
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural flexibility and mobility of coordination polymers on Cu(111)†
Waka Nakanishi‡
 *ab,
Masayuki Takeuchiabc and
Keisuke Sagisaka‡*d
*ab,
Masayuki Takeuchiabc and
Keisuke Sagisaka‡*d
aMolecular Design and Function Group, National Institute for Materials Science (NIMS), 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan. E-mail: NAKANISHI.Waka@nims.go.jp
bResearch Center for Autonomous Systems Materialogy (ASMat), Institute of Integrated Research (IIR), Institute of Science Tokyo (Science Tokyo), 4259 Nagatsuta-cho, Midori-ku, Yokohama 226-8501, Japan
cDepartment of Materials Science and Engineering, Faculty of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8577, Japan
dResearch Center for Advanced Measurement and Characterization, National Institute for Materials Science (NIMS), 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan. E-mail: SAGISAKA.Keisuke@nims.go.jp
First published on 28th April 2025
Abstract
Coordination polymers on surfaces have been investigated at the atomic level via scanning tunneling microscopy (STM), revealing a variety of structures and electronic states. However, analysis of their dynamic behaviour at low temperatures has been hampered due to the strong adsorption of organic ligands on metal surfaces. In this study, we designed 2,7-dicyano-9,9-dimethyl-9H-fluorene (DCF) as a ligand for coordination polymers in order to reveal its mobility at low temperatures. The dimethyl group attached to the ligand reduces the interaction with the metal surface and serves as an indicator of the ligand orientation. This enables individual tracking of DCF in the DCF–Cu polymer on Cu(111) after their coordination with copper adatoms. Upon heating from 4 K to 78 K, branched structures were less mobile, while linear structures and short polymers with free ends exhibited higher mobility. Partial cleavage, recombination, and insertion of the polymer chains were observed in some sections of the polymer chains. Density functional theory (DFT) calculations suggest that the flexibility of the coordination angle (180° ± 20°) facilitates such transformations. This study provides a direct observation of the motion differences attributed to polymer chain structures as proposed in real materials.
Introduction
Coordination polymers, including metal organic frameworks (MOFs), are supramolecular polymers1,2 composed of coordination bonds between metals and organic ligands. These systems have attracted extensive fundamental research interest due to their structural diversity and broad range of applications, such as gas storage and separation, catalysis, and energy storage. Recently, attention has shifted towards flexible coordination polymers, as their structural flexibility allows deformation in response to external stimuli, potentially leading to higher functionality.3–5 The overall flexibility of coordination polymers originates from the structural flexibility of individual organic ligands and the inherent flexibility of coordination bonds due to the relatively weak bonding interactions between metals and ligands.6–8 While the average properties of coordination polymers have been characterized by X-ray diffraction (XRD), nuclear magnetic resonance (NMR), differential scanning calorimetry (DSC), etc.,4,5 a detailed understanding of their flexibility at the molecular level remains elusive. In particular, the role of local distortions and strains in contributing to the overall flexibility has not been explored. Recently, transmission electron microscopy (TEM)9–11 and scanning tunneling microscopy (STM)12,13 have been used for observing and understanding structures at the molecular level. TEM offers excellent temporal resolution on the order of milliseconds to several seconds, making it suitable for analysing dynamics. On the other hand, STM provides outstanding height and lateral resolution on the picometer scale, which is ideal for detailed analysis of molecular structural changes. Both techniques are complementary to each other in elucidating the polymer structural properties at the molecular level.Coordination polymers on metal surfaces serve as ideal models for investigating dynamic molecular structures using STM. These polymers are synthesized through the reaction of adatoms with ligands on metal surfaces.14,15 Ligands with large π-conjugated planar structures, such as phthalocyanines and porphyrins, are commonly employed due to their stability on surfaces, facilitating STM analysis.16–19 However, these planar ligands exhibit strong adsorption onto the metal surface, typically requiring heating above 100 °C to synthesize the coordination polymer on the surface. Under these elevated temperatures, the coordination bonds on the metal are often cleaved. Consequently, research employing such large π-conjugated planar ligands has been unable to investigate the dynamic flexibility of metal–ligand coordination.
