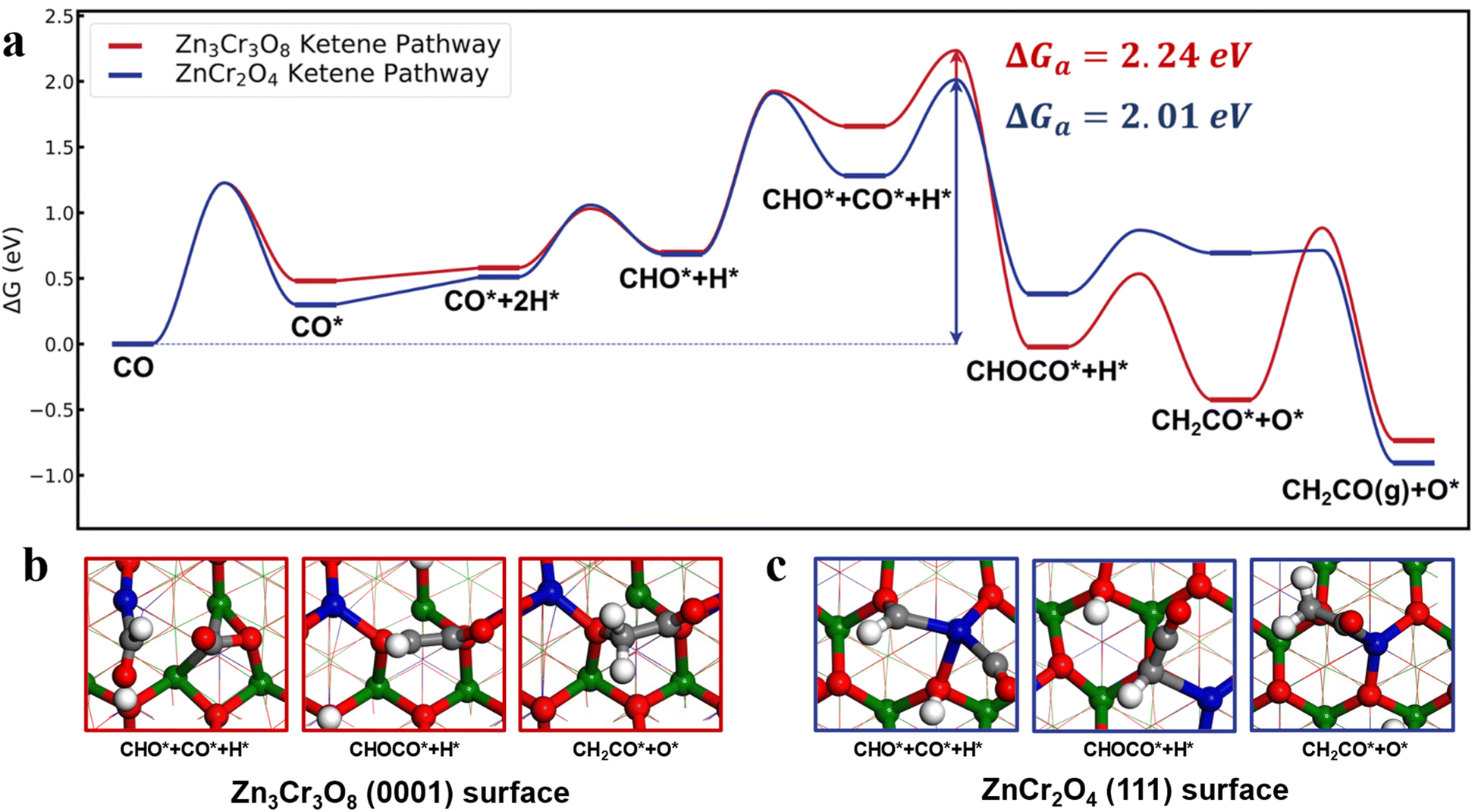 | ||
| Fig. 1 Syngas conversion mechanisms on the Zn3Cr3O8 (0001) and ZnCr2O4 (111) surfaces at 673 K and 2.5 MPa (H2/CO = 2.5). (a) Gibbs free energy profiles for the formation pathways of ketene on the two ZnCrOx surface. The red and blue lines denote ketene formation routes on the Zn3Cr3O8 (0001) and ZnCr2O4 (111) surfaces, respectively (* indicates surface-adsorbed species). (b) Surface structures of key intermediates on the Zn3Cr3O8 (0001) surface. (c) Surface structures of key intermediates on the ZnCr2O4 (111) surface. The blue, green, red, gray and white spheres represent Zn, Cr, O, C, and H atoms, respectively. | ||
Compared to the reaction on the Zn3Cr3O8 surface, the syngas conversion to ketene on ZnCr2O4 exhibits a lower total Gibbs free energy barrier. As shown in Fig. 1a (blue line), the major differences between the two catalysts lie in the adsorption energy of CO in the presence of CHO* species. The ZnCr2O4 surface can adsorb CO with an adsorption energy of 0.60 eV, which is 0.36 eV stronger than that on the Zn3Cr3O8 surface. Subsequently, the CHO*–CO* coupling free energy barrier measures 0.73 eV, resulting in a total Gibbs free energy barrier of 2.01 eV, 0.23 eV lower than that observed on the Zn3Cr3O8 surface. Further conversion of CHOCO* to ketene requires only a minimal Gibbs free energy barrier of 0.49 eV.
3.2 Ketene formation on SAPO-34
Next, we explore the ketene formation pathway on the zeolite. For SAPO-34 zeolite, we selected a medium-strength acid site with an NH3 temperature-programmed desorption peak at 737 K.40 The introduced Si atom into SAPO-34 is located at the corner of the 6- and 8-membered-ring (MR) structure, slightly distorted by the additional H atom (see ESI Fig. S5†). We found that in SAPO-34 zeolite, ketene formation proceeds through the methanol to pathway, followed by methoxy carbonylation, as illustrated in Fig. 2a. The surface
pathway, followed by methoxy carbonylation, as illustrated in Fig. 2a. The surface  species is identified to play a central role in both ketene formation and conversion. All the local minima and transition states involving
species is identified to play a central role in both ketene formation and conversion. All the local minima and transition states involving  are shown in Fig. S6.† The detailed reaction pathways are presented as follows.
are shown in Fig. S6.† The detailed reaction pathways are presented as follows.
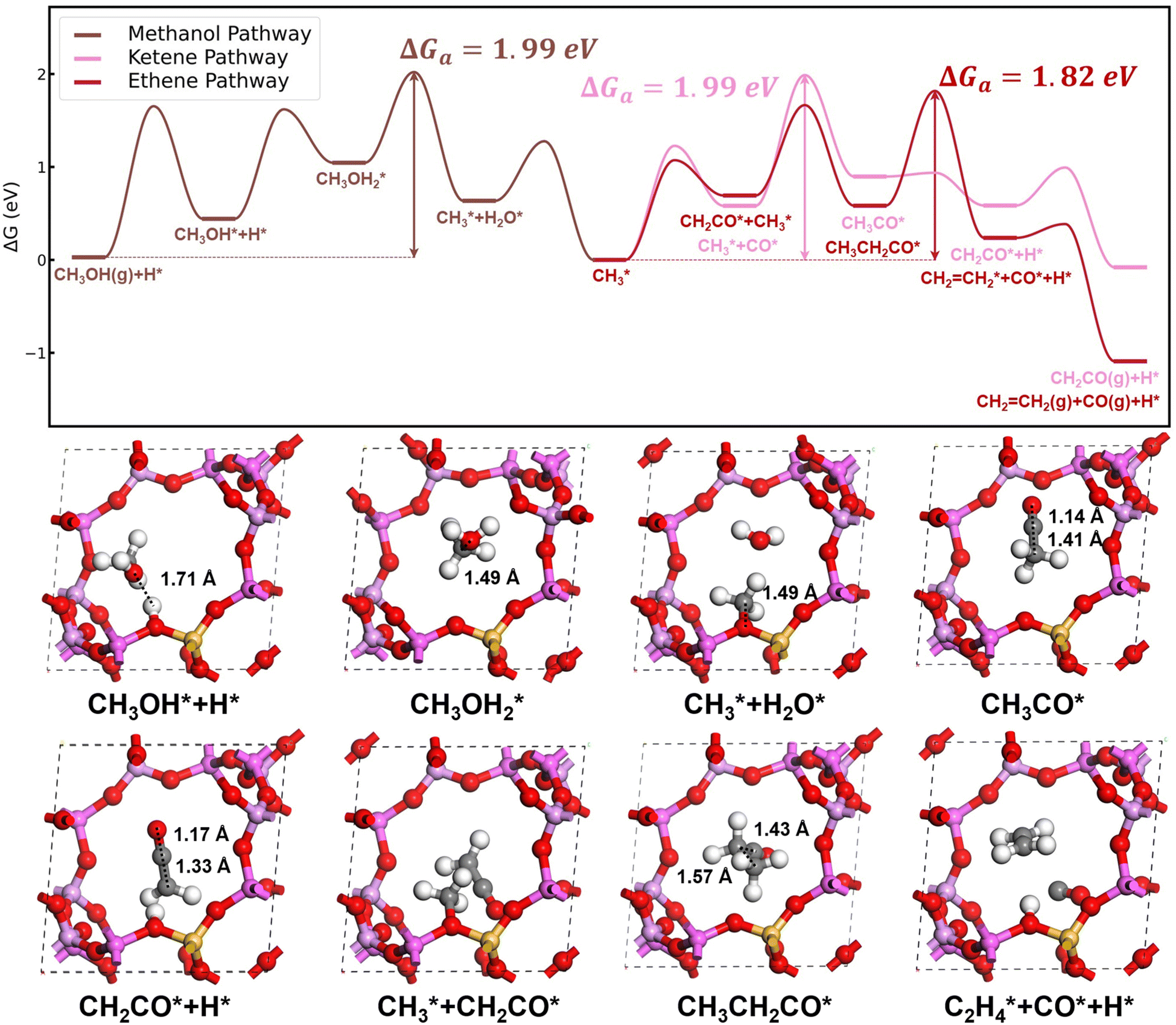 | ||
Fig. 2 Gibbs free energy profiles for C–C coupling pathways on H-SAPO-34 catalysts and surface structures of key intermediates in the C–C coupling pathways on the H-SAPO-34 inner surface. Methanol-to-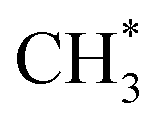 and and  conversion-to-ketene/ethene are plotted in brown, pink and red lines, respectively. Structures of surface-adsorbed and O-adsorbed species are denoted by *. Al, Si, P, O, C and H are represented by purple, yellow, pink, red, gray and white spheres, respectively. conversion-to-ketene/ethene are plotted in brown, pink and red lines, respectively. Structures of surface-adsorbed and O-adsorbed species are denoted by *. Al, Si, P, O, C and H are represented by purple, yellow, pink, red, gray and white spheres, respectively. | ||
 . In the methanol-to-
. In the methanol-to-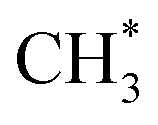 pathway, CH3OH diffuses into the 8-MR cages of H-SAPO-34 and anchors near acidic sites through hydrogen bonding with a length of 1.71 Å, as illustrated in Fig. 2. The adsorbed methanol (CH3OH*) is protonated by surface H* to form hydrogenated methanol
pathway, CH3OH diffuses into the 8-MR cages of H-SAPO-34 and anchors near acidic sites through hydrogen bonding with a length of 1.71 Å, as illustrated in Fig. 2. The adsorbed methanol (CH3OH*) is protonated by surface H* to form hydrogenated methanol  , whose C–O bond length measures 1.49 Å, as shown in Fig. 2. This step exhibits a Gibbs free energy barrier of 1.60 eV and a reaction free energy of 1.05 eV. Subsequently,
, whose C–O bond length measures 1.49 Å, as shown in Fig. 2. This step exhibits a Gibbs free energy barrier of 1.60 eV and a reaction free energy of 1.05 eV. Subsequently,  undergoes C–O dissociation to generate the key intermediate
undergoes C–O dissociation to generate the key intermediate  and water, with a Gibbs free energy barrier of 0.98 eV and an exothermic energy change of 1.05 eV. The length of the newly formed C–O bond in the key intermediate
and water, with a Gibbs free energy barrier of 0.98 eV and an exothermic energy change of 1.05 eV. The length of the newly formed C–O bond in the key intermediate 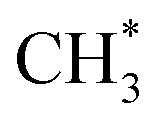 is 1.49 Å, similar to that of the
is 1.49 Å, similar to that of the 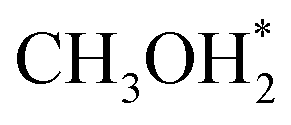 species (see Fig. 2). The total Gibbs free energy barrier for
species (see Fig. 2). The total Gibbs free energy barrier for  generation is 1.99 eV, consistent with literature reports.41–43
generation is 1.99 eV, consistent with literature reports.41–43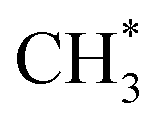 to CH2CO. The further transformation of
to CH2CO. The further transformation of  to ketene also suffers from a 3.3 energy barrier, as illustrated in Fig. 2 (pink line). The CO adsorbs near the
to ketene also suffers from a 3.3 energy barrier, as illustrated in Fig. 2 (pink line). The CO adsorbs near the  group with an energy need of 0.58 eV. Subsequently,
group with an energy need of 0.58 eV. Subsequently, 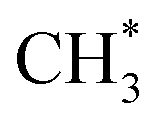 combines with CO* to form an adsorbed acetyl species (CH3CO*) with a C–C bond length of 1.41 Å and a C–O bond length of 1.14 Å (see Fig. 2), overcoming a Gibbs free energy barrier of 1.41 eV and requiring an energy input of 0.32 eV. This step is followed by dehydrogenation, which requires overcoming a Gibbs free energy barrier of 0.04 eV, resulting in the formation of CH2CO. The C–C bond shortens to 1.33 Å and the C–O bond elongates to 1.17 Å compared to CH3CO*, as shown in Fig. 2. The total Gibbs free energy barrier for this pathway is 1.99 eV. It is noteworthy that the reversion of this pathway, i.e. ketene transformation to