This study aims to elucidate the structural and temperature-responsive characteristics of metal-coordinated supramolecular polymers by using the 2,7-dicyano-9,9-dimethyl-9H-fluorene (DCF) ligand. This ligand is specifically designed to minimize interaction with a surface to facilitate enhanced movement on the substrate,20–26 and its asymmetric structure allows individual tracking. In DCF, the cyano groups have minimal steric hindrance and the ability to achieve various coordination numbers.14,15,20–22 Copper, which typically prefers di-coordination but can also accommodate tri-coordination, was chosen as the central metal. The STM study revealed the formation of dispersed, wavy linear di-coordinated polymers, accompanied by partially tri-coordinated branch networks, on the Cu(111) surface following mild annealing of DCF in the presence of Cu adatoms. Our analyses demonstrate that the local curvature of the coordination polymers is mainly governed by the combination of three distinct types of DCF trimers, each characterized by specific molecular orientations. This polymer exhibits three temperature-dependent behaviours: from 4 K to 40 K, the polymers remain static; at 48 K, their mobility varies depending on the polymer structure, with significant movement observed in the two-coordinated linear structure, while minimal movement occurs in the three-coordinated branched structure. At 71 K, the polymer chains exhibit greater movement, accompanied by processes such as chain scission, insertion, and recombination. The structures of polymer chains, such as linear and crosslinked configurations, play a crucial role in determining their dynamic behaviour and, consequently, the mechanical properties of polymeric materials. This study utilized STM to directly observe and analyse the structural changes of coordination polymer chains. The results demonstrate that coordination polymers on metal surfaces can serve as good models for understanding the flexibility and mobility of polymer materials at the molecular level.
Results and discussion
Molecular design
The ligand molecule DCF was designed and synthesized in order to reduce surface interactions and promote the formation of coordination polymers (Fig. 1): dimethyl group was attached to the fluorene structure to prevent the ligand from strong adsorption onto the metal surfaces.20–26 Furthermore, as will be explained in detail later, this dimethyl group serves as an indicator of the molecular orientation relative to the polymer chain direction, which is essential for tracking each ligand throughout the analysis process. Two cyano groups were introduced at both ends of the fluorene backbone to form the DCF–Cu polymer via coordination with copper adatom. Structure optimization conducted by DFT calculations confirmed that the two cyano groups form an angle of 156°. | ||
| Fig. 1 Optimized structure of DCF with the angle between the two cyano groups (left). Schematic view of DCF–Cu polymer on Cu(111), in which each of the two methyl groups assists in reducing adsorption onto the surface and in assignment of the orientation of the ligands (right). | ||
Synthesis of the DCF–Cu polymer on Cu(111)