combines with CO* to form an adsorbed acetyl species (CH3CO*) with a C–C bond length of 1.41 Å and a C–O bond length of 1.14 Å (see Fig. 2), overcoming a Gibbs free energy barrier of 1.41 eV and requiring an energy input of 0.32 eV. This step is followed by dehydrogenation, which requires overcoming a Gibbs free energy barrier of 0.04 eV, resulting in the formation of CH2CO. The C–C bond shortens to 1.33 Å and the C–O bond elongates to 1.17 Å compared to CH3CO*, as shown in Fig. 2. The total Gibbs free energy barrier for this pathway is 1.99 eV. It is noteworthy that the reversion of this pathway, i.e. ketene transformation to 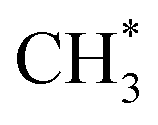 , has a total Gibbs free energy barrier of 2.07 eV, being higher than the other pathways reported in Fig. 2. This suggests that ketene once produced on ZnCrOx surfaces is very likely to diffuse to zeolite.
, has a total Gibbs free energy barrier of 2.07 eV, being higher than the other pathways reported in Fig. 2. This suggests that ketene once produced on ZnCrOx surfaces is very likely to diffuse to zeolite.3.3 Ketene conversion on SAPO-34
Once ketene is formed, its further conversion with surface to ethene is more facile, as illustrated by the red line in Fig. 2. Initially, ketene adsorbs near the surface
to ethene is more facile, as illustrated by the red line in Fig. 2. Initially, ketene adsorbs near the surface 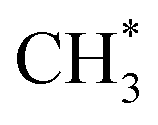 species with an energy requirement of 0.69 eV, as shown in Fig. 2. Subsequently, the adsorbed CH2CO species undergoes C–C coupling with
species with an energy requirement of 0.69 eV, as shown in Fig. 2. Subsequently, the adsorbed CH2CO species undergoes C–C coupling with 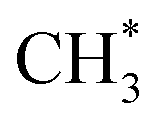 to form the propionyl species (CH3CH2CO*), with the newly formed C–C bond measuring 1.57 Å while the length of the first C–C bond elongates to 1.43 Å, as illustrated in Fig. 2. This step exhibits a moderate Gibbs free energy barrier of 0.97 eV and is exothermic by 0.11 eV. CH3CH2CO* then cleaves the C–H bond in the methyl group to regenerate the zeolite acid site and form the CH2CH2CO* species, which spontaneously decomposes through C–C bond cleavage to generate CO and ethylene molecules. This step presents a Gibbs free energy barrier of 1.23 eV and releases substantial energy (1.67 eV). The overall Gibbs free energy barrier for ketene conversion to ethene is 1.82 eV.
to form the propionyl species (CH3CH2CO*), with the newly formed C–C bond measuring 1.57 Å while the length of the first C–C bond elongates to 1.43 Å, as illustrated in Fig. 2. This step exhibits a moderate Gibbs free energy barrier of 0.97 eV and is exothermic by 0.11 eV. CH3CH2CO* then cleaves the C–H bond in the methyl group to regenerate the zeolite acid site and form the CH2CH2CO* species, which spontaneously decomposes through C–C bond cleavage to generate CO and ethylene molecules. This step presents a Gibbs free energy barrier of 1.23 eV and releases substantial energy (1.67 eV). The overall Gibbs free energy barrier for ketene conversion to ethene is 1.82 eV.
3.4 Byproduct pathways: methanol, methane, CO2, DME, and ester formation and methanol and ketene diffusion
Apart from ketene pathways, we also considered the formation of other byproducts, including methanol, methane, CO2, DME and two esters (methyl acetate and formate acetate) in order to establish a complete reaction network for microkinetic simulations. The exploration of the associate reaction pathways was conducted when it was necessary.For the formation of methanol and methane, our previous work has demonstrated that the Zn3Cr3O8 (0001) surface serves as the dominant active surface for methanol formation with a Gibbs free energy barrier of 1.69 eV, while the ZnCr2O4 (111) surface is responsible for methane formation with a Gibbs free energy barrier of 1.75 eV.
As for CO2 formation, during the deoxygenation process, the presence of CO leads to the formation of water, which is subsequently catalyzed to CO2 by ZnCrOx. We therefore further examined the water–gas shift (WGS) process on the ZnCrOx surface. As illustrated in Fig. S7,† this process follows the H2O → OH* → COOH* → CO2 pathway with a total Gibbs free energy barrier of <1.30 eV. This indicates the ease of the WGS process, where most H2O is expected to be transformed into CO2.