Coordination polymers composed of DCF and Cu adatoms were synthesized on a Cu(111) surface. The DCF–Cu polymer was formed through the annealing of pre-deposited DCFs and copper adatoms supplied by the Cu(111) surface. When DCF molecules were deposited onto a cold Cu(111) surface at 7 K, they predominantly remained as isolated entities (Fig. 2a). Upon mild heating to 323 K, beaded and meandering polymers were formed (Fig. 2b and c). The ligands typically appear as nearly triangular in STM images, while the coordinated metal remains dark and undetectable, as is common for coordination polymers on metal surfaces (Fig. 2b and c).25,26,29–33,37,41 It is reasonable to infer the formation of coordination bonds based on the observed inter-DCF distances of 1.5–1.6 nm (Fig. S12†). During the annealing process at 323 K, DCF molecules migrated across the surface and reacted with copper adatoms supplied by the Cu(111) substrate, leading to the formation of DCF–Cu polymers. In these polymers, a single copper atom predominantly coordinated with two DCF molecules (Fig. 2b). This process ultimately resulted in the formation of 91.1 ± 0.8% di-coordinate linear polymers and 8.8 ± 0.9% tri-coordinate branched structures (Fig. S13 and Table S1†).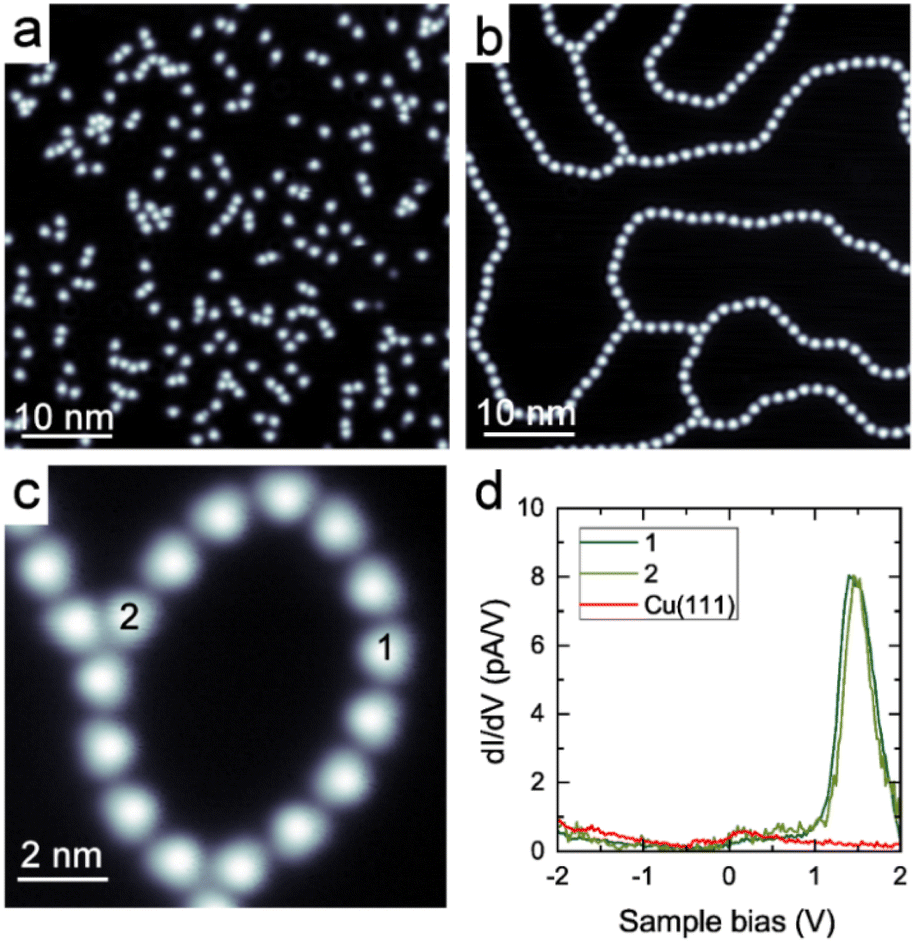 | ||
| Fig. 2 STM images of the DCF molecules on Cu(111) recorded at 4 K, (a) as deposited at 7 K, Vs = + 0.3 V, I = 10 pA. (b) After annealing at 323 K, Vs = +0.5 V, I = 10 pA. (c) A high resolution image of the DCF–Cu polymer. Vs = +1.0 V, I = 10 pA. (d) Differential conductance (dI/dV) spectra of the DCF–Cu polymer. Each spectrum was recorded by positioning the STM tip over the center of the DCF molecule, as indicated by the number in (c). Set point: Vs = +1.0 V, I = 50 pA. Lock-in detection settings: Vmod = 10 mV, f = 850 Hz. | ||
The electronic state of the DCF–Cu polymer was examined by measuring dI/dV spectra at multiple locations, including both linear and branched parts. As a result, we observed a prominent peak at +1.45 V at a linear di-coordinated part (location 1 in Fig. 2c) and a branched tri-coordinated part (location 2 in Fig. 2c). Our DFT calculations attribute this peak to the lowest unoccupied molecular orbital, which is primarily distributed over the DCF molecule (Fig. S10†). This observation is consistent with the previous result indicating that the peak position is independent of the coordination number.21,22 As a reference, we attempted to measure a spectrum on single isolated molecules before coordination. However, these molecules were too mobile, even on the cold surface at 4 K during sample bias-sweeping, to measure the dI/dV spectrum.
The adjacent polymer chains of the DCF–Cu polymer maintain a distinct separation, similar to other coordination polymers observed on metal surfaces.27–31 Bader charge analysis, performed on a trans-DCF–Cu–DCF–Cu polymer adsorbed on a Cu(111) slab, indicates that the central copper atom carries a charge of +0.5e, while the ligand holds a charge of −0.19e, attributed to the electron-withdrawing effect of the cyano groups (Fig. S11†). Consequently, the polymer exhibits an overall positive charge of +0.31e per DCF–Cu unit. This electrostatic repulsion likely accounts for the observed separation between polymer chains.