For DME formation, it involves the reaction of 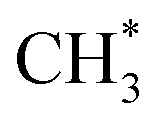 with adsorbed CH3OH*, forming protonated dimethyl ether
with adsorbed CH3OH*, forming protonated dimethyl ether 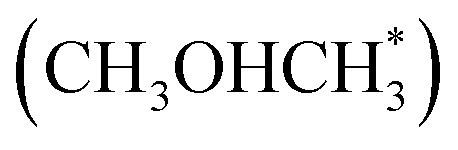 , which then deprotonates to produce DME (Fig. S8†). This pathway is dynamically favorable, with an overall Gibbs free energy barrier of 1.49 eV.
, which then deprotonates to produce DME (Fig. S8†). This pathway is dynamically favorable, with an overall Gibbs free energy barrier of 1.49 eV.
For the formation of the two esters, the processes proceed through C–O bond addition of CHO* and CH3CO* with CH3OH, followed by deprotonation, with a total Gibbs free energy barrier of approximately 1.5 eV and a reaction energy of −0.5 eV. Detailed reaction profiles and energy diagrams for these pathways are presented in Fig. S8.†
To complete the kinetics cycle, we also examined the diffusion kinetics of molecules, CH3OH and CH2CO, in the AlPO-34 zeolite by utilizing enhanced molecular dynamics via umbrella sampling.44–46 The diffusion barriers for both methanol and ketene are found to be below 0.6 eV (see Fig. S9†), indicating that the diffusion of molecules is not rate-determining and will not change the STO reaction mechanism.
3.5 Microkinetic simulation of ZnCrOx|SAPO-34 catalysts
Our microkinetic simulations were conducted based on the DFT reaction network data using CATKINAS software. Three scenarios were considered: ZnCrOx alone, SAPO-34 zeolite alone, and the coupled ZnCrOx and SAPO-34 zeolite composite, each involving 23, 19 and 42 elementary steps in building the microkinetic equations, respectively. In the coupled system, diffusion kinetics were neglected, with each gas molecule assumed to diffuse randomly.First, we assessed the product distribution of ZnCrOx in the absence of zeolite and the result on the product distribution is shown in Fig. 3 (Zn3Cr3O8 column). It shows that methanol is predominately formed on the Zn3Cr3O8 (0001) surface, consistent with previous reports that the main product of ZnCrOx with Zn:Cr = 1:1 is methanol.47 Methane and ketene production remains negligible, with partial pressures at least four orders of magnitude lower than that of methanol (see Fig. S10†). The CO conversion rate is 1.64 s−1, comparable to experimental results.37,47 For the ZnCr2O4 surface, the product distribution shows a notable difference, with 50% methane and 50% CO2 as the primary products. The methane synthesis pathway is both dynamically and thermodynamically more favorable compared to methanol and ketene formation (see Fig. S11†). The CO conversion rate is 0.28 s−1, accounting for one-sixth of that on the Zn3Cr3O8 surface, consistent with the lower reaction activity of the ZnCrOx catalyst with Zn:Cr = 1:2.2,47 In summary, in the absence of the zeolite, ketene formation is unattainable, aligning with experimental knowledge that only methane and methanol can be produced on the ZnCrOx catalysts.
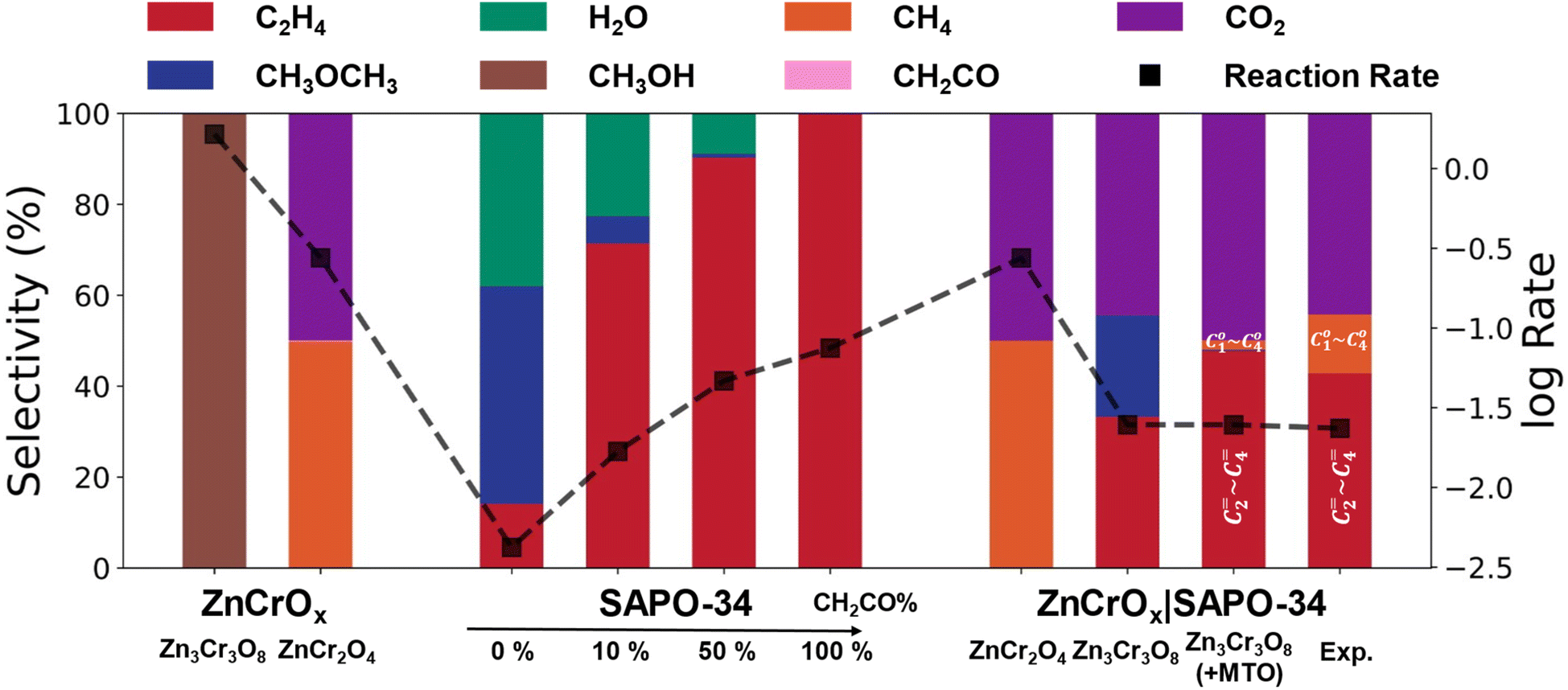 | ||
| Fig. 3 Conversion and selectivity results from microkinetic simulations at the steady state on ZnCrOx, SAPO-34 and ZnCrOx|SAPO-34 surfaces under different conditions. For ZnCrOx and ZnCrOx|SAPO-34, the temperature and pressure were maintained at 673 K and 2.5 MPa, with H2/CO = 2.5. For SAPO-34, an additional total pressure of methanol and ketene (1.0 MPa) was included under these conditions. Two columns in the ZnCrOx part show the results from Zn3Cr3O8 (0001) and ZnCr2O4 (111) surfaces. In the SAPO-34 part, four columns represent varying ratios of CH3OH (M) to CH2CO (K) in the feed gas, adjusted across four levels: all methanol, 9:1, 1:1 and all ketene (denoted by the percentage of CH2CO). The first two columns in the ZnCrOx|SAPO-34 part show results from SAPO-34 composited with the Zn3Cr3O8 (0001) and ZnCr2O4 (111) surfaces, respectively; the third column demonstrates the assistance of typical MTO results with Zn3Cr3O8|SAPO-34; the last column presents experimental results from ref. 1. The red and orange colors in the last two columns represent C2–4 light olefins and C1–4 light alkanes, respectively. Conversion is expressed using the common logarithm form of the total CO consumption rate, denoted by squares. | ||