Origins of polymer shape and structural flexibility
The DCF–Cu polymer exhibits characteristic meandering features. The winding of microscopic polymers is correlated with the flexibility and viscoelasticity of macroscopic polymer materials.32,33 In contrast, it is intriguing to understand what parameters dominate the meandering shape at the molecular scale. Before initiating a statistical analysis, we examined the orientations of all DCF molecules forming the polymers within an area of 30 nm × 30 nm (Fig. 3a). With a small sample bias (<0.1 V), the DCF molecule appears as a triangular shape with a bright spot, corresponding to one of the central dimethyl groups (Fig. 3b). This feature allows us to define the molecular orientation: the dimethyl group side of the molecule is indicated by a white triangle in Fig. 3a and b. Moreover, we measured the angle formed by three DCF molecules bonded in a row through Cu atoms. In particular, the three brightest points corresponding to the methyl group of each molecule were used to define the angle of the trimer (Fig. 3b). From Fig. 3a, we collected the angles for 64 sets of timers. We note that the fluctuations in the trimer angle primarily arise from changes in the two CN–Cu–CN bond angles within the trimer, and these changes are equivalent to the variations in the angle formed by the three methyl groups (see Fig. S9†). | ||
| Fig. 3 Coordination structures of the DCF–Cu polymer on Cu(111). (a) STM image of the linear DCF–Cu polymer recorded at 4 K. Imaging conditions: Vs = +0.02 V, I = 50 pA. (b) High resolution STM image of the DCF–Cu polymer. The molecular model of the DCF–Cu polymer is superimposed. Triangles in (a) and (b) denote the molecular orientation of the methyl group side. The angle of the trimer (θ) is defined as the angle made by two arrows starting at the methyl group of the central molecule toward the methyl groups in the two adjacent molecules. (c) Three optimized structures of the DCF–Cu trimer. (d) Distribution (dots) and the averaged (line) value of the trimer angles obtained from (a). | ||
Depending on the molecular orientation, the DCF–Cu trimer can be grouped into three types, as illustrated in Fig. 3c. Type A is a trimer with all three molecules oriented in the same direction. Type B has two adjacent molecules oriented in the same direction, with the third molecule oriented oppositely. Type C features alternating molecular orientations. The molecular configurations for each type were optimized using DFT calculations. The computed energies indicate negligible differences among the three types, with an average stabilization energy of 12.8 kcal mol−1 per cyano group coordination (Fig. S9†). The resulting CN–Cu–CN bonding angles are 180°, with the angles defined by the three methyl groups being 158° for Type A, 183° for Type B, and 206° for Type C (Fig. 3c and see also Fig. S7–S9†). Each example of these types is indicated in Fig. 3a.
Measured angles of the trimer from the STM image in Fig. 3a are plotted by type in Fig. 3d. The observed average angles for Type A, B, and C are 153°, 177°, and 200°, respectively, agreeing well with the theoretical predictions. These plots also reveal that the angles of the trimer are distributed approximately ±20° from the average values. DFT calculations resulted in the energy increase by only 2.3 kcal mol−1 when the CN–Cu–CN bond angle is deviated by ±20° from 180°, suggesting that the experimentally observed degree of flexibility is energetically plausible (Fig. S7b†). Additional DFT calculations predict that DCF molecules bonded through a Cu atom become twisted as the deviation of the CN–Cu–CN bond angle exceeds ±20° from 180° (Fig. S7a†). Although the exact valency of the adatom and coordinated state on metal surfaces remains a subject of debate,34–38 the calculation outcome remains consistent regardless of whether the Cu valency is 0, +1, or +2 (Fig. S8†). This result reasonably agrees with the experimental observations that the trimer angle varies within ±20° from 180°, as the planarity of the molecules is maintained due to constraints imposed by the substrate surface. From these observations, we conclude that the fundamental structure of the winding DCF–Cu polymer is governed by the combination of three distinct molecular configurations, dictated by the orientations of three consecutive DCF molecules. Each configuration is characterized by a specific alignment angle, but it retains its flexibility due to the variation of the CN–Cu–CN coordination bond angle, which can deviate by ±20° from 180°. This inherent flexibility plays a role in shaping the local structure of the polymers.
Temperature-induced motion and polymer structure
To investigate the structural fluctuation of the coordination polymers upon heating, STM observations were conducted while gradually increasing the sample temperature up to 71 K (Fig. 4). The relative orientation of DCF was utilized to track the movement of each DCF molecule within all the polymers individually (Fig. S14–S16†). Below 40 K, the DCF–Cu polymer remained static, allowing for stable STM observations during the heating of the surface. In this temperature range, no structural changes were observed. Above 40 K, on the other hand, the mobility of the polymers increased, disrupting STM imaging. As an alternative approach, the sample was heated to either 48 K or 71 K for 10 minutes and then cooled down to 4 K to quench the structural changes for STM imaging. Thus, the data for 48 K and 71 K represent snapshots at those temperatures. The STM image for 48 K shows subtle changes in the polymer's shape (Fig. 4b). Pronounced structural changes were observed for polymers with free ends. The branching points, where the three molecules converge, are mostly pinned to the surface, while the linear segments of the polymers exhibit slight displacement. These observations are attributed to the vibrational motion of polymer segments induced by thermal activation at 48 K.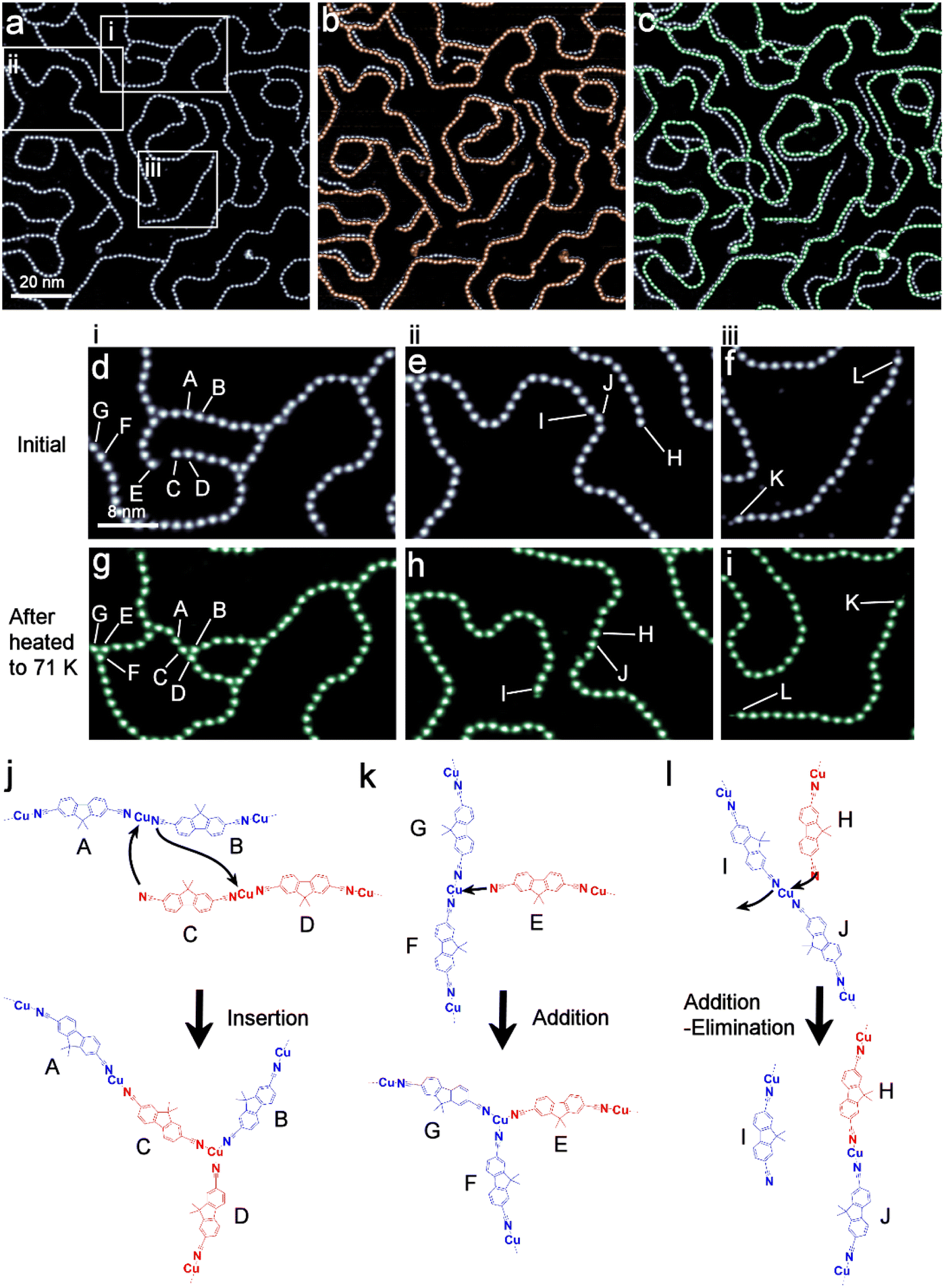 | ||
| Fig. 4 Change in the winding shapes of the DCF–Cu polymer on Cu(111) by heating. (a) Initial shapes of the polymers (gray), (b) after heating to 48 K (brown) and (c) 71 K (green). All STM images were recorded at 4 K. For comparison, initial polymers (gray) are superimposed in (b) and (c). (d)–(i) Zoomed images in areas i, ii, and iii indicated in (a). The uppercase letters refer to the positions where insertion (A–D), addition (E and F), addition–elimination (H–J) reactions of the coordination bonds, and locomotion (K and L) are observed. The reaction schemes for insertion (j), addition (k), and addition–elimination (l) reactions are also shown. | ||
Heating to 71 K resulted in further significant positional changes of the polymer, along with rearrangement of the polymer chains (Fig. 4c). In the region i (Fig. 4d and g), molecule C inserted between A and B to form a three-coordinated structure B–C–D (Fig. 4j), while E joined G and F to create another three-coordinated structure (Fig. 4k). In region ii (Fig. 4e and h), the addition–elimination reaction of the polymer chain is observed: H combined with J, whereas I dissociated (Fig. 4l). A thorough analysis of the trimer angles revealed that the trimer angle centred on molecule I deviated significantly (22°) from the average angle (Fig. S7 and Table S2†). This observation suggests that dissociation likely occurred during the temperature increase, as greater strain accumulated at molecule I than at others. In region iii (Fig. 4f and i), the K–L chain underwent a complete lateral rotation, reversing its position. It is noteworthy that none of the individual molecules flipped their orientation within any of the polymers. This temperature supplied sufficient energy to enable lateral motion of the polymers, such as the lateral rotation of a single chain of polymer with free ends (Region iii), but it is not effective enough to induce twisting of the polymer chains on the surface.