Next, we investigated the product distribution of SAPO-34 in the absence of ZnCrOx. Considering that both ketene and methanol are proposed as the transfer intermediates in the ZnCrOx|SAPO-34 system, we directly introduced the ketene and methanol into the feed gas with varying methanol:ketene (M:K) ratios, as shown in all SAPO-34 columns of Fig. 3. All the experiments were conducted at a temperature of 673 K, with a syngas pressure of 25.0 bar (H2/CO = 2.5), and methanol and ketene pressures of 10.0 bar at varying M:K ratios. When only pure methanol was added to the feed gas, the reaction rate was 0.004 s−1, with a product distribution of 34% ethene, 21% CH3OCH3 and 45% H2O release. Upon the introduction of ketene into the feed gas, ethene selectivity significantly improves to >78% with <3.0% CH3OCH3 production when the M:K ratio is <9:1. The conversion rate also increases to >0.01 s−1. This indicates that the presence of ketene can indeed enhance the ethene selectivity and reaction rate.
Finally, we take both ZnCrOx and SAPO-34 into consideration with the possible products reaching eight species, including CO2, CH3OH, CH2CO, CH4, CH2CH2, CH3COOCH3, HCOOCH3 and CH3OCH3. As illustrated in Fig. 3 and S12,† in the ZnCr2O4|SAPO-34 system, we observed that once CH4 is generated on ZnCr2O4, it becomes too stable to convert into any other product, resulting in CH4 remaining as the final product even with the addition of SAPO-34. In contrast, in the Zn3Cr3O8|SAPO-34 system, we found that the CO conversion rate is 0.025 s−1 and the main products are CO2, ethene and DME with selectivities of 44.4%, 33.3% and 22.3%, respectively, as shown in the Zn3Cr3O8|SAPO-34 column of Fig. 3. When considering the DME conversion via the typical MTO process, based on the experimental results indicating that the MTO process normally generates ∼80% C2–C4 olefins and ∼10% light alkanes (<C4),48 we find that the final product selectivity of CO2, light olefin and alkane is 49.9%, 47.8% and 2.1%, respectively (Zn3Cr3O8|SAPO-34 with the MTO column in Fig. 3). This result, not only the evaluated CO conversion rate but also the product distribution, is well consistent with the experimental results for the products of the ZnCrOx|SAPO-34 system reported by Jiao et al. (see the ZnCrOx|SAPO-34 column in Fig. 3).1
Microkinetic simulations allow us to evaluate quantitatively the contribution of each reaction pathway in the Zn3Cr3O8|SAPO-34 catalyst. As illustrated in Fig. 4, we observed that approximately 95.8% of the CHO* species undergo stepwise hydrogenation to produce CH3OH, while a minor fraction of 4.2% of CHO* species follows the C–C coupling pathway to form CH2CO on the Zn3Cr3O8 (0001) surface. The gas mixture of CH3OH and a small amount of CH2CO then diffuses into the zeolite cages. Within the zeolite framework, all the methanol is converted into methyl groups, of which 46.3% undergo further carbonylation to form ketene, while the other half remains and couples with the generated ketene to form ethene. Therefore, 86.1% of ketene can be derived from CH3OH within the zeolite cage. This in situ generated ketene within the zeolite, along with 13.9% of ketene diffusing from the ZnCrOx surface, contributes to the production of ethene.
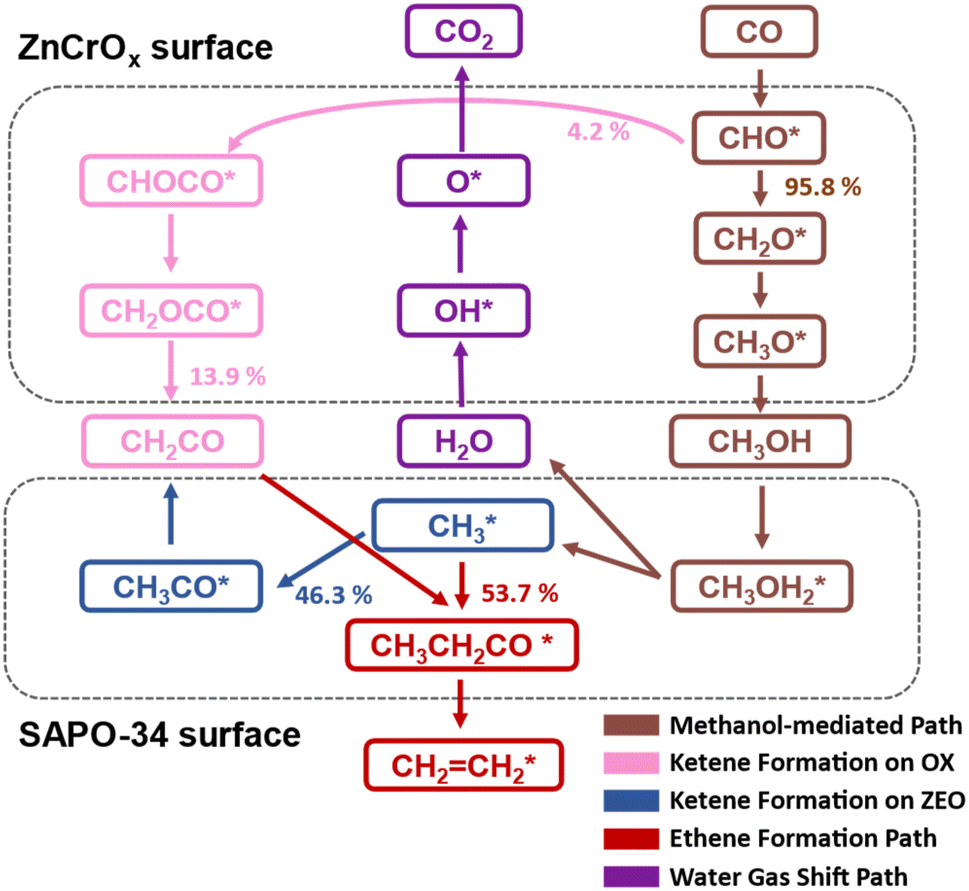 | ||
| Fig. 4 Key reaction pathways extracted from the STO conversion on the Zn3Cr3O8|SAPO-34 catalyst. Species on the surfaces of ZnCrOx and SAPO-34 are enclosed in upper and lower frames, respectively. The methanol-mediated pathway is depicted in brown; the ketene formation pathways on ZnCrOx and SAPO-34 surfaces are plotted in pink and blue, respectively; the ethene formation and water gas shift pathways are shown in red and purple, respectively. | ||
We are now in a position to clarify the roles of ketene and methanol as reaction intermediates in the OX–ZEO system. Our results reveal that methanol serves as the primary transport intermediate between the metal oxide and zeolite, while ketene acts as the dominant intermediate for ethene formation. This explains why both ketene1,7–13,18 and methanol14,15 are observed in experiments. Moreover, the zeolite component actively participates in ketene generation rather than merely receiving it from ZnCrOx, with the methanol carbonylation step in zeolite being the rate determining step. Consequently, optimizing the activity of the zeolite part should be the correct direction for improving the performance of STO catalysts, which has been validated by recent experimental results showing CO conversion exceeding 80% through zeolite modification by Ge.4
4. Conclusions
By using G-NN based large-scale atomic simulations, this work unraveled STO reaction pathways on ZnCrOx|SAPO-34 composite catalysts with particular focus on ketene generation and transformation pathways, including the formation of ketene on ZnCrOx via CO hydrogenation and on SAPO-34 via methanol carbonylation, and the subsequent conversion of ketene to products (ethene) in a zeolite. We show that methanol serves as the primary transport intermediate between metal oxides and zeolites, while ketene acts as the dominant intermediate for ethene formation in the OX–ZEO system. Our main findings are highlighted as follows.On the ZnCrOx surface, ketene formation requires a higher Gibbs free energy barrier (>2.0 eV) compared to methanol and methane synthesis (<1.8 eV), indicating the difficulty of ketene formation when using the ZnCrOx catalyst alone. Within the SAPO-34 zeolite, ketene can be formed through the methanol carbonylation pathway with a Gibbs free energy barrier of 1.99 eV. The subsequent conversion of ketene to ethylene follows the CH2CO* → CH3CH2CO* → CH2CH2 pathway, requiring a Gibbs free energy barrier of 1.82 eV. Taking both ZnCrOx and SAPO-34 into consideration, microkinetic simulations prove that ketene generation follows a dual-site mechanism: the majority of ketene (86.1%) originates from STM on the ZnCrOx surface coupled with methanol carbonylation within the zeolite, while the remaining ketene (13.9%) is derived from CHO*–CO coupling on the ZnCrOx surface. The introduction of the ketene pathway into the STO process significantly alters the reaction performance with a higher ethene selectivity, distinct from the typical MTO process. The methanol carbonylation step in zeolite is the rate-determining step and thus decides the activity of STO catalysts, implying that the tuning of the zeolite properties should be an effective measure to improve the activity of the composite catalyst.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Z.-P. L. and S. M. conceived the project and contributed to the design and analyses of the data. Z.-Y. Y. carried out most of the calculations and wrote the draft of the paper. All the authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2024YFA1509600), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB1180000), the National Science Foundation of China (22422208, 12188101, 22203101, and 22033003), the Youth Innovation Promotion Association CAS (No. 2023265), the Fundamental Research Funds for the Central Universities (20720220011), and the Science & Technology Commission of Shanghai Municipality (23ZR1476100). The numerical calculations in this study were partially carried out on the ORISE supercomputer.References
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Selective Conversion of Syngas to Light Olefins, Science, 2016, 351(6277), 1065–1068, DOI:10.1126/science.aaf1835.
- K. Cheng, B. Gu, X. Liu, J. Kang, Q. Zhang and Y. Wang, Direct and Highly Selective Conversion of Synthesis Gas into Lower Olefins: Design of a Bifunctional Catalyst Combining Methanol Synthesis and Carbon-Carbon Coupling, Angew. Chem., Int. Ed., 2016, 55(15), 4725–4728, DOI:10.1002/anie.201601208.
- G. Raveendra, C. Li, Y. Cheng, F. Meng and Z. Li, Direct Transformation of Syngas to Lower Olefins Synthesis over Hybrid Zn-Al2O3/SAPO-34 Catalysts, New J. Chem., 2018, 42(6), 4419–4431, 10.1039/C7NJ04734G.
- F. Jiao, B. Bai, G. Li, X. Pan, Y. Ye, S. Qu, C. Xu, J. Xiao, Z. Jia, W. Liu, T. Peng, Y. Ding, C. Liu, J. Li and X. Bao, Disentangling the Activity-Selectivity Trade-off in Catalytic Conversion of Syngas to Light Olefins, Science, 2023, 380(6646), 727–730, DOI:10.1126/science.adg2491.
- Z.-Y. Yao, S. Ma and Z.-P. Liu, Active Sites and Reaction Mechanisms of COx Hydrogenation on Zinc-Based Mixed Oxide Catalysts, Sci. China: Chem., 2024, 67 DOI:10.1007/s11426-024-2212-0.
- F. Jiao, X. Pan, K. Gong, Y. Chen, G. Li and X. Bao, Shape-Selective Zeolites Promote Ethylene Formation from Syngas via a Ketene Intermediate, Angew. Chem., Int. Ed., 2018, 57(17), 4692–4696, DOI:10.1002/anie.201801397.