Based on our observations, the structural changes of the polymer chains upon heating can be understood in terms of the following characteristic properties of the polymer:
1. Affinity between Cu adatoms and the cyano-group of the molecules: this interaction, estimated to be average 12.8 kcal mol−1 in vacuum, facilitates the formation of coordination complexes and contributes to the thermal stability of the polymer. Upon heating to 71 K, polymers with free termini exhibit enhanced mobility and reactivity compared to closed structures. In some cases, a free-terminal polymer coordinates with a Cu atom within a linear polymer, resulting in a tri-coordinated configuration that lowers the overall system energy (Fig. 4d and g). Moreover, addition–elimination reactions (Fig. 4e and h) proceed via the transient formation of a tri-coordination structure, which subsequently dissociates into a linear polymer and a free-terminal polymer, with no net change in system energy. These structural transformations are determined by the energy landscape of the system and the reaction probabilities associated with the highly reactive free-terminal polymers.
2. Flexibility of the coordination angle: this property determines the overall polymer shape and constrains the extent of polymer motion upon heating. With sufficient thermal energy, the flexibility allows the polymers to adjust its bonding angle with minimal energy cost.
3. Interpolymer interaction (repulsion): the polymers carry a slight positive charge, leading to mutual repulsion that prevents polymer bundling. This effect ensures that polymers maintain a characteristic separation distance. While thermal vibrations allow for occasional collisions and separations, the majority of interpolymer distance remains relatively constant.
As a result of these synergistic effects, certain regions of the polymer structure undergo significant rearrangements upon heating, while others exhibit only minor changes. Polymer chains with free ends located on relatively open surface areas experience greater displacement, which facilitates the formation of branched structures and promotes addition–elimination reactions. In contrast, initially stable closed structures are constrained by interpolymer repulsion, which limits their accessible surface area for movement; as a result, these structures predominantly exhibit localized vibrational motion without substantial displacement.
Conclusions
In this study, by designing and synthesizing the organic ligand DCF with minimized surface interactions and an asymmetric structure with respect to the coordination direction, it became possible to observe the movement of the DCF–Cu polymer on the Cu(111) surface while maintaining coordination bonds, and to track individual ligands. This approach enables a molecular-level understanding of temperature-dependent structural changes using STM. The design principle is particularly applicable to analysing the dynamic behaviour of organic molecules that strongly adsorb onto metal surfaces, especially those forming supramolecular polymers through weak non-chemical bonds that are cleaved at high temperatures. This method provides the potential to observe and analyse the structure, cleavage, recombination tendencies, and mobility characteristics of supramolecular polymers at the molecular level.The DCF–Cu polymer predominantly exhibits a linear di-coordinated structure with occasional tri-coordinated branches, and the structure resembles cross-linked polymer materials. Through the detailed analysis of STM, the CN–Cu–CN coordination bond angle, while typically close to 180°, was revealed to have a flexibility of ±20°, which contributes to the overall flexibility of the polymers. This polymer exhibits three distinct temperature-dependent behaviours: up to 40 K the polymers remain static; at 48 K the polymer chains with free ends exhibit mobility; at 71 K movements of linear segments in the polymers are detected. In contrast, the branched polymer structures exhibit minimal motion throughout the entire temperature range. Moreover, the observed strain accumulation and subsequent chain cleavage are directly linked to critical mechanical properties, including stiffness and durability. Although the topology of the polymer chains significantly affects the physical properties,39–42 yet comprehensive understanding at the molecular level is lacking. Our insights into the polymer structure and molecular-level motion illuminate the connections between molecular-level phenomena and the macroscopic mechanical properties of polymers.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
W. N. and K. S. designed the project, performed the experiments (organic synthesis: W. N.; STM: K. S.), conducted theoretical calculations, and analysed the data. All authors contributed to writing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A part of calculations in this study were performed on a Numerical Materials Simulator at NIMS. This work was partially supported by JSPS KAKENHI Grant Numbers, 22K05088 and 22K04860. This study is also supported by a MEXT “NIMS Molecule and Material Synthesis Platform” program. A part of this work was supported by “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT). Proposal Number JPMXP1224NM5109.Notes and references
- T. Aida and E. W. Meijer, Isr. J. Chem., 2020, 60, 33–47 Search PubMed.