- D. B. Rasmussen, J. M. Christensen, B. Temel, F. Studt, P. G. Moses, J. Rossmeisl, A. Riisager and A. D. Jensen, Ketene as a Reaction Intermediate in the Carbonylation of Dimethyl Ether to Methyl Acetate over Mordenite, Angew. Chem., Int. Ed., 2015, 54(25), 7261–7264, DOI:10.1002/anie.201410974.
- B. Li, J. Xu, B. Han, X. Wang, G. Qi, Z. Zhang, C. Wang and F. Deng, Insight into Dimethyl Ether Carbonylation Reaction over Mordenite Zeolite from In-Situ Solid-State NMR Spectroscopy, J. Phys. Chem. C, 2013, 117(11), 5840–5847, DOI:10.1021/jp400331m.
- K. Vikulov, S. Coluccia and G. Martra, Fourier-Transform Infrared Spectroscopic Studies of the Adsorption of Ketene on Silica, Faraday Trans., 1993, 89(7), 1121, 10.1039/ft9938901121.
- C.-K. Ni, E. A. Wade, M. V. Ashikhmin and C. B. Moore, Infrared Spectroscopy of Ketene by Two-Step Photodissociation, J. Mol. Spectrosc., 1996, 177(2), 285–293, DOI:10.1006/jmsp.1996.0142.
- Y. Zhang, P. Gao, F. Jiao, Y. Chen, Y. Ding, G. Hou, X. Pan and X. Bao, Chemistry of Ketene Transformation to Gasoline Catalyzed by H-SAPO-11, J. Am. Chem. Soc., 2022, 144(40), 18251–18258, DOI:10.1021/jacs.2c03478.
- M. Xie, X. Fang, H. Liu, Z. Chen, B. Li, L. Yang and W. Zhu, Cyclic Oxygenate-Based Deactivation Mechanism in Dimethyl Ether Carbonylation Reaction over a Pyridine-Modified H-MOR Catalyst, ACS Catal., 2023, 13(21), 14327–14333, DOI:10.1021/acscatal.3c04344.
- Y. Ye, E. Abou-Hamad, X. Gong, T. B. Shoinkhorova, A. Dokania, J. Gascon and A. D. Chowdhury, Mapping the Methanol-to-Gasoline Process Over Zeolite Beta, Angew. Chem., Int. Ed., 2023, 62(24), e202303124, DOI:10.1002/anie.202303124.
- X. Liu, W. Zhou, Y. Yang, K. Cheng, J. Kang, L. Zhang, G. Zhang, X. Min, Q. Zhang and Y. Wang, Design of Efficient Bifunctional Catalysts for Direct Conversion of Syngas into Lower Olefins via Methanol/Dimethyl Ether Intermediates, Chem. Sci., 2018, 9(20), 4708–4718, 10.1039/C8SC01597J.
- Z. Li, J. Wang, Y. Qu, H. Liu, C. Tang, S. Miao, Z. Feng, H. An and C. Li, Highly Selective Conversion of Carbon Dioxide to Lower Olefins, ACS Catal., 2017, 7(12), 8544–8548, DOI:10.1021/acscatal.7b03251.
- S. Ma, S.-D. Huang and Z.-P. Liu, Dynamic Coordination of Cations and Catalytic Selectivity on Zinc–Chromium Oxide Alloys during Syngas Conversion, Nat. Catal., 2019, 2(8), 671–677, DOI:10.1038/s41929-019-0293-8.
- Z. Lai, N. Sun, J. Jin, J. Chen, H. Wang and P. Hu, Resolving the Intricate Mechanism and Selectivity of Syngas Conversion on Reduced ZnCr2Ox: A Quantitative Study from DFT and Microkinetic Simulations, ACS Catal., 2021, 11(21), 12977–12988, DOI:10.1021/acscatal.1c03579.
- C.-M. Wang, Y.-D. Wang and Z.-K. Xie, Methylation of Olefins with Ketene in Zeotypes and Its Implications for the Direct Conversion of Syngas to Light Olefins: A Periodic DFT Study, Catal. Sci. Technol., 2016, 6(17), 6644–6649, 10.1039/C6CY01095D.
- S. Huang, C. Shang, P. Kang, X. Zhang and Z. Liu, LASP: Fast Global Potential Energy Surface Exploration, WIREs Comput. Mol. Sci., 2019, 9(6) DOI:10.1002/wcms.1415.
- S.-D. Huang, C. Shang, X.-J. Zhang and Z.-P. Liu, Material Discovery by Combining Stochastic Surface Walking Global Optimization with a Neural Network, Chem. Sci., 2017, 8(9), 6327–6337, 10.1039/C7SC01459G.
- P.-L. Kang, Z.-X. Yang, C. Shang and Z.-P. Liu, Global Neural Network Potential with Explicit Many-Body Functions for Improved Descriptions of Complex Potential Energy Surface, J. Chem. Theory Comput., 2023, 19(21), 7972–7981, DOI:10.1021/acs.jctc.3c00873.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186, DOI:10.1103/PhysRevB.54.11169.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979, DOI:10.1103/PhysRevB.50.17953.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775, DOI:10.1103/PhysRevB.59.1758.
- J. P. Perdew and Y. Wang, Accurate and Simple Analytic Representation of the Electron-Gas Correlation Energy, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45(23), 13244–13249, DOI:10.1103/PhysRevB.45.13244.
- A. N. Yaresko, Electronic Band Structure and Exchange Coupling Constants in ACr2X4 Spinels (A = Zn, Cd, Hg; X = O, S, Se), Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77(11), 115106, DOI:10.1103/PhysRevB.77.115106.
- J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D. Landis, J. K. Nørskov, T. Bligaard and K. W. Jacobsen, Density Functionals for Surface Science: Exchange-Correlation Model Development with Bayesian Error Estimation, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85(23), 235149, DOI:10.1103/PhysRevB.85.235149.
- X.-J. Zhang, C. Shang and Z.-P. Liu, Double-Ended Surface Walking Method for Pathway Building and Transition State Location of Complex Reactions, J. Chem. Theory Comput., 2013, 9(12), 5745–5753, DOI:10.1021/ct4008475.
- C. Shang and Z.-P. Liu, Stochastic Surface Walking Method for Structure Prediction and Pathway Searching, J. Chem. Theory Comput., 2013, 9(3), 1838–1845, DOI:10.1021/ct301010b.
- J. Chen, M. Jia, P. Hu and H. Wang, CATKINAS: A Large-scale Catalytic Microkinetic Analysis Software for Mechanism Auto-analysis and Catalyst Screening, J. Comput. Chem., 2021, 42(5), 379–391, DOI:10.1002/jcc.26464.
- Y. Chen, S. Han, X. Pan, F. Jiao, W. Liu, Y. Pan and X. Bao, Visualization of the Active Sites of Zinc–Chromium Oxides and the CO/H2 Activation Mechanism in Direct Syngas Conversion, J. Am. Chem. Soc., 2024, 146(3), 1887–1893, DOI:10.1021/jacs.3c07332.
- R. Dumitru, F. Manea, C. Păcurariu, L. Lupa, A. Pop, A. Cioablă, A. Surdu and A. Ianculescu, Synthesis, Characterization of Nanosized ZnCr2O4 and Its Photocatalytic Performance in the Degradation of Humic Acid from Drinking Water, Catalysts, 2018, 8(5), 210, DOI:10.3390/catal8050210.
- G. D. Piero, F. Trifiro and A. Vaccari, Non-Stoicheiometric Zn-Cr Spinel as Active Phase in the Catalytic Synthesis of Methanol, J. Chem. Soc., Chem. Commun., 1984,(10), 656–658, 10.1039/C39840000656.
- M. Bertoldi, B. Fubini, E. Giamello, G. Busca, F. Trifirò and A. Vaccari, Structure and Reactivity of Zinc–Chromium Mixed Oxides. Part 1.—The Role of Non-Stoichiometry on Bulk and Surface Properties, J. Chem. Soc., Faraday Trans. 1, 1988, 84(5), 1405, 10.1039/f19888401405.
- A. Riva, F. Trifirò, A. Vaccari, L. Mintchev and G. Busca, Structure and Reactivity of Zinc–Chromium Mixed Oxides. Part 2.—Study of the Surface Reactivity by Temperature-Programmed Desorption of Methanol, J. Chem. Soc., Faraday Trans. 1, 1988, 84(5), 1423, 10.1039/f19888401423.
- E. Giamello, B. Fubini, M. Bertoldi, G. Busca and A. Vaccari, Structure and Reactivity of Zinc–Chromium Mixed Oxides. Part 3.—The Surface Interaction with Carbon Monoxide, J. Chem. Soc., Faraday Trans. 1, 1989, 85(2), 237, 10.1039/f19898500237.
- E. Errani, F. Trifiro, A. Vaccari, M. Richter and G. Del Piero, Structure and Reactivity of Zn-Cr Mixed Oxides. Role of Non-Stoichiometry in the Catalytic Synthesis of Methanol, Catal. Lett., 1989, 3(1), 65–72, DOI:10.1007/BF00765056.
- L. Tan, G. Yang, Y. Yoneyama, Y. Kou, Y. Tan, T. Vitidsant and N. Tsubaki, Iso-Butanol Direct Synthesis from Syngas over the Alkali Metals Modified Cr/ZnO Catalysts, Appl. Catal., A, 2015, 505, 141–149, DOI:10.1016/j.apcata.2015.08.002.
- S. Tian, S. Wang, Y. Wu, J. Gao, P. Wang, H. Xie, G. Yang, Y. Han and Y. Tan, The Role of Potassium Promoter in Isobutanol Synthesis over Zn–Cr Based Catalysts, Catal. Sci. Technol., 2016, 6(12), 4105–4115, 10.1039/C5CY02030A.
- M. Sicong and L. Zhi-Pan, Zeolite Acidity database of NH3-TPD, http://www.lasphub.com/zeolite/#/zeoacidity.
- S. R. Blaszkowski and R. A. van Santen, Theoretical Study of the Mechanism of Surface Methoxy and Dimethyl Ether Formation from Methanol Catalyzed by Zeolitic Protons, J. Phys. Chem. B, 1997, 101(13), 2292–2305, DOI:10.1021/jp962006+.
- A. Sierraalta, R. Añez, D. S. Coll and P. Alejos, Conversion of Methanol to Dimethyl Ether over Silicoaluminophosphates: Isolated Acid Sites and the Influence of Silicon Islands. A DFT-ONIOM Study, Microporous Mesoporous Mater., 2020, 292, 109732, DOI:10.1016/j.micromeso.2019.109732.
- P. Huber, F. Studt and P. N. Plessow, Reactivity of Surface Lewis and Brønsted Acid Sites in Zeolite Catalysis: A Computational Case Study of DME Synthesis Using H-SSZ-13, J. Phys. Chem. C, 2022, 126(13), 5896–5905, DOI:10.1021/acs.jpcc.2c00668.
- J. Kästner and W. Thiel, Bridging the Gap between Thermodynamic Integration and Umbrella Sampling Provides a Novel Analysis Method: “Umbrella Integration.”, J. Chem. Phys., 2005, 123(14), 144104, DOI:10.1063/1.2052648.
- L.-H. Luo, S.-D. Huang, C. Shang and Z.-P. Liu, Resolving Activation Entropy of CO Oxidation under the Solid–Gas and Solid–Liquid Conditions from Machine Learning Simulation, ACS Catal., 2022, 12(10), 6265–6275, DOI:10.1021/acscatal.2c01561.
- Z.-Y. Hu, L.-H. Luo, C. Shang and Z.-P. Liu, Free Energy Pathway Exploration of Catalytic Formic Acid Decomposition on Pt-Group Metals in Aqueous Surroundings, ACS Catal., 2024, 14(10), 7684–7695, DOI:10.1021/acscatal.4c00959.
- H. Song, D. Laudenschleger, J. J. Carey, H. Ruland, M. Nolan and M. Muhler, Spinel-Structured ZnCr2O4 with Excess Zn Is the Active ZnO/Cr2O3 Catalyst for High-Temperature Methanol Synthesis, ACS Catal., 2017, 7(11), 7610–7622, DOI:10.1021/acscatal.7b01822.
- S.-G. Lee, H.-S. Kim, Y.-H. Kim, E.-J. Kang, D.-H. Lee and C.-S. Park, Dimethyl Ether Conversion to Light Olefins over the SAPO-34/ZrO2 Composite Catalysts with High Lifetime, J. Ind. Eng. Chem., 2014, 20(1), 61–67, DOI:10.1016/j.jiec.2013.04.026.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01651g |
| This journal is © The Royal Society of Chemistry 2025 |