- F. Xu and B. L. Feringa, Adv. Mater., 2023, 35, 2204413 CrossRef CAS PubMed.
- T. Kundu, B. B. Shah, L. Bolinois and D. Zhao, Chem. Mater., 2019, 31, 2842–2847 CrossRef CAS.
- A. Halder and D. Ghoshal, CrystEngComm, 2018, 20, 1322–1345 Search PubMed.
- Z. Chang, D.-H. Yang, J. Xu, T.-L. Hu and X.-H. Bu, Adv. Mater., 2015, 27, 5432–5441 Search PubMed.
- E. V. Shaw, A. M. Chester, G. P. Robertson, C. Castillo-Blas and T. D. Bennett, Chem. Sci., 2024, 15, 10689–10712 RSC.
- X. Liu, A. A. L. Michalchuk, B. Bhattacharya, N. Yasuda, F. Emmerling and C. R. Pulham, Nat. Commun., 2021, 12, 3871 Search PubMed.
- Z. Lin, J. J. Richardson, J. Zhou and F. Caruso, Nat. Rev. Chem, 2023, 7, 273–286 CrossRef CAS PubMed.
- S. T. Skowron, T. W. Chamberlain, J. Biskup, U. Kaiser, E. Besley and A. N. Khlobystov, Acc. Chem. Res., 2017, 50, 1797–1807 CrossRef CAS PubMed.
- K. Harano, T. Nakamuro and E. Nakamura, Microscopy, 2024, 73, 101–116 CrossRef CAS PubMed.
- N. de Jonge and F. M. Ross, Nat. Nanotechnol., 2011, 6, 695–704 CrossRef CAS PubMed.
- L. Grill and S. Hecht, Nat. Chem., 2020, 12, 115–130 CrossRef CAS PubMed.
- H.-Y. Gao, Phys. Chem. Chem. Phys., 2024, 26, 19052–19068 RSC.
- Y.-F. Geng, P. Li, J.-Z. Li, X.-M. Zhang, Q.-D. Zeng and C. Wang, Coord. Chem. Rev., 2017, 337, 145–177 CrossRef CAS.
- J. Li, C. Wäckerlin, S. Schnidrig, E. Joliat, R. Alberto and K.-H. Ernst, Helv. Chim. Acta, 2017, 100, e1600278 CrossRef.
- F. Bischoff, Y. He, K. Seufert, D. Stassen, D. Bonifazi, J. V. Barth and W. Auwärter, Chem.–Eur. J., 2016, 22, 15298–15306 CrossRef CAS PubMed.
- M. Lepper, T. Schmitt, M. Gurrath, M. Raschmann, L. Zhang, M. Stark, H. Hölzel, N. Jux, B. Meyer, M. A. Schneider, H.-P. Steinrück and H. Marbach, J. Phys. Chem. C, 2017, 121, 26361–26371 CrossRef CAS.
- R. Adhikari, G. Siglreithmaier, M. Gurrath, M. Meusel, J. Kuliga, M. Lepper, H. Hölzel, N. Jux, B. Meyer, H.-P. Steinrück and H. Marbach, Chem.–Eur. J., 2020, 26, 13408–13418 CrossRef CAS PubMed.
- M. Lepper, J. Köbl, L. Zhang, M. Meusel, H. Hölzel, D. Lungerich, N. Jux, A. de Siervo, B. Meyer, H.-P. Steinrück and H. Marbach, Angew. Chem., Int. Ed., 2018, 57, 10074–10079 CrossRef CAS PubMed.
- V. J. Langlais, R. R. Schlittler, H. Tang, A. Gourdon, C. Joachim and J. K. Gimzewski, Phys. Rev. Lett., 1999, 83, 2809–2812 CrossRef CAS.
- W. Nakanishi, A. Nakata, P. Perez, M. Takeuchi, C. Joachim and K. Sagisaka, J. Phys. Chem. C, 2021, 125, 9937–9946 CrossRef CAS.
- W. Nakanishi, Y. Matsushita, M. Takeuchi and K. Sagisaka, Phys. Chem. Chem. Phys., 2023, 25, 13702–13707 RSC.
- C. Joachim, H. Tang, F. Moresco, G. Rapenne and G. Meyer, Nanotechnology, 2002, 13, 330–335 Search PubMed.
- L. Lafferentz, F. Ample, H. Yu, S. Hecht, C. Joachim and L. Grill, Science, 2009, 323, 1193–1197 CrossRef CAS PubMed.
- C. Bombis, F. Ample, L. Lafferentz, H. Yu, S. Hecht, C. Joachim and L. Grill, Angew. Chem., Int. Ed., 2009, 48, 9966–9970 CrossRef CAS PubMed.
- W.-H. Soe, Y. Shirai, C. Durand, Y. Yonamine, K. Minami, X. Bouju, M. Kolmer, K. Ariga, C. Joachim and W. Nakanishi, ACS Nano, 2017, 11, 10357–10365 CrossRef CAS PubMed.
- S. L. Tait, A. Langner, N. Lin, S. Stepanow, C. Rajadurai, M. Ruben and K. Kern, J. Phys. Chem. C, 2007, 111, 10982–10987 CrossRef CAS.
- N. Lin, S. Stepanow, M. Ruben and J. V. Barth, Top. Curr. Chem., 2009, 287, 1–44 CAS.
- D. Heim, D. Écija, K. Seufert, W. Auwärter, C. Aurisicchio, C. Fabbro, D. Bonifazi and J. V. Barth, J. Am. Chem. Soc., 2010, 132, 6783–6790 CrossRef CAS PubMed.
- W. Wang, X. Shi, S. Wang, J. Liu, M. A. Van Hove, P. N. Liu, R.-Q. Zhang and N. Lin, Phys. Rev. Lett., 2013, 110, 046802 CrossRef PubMed.
- T. A. Pham, F. Song, M. N. Alberti, M.-T. Nguyen, N. Trapp, C. Thilgen, F. Diederich and M. Stöhr, Chem. Commun., 2015, 51, 14473–14476 RSC.
- L. Sangroniz, M. Fernández and A. Santamaria, Polymer, 2023, 271, 125811 CrossRef CAS.
- P.-G. de Gennes, Scaling Concepts in Polymer Physics, Cornell University Press, 1979 Search PubMed.
- Y. Li, J. Xiao, T. E. Shubina, M. Chen, Z. Shi, M. Schmid, H.-P. Steinrück, J. M. Gottfried and N. Lin, J. Am. Chem. Soc., 2012, 134, 6401–6408 Search PubMed.
- M. N. Faraggi, N. Jiang, N. Gonzalez-Lakunza, A. Langner, S. Stepanow, K. Kern and A. Arnau, J. Phys. Chem. C, 2012, 116, 24558–24565 CrossRef CAS.
- C. Krull, M. Castelli, P. Hapala, D. Kumar, A. Tadich, M. Capsoni, M. T. Edmonds, J. Hellerstedt, S. A. Burke, P. Jelinek and A. Schiffrin, Nat. Commun., 2018, 9, 3211 Search PubMed.
- W.-H. Soe, C. Manzano, R. Robles, N. Lorente and C. Joachim, Nano Lett., 2020, 20, 384–388 CrossRef CAS PubMed.
- F. Queck, O. Krejčí, P. Scheuerer, F. Bolland, M. Otyepka, P. Jelínek and J. Repp, J. Am. Chem. Soc., 2018, 140, 12884–12889 Search PubMed.
- F. M. Haque and S. M. Grayson, Nat. Chem., 2020, 12, 433–444 CrossRef CAS.
- H. Sato, D. Aoki, H. Marubayashi, S. Uchida, H. Sogawa, S. Nojima, X. Liang, K. Nakajima, T. Hayakawa and T. Takata, Nat. Commun., 2021, 12, 6175 CrossRef PubMed.
- T. Fujiyabu, N. Sakumichi, T. Katashima, C. Liu, K. Mayumi, U. Chung and T. Sakai, Sci. Adv., 2022, 8, eabk0010 Search PubMed.
- S. Jiang, A. B. Dieng and M. A. Webb, npj Comput. Mater., 2024, 10, 139 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Conditions for synthesis, theoretical calculations, and STM experiments. See DOI: https://doi.org/10.1039/d5sc01949d |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |