
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Strategically designed catalysts for ammonia synthesis under mild conditions: recent advances and challenges
Sai Sundara Sandeep Ganti†
,
Pintu Kumar Roy† ,
Nayonay Wagh,
Kona Naga Surya Siva Sai and
Sushant Kumar*
,
Nayonay Wagh,
Kona Naga Surya Siva Sai and
Sushant Kumar*
Gas-Solid Interaction Laboratory, Department of Chemical and Biochemical Engineering, Indian Institute of Technology Patna, Bihta, Patna, 801106, Bihar, India. E-mail: sushantkumar@iitp.ac.in
First published on 22nd March 2025
Abstract
The role of ammonia would continue to be significant in the changing energy landscape with focus on mitigating the carbon footprint per unit of ammonia produced. Since ammonia is a zero-carbon molecule and increasingly considered an important hydrogen energy carrier for future energy systems, its generation under mild conditions and subsequent industrial acceptance are critical. Therefore, the recent challenges include designing and engineering alternative greener methods that can generate ammonia at low input energy and facilitate inexpensive, localized, and renewable energy-coupled ammonia generation. This review underscores the recent developments in the design strategies of novel catalysts, particularly emphasizing the recent advances in different classes of thermal, electrochemical, and non-thermal plasma catalysis that can generate ammonia under mild conditions. Hence, this article can serve as a comprehensive work for engineering novel catalysts and methods, contributing to the generation of sustainable and cost-efficient solutions for expanding the landscape of ammonia applications.
1. Introduction
With the massive increase in the human population following the industrial revolution in the 19th century, there was a fear that mankind would suffer from an acute shortage of food. Fixed nitrogen was found to be a crucial component of fertilisers that promoted healthy crop growth, and most of the nitrogen-containing fertilisers were obtained from nitrate reserves that were limited. Amidst rising concerns of the complete exhaustion of these reserves, there was a pressing need to come up with a scalable method for nitrogen fixation. A major breakthrough occurred in the year 1908, when German chemist Fritz Haber invented a method for ammonia synthesis at high temperature and pressure using a closed circulatory system, which was later industrialised by Carl Bosch. Both scientists received a Nobel Prize for their contribution in developing the process, which is now known as the Haber–Bosch (HB) method for ammonia synthesis and regarded by many as the greatest invention of the 20th century.The development of this large-scale ammonia production method made fertilisers affordable and readily available, exponentially increasing the crop production. Consequently, Haber's invention is credited to have saved the lives of millions of people, playing a pivotal role in the population growth. To this day, the HB process remains the dominant mode of production of ammonia. Ammonia is the second largest commercially manufactured chemical in the world at an annual production rate of almost 200 million metric tons, with the capacity reaching nearly 240 million metric tons.1,2 Roughly 80% of the ammonia produced is used for making fertilisers, and ammonia-based nitrogenous fertilisers, indirectly, feed almost 70% of the global population.3 It is estimated that nearly half of the nitrogen in our body comes from ammonia synthesized in plants.4
In addition to fe rtilizers, ammonia is also a building block for the production of many pharmaceuticals and commercial chemicals, such as nitric acid, hydrazine, cyanides and amino acids.5 It is also commercially used as a refrigerant in large freezing and refrigeration plants because it has high volatility and a high latent heat of vaporisation during phase change from liquid state to gaseous state.6 The development of CFCs (chlorofluorocarbons) in the early 20th century resulted in a decline in the use of ammonia as a refrigerant, but upon the subsequent discovery of the contribution of CFCs to ozone depletion, a global phase-out of these CFCs was called for under the Montreal Protocol in the year 1989. This shifted the focus back to ammonia, as it is highly energy efficient and also poses the advantage of having a zero rating for both Ozone Depletion Potential (ODP) and Global Warming Potential (GWP), apart from being less expensive and more efficient than CFCs.6,7 Another common use of ammonia is as a cleaning agent to remove stains because of its ability to dissolve dirt and grime. It evaporates rapidly after application, not leaving behind any streaks on glass surfaces.8
Lately, ammonia has gained widespread attention due to its ability to act as an efficient hydrogen carrier. Storing hydrogen as a liquid requires a cryogenic temperature of −253 °C, and compressed hydrogen requires vessels capable of withstanding a pressure of 70 MPa at ambient temperature. Storing and transporting hydrogen is also risky due to the high flammability and low ignition energy of hydrogen gas.9 However, ammonia can be liquefied at −33 °C (at atmospheric pressure) or at 0.8–1.0 MPa (at 20 °C), making it much easier to store and transport. Liquid ammonia has a greater volumetric hydrogen density than liquid hydrogen. At −253 °C and 0.1 MPa, liquid hydrogen has a density of 70 kgH2 m−3, whereas liquid ammonia at 27 °C and 1.0 MPa has nearly 106 kgH2 m−3.10 Liquefied ammonia also has a greater energy density (3.83 MW h m−3) than that of liquid hydrogen (2.64 MW h m−3).11 Therefore, ammonia can easily be transported using the current infrastructure of fossil fuels, giving rise to a broader supply chain, where it can be catalytically cracked to produce hydrogen gas at a site of distribution and use.12,13
Due to the above-mentioned reasons, ammonia is being extensively studied for use in internal combustion engines as it seems to be a promising carbon-free fuel. Ammonia has a high octane rating (110–130 versus gasoline at 86–93) and a narrow flammability range, and it can increase the engine's compression ratio, resulting in a higher cycle thermal efficiency.14 However, due to its high ignition energy and low calorific value, it is challenging to directly use pure ammonia as a fuel.15 Therefore, it is often blended with other fuels, which helps combat the problems of low burning velocity and narrow the flammability range of ammonia, thus enhancing its combustion characteristics.16 When pure ammonia is ideally combusted (2NH3 + 3/2O2 → N2 + 3H2O), it undergoes an exothermic reaction releasing N2 and H2O.17 However, it was found that ammonia practically undergoes incomplete combustion to release NOx (nitrogen oxides), which cause even more environmental damage than CO2.15 The NOx emission could be mitigated using selective catalytic reduction and recirculation of exhaust gas in the combustion chamber.18
It was found that ammonia could be used as a fuel in solid oxide fuel cells (SOFCs), where NOx formation could be avoided whilst achieving a higher efficiency than that of internal combustion engines.19,20 SOFCs do not require external cracking of ammonia, and the ammonia decomposition reaction (endothermic) could be combined with the electrochemical reaction (exothermic), eliminating the need for a complex design to increase the thermodynamic efficiency.21 Thus, due to its vast potential to act as an alternative clean fuel, ammonia-fueled ships have become a reality and its applications as a fuel could soon be expanded to the aviation sector by using it to power aircrafts.17,22 Ammonia could therefore be used as an energy source across multiple sectors, replacing conventional fossil fuels in any application. The combustion of this “replacement fossil fuel” produces only nitrogen and water, which could again be reused for ammonia production and hydrogen generation, resulting in a sustainable circular economy. Fig. 1 depicts the concept of a circular ammonia economy, envisioned by MacFarlane et al. in their article.12
 | ||
| Fig. 1 “Ammonia economy”: a model system in which ammonia acts as the central currency for all energy vectors or uses. Adapted with permission from ref. 12. Copyright 2020, Elsevier. | ||
1.1 Thermodynamics
| N2 + 3H2 ⇌ 2NH3; ΔH° = −92 kJ mol−1 | (1) |
 | ||
| Fig. 2 Temperature- and pressure-dependent production of ammonia with the corresponding yields (%). Adapted from the work of Hattori et al.,26 licensed under CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). | ||
To optimize the operating conditions, a detailed mechanistic understanding of catalytically produced ammonia from nitrogen and hydrogen is essential. Based on extensive surface science experiments, it is widely concluded that the dissociative adsorption of N2 is the rate-determining step.27 Interesting, Ertl et al. investigated nitrogen interaction on both Fe(100) and Fe(111) surfaces in a wide temperature region (−133–727 °C) using numerous techniques such as Auger electron spectroscopy, thermal desorption spectroscopy, work function measurements, UV photoelectron spectroscopy, and low-energy electron diffraction and found that nitrogen adsorption is the rate-limiting step.28 Even though numerous experiments have been reported, there is still unclear understanding of the exact mechanistic details for ammonia synthesis. In particular, it is unclear whether N2 dissociates directly over the catalyst surface or on some intermediates. The experimental observations vary with metals and crystal faces and under different conditions.29,30 However, under industrial conditions (i.e., high temperature and pressure), such difference in mechanism is most notable.
In one of the previous works of our research group, the expression for the rate-determining step (RDS) for the synthesis of ammonia was determined by fitting the modelled rate equations to observed reaction rates.31 The rate equations were generated assuming the working mechanism as the Langmuir–Hinshelwood mechanism.32–34 The following sequence of elementary steps is involved in the overall reaction:
| H2(g) → H2(ads) | (2) |
| N2(g) → N2(ads) | (3) |
| H2(ads) → 2H(ads) | (4) |
| N2(ads) → 2N(ads) | (5) |
| N(ads) + H(ads) → NH(ads) | (6) |
| NH(ads) + H(ads) → NH2(ads) | (7) |
| NH2(ads) + H(ads) → NH3(ads) | (8) |
| NH3(ads) → NH3(g) | (9) |
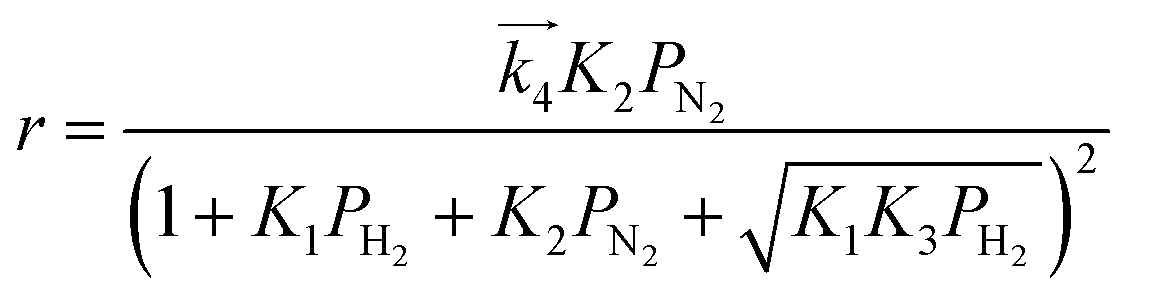 | (10) |
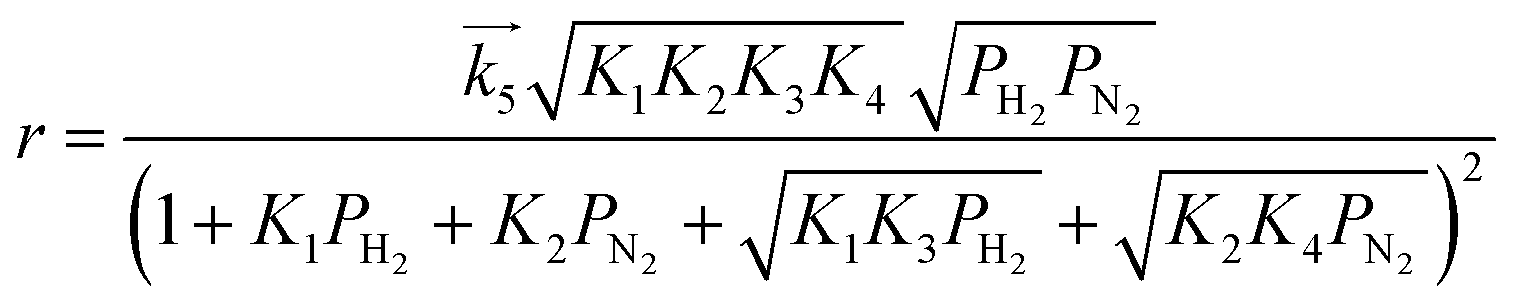 | (11) |
 | (12) |
 | (13) |
 are the rate constants for forward reactions (5)–(8), and Ki is the equilibrium constant in step i. Eqn (10)–(13) are generated assuming that the steps in (5)–(8) are the rate-determining steps (RDSs), respectively. To identify the RDS for ammonia synthesis, the derived equations are fitted into observed experimental rates using a least squares method. Finally, the procedure enables determining which equation best fits and explains the experimental rates.
are the rate constants for forward reactions (5)–(8), and Ki is the equilibrium constant in step i. Eqn (10)–(13) are generated assuming that the steps in (5)–(8) are the rate-determining steps (RDSs), respectively. To identify the RDS for ammonia synthesis, the derived equations are fitted into observed experimental rates using a least squares method. Finally, the procedure enables determining which equation best fits and explains the experimental rates.
1.2 Challenges in industrial ammonia production
Over the past few decades, various catalysts were extensively studied and developed in efforts to optimise the HB process. Fig. 3 outlines the timeline of different types of catalysts developed over the last few decades. | ||
| Fig. 3 Timeline showing the development of various catalysts for ammonia synthesis. | ||
Iron catalyst is used in the present-day HB process, with operating conditions (high temperature) going against Le Chatelier's principle to make commercial production more viable. Operating under such harsh conditions consumes enormous energy, owing to both heating and pressurisation, and despite all of this, the resulting yield is nearly 30–40%.25,26 The low conversion rate of this process calls for an additional recirculation system and a larger reactor, with the unreacted hydrogen and nitrogen being recycled and mixed with the new feedstock, leading to a higher capital and operational cost.38 Therefore, significant efforts have been made in catalyst development for ammonia synthesis over the past few decades, yet ammonia production accounts for about 2% of the global energy consumption.39,40
Alternative methods of ammonia synthesis such as electrochemical and NTP methods, which are discussed in detail in the later part of this paper, may seem as promising alternatives to the HB process as they are completely carbon free and could also be driven by renewable energy sources such as solar and wind energies. They also facilitate the production of ammonia at room temperature and atmospheric pressure, and are also compatible with on-demand, on-site production. However, they are unlikely to achieve the necessary scale and cost to compete with the conventional method as of yet, and hence the HB process is still going to be the dominant method of ammonia production in the foreseeable future.41
Considering the aforementioned challenges, it is obvious that new catalysts that can work under mild conditions in a given ammonia synthesis convertor could address some of the prevalent issues such as higher compressor duty and expensive construction materials for ammonia synthesis process. Over these years, several different convertors have been designed and can be broadly categorized as tube-cooled convertors and quench convertors. In general, the ammonia synthesis loop consists of a catalytic ammonia converter; a compressor to attain the desired operating pressure and to recycle the stream before injection into the converter and a condenser to separate the produced ammonia.
Expectedly, the synthesis loop at lower pressures will remarkably reduce the compressor duty, and lower-pressure-rated design materials can also be employed as construction materials. However, once ammonia is condensed and separated, the synthesis loop pressure is reduced, and therefore, the refrigerator power has to be increased to attain the lower temperatures necessary for separation. To operate at low ammonia synthesis loop pressure (ca. <10.0 MPa) and achieve the required conversion per pass, it has been suggested to place an ammonia absorption reactor consisting of crystalline salts before the condensation step. The absorption of ammonia should be performed at a partial pressure as low as 0.0002 MPa at moderate temperatures.42–44
Similarly, at a lower temperature, the theoretical maximum conversion for ammonia is increased (see Fig. 2). However, convertors operating at relatively low temperatures should be equipped with a better heat removal system, as the ammonia synthesis is an exothermic reaction. Importantly, there has a been significant reduction in operating pressure while maintaining viable condenser temperatures and also a remarkable increase in conversion at a lower temperature has been reported that could have limitation on convertor heat removal efficiency. Therefore, the implementation of active catalysts that exhibit improved activity at lower pressures and temperatures would need modifications in the loop designs. Herein, we summarize the activity of catalysts operating at lower temperatures and pressures in a categorical manner, including conventional thermal catalysts and emerging catalysts for electrochemical and NTP processes.
In the future, ammonia economy is expected to grow in line with the increasing global energy consumption and ammonia demand, especially for fertilizers and shipping fuel. Ammonia production will remain dominated by current HB technology, transitioning to Gen 1 with CO2 sequestration in the next decade. Gen 2, involving green hydrogen, will become commercially viable around 2030, gradually replacing existing methods. Gen 3, fully electrochemical and renewable-powered, will emerge by the end of the decade, initially for ammonia as a maritime fuel followed by its application in energy storage, becoming the preferred technology by the 2040s. Fig. 4 describes the roadmap of ammonia economy.
 | ||
| Fig. 4 Forecasted shares of the current and future ammonia production technologies. Adapted with permission from ref. 12. Copyright 2020, Elsevier. | ||
2. Overview
The rising focus on developing catalysts for ammonia synthesis is evident from the notable rise in the number of publications in this area over the last two decades (Fig. 5). This clearly indicates the increasing realisation of the immense value that ammonia holds, and hence developing a sustainable mode of production of ammonia is of utmost importance to utilise it as a truly “green fuel” or for various other purposes discussed earlier. | ||
| Fig. 5 Plot showing the number of publications per year for articles pertaining to ammonia synthesis. | ||
Recently, several review articles have attempted to provide an overview of catalyst development for ammonia synthesis across different modes of production. Tian et al. delved into the advancements in thermal catalysts for ammonia synthesis, categorising them based on their constituent elements to discuss notable works in each category.45 In another review article, Tian et al. provided a comprehensive understanding of different modes of ammonia production, including photocatalytic and chemical looping methods.46 Gharahshiran et al. provided an in-depth review of the reactor and catalyst development for NTP-assisted ammonia synthesis.47 Mu et al. in their review article discussed important theories behind catalyst design and the underlying reaction mechanisms to address the existing challenges in electrochemical ammonia synthesis.48 Long et al. particularly focused on the evolution of single-atom catalysts for electrochemical nitrate reduction reaction in their detailed work.49
In this review article, we particularly focus on thermal catalysts for low-temperature ammonia synthesis, while also discussing the progress in catalysts for NTP-assisted and electrochemical ammonia synthesis, as we believe they are highly likely to shape the future of ammonia production. Our aim is to provide the reader with a concise summary of recent research pertaining to low-temperature ammonia synthesis. A key aspect of this work is the extensive summary of catalysts for each category, and the holistic review of the dominant modes of ammonia production.
We begin with thermal catalysts, broadly categorised in this work as nitrides, hydrides, intermetallic compounds, electrides and oxides. Under each category, the catalysts are further grouped based on similarities in their composition or structure. We then proceed to discuss the catalyst developments in NTP-assisted ammonia synthesis, specifically for dielectric barrier discharge (DBD) as it is the most commonly used reactor configuration. Progress in electrocatalysts for ammonia synthesis is then discussed separately for the nitrogen reduction reaction and nitrate reduction reaction. We conclude by providing our perspective for the rational catalyst design for future research and offer insights into the outlook of green ammonia synthesis.
3. Thermal catalysts
3.1 Nitride and oxy-nitride-based catalysts
Nitride catalysts are highly efficient for ammonia synthesis, offering excellent catalytic activity and stability under reaction conditions. The presence of nitrogen vacancies facilitates the associative pathway, enabling the activation of stable N2 bond under mild conditions. Additionally, nitride catalysts are prone to oxidation, leading to the formation of oxynitrides during the reaction. Interestingly, this transformation often enhances catalytic activity, as oxynitrides possess a lower work function that promotes efficient N2 dissociation. Another key factor contributing to the high activity of nitride catalysts is the operation of the Mars-van Krevelen mechanism, wherein lattice nitrogen participates in the reaction as an active species, creating a dynamic cycle of nitrogen replenishment that significantly boosts catalytic performance.Till date, both binary nitrides (based on cerium,50 uranium,51 vanadium,52 molybdenum,53 and rhenium54) and ternary nitrides (based on Co–Mo, Ni–Mo, and Fe–Mo55–58) have been extensively examined and the basis of their design can be understood from the volcano-type plot generated from the turn over frequency (TOF) of various ammonia synthesis catalysts, calculated using density functional theory (DFT). Jacobsen et al.59 calculated the TOF of these catalysts through a combination of a microkinetic model for ammonia synthesis and the linear relation between the nitrogen dissociation potential energy and activation energy. The obtained volcano plot of the TOF of various elements against the nitrogen adsorption energy is shown in Fig. 6.
 | ||
| Fig. 6 Calculated TOF (s−1) for ammonia synthesis with variation in nitrogen adsorption energy (400 °C, 5.0 MPa). Adapted with permission from ref. 59. Copyright 2001, the American Chemical Society. | ||
Fig. 6 exhibits a characteristic volcano-shaped profile, indicating the presence of an optimal nitrogen adsorption energy. This behaviour can be explained through DFT calculations conducted by Jacobsen et al., which demonstrate a linear relationship between the activation energy for nitrogen dissociation and the binding energy of atomic nitrogen across various transition metal surfaces, like the Brønsted–Evans–Polanyi (BEP) relation.60 Accordingly, the curve identifies two distinct pathways to achieve high catalytic activity: (1) reducing the activation barrier for nitrogen dissociation, which necessitates strong metal–N interactions, or (2) minimizing the surface coverage of adsorbed atomic nitrogen during NH3 synthesis, which requires weaker metal–N interactions. For instance, the combination of molybdenum (Mo) and cobalt (Co), forming the CoMo system, has demonstrated optimal catalytic activity for ammonia synthesis. Molybdenum exhibits strong nitrogen adsorption, facilitating efficient N2 dissociation. However, this strong adsorption makes the desorption of nitrogen species from the surface a challenge. In contrast, cobalt, with its weaker nitrogen adsorption, promotes easier desorption. The synergy between these two metals results in an optimal nitrogen adsorption energy, significantly enhancing ammonia synthesis activity compared to the performance of each metal individually. This principle has guided the design of various material combinations for ammonia synthesis reactions.
The synthesis of nitride-, nitride-hydride- and oxynitride-based catalysts involves a wide range of methods to achieve specific structural and chemical properties, which enhance the rate of ammonia synthesis. For instance, Ca3CrN3 (ref. 61) was synthesized by a topochemical method, which involves chemical transformation of a solid while preserving the key structural motifs of the parent material. Synthesis using this method allows precise control over the arrangement of atoms in the lattice. In this method, Ca3CrN3 orthorhombic nitride was heated under hydrogen flow at a temperature of 450 °C for 2 h and then thermally treated in a N2/H2/Ar mixture at 400 °C for 24 h. This thermal treatment leads to rotational structural transformation to form a hexagonal nitride hydride, h-Ca3CrN3H.
Zhang et al. observed that Ru/ZrO2–N62 prepared by ammonia-treated ZrO2 (obtained from ZrCl4) exhibited 4 times higher rate than that prepared using zirconium nitrate. Here, ZrO2–O and ZrO2–N were synthesized using a ZrCl4 precursor via a UiO-66 route. The UiO-66 route forms nitrogen-modified ZrO2 (ZrO2–N) post ammonia treatment which enhances Ru dispersion and electron density. This improves N2 activation and boosts ammonia synthesis efficiency. For the preparation of UiO-66, ZrCl4 and H2BDC were dissolved in DMF. This solution was sonicated for 30 minutes followed by heating in an oven at 120 °C for 24 h. Sonication improves precursor dispersion, enhances mixing, and promotes uniform nucleation, which improves the catalyst quality. After sonication at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 minutes, the product was washed with methanol and DMF and finally dried at 90 °C to obtain UiO-66. It was then calcined in an NH3 atmosphere at a temperature of 600 °C for 3 h to form ZrO2. The TEM images for Ru/ZrO2–O and Ru/ZrO2–N are shown in Fig. 7g–j.
000 rpm for 5 minutes, the product was washed with methanol and DMF and finally dried at 90 °C to obtain UiO-66. It was then calcined in an NH3 atmosphere at a temperature of 600 °C for 3 h to form ZrO2. The TEM images for Ru/ZrO2–O and Ru/ZrO2–N are shown in Fig. 7g–j.
 | ||
| Fig. 7 Various synthesis pathways for nitride and oxy-nitride catalysts. SEM images of (a) γ-Mo2N derived from MoO3 powder; (b) nanorods of γ-Mo2N; (c) β-Mo2N0.78 derived from MoO3 powder; and (d) β-Mo2N0.78 derived from MoO3 nanorods prepared via a precursor-driven nitridation method. Adapted with permission from ref. 55. Copyright 2007, Elsevier (e) XRD crystallographs of BaTiO3−x, BaTiO3−xHx, BaTiO3−xNy, and BaTiO2.88 synthesized via a solid-state reaction method. Adapted from the work of Miyazaki et al.,63 licensed under CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). (f) TGA curves showing mass increase% for Ni2Mo3N, Ni1.7Cu0.3Mo3N, Ni1.8Cu0.2Mo3N and Ni1.9Cu0.1Mo3N catalysts synthesized at 900 °C under ammonia (prepared via a citrate gel-assisted ammonolysis method). Adpated from the work of Al Sobhi et al.,64 licensed under CC BY 3.0 (https://creativecommons.org/licenses/by/4.0/). TEM images of (g and h) Ru/ZrO2–O and (i and j) Ru/ZrO2–N prepared via a UiO-66 route. Adapted with permission from ref. 62. Copyright 2022, Elsevier. | ||
The solid-state reaction method used for the synthesis of BaTiO3−xHx63 enables high-temperature-driven phase formation and provides better stoichiometric control. This method is used for the synthesis of complex oxides, nitrides, and hydrogen-substituted materials. BaH2 and TiO2 were heated at 800 °C for a period of 20 h under hydrogen flow to obtain BaTiO3−xHx. It was then heated at 600 °C for 12 h in a nitrogen atmosphere, where it undergoes hydride substitution to obtain BaTiO3−xNy oxy-nitride. Fe, Ru, Co and Ni were loaded on the support by mixing their respective metal precursors with the support in an agate mortar in an Ar environment. The mixture was then treated thermally under hydrogen flow at a temperature of 300 °C or 400 °C for 2 h to obtain the catalysts in their final forms. XRD crystallographs for BaTiO3−x, BaTiO3−xHx, BaTiO3−xNy, and BaTiO2.88 are shown in Fig. 7e.
In another study, ball milling method was used to synthesize Ru/ZrH2, Ru/ZrN, Ru/ZrO2, Ru/TiH2, Ru/TiN, and Ru/TiO2 (ref. 65) catalysts. This method promotes uniform metal dispersion across various supports due to the high-energy impact during milling. ZrH2 along with ruthenium acetylacetone was taken in an agate jar and then ball milled for a period of 2 h at 300 rpm to obtain Ru/ZrH2. For the synthesis of K-, Ba- and Cs-promoted Ru/ZrH2 catalysts, the usual wetness impregnation method was used where nitrate precursors of the respective promoters were dissolved in DI water and then added dropwise to the Ru/ZrH2 followed by heat treatment at a temperature of 400 °C for 2 h under a 10 vol% H2/Ar atmosphere.
Precursor-driven nitridation, utilized for synthesizing Co3Mo3N, Fe3Mo3N, and Ni2Mo3N,55 emphasizes precise precursor preparation followed by high-temperature ammonolysis to form metal nitrides with defined compositions and catalytic properties. This method involves breaking metal–oxygen or metal–halide bonds in the precursor, allowing nitrogen from NH3 to diffuse and replace oxygen, forming metal–nitrogen bonds. This stabilizes the nitride phase, tuning electronic and catalytic properties. Cobalt molybdate hydrate (CoMoO4·nH2O) was prepared by mixing aqueous solutions of Co (NO)3·6H2O and (NH4)6Mo7O24·4H2O at 80 °C followed by vacuum filtration to obtain a purple precipitate. It was then washed with DI water and ethanol and then dried at 150 °C followed by calcination at a temperature of 500 °C. Nitridation of this powder was done in a vertical quartz reactor with 94 mL min−1 NH3 flow. The powder underwent three stages of thermal treatment with the increase in temperature to 357 °C at a ramp rate of 5.6 °C min−1 followed by heating to 447 °C at 0.5 °C min−1 and finally reaching 785 °C at 2.1 °C min−1 with a dwell time of 5 h. The nitrided powder thus obtained was allowed to cool under ammonia flow followed by nitrogen flushing at 100 mL min−1 for purging the reactor. Fig. 7a–d shows the SEM images of γ-Mo2N, nanorods of γ-Mo2N, β-Mo2N0.78, and β-Mo2N0.78 derived from MoO3 nanorods.
The Pechini method is used for the synthesis of highly pure metal oxides using citric acid and ethylene glycol to form a gel. This is followed by heat treatment to get the final product. Certain modifications in optimizing precursors, polymerization and nitridation improve the effectiveness of this method. The modified Pechini method used for bimetallic nitride synthesis results in highly pure nitrides such as Ni2Mo3N66 and CoNiMo3N. NiMoO4 oxide precursors were prepared by dissolving (NH4)6Mo7O24·4H2O and M(NO3)2·6H2O (M = Ni or Co) in aqueous HNO3 and then in citric acid monohydrate to form a solution. The solution was then evaporated at 70 °C under constant stirring till a light green (Ni–Mo system) or wine-red (Co–Ni–Mo system) gel was obtained. This gel was then calcined in air at 500 °C for a period of 2 h which led to the formation of greyish foams as the final oxide precursor. These oxide precursors were then subjected to ammonolysis in a quartz furnace tube at a temperature of 700 °C under ammonia flow. This method led to the production of high-ceramic yield nitride products.
In a similar method, citric acid was used as a chelating agent in the citrate gel-assisted high-temperature ammonolysis method, which led to the formation of nanocrystalline metal nitrides such as Ni1.1Fe0.9Mo3N.64 (NH4)6Mo7O24·4H2O, along with either Cu(NO3)2·4H2O or Fe(NO3)2·6H2O, was dissolved in HNO3, which led to the formation of a green solution. Citric acid monohydrate was added to this solution followed by evaporation at 70 °C to form a gel, which was then thermally treated in an ashing furnace at 500 °C for 2 h. Finally, the furnace tube was purged with N2 and then passivation of the sample surfaces was done by letting air to diffuse slowly to finally obtain a black crystalline product. Fig. 7f displays the TGA curves showing mass increase% for Ni2Mo3N, Ni1.7Cu0.3Mo3N, Ni1.8Cu0.2Mo3N and Ni1.9Cu0.1Mo3N catalysts synthesized at 900 °C under ammonia.
Recently, a number of metal nitride phases have been reported, which exhibit excellent catalytic activity for ammonia synthesis under mild conditions. Often, metal nitrides offer high stability, greater catalytic activity, and high yield, comparing traditional metals (Fe, Ru, and Co)-catalysts.
In addition to nitrides, ternary amides of transition and alkali metals have been reported. For instance, Cao et al. prepared Rb2[Mn(NH2)4] and K2[Mn(NH2)4] via mechanical milling of the respective metals at room temperature under 0.7 MPa of ammonia.23 These catalysts showed better performance than the best performing ternary nitride (i.e., Co3Mo3N). Ammonia synthesis rates of 11 mmol g−1 h−1 and 6 mmol g−1 h−1 were observed for K2[Mn(NH2)4] and Rb2[Mn(NH2)4], respectively, at 400 °C and 1.0 MPa. Notably, under similar conditions, the synthesis rate was merely 5.3 mmol g−1 h−1over the Co3Mo3N catalyst.58 Therefore, ternary amides could be a better choice than nitrides for use as ammonia synthesis catalysts. The reported catalytic activities for nitride-based catalysts are provided in Table 1.
| S. no. | Catalyst | Temp. (°C) | Pressure (MPa) | Rate (mmol g−1 h−1) | Activation energy (kJ mol−1) | Metal % | WHSV | N2 order | H2 order | NH3 order | Surface area (m2 g−1) | Turnover frequency (TOF) | Catalyst synthesis method | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Ca3CrN3H | 400 | 5 | 3.8 | 75 ± 2 | — | 66000 |
1.13 ± 0.02 | 2.45 ± 0.29 | −0.48 ± 0.07 | 1 | 2.1 | Topo-chemical method | 61 |
| 2 | Ru/ZrO2–N | 400 | 1 | 10.26 | — | 5.37% Ru | 36000 |
— | — | — | 53 | 6.1 × 10−3 | Sol–gel, hydrothermal method | 62 |
| 3 | Fe/BaTiO3−xNy | 400 | 0.9 | 8.3 | 41.7 | 1.99% Fe | 36000 |
0.88 | 1.51 | −1.03 | 3 | — | Solid-state reaction method | 63 |
| 4 | Ru/ZrN | 400 | 1 | 2.61 | 108 ± 4 | 1.95% Ru | 60000 |
0.48 | 0.12 | −2.63 | 10 | 3.8 × 10−3 | Ball milling method | 65 |
| 5 | Ru/N–C | 400 | 1 | 1.5* | 79 | 1.34% Ru | 60000 |
0.41 | 0.95 | — | 88 | 2.76 × 10−3 | Solution-based thermal polymerization method | 67 |
| 6 | Co–N2 | 300 | 1 | 2.73 | — | 3.20% Co | 60000 |
0.45 | 0.27 | −0.89 | 230.4 | 1.41 × 10−3 | Template-assisted nitrogen-doped carbonization method | 68 |
| 7 | Co–N3 | 300 | 1 | 2.23 | — | 3.24% Co | 60000 |
— | — | −1.04 | 221.1 | 1.09 × 10−3 | Template-assisted nitrogen-doped carbonization method | 68 |
| 8 | Co–N4 | 300 | 1 | 1.42 | — | 3.66% Co | 60000 |
— | — | −1.44 | 191.3 | 0.76 × 10−3 | Template-assisted nitrogen-doped carbonization method | 68 |
| 9 | γ-Mo2N | 400 | 0.1 | 0.034 | — | — | 9000 | — | — | — | — | — | Controlled ammonolysis nitridation method | 55 |
| 10 | β-Mo2N0.78 | 400 | 0.1 | 0.035 | — | — | 9000 | — | — | — | — | — | In situ fixed-bed nitridation method | 55 |
| 11 | δ-MoN | 400 | 0.1 | 0.004 | — | — | 9000 | — | — | — | — | — | Controlled high-temperature ammonolysis nitridation method | 55 |
| 12 | Co3Mo3N | 400 | 0.1 | 0.167 | — | — | 9000 | — | — | — | — | — | Precursor-driven nitridation method | 55 |
| 13 | Co3Mo3N | 400 | 0.1 | 0.652 | — | — | 9000 | — | — | — | — | — | Nitridation method | 69 |
| 14 | Fe3Mo3N | 400 | 0.1 | 0.095 | — | — | 9000 | — | — | — | — | — | Molybdate nitridation method | 55 |
| 15 | Ni2Mo3N | 400 | 0.1 | 0.029 | — | — | 9000 | — | — | — | — | — | Molybdate nitridation method | 55 |
| 16 | Ni2Mo3N | 400 | 0.1 | 0.395 | — | — | 9000 | — | — | — | — | — | Modified Pechini method- | 57 |
| 17 | Co3Mo3N Cs promoted | 400 | 0.1 | 0.869 | — | — | 9000 | — | — | — | — | — | Promoter-enhanced nitridation method | 69 |
| 18 | Co3Mo3N K promoted | 400 | 0.1 | 0.986 | — | — | 9000 | — | — | — | — | — | Promoter-enhanced nitridation method | 69 |
| 19 | NiCoMo3N | 400 | 0.1 | 0.166 | — | — | 12000 |
— | — | — | — | — | Modified Pechini method | 66 |
| 20 | Ni–LaN | 400 | 0.1 | 5.543 | — | — | 36000 |
— | — | — | — | — | Nitridation method | 70 |
| 21 | Ni1.1Fe0.9Mo3N | 500 | 0.1 | 0.354 | — | — | 5200 | — | — | — | — | — | Citrate gel-assisted ammonolysis method | 64 |
| 22 | CeN NPs | 400 | 0.9 | 1.450 | — | — | 36000 |
— | — | — | — | — | Hydride-based nitridation | 71 |
| 23 | K2[Mn(NH2)4] | 400 | 1 | 11.141 | — | — | 6000 | — | — | — | — | — | Mechanochemical ammoniation method | 23 |
| 24 | γ-Mo2N | 400 | 10 | 30 (mL g−1 h−1) | — | — | — | — | — | — | — | — | Controlled ammonolysis method | 58 |
| 25 | Co3Mo3N | 400 | 10 | 120 (mL g−1 h−1) | — | — | — | — | — | — | — | — | Controlled ammonolysis method | 58 |
| 26 | Fe3Mo3N | 400 | 10 | 90 (mL g−1 h−1) | — | — | — | — | — | — | — | — | Controlled ammonolysis method | 58 |
| 27 | Ni2Mo3N | 400 | 10 | 80 (mL g−1 h−1) | — | — | — | — | — | — | — | — | Controlled ammonolysis method | 58 |
| 29 | Co3Mo3N (5% Cs) | 400 | 10 | 1040 (mL g−1 h−1) | — | — | — | — | — | — | — | — | Controlled ammonolysis method | 58 |
| 29 | Fe3Mo3N (5% Cs) | 400 | 10 | 440 (mL g−1 h−1) | — | — | — | — | — | — | — | — | Controlled ammonolysis method | 58 |
| 30 | Ni2Mo3N (5% Cs) | 400 | 10 | 530 (mL g−1 h−1) | — | — | — | — | — | — | — | — | Controlled ammonolysis method | 58 |
Because of the low binding energy for N2, Ni metal has illustrated poor performance towards ammonia synthesis, even if the metal was used with excellent supports such as C12A7:e− and MgO. Based on the volcano plot (Fig. 6), our group has demonstrated ammonia synthesis reaction using bi-metal catalysts such as Co/Fe, Ru/Fe, and Ni/Fe.31 All the tested catalysts have shown different trends for nitrogen dissociation and ammonia synthesis activity. In comparison to Ru/Fe and Co/Fe, the catalytic activity of Ni/Fe was very poor even under high-temperature reaction conditions, which is in line with the Jacobsen volcano plot for transition metals.59 Ru/Fe and Co/Fe catalysts have shown NHx formation as the rate-determining step while for Ni/Fe, the rate-determining step was N2 dissociation. This observation clearly indicates that both Ru/Fe and Co/Fe catalysts can efficiently break N2 molecules, whereas the Ni/Fe catalyst has difficulty to break it. On the contrary, Ye et al. synthesized a Ni–LaN catalyst and found an excellent rate of 5.543 mmol g−1 h−1 at 400 °C and 0.1 MPa.70 In this exemplary work, surface nitrogen vacancies present in the LaN catalyst facilitate the weakening of the N triple bond and the hydrogen dissociation over the Ni surface. In a similar work by the same group, Ni–CeN was used as a novel catalyst and displayed an activity of 6.5 mmol g−1 h−1 at 400 °C and 0.9 MPa.71 Under these conditions, the effluent NH3 concentration was equal to the thermodynamic equilibrium value (% effluent concentration NH3 = 0.45 vol%). In addition to CeN, other nitrides such as YN and LaN were investigated. The catalytic performance and reaction kinetics of all the catalysts used in this work are summarised in Fig. 8.
 | ||
| Fig. 8 (a) Catalytic performance of Ni-loaded nitride catalysts at 400°Cand 0.1 MPa (b) Arrhenius plots of Ni/CeN, Ni/LaN bulk and nanoparticles (NPs) at 0.1 MPa. (c) Temperature-dependence of ammonia synthesis activity over NPs and bulk particles of Ni/CeN at 0.1 and 0.9 MPa (d) time course of ammonia synthesis over Ni/CeN NPs at 340 °C and different pressures (0.1 and 0.9 MPa). Other reaction conditions: 0.1 g catalyst, N2:H2 = 15:45 mL min−1, 150–400 °C. Adapted with permission from ref. 71. Copyright 2020, the American Chemical Society. | ||
In this work, ammonia was produced from the reaction between lattice N in the CeN (validated by in situ experiment with 15N2 and H2 using the CeN catalyst) and adsorbed hydrogen. Hence, nitrogen vacancy containing nitride can activate the stable N2 bond easily under low-temperature and low-pressure reaction conditions. Such works successfully demonstrate the possible application of Ni for ammonia synthesis, as more expensive Ru catalysts have.
In an interesting article by Zhou et al., both associative and dissociative routes were utilized for enhancing the catalytic activity of Ru for the generation of ammonia under mild conditions.72 In particular, the authors ball milled desired amounts of ZrH2, TiCN, and ruthenium acetylacetone to synthesize TiCN-promoted Ru/ZrH2. The as-developed catalyst demonstrates a rate of 25.6 mmol g−1 h−1 at 400 °C and 1.0 MPa. This study disclosed that TiCN donates electrons to Ru clusters, which not only readily dissociate the N2 bond via the dissociative mechanism but also prohibit Ru NPs from sintering. Additionally, the nitrogen vacancies on TiCN favor the N2 hydrogenation to NNH via the associative pathway. Both the associative and dissociative pathways are depicted in Fig. 9a and b.
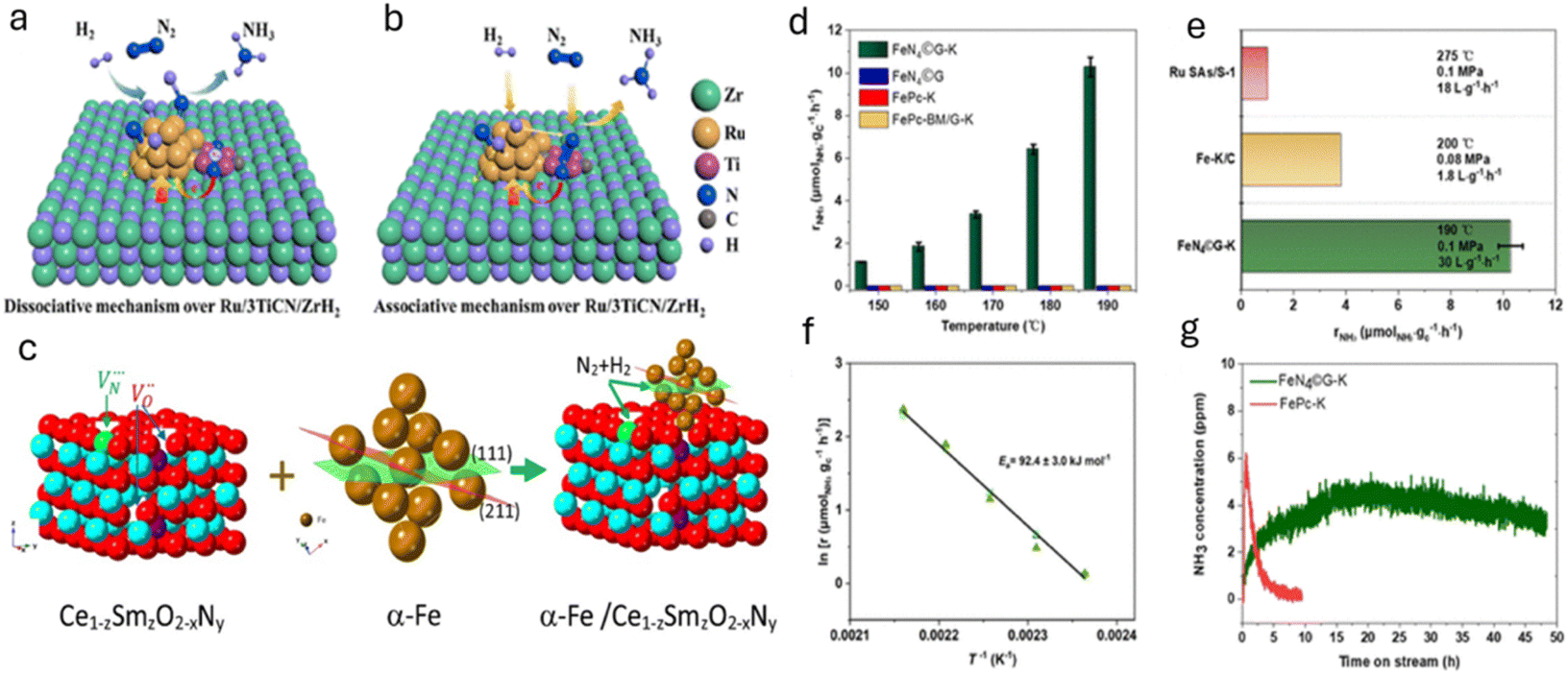 | ||
| Fig. 9 (a) Dissociative and (b) associative mechanisms of NH3 synthesis on Ru/3TiCN/ZrH2. Atoms are coloured as follows: Ru (gold), Zr (green), Ti (purple), N (dark blue), C (grey), and H (light blue). Adapted with permission from ref. 72. Copyright 2022, the American Chemical Society. (c) Illustration showing the interaction of anion vacancies with α-Fe particles and reactants H2 and N2. The (111) plane in α-Fe is shown in green, and the (211) plane in red. Adapted with permission from ref. 73. Copyright 2021, Elsevier. (d) Ammonia production rates for FeN4©G-K, FeN4©G, FePc-K, and FePc-BM/G-K at different temperatures at 0.1 MPa. (e) Comparison of FeN4©G-K ammonia production rate with Fe-K/C and Ru SAs/S-1 under similar conditions. (f) Arrhenius plot showing the ammonia synthesis activity of FeN4©G-K from 150 to 190 °C at 0.1 MPa. (g) Stability test of FeN4©G-K versus FePc-K at 170 °C and 0.1 MPa. Reaction conditions: N2/H2 = 1/3, WHSV = 30 L gcat−1 h−1, unless otherwise noted. Adapted with permission from ref. 74. Copyright 2023, the American Chemical Society. | ||
Since nitrides offer excellent properties as ammonia synthesis catalysts, the stability of these nitrides has to be evaluated in the presence of oxygenates such as O2, CO, and H2O at high temperatures. As is known, at high temperatures, nitrides are prone to oxidation and can transform to oxynitrides or oxides in the presence of oxygenates.75 Recently, Prof. Hosono and his group have remarkably enhanced the performance of Ru by using barium oxynitride as a promoter. In this work, an electride (BaOxNy:ez−) was used, which increased the ammonia synthesis activity by 40–100 times, comparing the conventional BaO promoter at 400 °C and 0.1 MPa.23 Under this condition, the ammonia formation rate of Ru supported with BaOxNy was 2.05 mmol g−1 h−1.
To assess the high catalytic activity for the production of ammonia, barium oxynitride was generated by the solid-state reaction of Ba2N and BaO. Specifically, a mixture consisting of a definite molar ratio of Ba2N and BaO was sealed in Ar-filled stainless-steel tubes and were heated at 750 °C for 24 h. This results in the formation of dark-brown BaOxNy, and Ru nanoparticles (precursor: trirutheniumdodecacarbonyl, Ru3(CO)12, 99%, Sigma-Aldrich) were loaded onto this support via a chemical vapor deposition method. It is reported that the inclusion of N3− into the O2− site of BaOxNy:ez− led to an increase in the concentration of low-work-function electrons in the lattice sites, which strengthen the electron donating ability of the BaOxNy:ez− catalyst.
In the same line, Humphreys et al. reported the use of oxynitrides as promoters for the low-cost Fe catalyst for ammonia synthesis.73 The authors introduced a large amount of anion vacancies in CeO2 by incorporating cations (Sm3+ ions) and anions (N3− ions), which significantly increased the nitrogen vacancies. In particular, 80 wt% Fe-20 wt% Ce1−zSmzO2−xNy (z ≤ 0.5) was synthesized using a facile one-step combustion synthesis method and has demonstrated the highest ammonia synthesis rate of 18.8 mmol g−1 h−1 at a temperature of 400 °C and a pressure of 3.0 MPa. It is deduced that, during the reaction and high concentration of hydrogen, the cerium oxynitride could resemble BaCeO3−xNyHz, suggesting the formation of hydride intermediates. The apparent activation energy of the as-prepared catalyst with z ≥ 0.3 was 45 kJ mol−1, the lowest activation energy for all reported ammonia synthesis catalysts. It is believed that the higher catalytic activity is directly related to the larger lattice volume of the support that allows large mobility of the N3−/H− ions. Here, the reaction proceeds through the Mars-van Krevelen mechanism mediated by the anion vacancies. The high concentration of anion vacancies in Ce1−zSmzO2−xNy will facilitate the nesting/anchoring of Fe particles, resulting in strong metal support interaction (SMSI). This SMSI can prohibit the sintering of Fe particles even under a strong oxidising environment. Therefore, a large amount of anion vacancies in cation-doped cerium oxynitride improved both the stability and catalytic activity for ammonia synthesis. High N3− ion conductivity can expand the ammonia synthesis reaction zone by allowing N3− ions to form anywhere on the surface and quickly move through oxynitride particles to active sites. These sites, where α-Fe contacts Ce1−zSmzO2−xNy, complete the reaction, boosting the catalytic activity. This is depicted in Fig. 9c.
The development of heterogeneous catalysts by replicating the nitrogenase structure has attracted wide attention, as it can enable nitrogen fixation under mild conditions. Hence, nanomaterials with enzyme-like characteristics have been recently used for ammonia production. For instance, Zhang et al. performed hydrogenation of a thiolate-bridged FeIVFeIVμ-nitrido complex at 2.0 MPa and room temperature to generate ammonia.76 Similarly, Chen et al. reported the catalytic performance of an iron-single-site catalyst (FeN4©G-K) confined within the graphene matrix, synthesized by milling iron phthalocyanine with graphene for 2 h at 450 rpm in ethanol and promoted by alkali metals such as K.74 Herein, an activity of 1.1μmol g−1 h−1 at 150 °C and 10.3 μmol g−1 h−1 at 190 °C was observed using the as-prepared catalyst. The reaction intermediates such as  were identified by TOF-MS, which clearly indicates that ammonia synthesis process follows an associative hydrogenation mechanism. Moreover, this study also finds that alkali metal, K, significantly reduces the hydrogenation barriers of
were identified by TOF-MS, which clearly indicates that ammonia synthesis process follows an associative hydrogenation mechanism. Moreover, this study also finds that alkali metal, K, significantly reduces the hydrogenation barriers of  and, therefore, accelerates the reaction rate.
and, therefore, accelerates the reaction rate.
The catalytic performance of FeN4©G-K for ammonia synthesis was analyzed with a gas mixture of 25% N2–75% H2 at a weight hourly space velocity (WHSV) of 30 L gcat−1 h−1. The results show FeN4©G-K to be active at atmospheric pressure in the temperature range of 150–190 °C, with the NH3 formation rate increasing from 1.1 μmol NH3 gc−1 h−1 at 150 °C to 10.3 μmol NH3 gcat−1 h−1 at 190 °C. This contrasts sharply with FePc-K and FePc-BM/G-K, which showed no activity despite possessing FeN4 sites and K promotion. This difference highlights the importance of the FeN4 sites embedded in a graphene lattice and the role of K in promoting the reaction. Compared with previous catalysts, FeN4©G-K demonstrates superior performance; its NH3 formation rate at 0.1 MPa and 190 °C is about 2.5 times higher than that of Fe–K/C and significantly outperforms Ru SAs/S-1. This improvement is attributed to the unique structure of FeN4©G-K and its higher Fe loading. Additionally, the catalyst exhibits a relatively low activation energy (92.4 kJ mol−1), indicating efficient N2 dissociation. Stability tests reveal FeN4©G-K to remain active over 48 hours at 170 °C and 0.1 MPa, while FePc-K quickly deactivates, likely due to hydrogenation of FeN4 sites. This distinction underscores the stability and effectiveness of FeN4©G-K, making it a promising catalyst for ammonia synthesis (Fig. 9d–g).
Nitride catalysts therefore present several benefits such as the operation of the Mars-van Krevelen mechanism and the ability to form oxynitride phases, which play a pivotal role in enhancing the ammonia synthesis rate. To fully exploit these advantages, future efforts could focus on controlling oxynitride formation and optimizing nitrogen vacancy concentrations, thereby maximizing the synergy between these mechanisms and further enhancing catalyst efficiency for ammonia synthesis.
3.2 Hydride-based catalysts
As discussed earlier, the dissociation of strong nitrogen bonds is one of the difficult steps for the ammonia synthesis, and conventional catalysts produce ammonia through a relatively energy-expensive pathway. Recently, hydrides have emerged as potential candidates to break nitrogen bonds under mild conditions, and found to dissociate nitrogen bonds in two ways: (i) by reacting hydrogen with an adsorbed N-atom from the transition metal to form ammonia, (ii) by splitting hydrogen into electrons and protons; where, electrons transfer to the anti-bonding π-orbital of nitrogen molecules and readily break the nitrogen bonds (see Tables 2–4 for detailed summary of hydride-based catalysts).| S. no. | Catalyst | Temp. (°C) | Pressure (MPa) | Rate (mmol g−1 h−1) | Activation energy (kJ mol−1) | Metal% | WHSV | N2 order | H2 order | NH3 order | Surface area (m2 g−1) | Turnover frequency (TOF) | Catalyst synthesis method | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1Ru/ZrH2 | 400 | 0.1 | 0.056 | 72.5 | 1% Ru | — | 0.79 | 0.74 | −0.65 | — | — | Dispersion and deposition method | 77 |
| 2 | 7Ru/ZrH2 | 400 | 0.1 | 0.574 | 70.3 | 7% Ru | — | 1.63 | 0.98 | −1.70 | 0.51 | — | Dispersion and deposition method | 77 |
| 3 | 7Ru–0.2K/ZrH2 | 400 | 0.1 | 0.698 | 64.5 | 7% Ru | — | 0.75 | 0.31 | −0.26 | — | — | Dispersion and deposition method | 77 |
| 4 | Ru/ZrH2 | 400 | 1 | 8.98 | 70 ± 1 | 1.93% Ru | 60000 |
0.51 | 0.04 | −1.50 | 4 | 13.1 × 10−3 | Ball milling method | 65 |
| 5 | Ru/ZrH2 | 400 | 1 | 11.6 | 97 | 1.70% Ru | 60000 |
— | — | — | 2.0 | 0.019 | Ball milling method | 72 |
| 6 | VH0.39 | 400 | 5 | 3.2 | 118 ± 6 | — | 66000 |
0.39 ± 0.04 | 1.04 ± 0.06 | −1.2 ± 0.2 | — | — | Ball milling method | 78 |
| S. no. | Catalyst | Temp. (°C) | Pressure (MPa) | Rate (mmol g−1 h−1) | Activation energy (kJ mol−1) | Metal% | WHSV | N2 order | H2 order | NH3 order | Surface area (m2 g−1) | Turnover frequency (TOF) | Catalyst synthesis method | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Ca2RuH6/MgO | 300 | 0.1 | 8.6 | 75.4 ± 2.4 | 9% | 60000 |
0.94 | 0.26 | −0.57 | — | 8.0 × 10−3 | Ball milling-calcination method | 79 |
| 2 | Na4RuH6/MgO | 300 | 0.1 | 8.3 | 118.8 ± 1.7 | 7.6% | 60000 |
0.92 | −0.32 | −0.29 | — | 7.9 × 10−3 | Ball milling-calcination method | 79 |
| 3 | K3RuH7/MgO | 300 | 0.1 | 15.7 | 139.7 ± 6.8 | 8.2% | 60000 |
1.17 | −0.53 | −0.36 | — | 13.7 × 10−3 | Ball milling-calcination method | 79 |
| 4 | Li4RuH6/MgO | 300 | 0.1 | 9.1 | 71.2 | 8.0% | 60000 |
0.91 | 0.30 | −0.59 | — | 8.6 × 10−3 | Ball milling-calcination method | 79 |
| 5 | Ba2RuH6/MgO | 300 | 0.1 | 13.8 | 63.9 | 5.0% | 60000 |
0.92 | 1.00 | −0.63 | — | 24.2 × 10−3 | Ball milling-calcination method | 79 |
| 6 | Li4RuH6/MgO | 225 | 0.1 | — | 102.8 ± 3.3 | 8% Ru | 60000 |
0.82 ± 0.02 | −0.47 ± 0.01 | −0.18 ± 0.02 | — | Ball milling-calcination method | 79 | |
| 7 | Li4RuH6/MgO | 300 | 0.1 | — | 71.2 ± 1.5 | 8% Ru | 60000 |
0.91 ± 0.03 | 0.30 ± 0.03 | −0.59 ± 0.01 | — | 6 × 10−3 | Ball milling-calcination method | 79 |
| 8 | LiH/Co–Mg–O | 300 | 1 | 19 | 55.8 ± 2.2 | 21.3% (Co), 17.2% (Li) | 60000 |
0.76 ± 0.06 | 1.19 ± 0.06 | 0.52 ± 0.02 | 96 | — | Ball milling method | 79 |
| 9 | LiH–Ir/MgO | 400 | 1 | 0.99 | 53.4 ± 2.6 | 12.9% Ir | 60000 |
1.27 ± 0.06 | −0.36 ± 0.02 | −0.65 ± 0.02 | — | — | Ball milling method | 80 |
| 10 | Ba2RuH6/MgO | 200 | 0.1 | 0.5 | 90.3 ± 2.1 | 5% Ru | 60000 |
0.92 ± 0.01 | −0.49 ± 0.02 | −0.39 ± 0.01 | — | 1 × 10−3 | Impregnation–reduction method | 81 |
| 11 | Ba2RuH6/MgO | 300 | 0.1 | 12.6 | 63.9 ± 4.5 | 5% Ru | 60000 |
0.83 ± 0.03 | 1.00 ± 0.01 | −0.63 ± 0.02 | — | 0.017 | Impregnation–reduction method | 81 |
| S. no. | Catalyst | Temp. (°C) | Pressure (MPa) | Rate (mmol g−1 h−1) | Activation energy (kJ mol−1) | Metal% | WHSV | N2 order | H2 order | NH3 order | Surface area (m2 g−1) | Turnover frequency (TOF) | Catalyst synthesis method | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | BaH2–BaO/Fe/CaH2 | 300 | 0.9 | 5.5 | 40 ± 5 | — | 36000 |
1.0 | 1.5 | −1.1 | 7 | 12.2 | Impregnation–reduction method | 26 |
| 2 | Ru/BaH2–BaO | 300 | 0.9 | 16.5 | 79 ± 5 | 10% | 36000 |
1.4 | −1.6 | −1.7 | 20 | 3.5 × 10−3 | Impregnation–reduction method | 26 |
| 3 | BaH2/Ni–MgO | 300 | 1 | 1.76 | 87 ± 6 | 20.5% (Ni) | 60000 |
0.79 ± 0.04 | −0.98 ± 0.09 | 0.83 ± 0.03 | 45.3 | — | Co-precipitation–impregnation method | 82 |
| 4 | Ru/Ba(OH)2–TiH2 (10% Ba(OH)2) | 400 | 0.1 | 3.05 | — | 1% Ru | 24000 |
0.79 | 0.15 | −0.36 | 1.65–1.91 | 6 × 10−2 | Wetness impregnation method | 83 |
| 5 | Fe/BaTiO3−xHx | 400 | 0.9 | 4.8 | 42.3 | 1.83% Fe | 36000 |
— | — | — | 2.6 | — | Solid-state reaction method | 63 |
| 6 | BaH2–BaO/Fe | 300 | 0.9 | 5.5 | 40 | — | 36000 |
0.77 | 0.47 | −1.13 | 7 | 12.2 | Impregnation–reduction method | 84 |
| 7 | Ru/BaO–BaH2 | 300 | 0.9 | 16.5 | 69 | 10% Ru | 36000 |
0.70 | −0.79 | −1.65 | 20 | 3.5 × 10−3 | Reduction–deposition method | 84 |
The unique ability of these catalysts to exhibit exceptional ammonia synthesis activity under low-temperature conditions, where the rate-determining step shifts from N2 dissociation to NHx formation, makes them highly promising for efficient ammonia production.
The process of synthesizing hydride-based catalysts for ammonia synthesis entails the preparation of metal hydrides that facilitate catalytic activity through rapid hydrogen release. Generally, preparation of these catalysts is carried out by reducing metals in hydrogen-rich atmospheres. Through careful engineering, the preparation has been optimized to favor metal dispersion for hydrogen storage and electronic characteristics of the catalyst. Because of their high reactivity, hydride-based catalysts can make efficient use of hydrogen in ammonia synthesis, thus posing an important avenue for improving catalytic processes in ammonia production.
Vacuum-assisted metal deposition has been employed in Ru-based catalyst deposition on TiMn2, ZrH2,77 MgO, and carbon substrates. Here Ru atoms diffuse on the substrate surface and then transit to stable sites. This is followed by their nucleation into clusters and then adhesion via chemical bonding, van der Waals forces or mechanical interlocking, which ensures better dispersion and strong attachment. TiMn2 and ZrH2 powders were prepared by hand milling in an alumina mortar and pestle. Ruthenium(III) acetylacetonate (Ru(acac)3) along with TiMn2 and ZrH2 powder, was dispersed in tetrahydrofuran (THF) and then the resulting solution was evaporated at a high temperature with constant stirring under vacuum. In this technique, evaporation under vacuum pressure ensures that the metal loading and dispersion are completely uniform. This is particularly beneficial in hydrogenation reactions because it allows modulation in the electron-donating and hydrogen-storing abilities, which, in turn, helps to boost the activity of the catalyst.
Ball-milling synthesis is another more widely used method applied for Ru/ZrH2,72 Ru/ZrN, Ru/ZrO2, Ru/TiH2, Ru/TiN, and Ru/TiO2 as catalysts. Here, high-energy mechanical impacts ensure uniform distribution of metals with the stability and reactivity of the catalyst. By impregnating with alkali metal promoters K, Ba, and Cs, the catalytic properties are subsequently modified by changing electronic interactions and adsorption energies in Ru/ZrH2 systems. ZrO2 was prepared by dissolving a Zr(NO3)2 precursor in DI water followed by the addition of a NaOH solution to tune the pH to 9.5. After continuing the stirring for 1 h, the resulting sediment was washed with DI water till the pH turned 7. Thereafter, it was dried at 60 °C for a period of 12 h and lastly calcined at 550 °C for 4 h to form a ZrO2 support. Ruthenium was then added to these supports by a ball-milling method in which support (ZrH2/ZrO2) along with ruthenium acetylacetone was taken in an agate jar and ball-milled at 300 rpm for a 2 h period. For the synthesis of promoted Ru/ZrH2 catalyst, nitrate precursors of K, Ba and Cs were dissolved in DI water and then this solution was added dropwise to the ZrH2 support followed by drying using an infrared lamp. At last, these powders were treated thermally at a temperature of 400 °C for 2 h in a 10 vol% H2/Ar atmosphere to obtain the promoted Ru/ZrH2 catalysts.
Similarly, several routes for the synthesis of metal hydride catalysts with oxide support catalysts are developed for specific properties of the material. Examples include calcination of the ball-milled mixture of LiH (or LiD) or BaH2 with Ru powder in H2 (or D2) at elevated temperatures and pressures to give Li4RuH6 and Ba2RuH6 (ref. 79) samples. Ball milling ensures fine mixing, and calcination at high temperatures promotes the diffusion of hydrogen for the formation of desired hydrides through solid-state reactions. For this, hydrides of Li, Na, K, Ba and Ca were milled with Ru powder at a high temperature (700 °C) and pressure (50 bar). However, under these conditions, only partial synthesis of Na4RuH6 and K3RuH7 is possible and some amount of NaH/KH and Ru remain unreacted.
In another method of preparation of Li4RuH6/MgO catalysts, reduced Ru/MgO was impregnated in a lithium–ammonia solution (Li:Ru ≈ 4:1), followed by hydrogenation at 300 °C to form Li4RuH6. In this method, ammonia stabilizes metal ions ensuring uniform precursor distribution, while metallic Li or Ba reacts with ammonia to form metal amides. Subsequent hydrogenation at 300 °C facilitates the conversion of these intermediates into hydride compounds (Li4RuH6 or Ba2RuH6) on the MgO support. The Ba2RuH6/MgO81 catalyst was prepared similarly by an ammonia-mediated impregnation method. Using ammonia helps in stabilizing metal ions and controls precursor distribution. A barium–ammonia solution (molar ratio: Ba/Ru = 4/1) was used for the impregnation of Ru/MgO, where metallic Ba was converted into barium amide (Ba(NH2)2) in the presence of Ru. Then excess of ammonia was removed followed by hydrogenation at a temperature of 300 °C, which led to the formation of Ba2RuH6 on MgO. Fig. 10 shows the rate of ammonia synthesis for Ca2RuH6-based catalysts at 300 °C and apparent activation along with STEM and elemental mapping images of the Ca2RuH6/MgO hydride catalyst. It also shows the FT-IR spectra of Ca2Ru(H/D)6, Ca2Ru(H/D)6/MgO, and post-reaction Ca2Ru(H/D)6/MgO catalysts (reaction conditions: N2/(H2/D2) = 1:3, 20 bar, 400 °C).
 | ||
| Fig. 10 Various synthesis pathways for hydride and oxy-hydride catalysts. (a) Rate of ammonia synthesis for Ca2RuH6-based catalysts prepared via an ammonia-mediated impregnation method and corresponding activation energies. (b) STEM and elemental mapping images of the Ca2RuH6/MgO hydride catalyst. (c) FT-IR spectra of Ca2Ru(H/D)6, Ca2Ru(H/D)6/MgO, and post-reaction Ca2Ru(H/D)6/MgO catalysts (reaction conditions: N2/(H2/D2) = 1:3, 20 bar, 400 °C). Adapted with permission from ref. 81. Copyright 2022, the American Chemical Society. | ||
A LiH–Ir80 composite was synthesized by a ball milling method by reacting IrCl3 with LiH at a molar ratio of 18/1, followed by ball-milling at 200 rpm for 3 h, and washing with THF to remove LiCl, resulting in a composite with a molar ratio of 15/1. The BaO–Ru/MgO catalyst was prepared by impregnating Ba(NO3)2 on Ru/MgO, followed by reduction at 400 °C. All these methods guarantee controlled incorporation of Ba and dispersion of Ru with great significance regarding catalytic activity.
BaH2–BaO/Fe/CaH2 (ref. 26)-based oxyhydride was formed using the impregnation–reduction method for uniform depositions of transition metal nanoparticles on the BaH2–BaO phase. The reduction step is essential to convert precursors into active metal nanoparticles. Fe (NO3)3·9H2O was dissolved in DI water followed by evaporation at 70 °C. The resulting red solid was heated at a temperature of 300 °C for a period of 10 h to obtain α-Fe2O3. BaH2–BaO/Fe/CaH2 was prepared by dissolving α-Fe2O3 and Ba (NO3)2 in DI water followed by evaporation. Ba (NO3)2-impregnated α-Fe2O3 thus formed was heated to 300 °C for a 10 h period in air. At last, it was mixed with CaH2 in an alumina mortar to obtain the final catalyst.
In co-precipitation of Ni–Mg–O83 solid solutions, Ni and Mg precursors with oxalic acid were mixed homogeneously followed by controlled aging and filtration, drying, and reduction in hydrogen, facilitating uniform metal dispersion and catalytic stabilization. Oxalic acid and magnesium acetate were dissolved in DI water to prepare their solutions. The magnesium acetate solution was then added dropwise to the oxalic acid solution and allowed to age at 30 °C for 10 h. The solid thus obtained was dried for 10 h at a temperature of 100 °C. Finally, it was reduced at 420 °C for 5 h in a hydrogen atmosphere in a Ni–Mg–O solid solution.
In the case of BaH2/Ni–Mg–O,82 BaH2 was dispersed well on the Ni–Mg–O support through modification of Ba ammonium solution, and this was followed by hydrogenation under high-temperature and -pressure conditions, which further optimized catalytic activity in the second step. In another work, BaTiO3−xNy63 was prepared by a solid-state reaction method, which involved high-temperature hydrogen treatment (800 °C), hydride substitution at 600 °C, and oxidation at 900 °C essential to get the desired composition of oxyhydride.
Catalytic materials frequently involve some transition metals, which have strong nitrogen dissociation energy. This implies that transition metals can easily break the nitrogen molecules but have challenges in desorbing them from the surface. Hence, most of the active sites are unavailable for the subsequent nitrogen activation, which severely impedes the ammonia formation rate. To address this issue, Wang et al. have recently provided a potential solution to the long-standing scaling relationship between the apparent activation energy and the nitrogen binding energy.85
They mixed LiH to early and late 3d transition metals (TM) such as Mn, Fe, Co, V, Cr and Ni to offer another catalytic site for nitrogen molecules. In particular, negatively charged hydrogen atoms of LiH reduces activated nitrogen atoms from the transition metal or its nitride, and combines with nitrogen atoms to synthesize lithium amide. Subsequently, the lithium amide splits into H2 to release NH3 and regenerates LiH. The following are the reactions involved:
| N2 + 2* → 2N* | (14) |
| N* + LiH → * + [LiNH] | (15) |
| [LiNH] + H2 → LiH + NH3 | (16) |
Moreover, at a lower temperature, the rate-determining step shifts from nitrogen dissociation to the hydrogenation of surface-adsorbed nitrogen atoms. At 300 °C and under 1.0 MPa, the activities were at least four (Cr–, Mn– and Co–LiH), three (V–LiH), two (Ni–LiH), and one (Fe–LiH) orders of magnitude greater than that of the pristine TM. The reported activity for Fe–LiH and Co–LiH were ca. 11 mmol g−1 h−1, at 350 °C and 1.0 MPa pressure. Importantly, the apparent activation energies for these selected catalysts were close to 50–60 kJ mol−1. In the same line, Yan et al. synthesized MgO-supported LiH–Ir catalyst, which exhibits an ammonia production rate of 7.7 mmol g−1 h−1 at 400 °C and 1.0 MPa. This rate was ca. 100-fold higher comparing the bulk LiH–Ir composite and the MgO-supported Ir metal catalyst. The study disclosed that the in situ formation of Li–Ir complex hydride species led to the activation and hydrogenation of nitrogen to ammonia.86
Inoue et al. reported ammonia synthesis over flat-shaped Ru NPs decorated on calcium amide, Ca(NH2)2.87 The catalyst was able to fix N2 to ammonia at a temperature as low as 200 °C under 0.1 MPa. The ammonia production using Ca(NH2)2 was primarily via its decomposition reaction (Ca (NH2)2 → CaNH + NH3). However, 10 wt% Ru/Ca(NH2)2 acted as a stable catalyst and produced ca. 37.8 mmol ammonia within 45 h, at 340 °C and 0.1 MPa. The rate significantly reached to a maximum of 9.6 mmol g−1 h−1 at 350 °C. Such high activity was attributed to the high electron promotion effect of Ca(NH2)2. While Ca(NH2)2 alone shows a rapid decline in ammonia production over time (Fig. 11a), Ru/Ca(NH2)2 remains highly efficient, with a substantial increase in ammonia production compared to the decomposition of Ca(NH2)2. The catalytic activity of Ru/Ca(NH2)2 is shown to be superior to that of Cs–Ru/MgO across the entire temperature range, with ammonia production starting as low as 473 K (200 °C), whereas Cs–Ru/MgO shows no detectable ammonia production below 523 K (250 °C) (Fig. 11b). The time-course of ammonia synthesis from 15N2 and H2 at 473 K (200 °C) further confirms that ammonia is synthesized directly from the gas phase, and the signals for 15NH3 and 15NH2 increase over time, providing clear evidence of N2 dissociation and NH bond formation (Fig. 11c). The ammonia synthesis rate over Ru/Ca(NH2)2 is shown to increase with reaction pressure, with the catalyst remaining unaffected by hydrogen poisoning, unlike Cs–Ru/MgO. These findings demonstrate that Ru/Ca(NH2)2 offers a highly efficient, stable, and low-temperature alternative for ammonia synthesis (Fig. 11d).
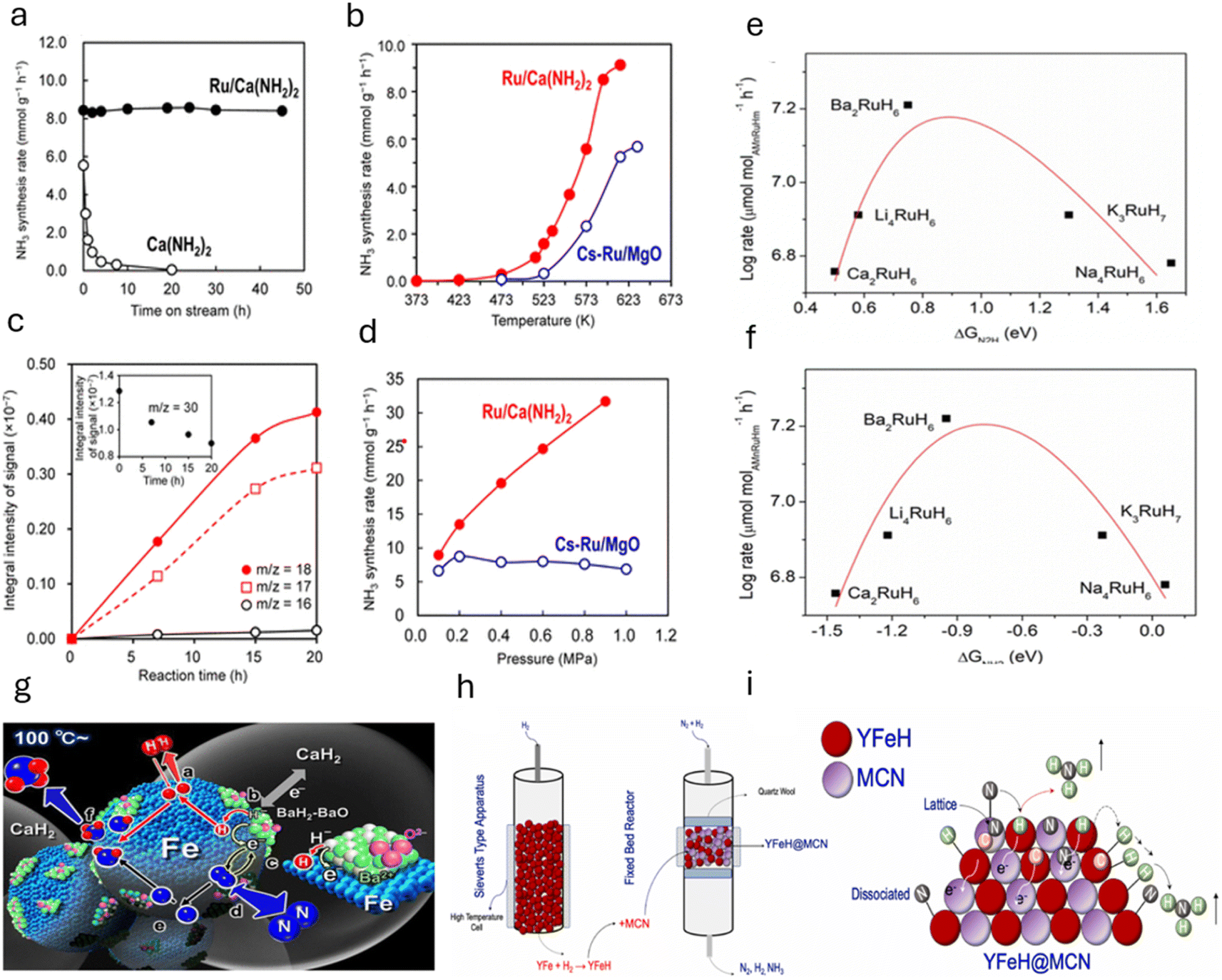 | ||
| Fig. 11 (a) NH3 synthesis over Ca(NH2)2 and 8 wt% Ru/Ca(NH2)2 at 613 K (340 °C) and 0.1 MPa over time. (b) NH3 synthesis rate versus temperature for 10 wt% Ru/Ca(NH2)2 and 10 wt% Ru–Cs/MgO at 0.1 MPa. (c) NH3 synthesis over Ru/Ca(NH2)2 from 15N2 and H2 at 473 K (200 °C) and 45 kPa; the inset shows 15N2 concentration over time. (d) NH3 synthesis rate at 613 K (340 °C) for 10 wt% Ru/Ca(NH2)2 and 10 wt% Ru–Cs/MgO at different pressures. Adapted with permission from ref. 87. Copyright 2016, the American Chemical Society. (e) Volcano-type plot for ammonia synthesis reactions over AMn[RuHm] (AM = Li, Na, K, Ca, and Ba) catalyst series at 300 °C and 0.1 MPa with the descriptor of the interaction strength between AM cations and NxHy species as the N2H adsorption energy and (f) NH2 adsorption energy. Adapted with permission from ref. 79. Copyright 2023, American Chemical Society. (g) Illustration of the reaction mechanism for ammonia synthesis on BaH2–BaO/Fe/CaH2. Adapted from the work of Hattori et al.,26 licensed under CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). (h) YFeH is synthesized in a Sieverts-type apparatus and then mixed with MCN to form YFeH@MCN, which subsequently reacts with N2 and H2 to produce ammonia. (i) Schematic representation of ammonia formation on YFeH@MCN catalyst, which contains both dissociated and lattice nitrogen, which reacts with hydrogen from YFeH or dissociated hydrogen in sequential steps to form ammonia. Adapted with permission from ref. 88. Copyright 2024, Elsevier. | ||
This study also unveils the role of Ba-doping into the catalytic system that led to no degradation in the activity profile after ca. 700 h of operations, since most of the conventional catalysts have a dissociative path for ammonia synthesis, which leads to a lower ammonia yield under low-temperature conditions.
Keeping this in mind, Wang et al. examined the catalytic performance of strategically designed ternary ruthenium complex hydrides, Li4RuH6 and Ba2RuH6, consisting of electron- and hydrogen-rich [RuH6] anionic centers.89 Herein, it is illustrated that the reduction of nitrogen occurs through the dual effect of multiple electron-rich sites and lattice hydridic hydrogen (acting as a proton and electron carrier). This resulted in an activity of 150 and 120 μmol g−1 h−1 observed for the bulk phase of Li4RuH6 and Ba2RuH6, respectively, at (<300 °C, 0.1–1.0 MPa). In the as-designed catalysts, Ru is in the ionic state and coordinated with six hydridic hydrogens, which forms electron-rich anions ([RuH6]4−) spatially located in the K4CdCl6 (or K2PtCl6)-type framework of Li (or Ba) cations. Such configuration promotes the associative hydrogenolysis of nitrogen through the support of [RuH6]4− centers, lattice hydridic hydrogens, and nearby Li or Ba cations. Additionally, the intermediates NxHy were enhanced by the Li/Ba cations, which positively impact the effectiveness of the catalyst. Hence, this work successfully exhibits that a transition-metal complex can reduce nitrogen via a non-dissociative pathway. As a follow-up work, the authors performed both experimental and theoretical works to gain more insights into the mechanistic details for ammonia formation over Ba2RuH6. The work unveils that Ba2RuH6 offers multifunctional and redox-active catalytic center for non-dissociative N2 reduction. Herein, Ba provides the site for N2 activation and H2 dissociation in synergy with Ru metals. Hence, at 300 °C and 0.1 MPa, the production rates of NH3 over Ba2RuH6 were ca. 25, 6.3 and 9.5 times greater than that of Ru/MgO, Cs2O–Ru/MgO, and BaO–Ru/MgO catalysts and reached the thermodynamic limit at 325 °C under a space velocity of 7500 mL g−1 h−1. Importantly, the catalyst displayed a positive reaction order (=1.0) with respect to H2 at 300 °C. The findings suggest that barium donates electrons and bonds with NxHy species.
In a similar study by Prof. Chen's group, the performance of a series of ruthenium complex hydrides AMn[RuHm] (AM = Li, Na, K, Ca, or Ba) was examined to compare the catalytic performance with Li4RuH6 and Ba2RuH6 catalysts.79 In comparison, under a total pressure of 1.0 MPa and at temperatures above 300 °C, the activities of supported Ru complex hydride catalysts were following this trend: Ba2RuH6/MgO ≈ K3RuH7/MgO > Li4RuH6/MgO > Na4RuH6/MgO > Ca2RuH6/MgO, whereas, at temperatures below 300 °C, the activity order was Ba2RuH6/MgO > Li4RuH6/MgO > Ca2RuH6/MgO > Na4RuH6/MgO > K3RuH7/MgO. In particular, the Ca2RuH6 sample demonstrated an activity of ca. 0.22 mmol g−1 h−1 at 300 °C and 0.1 MPa. The authors also tested the kinetic parameters and found that in general, Ca2RuH6/MgO, Li4RuH6/MgO, and Ba2RuH6/MgO catalysts exhibited low values of apparent activation energies than the Ru metal catalysts, indicating their high activity at lower temperatures. Interestingly, Ru-metal-based catalysts exhibit negative H2 orders, which lead to notorious hydrogen poisoning of the catalysts; however, Ca2RuH6/MgO, Li4RuH6/MgO, and Ba2RuH6/MgO have positive reaction orders with respect to H2, suggesting that these catalysts alleviate the issue of hydrogen poisoning. Insights into the mechanistic details reveal that the bonding strengths between AM cations and the reacting NxHy species govern the excellent features of AM-promoted Ru metal catalysts.
The N2H and NH2 adsorption energies of these hydrides were plotted against their respective activities, and a volcano-shaped plot was observed in both cases (Fig. 11e and f). Ca2RuH6 and Li4RuH6 work well at lower temperatures due to their low NxHy adsorption energies. This favors the activation of N2 over the formation and desorption of NH3. Ba2RuH6 has a moderate NxHy adsorption energy (exactly in between both plots), indicating that it would enhance ammonia synthesis for a wide range of temperatures. K3RuH7 and Na4RuH6, however, have high NxHy adsorption energies owing to which NH3 formation is preferred over N2 activation, resulting in good ammonia production at higher temperatures.
Next, Fe-based catalysts are considered first-generation classical transition metals for ammonia synthesis due to their significantly high rate under high-temperature and high-pressure reaction conditions. However, these catalysts have low activity at low temperatures. Using support materials with Fe can enhance the activity of the catalyst under mild reaction conditions. Hattori et al. discovered that metallic iron particles can help make ammonia from hydrogen (H2) and nitrogen (N2) at 100 °C when combined with BaH2. Iron works much better than other metals such as Ru, Co, and Ni because it releases hydrogen atoms (H adatoms) as H2 molecules at low temperatures (<100 °C). Other metals such as Ru, Co, and Ni hold onto hydrogen atoms more strongly, which can poison the catalyst and make it less effective at higher temperatures (>150–200 °C). Using support materials with Fe can enhance the activity of the catalyst under mild reaction conditions. For instance, in 2024, Michikazu Hara's group reported that Fe-based catalysts can produce ammonia via low-temperature reactions when supported with BaH2. An ammonia synthesis rate of 720 μmol g−1 h−1 was obtained at a temperature of 200 °C and a pressure of 0.9 MPa for the catalyst BaH2/BaO/Fe/CaH2. Such significantly high rate observed at low temperatures was attributed to the strong electron-donating nature of Fe in the presence of BaH2. Moreover, it was observed that the hydrogen poisoning effect was not happening under low-temperature reaction conditions. A plausible reaction mechanism of ammonia synthesis on BaH2–BaO/Fe/CaH2 is shown in Fig. 11g.
Recently, our group has shown that combining yttrium hydride with molybdenum carbonitride can play a pivotal role in low-temperature ammonia synthesis reactions. As reported in one of our work,88 molybdenum-based carbide can give ammonia under ambient pressure conditions; however, the reaction occurs at high temperatures. This work succeeds a previous work by our group on enhancing ammonia synthesis using a transition metal (Fe and Co)-modified Mo2C catalyst.90 Mixing it with yttrium hydride has started producing ammonia even at 200 °C. The ammonia synthesis activity 232 μmol g−1 h−1 was given at 200 °C and 0.1 MPa for the catalyst composition 10:90 (MCN:YH). YFeH was synthesized in a Sieverts-type apparatus followed by mixing with MCN to form YFeH@MCN. The synthesis method is depicted in Fig. 11h. Moreover, the ammonia synthesis rate was increased after changing the catalysts' weight ratio and temperature. For example, the catalyst MCN:YH (5:95) has shown a rate of 448 μmol g−1 h−1 at a temperature of 255 °C and a pressure of 0.1 MPa. Such high activity for ammonia synthesis under low-temperature reaction conditions could be possible due to the presence of hydride support, as it is found that the hydride-based materials under ammonia synthesis reaction conditions have the ability to generate electrons by splitting into protons and electrons. Moreover, in situ generated hydride directly participates in an ammonia synthesis reaction by following M–H pathways (M = metals), which plays an essential role in high ammonia generation rate. From our experimental findings, we observed that yttrium hydride enhanced the electron density, which was used by MCN to dissociate nitrogen molecules efficiently under low-temperature reaction conditions. The schematic mechanism of ammonia formation over YFeH@MCN is shown in Fig. 11i.
The low stability of hydride-based catalysts under reaction conditions remains a critical challenge, as they tend to degrade during prolonged exposure to synthesis environments. One effective strategy to address this issue is the implementation of chemical looping reactions, which can not only prevent stability issues but also enhance ammonia synthesis activity by maintaining the catalyst's structural integrity. Furthermore, designing hydrides with improved thermal and chemical stability, along with exploring novel hydride compositions, could further augment their durability and broaden their applicability in industrial ammonia synthesis.
3.3 Intermetallic-based catalysts
In recent years, a significant shift in ammonia synthesis methods has been underway, with a growing consensus to utilize intermetallic compounds. Intermetallic catalysts are composed of two or more metals that form a crystalline structure, offering unique properties beneficial for ammonia synthesis. These catalysts are highly active due to the synergy between the metal components, optimizing electronic properties and enhancing catalytic sites for nitrogen activation. In particular, these compounds consist of a rare earth metal and an active transition metal, and therein, the rare earth metal transfers electrons to the transition metal that facilitates dissociation of nitrogen molecules by lowering the activation barrier of ammonia synthesis.91,92 Their ability to reversibly store hydrogen prevents hydrogen poisoning, maintaining active catalytic sites and improving overall efficiency.93–95 Table 5 lists the ammonia synthesis activity of intermetallic-based catalysts under low-pressure and low-temperature conditions.| S. no. | Catalyst | Temp. (°C) | Pressure (MPa) | Rate (mmol g−1 h−1) | Activation energy (kJ mol−1) | Metal% | WHSV | N2 order | H2 order | NH3 order | Surface area (m2 g−1) | Turnover frequency (TOF) (s−1) | Catalyst synthesis method | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Ru/Ti0.04–Ce | 400 | 1 | 11.009 | — | 1.9% Ru, 5.1% TiO2 | 36000 |
— | — | — | 62 | 10.8 × 10−3 | Hydrothermal; co-precipitation method | 96 |
| 2 | Ru/Ti0.13–Ce | 400 | 1 | 15.2 | — | 2.2% Ru, 14.8% TiO2 | 36000 |
0.27 | 0 | −0.56 | 61 | — | Hydrothermal; co-precipitation method | 96 |
| 3 | Ru/Ti0.18–Ce | 400 | 1 | 18.912 | — | 2.1% Ru, 19.4% TiO2 | 36000 |
0.5 | 0.47 | −1.07 | 65 | 18.7 × 10−3 | Hydrothermal; co-precipitation method | 96 |
| 4 | Ru/Ti0.25–Ce | 400 | 1 | 13.5 | — | 2.2% Ru, 25.3% TiO2 | 36000 |
0.61 | 0.20 | −1.14 | 64 | — | Hydrothermal; co-precipitation method | 96 |
| 5 | Ru/Ti0.35–Ce | 400 | 1 | 9.773 | — | 2.2% Ru, 32.0% TiO2 | 36000 |
— | — | — | 62 | 9.6 × 10−3 | Hydrothermal; co-precipitation method | 96 |
| 6 | Ru/Ti0.98-Ce | 400 | 1 | 7.120 | — | 2.0% Ru, 57.2% TiO2 | 36000 |
0.84 | −0.17 | −1.53 | 55 | 7.0 × 10−3 | Hydrothermal; co-precipitation method | 96 |
| 7 | LaNi5 (bulk) | 400 | 0.1 | 0.25 | 50.1 | 67.269 (Ni) | 36000 |
— | — | — | 0.4 | — | Arc melting method | 97 |
| 8 | LaNi5 NPs | 400 | 0.1 | 4.487 | 53.8 | 67.235 (Ni) | 36000 |
1.28 | 1.60 | −1.74 | 38.7 | — | Ar/H2 arc evaporation method | 97 |
| 9 | Ru/LaCoSi | 400 | 0.1 | 3.4 | 50 | 4.3 (Ru) | 18000 |
1 | 1.17 | −1.06 | 2.6 | 0.03 | Arc melting; CVD method | 98 |
| 10 | Ru/LaFeSi | 400 | 0.1 | 2.8 | 56 | 4.4 | 18000 |
0.99 | 0.94 | −1 | 3.1 | 0.027 | Arc melting; CVD method | 98 |
| 11 | Ru/LaMnSi | 400 | 0.1 | 1.1 | 57 | 4.7 | 18000 |
0.98 | 1.10 | −0.93 | 3.9 | 0.012 | Arc melting; CVD method | 98 |
| 12 | Ru1.4/LaScSi | 400 | 0.1 | 3.9 | — | 1.4 | 36000 |
— | — | — | 1 | 2 | Arc melting and annealing method | 99 |
| 13 | Ru1.7/LaScSi | 400 | 0.1 | 2.33 | — | 1.7 | 36000 |
— | — | — | 1 | 2.4 | Arc melting and annealing method | 99 |
| 14 | Ru1.7/CeScSi | 400 | 0.1 | 2.25 | — | 1.7 | 36000 |
— | — | — | 1 | 1.9 | Arc melting and annealing method | 99 |
| 15 | Ru1.7/PrScSi | 400 | 0.1 | 2.2 | — | 1.7 | 36000 |
— | — | — | 1 | 2.6 | Arc melting and annealing method | 99 |
| 16 | Ru1.7/NdScSi | 400 | 0.1 | 2 | — | 1.7 | 36000 |
— | — | — | 1 | 1.4 | Arc melting and annealing method | 99 |
| 17 | Ru1.5/SmScSi | 400 | 0.1 | 1.2 | — | 1.5 | 36000 |
— | — | — | 1 | 1.9 | High-frequency induction melting method | 99 |
| 18 | Ru1.3/GdScSi | 400 | 0.1 | 0.5 | — | 1.3 | 36000 |
— | — | — | 1 | 1 | Arc melting and annealing method | 99 |
Optimally tuned electronic and geometric structures conferred by the intermetallic catalyst, which is a well-defined ordered metal alloy, provide better catalytic performance for ammonia synthesis. Optimizing metal–metal interactions promotes nitrogen activation by decreasing the energy barrier for N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond cleavage.
N bond cleavage.
Arc melting method was used to synthesize LaNi5 bulk.97 For this preparation, lanthanum and nickel in their elemental form were taken in a copper hearth and then arc melted in an inert (Ar) atmosphere, which resulted in the formation of silver-coloured ingots. The intense heat during arc melting allows proper mixing and alloying of the metals, which leads to the formation of homogeneous LaNi5 ingots upon solidification. These ingots were then ground in an agate mortar. For the preparation of LaNi5 nanoparticles (NPs), the obtained LaNi5 ingots were treated in an Ar/H2 arc evaporation system, where the temperature goes as high as 10000 °C, which vaporizes the base materials. In this process, the partial pressures of Ar and H2 were set to 0.03 and 0.03 MPa, respectively, at an air flow rate of 60 mL min−1. To avoid exposing LaNi5 NPs to air, the filter room was detached from the evaporation system and NPs were directly collected from there.
The chemical vapour deposition method incorporates Ru-loaded catalysts on La–TM–Si98 intermetallic compounds in which Ru3(CO)12 was deposited through a programmable temperature ramp method. In the CVD method, the metal precursors vaporize and decompose at controlled temperatures allowing Ru atoms to deposit on the La–TM–Si surface, which allows a uniform Ru catalyst layer. La rods, TM granules and Si pieces were taken in stoichiometric amounts and arc melted in an argon atmosphere. The ingots thus obtained were annealed for a duration of 5 days at a temperature of 1000 °C and then cooled down to 800 °C and held for 10 more days. The final step was grounding in a glovebox to obtain fine powders. Ru was loaded on La–TM–Si by a chemical vapour deposition method using a Ru3(CO)12 precursor. Both the powders were taken in a silica tube, which was temperature programmed to heat to 70 °C, held for 1 h and then to 120 °C for another 2 h and finally to 250 °C for last 2 h followed by cooling to room temperature to obtain the catalyst in its final form. The SEM images of LaCoSi, Ru/LaCoSi, Ru/LaFeSi and Ru/LaMnSi can be seen in Fig. 12c–f, respectively.
 | ||
| Fig. 12 (a) Lattice parameters and TiO2 content variation with titanium butoxide volumes. (b) XRD crystallographs of Ti-doped CeO2 prepared via a combination of hydrothermal and co-precipitation methods. Adapted with permission from ref. 96. Copyright 2022, Elsevier. SEM images of (c) LaCoSi, (d) Ru/LaCoSi, (e) Ru/LaFeSi and (f) Ru/LaMnSi catalysts prepared via a combination of arc-melting and CVD methods. Adapted with permission from ref. 98. Copyright 2022, Elsevier. | ||
In a similar work, RScSi99 with R = La, Ce, Pr, Nd, Sm, and Gd were synthesized by melting the metallic powders beginning with rare earth ingots, Sc pieces and Si lumps. A high-frequency furnace was used to vaporize volatile oxides while melting the rare earth metals. The metal samples were arc melted for 5 times in an argon atmosphere followed by annealing at a temperature of 900 °C for a period of 4 weeks. The buttons so obtained were crushed and sieved before depositing Ru NPs. Ru was loaded on RScSi pristine phases by a chemical vapor deposition method using Ru3(CO)12 as a Ru precursor.
The combination of hydrothermal and co-precipitation methods was used to synthesize a Ru/Tix–Ce96 catalyst. Cerium nitrate was dispersed in ethanol, added to an aqueous NaOH solution, and stirred for 30 minutes followed by hydrothermal treatment at 100 °C for 24 h. During hydrothermal treatment, the nitrate precursors react with NaOH under heat and pressure, which leads to phase transformation and controlled crystallization in a liquid medium. The resulting precipitate was washed with DI water and ethanol to make the pH 7, which yielded a gel. This gel was dried and calcined at a temperature of 550 °C for 4 h to obtain CeO2. Now titanium butoxide and ethanol mixed solution was added to this gel followed by pH adjustment to 10 using ammonia solution and then the mixture was heated at 80 °C for 2 h. In the co-precipitation method, simultaneous precipitation of multiple components occurs by adjusting pH, ensuring uniform composition and fine particle dispersion. Finally, after drying and calcination, ruthenium was loaded on Tix–Ce by the usual wetness impregnation method using ruthenium nitrosyl nitrate as the precursor. Fig. 12a shows the variation of lattice parameters and TiO2 contents with titanium butoxide volumes.
LaNi5 is a famous and well-investigated hydrogen storage material, which can store up to 1.40 wt% H2 at room temperature.100 Therefore, LaNi5 has attracted attention for designing ammonia synthesis catalysts in combination with inexpensive transition metals. For instance, Ye et al. designed a Ni–LaN core–shell structure by mixing LaNi5 (prepared from arc-melting method) and Ni (from thermal decomposition of nickelocene (Ni(C5H5)2)) in an Ar-filled glovebox. The mixture was subsequently heated to 250 °C for 1.5 h in pure H2 (flow rate = 2.5 mL min−1; pressure = 0.1 MPa).97 The catalytic activity of the as-prepared Ni–LaN catalyst was as high as 4.5mmol g−1 h−1 at 400 °C and 0.1 MPa with low activation energy (ca.50 kJ mol−1), which was about 20-fold greater than that of the LaNi5 bulk particles. However, one of the issues with LaNi5 is its phase separation of LaHx and Ni metal after reversible H2 storage, even at room temperature.101–103 Herein, during ammonia synthesis reaction, high temperatures readily induced such phase separation of LaNi5 to LaHx, which combined with N2 to form LaN (i.e., LaNi5 + 1.5H2 + N2 → LaN + 5 Ni + NH3). Notably, these thin layers of LaN further prevent the decomposition of the internal LaNi5, providing stability to the catalytic performance. The decomposition process of LaNi5 in the bulk and nanoparticle samples is represented in Fig. 13a and b.
 | ||
| Fig. 13 (a) Illustrations of the decomposition process during ammonia synthesis reaction over LaNi5 bulk and (b) LaNi5 nanoparticles. Adapted with permission from ref. 97. Copyright 2020, the American Chemical Society. (c) Ammonia synthesis mechanism over La–TM–Si (TM = Co and Fe) and (d) LaMnSi. Colour scheme of elements is as follows-light blue: La; red: TM; tawny: Si; dark blue: N; white: H. Adapted with permission from ref. 104. Copyright 2023, John Wiley and Sons. | ||
Therefore, this work illustrates the excellent application of Ni by combining with early transition metal La, which is supposed to have low activity for ammonia synthesis, obeying the scaling rules of pure metals predicted from the volcano plot.
The application of intermetallic compounds for ammonia synthesis is not limited to the combination of two metals. In the absence of Ru metals, a dual-site mechanism was reported for LaCoSi and LaFeSi in a study by Li et al.104 The ammonia synthesis rates of LaCoSi, LaFeSi and LaMnSi were 1.22, 0.62, and 0.12 mmol g−1 h−1, respectively, at 0.1 MPa and 400 °C. The activation energies were 42, 43, and 54 kJ mol−1 for LaCoSi, LaFeSi and LaMnSi, respectively. The reaction order for H2 was positive, suggesting the absence of the hydrogen poisoning effect. Further findings indicate that N2 activation occurs on the transition metals site and N hydrogenation happens on the La site. However, in the case of La–Mn–Si, the mobility of N from Mn to La was inhibited, which led to poor hydrogenation of N atoms on the Mn site, and therefore, low activity was observed for this catalyst system. The illustration for the same can be found in Fig. 13c and d.
Gong et al. synthesized La–TM–Si (TM = Co, Fe, and Mn) by a conventional arc-melting method. Further, these powders were mixed with an Ru precursor (i.e., Ru3(CO)12), sealed in a silica tube, and heated. The as-prepared catalysts were then examined for their ability to shift the rate-limiting step from the slow nitrogen dissociation to NHx formation.93 With 2 wt% Ru loading, these catalysts exhibited ammonia synthesis rate as high as 1.28, 3.88, and 5.23 mmol g−1 h−1 for Ru/LaMnSi, Ru/LaFeSi, and Ru/LaCoSi, respectively. Despite the fact that Ru/LaMnSi and Ru/LaFeSi have identical work function values, the latter showed a much higher activity, which clearly demonstrate that the activity is not solely dependent on the electron donation ability of the support (or electride materials). However, the activity was observed to be dependent on Ru-loading but independent of the surface area of the catalysts.
Fig. 14a highlights the superior catalytic performance of Ru/La–TM–Si catalysts, where increased Ru loadings and favourable reaction kinetics result in enhanced ammonia synthesis. The low activation energies (Fig. 14b) confirm a distinct reaction pathway that differs from conventional catalysts, with efficient N2 activation, as shown by the N2 reaction orders (near-unity) (Fig. 14c). Additionally, the positive H2 reaction orders (Fig. 14d) imply resistance to hydrogen poisoning, which is typically problematic for other catalysts. The synthesis rate consistently rises with increasing pressure, underscoring the catalyst's robust performance under different conditions, particularly for Ru/LaCoSi (Fig. 14e and f). This highlights the role of La–TM–Si supports in enabling effective ammonia production by providing reversible hydrogen storage and minimizing adverse interactions with H2.
 | ||
| Fig. 14 (a) Catalytic activity of Ru/La–TM–Si with varying Ru loadings at 0.1 MPa, 400 °C, and N2/H2 = 15:45 mL min−1. (b) Arrhenius plots for ammonia synthesis over Ru/La–TM–Si. (c) Reaction orders for N2 and (d) H2 calculated using the power law equation at 0.1 MPa. (e) Effect of pressure on ammonia synthesis for 5 wt% Ru/La–TM–Si. (f) Ammonia production over time for Ru/La–TM–Si at 400 °C, 0.1 MPa, with N2/H2 = 15:45 mL min−1. Each experiment used 0.1 g of catalyst. Adapted with permission from ref. 98. Copyright 2022, the American Chemical Society. (g) Nitrogen isotopic distribution during the homomolecular exchange of 15N2/14N2 at 500 °C for Ru/LaScSiH1.5 and (h) partially dehydrogenated Ru/LaScSiHx, treated under Ar at 500 °C for 2 hours. The dashed line indicates the change in H2 partial pressure (m/z = 2 signal). Adapted with permission from ref. 105. Copyright 2023, Elsevier. | ||
Croisé et al. used Ru/RScSi (R = La, Ce, Pr, Nd, Sm, and Gd) to investigate the activity of various rare earth metals for ammonia synthesis at 300–450 °C and under 0.1–0.5 MPa.99 The activity trend from La to Gd lanthanide series is as follows: Ru1.7/LaScSi = Ru1.7/CeScSi = Ru1.7/PrScSi > Ru1.7/NdScSi > Ru1.5/SmScSi > Ru1.3/GdScSi, which was found to be strongly dependent on their respective hydride phases. These catalysts were arc-melted under a pure argon atmosphere. Notably, Ru1.7/CeScSi displayed catalytic activity of 0.58 mmol g−1 h−1 at 300 °C and 0.1 MPa, which is closely related to its reversible hydrogen-storage properties. Moreover, this was ascribed to the prevention of hydrogen poisoning of the Ru surface, and therefore, it is concluded that the electron-donating effect of such electrides can be tuned via partial hydrogenation of the RScSi materials. Through extensive experiments, it was evident that the RScSi support facilitates the nitrogen dissociation with possible storage of N-species and the hydrogen present in the formed ammonia stems from hydrogen gas and not from the hydride phase. Importantly, it was speculated that the formation/desorption of NHx species could be the rate-limiting step.
In a follow-up work, Croisé et al. investigated LaScSi and CeTiGe using DFT calculations and 15N/14N homomolecular exchange experiments to establish a correlation between the electride-type material characteristics, partial hydrogenation of the structure and the efficiency for dissociative nitrogen activation (Fig. 14g and h).105 It was found that Ru/LaScSi was relatively more active, owing to their electride-like properties, which were substantially increased when the catalyst was partially hydrogenated, and subsequently, led to a greater activity in a nitrogen equilibration reaction. Although no electride-like properties are present in the Ru/CeTiGe catalyst, this article unveils that the catalyst follows the associative mechanism to produce ammonia.
While the high efficiency of intermetallic catalysts might seem promising, they often rely on rare-earth metals, which are expensive and limited in supply, making them economically unfeasible for large-scale use. Additionally, under high-temperature and high-pressure conditions, these catalysts may suffer from sintering or phase changes, leading to reduced performance. The synthesis of intermetallic is also complex, requiring precise control of composition and processing conditions. To overcome these challenges, research should focus on finding cost-effective alternatives to rare-earth metals, improving catalyst stability under harsh conditions, and exploring advanced synthesis methods to enhance their performance and scalability for industrial ammonia synthesis.
3.4 Electride-based catalysts
Electrides are crystals in which cavity-confined electrons act as anions, and were first prepared by J. L. Dye in 1983 using crown ethers.106,107 Generally, these electrons are trapped in an anion vacancy site, known as F-center (a point defect). The high electron concentration in the F-center makes electrides very active. These trapped electrons play a pivotal role in catalysis by donating electrons to transition metals. The transition metals, in turn, can back-donate these electrons to the nitrogen molecules, facilitating the activation of N2. This electron transfer mechanism significantly enhances the catalytic activity of electrides, making them highly efficient for N2 activation in ammonia synthesis. As a result, such materials with unique electronic structures and composed of oxides (such as CaO, and Al2O3) offer a low work function, compared to alkali metals. Moreover, electrides are chemically inert and thermally stable. Consequently, this material finds huge application in ammonia synthesis under mild conditions because these surface electrons can readily be transferred to the transition metals via an energetically favorable step. The performances of various electride-based catalysts and their corresponding synthesis methods are shown in Table 6.| S. no. | Catalyst | Temp. (°C) | Pressure (MPa) | Rate (mmol g−1 h−1) | Activation energy (kJ mol−1) | Metal% | WHSV | N2 order | H2 order | NH3 order | Surface area (m2 g−1) | Turnover frequency (TOF) (s−1) | Catalyst synthesis method | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Ru/BaAl2O4−xHy | 340 | 0.9 | 13.304 | 65.7 | 2 | 36000 |
0.72 | 1.02 | −1 | — | 5.7 × 10−2 | Solid state reaction method; hydrogenation method | 108 |
| 2 | Ru/BaAl2O4 | 340 | 0.9 | — | — | 2 | 36000 |
1.29 | 0.30 | −0.66 | — | — | Solid state reaction method | 108 |
| 3 | LaRuSi | 400 | 0.1 | Activity/surface area = 1433 μmol m−2 h−1 | 42 | — | — | — | — | — | 1.2 | — | Arc melting method | 109 |
| 4 | CaRuSi | 400 | 0.1 | 35 μmol m−2 h−1 | — | — | — | — | — | — | 1.5 | — | Solid-state reaction method | 109 |
| 5 | Ru/C12A7:O2− | 400 | 0.1 | — | — | 1.60 | 30 cm3 min−1 | — | — | — | — | 0.015 | Modified Pechini method | 110 |
| 6 | Ru/C12A7:e− | 400 | 0.1 | 1.5 | 72 | 2 | 30 cm3 min−1 | 0.7 | 0.3 | — | — | 0.087 | Aluminothermic reduction method | 110 |
| 7 | Ru/BaOxNy e−z (x = 0.5) | 400 | 0.1 | 2.050 | 62 | 1.8 | 36000 |
— | — | — | 0.7 | 0.292 | Solid-state reaction method | 76 |
Electride-based catalysts for ammonia synthesis enhance the activation of nitrogen sites by providing strong sites characterized by highly active electron donation important for the cleavage of the NN bond. The method of preparation of these catalysts emphasizes the optimization of their electronic properties.
BaAl2O4−xHy and BaOxNy108 were synthesized by a solid-state reaction method to obtain the desired phase purity and to carry out high-temperature processing for controlled composition. The high temperature in this method induces diffusion-driven chemical reactions between solid reactants, leading to phase formation. γ-Al2O3 was made to react with BaH2 with the addition of BaCO3 under H2 flow. The purpose of addition of BaCO3 was to increase the purity of BaAl2O4−xHy. BaH2 was synthesized by the reaction of metallic Ba and H2 at a temperature of 450 °C for a period of 20 h. Then it was mixed with γ-Al2O3 and BaCO3 in an agate mortar in an argon environment. This was followed by annealing in a hydrogen atmosphere at a temperature of 800 °C for 20 h to get the support in final form. Al(OH)3 and BaCO3 underwent solid-state reaction to form BaAl2O4. They were mixed thoroughly in an agate mortar followed by calcination at a temperature of 1000 °C for 20 h in air. For loading Ru on the support, chemical vapour deposition method was used. For Co loading, Co2(CO)8 along with the support was ground in an agate mortar in an Argon environment followed by heating in a quartz tube at 200 °C for 2 h in a mixture of N2 and H2 gases. Finally, the mixture was thermally treated at 500 °C for 1.5 h to obtain the cobalt-loaded catalyst.
The modified Pechini method was used for the synthesis of C12A7:O2−.110 The modified Pechini method is based on the sol–gel and polymeric precursor approach. Metal ions are chelated using citric acid and EDTA to form a stable complex, which ensures uniform mixing at the molecular level. Upon heating, polymerization occurs, creating a gel that prevents phase separation. Subsequent calcination decomposes organics, leading to controlled crystallization of the desired oxide phase with high purity and homogeneity. Calcium and aluminium nitrates were dissolved in EDTA and citric acid solution with an EDTA/citric acid/metal ion ratio of 1.5/1.5/1. This was followed by the addition of ammonium hydroxide in the slurry till a clear solution was formed. Water was evaporated off this solution to obtain a claybank gel which was placed in an oven and dried at a temperature of 150 °C for 48 h period to yield charcoal-like powder. Mayenite in its pure form (C12A7) was obtained by calcination of this powder at a temperature of 1200 °C for 10 h. C12A7:e− electride was obtained by an aluminothermic method.
Aluminum acts as a strong reductant and thus donates electrons to reduce CaO and Al2O3, resulting in the formation of metallic aluminum oxide by-products. During this process, oxygen atoms are extracted from the lattice which creates anionic vacancies that trap free electrons, stabilizing them within the C12A7 framework. This results in the formation of an electride, where these trapped electrons act as anions, giving the material its unique electronic properties. In this method, CaO, Al2O3, and Al powders were mixed in an agate mortar followed by calcination in a tubular furnace at a temperature of 1200 °C for 8 h in an argon atmosphere. Ru was loaded onto these supports by the chemical vapour deposition method to obtain the catalyst in its final form.
Arc-melting was primarily used to synthesize LaRuSi109 and CaRuSi electrides, promoting uniformity via repeated melting and annealing. LaRuSi was synthesized by arc melting La, Ru, and Si (1:1:1) under an Ar atmosphere, with repeated melting for homogeneity. The ingot was annealed for two weeks and then ground under an Ar atmosphere for use as a catalyst.
CaRuSi was prepared via a solid-state route. The RuSi precursor was first synthesized by arc melting, then mixed with Ca ingots (5% excess) and sealed in a Ta crucible inside an Ar-filled quartz tube. Heat treatment was carried out at 850 °C for 10 h, followed by 800 °C for 2 days to obtain the pure phase.
In 2012, Kitano et al. reported the activity of a stable electride, C12A7:e−, synthesized by a solid-state reaction that efficiently donates electrons to the Ru catalyst. Ru/C12A7:e− displayed substantially superior activity than that of Ru–Cs/MgO and Ru–Ba/AC.111 For instance, the reported turnover frequency (TOF) for Ru/C12A7:e− at 1.0 MPa was 0.98 s−1, which is significantly higher than those of Ru–Ba/AC and Ru–Cs/MgO (0.05–0.28 s−1), respectively, at elevated pressures (2.0–6.3 MPa).111–114 Such results also demonstrate that the catalytic activity is limited for conventional oxide-supported Ru catalysts, because the dissociative adsorption of hydrogen on Ru hinders the cleavage of nitrogen molecules at elevated pressures. To get an insight into the mechanistic details, isotopic FT-IR spectra using 14N2/15N2 were recorded. The article concludes that electrons trapped in C12A7:e− transferred to Ru metal, which considerably lowered the work function of Ru by increasing the Fermi level. Consequently, back donation from Ru with excess electron density to the π*-antibonding orbital of nitrogen increased, which weakened the NN bond of adsorbed nitrogen molecules. The surface adsorbed dissociated hydrogen molecules spilled over to C12A7:e− and were incorporated into the cages as H− ions, and prevented hydrogen poisoning of the Ru surface. Further, the activated nitrogen species combine with H− ions (inside the C12A7 nanocages) to form ammonia.
Within the literature, it is established that electron-rich supports with defects consisting of electrons lower the work function and remarkably enhance the catalytic ammonia synthesis. However, the mechanism of such promotional effect and the exact reason for the suppression of hydrogen poisoning in the presence of electride-supported Ru catalysts were unclear. To address this issue, Kammert et al. examined the hydride species in the C12A7:e− electride during ammonia production using techniques such as neutron diffraction and inelastic neutron scattering.110 With a Ru loading of 2 wt%, the ammonia synthesis rate was 1.5 mmol g−1 h−1 at 360 °C and the apparent activation energy was 72 kJ mol−1. The comparative TOF values of previously published reports and different forms of C12A7 are illustrated in Fig. 15a and b.
 | ||
| Fig. 15 (a) Comparative turnover frequency (TOF) of ammonia synthesis at 400 °C in 0.1 MPa (3/1 = H2/N2). (b) Variation of TOF with hydrogen pressure over the electride and oxygen forms of Ru/C12A7 catalysts. Adapted with permission from ref. 110. Copyright 2020, the American Chemical Society. (c) Performance of the Co/BaAl2O4−xHy catalyst for ammonia synthesis, and comparison of different Co-based catalysts at different temperatures under 0.90 MPa pressure. (d) Stability analysis of Co (4.7 wt%)/BaAl2O4−xHy at 400 °C and 0.90 MPa over time. (e) Ammonia synthesis rates and activation energies for various Co catalysts at 300 °C and 0.90 MPa. (f) Illustration of Co/BaAl2O4−xHy's activity changes after exposure to air and improvement after treatment at 600 °C in H2 flow for 6 hours. All tests were performed at a flow rate of 36000 mL g−1 h−1. Adapted with permission from ref. 108. Copyright 2023, the American Chemical Society. | ||
The orders of the reaction with respect to N2 and H2 were 0.7 and 0.3, respectively. Further, this study found that the confined hydrides are stable and not readily exchanged in the C12A7:e− framework, contrary to the previous reports. However, these hydride species could exchange with the surface species which limits their kinetic role in the production and mechanism of ammonia production. Therefore, this study concludes that hydrogen species adsorbed over the Ru surface predominantly contribute to ammonia synthesis. Importantly, the coverage of reactive intermediates and nature of adsorbates on the Ru surface could be the reason that hydrogen poisoning was suppressed during ammonia production by the electride-supported Ru.
Jiang et al. reported that BaAl2O4−xHy, a novel oxyhydride electride with a tridymite-like structure, significantly enhances ammonia synthesis over Co catalysts, achieving 500 mmol gCo−1 h−1 at 340 °C and 0.90 MPa with a low activation energy of 49.6 kJ mol−1.108 Its structure contains interstitial cage sites that host anionic electrons, with low work functions (1.7–2.6 eV) that strongly donate electrons to Co, thus accelerating N2 reduction into ammonia using lattice H− ions. The stability of BaAl2O4−xHy and its resistance to oxidation give it robust reusability, making it superior to traditional hydride-based catalysts. Fig. 15c–f show performance of the Co/BaAl2O4−xHy catalyst for ammonia synthesis and compare different Co-based catalysts at different temperatures under 0.90 MPa pressure.
In an interesting article by Hattori et al., a novel approach to produce ammonia at low temperatures (50 °C) using a stable electron-donating catalyst, CaFH, was reported.25 This work used appropriate amounts of CaF2 and CaH2 and heated to 550 °C under argon flow for 20 h, resulting in a solid solution of cubic CaFH catalyst. Ru(acac)3, a precursor, was heated under hydrogen flow at high temperature to synthesize a Ru/CaFH catalyst with a loading of 12 wt% Ru. The as-prepared Ru/CaFH catalyst produced ammonia at rates of 50, 75, 120, and 190 μmol g−1 h−1 at 50, 75, 100, and 125 °C, respectively, with an extremely small activation energy of 20 kJ mol−1, calculated in the range of 50–150 °C. The FTIR spectra for N2 adsorption over this catalyst is shown in Fig. 16a. At room temperature, stretching bands of adsorbed nitrogen on Ru/CaH2 at 2100–2250 cm−1 were observed, which are lower than that of nitrogen gas (2744 cm−1). This result implies that the electron donation from the catalyst to the π*-antibonding orbitals of adsorbed nitrogen via Ru d-orbitals facilitates the dissociation of nitrogen bond. Comparing Ru/CaH2, a large shift in the wavenumber was observed for Ru/CaFH, which could lead to high activity even at room temperature. In the end, the findings from H2-TPD and DFT experiments further help to corroborate that nitrogen molecules were readily dissociated into N adatoms, which combine with H from CaFH to generate ammonia at room temperature. This mechanism is illustrated in Fig. 16b. Herein, the resulting CaFH acts as a surface electride with a lower work function value (ϕ = 2.2 eV) than that of CaH2 (ϕ = 2.7 eV).
 | ||
| Fig. 16 (a) FT-IR spectra for nitrogen adsorption on Ru/CaFH and Ru/CaH2 after ammonia generation at 340 °C and subsequent cooling to 20 °C. (b) Plausible reaction mechanism. Adapted with permission from ref. 25. Copyright 2020, Springer Nature. (c) Energy profiles for ammonia synthesis calculated for LaRuSi and (d) CaRuSi surfaces. Adapted with permission from ref. 109. Copyright 2022, the American Chemical Society. | ||
In another work, Gong et al. demonstrated that the LaRuSi electride enhances NH3 synthesis by disrupting scaling relations through two key mechanisms: it achieves low N2 activation energy on a Ru-terminated surface without overly strong nitrogen adsorption, and the La site facilitates NHx formation, further lowering reaction barriers.109 Energy profiles of NH3 synthesis on LaRuSi and CaRuSi surfaces are shown in Fig. 16c and d, respectively. The activation energies (Ea) for NH, NH2, and NH3 formation on LaRuSi are 1.07, 1.45, and 1.27 eV, respectively—significantly lower than on LaRuSi(001)–Ru. NH3 desorption from LaRuSi(001)–La requires only 0.57 eV, breaking traditional Ru catalyst scaling relations, with NH2 formation as the rate-determining step (RDS). For CaRuSi, NHx formation steps are more favorable on Ca-terminated surfaces compared to Ru-terminated, with NH3 desorbing more easily from CaRuSi(001)–Ca. The NH2 formation step is the RDS for NH3 synthesis on both LaRuSi and CaRuSi.
Despite their remarkable activity, electride catalysts face several challenges. One of the primary concerns is the limited activity with certain transition metals, such as ruthenium, which restricts their scalability and cost-effectiveness. The reliance on specific metals for optimal performance makes these catalysts less versatile in industrial applications. Additionally, maintaining the stability of electrides under reaction conditions can be challenging due to the high reactivity of the trapped electrons, which may degrade over time, especially under harsh reaction environments. The high sensitivity of electrides to moisture and oxygen can also further decrease their performance. To address these challenges, future research should focus on identifying alternative transition metals that can work effectively with electrides to expand their applicability. Strategies to stabilize the electron density in electrides, such as doping with stabilizing elements or optimizing the crystal structure, could enhance their longevity and resistance to degradation. Furthermore, the development of electride systems with greater stability in the presence of moisture and oxygen would increase their practical feasibility for industrial-scale ammonia synthesis.
3.5 Oxide-based catalysts
Another class of catalysts which hold the potential for ammonia synthesis under mild conditions are the oxide-based catalysts. Mixed metal oxides or perovskite-structure-based oxide catalysts have also demonstrated significant activity for ammonia synthesis (see Tables 7 and 8). Oxide catalysts are widely recognized for their basic nature, high stability, and strong metal–support interactions, which play a critical role in enhancing the catalytic performance for ammonia synthesis. Oxygen vacancies intrinsic to oxide supports act as active sites for N2 activation by facilitating the adsorption and dissociation of nitrogen molecules. The strong metal–support interactions not only stabilize the active metal species but also enhance electron transfer between the support and the metal, further improving catalytic activity. In the case of mixed oxides, the synergistic interplay between multiple metal components leads to an enriched concentration of oxygen vacancies, amplifying the catalyst's activity. As is known, the enhanced electron donation properties and active site availability facilitate the activation of nitrogen, and also, the hydrogenation process. Therefore, oxide-based catalysts are of particular interest for ammonia synthesis under mild conditions.| S. no. | Catalyst | Temp. (°C) | Pressure (MPa) | Rate (mmol g−1 h−1) | Activation energy (kJ mol−1) | Metal% | WHSV | N2 order | H2 order | NH3 order | Surface area (m2 g−1) | Turnover frequency (TOF) | Catalyst synthesis method | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Exact rate of ammonia production is not given in the source paper; listed values are closest approximates obtained from the plot in the source paper. | ||||||||||||||
| 1 | Ru/La2O3 | 400 | 1 | 32.14 | 118 | 5% Ru | 24000 |
1.00 | −0.46 | −0.57 | 61.1 | — | Deposition–precipitation method | 115 |
| 2 | Co/MgO–Nd2O3 (Mg/Nd molar ratio – 2:1) |
400 | 6.3 | 23.4 | 58.9 | 10% Co | 140000 |
— | — | — | 22.9 | 3.9 × 102 | Co-precipitation method | 116 |
| 3 | Co/MgO–Nd2O3 (Mg/Nd molar ratio – 4:1) |
400 | 6.3 | 29.9 | 55.3 | 10% Co | 140000 |
— | — | — | 29.3 | 3.8 × 102 | Co-precipitation method | 116 |
| 4 | Co/MgO–Nd2O3 (Mg/Nd molar ratio – 7:1) |
400 | 6.3 | 33.2 | 53.9 | 10% Co | 140000 |
— | — | — | 37.8 | 3.6 × 102 | Co-precipitation method | 116 |
| 5 | Co/MgO–Nd2O3 (Mg/Nd molar ratio – 10:1) |
400 | 6.3 | 39.3 | 53.7 | 10% Co | 140000 |
— | — | — | 49.5 | 3.5 × 102 | Co-precipitation method | 116 |
| 6 | Co/MgO–Nd2O3 (Mg/Nd molar ratio – 12:1) |
400 | 6.3 | 35.5 | 54.6 | 10% Co | 140000 |
— | — | — | 46.9 | 3.6 × 102 | Co-precipitation method | 116 |
| 7 | Ru/Al2O3 | 400 | 1 | 1.432 | 82 | 3% Ru | 24000 |
1.0 | −0.1 | −1.1 | 169 | 2.6 × 10−3 | Co-precipitation method | 117 |
| 8 | Ce–Ru/Al2O3 (1:1 molar ratio of Ce/Ru) |
400 | 1 | 14.352 | 75 | 3% Ru, 4.1% Ce | 24000 |
1.0 | 0.8 | −0.2 | 169 | 0.029 | Co-precipitation method | 117 |
| 9 | Ru/CeO2 (cycle) | 200 | 0.1 | 0.65 | 60 | 5% Ru | 36000 |
— | — | — | — | — | Impregnation method | 118 |
| 10 | Ru/CeO2 (steady state) | 200 | 0.1 | 0.12 | 60 | 5% Ru | 36000 |
— | — | — | — | — | Impregnation method | 118 |
| 11 | Ru/CeO2 (cycle) | 300 | 0.1 | 1.87 | 60 | 5% Ru | 36000 |
— | — | — | — | — | Impregnation method | 118 |
| 12 | Ru/CeO2 (steady state) | 300 | 0.1 | 2.23 | 60 | 5% Ru | 36000 |
— | — | — | — | — | Impregnation method | 118 |
| 13 | Ru/CeO2-w | 400 | 1 | 22.62 | 51.5 | 3% Ru | 36000 |
0.96 | 1.01 | −0.75 | 38 | 21.8 × 10−3 | Precipitation method | 119 |
| 14 | Ru/CeO2-e | 400 | 1 | 4.582 | 58.3 | 3% Ru | 36000 |
0.80 | 0.43 | −0.66 | 49 | 4.4 × 10−3 | Precipitation method | 119 |
| 15 | Ru/MgO + BaO/MgO | 400 | 1 | 1.25 | 93.4 | 2.5% Ru, 2.5% Ba | 80000 |
0.92 | −0.40 | −0.32 | 223 | — | Hydrothermal synthesis method | 120 |
| 16 | Ba–Ru/MgO-20 | 400 | 1 | 24.72 | 83.9 | 4.8% Ru, 4.9% Ba | 80000 |
0.86 | 0.35 | −0.78 | 191 | — | Hydrothermal impregnation-reduction method | 120 |
| 17 | Ba–Ru/MgO-10 | 400 | 1 | 28.2 | 75.2 | 4.7% Ru, 4.8% Ba | 80000 |
0.86 | 0.43 | −0.75 | 189 | — | Hydrothermal impregnation–reduction method | 120 |
| 18 | Ba–Ru/MgO-2 | 400 | 1 | 38.4 | 69.7 | 4.8% Ru, 4.8% Ba | 80000 |
0.86 | 0.52 | −0.82 | 195 | — | Hydrothermal impregnation–reduction method | 120 |
| 19 | Ba–Ru/MgO–2H | 400 | 1 | 45.36 | 64.3 | 4.8% Ru, 4.8% Ba | 80000 |
0.84 | 0.59 | −0.78 | 185 | — | Hydrothermal impregnation–reduction method | 120 |
| 20 | Co(10)/Nd2O3 | 470 | 6.3 | 59.41 | — | 9.9% Co, 77.2% Nd | — | — | — | — | 27.3 | 0.085 | Deposition–precipitation method | 121 |
| 21 | Co(19)/Nd2O3 | 470 | 6.3 | 81.76 | — | 19.4% Co, 65.6% Nd | — | — | — | — | 32.0 | 0.130 | Deposition–precipitation method | 121 |
| 22 | Co(31)/Nd2O3 | 470 | 6.3 | 74.70 | — | 30.8% Co, 59.6% Nd | — | — | — | — | 36.5 | 0.115 | Deposition–precipitation method | 121 |
| 23 | Co(39)/Nd2O3 | 470 | 6.3 | 83.52 | — | 38.6% Co, 46.8% Nd | — | — | — | — | 39.6 | 0.110 | Deposition–precipitation method | 121 |
| 24 | Co(50)/Nd2O3 | 470 | 6.3 | 107.06 | — | 49.6% Co, 39.5% Nd | — | — | — | — | 41.8 | 0.135 | Deposition–precipitation method | 121 |
| 25 | Ru/MgO–CeO2 Ce/Mg = 1 | 400 | 0.1 | 3000 ppm | 41 | 3% Ru | — | 1.21 | 0.36 | −0.65 | 64.44 | — | Co-precipitation method | 122 |
| 26 | Ru/CeO2 | 400 | 0.1 | 2850 ppm | 38 | 3% Ru | — | 1.14 | 0.55 | −0.67 | 102.77 | — | Precipitation method | 122 |
| 27 | Ru/MgO | 450 | 0.1 | 1480 ppm | 115 | 1% Ru | — | 1.35 | −1.41 | −0.37 | 25 | — | Precipitation method | 123 |
| 28 | Ce/Mg = 0.01 | 450 | 0.1 | 1890 ppm | 78 | 1% Ru | — | 1.07 | −0.91 | −0.08 | 58 | — | Impregnation–precipitation method | 123 |
| 29 | Ce/Mg = 0.03 | 400 | 0.1 | 1942 ppm | 60 | 1% Ru | — | 0.74 | −0.37 | −0.08 | 108 | — | Impregnation–precipitation method | 123 |
| 30 | Ce/Mg = 0.05 | 400 | 0.1 | 2192 ppm | 53 | 1% Ru | — | 0.81 | −0.34 | −0.17 | 102 | — | Impregnation–precipitation method | 123 |
| 31 | Ce/Mg = 0.1 | 400 | 0.1 | 2492 ppm | 44 | 1% Ru | — | 0.84 | 0.19 | −0.35 | 116 | — | Impregnation–precipitation method | 123 |
| 32 | Ce/Mg = 0.3 | 400 | 0.1 | 2458 ppm | 44 | 1% Ru | — | 0.89 | 0.15 | −0.23 | 116 | — | Impregnation–precipitation method | 123 |
| 33 | Ce/Mg = 0.5 | 450 | 0.1 | 1848 ppm | 62 | 1% Ru | — | 0.64 | −0.08 | −0.73 | 102 | — | Impregnation–precipitation method | 123 |
| 34 | Ru/CeO2 | 400 | 0.1 | 2014 ppm | 58 | 1% Ru | — | 0.96 | 0.01 | −0.35 | 95 | — | Precipitation method | 123 |
| 35 | 3 Ru/Y2O3-m | 400 | 1 | 2.5 | 108 | 3% Ru | 24000 |
— | — | — | 44.9 | 5 × 10−3 | Hydrothermal and milling method | 124 |
| 36 | 3 Ru/Y2O3-p | 400 | 1 | 9 | 99 | 3% Ru | 24000 |
— | — | — | 44.8 | 0.014 | Hydrothermal and milling method | 124 |
| 37 | 5 Ru/Y2O3-m | 400 | 1 | 4.378 | 104 | 5% Ru | 24000 |
1.1 | −1.3 | −0.7 | 45.8 | 5 × 10−3 | Hydrothermal and milling method | 124 |
| 38 | 5 Ru/Y2O3-p | 400 | 1 | 21.12 | 97 | 5% Ru | 24000 |
1.1 | −0.9 | −0.5 | 47.6 | 0.019 | Hydrothermal and milling method | 124 |
| 39 | Ru/LCO | 400 | 1 | 10.8 | — | 5% Ru | 54000 |
— | — | — | 53 | — | Co-precipitation method | 125 |
| 40 | Ru/4Ba/LCO | 400 | 1 | 23.8 | — | 5% Ru, 4% Ba | 54000 |
— | — | — | 38 | — | Co-precipitation–impregnation method | 125 |
| 41 | Ru/4Cs/LCO | 400 | 1 | 27.1 | — | 5% Ru, 4% Cs | 54000 |
— | — | — | 11 | — | Co-precipitation–impregnation method | 125 |
| 42 | Ru/4K/LCO | 400 | 1 | 35.9 | — | 5% Ru, 4% K | 54000 |
— | — | — | 17 | — | Co-precipitation–impregnation method | 125 |
| 43 | Ru/4Li/LCO | 400 | 1 | 6.6 | — | 5% Ru, 4% Li | 54000 |
— | — | — | 6 | — | Co-precipitation–impregnation method | 125 |
| 44 | Ru/Sm2O3 | 400 | 1 | 13.596 | — | 5% Ru | 24000 |
1.08 | −0.72 | −0.92 | 54.0 | — | Hydrothermal, deposition–precipitation method | 126 |
| 45 | Ru/Sm2O3 (activated) | 400 | 1 | 32.214 | — | 5% Ru | 24000 |
1.08 | −0.72 | −0.92 | 54.0 | — | Hydrothermal, deposition–precipitation method | 126 |
| 46 | Ru/CeO2 | 400 | 1 | 4 | — | 2.1% Ru | 36000 |
0.81 | −0.57 | −1.29 | 71 | 3.9 × 10−3 | Hydrothermal method | 96 |
| 47 | Ru/CeO2 + Ru/TiO2 | 400 | 1 | 4.120 | — | — | 36000 |
— | — | — | — | 3.9 × 10−3 | Hydrothermal method | 96 |
| 48 | Ru/Cs+(0.35)/CeO2 | 350 | 0.1 | 4.62 | — | 2% Ru | 36000 |
0.6 | 0.2 | −0.8 | 23.4 | 34.5 × 10−3 | Impregnation method | 127 |
| 49 | Cs+(0.35)/Ru/CeO2 | 350 | 0.1 | 2.11 | — | 2% Ru | 36000 |
0.8 | −0.3 | −0.9 | — | 20.9 × 10−3 | Impregnation method | 127 |
| 50 | Cs/Ru/MgO | 350 | 0.1 | 1.01 | — | 2% Ru | 36000 |
0.7 | −0.7 | 0 | — | — | Impregnation method | 127 |
| 51 | Ru/Cs+(0.35)/CeO2 | 350 | 0.1 | 2.69 | — | 1% Ru | 36000 |
— | — | — | — | 15.4 × 10−3 | Impregnation method | 127 |
| 52 | Ru/Cs+(0.35)/CeO2 | 350 | 0.1 | 4.27 | — | 5% Ru | 36000 |
— | — | — | — | 46.2 × 10−3 | Impregnation method | 127 |
| 53 | Ru/Cs+(0.2)/CeO2 | 350 | 0.1 | 0.96 | — | 2% Ru | 36000 |
— | — | — | 65.5 | 7.1 × 10−3 | Impregnation method | 127 |
| 54 | Ru/CeO2 | 350 | 0.1 | 0.31 | — | 2% Ru | 36000 |
— | — | — | 112.3 | 0.69 × 10−3 | Impregnation method | 127 |
| 55 | Ru/Cs+(0.6)/CeO2 | 350 | 0.1 | 0.94 | — | 2% Ru | 36000 |
— | — | — | 3.7 | 22.4 × 10−3 | Impregnation method | 127 |
| 56 | Mo/CeO2 | 400 | 1 | 0.438 | 60 | Mo:Ce mass ratio = 0.35 |
36000 |
0.68 | 0.14 | −0.7 | 8 | Co-precipitation method | 128 | |
| 57 | Co/La2O3 | 350 | 1 | 2.8 | — | 20% Co | 72000 |
0.97 | 0.32 | −0.51 | 25.8 | 0.051 | Precipitation–impregnation method | 129 |
| 58 | Co/Ba/La2O3 500 | 350 | 1 | 2.4 | 73.1 | 20% Co | 72000 |
— | — | — | 37.5 | 0.019 | Precipitation–impregnation method | 129 |
| 59 | Co/Ba/La2O3 700 | 350 | 1 | 19.3 | 45.7 | 20% Co | 72000 |
0.85 | 0.43 | −0.17 | 24.9 | 0.223 | Precipitation–impregnation method | 129 |
| 60 | Co/Ba/La2O3 800 | 350 | 1 | 17.1 | — | 20% Co | 72000 |
— | — | — | 10.1 | 0.304 | Precipitation–impregnation method | 129 |
| 61 | 1.4Ru/CeO2 | 400 | 1 | 3.8a | 72 | 1.4% Ru | 72000 |
0.62 | 0.27 | −0.39 | 92 | 2.028a | Solvothermal method | 130 |
| 62 | 2.8Ru/CeO2 | 400 | 1 | 10.4 | 64 | 2.8% Ru | 72000 |
0.58 | 0.33 | −0.47 | 103 | 2.78a | Solvothermal method | 130 |
| 63 | 5.3Ru/CeO2 | 400 | 1 | 5a | 87 | 5.3% Ru | 72000 |
1.30 | −0.35 | −0.64 | 64 | 0.56a | Solvothermal method | 130 |
| 64 | Ru/ZrO2 | 400 | 1 | 5.44 | 125 ± 4 | 1.92% Ru | 60000 |
0.69 | −0.58 | −0.73 | 29 | 8 × 10−3 | Precipitation-milling method | 65 |
| 65 | Ru(2)/Sm2O3 | 400 | 1 | 13.7a | 106.3 | 2% Ru | 24000 |
— | — | — | 41.0 | 0.0247 | Hydrothermal, ion-adsorption method | 131 |
| 66 | Ru(3)/Sm2O3 | 400 | 1 | 17.5a | 109.9 | 3% Ru | 24000 |
— | — | — | 49.6 | 0.0221 | Hydrothermal, ion-adsorption method | 131 |
| 67 | Ru(4)/Sm2O3 | 400 | 1 | 22.570 | 104.8 | 4% Ru | 24000 |
— | — | — | 54.3 | 0.0229 | Hydrothermal, ion-adsorption method | 131 |
| 68 | Ru(5)/Sm2O3 | 400 | 1 | 23.040 | 105.1 | 5% Ru | 24000 |
— | — | — | 56.2 | 0.0135 | Hydrothermal, ion-adsorption method | 131 |
| 69 | Ru/Pr2O3 | 400 | 1 | — | — | — | — | 1.11 | 0.02 | −0.55 | 24 | 0.24 | Precipitation–impregnation method | 132 |
| 70 | Ru/CeO2 | 400 | 1 | — | — | — | — | 0.85 | 0 | −0.24 | 34 | 0.09 | Precipitation–impregnation method | 132 |
| 71 | Ru/La2O3 | 400 | 1 | — | — | — | — | 1.10 | −0.03 | −0.57 | 22 | 0.24 | Precipitation–impregnation method | 132 |
| 72 | Ru/Nd2O3 | 400 | 1 | — | — | — | — | 1.25 | −0.29 | −0.61 | 22 | 0.09 | Precipitation–impregnation method | 132 |
| 73 | Ru/Sm2O3 | 400 | 1 | — | — | — | — | 1.11 | −0.11 | −0.47 | 17 | 0.08 | Precipitation–impregnation method | 132 |
| 74 | Ru/Gd2O3 | 400 | 1 | — | — | — | — | 1.12 | 0 | −0.23 | 12 | 0.05 | Precipitation–impregnation method | 132 |
| 75 | Ru/Yb2O3 | 400 | 1 | — | — | — | — | — | — | — | 4 | — | Precipitation–impregnation method | 132 |
| 76 | Ru/Ho2O3 | 400 | 1 | — | — | — | — | — | — | — | 3 | — | Precipitation–impregnation method | 132 |
| 77 | Ru/Er2O3 | 400 | 1 | — | — | — | — | — | — | — | 4 | — | Precipitation–impregnation method | 132 |
| 78 | Ru/Tb2O3 | 400 | 1 | — | — | — | — | — | — | — | 3 | — | Precipitation–impregnation method | 132 |
| 79 | Cs–Co/MgO | 300 | 1 | 0.48 | — | 3.20% Co | 60000 |
— | — | — | 0.24 × 10−3 | Impregnation method | 68 | |
| 80 | Co(10)/Mg–La | 400 | 6.3 | 16.0 | 62 | 10% Co | 140000 |
— | — | — | 25 | 0.025 | Co-precipitation, impregnation method | 133 |
| 81 | Co(20)/Mg–La | 400 | 6.3 | 24.2 | 58 | 20% Co | 140000 |
— | — | — | 25 | 0.023 | Co-precipitation, impregnation method | 133 |
| 82 | Co(30)/Mg–La | 400 | 6.3 | 31.8 | 57 | 30% Co | 140000 |
— | — | — | 30 | 0.025 | Co-precipitation, impregnation method | 133 |
| 83 | Co(40)/Mg–La | 400 | 6.3 | 40.3 | 59 | 40% Co | 140000 |
— | — | — | 33 | 0.025 | Co-precipitation, impregnation method | 133 |
| 84 | Co(50)/Mg–La | 400 | 6.3 | 38.2 | 60 | 50% Co | 140000 |
— | — | — | 33 | 0.021 | Co-precipitation, impregnation method | 133 |
| 85 | Ru/CeO2 | 400 | 1 | 16.867 | 75 | 2.7% Ru | 36000 |
0.48 | 0.66 | 56 | 16.2 × 10−3 | Calcination–impregnation method | 134 | |
| 86 | Mo/CeO2 | 400 | 1 | 0.438 | 60 | Mo:Ce mass ratio = 0.35 |
36000 |
0.68 | 0.14 | −0.7 | 8 | — | Co-precipitation/impregnation/physical mixing | 128 |
| 87 | Ru/CeO2–H | 400 | 1 | 4.064 | 60 | 1% Ru | 36000 |
1.0 | −0.53 | −0.72 | 86 | 0.058 | Hydrothermal, reduction in hydrogen | 135 |
| 88 | Ru/CeO2–C250H | 400 | 1 | 8.033 | 72 | 1% Ru | 36000 |
1.2 | 0.3 | −0.75 | 82 | 0.123 | Hydrothermal, reduction, CO treatment | 135 |
| 89 | Ru/CeO2–C500H | 400 | 1 | 9.244 | 67 | 1% Ru | 36000 |
1.3 | 0.36 | −0.79 | 81 | 0.143 | Hydrothermal, reduction, CO treatment | 135 |
| 90 | Ru/BTO | 400 | 0.1 | 1.506 | 69.4 | 2% Ru | 36000 |
1.38 | −0.47 | −0.27 | — | 5.3 × 10−3 | Hydrothermal method | 136 |
| 91 | Ru/CaBTO600 | 400 | 0.1 | 4.944 | 72.7 | 2.2% Ru | 36000 |
1.07 | 0.33 | −0.32 | — | 0.0231 | Hydrothermal, thermal modification with CaH2 | 136 |
| S. no. | Catalyst | Temp. (°C) | Pressure (MPa) | Rate (mmol g−1 h−1) | Activation energy (kJ mol−1) | Metal% | WHSV | N2 order | H2 order | NH3 order | Surface area (m2 g−1) | Turnover frequency (TOF) | Catalyst synthesis method | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CoCe-0.05 | 400 | 5 | 2.35 | 196 | 18000 |
— | — | — | 18.4 ± 0.06 | — | Co-precipitation method | 137 | |
| 2 | 5Ba/CoCe-0.01 | 400 | 5 | 2.94 | 99 | 5% Ba | 18000 |
— | — | — | 3.05 ± 0.10 | — | Co-precipitation–impregnation method | 137 |
| 3 | 5Ba/CoCe-0.05 | 400 | 5 | 14.94 | 57 | 5% Ba | 18000 |
— | — | — | 7.61 ± 0.13 | — | Co-precipitation–impregnation method | 137 |
| 4 | 5Ba/CoCe-0.20 | 400 | 5 | 5.29 | 113 | 5% Ba | 18000 |
— | — | — | 13.9 ± 0.20 | — | Co-precipitation–impregnation method | 137 |
| 5 | 5Ba/CoCe-0.50 | 400 | 5 | 3.94 | 103 | 5% Ba | 18000 |
— | — | — | 15.3 ± 0.23 | — | Co-precipitation–impregnation method | 137 |
| 6 | Ru/BNO | 400 | 0.1 | — | — | — | 36000 |
1.01 | −0.27 | −0.62 | 18 | — | Hydrothermal, physical grinding method | 138 |
| 7 | Ru/0.20CaBNO | 400 | 0.1 | 4.012 | — | 4% Ru | 36000 |
0.89 | 1.12 | −0.54 | 12 | 6.4 × 10−3 | Hydrothermal, physical grinding method | 138 |
| 8 | 3Cs–Ru/0.20CaBNO | 400 | 0.1 | 5.369 | — | 4% Ru | 36000 |
0.79 | 1.24 | −0.89 | 9 | 9.8 × 10−3 | Hydrothermal, physical grinding method | 138 |
| 9 | Ru1.1/BaCeO3 | 400 | 1 | 19.4 | 72 | 0.44% Ru | 60000 |
0.72 | 0.13 | −0.77 | 4.5 | 0.124 | Sol–gel method | 139 |
| 10 | Ru1.5/BaCeO3 | 400 | 1 | 17.2 | 93 | 0.42% Ru | 60000 |
— | — | — | 4.9 | 0.115 | Sol–gel method | 139 |
| 11 | Ru2.0/BaCeO3 | 400 | 1 | 5.5 | 112 | 0.44% Ru | 60000 |
— | — | — | 3.6 | 0.036 | Sol–gel method | 139 |
| 12 | Ru3.0/BaCeO3 | 400 | 1 | 3.4 | 127 | 0.46% Ru | 60000 |
1.07 | −1.20 | −2.60 | 4.0 | 0.021 | Sol–gel method | 139 |
| 13 | Ru/BaCe0.9Y0.1O3−δ (Ru is exsolved) | 450 | 0.1 | 3.68 | 160 | 0.95% Ru | 360000 |
— | — | — | 3.08 | 0.19962 | Glycine nitrate process (GNP) | 140 |
| 14 | Ru/BaCe0.9Y0.1O3−δ (Ru is impregnated) | 450 | 0.1 | 0.45 | 5.28 | 0.95% Ru | 144000 |
— | — | — | 3.33 | 3.5 × 10−3 | Glycine nitrate process (GNP) | 140 |
Oxide-supported catalysts based on stabilization and tunability of active sites favor effective nitrogen activation in ammonia synthesis. Generally, these catalysts are transition metal oxides (for example, CeO2 and TiO2) or perovskite-type oxides, and their improvement of catalytic performance occurs through the modification of metal–support interactions and electronic properties. Various synthesis strategies were employed to modify the properties of oxide-based catalysts.
The precipitation method allowed the regulation of nucleation during the synthesis of La(OH)3,115 which was followed by hydrothermal treatment to enhance crystallinity followed by a deposition–precipitation method for uniform dispersion of Ru within La2O3. During precipitation, La(OH)3 nuclei form as KOH increases the pH, which leads to the controlled growth of hydroxide particles. Hydrothermal treatment enhances crystallinity, while deposition–precipitation ensures uniform Ru dispersion by controlled nucleation and growth of Ru species on La2O3 using urea as a precipitating agent. Lanthanum nitrate was dissolved in DI water to which 20% KOH was added till the pH became 14 and a white precipitate was formed. The mixture was continuously stirred for 30 minutes and then transferred to a Teflon-lined stainless-steel autoclave in which the mixture was made to undergo hydrothermal treatment at a temperature of 180 °C for 12 h. Finally, the mixture was washed several times with DI water to remove any remaining ions and then dried at a temperature of 80 °C for 12 h. Ru was loaded on this support by a deposition–precipitation method. La(OH)3 was added to an aqueous solution of RuNO(NO3)3 followed by addition of urea in a Ru-to-urea molar ratio of 1/200. The mixture was then thermally treated at 80 °C for 8 h and then aged at ambient temperature for a period of 12 h. This was followed by filtration and washing with DI water and finally drying at 80 °C for 6 h.
A combination of milling method and reduction were used for Y2O3-based catalysts,124 while calcination under controlled conditions gave stability and phase formation in different supports such as La2O3 and CeO. The Y2O3 support was synthesized hydrothermally. RuO2 NPs were prepared by adding a RuCl3 precursor to a KOH solution under constant stirring, followed by filtration, washing, and drying at a temperature of 80 °C for a period of 8 h. Ru/Y2O3-m were prepared by milling RuO2 and Y2O3 in a mortar for 10 min and then reducing at 400 °C for 2 h under 75% H2/N2. Fig. 17 shows the TEM images along with Ru particle size distributions for 3% Ru/Y2O3-m (a and e), 5% Ru/Y2O3-m (b and f), 3% Ru/Y2O3-p (c and g), and 5% Ru/Y2O3-p (d and h) catalysts, synthesized using the above method.
 | ||
| Fig. 17 Various synthesis strategies for oxide-based catalysts. TEM images along with Ru particle size distributions for 3% Ru/Y2O3-m (a and e), 5% Ru/Y2O3-m (b and f), 3% Ru/Y2O3-p (c and g), and 5% Ru/Y2O3-p (d and h) synthesized via a combination of hydrothermal, milling and reduction methods. Adapted with permission from ref. 124. Copyright 2023, the Royal Society of Chemistry. | ||
The co-precipitation method enabled the formation of homogeneous MgO–Nd2O3 (ref. 116) supports, allowing fine tuning of the Mg/Nd ratio, followed by wet impregnation for efficient loading of Co. Magnesium and neodymium nitrates were dissolved in water at 30 °C, followed by dropwise addition of K2CO3 and KOH under stirring until the pH became 11. The precipitate was aged at 65 °C for 18 h, washed, dried (120 °C, 18 h), and calcined (450 °C, 18 h).
Co (10 wt%) was loaded on these supports via wet impregnation using cobalt nitrate, followed by drying (120 °C, 18 h) and calcination (500 °C, 18 h). Similarly, benchmark Co/MgO and Co/Nd2O3 catalysts were also prepared. Before catalytic testing, all samples were reduced in H2 at 550 °C to ensure complete Co precursor reduction.
CeO2 (ref. 119) was synthesized using the precipitation method with ethanol (CeO2-e) and water (CeO2-w) as solvents. For CeO2-e synthesis, cerium nitrate was dissolved in ethanol, followed by the dropwise addition of 28 wt% ammonia solution and dilution with ethanol. The mixture was stirred for 2 h, then the precipitate was filtered, washed with ethanol, dried at 60 °C overnight, and finally calcined at 500 °C for 2 h. CeO2-w was similarly prepared, using water as the solvent instead of ethanol. Ru was then incorporated onto these supports using the usual wetness impregnation method with ruthenium nitrosyl nitrate as the precursor.
In the synthesis of Ru/MgO,120 a hydrothermal method facilitated the development of hexagonal nanoplates having better porosity and dispersion, while Ba impregnation changed Ru/MgO interfaces, favouring the optimization of catalytic sites. RuCl3 and NaOH mixed with MgO in water and thermally treated at 160 °C. This resulted in the formation of Ru-modified Mg(OH)2 with a hexagonal nanoplate morphology, which transformed into mesoporous MgO after calcination at 500 °C. Ru4+ was atomically dispersed on MgO, and subsequent H2 reduction at a temperature of 500 °C formed Ru particles. Ba–Ru/MgO catalysts (Ba/Ru molar ratio = 0.75) were prepared by impregnating Ru/MgO with Ba (NO3)2, followed by H2 treatment at 500 °C. Ru catalyzed Ba(NO3)2 decomposition at a lower temperature (225 °C), forming BaO–Ru interfaces. To control BaO–Ru interactions, the temperature ramp was varied (2–20 °C min−1), with the slowest ramp (2 °C min−1) in humid H2 producing “Ba–Ru/MgO–2H.” A physical mixture of Ru/MgO and BaO/MgO was also prepared as a control sample.
The solvothermal method was used to prepare Ru–CeO2 (ref. 130) composites, conducive to high Ru dispersion. Here, the Ru–Ce–BTC MOF forms through coordination between cerium, Ru, and BTC in DMF, creating a porous structure. Calcination converts the MOF into RuOx–CeO2, and subsequent hydrogen reduction at 450 °C generates highly dispersed Ru–CeO2 catalysts with controlled Ru loading. The Ru–Ce–BTC MOF was synthesized by stirring cerium nitrate, Ru(acac)3, and benzene-1,3,5-tricarboxylate (BTC) in DMF at 130 °C for 20 h. This was followed by washing the solid, drying at 80 °C, and calcination at a temperature of 450 °C for 3 h in air to obtain RuOx–CeO2. Finally, the solids were reduced at 450 °C under 10% H2/N2 to yield Ru–CeO2 catalysts with Ru contents of 1.4–5.3 wt%. For comparison, the Ce–BTC MOF was synthesized without Ru, followed by calcination to obtain CeO2. An impregnated Ru/CeO2 catalyst was prepared by adding Ru(acac)3 in ethanol to CeO2, drying at 100 °C, calcining at 450 °C, and reducing under 10% H2/N2. Fig. 18 shows the schematic of this preparation method.
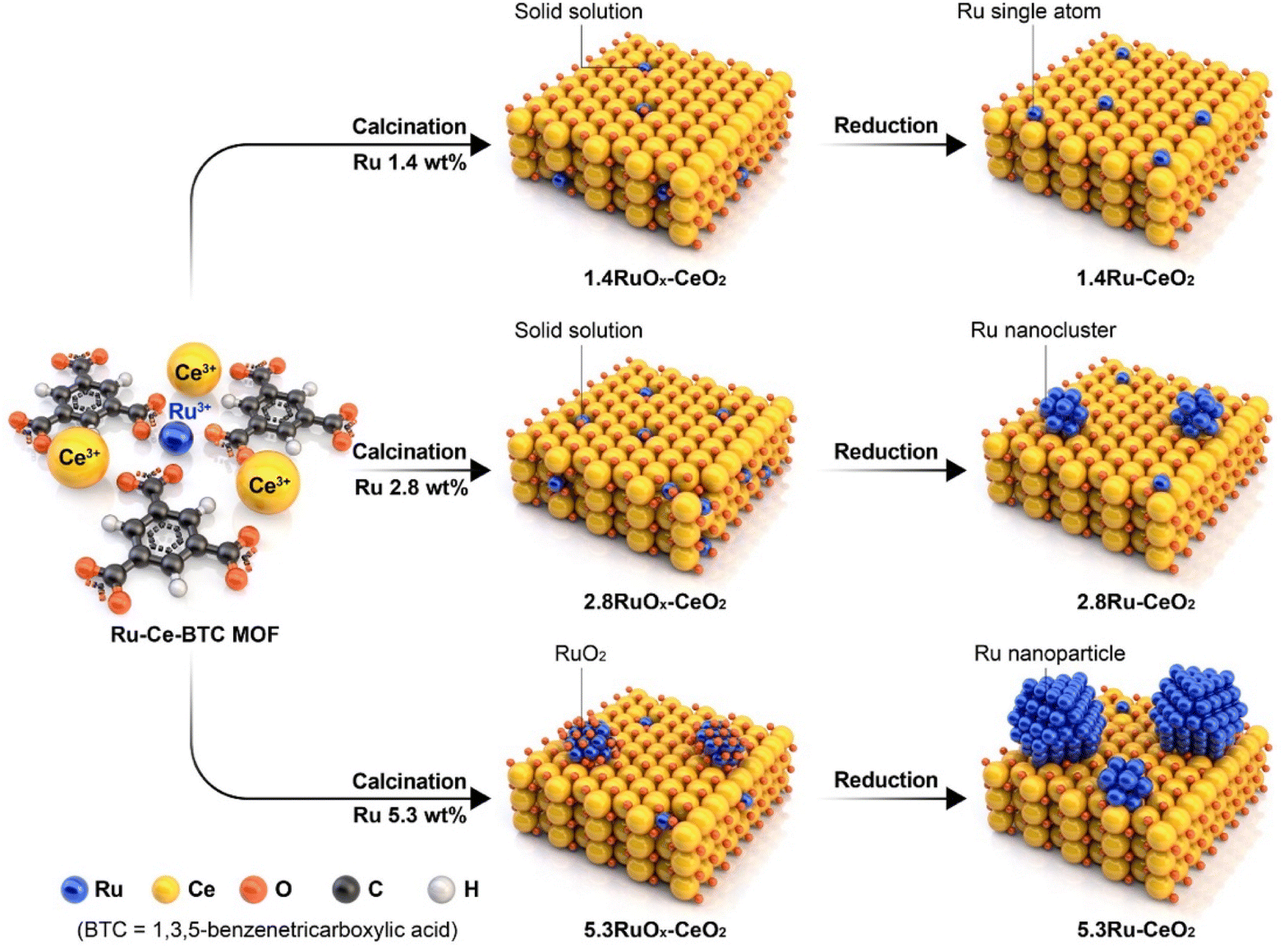 | ||
| Fig. 18 Method of synthesis of Ru–CeO2 catalysts. Here Ru–Ce–BTC shows the raw form, which is followed by calcination to form RuOx–CeO2 and then Ru–CeO2 via the reduction method. Adapted with permission from ref. 130. Copyright 2022, Elsevier. | ||
The hydrothermal method was also used for the development of nanostructured supports such as Y2O3 and Sm2O3,131 and thereafter, deposition and reduction to obtain the final catalysts. Fig. 19 illustrates this synthesis method in detail. Sm(OH)3 was prepared by dissolving samarium nitrate in DI water followed by adjusting the pH to ∼13 with 20 wt% KOH and stirring for 15 min. The mixture was then hydrothermally treated at 180 °C for 12 h, filtered, washed, and dried at 80 °C. Finally, Sm2O3 was obtained by calcining Sm(OH)3 at 500 °C for 2 h. Ru/Sm2O3 catalysts (2–5 wt% Ru) were synthesized via an ion adsorption method by dispersing Sm(OH)3 support in water, adding a RuCl3 precursor solution, and stirring for 6 h. The precipitate was filtered, washed, dried at 80 °C, and reduced in 75% H2/N2 at 400 °C for 2 h. Ru/Sm(OH)3 reduced at 400 °C and 1.0 MPa for a period of 1 h was termed the “reduced catalyst,” and another sample reduced for 12 h was termed the “activated catalyst.” As a reference, 4% Ru NPs/Sm2O3 was prepared by an impregnation method, dried at 80 °C, calcined at 550 °C in Ar for 2 h, and reduced at 400 °C in 75% H2/N2 to obtain the final catalyst.
 | ||
| Fig. 19 (a) Schematic of the synthesis method of Ru(cluster)/Sm2O3. (b) Green ion adsorption method for the synthesis of Ru/Sm2O3 catalyst. (c) TEM image of the prepared Sm(OH)3 support. (d) TEM image of 4% Ru/Sm2O3 catalyst. (e) HAADF-STEM image of 4% Ru/Sm2O3 and EDX elemental mappings for O, Ru and Sm. Adapted with permission from ref. 131. Copyright 2022, the Royal Society of Chemistry. | ||
The repetitive deposition–precipitation method allowed for the fine loading of Co onto Nd2O3 (ref. 121) through multiple cycles for enhanced metal–support interaction.
Nd2O3 powder was calcined at 800 °C for 16 h to remove hydroxides and carbonates. Co catalysts were prepared via recurrent deposition–precipitation. CoCO3 was precipitated using K2CO3 from cobalt nitrate, on an Nd2O3 suspension at a temperature of 85 °C at a rotation speed of 500 rpm, until the pH became 9, and then aged for 1 h. The precipitate was filtered, washed till the pH turned 7 and then dried at 120 °C for 24 h. Finally, it was calcined at 500 °C for a period of 5 h. Higher Co loadings were achieved by sequential deposition–calcination cycles. The final catalysts (Co(X)/Nd2O3) were tabletted, crushed (0.2–0.63 mm), and reduced in situ before use.
Precipitation and coprecipitation methods are important for the precise control of the composition of CoCe-m perovskite catalysts,137 which guarantees uniform active site distribution, increasing the catalytic efficiency. For Ba incorporation, the impregnation method ensures good loading on the Co catalyst and enhances thermal stability and dispersion of the active components, which is vital for high-performance reactions. The sol–gel method used for the synthesis of BaCeO3 (ref. 139) serves to promote phase purity and homogeneity, which are needed for stability and activity of this compound in high-temperature reactions. The modified alkaline ethylene glycol reduction method for making Ru nanoparticles allows for size precision control helpful in maximizing surface area and active sites from the perspective of catalytic activity in hydrogenation and calcination reactions. Finally, the glycine nitrate process (GNP)140 for Ru-doped BCY enhances the exsolution and stability of catalysts under reducing conditions, making it potential for difficult catalytic processes such as fuel cells and ammonia synthesis. All these synthesis methods are optimizing the performance of individual catalysts, as well as ensuring their robustness and stability across multiple industrial applications worldwide.
Recently, Sato et al. have published a comprehensive review for the catalytic role of basic oxide-Ru interface for generating ammonia under mild conditions.141 Within the literature, cobalt-based catalysts are generally employed with the support materials to increase the catalytic activity. Hence, several supports such as activated carbon,142,143 cerium oxide,144 magnesium oxide,145 or mixed magnesium–lanthanum oxides146,147 are frequently used. Moreover, it is found that catalyst supports such as electrides, hydrides, and rare earth metal nitrides rapidly lose their activity with time, primarily because of the tendency to agglomerate the active metal species.
In this regard, Ronduda et al. reported a detailed study consisting of cobalt catalysts supported on mixed MgO–Ln2O3 (Ln = La, Nd, and Eu), and studied the effect of metallic cobalt loading for the activity towards ammonia synthesis.116,148 Mixing magnesium oxide with lanthanide oxide provides the possibility to reduce the price, and exploit the advantages of both oxides. This series of works showed that tailoring the support composition by selecting lanthanide elements differing in electronegativity values remarkably alter active phase dispersion, access of active sites for hydrogen and density of basic sites on the surface. Consequently, this results in a better activity than the other studied catalysts. For instance, a series of Co/MgO–Nd2O3 catalysts with varying molar ratios of Mg/Nd (2, 4, 7, 10, and 12) resulted in a gradual change in the adsorption as well as textural and structural properties.116 The variation in the catalytic activity of Co/MgO–Nd2O3 was observed due to the difference in adsorption properties. Mg/Nd was reported to have the maximum number of basic sites on the surface and exhibited the highest Co dispersion, which resulted in the best distribution of Co particles. All this led to enhanced catalytic activity of the Co/10MgO–Nd2O3 catalyst over other conventional catalysts. As the binary composite support consisting of MgO and Nd2O3 was used, the resulting catalyst demonstrated a greater activity than a single-composite support in aspects of catalyst activity, stability and cost of synthesis. Ammonia synthesis activity of Co/Mg–Nd catalysts with varying Mg/Nd ratios and comparison with different catalysts are shown in Fig. 20a–c. Moreover, the activity was closely related to the molar ratio of Mg/Nd, and exhibited a rate of 23.4 mmol g−1 h−1, which increased to 39.3 mmol g−1 h−1 when Co/10Mg–Nd was employed. This also led to a significant decrease in the activation energy from 58.9 kJ mol−1 (Co/2Mg–Nd) to 53.7 kJ mol−1 (Co/10Mg–Nd).
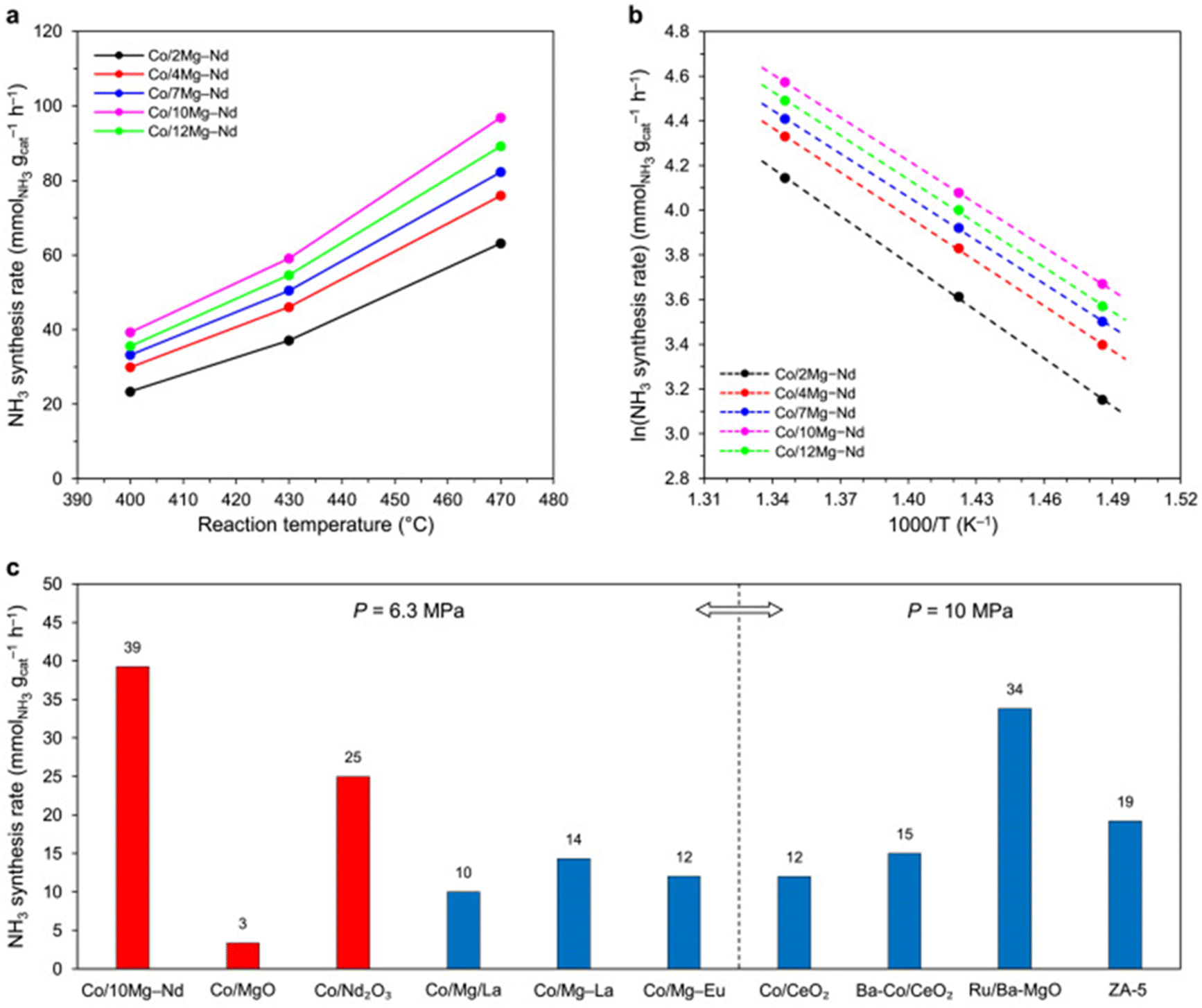 | ||
| Fig. 20 Ammonia synthesis activity of Co/Mg–Nd catalysts with varying Mg/Nd ratios. (a) NH3 synthesis rates as a function of temperature at 6.3 MPa. (b) Arrhenius plots under the same pressure. (c) Comparison of the NH3 synthesis rates at 400 °C across different catalysts, including Co/10Mg–Nd, Co/MgO, and Co/Nd2O3. Adapted with permission from ref. 116. Copyright 2022, Elsevier. | ||
It was thus concluded that different surface adsorption properties, which were tuned by the change in the molar ratio of Mg/Nd, were responsible for the enhanced catalytic activity of the Co/Mg–Nd catalyst. The nitrogen dissociation rate was increased due to the large number of basic sites and greater concentration of oxygen species with electron donating properties. Increased number of basic sites could facilitate strong interaction between magnesium and neodymium oxides.
In another work of Ronduda et al., authors investigated the threshold value for cobalt loading, which could provide high catalytic activity and maximize the utilization of active cobalt surface sites for participating in the ammonia synthesis reaction.133 This study is important for the development of cost-effective and efficient catalysts. Specifically, the authors studied the effect of metal loading on the catalytic activity of Co catalysts supported on MgO–La2O3 oxides (prepared by the wetness impregnation method) for the synthesis of ammonia. A relationship was developed between the amount of Co loading and the rate of ammonia synthesis. The rate reached a maximum at 40 wt% Co deposition. It was noted that changing the Co loading amount does not show any significant change in the surface-specific activity (TOF). The study also shows that ammonia synthesis rate for the given catalyst does not depend on the structural characteristics or size of Co NPs but is a strong function of Co loading amount. At 400 °C and 6.3 MPa, the ammonia synthesis rate of Co (10)/Mg–La was 15.7 mmolNH3 gcat−1 h−1, which was increased to 40.3 mmolNH3 gcat−1 h−1 for Co(50)/Mg–La. However, a further increase in the Co loading from 40 to 50 wt% led to a slight decrease in the rates, owing to the decrease in the number of basic sites. Moreover, the rates were increased by only 20%, when the pressure was increased from 6.3 to 9.0 MPa. Importantly, the catalysts were thermally stable and showed high resistance towards sintering and thus, a negligible decrease in the activity was noticed even after overheating at 600 °C for 75 h. The findings clearly demonstrate that the ammonia synthesis rate was dependent on the amount of Co loading.
Next, researchers have co-incorporated Ba and Ce by a coprecipitation method to prepare (Ba/CoCe) and examine their activity.137 The striking observation was that the BaCeO3 perovskite boosts the ammonia synthesis kinetics. Moreover, the in situ DRIFTS analysis (shown in Fig. 21a) revealed that the reaction at the Co–BaCeO3 interface follows the associative pathway, confirmed by the formation of hydrazine intermediate (–N2H4) and subsequent homolytic dissociation, which generates ammonia under mild conditions. In particular, the catalytic activity of Co, CoCe-m, nBa/Co, and nBa/CoCe-m catalysts were evaluated at a temperature of 400 °C and a pressure of 5.0 MPa. The catalytic performance was compared in terms of weight time yield (WTY, gNH3 gcat−1 h−1) of NH3 over catalysts. The pure Co catalyst exhibited very low activity, but doping Co with Ce up to 0.05 M ratio led to an increase in activity irrespective of the change in Ba content (0, 2, and 5 wt%). A further increase in the Ce amount had negative effects, which suggest a volcano trend in the catalytic activity curve. WTYNH3 (gNH3 gcat−1 h−1) for 5Ba/CoCe-0.05, Ba-5 wt% Ru/CB, Ba-2.5 wt% Ru/CB, and Co@Ba/MgO were 0.254, 1.359, 0.458 and 0.040, respectively. Upon optimizing the content of Ba, it was found that 5Ba/CoCe-0.05 shows the highest activity (WTY = 0.254 gNH3 gcat−1 h−1) among all the catalysts. It was seen that the co-incorporation of a small quantity of Ba and Ce on Co can increase the activity of the Co catalyst to a maximum of 92-folds, comparing pristine Co. To determine the size of reactor and also the process variables, the volume-normalized catalytic activity was determined. To judge how effective the Ba/CoCe catalyst is, a comparison between its space time yield of NH3 (STYNH3, gNH3 cmcat−3 h−1) was made with other benchmark catalysts. Then 5 Ba/CoCe-0.05 exhibited much higher STYNH3 (0.309 gNH3 cmcat−3 h−1) when compared to the Ba-promoted Co catalyst (Co@Ba/MgO, STYNH3 = 0.024 gNH3 cmcat−3 h−1).
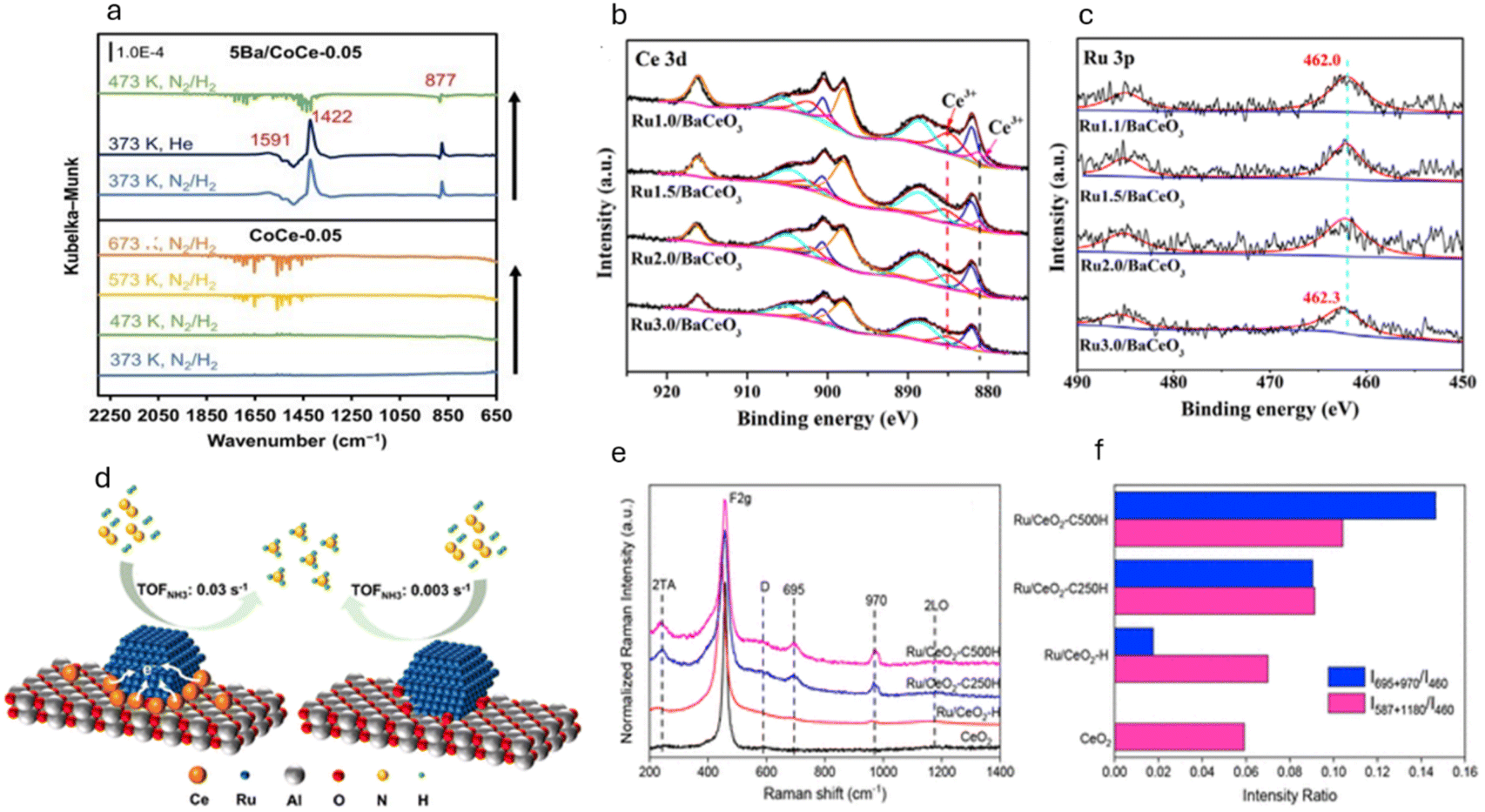 | ||
| Fig. 21 (a) In situ DRIFT spectra for ammonia synthesis over CoCe-0.05 and 5Ba/CoCe-0.05 catalysts examined at 373–673 K (100–400 °C) and 0.1 MPa. Adapted with permission from ref. 137. Copyright 2023, Elsevier. (b) Ce 3d XPS spectra and (c) Ru 3p XPS spectra of the Rux/BaCeO3 catalyst. Adapted with permission from ref. 139. Copyright 2021, Elsevier. (d) Schematic of the ammonia formation over Ce–Ru/Al2O3 and Ru/Al2O3 catalysts. Adapted with permission from ref. 117. Copyright 2023, the Royal Society of Chemistry. (e) Raman spectra of different samples. (f) I695+970/I460 and I587+1180/I460 intensity ratios. Adapted with permission from ref. 135, Copyright 2021, the American Chemical Society. | ||
On evaluation of activation energies for different catalysts, it was found that the activation energy for Co was 141 kJ mol−1 and those for 5Ba/Co and CoCe-0.05 were 157 and 196 kJ mol−1, respectively. The high value of activation energy is in contrast to the fact that they exhibited a higher catalytic activity than that of pure Co. It was thus concluded that singly doped Ba or Ce do not show any reduction in activation energy barrier for the synthesis of ammonia. An apparent activation energy of 57 kJ mol−1 for 5Ba/CoCe-0.05 is the least among all the examined catalysts. These studies show that the presence of Ba and Ce tunes the active sites in such a way that the rate-determining step shifts towards a more kinetically favourable step.
The reason behind high activity of Co/BaCeO3−xNyHz was the progression of the reaction via the Mars-van Krevelen (MvK) mechanism, while the high activation barrier in the case of Co/BaCeO3 was due to reaction progression via the Langmuir–Hinshelwood mechanism. In situ DRIFTS studies were performed to get an insight into the reaction mechanism and the obtained spectra can be found in Fig. 21a.
As Fig. 21a depicts, the 5Ba/CoCe-0.05 catalyst demonstrated peaks at 1591 cm−1 (deformation of –NH2), 1422 cm−1 (wagging of –NH2) and 877 cm−1 (rocking of –N2H4).149–151 Hence, the observation of –N2H4 proved that the reaction proceeds via associative mechanism. When the gas flow was switched to He, peaks with similar intensities were observed, which indicates the presence of strongly chemisorbed species. At 200 °C, the peak intensity diminished, indicating the rapid desorption rate, which is also confirmed from TPD MS studies. From the results of in situ DRIFTS, it was observed that ammonia synthesis at the Co–BaCeO3 interface is mediated by the generation of hydrazine, which dissociated thereafter (i.e. associative mechanism).
In another work, Zhou studied the ammonia synthesis ability of Rux/BaCeO3 catalysts with Ru sizes of 1.1–3.0 nm. It was found that reducing the Ru size significantly boosts NH3 synthesis.139 Ru1.1/BaCeO3 achieved 19.4 mmol gcat−1 h−1 at 400 °C and 1 MPa, 5.7 times the rate of Ru3.0/BaCeO3, outperforming many Ru-based catalysts. Smaller Ru particles enhance Ce3+ and oxygen vacancy formation in BaCeO3, facilitating electron transfer to Ru and improving N2 dissociation. Additionally, smaller Ru size promotes hydrogen spillover to BaCeO3, reducing hydrogen poisoning and enhancing catalytic efficiency. The Ce 3d XPS spectra of Rux/BaCeO3 (x = 1.1–3.0) catalysts reveal ten characteristic peaks (Fig. 21b and c). Ce3+ species are identified at 881.1 and 884.8 eV, while six other peaks correspond to Ce4+. The Ce3+ proportions (30%, 24%, 17%, and 14%) correlate with the increasing x values. The formation of Ce3+ creates oxygen vacancies (Ov) in BaCeO3, with concentrations estimated at 5.0%, 4.0%, 2.8%, and 2.3%. These Ce3+ and Ov species act as electron donors, altering the Ru valence. The Ru 3p XPS spectra show positively charged Ru at higher x values (462.3 eV for Ru3.0) and metallic Ru at lower x values (462.0 eV for Ru1.1). Ru L-edge NEXAFS confirms changes in Ru's electronic structure due to Ce3+ and Ov presence.
Previous works illustrated that the interfacial synergy between metal nanoparticles and the metal oxide significantly influence the catalytic performance of Ru-based catalysts. Hence, the major issue is to precisely construct the interface structure at the nanoscale of the promoter-decorated Ru nanoparticles. This challenge was addressed by Feng et al., who prepared the Ce–Ru/Al2O3 catalyst using a co-precipitation method, and the catalyst consists of Ru nanoparticles supported on γ-Al2O3 with sub-nanometer ceria (CeO2) species.117 A ten-fold increase in the catalytic activity was noticed when compared to Ru/Al2O3 at a temperature of 400 °C and 1.0 MPa. The reason behind this was explained to be the aggregation of sub-nanometre CeO2 species on the Ru nanoparticles, leading to efficient electron transfer from CeO2 to Ru NPs. The presence of CeO2 species shows multiple positive effects as it diminishes hydrogen poisoning, facilitates N2 adsorption and supports hydrogenation of dissociated N* atoms.
In the same work of Feng et al., it was observed that Ru/Al2O3 showed very low activity for ammonia synthesis at 350 °C.117 Even on increasing the temperature to 400 °C, it exhibited a rate of only about 1.435 mmol gcat−1 h−1. However, when the Ru NPs were decorated with CeO2 species, a dramatic increase in its catalytic activity was observed under identical reaction conditions. As the content of CeO2 increased in the Ce–Ru (x:1)/Al2O3 catalysts, the ammonia synthesis rate of Ce–Ru(0.125:1)/Al2O3, Ce–Ru(0.25:1)/Al2O3, Ce–Ru(0.5:1)/Al2O3 and Ce–Ru(1:1)/Al2O3 catalysts enhanced to 3.123, 7.925, 14.040 and 14.350 mmol gcat−1 h−1 at 400 °C, respectively. The synthesis rate of 14.350 mmol gcat−1 h−1 was obtained for Ce–Ru(1:1)/Al2O3 at a temperature of 400 °C, which is about ten-times higher than that of Ru/Al2O3. Comparing the activation energies for Ru/Al2O3 and Ce–Ru(1:1)/Al2O3 catalysts, it was observed that they exhibited similar activation energies of 82 kJ mol−1 and 75 kJ mol−1, respectively, which could be because of a similar mechanism pathway for both the catalysts. Additionally, turnover frequencies (TOF) were calculated based on the metal dispersion values from CO chemisorption results. The Ce–Ru (1:1)/Al2O3 catalyst exhibited much greater values of TOF than the Ru/Al2O3 sample, which is attributed to the outstanding intrinsic activity of the Ce–Ru (1:1)/Al2O3 catalyst. Fig. 21d illustrates a plausible mechanism for ammonia synthesis over Ce–Ru/Al2O3 and Ru/Al2O3 catalysts. In the Ru/Al2O3catalyst, the Ru nanoparticles are entirely covered by the dissociated H* atoms, leading to poor access of the nitrogen molecules to the active Ru nanoparticles and subsequent interaction with nitrogen molecules. On the other hand, in Ce–Ru/Al2O3, the strong interaction between sub-nanometer Ce species and Ru nanoparticles can facilitate nitrogen adsorption and hydrogen activation. Therefore, abundant Ce species on Ru nanoparticles can promote ammonia synthesis via the efficient formation of NHx (x = 1, 2 and 3) species from dissociated H* and N* adatoms on Ru–CeO2 interfaces.
In another work, Lin discovered that in Ru/CeO2 catalysts, strong metal–support interactions (SMSI) reduce the availability of metallic sites, hindering the performance for reactions requiring such sites.135 However, CO activation of Ru/CeO2 enhances the reduction and exposure of Ru species, increases Ce3+ concentration, and creates oxygen vacancies (Ov), leading to electron-rich Ruδ− species and Ruδ−–Ov–Ce3+ sites. This CO-activated Ru/CeO2 catalyst demonstrates high activity in ammonia synthesis and effectively minimizes hydrogen poisoning. H2-TPR, DRIFTS, XPS, and Raman analysis (Fig. 21e and f) indicate that the CO activation of Ru/CeO2 notably increases the reducibility and exposure of Ru species, enhancing Ce3+, oxygen vacancies, and Ruδ−–Ov–Ce3+ concentrations.
Since electrides and metallic hydrides are unstable in air, alternative materials could provide solution for the industrial-scale production of ammonia. In a recent work of Zhao et al., Ru catalysts were supported on perovskites of varying valence transition metals (e.g., Nb and Ta).138 In prior works, the respective authors synthesized Ru/Sr2Nb2O6,152 Ru/Sr2Ta2O7 (ref. 153) and Ru/Ba5Ta4O15 (ref. 154) and reported their excellent catalytic activity for ammonia synthesis. These valence transition metals can be reduced to generate oxygen vacancies (VO), and the electrons confined in these VO can be easily donated to Ru and further accelerate the dissociation of surface nitrogen molecules. Additionally, the VO can accept the hydrogen atoms from Ru (via spillover mechanism) to avoid hydrogen poisoning of the Ru atoms. To generate more VO in Ba5Ta4O15, Zhao et al. pretreated the support with CaH2 in different atmospheres and exploited its anionic defects for the synthesis of ammonia.138 Importantly, the electron paramagnetic resonance (EPR) spectra was acquired to confirm the formation of anionic defects  in CaH2-reduced BNO supports (Fig. 22). It was seen that supporting Ru on 0.20CaBNO600 results in a decrease in the EPR signal of oxygen vacancies, which could be due to the partial transfer of electrons in oxygen vacancies to Ru NPs. This was also attributed to the formation of H atoms on Ru particles spilled over the support and reactions with the electrons trapped in oxygen vacancies, which result in the conversion of oxygen vacancies to H−
in CaH2-reduced BNO supports (Fig. 22). It was seen that supporting Ru on 0.20CaBNO600 results in a decrease in the EPR signal of oxygen vacancies, which could be due to the partial transfer of electrons in oxygen vacancies to Ru NPs. This was also attributed to the formation of H atoms on Ru particles spilled over the support and reactions with the electrons trapped in oxygen vacancies, which result in the conversion of oxygen vacancies to H−  . The oxygen vacancies with trapped electrons are represented as
. The oxygen vacancies with trapped electrons are represented as  . The addition of Cs promoter led to an increase of all the EPR signals. Importantly, the electron paramagnetic resonance (EPR) spectra was acquired to confirm the formation of anionic defects
. The addition of Cs promoter led to an increase of all the EPR signals. Importantly, the electron paramagnetic resonance (EPR) spectra was acquired to confirm the formation of anionic defects  in CaH2-reduced BNO supports (Fig. 22).
in CaH2-reduced BNO supports (Fig. 22).
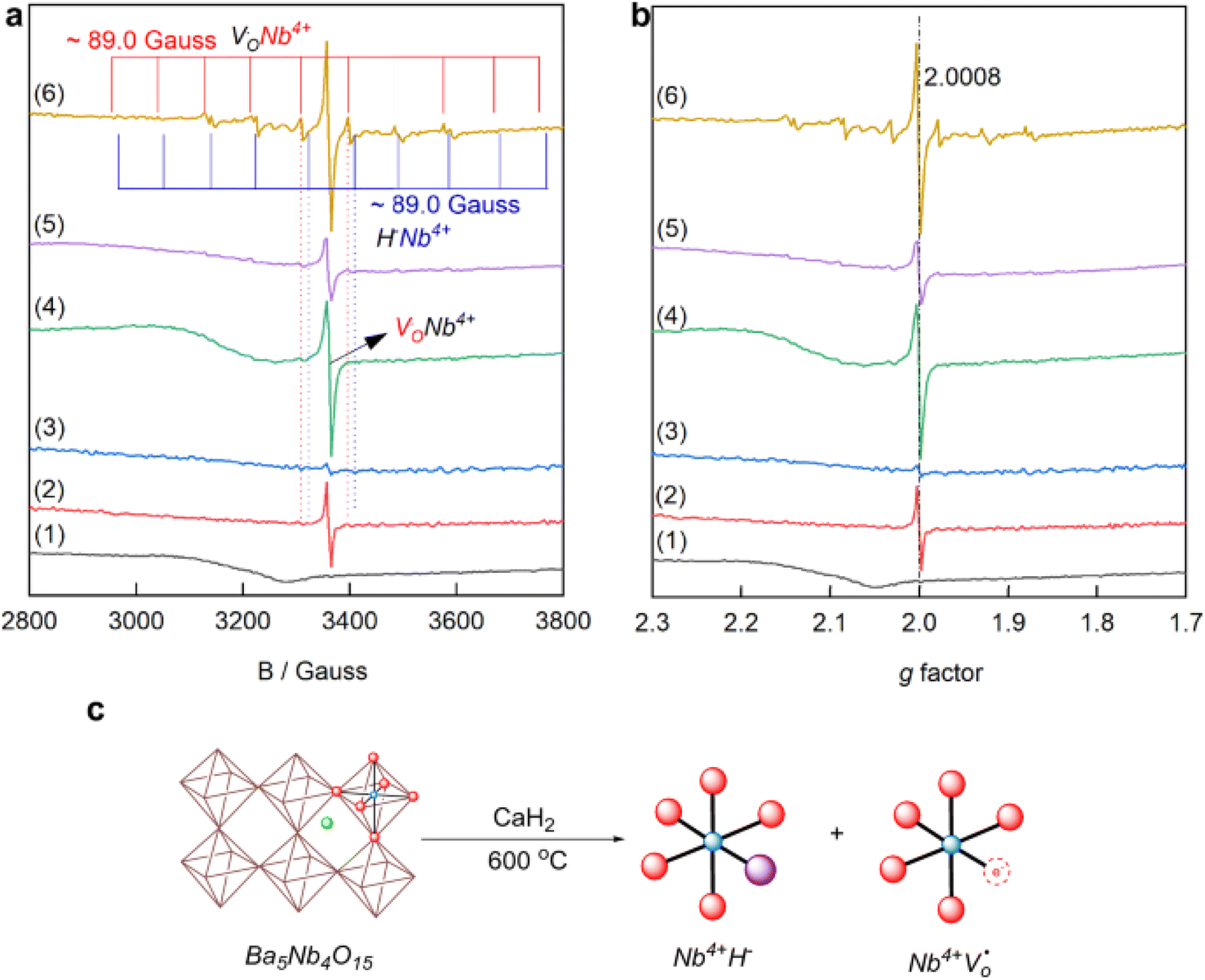 | ||
| Fig. 22 EPR spectra ((a) magnetic field (b) g-factor) of (1) pristine BNO (Ba5Nb4O15), (2) BNO, (3) Ru/BNO, (4) 0.20CaBNO600, (5) Ru/0.20CaBNO600, and (6) 3Cs–Ru/0.20CaBNO600. All samples other than pristine BNO have been reduced under H2 for 3 h at 400 °C prior to collecting the EPR spectra. (c) Schematic model demonstrating the generation of two types of distorted octahedra (NbO6) connected to H- or oxygen vacancy. Adapted with permission from ref. 138. Copyright 2023, Springer Nature. | ||
On optimizing the Ru loading, it was found that Ru/0.20CaBNO500 shows an ammonia synthesis rate of 2.6 mmol g−1 h−1, which was the best among all the tested catalysts. 0.20CaBNO600 support was then reduced at 600 °C for a period of 12 h under argon flow and the best activity achieved was 4.0 mmol g−1 h−1 at 400 °C and 0.10 MPa. For further enhancement, Cs promoter was added to Ru/0.20CaBNO600. It was observed that 3Cs–Ru/0.20CaBNO600, which was activated at a temperature of 400 °C for 3 h in syngas, exhibited an optimum activity of 5.3 mmol g−1 h−1. The same catalyst was tested for a period of 72 h and a 10% loss of activity was observed after a period of 40 h, which can be due to the loss of specific surface area of the support. Hence, Cs-addition helped to form the anionic defects in the support under the reaction conditions.
Of several oxides, MgO is an interesting material mainly because of its basicity, which increases high electron density in supported metal particles. Moreover, MgO is inexpensive and thermally stable. In particular, the MgO(111) facets have high electrostatic polarity compared to other facets of MgO, which helps to suppress hydrogen poisoning of Ru particles by capturing H+. Often, the Ru/BaO/MgO composite has been employed for ammonia synthesis under mild conditions155–158 and the Ru–BaO interfacial area is primarily tailored to increase the activity.159 This interface promotes the splitting of H atoms into H+/e− pairs; where BaO holds H+ (as it has strong basic characteristics and is non-reducible) and Ru holds e−. Expectedly, increased electron density on Ru facilitates the adsorption of nitrogen on the surface and also inhibits hydrogen poisoning. For instance, Y. Baik et al. synthesized a series of Ba–Ru/MgO catalysts, with ∼2.3 nm Ru particle size and configured the BaO–Ru interfacial structures to obtain a synthesis rate of 945 mmol g−1 h−1 under mild conditions (400 °C and 1.0 MPa).159 Moreover, identical phenomena were also noticed for a Cs-promoted Ru/MgO catalyst; however, CsxOy–Ru interfaces were less efficient in storing H+/e− pairs than the BaO–Ru interfaces.
Recently, Lee et al. have investigated the role of MgO as a support for Ru–BaO in the synthesis of ammonia.160 A comparative study using pristine MgO and MgO–Al2O3 mixed oxide (MgAlOx) with varying MgO contents was performed. The study revealed that MgO imparts strong basicity to BaO, which acts as an electronic promoter to Ru. In specific, the authors first prepared MgOn, where n is the MgO/(MgO + Al2O3) wt% in the precursor (n = 5, 30, 70, and 100) and next, wet-impregnation method was used to include Ba, and Ru to synthesize Ru–Ba/MgOn catalysts. The weight-time-yield (WTY) of ammonia over the different catalysts, along with various other kinetic parameters, is given in Fig. 23.
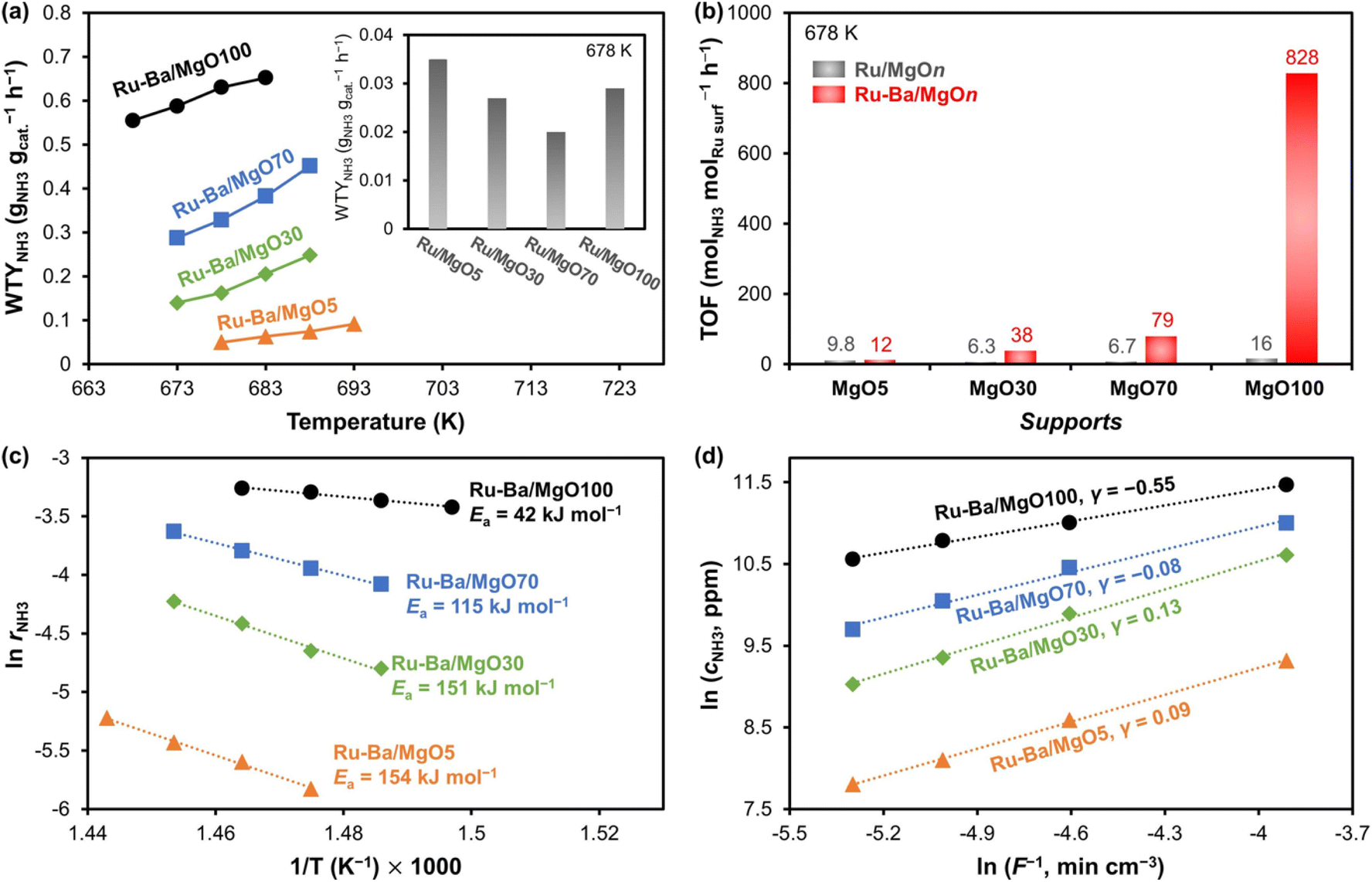 | ||
| Fig. 23 (a) Temperature-dependence ammonia synthesis rates over different catalysts. (b) TOF at 405 °C for Ru/MgOn and Ru–Ba/MgOn catalysts. (c) Arrhenius plot for activation energy determination. (d) Reaction order analysis for ammonia over Ru–Ba/MgOn catalysts. Adapted with permission from ref. 160. Copyright 2024, Elsevier. | ||
A clear correlation between the catalytic activity and the MgO content of the Ru–Ba/MgOn catalysts was noticed (Ru–Ba/MgO5 < Ru–Ba/MgO30 < Ru–Ba/MgO70 < Ru–Ba/MgO100), indicating the superior role of MgO. Notably, the Ru–Ba/MgO100 catalyst disclosed a TOF that is two orders of magnitude higher than that of Ba-free catalysts. Additionally, the apparent activation energy decreased from 154 kJ mol−1 to 42 kJ mol−1 with the increasing content of MgO in the catalysts. Importantly, a low activation energy of 42 kJ mol−1 unveils the high potential of Ru–Ba/MgO100 for low-temperature applications. However, the reaction order for ammonia was a huge negative value (−0.55) for Ru–Ba/MgO100, implying a strong interaction between ammonia and the catalyst surface. Additionally, this work includes diffusion limitation studies, which disclose that the Ru–Ba/MgO catalyst can have a pellet radius of few mm, and the pore size has negligible effects on the diffusion limitation. Such results are critical for the rational catalyst design.
Other oxides such as Al2O3,161–163 MgO,164,165 MgAl2O4,166 Pr2O3,167 La2O3,168 CeO2,135 La0.5Ce0.5O1.75,169 Ru/La0.5Pr0.5O1.75,170 and barium hexa-aluminate171,172 were employed as supports for Ru-based catalysts. The role of oxide supports is not only to disperse the Ru nanoparticles but also to provide stability. Moreover, these supports strengthen the interface phenomena through metal–support interactions and this contributes to increase the catalytic performance of Ru nanoparticles on oxide supports. Yttrium oxide (Y2O3) is one such material, which offers unique properties such as high chemical stability, high melting point, and low volatility. Feng et al. prepared Y2O3-supported Ru via a milling method (referred to as Ru/Y2O3-m) or precipitation method (denoted as Ru/Y2O3-p), and subsequently, reduced these samples at 400 °C for 2 h in a 75% H2/N2 stream.124 The activity tests were evaluated at 1.0 MPa. Under this condition, the given catalyst was heated to a temperature of 400 °C at a ramp rate of 5 °C min−1 under a flow of 75% H2/N2 mixture and made to dwell at 400 °C until the activity reached a steady-state. Notably, the bare Y2O3 support exhibited negligible activity for ammonia synthesis, whereas Ru/Y2O3-m and Ru/Y2O3-p samples exhibited much higher activity under the same conditions; 5% Ru/Y2O3-p catalyst exhibited ammonia synthesis rate of 21.120 mmol gcat−1 h−1 at a temperature of 400 °C with a WHSV of 24000 mL gcat−1 min−1, which was greater than both references, i.e., 5% Ru/γ-Al2O3-p (6.5 mmol gcat−1 h−1) and Ru/Y2O3-m (4.378 mmol gcat−1 h−1). Keeping the same conditions, the activity of the 5% Ru/Y2O3-p (18.456 mmol gcat−1 h−1) catalyst was close to those of many conventionally used catalysts such as Ba–Ru/AC, Cs–Ru/MgO and Ru/CeO2, proving its superior catalytic performance.
Of note, the structures of active sites in different Ru/Y2O3-m and Ru/Y2O3-p catalysts were identical, as confirmed by their identical activation energies. For estimating the intrinsic activity of different catalysts, their TOFs were compared. The 5% Ru/Y2O3-p showed a TOF of 0.019 s−1 which is greater than that of 3% Ru/Y2O3-m (0.005 s−1) and 5% Ru/Y2O3-m (0.005 s−1) samples, at 400 °C. It should be noted that the TOF of 5% Ru/Y2O3-p (0.011 s−1) at a temperature of 350 °C was 7.9 and 8.5 times greater, as compared to that of the 3% Ru/Y2O3-m (0.0014 s−1) and 5% Ru/Y2O3-m (0.0013 s−1) catalysts, respectively. Such increased values of TOFs were further examined to get an insight into the mechanistic details. It was thus concluded that the modulated electronic structures enhanced the activity of Ru nanoparticles in Ru/Y2O3-p, owing to the facilitated dissociation process of nitrogen and hydrogen molecules. Moreover, the metal support interaction in Ru/Y2O3-p catalysts can regulate the electronic structures of Ru nanoparticles, which then facilitates activation and dissociation to form ammonia.
Previous reports suggest that the adsorption ability of hydrogen can be tailored by the spatial arrangements of Ti species for Ru/CeO2 catalysts, and thus, the as-prepared catalysts can demonstrate a superior catalytic activity.173 In this regard, Li et al. further investigated the effect of Ti-species on CeO2-supported Ru catalysts to engineer an efficient ammonia synthesis catalyst.96 Fig. 24a and b illustrate the enhancement of hydrogen desorption on the Ru/CeO2 catalyst before and after the introduction of Ti species.
 | ||
| Fig. 24 (a) Schematic of the influence of Ti species on ceria-supported Ru catalysts Ru/CeO2 and (b) Ru/Ti0.18–Ce. Adapted with permission from ref. 96. Copyright 2022, Elsevier. (c) Illustration of the surface morphology of Ru/Ba/LaCeOx_500red, (d) Ru/Ba/LaCeOx_700red, (e) nitrogen activation steps on Ru/Ba/LaCeOx_700red, and (f) Ru/LaCeOx_650red. Adapted with permission from ref. 174. Copyright 2020, the American Chemical Society. | ||
In this work, a solid solution of Ti and CeO2 (CeTiO2−x) was synthesized and characterized by XRD and Raman spectroscopy. The detailed study found that the solid-solution was conductive for higher dispersion of Ru particles (validated by Raman and UV-vis results) and hydrogen adsorption. CO-TPR and 1H NMR indicates that Ti in Ru/CeO2 decreased the number of the active oxygen species, and therefore, the catalysts led to an increase in the hydrogen species desorbed at low temperatures, which facilitates nitrogen dissociation and alleviates hydrogen poisoning. This study illustrates that the ammonia synthesis rates of Ru catalysts follow a volcanic trend with the increase in titanium butoxide volume. Comparing Ru/CeO2 and Ru/TiO2, the formation of CeTiO2−x solid was essential to increase the activity of Ru/Ti0.18–Ce (18.9 mmol g−1 h−1). Moreover, in the absence of heat and mass transfer effects, the nitrogen reaction order was below 1.0 and the hydrogen reaction order was 0.47, confirming the role of Ti in increasing the nitrogen dissociation, and suppressing hydrogen poisoning, and thereby, enhancing the ammonia synthesis rates. As shown in Fig. 24b, CeTiO2−x controls the amount of active oxygen species, which could be used for hydrogen spillover. Below 300 °C, the desorption of hydrogen species from the Ru sites of Ru/Ti0.18–Ce was higher because of the Ti species and this resulted in the hydrogen-assisted nitrogen activation pathway instead of direct dissociation. Therefore, Ru/Ti0.18–Ce was found to be an efficient ammonia synthesis catalyst.
In an interesting work of Sato et al., Ru/Ba/LaCeOx, pre-reduced at 700 °C, demonstrated an activity of 52.3 mmol g−1 h−1 at 350 °C, and 1.0 MPa.174 The TOF at this temperature was more than 8 times that of the Cs+/Ru/MgO catalyst. Moreover, hydrogen poisoning, a typical drawback of oxide-supported Ru catalyst, was efficiently suppressed by the Ru/Ba/LaCeOx catalyst. The characterization results revealed that Ru particles were surrounded by low-crystalline, oxygen-deficient nanofraction including Ba2+, Ce3+, and La3+. A pictorial representation of the same can be found in Fig. 24c–f for better understanding.
To study the effect of Ba2+, a comparison of the physicochemical properties of Ru/Ba/LaCeOx_700red with those of Ru/LaCeOx_650red was performed. Comparing the later catalyst, the former had a smaller specific surface area and larger Ru particles. The TOF for Ru/Ba/LaCeOx_700red (0.248 s−1) was about five-times that for Ru/LaCeOx_650red (0.049 s−1). It was therefore concluded that addition of Ba2+ had a positive effect on the TOF and synthesis rate of ammonia. Moreover, the effect of catalyst pre-reduction temperature on the ammonia generation rate of Ru/Ba/LaCeOx was explored. With the increase in pre-reduction temperature, the ammonia synthesis rate was also found to considerably increase. However, beyond 800 °C, this rate was decreased. Herein, the catalysts' structure was obtained by using the alkaline earth compounds and rare earth oxides which contain redox-active atoms. This compound showed greater electron-donating capacity owing to the strong basicity of the incorporated cations, removal of carbonate, and formation of oxygen deficient sites that remarkably avoided the electron withdrawing O2− anions from the Ru-nanofraction interface. These electrons were then efficiently donated to antibonding orbitals of nitrogen molecules via Ru, which next cleaved the strong nitrogen triple bond and promoted the ammonia synthesis.
In a similar study by Sato et al., Co nanoparticles were encapsulated with BaO, which increased the activity of Co, and in particular, Co@BaO/MgO-700red displayed a superior rate of ammonia synthesis.175 For instance, a rate of 24.6 mmol g−1 h−1 and TOF (0.246 s−1) of the catalyst were observed at 350 °C and 1.0 MPa. This result was about 80 and 250 times greater, respectively, than those of the non-doped pristine catalyst. With increase in pressure to 3.0 MPa, the rate was enhanced to 48.4mmol g−1 h−1, which was significantly higher than that of active Ru-based catalysts. Based on the experimental findings, the underlying mechanism was proposed. The details suggest that the adsorption of nitrogen on Co atoms at the catalyst surface weakened the strong bond of nitrogen molecules by the electron donation from Ba2+ of BaO via adjacent Co atoms. Such steps accelerate the breaking of the triple bond and enhance the rates.
A major limitation of oxide and mixed-oxide-based catalysts is the requirement for high temperatures to activate N2, which can limit their energy efficiency. Additionally, maintaining the stability of oxygen vacancies under prolonged reaction conditions can be challenging, as vacancy annihilation may reduce catalytic activity over time. Furthermore, sintering of active metals, despite strong metal–support interactions, can still occur under harsh conditions, leading to deactivation. To address these challenges, future research should focus on developing oxide and mixed oxide catalysts with improved low-temperature activity by fine-tuning the oxygen vacancy concentration and electronic properties. Advanced synthesis techniques should be employed to achieve uniform metal dispersion and robust metal–support interfaces. Exploring novel oxide systems and doping strategies can also help enhance stability, reduce sintering, and maintain high activity over prolonged operation.
4. Observation for catalyst development
Next, a detailed analysis for potential industrial catalysts reveals that the catalysts should meet set criteria such as product selectivity (>70%), product concentration (>3 wt%), catalyst activity (0.1–10 tonneproduct per mreactor3 per h), and catalyst consumption (0.1–10 kgcat per tonneproduct).176,177 Of these, criteria such as catalyst activity and their consumption are of great interest. Considering a catalyst density of 1 tonne per m3 for a catalytic fixed bed reactor, the threshold activity of the catalyst should be greater than 6 mmol g−1 h−1. Interestingly, most of the newer-generation catalysts demonstrate higher rates. However, it should be noted that no more than 1 kg of catalyst should be consumed to generate 1 tonne of ammonia, and therefore, such criteria can only be met if the catalyst remains stable on stream for at least 6 months. To the best of our knowledge, such stability reports for the catalyst are missing, and hence, nothing conclusive can be stated on their potential as an industrial catalyst. Additionally, some of these catalysts such as electride-supported and amide catalysts are prone to deterioration under moisture and could also agglomerate, which adversely affects the catalytic activity.Fig. 25 summarizes the TOF of different classes of thermal catalysts. The TOF value describes the number of catalytic cycles per active site and time. It is determined under conditions for true kinetic control. The variation in TOF also indicates the structure sensitivity of ammonia synthesis. Fig. 25 shows that the TOFs reported with hydrides such as BaH2–BaO/Fe/CaH2 catalysts are significantly greater (12.2 s−1) than those of the other reports. Moreover, the ammonia synthesis rates are 5.5 mmol g−1 h−1, under 0.9 MPa and 300 °C. Under such mild reaction conditions, BaH2–BaO/Fe/CaH2 has positive orders for N2 and H2, possibly indicating relatively high efficiencies at lower temperatures.26,84 Similarly, other nitrides such as Ca3CrN3H catalysts also exhibit higher TOF values.44 In the same line, some of the Ru-supported RScSi silicides (R = La, Ce, Pr, Nd, Sm, and Gd) exhibit TOF > 2.0, which was attributed to the easy dissociation of the nitrogen molecules with possible N-species storage by the RScSi support and hydrogen in NH3 comes from the gas-phase H2 and not particularly from the hydride phase.99
 | ||
| Fig. 25 Scatter plot showing the TOF of the thermal catalyst as a function of temperature for ammonia synthesis. Here, the maximum pressure considered is 0.9 MPa. | ||
Moreover, under other conditions with less efficient catalysts, the TOF values are one or even three orders of magnitude less than 1.0 and the corresponding order of NH3 is often negative. Notably, any enhancement in efficiency can be possible by altering the synthesis procedures to improve the density, dispersion, and accessibility of active sites in these catalytic materials. In contrast, other catalysts such as oxides and intermetallics displayed mediocre to high efficiency at 400 °C. However, at elevated temperatures (>400 °C), the efficiencies of oxide catalysts are significantly lower. Interestingly, under such conditions, a considerable value of TOF (>2.0 s−1) has been reported, which appreciates the development in the strategies to facilitate nitrogen dissociation and readily enable adsorption–desorption of hydrogen molecules. It should be emphasized that scalable synthesis methods for catalysts are vital to maximize and retain the contact between the catalyst and co-catalyst/support for prolonged lifetime in pursuit of more efficient catalysts. Hence, focus should be driven to explore porous, high surface area catalyst supports and synthesis strategies to enhance the metal–support interactions that would render a substantial increase in the catalytic efficiency. Therefore, the proliferation of small-scale mild-conditions ammonia synthesis facilities at a localized site can be possible only after the detailed techno-economic analysis of the developed catalysts and their performance as well.
5. Non-thermal plasma (NTP)-assisted ammonia synthesis
5.1 Introduction
With the advent of new technologies in the field of ammonia synthesis, synthesis via NTP has gained significant attention. Mild conditions with low-cost input as well as low energy consumption make non-thermal plasma an attractive route for the production of ammonia. Well known as the “fourth state of matter”, plasma is an ionized gas which possesses electrical neutrality. In NTP, the temperature of the gas is nearly equal to the room temperature while that of the electrons reaches values as high as 10000–100000 K (1–10 eV), unlike thermal plasma in which the gas and electron temperatures are identical. In the case of NTP, input energy is transferred to the electrons selectively. These electrons collide with the molecules of the reacting gases to produce new species (excited state atoms, reactive radicals, ozone, etc.). These species can assist the dissociation of molecules and can thus initiate physical and chemical reactions under mild conditions. Thus, NTP holds potential to be an effective technique for ammonia synthesis.
Direct production (without the use of catalyst) of ammonia using NTP is possible as it dissociates the reactants, N2 and H2, which sequentially combined to produce ammonia. On employing a catalyst, the effect of interaction of the active species produced during the dissociation with the catalyst can lead to promotion of ammonia synthesis. In case of NTP, the initial step for ammonia synthesis is the dissociation and ionization of nitrogen and hydrogen. Followed by this is the generation of radicals due to collision of the elements present in the plasma region.178 It should be noted that the dissociation of N2 is the rate-determining step here. The NH radicals formed in the process act as precursors for ammonia synthesis. The electron-as well as vibration-excited reactions along with the radicals absorbed on the surface play an important role in the NTP-assisted ammonia synthesis. The reactions occurring in the gas phase along with the those in the surface adsorption phase are as follows:178
| e + N2 → e + 2N | (17) |
| N + H2(ν) → H + NH | (18) |
| N + surf → N(s) | (19) |
| H + surf → H(s) | (20) |
| NH + H(s) → NH2(s) | (21) |
| NH2(s) +H(s) → NH3 | (22) |
| NH + H(s) → NH2(s) | (23) |
| NH2(s) + H(s) → NH3 | (24) |
The reactions occurring in the gas phase are the source of excited state molecules which then lead to surface adsorption reactions for the formation of NH3 (or NOx). The N, H and NHx radicals generated in the process stay on the catalyst surface. While in the gas phase, N2 dissociates on electron excitation and forms atomic nitrogen, which then reacts with the hydrogen molecules excited by vibration to generate NH radicals. These NH radicals react with H atoms present in the gas phase or those on the surface of the catalyst to produce ammonia.
NTP can also be used for pre-treatment of the catalyst used for ammonia synthesis. Plasma can be used to generate doping and defects on the surface of the material keeping the nanostructures unaffected. These defects can be created by generating nitrogen and oxygen vacancies, by etching of the catalyst or by modification of the substrate.
5.2 Types of plasma discharge
Within NTP, there are various types of discharges that have been employed to facilitate the ammonia synthesis reaction, namely:(i) Glow discharge
(ii) Gliding arc discharge
(iii) Microwave discharge
(iv) Radiofrequency discharge
(v) Dielectric barrier discharge (DBD)
Each one of the above-mentioned discharges calls for a unique reactor setup with many innovative changes having been made over the past few decades. The history and development of catalysts for the aforementioned types of discharges have been discussed in detail in a recent review article.179 However, the most commonly used reactor in the recent times, not just for ammonia synthesis but also for most of the NTP-assisted catalytic reactions, is the DBD reactor which we will discuss about in the subsequent section.
5.3 Dielectric barrier discharge
A dielectric barrier discharge (DBD) is the most widely used type of plasma discharge with most of the recent studies on plasma assisted ammonia synthesis being conducted in a DBD reactor. It was first used by Siemens in 1857 for ozone generation and had long been known as ozone production discharge. It is also called a silent discharge because it is inaudible.In a DBD, electric discharge is generated between two electrodes separated by at least one layer of an insulating dielectric barrier, upon application of a high AC voltage. One of the electrodes could also be covered in the insulating dielectric material so as to achieve this separation. The purpose of the dielectric is to limit the current and prevent rapid or excessive discharge, resulting in a discharge that is arc-free, preventing the heating of the gas.180 A few common materials that are used as dielectrics are glass, quartz, ceramics and polymers such as polycarbonate (PC) and polytetrafluoroethylene (PTFE).181 Discharges are produced when the electric field around the electrodes is high enough, but they get accumulated on the dielectric material that is separating the electrodes. The accumulation of charge carriers on the dielectric generates an electric field that opposes the electric field induced by the external AC voltage. Hence, the discharge is observed as many small plasma filaments of duration in the order of nanoseconds.182,183 Most of the chemical reactions associated with a plasma discharge occur in these filaments or “micro-discharges”.180 The schematic of a DBD reactor for ammonia synthesis and the corresponding reaction mechanism are shown in Fig. 26.
 | ||
| Fig. 26 Schematic of a DBD reactor and the underlying mechanism for ammonia synthesis using NTP. | ||
While a lot of geometric modifications of DBD have been done, they can broadly be classified as volume DBD (VDBD) and surface DBD (SDBD).182 In a VDBD, discharge occurs in the space between the dielectric and the grounded electrode, i.e. the gap between the electrodes is not entirely filled with the dielectric material. In the case of SDBD, the gap between the electrodes is filled with the dielectric, the discharge is limited to the surface of the dielectric and is generally dense as compared to VDBD. DBD is useful for the treatment of temperature sensitive materials since the temperature of the gas and ions is near ambient.183 An arc discharge is hard to control and may also damage the electrode. DBD, however, can easily be operated at atmospheric pressure, has the potential to be scaled up to industrial scale and also offers a lot of versatility with regard to its configuration, enabling us to tailor the design to suit our needs.184 In 1969, Eremin et al. published the first article on the use of DBD for ammonia synthesis.185 Since then, a number of studies have been conducted in this domain, a few of which have been talked about in the subsequent section.
Below is the updated table adapted from the review paper of Gharahshiran et al.,47 which outlines the major breakthroughs in catalytic ammonia synthesis using a DBD reactor:
Various routes of catalyst synthesis were used to achieve the optimization of metal dispersion, stability, and catalytic efficiency. γ-Al2O3,186 MCM-41,187 and Ru/L-MgO188 were synthesized using an incipient wetness impregnation technique, in which the metal precursor salts were deposited onto supports for precise control of metal loading and uniform metal distribution.
Synthesis of SBA-15 (ref. 189) by a sol–gel-assisted synthetic method provided control over the pore structure and a high surface area. Non-porous silica (NP-SiO2) and fumed silica (EH-5) were used as comparative supports. The sol–gel method involves hydrolysis and condensation of a silica precursor in the presence of a templating agent under acidic or basic conditions. The process begins with the formation of a colloidal sol, where silica nanoparticles are dispersed in a liquid. As the reaction proceeds, these nanoparticles undergo condensation, forming a gel-like network. The template is removed by controlled drying and calcination, resulting in a porous silica structure such as SBA-15, which has a high surface area and tunable pore sizes.
The anodization and impregnation method was used to design Ru/alumina tubular membrane catalysts190 that had enhanced porosity and sufficiently high gas permeability. In this method, an aluminum substrate undergoes electrochemical oxidation (anodization) in an acidic electrolyte, resulting in the formation of a porous alumina (Al2O3) layer with well-defined nanochannels. This enhances the surface area and porosity. The porous alumina is then impregnated with a ruthenium (Ru) precursor solution, allowing Ru to be uniformly distributed within the pores. Subsequent drying, calcination, and reduction steps convert the precursor into active Ru nanoparticles, resulting in a Ru/alumina tubular membrane catalyst with improved gas permeability and catalytic performance.
In another work, Ni/MCM-41 (ref. 187) catalysts were synthesized by an impregnation method, which allowed the adjustment of Ni on the surface thus affecting surface accessibility and stability. Ni was loaded on MCM-41 and SiO2 supports using a variety of deposition methods. MCM-41 was prepared by a sol–gel process with CTAC, TEOS, and NH3 H2O, and calcined at a temperature of 550 °C. After that Ni was deposited at three different locations: within the mesopores (Ni/MCM-in), outside the framework (Ni/MCM-out), and integrated in the framework (Ni/MCM-both). Ni/MCM-in was prepared by impregnating preformed MCM-41 with a solution of Ni(NO3)2. All these catalysts were calcined at 550 °C and reduced in Ar/H2 at 750 °C before use for plasma-catalytic ammonia synthesis.
Hydrothermal synthesis was used for the production of Ni/LaOF191 catalysts with high crystallinity and uniform morphology. LaOF was synthesized by dissolving LaCl3·7H2O, urea, and NH4F in DI water, stirring for 30 min, and heating in an autoclave at 180 °C for 12 h. The resulting white powder was washed, dried at 60 °C, and calcined at 800 °C under argon flow.
SBA-15 modified by MgO was synthesized via in situ doping192 and impregnation with Ni-loaded variants to enhance Ni–MgO interactions and catalytic performance. In situ doping involved dissolving the P123 polymer and magnesium acetate in HCl, adding tetraethyl orthosilicate (TEOS), stirring overnight, and hydrothermally treating at 100 °C for 24 h, followed by washing, drying, and calcination at 550 °C. SBA-15 was also modified with MgO using the impregnation method. Ni–Mg/SBA-15 catalysts (5 wt% Ni) were then prepared by impregnating Mg/SBA-15 with Ni(NO3)2, followed by drying and calcination at 550 °C for 6 h.
As can be observed in Table 9, γ-Al2O3 has been explored as a support for catalysts in NTP-assisted ammonia synthesis in several studies. Notably, Wang et al. were among the first to compare different transition metals supported on alumina for their catalytic activity in NTP-assisted ammonia synthesis. Ni/Al2O3 gave the best synthesis rate of 390 μmol g−1 h−1 and a yield of 0.77%, and Fe barely added to the activity of alumina alone, with Cu giving an intermediate activity. The low activity of Fe was attributed to the fact that it had stronger adsorption of N2 species, which increased the barrier for hydrogenation, thereby resulting in a lower rate of ammonia synthesis. The lower metal–N bond strength in case of Ni resulted in an increased ammonia yield, as a weaker M–N bond strength indicates a lower rate of ammonia dissociation.193 Zhu et al. investigated the effect of pH on the activity of γ-Al2O3 pellets in a packed bed dielectric barrier discharge reactor and found that ammonia synthesis followed the order of alkaline γ-Al2O3 > neutral γ-Al2O3 > acidic γ-Al2O3 > blank tube.194
| S. no. | Catalyst | Plasma conditions | H2/N2 ratio | Total flow rate (mL min−1) | Ammonia yield (%) | Ammonia energy efficiency (g-NH3 kW−1 h−1) | Energy consumption (MJ mol−1) | Catalyst synthesis method | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Al2O3 | 18.3 kHz, 0.42 kV, 1 W | 0.5 | 22.5 (20 vol% He) | 0.03 | N/A | N/A | Incipient impregnation method | 186 |
| 2 | Al2O3 | 50 kHz, 6 kV | 1 | 100 | 1.2 | N/A | N/A | Conventional impregnation method | 195 |
| 3 | γ-Al2O3 | 21 kHz, 46.7 W | 3 | 400 | 0.54 | 1.05 | N/A | — | 196 |
| 4 | MgO | 21 kHz, 46.7 W | 3 | 400 | 0.48 | 0.94 | N/A | — | 196 |
| 5 | Alkaline γ-Al2O3 | 24.25 W | 3 | 100 | N/A | 6.58 | 9.3 | — | 194 |
| 6 | SBA-15 | 20–23 kHz, 8.6 kV, 15 W | 3 | 25 | N/A | 4.6 | N/A | Sol–gel method | 189 |
| 7 | Zeolite 5A | 7.5 kV, 13.3 W | 1 | 25 | N/A | 15.5 | 3.95 | — | 197 |
| 8 | PZT | 3 kV | 1 | 38.3 | 2.8 | 0.9 | 136 | — | 198 |
| 9 | PZT | 5.5 kV | 3 | 11.5 | 0.5 | 0.75 | 81.6 | — | 199 |
| 10 | ZIF-8 | 20–23 kHz, 8.6 kV, 15 W | 3 | 25 | N/A | 1.9 | N/A | — | 189 |
| 11 | Cu wool | 5 kV | 1 | 100 | 3.5 | 3.3 | 18.5 | Electroless plating method | 200 |
| 12 | Au wool | 128.7 W | 1 | 100 | N/A | 0.58 | N/A | — | 201 |
| 13 | CoLa/Al2O3 | 23.5 kHz, 51 W | 1 | 100 | 0.9 | N/A | 77 | Wet impregnation method | 202 |
| 14 | Rh/γ-Al2O3 | 8 kV, 24 W | 0.5 | 100 | 1.43 | 0.94 | 65 | Wet impregnation method | 203 |
| 15 | Ru/alumina membrane | 4.5 kV, 127 W | 3 | 40 | 4.36 | 0.37 | 163.9 | — | 204 |
| 16 | Ru/alumina membrane | 4.5 kV, 127 W | 3 | 30 | 4.62 | 0.4 | 154.7 | — | 190 |
| 17 | Ru/alumina | 7.5 kV | 3 | 1000 | 0.05 | 1.9 | 32.2 | — | — |
| 18 | Ru/L-MgO | 9.2 kHz, 60 W | 0.5 | 300 | 4 | 1.29 | N/A | Incipient impregnation method | 188 |
| 19 | Ru/activated carbon | 13.3 W | 3 | 100 | N/A | 0.72 | 85 | Ultrasonic-enhanced impregnation method | 205 |
| 20 | Cs–Ru/MgO | 6 kV | 3 | 4000 | 2.41 | 2.3 | 26.6 | Impregnation method | 206 |
| 21 | Ru–Mg/alumina | 5.4 kV | 4 | 2000 | 2.55 | 35.7 | 1.7 | Impregnation method | 207 |
| 22 | Ru/Si-MCM-41 with Cs, K, Ba as promoters | 5 kV | 1 | N/A | N/A | 1.7 | 3.6 | Liquid impregnation method | 208 |
| 23 | Ni/Al2O3 | 12 kV, 25.1 W | 2 | 56 | N/A | 0.56 | N/A | Incipient impregnation method | 209 |
| 24 | Ni/Al2O3 | 10 W | 2 | 100 | 2 | 0.89 | 68.9 | — | 210 |
| 25 | Ni/MCM-41 | 9.2 kHz, 9.7 kV, 40 W | 3 | 40 | 5.3 | 1.2 | N/A | Impregnation method | 187 |
| 26 | Ni/LaOF | 7.87 kHz, 13 W | 3 | 25 | N/A | 2.7 | N/A | Hydrothermal method | 191 |
| 27 | Co–Ni/Al2O3 | 30.81 W | 1 | 200 | N/A | 0.83 | N/A | Incipient impregnation method | 211 |
| 28 | Ni–Mg0.02/SBA-15 | 9.5 kV | 1 | 20 | N/A | 1.05 | N/A | Doping and incipient impregnation method | 192 |
| 29 | Ni/SiO2 | 13.75 kV, 13.79 W | 1 | 50 | 0.49 | 8.08 | N/A | Incipient wetness impregnation | 212 |
Iwamoto et al. compared the catalytic activities of Ni, Fe and Ru supported on alumina, and also tested alumina alone. Conventional wetness impregnation was used to prepare the metal loaded catalysts. Surprisingly, it was found that alumina by itself gave a better ammonia yield as compared to alumina loaded with Fe and Ru, indicating that these metals decreased the activity of alumina. Ni/γ-Al2O3 had the best catalytic activity with an ammonia yield of 6.3% at an applied voltage of 6.0 kV, a residence time of reactant gases of 0.12 min, and PH2/PN2 = 1. It was found that NiO and NiAl2O4 were generated on Al2O3, and the NiO species readily reduced to Ni in the plasma reaction, which served as active sites.195 Winter et al. studied the catalytic surface reactions on the γ-Al2O3-supported Ni and Fe, and found that the gas phase ammonia production varied as Al2O3 < Fe < Ni. The higher activity of Ni was attributed to the fact that it had a higher concentration of adsorbed species. It was also found that Ni favoured the adsorption of NHx while Fe favoured the adsorption of N2Hy.186 Patil et al. also tested various catalysts supported on alumina, and found that 2% Rh/Al2O3 was the most efficient among noble metals and 10% Ni/Al2O3 was the most efficient among transition metals.203
Despite obtaining a better rate with noble metals, Ni has gained limelight due to its lower cost. A number of studies for plasma-catalytic ammonia synthesis hence used Ni-based catalysts. Gorky et al. studied the effect of the size of Ni nanoparticles on the catalytic activity. Silica was chosen as the support over alumina due to its inert nature, so that the actual effect of size of NPs could be studied without having to worry about the interaction with the support. Two sets of catalysts were prepared, one using wetness impregnation and the other using surface organometallic grafting (SOG). The SOG catalyst showed a higher rate, with the smallest size of nanoparticles giving the highest rate of ammonia production.212 One of the major challenges in NTP-assisted ammonia synthesis is the plasma-assisted decomposition of the desorbed ammonia. Shah et al. used zeolite 5A as a support based on the hypothesis that its surface negative charge might enhance nitrogen dissociation. Using such inert porous packing improved the ammonia formation rate due to the in situ adsorption of ammonia by the packing. Zeolite 5A also promoted the formation of micro discharges and changed the reactor's voltage–current characteristics, which further helped in improving the ammonia yield.197
Although the use of such catalysts improved the ammonia synthesis rate, reverse reaction, i.e. ammonia decomposition remains a challenge. To address this issue, Wang et al. proposed a mesoporous MCM-41 support with a controlled deposition of nickel nanoparticles, with the aim of developing a “shielding protection” catalyst.187 Ni was deposited on the external surface of MCM-41 to expose the catalyst surface to plasma-activated reactant gases which enhanced the plasma-catalytic interactions and improved the ammonia production rate. An illustration of the proposed mechanism can be found in Fig. 27.
 | ||
| Fig. 27 Schematic of the “shielding” mechanism of MCM-41 and the reactions involved. Adapted from the work of Wang et al.,187 licensed under CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). | ||
After the addition of hydrogen to the nitrogen atom, the resulting NH3 molecule desorbed from the surface of Ni catalyst and diffused from the external surface of MCM-41 (where there is a relatively high concentration of ammonia) to its mesopores (low-concentration channels). Due to the absence of plasma in these mesopore channels, the reverse reaction, i.e. decomposition of NH3, was inhibited, resulting in a higher ammonia yield. Moreover, the Ni NPs dispersed over the external surface of MCM-41 provided good accessibility and enhanced the activation of N2 molecules, resulting in an exceptional NH3 yield of >5%.
Ru is an excellent thermal catalyst for ammonia synthesis, and therefore, it has also been widely studied for NTP-assisted ammonia synthesis. Cheaper Ni based catalysts have come into the picture recently, but Ru has been a material of interest in this regard even before. The work by Kim et al. is one of the earliest ones to report a Ru-based catalyst supported on alumina. They have reported a Mg-promoted Ru/γ-Al2O3 catalyst and performed the experiment under N2-rich conditions, perhaps due to the fact the Ru faces the issue of hydrogen poisoning. The presence of RuO2 was found to be beneficial for ammonia formation, but it was getting reduced by H radicals, due to which base metals were introduced as promoters to protect RuO2 from rapid reduction.207 Peng et al. reported Cs promoted Ru supported on activated carbon, again in a nitrogen-rich environment, confirming that a N2-rich environment is more suitable for ammonia synthesis using Ru-based catalysts due to the relative difficulty in nitrogen dissociation.206 Xie et al. studied the effect of various parameters on the rate of ammonia formation using a Ru/L-MgO catalyst. They found that the ammonia yield increased with the increase in the total gas flow rate, discharge power and discharge temperature.188 Hu et al. also conducted a study on the catalytic activity of a series of metals supported on activated carbon. They found that the order of NH3 concentration is as follows: Ru/AC > Co/AC > Fe/AC > Ni/AC. This finding was contrary to the high activity of Ni as compared to Fe and Co, when alumina support was used. The enhancement in rate was attributed to the basicity of the metal/AC catalysts that occurred as a result of the incorporation of the active metal onto the activated carbon support.205
Recently, our group has investigated the use of transition metal-loaded boron-doped graphitic carbon nitride (BCN) for NTP-assisted ammonia synthesis under near ambient conditions. The empty orbitals in the boron atom aid in the activation of the inert nitrogen molecule, while the surface vacancies in graphitic carbon nitride attract electrons of surrounding carbon atoms, forming electron-deficient sites which further enhance N2 activation. The strong acidic sites in BCN impede the desorption of ammonia gas which is basic in nature, hence Fe and Co were loaded onto BCN to weaken these acidic sites. Among the tested catalysts, Co–BCN demonstrated the highest ammonia synthesis rate of 2.48 mmol g−1 h−1, corresponding to an energy efficiency of 2.09 g-NH3 kW h−1, which was around 2.89 times higher than that of plasma alone. It was also found that the ammonia formation reaction occurred via a dual mechanism involving both Langmuir–Hinshelwood (L–H) and Mars van Krevelen (MvK) pathways. The existence of MvK mechanism was confirmed by subjecting the Co–BCN catalyst to pure N2 and H2 flows and passing the effluent gas in a solution of Nessler's reagent. No colour change was detected under pure N2; however, under pure H2 the solution formed a dark-brown precipitate, indicating that the lattice nitrogen atom in BCN underwent hydrogenation to form ammonia.213
Non-thermal plasma-assisted ammonia synthesis therefore represents a promising alternative to traditional methods, primarily due to its ability to produce ammonia under ambient temperature and atmospheric pressure. Unlike conventional thermal catalysis, which typically requires high temperatures (around 400–500 °C) and pressures (15–30 MPa) to activate nitrogen molecules, plasma-assisted synthesis operates under non-equilibrium reaction conditions. This allows for the activation of nitrogen N2 at much lower temperatures, where the energy input is directed towards exciting electrons, ions, and radicals, creating a highly reactive environment that facilitates N2 dissociation. Consequently, plasma-assisted ammonia synthesis can achieve higher ammonia yields compared to thermal catalysis, making it a potentially more energy-efficient method for ammonia production, especially if scaled up. The non-equilibrium nature of the plasma also allows for a broader range of reaction pathways that can enhance nitrogen activation, offering flexibility in optimizing ammonia synthesis. The plasma state provides a high density of reactive species such as electrons, ions, and excited molecules, which are highly effective in overcoming the strong triple bond of nitrogen, making the process more efficient in terms of both energy and reactant utilization.
However, despite these advantages, non-thermal plasma-assisted ammonia synthesis faces several challenges that need to be addressed before it can be commercially viable. A primary concern is the high-power consumption associated with generating and maintaining the plasma. The energy required to sustain the plasma can significantly increase operational costs, particularly when scaling up the process for industrial-scale ammonia production. Additionally, the design of plasma reactors capable of continuously producing ammonia remains a significant hurdle. Current reactor designs, while effective at the laboratory scale, may not be optimized for continuous operation, limiting the potential for sustained, large-scale ammonia synthesis. Furthermore, the plasma reactor must ensure a homogeneous distribution of plasma across the reaction zone to maintain high reaction rates, which can be difficult to achieve with current designs.
To overcome these challenges, further research is required to improve the energy efficiency of plasma generation, such as optimizing power supply systems and plasma reactor configurations. Additionally, the development of new reactor designs that allow for continuous operation while maintaining plasma stability and effective heat management will be crucial for scaling up this technology. Advances in plasma diagnostics and monitoring will also aid in fine-tuning the process conditions to maximize ammonia yield while minimizing power consumption. With these improvements, non-thermal plasma-assisted ammonia synthesis has the potential to revolutionize ammonia production, offering a more sustainable and energy-efficient alternative to traditional Haber–Bosch processes.
6. Electrochemical ammonia synthesis
6.1 Introduction
The compulsion to enhance the energy efficiency of the industrial ammonia production process, and the necessity to make it environmentally compatible by using renewable energy sources and eliminating the use of fossil fuels as H2 sources, has directed the attention of researchers towards electrochemical ammonia synthesis. Electrochemical ammonia synthesis involves the use of an electrochemical cell consisting of a cathode, an anode and an electrolyte to produce ammonia using a nitrogen source (nitrogen, nitrate or nitric oxide) and a hydrogen source (water). Apart from the carbon neutrality of this method, the modularity of the apparatus involved and the ability to control the reactions by adjusting external parameters (such as applied voltage) particularly distinguish it from the conventional mode of ammonia synthesis.Lately, the HB process is being integrated with a water electrolyser to reduce the dependence on fossil fuels for sourcing hydrogen. The direct N2 reduction process using an electrochemical cell has lower capital costs as compared to this combined system. Therefore, due to a lower capital cost (for green ammonia production) and its modular setup, electrochemical ammonia synthesis is conducive to small scale operations, facilitating on-demand, on-site NH3 production.214 While Haber's discovery in the early 20th century brought light to ammonia, it was in fact in 1807 that ammonia was first synthesised, electrochemically, by Humphry Davy. Davy is well known in the field of electrolysis due to his discovery of most of the alkali and alkaline earth elements. When he electrolysed distilled water in the presence of air (of which N2 is a major constituent), he found small traces of ammonia. The same experiment when repeated in vacuum (no nitrogen source) produced only oxygen and hydrogen. Not much was made of this result, until much later in 1922 when Friedrich Fichter and Richard Suter repeated his experiments by electrolysing dilute sulphuric acid using platinum electrodes at a pressure of 200 atm in a nitrogen rich environment. The formation of ammonia was confirmed, however only at a rate of 0.3 mg min−1, hence it was believed that the electrochemical synthesis of ammonia was not going to be economically viable.215 It was only in the late 20th century, when the increasing awareness about the negative impact of the HB process on our planet led to the exploration of alternative pathways for ammonia synthesis, that the electrochemical method gained limelight. Since then, this field has seen a massive development, with the discovery of various electrocatalysts, electrolytes and improvement in the electrochemical cell design. These efforts have significantly improved the reaction rate and selectivity, making the entire process more scalable and cost-effective.
However, there remain certain challenges that we still need to overcome to fully unlock the potential of electrochemical ammonia synthesis. In this section of the paper, we will briefly discuss different routes for the electrochemical synthesis of ammonia and the catalytic advancements made in this field over the recent years.
6.2 Pathways of electrochemical ammonia synthesis
| Anode: 3H2O → 3/2O2 + 6H+ + 6e− | (25) |
| Cathode: N2 + 6H+ + 6e− → 2NH3 | (26) |
| Overall: N2 + 3H2O ⇄ 2NH3 + 3/2O2 | (27) |
 | ||
| Fig. 28 Schematic of the electrochemical setup for NRR. | ||
The first step in the NRR is (i) the diffusion of N2 molecules from bulk to the interface of the electrode (catalyst) and the electrolyte. (ii) This is followed by the adsorption of N2 onto the surface of the catalyst, where it is activated and protonated to yield NH3. Byproducts such as hydrazine (N2H4) and diazene (N2H2) are also formed in small amounts; a detailed discussion on this is provided in one of the subsequent sections. (iii) These products desorb from the catalyst surface and diffuse back into the electrolyte.214,216
Limitation in any of these steps could disrupt the desired reaction, making it necessary to design a well-suited electrocatalytic system to ensure that all the challenges associated with the above steps could be overcome. The bottleneck of NRR is the highly inert nature of the N2 molecule. The triple bond of nitrogen exhibits a very high bond energy of roughly 941 kJ mol−1, making it very hard to dissociate. The HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) energy gaps of nitrogen are also very high at 10.82 eV.4 This energy gap represents the lowest energy required for the excitation of electrons in a molecule; therefore, a higher HOMO–LUMO gap would make the compound less reactive. Nitrogen also has a low proton and electron affinity, limiting the proton–electron transfer, which makes hydrogenation very challenging. The non-polarity of nitrogen also adds to its limited solubility in electrolytes.4
Another obstacle is the parasitic HER (hydrogen evolution reaction), which competes with NRR and favours the reduction of H+ over N2.
| Hydrogen evolution reaction: 2H+ + 2e− → H2 | (28) |
Both the HER and the NRR occur at similar potential values; however, the HER has faster kinetics mainly because it requires only 2e− but NRR requires 6e−. Moreover, the inert nature of nitrogen molecule makes HER more favourable despite similarity in the thermodynamic potentials of both the reactions. Therefore, the HER consumes protons and electrons, reducing their availability for the NRR, thus resulting in lower ammonia production and faradaic efficiency. Faradaic efficiency (FE) is an important parameter in electrochemistry, which gives an idea about the overall selectivity of an electrochemical process. It is a ratio of the number of electrons consumed by a given electrode to the total amount of charge passed during the process. This gives us the amount of product formed relative to what would have formed if the total charge passed during the electrolysis was utilised for the desired reaction.217 While a higher FE is a good indicator of reaction selectivity, it cannot be relied upon as the sole indicator of catalyst performance. An increase in the FE does imply that the catalyst has suitable properties to inhibit the HER, but it does not signify a higher ammonia production rate. Hence, the ammonia production rate is separately measured, normalised to the amount of catalyst used or the electrochemical surface area, in order to gain further insights into the catalyst performance.214
While eqn (27) gives an overview of the overall reactions, it is also necessary to understand the exact mechanism in which N2 gets reduced to NH3 for innovation in catalyst development and enhancing the reaction selectivity. Theoretical investigations have proposed various mechanisms to understand the NRR, among which the most widely accepted are the dissociative, associative, Mars-van Krevelen (MvK) and Li-mediated pathways. The dissociative mechanism involves the cleavage of the N2 bond which, as discussed earlier, requires a large amount of energy. Therefore, this mechanism is often associated with processes involving harsh reaction conditions and is most commonly followed in the HB process. After adsorption on the catalyst surface, the N2 molecule dissociates into two adsorbed N atoms (represented as *N). The *N atoms then undergo sequential hydrogenation, forming *NH, *NH2 and subsequently NH3. Since electrochemical ammonia synthesis is seen as a low temperature alternative to the HB process, this mechanism does not fit the purpose of electrochemical NRR as much as the other mechanisms under consideration.
Contrary to the dissociative mechanism, where the N2 molecule is broken down into *N atoms immediately after adsorption, the associative pathway is characterised by nitrogen bond cleavage during the protonation process. Associative pathway is further categorised into distal, alternating and enzymatic pathways. In distal and alternating pathways, the nitrogen molecule is adsorbed onto the catalyst surface in the end-on mode, where only one N atom directly interacts with the active site, while the other N atom extends away from the catalyst surface. In the enzymatic pathway, the nitrogen molecule is adsorbed in the side-on mode, where both the N atoms interact with the catalyst surface (see Fig. 29).
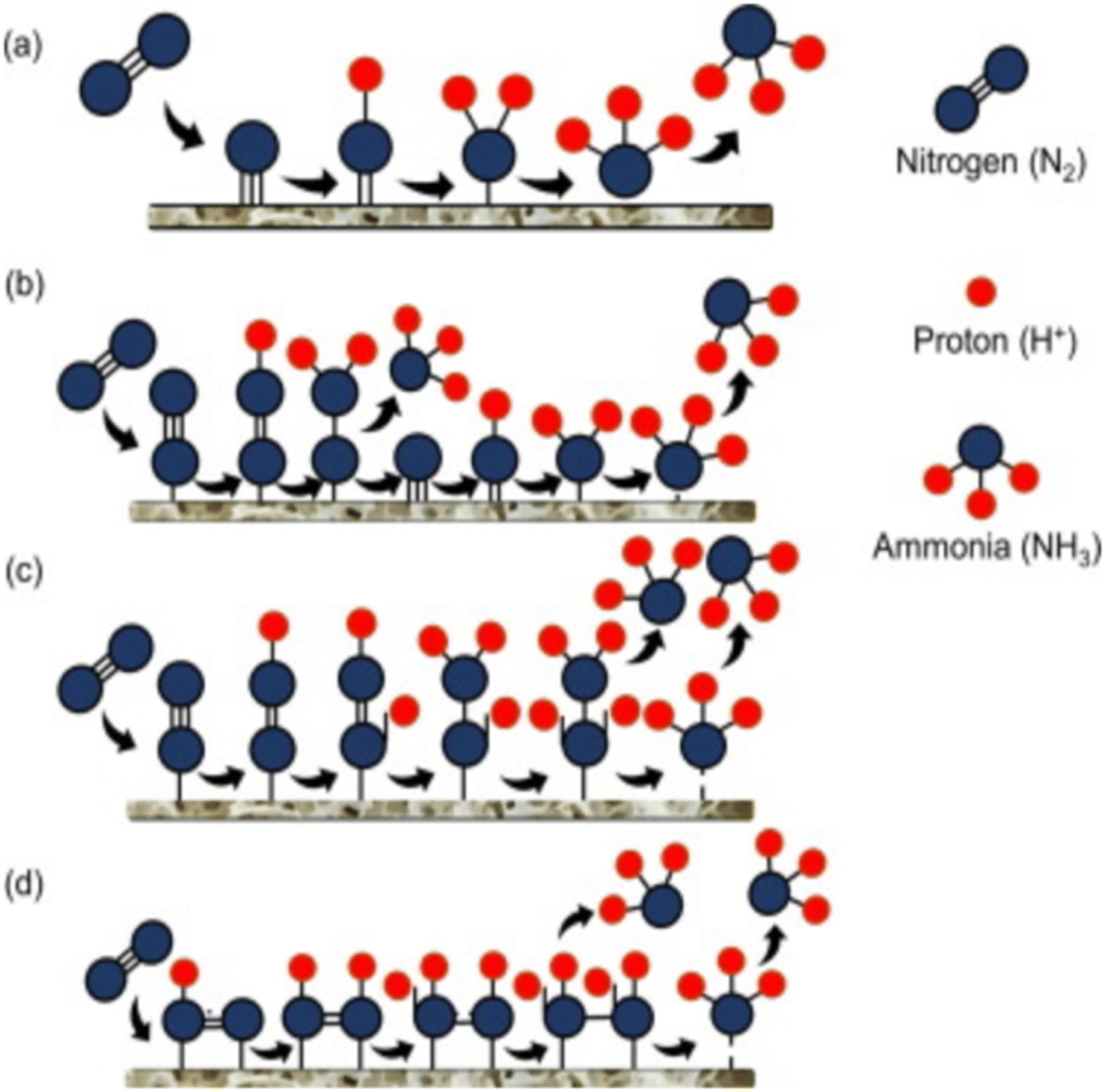 | ||
| Fig. 29 Illustration of the (a) dissociative mechanism, (b) distal associative mechanism (end-on binding), (c) alternate associative mechanism (end-on binding) and (d) enzymatic associative mechanism (side-on binding). Adapted with permission from ref. 216. Copyright 2024, Elsevier. | ||
In the distal associative pathway, hydrogenation first occurs on the nitrogen atom that is away from the catalyst surface. This gives the first NH3 molecule, which gets desorbed from the catalyst surface, followed by the hydrogenation of the proximal nitrogen atom to give the second NH3 molecule. After the desorption of the second NH3 molecule, another N2 molecule gets adsorbed in the vacant site on the catalyst surface and the same process repeats. In the alternate associative pathway, sequential hydrogenation occurs simultaneously in both the nitrogen atoms, resulting in the formation of two NH3 molecules at the same time.
The enzymatic pathway is similar to the alternate pathway, except for the way the N2 molecule is adsorbed onto the catalyst surface (side-on in the case of enzymatic vs. end-on in the case of alternate). Fig. 29 provides a good understanding of these mechanisms, along with the orientation of N2 adsorption on the catalyst surface.
As discussed earlier, while NH3 is the main product of the NRR, the partial protonation of nitrogen also yields diazene (N2H2) and hydrazine (N2H4) as byproducts. Diazene is unstable and decomposes to nitrogen and ammonia in the electrolyte, hence it is present in very little quantities.214 In the case of hydrazine, its desorption from the catalyst surface is a highly endothermic process, therefore it generally remains attached to the catalyst surface and undergoes further protonation to desorb as ammonia and is only detected in small amounts as hydrazine in the final product.216
The formation of these byproducts, however, can be used to predict the mechanism of the NRR. Both hydrazine and diazene can form as intermediates during the associative pathway of nitrogen reduction218 (Fig. 29b–d). Typically, each nitrogen atom undergoes protonation thrice before desorbing as ammonia; however, it is possible that the N2 molecule that attaches onto the catalyst surface (via associative pathway) undergoes partial protonation only, resulting in the formation of hydrazine or diazene. Thus, while quantification of these byproducts is not generally necessary owing to their low concentration, the detection of these byproducts may provide useful insights on the pathway followed by the catalyst for the NRR.
The Mars-van Krevelen (MvK) mechanism is perhaps one of the most effective NRR mechanisms. In the context of ammonia synthesis, it is applicable when metal nitride catalysts are used. The nitrogen atom present on the surface of the transition metal nitride catalyst acts as a reactant and undergoes reduction to NH3. When this molecule is desorbed, it creates a nitrogen vacancy on the catalyst surface which is replenished by the feed N2 gas, after which further NRR occurs on that site. Since the first NH3 molecule is formed without using N2 from the feed, it eliminates the necessity to cleave the nitrogen triple bond. The vacant nitrogen site after desorption of this molecule promotes N2 adsorption and facilitates nitrogen reduction.216 The schematic representation of MvK mechanism can be found in Fig. 30a.
 | ||
| Fig. 30 (a) Schematic of the Mars van Krevelen mechanism for NRR. Adapted with permission from ref. 219. Copyright 2023, John Wiley and Sons. (b) Variation of NH3 yield rates and faradaic efficiency (blue dashed line) with the potential for MV-MoN@NC. (c) Ammonia yield at −0.2 V vs. RHE for different catalysts used in the study. (d) Polarization curves for MoN, MoN@NC and MV-MoN@NC. (e) Gibbs free energy evolved for nitrogen reduction reactions at different reaction coordinates for MoN and MV-MoN. Adapted with permission from ref. 220. Copyright 2020, Elsevier. (f) Sublayer lattice nitrogen diffusion during (i) initial stage and (ii) final stage (downward green triangle: surface N vacancy; upward green triangle: sublayer N vacancy; magenta: Mn atoms; blue: N atoms). (g) Potential energy surfaces for the diffusion of nitrogen onto the surface from the sublayer (magenta: Mn; gold: Fe; green: Ni; blue: N). Adapted with permission from ref. 221. Copyright 2018, American Chemical Society. (h) Schematic representation of the mechanism of lithium-mediated ammonia synthesis. Adapted from the work of Krishnamurthy et al.,222 licensed under CC-BY-NC-ND 4.0 (https://creativecommons.org/licenses/by-nc-nd/4.0/)/cropped from original. | ||
It is worth noting that the doping of metal nitrides with heteroatoms results in improvement of eNRR activity and ammonia production. Li and co-workers found out that Fe-doped MoN2 remarkably increased the NRR performance. The Fe atom doping weakened the metal–N bond, which helped in the activation of N2 and NH3 production, while the high spin state of Fe also contributed to the improvement of eNRR performance.221 The sub layer diffusion of N onto the surface also plays a crucial role in the continuous NRR (Fig. 30f). DFT calculations revealed that the diffusion energy barrier decreased by doping Fe within the sublayer (Fes@Mn4N) from 1.12 to 0.56 eV, and there is no significant change in doping on the surface (Fet@Mn4N) (1.12 to 1.09 eV) (Fig. 30g). This is because Fe forms weaker bonds compared to Mn with N, resulting in low diffusion energy barrier.
Based on the success of oxygen evolution on cation-vacancy oxide catalysts, cation vacancy-engineered TMN electrocatalysts may lead to advances in NRR. Yang's group developed a nitrogen-doped carbon-supported MoN electrocatalyst (MV-MoN@NC).220 They reported that varying the density of Mo vacancies resulted in the improvement of NRR performance. It is observed from Fig. 30d that MV-MoN@NC results in a higher current density than that of MoN@NC, which confirms that Mo vacancies contribute to the improvement of NRR performance. Moreover, when compared with the non-defect samples such as MoN, NC, and MoN@NC, it is observed that MV-MoN@NC showed a higher NH3 yield (Fig. 30b and c). The effect of Mo vacancies has been studied by DFT calculations, and it was seen that MV-MoN@NC decreases the barrier height for the potential-determining step from 1.40 to 0.61 eV (Fig. 30e).
Lithium-mediated ammonia synthesis is an indirect method for the electrochemical synthesis of ammonia. Lithium has a large negative potential of −3.05 vs. SHE, which makes it a strong reducing agent. A solution of lithium salt is electrolysed to produce Li+ ions, which get deposited as Li on the cathode. Typically, polar aprotic solvents are used to dissolve Li salts because aqueous electrolytes suffer from the competing HER and the evolution of oxygen at the cathode also causes high overpotentials due to slow reaction kinetics, impeding the NRR.214 The highly reducing Li reacts with N2 gas to form Li3N, which then reacts with a proton source (such as alcohols) present in the electrolyte to form NH3 and Li+ (which can be cycled back to establish a continuous process).222 The schematic representation of this process can be found in Fig. 30h. The following reactions are involved in Li-mediated ammonia synthesis:
| 3Li+ + 3e− → 3Li | (29) |
| 3Li + ½N2 → Li3N | (30) |
| Li3N + 3H+ → NH3 + Li+ | (31) |
The electrochemical nitrate reduction reaction can occur in two mechanisms:224
(i) The indirect pathway – the indirect nitrate reduction process occurs at a higher nitrate concentration (>1 M) in a highly acidic medium. In this process, nitrate itself does not engage in electron transfer, rather it is the electroactive intermediates such as 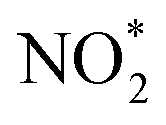 and NO+ that participate in an autocatalytic cycle.
and NO+ that participate in an autocatalytic cycle.
(ii) The direct pathway – most of the studies are carried out at lower nitrate concentrations (<1 M) following the direct nitrate reduction process, which is further divided into two pathways: electron-mediated reduction and active adsorbed hydrogen reduction.
During electron-mediated reduction, N2 and NH3 are the main products while NO2, NO, N2O and NO2− are formed as intermediates. The first step is the adsorption of nitrate on the electrode surface, followed by its reduction to nitrite (NO2−), which is the rate-determining step. The intermediate NO2− is highly reactive and forms NO, which has an important role in determining the product distribution. It can either undergo further reduction to give NH4+ as the end product or it can desorb from the electrode surface to form aqueous NO. NOaq forms a weakly adsorbed dimer, which gives rise to N2O, which is further reduced to N2.225,226
The following are all the reactions involved:
| NO3(ads)− + 2H+ + 2e− → NO2(ads)− + H2O | (32) |
| NO2(ads)− + 2H+ + e− → NO(ads) + H2O | (33) |
| NO(ads) + 6H+ + 5e− → NH4+ + H2O | (34) |
| NO(ads) → NO(aq) | (35) |
| NO(ads) + NO(aq) + 2H+ + 2e− → N2O(ads) + H2O | (36) |
| N2O(ads) + 2H+ + 2e− → N2 + H2O | (37) |
The overall reaction can be written as follows:
| 2NO3− + 12H+ + 10e− → N2 + 6H2O, E° = 1.17 V vs. SHE | (38) |
| NO3− + 9H+ + 8e− → NH3 + 3H2O, E° = −0.12 V vs. SHE | (39) |
During active adsorbed hydrogen reduction, the highly reducing atomic hydrogen is used to reduce the adsorbed NO3− and the intermediates (NO2− and NO in this case) to form N–H bonds yielding NH3 as the end product.225,226 The reactions involved are as follows:
| NO3(ads)− + 2H(ads) → NO2(ads)− + H2O | (40) |
| NO2(ads)− + H(ads) → NO (ads) + OH− | (41) |
| NO(ads) + 2H(ads) → N(ads) + H2O | (42) |
| N(ads) + H(ads) → NH(ads) | (43) |
| NH(ads) + H(ads) → NH2(ads) | (44) |
| NH2(ads) + H(ads) → NH3(ads) | (45) |
Noble metals such as Pd and Pt have a high affinity for hydrogen adsorption and are hence used as catalysts for this reaction.
Long et al. made thermodynamic estimations based on DFT calculations, suggesting that the NO reduction reaction (NORR) is more effective than the NRR. They have considered two different routes for the hydrogenation process – a Tafel type route and Heyrovsky type route, in both dissociative and associative pathways. The N* binding energy was chosen as a descriptor for the NORR as the adsorption energies of all the involved intermediates showed good scaling relations with it. Among 10 transition metals, copper was found to be the most optimal in the Heyrovsky mechanism. This finding was experimentally validated by using a Pt foil, Cu foil and Cu foam as electrodes. An electrochemical ammonia synthesis rate of 517.1 μmol h−1 cm−2 with an FE of 93.5% was achieved at −0.9 V vs. RHE over a Cu foam, which also showed a long-term stability for 100 hours run.227
Wan et al. also performed DFT studies to determine the selectivity and activity of NORR over different catalysts.50 Their finding was similar to that of Long et al.,227 with Cu being the most active metal for NO reduction to NH3. The hydrogenation of adsorbed NO species was predicted to be the rate determining step. At a potential of 0–0.3 V vs. RHE, the current density was lesser than that at a lower potential due to the limiting accessibility of adsorbed *H species; however, NH3 was still the major product and Cu had a higher current density than other transition metals under the same conditions. At a higher potential (>0.3 V vs. RHE), a low barrier for N coupling with NO resulted in the formation of N2O. Niu et al. screened different transition metals embedded in C2N (N doped graphene) to come up with an efficient single-atomic catalyst for NO reduction to NH3.228 Based on DFT calculations, out of 23 transition metals from Ti to Au, Zr–C2N was found to be the most promising catalyst for the NORR.
Subsequently, many studies have been conducted across the world to test a wide range of catalysts for the conversion of nitric oxide into ammonia.229–233
Based on the choice of electrolyte, we categorised electrochemical catalysts for ammonia synthesis into two groups. Electrolytes containing the nitrate ion (NO3−) or nitrate salts correspond to the nitrate reduction pathway and have been grouped into one table (Table 11).
The remaining studies have generally used Na2SO4 as an electrolyte as it does not interfere with the electrodes and also improves the conductivity of water, facilitating electrolysis. These studies focus on the nitrogen reduction reaction (Table 10).
| S. no. | Working electrode | Counter electrode | Reference electrode | Electrolyte | Applied voltage range (V) | Yield rate (μg h−1 mgcat−1) | Voltage at maximum yield (V) | Faradaic efficiency (%) | Voltage at maximum faradaic efficiency (V) | Catalyst synthesis method | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | TiO2/Ti3C2TxMXene | Graphite rod | Ag/AgCl | 0.1 M Na2SO4 | −0.75 to −1 | 44.17 | −0.95 | 44.68 | −0.75 | Hydrothermal oxidation | 234 |
| 2 | np-CuMn | — | — | 0.1 M Na2SO4 | — | 28.9 μg h−1 cm−2 | −0.3 vs. RHE | 9.83 | −0.3 vs. RHE | Arc melting followed by chemical dissolution | 235 |
| 3 | Mn–TiO2/CP | Graphite rod | Ag/AgCl electrode | 0.1 M Na2SO4 | −0.45 to −0.65 | 20.05 | −0.5 | 11.93 | −0.5 | Normal mixing | 236 |
| 4 | TiO2/JE-CMTs/CP | Graphite rod | Ag/AgCl/saturated KCl | 0.1 M Na2SO4 | −0.45 to −0.65 | 20.03 | −0.5 | 10.76 | −0.5 | — | 237 |
| 5 | 1T′′′ MoS2/CC | Graphite rod | Saturated calomel electrode (SCE) | 0.1 M Na2SO4 | −0.1 to −0.5 | 9.09 | −0.3 | 13.6 | −0.3 | Solid state reaction | 238 |
| 6 | C18@CoP/TM | Graphite rod | Ag/AgC | 0.1 M Na2SO4 | −0.1 to −0.5 | 1.44 × 10−10 mol s−1 cm−2 | −0.2 V | 14.03 | −0.2 V | — | 239 |
| 7 | C18@Fe3P/CP | Graphite rod | Ag/AgCl | 0.1 M Na2SO4 | −0.45 to −0.25 | 1.80 × 10−10 mol s−1 cm−2 | −0.3 V | 11.22 | −0.3 V | Phosphidation followed by surface functionalization | 240 |
| 8 | Cu3P NRs | — | — | 0.1 M HCl | −0.1 to −0.5 | 18.9 | −0.2 V | 37.8 | −0.2 V | Solvothermal synthesis | 241 |
| 9 | CuS-CPSs/CP | Graphite rod | Ag/AgCl | 0.1 M HCl | −0.25 to −0.05 | 18.18 | −0.15 V | 5.63 | −0.15 V | Hydrothermal | 242 |
| 10 | PdFe3 | Platinum foil | Ag/AgCl electrode | Saturated KCl electrolyte | −0.1 to −0.25 | 29.07 μg h−1 mgcat−1 | −0.2 V vs. RHE | 22.8 | −0.2 V vs. RHE | Solvothermal followed by thermal reduction | 243 |
| 11 | ZnCo2O4/MgO/BiVO4 | Platinum | Ag/AgCl electrode | 0.1 M KOH | 0.8 to 1.2 V | 35.54 μmol h−1 gcat−1 | 1.0 V vs. RHE | 30.99 | 1.0 V vs. RHE | Hydrothermal | 244 |
| 12 | MnCo2O4/MgO/BiVO4 | Platinum | Ag/AgCl electrode | 0.1 M KOH | 0.8 to 1.2 | 34.03 μmol h−1 gcat−1 | 1.1 V vs. RHE | 67.46 | 1.1 V vs. RHE | Hydrothermal | 244 |
| 13 | TiB2 nanosheets | Platinum wire | Ag/AgCl electrode | 0.1 M Na2SO4 (pH = 10.5) | −0.15 to −0.5 | 318 μg h−1 cm−2 | −0.2 V vs. RHE | 57 | −0.2 V vs. RHE | Surfactant-assisted exfoliation | 245 |
| 14 | ZrO2 nanofibers | Graphite plate | Ag/AgCl electrode | 0.1 M Na2SO4 (pH = 7) | −0.5 to −0.9 | 9.63 mg h−1 mgcat−1 | −0.7 V vs. RHE | 12.1 | −0.7 V vs. RHE | Electrospinning-assisted thermal decomposition | 246 |
| 15 | PdH0.43NRs | Graphite rod | Ag/AgCl | 0.1 M Na2SO4 | −0.1 to −0.5 | 17.53 | −0.2 V | 23.78 | −0.1 V | Hydrothermal followed by solvothermal | 247 |
| 16 | NiPC | Platinum wire | Ag/AgCl | 0.1 M HCl | −0.2 to −0.6 | 85 | −0.3 V | 25 | −0.3 V | Solvothermal | 248 |
| 17 | BiOCl@Ti3C2Tx/CC | Graphite rod | Ag/AgCl | 0.1 M HCL | 0 to −0.2 | 4.06 μmol h−1 cm−2 | −0.1 V | 11.9 | −0.1 V | Hydrothermal | 249 |
| 18 | Au–Bi2Te3 NSs | Carbon rods | Ag/AgCl | 0.1 M Na2SO4 | −0.2 to −0.6 | 32.73 | −0.4 V | 20.39 | −0.4 V | — | 250 |
| 19 | Fe2O3 NR's | Platinum foil | Ag/AgCl | 0.1 M Na2SO4 | −0.3 to −0.6 | 78.02 μmol h−1 cm−2 | −0.6 V | 86.73 | −0.4 V | Hydrothermal | 251 |
| 20 | MoS2 | Graphite rod | Ag/AgCl | 0.1 M Na2SO4 | −0.4 to −0.7 | 8.08 × 10−11 mol s−1 cm−2 | −0.5 V | 1.17 | −0.5 V | Hydrothermal | 252 |
| 21 | CuS/C | Graphite rod | Hg/HgCl2 | 0.1 M KOH saturated with N2 | 0 to −0.6 | 0.9 ± 0.1 μmol h−1 cm−2 | −0.2 V vs. RHE | 11 ± 1 | −0.2 V vs. RHE | Dissolution | 253 |
| 22 | CoPc NTs | Platinum wire | Ag/AgCl | 0.1 M HCl N2, Ar saturated | −0.2 to −0.6 | 105.52 | −0.3 V vs. RHE | 27.19 | −0.3 V vs. RHE | — | 254 |
| 23 | MoB2 | Platinum plate | Ag/AgCl | 0.1 M Na2SO4 | −0.2 to −0.7 | 40.94 ± 0.97 | −0.4 V vs. RHE | 30.84 ± 0.81 | −0.3 V vs. RHE | — | 255 |
| 24 | CoP/TM | Platinum foil | Ag/AgCl | 0.2 M Na2SO4 | 0 to −0.4 | 47.22 μmol h−1 cm−2 | −0.2 V vs. RHE | 88.3 | −0.2 V vs. RHE | Hydrothermal followed by phosphidation | 256 |
| 25 | TiO2/Ti3C2Tx MXene | Graphite rod | Ag/AgCl | 0.1 mol L−1 Na2SO4 | −0.75 to −1 | 44.17 | −0.95 | 44.68 | −0.75 | Hydrothermal oxidation | 234 |
| 26 | α-VSe2−x | Graphite rod | Ag/AgCl (saturated KCl) | 0.5 M LiClO4 | −0.2 to −0.6 | 65.7 | −0.4 | 16.3 | −0.4 | Solvothermal | 257 |
| 27 | FeOOH QDs-GS/CP | Graphite rod | Ag/AgCl/saturated KCl | 0.1 M LiClO4 | −0.3 to −0.7 | 27.3 | −0.4 | 14.6 | −0.4 | Mechanical mixing | 258 |
| 28 | MoS2/C3N4 | — | — | 0.1 M LiClO4 | −0.2 to −0.6 | 18.5 | −0.3 | 17.8 | −0.3 | Solvothermal | 259 |
| 29 | W-NO/NC | Graphite rod | Ag/AgCl | 0.5 M LiClO4 | −0.6 to −0.85 | 12.62 | −0.7 V | 8.35 | −0.7 V | Solvothermal | 260 |
| 30 | La-doped TiO2 nanorods (https://www.sciencedirect.com/topics/chemical-engineering/nanorod) | Graphite rod | Ag/AgCl electrode | 0.1 M LiClO4 | −0.65 to −0.85 | 23.06 μg h−1 mgcat−1 | −0.7 V vs. RHE | 14.54 | −0.7 V vs. RHE | Hydrothermal | 185 |
| S. no. | Working electrode | Counter electrode | Reference electrode | Electrolyte | Applied voltage range (V) | Yield rate (μg h−1 mgcat−1) | Voltage at maximum yield (V) | Faradaic efficiency (%) | Voltage at maximum faradaic efficiency (V) | Catalyst synthesis method | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Fe(TCNQ)2/CF | Graphite rod | Ag/AgCl | 0.2 M NaNO3 + 0.1 M Na2SO4 | −0.6 to −1.1 | 11351.6 μg h−1 cm−2 |
−1.1 | 85.2 | −1.1 | Cation exchange | 261 |
| 2 | CoP NAs/CFC | Graphite rod | SCE | 1.0 M NaNO3 and 1.0 M NaOH | −0.1 to −0.5 | 15.44 mol h−1 m−2 | −0.5 V vs. RHE | 100 | −0.4 V vs. RHE | Electrodeposition followed by phosphorization | 262 |
| 3 | Cu@C | Graphite rod | Ag/AgCl | 0.1 M KOH + 1 mM NO3− | −0.1 to −0.9 | 469.5 μg h−1 cm−2 | −0.9 V vs. RHE | 72.0% | −0.3 V vs. RHE | Solvothermal followed by pyrolysis | 263 |
| 4 | BC2N/Pd | Platinum wire | Hg/HgO | 0.1 M KOH + 250 mM KNO3 | −0.3 to −0.7 | 4325 | −0.7 V | 97.42 | −0.3 V | Annealing | 264 |
| 5 | Amorphous Ru nanoclusters | Platinum disc | Ag/AgCl | 5 mM Cs2CO3 + 500 ppm NO3− | −0.1 to −0.5 | 145.1 | −0.2 V | 80.62 | −0.2 V | Mechanical mixing | 265 |
| 6 | Pd/Cu2O octahedra | Platinum foil | Hg/HgCl2 | 0.5 M K2SO4 + 50 ppm NO3− | −1.0 to −1.5 | 925.11 | −0.645 V | 96.56 | −0.645 V | Mechanical mixing | 266 |
| 7 | Pd/Co3O4 | Platinum foil | Hg/HgCl2 | 0.5 M K2SO4 + 200 mg L−1 NO3−–N | −1.1 to −1.5 | 0.204 mmol h−1 m−2 | −1.3 vs. SCE | 88.6 | −1.3 V vs. SCE | Hydrothermal followed by annealing | 267 |
| 8 | CuPd (3:1) aerogels |
Platinum foil | Hg/HgCl2 | 0.5 M K2SO4 + 50 mg L−1 NO3−–N | 0 to −1.2 | 784.37 | −0.46 vs. RHE | 90.02 | −0.46 V vs. RHE | Mixing | 268 |
| 9 | PdBP NAs/NF | Platinum plate | Hg/HgCl2 | 0.5 M K2SO4 + 100 mg L−1 NO3−–N | −0.46 to −0.86 | 0.109 mmol h−1 cm−2 | −0.66 vs. RHE | 64.73 | −0.66 vs. RHE | Chemical reduction | 269 |
| 10 | NiCo2O4/CC | Graphite rod | Hg/HgO | 0.1 M NaOH and 0.1 M NaNO3 | −0.1 to −0.6 | 973.2 μmol h−1 cm−2 | −0.6 V | 99 | −0.3 V | Hydrothermal followed by annealing | 270 |
| 11 | Fe SAC | Platinum foil | SCE | 0.1 M K2SO4 and 0.5 M KNO3 | −0.5 to −0.85 | 20000 |
−0.85 V | 75 | −0.66 V | TM-assisted carbonization method | 271 |
| 12 | Polycrystalline copper | Platinum plate | SCE | 0.5 M Na2SO4 + 0.1 M KNO3 | −0.066 to −0.466 V | 101.4 μmol h−1 cm−2 | −0.266 V vs. RHE | 93.91 | −0.26 V vs. RHE | Electro-deposition | 272 |
| 13 | FePc/TiO2 NSs | Platinum foil | Ag/AgCl electrode | 0.1 M HNO3/0.4 M KNO3 | 0 to −0.8 V | 17.4 mg h−1 cm−2 | −0.2 V vs. RHE | 90.6 | −0.2 V vs. RHE | — | 273 |
| 14 | NiCoO2@Cu | Platinum foil | Ag/AgCl electrode | 0.1 M Na2SO4 + 0.1 M NaNO3 | −0.5 to −0.9 V | 5940.73 μg h−1 cm−2 | −0.7 V vs. RHE | 94.2 | −0.7 V vs. RHE | Chemical deposition | 274 |
| 15 | Ru–Fe2O3 | Stone grinding rod | Ag/AgCl electrode | 0.1 M NaNO3 + 0.5 M Na2SO4 | −0.5 to −0.9 V | 329 μmol cm−2 h−1 | −0.9 V vs. RHE | 72.8 | −0.9 V vs. RHE | Impregnation | 275 |
| 16 | B–MoS2 | Platinum foil | Saturated Hg/HgO | 0.5 M Na2SO4 + 0.1 M NaNO3 | −0.3 to −0.8 V | 10.8 mg h−1 cm−2 | −0.7 V vs. RHE | 92.3 | −0.7 V vs. RHE | Hydrothermal | 276 |
| 17 | NiCo2O4 nanowire | Graphite rod | Hg/HgO | 0.1 M KOH + 0.1 M NaNO3 | −0.1 to −0.6 | 973.2 μmol h−1 cm−2 | −0.6 V | 99 | −0.3 V | Hydrothermal followed by annealing | 270 |
| 18 | ZnCo2O4 nanoarray | Carbon rod | Hg/HgO | 0.1 M KOH + 0.1 M NaNO3 | −0.8 to −0.2 | 634.74 mmol h−1 cm−2 | −0.8 V | 99 | −0.6 V | Hydrothermal followed by annealing | 277 |
| 19 | BCDs/NiCo2O4 nanowire | Pt foil | Saturated calomel electrode (SCE) | 0.5 M K2SO4 + 200 ppm NO3− | −0.7 to −0.1 | 173.9 μmol h−1 cm−2 | −0.55 V | 100 | −0.55 V | Hydrothermal followed by annealing | 278 |
| 20 | Co-doped Fe/Fe2O3 | Platinum wire | Saturated calomel electrode (SCE) | 0.1 M Na2SO4 + 50 ppm NO3− | −0.5 to 1.0 | 1505.9 μg h−1 cm−2 | −0.95 V | 85.2 | −0.95 V | — | 279 |
| 21 | CoTiO3−x nanofibers | Carbon rod | Hg/HgO | 0.1 M NaOH + 0.1 M NaNO3 | −0.6 to −1.1 | 30.4 mg h−1 mgcat−1 | −1.1 V | 92.6 | −1 V | Hydrogen plasma etching treatment of electrospun CoTiO3 precursor | 280 |
| 22 | RuOx/Pd | — | — | 1 M KOH + 0.1 M KNO3 | 0 to −0.7 V | 23.5 mg h−1 cm−2 | −0.5 V vs. RHE | 98.6 | −0.5 V vs. RHE | — | 281 |
| 23 | Co1–P/NPG | Platinum foil | Ag/AgCl electrode | 0.5 M K2SO4 + 0.1 M KNO3 | −0.5 to −0.9 V | 8.6 mg h−1 mgcat−1 | −0.7 V vs. RHE | 93.8 | −0.7 V vs. RHE | Pyrolysis | 282 |
| 24 | Cu@Cu2+1O nanowires | Platinum foil | SCE | 0.5 M K2SO4 + 50 mg L−1 NO3− | −1.1 to −1.5 | 576.53 | −0.545 vs. RHE | 87.7 | −0.545 vs. RHE | — | 283 |
| 25 | CuOx/TiO2 | Graphite rod | Ag/AgCl | 0.5 M Na2SO4 + 100 ppm NO3− | −0.45 to −0.95 | 1241.81 μg h−1 cm−2 | −0.75 vs. RHE | 92.34 | −0.75 vs. RHE | Electrodeposition and calcination | 281 |
| 26 | Co-doped TiO2 nanosheet | Graphite rod | Hg/HgO | 0.1 M NaOH + 0.1 M NO3− | −0.2 to −0.9 | 1127 μmol h−1 cm−2 | −0.9 vs. RHE | 98.2 | −0.5 vs. RHE | Solvothermal followed by ion exchange and annealing | 284 |
| 27 | Co@TiO2 | Carbon rod | Ag/AgCl | 0.1 M PBS + 0.1 M NO3− | −0.3 to −1.0 | 800 μmol h−1 cm−2 | −1.0 vs. RHE | 96.7 | −0.7 vs. RHE | Hydrothermal and ion exchange followed by calcination | 285 |
| 28 | FeS2@TiO2 | Pt | Hg/HgO | 0.1 M NaOH + 0.1 M NaNO3 | −0.2 to −0.7 | 860.3 μmol h−1 cm−2 | −0.7 vs. RHE | 97 | −0.4 vs. RHE | Hydrothermal and ion exchange followed by calcination | 286 |
| 29 | Cu-doped Co3O4 nanowire | Pt | Ag/AgCl | 0.1 M Na2SO4 + 500 ppm NO3− | −0.3 to −0.7 | 36.71 mmol h−1 g−1 | −0.6 vs. RHE | 86.5 | −0.6 vs. RHE | Hydrothermal and annealing | 287 |
| 30 | Co3O4 nanosheets with Co vacancies | Carbon rod | Hg/HgO | 0.1 M NaOH + 0.1 M NaNO3 | −0.1 to −0.6 | 517.5 μmol h−1 cm−2 | −0.6 vs. RHE | 97.2 | −0.4 vs. RHE | Hydrothermal, annealing followed by etching | 288 |
| 31 | Co0.5Cu0.5 | — | — | 1 M KOH + 50 mM KNO3 | 0.2 to −0.3 | 4250 | −0.25 V vs. RHE | 95 | −0.03 V vs. RHE | Dip-coating and annealing | 289 |
| 32 | Ag@NiO/CC | Graphite plate | Hg/HgO | 0.1 M NaOH with 0.1 M NO2− | −0.2 to −0.7 | 57510 |
−0.7 V vs. RHE | 97.7 | −0.4 V vs. RHE | Magnetron sputtering | 290 |
| 33 | Pd/TiO2 | Platinum plate | Ag/AgCl electrode | 1 M LiCl or 1 M LiCl + 0.25 M LiNO3 | −0.3 to −0.8 V | 1.12 mg cm−2 h−1 | −0.7 V vs. RHE | 92.1 | −0.7 V vs. RHE | Ion exchange followed by hydrothermal and annealing | 291 |
The effectiveness of the electrocatalytic NRR relies largely upon adequately electrocatalyzing the activation of the very strong triple bond of nitrogen molecules for reduction to ammonia. Such electrocatalysts, which generally comprise transition metals or metal alloys, seek to promote adsorption and activation of nitrogen while suppressing side reactions. Recent advancements in the synthesis of electrocatalysts are targeted toward upgrading catalytic activity, selectivity, and stability under reaction conditions with an emphasis on nanostructuring, doping, and modification of the electronic properties of catalysts for performance enhancement. HF etching is a method that allows the preparation of Ti3C2Tx MXenes234 by dissolving Ti3AlC2 in a 40% HF solution, subsequent treatments with NaBF4, and then heating to 160 °C, so that the TiO2@Ti3C2Tx composites are formed, providing stability and improved catalytic performance toward photocatalysis. The arc melting and selective dealloying methods allow for the preparation of Cu15Mn85 (ref. 235) alloys, where upon melting pure Cu and Mn, alloy ribbons are formed and then dealloying in HCl generates porous CuMn alloys, thus allowing fine composition control of the alloy, optimizing for catalytic reactions such as hydrogenation and oxidation. High-temperature treatment and chemical oxidation produce 1T′′′ MoS2 238 crystals upon heating the KMoS2 precursors at 1000 °C, and chemical oxidation in H2SO4 collectively enhances the catalytic activity via phase control, increasing the conductivity for hydrogen evolution reactions. 1T is the octahedral phase of MoS2 , and the 1T′ and 1T′′′ phases are formed by distortion of charge density waves of 1T MoS2. The preparation of Fe2O3/CP and Fe3P/CP240 involves reactions between FeCl3·6H2O and carbon paper under high-pressure conditions, yielding highly active catalysts for electrochemical applications, such that uniform depositions of iron oxide and phosphide on carbon paper are achieved for better performances in hydrogen evolution. The solvothermal method is used to synthesize PdFe3@G243 bimetallic alloys from the solvent-based treatment of metal precursors under high pressures, allowing excellent dispersion and stability of the active phase. The hydrothermal method was used to synthesize MnCo2O4/MgO/BiVO4 catalysts, which consist of reactions of nitrates with MgO/BiVO4 electrodes in a Teflon autoclave, allowing precise control over the crystal structure.
Various electrocatalysts for the NO3RR have been designed to enhance the catalytic activity towards ammonia synthesis, and CuTCNQ/CF261 is produced by keeping copper foam immersed in TCNQ-acetonitrile solvent for surface functionalization, whereas Fe(TCNQ)2/CF employs cationic exchange to form iron-based nanorods on copper foam. Electrodeposition is used for Co-based nanostructures such as Co(OH)2 NAs/CFC, CoP NAs/CFC, and Co NAs/CFC,262 allowing for the utmost degree of control over size, shape, and dispersion, which is critical for electrocatalytic performance. Metal boron organic polymers (BOPs/Pd)264 are synthesized by reacting boron clusters with palladium precursors, yielding highly dispersed metal sites that are a prerequisite for catalytic activity. The synthesis of single-atom catalysts (SACs) such as Fe SAC271 involves pyrolysis of a mixture of iron chloride and organic precursors with SiO2, which leads to the isolation of iron atoms that show enhanced catalytic activity. CoTiO3−x280 nanofibers were synthesized by an electrospinning method with hydrogen plasma etching, producing defects that lead to increased catalytic activity. The NiCoO2@Cu274 catalysts are synthesized by growing cobalt-based MOFs on the scaffold of copper foams, while Ag@NiO/CC and Ag/CC290 catalysts are fabricated using magnetron sputtering for precise metal loading to improve uniformity and catalytic performance. Each method presents particular advantages for controlling the composition and structure of the catalyst, which is directly correlated with enhancing catalytic performance during challenging reactions.
As discussed, the NRR faces the issue of low faradaic efficiency due to the competing HER, and the sluggish kinetics of N2 dissociation also result in a low ammonia synthesis rate. An ideal electrocatalyst must possess an active site that has empty d-orbitals to accommodate lone pair of electrons of N2 for adsorption. For the dissociation of the adsorbed N2 molecule, the catalyst must be able to donate the d-orbital electrons to the anti-bonding orbitals of nitrogen. This would aid in overcoming the barrier of the NN triple bond dissociation. To overcome the issue of HER, the electrocatalyst must favour the adsorption of nitrogen over protons. Additionally, an electrolyte with a low proton donor activity can help solve this problem.
With respect to the NRR, we find that a lot of studies involve the use of titanium-based compounds as electrocatalysts due to its high redox activity and low toxicity. Qian et al.234 used a titanium-based MXene due to its unique layered structure, hydrophilicity, flexibility and high conductivity. The MXene by itself however reduces the specific surface area and diffusivity of ions between layers due to its lamellar structure, which makes stacking very easy. In order to prevent this stacking issue, the authors used the edge Ti atoms as nucleation sites to generate stable TiO2, which prevented microstacking and enhanced the specific surface area.
The catalyst was found to be electrochemically durable after checking the ammonia yield and FE for six consecutive cycles, which were found to be almost similar throughout (Fig. 31a). The chronoamperometry curve (Fig. 31b) shows that there was minimal current density loss after continuous electrolysis for 12 h, which further confirms that the catalyst is highly stable during the NRR. The ammonia yield remained unaffected despite varying the flow rate of N2 (Fig. 31c), which shows that the N2 flow does not affect the rate of the reaction. The 1H NMR spectrum shows a specific double peak for 15NH4+ and three 14NH4+signals (Fig. 31d) corresponding to the 14N2 and 15N2 isotopes, respectively, indicating that NH3 was generated through electrocatalytic N2 reduction. This catalyst achieved a high faradaic efficiency (FE) of 44.68% at −0.75 V vs. RHE and a large NH3 yield of 44.17 μg h−1 mgcat−1 at −0.95 V.234 Wang et al. also used a similar Ti-based MXene modified by BiOCl. The halide of bismuth is a well-known photocatalyst with excellent stability; however, its use as an electrocatalyst was not explored. This modified Ti MXene achieved an ammonia synthesis rate of 4.06 μg h−1 cm−2 at −0.10 V (vs. RHE) with a faradaic efficiency of 11.98%, which was higher than that of BiOCl and Ti MXene individually.249 Titanium dioxide has also attracted significant attention as a promising NRR catalyst due to its abundance, non-toxicity and high thermal stability. It is also widely known as a photocatalyst and has been used in numerous photochemical reactions after specific modifications. However, it faces two challenges as a catalyst for the NRR: the relatively low conductivity, and the low N2 adsorption. In order to overcome these challenges, Chen et al. synthesized a Mn-doped TiO2 catalyst which gave an ammonia yield rate of 20.05 μg h−1 mgcat−1 and a faradaic efficiency of 11.93%, which was much higher than that of undoped TiO2. The enhancement was attributed to the fact that Mn (after doping) introduced oxygen vacancy concentration, which enhanced N2 activation.236 They have also conducted another study where TiO2 was combined with carbon microtubes derived from Juncus effusus (a plant commonly known as soft rush, generally found near wetlands and marshes) for the NRR. Biomass carbon was used as the multi-scale porous structure that easily disperses nanoparticles and enhances the diffusion of electrolyte. The 3D cross-linked hollow tubular structure of the catalyst resulted in an ammonia yield of 20.03 μg h−1 mgcat−1 and a FE of 10.76%.237
 | ||
| Fig. 31 (a) Ammonia yield and faradaic efficiency of the TiO2/Ti3C2Tx MXene at −0.95 V during six consecutive cycling tests. (b) Chronoamperometry curve of the TiO2/Ti3C2Tx MXene composites after electrolysis at −0.95 V for 12 h. (c) Ammonia yield and faradaic efficiency of the TiO2/Ti3C2Tx MXene composites under different N2 flow rates. (d) 1H NMR spectra of 14NH4+ and 15NH4+ yielded from the nitrogen reduction reaction at −0.95 V using 14N2 and 15N2 as the N2 source, respectively. Adapted with permission from ref. 234. Copyright 2022, Elsevier. | ||
A remarkable study was conducted by Rasyotra et al. where titanium diboride (TiB2) was used as an electrocatalyst for the NRR. The unique electronic structure of boron enhances N2 adsorption and reduces the binding strength with proton, thus inhibiting the HER. A high ammonia yield rate of 318 μg h−1 cm−2 (−0.2 V vs. RHE) was achieved, with an FE close to 57%. Using DFT, the authors found the presence of electron-deficient and electron-rich sites which function as “frustrated” Lewis pairs. The Ti and B atoms act as a Lewis base and Lewis acid, respectively, where the electron-rich Ti pushes the electron density towards N2 molecules from where the electron-deficient B pulls it. This synergy weakens the triple bond and activates the N2 molecule.245 Zhou et al. also studied a transition metal diboride for the NRR. Using DFT calculations, they proposed the use of MoB2 due to reasons similar to the previous study. Upon experimental validation, an ammonia synthesis rate of 40.94 ± 0.97 μg h−1 mg−1 (–0.4 V vs. RHE) and an FE of 30.84 ± 0.91% (–0.3 V vs. RHE) were achieved.255
Liu et al. presented an interesting article where they emphasized that the morphology of the catalyst has a huge impact on the catalyst performance. They used Cu3P nanoribbons as catalysts for the NRR and achieved a rate of 18.9 μg h−1 mgcat−1 with an FE of 37.8% at −0.2 V in an acid electrolyte, which was four times higher than those of Cu3P nanoparticles. The nanoribbon material had the flexibility of one-dimensional nanoparticles and the high surface area of two-dimensional nanoparticles, which aided in the improvement of ammonia formation.241
The nitrate reduction reaction has garnered a lot of attention due to its potential to mitigate the harmful effects of nitrate pollution in water, as discussed earlier. The development of catalysts to convert nitrate ions into the economically valuable ammonia has been a hot topic and may even have attracted more number of studies as compared to the NRR. Mu et al. and Li et al. studied the use of Pd metallene to support sub-nm RuOx clusters, so that they can be used as the catalyst for nitrate reduction reaction (NO3RR). Using this as a working electrode, gave a very high ammonia yield rate of 23.5 mg h−1 cm−2 and a high faradaic efficiency of 98.6% at −0.5 V vs. RHE. This is due to the low adsorption energy of NO3−, achieved by a synergistic effect between RuOx clusters and Pd metallene. Pd is responsible for dissociation of H2O to H+, and RuOx clusters facilitate the adsorption of NO3−. The adsorbed electron flow over the surface of Pd to the interface of RuOx/Pd and enable the reduction of nitrate.224,292
Single-atom catalysts (SAC) have been studied extensively for their use as electrocatalysts for the NO3 RR over metal-based catalysts due to the lack of active adjacent sites. This ensures that the coupling of N–N is prevented during the reduction of NO3−. Wu et al. reported that the Fe SAC gave a maximum faradaic efficiency of 75% at −0.66 V vs. reversible hydrogen electrode (RHE) and an impressive NH3 yield rate of ∼20000 μg h−1 mgcat−1.271 Recent studies have shown that the results obtained with a metal-nonmetal catalytic pair are better than those achieved using SACs. This is due to the sluggish dissociation of water over the surface of transition metals SACs. The Co1–P/NPG (Co–P catalytic pair with strong electronic interaction with N,P-doped graphitic carbon) exhibits extraordinary NO3− reduction with a maximum ammonia yield rate of 8.6 mgNH3 h−1 mgcatalyst−1 and a faradaic efficiency of 93.8% at −0.7 V vs. RHE. The Co site acts as an adsorption site for NO3− and the P site is responsible for the generation of H+ by water dissociation.282 Hai et al. studied the bimetallic synergistic effect in the NiCoO2@Cu catalyst, which showed a high faradaic efficiency of 94.2% at −0.7 V vs. RHE and an average ammonia yield rate of 5940.73 μg h−1 cm−2. The adsorption of NO3− occurs through bimetallic sites of Ni and Co, where each site adsorbs one oxygen, whereas in Co3O4, the two oxygen atoms are adsorbed on Co. Adsorption with NiCoO2 is more stable than adsorption with Co3O4 (Eads of −2.29 eV at Co adsorption site on NiCoO2 and Eads of −0.69 eV at Co adsorption site on Co3O4).274
Some studies have shown that Fe2O3 possesses a good adsorption rate of nitrate ions. The synergetic effect of these two could achieve extraordinary results. This was confirmed by Luo et al. through their study of Ru nanoparticles loaded on Fe2O3 for ammonia synthesis, which achieved a faradaic efficiency of 72.8% and an ammonia yield rate of 329 μmol cm−1 h−1.275 Luo et al. also studied boron-doped MoS2 catalyst, which gave a NH3 yield rate of 10.8 mg h−1 cm−2 at −0.7 V vs. RHE, with a faradaic efficiency of 92.3%. The introduction of boron as a dopant increased the binding energy of the 3-d orbital of Mo and 2-p orbital of S by 0.2 eV and 0.15 eV, respectively. As a result, boron extracts electrons from MoS2 to the doping site and makes the surface electron deficient. This resulted in an increased adsorption of NO3− ions. Since boron is electron rich, it can donate electrons to the antibonding orbital of nitrate ions and help in its activation. Moreover, B–MoS2 also exhibits high adsorption energy of H, which inhibits the occurrence of HERs, thereby resulting in higher efficiency.276
While electrochemical ammonia synthesis is an exciting and sustainable approach, overcoming its current limitations requires a multi-faceted strategy involving catalyst innovation, reactor design improvements, and process optimization. Research must focus on the development of advanced catalysts that enhance nitrogen activation, improve faradaic efficiency, and suppress the HER. Transition metal-based materials, single-atom catalysts, and surface-modified electrodes hold promise for addressing these challenges. Additionally, optimizing electrolytes by using non-aqueous systems or ionic liquids can help regulate proton availability and improve selectivity toward ammonia formation. Beyond materials development, reactor engineering is crucial for scaling up the process. Innovations such as flow-cell reactors, membrane-based systems, and high-surface-area electrodes could enhance mass transport and charge transfer, making large-scale operation feasible. Furthermore, integrating external stimuli such as thermal or photo-assisted techniques could accelerate reaction kinetics and improve ammonia production rates. By addressing these challenges, this technology can transition toward higher technology readiness level (TRL) and become a viable alternative for green ammonia production in the future.
7. Conclusion
Ammonia has long been essential as a fertilizer and is now gaining attention for its hydrogen storage and fuel potential. However, industrial ammonia synthesis is highly energy- and carbon-intensive, contributing to 2% of global energy use and 1.44% of CO2 emissions. Therefore, much of the ongoing research on ammonia is focused on developing catalysts for its sustainable production.In this review paper, we summarised the three major routes for low-temperature ammonia synthesis (thermochemical, electrochemical and non-thermal plasma-assisted synthesis) with a comprehensive discussion on the catalytic advancements made in each of these methods. The high cost of renewable energy coupled with its intermittency has resulted in a very high cost of ammonia produced from the electrochemical and NTP methods. Moreover, their modularity makes them more suitable for small-scale ammonia production, where it could be produced on demand to meet the regional needs. From the standpoint of large-scale ammonia production, the technology readiness level (TRL) of these methods is very low, and hence, the HB process is still going to sustain the global ammonia demand for the next few decades. Therefore, while this paper presents a detailed understanding of different modes of ammonia production and the corresponding catalytic developments, it is primarily focused on low-temperature catalysts for the production of ammonia following the HB route.
Several classes of catalysts have been studied for their ability to perform at low temperatures, with some materials such as hydrides and electrides yielding a decent ammonia production rate at temperatures as low as 250–300 °C. Optimising the cost of the catalyst has been the main challenge that researchers are trying to overcome. For instance, ruthenium is an excellent catalyst if we look at the rate of ammonia produced. However, due to its very high cost, it is not feasible for industrial applications. While obtaining a good ammonia formation rate is important, it is how we are cutting down the catalyst cost efficiently, which plays a pivotal role in transforming the HB process. Maximising the TOF can be useful in this regard; a higher TOF would mean that the catalyst converts more reactants per active site per unit time, resulting in a lesser amount of catalysts being used to produce a given amount of ammonia. The ammonia production rate, while seeming to be a good metric to judge the performance of the catalyst, masks two very necessary points that we must consider.
(i) First, it is necessary to understand that these studies are on a laboratory scale, and only the inspection of catalytic activity for large-scale production would give an idea of how readily the material can be adopted for industrial ammonia production.
(ii) Second, it is necessary to analyse the viability of implementing that catalyst for industrial use. While the initial cost of catalyst is important to consider, it is also necessary that the spent catalyst withstands a large number of cycles, so that it does not have to be continually replaced with a fresh catalyst. Often, complex formulations with very precise synthesis techniques are reported, but if this compromises the ease of operation, then it might not be feasible to use that material for large-scale production.
For future research on catalysts, it is recommended to formulate quantitative parameters which can include catalyst cost, lifetime, activity, and consumption in the perspective of ammonia manufacture. Currently, the activities of newer catalysts are compared with the conventional HB catalysts, which does not provide clarity over the several complexities in the catalyst design. This includes varying costs of transition metals, intricacies in the synthesis procedures and the important role of ammonia synthesis loop. Hence, it is vital for future studies on the design and development of catalysts to consider:
(i) Tailoring active sites of the catalysts to address the issues related to nitrogen activation, hydrogen activation, and hydrogenation of adsorbed nitrogen molecules, and desorption of NHx species.
(ii) Developing facile synthesis procedures for such catalysts with high intrinsic activity and ability to maximize the utility of available active sites.
(iii) Addressing the issue to prolong the lifetime of catalysts, particularly to match the scale and intermittency when coupled with renewable energy sources.
Moreover, the commercial catalyst has only seen a few modifications despite decades of operation, indicating that it is also necessary to investigate other ways to optimize the HB process. Designing an efficient reactor configuration to reduce the heat and compressor duty would synergise the enhancement offered by the catalyst and enable us to synthesize ammonia under optimal conditions at an optimal cost. While the above-mentioned changes can help alleviate the environmental concerns associated with the HB process, it is the decentralised production of green ammonia that could bring the concept of “circular ammonia economy” to reality. Small-scale plants are not much affected by the intermittency of renewable energy sources and are perfect to implement lab-scale studies on green ammonia production on a relatively large scale. Renewable energy could be used to drive the water splitting reaction to produce hydrogen on-site. This could then be used by the small-scale ammonia production plant to produce green ammonia, which can then be used for various purposes based on the location of the plant. Technologies with a low TRL or those that cannot withstand the harsh requirements of the industrial process can be implemented in these small-scale plants. Electrochemical and NTP methods can particularly be used in this scenario, as it is assumed that the ammonia supply chain in the future will be driven by an entirely carbon-free process. We can also obtain insights into the ground-level problems faced in the implementation of these methods. Hence, while the optimized HB process can meet the larger demands, increasing the number of these decentralised ammonia production plants would not only enrich the local supply chain of ammonia, but would also ease the transition to 100% green ammonia production in the future. Ammonia has a vast potential to mitigate carbon emissions due to its ability to act as an alternative to fossil fuels. The proposed decentralised model for ammonia production would not only benefit the billions of people dependent on ammonia but also push us closer to our sustainable development goals.
Data availability
The data presented in this article are gathered from the literature and are cited as per the reference style of the journal.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. K. extends gratitude to Central Power of Research Institute (CPRI) for providing fund grants to conduct the work (CPRI/R& D/TC/GDEC/2024). Authors thank the Indian Institute of Technology Patna (IIT Patna) for providing the required facilities to conduct the literature reviews. P. K. Roy and N. Wagh acknowledge IIT Patna and CPRI for their fellowships, respectively.References
- S. Giddey, S. P. S. Badwal, C. Munnings and M. Dolan, ACS Sustain. Chem. Eng., 2017, 5, 10231–10239 CrossRef CAS.
- Global ammonia annual production capacity|Statista, https://www.statista.com/statistics/1065865/ammonia-production-capacity-globally/, accessed 23 August 2024.
- A. E. Yüzbaşıoğlu, C. Avşar and A. O. Gezerman, Curr. Res. Green Sustainable Chem., 2022, 5, 100307 CrossRef.
- B. Yang, W. Ding, H. Zhang and S. Zhang, Energy Environ. Sci., 2021, 14, 672–687 RSC.
- Ammonia – American Chemical Society, https://www.acs.org/molecule-of-the-week/archive/a/ammonia.html, accessed 23 August 2024.
- Ammonia as a Refrigerant: Efficient and Eco-Friendly Choice, https://ammoniagas.com/ammonia-as-a-refrigerant/,accessed 22 August 2024.
- Benefits of Ammonia Use in Refrigeration|Danfoss, https://www.danfoss.com/en-in/markets/refrigeration-and-air-conditioning/dcs/industrial-refrigeration/ammonia-in-industrial-refrigeration/why-ammonia/, accessed 23 August 2024.
- Top Reasons to Avoid Ammonia Cleaners, https://www.freshwaveworks.com/blogs/tips-how-to-use/top-reasons-to-avoid-ammonia-cleaners, accessed 23 August 2024.
- Z. Wan, Y. Tao, J. Shao, Y. Zhang and H. You, Energy Convers. Manage., 2021, 228, 113729 CrossRef CAS.
- A. Valera-Medina, F. Amer-Hatem, A. K. Azad, I. C. Dedoussi, M. de Joannon, R. X. Fernandes, P. Glarborg, H. Hashemi, X. He, S. Mashruk, J. McGowan, C. Mounaim-Rouselle, A. Ortiz-Prado, A. Ortiz-Valera, I. Rossetti, B. Shu, M. Yehia, H. Xiao and M. Costa, Energy Fuels, 2021, 35, 6964–7029 CrossRef CAS.
- Ammonia’s Role in a Net-Zero Hydrogen Economy – Kleinman Center for Energy Policy, https://kleinmanenergy.upenn.edu/research/publications/ammonias-role-in-a-net-zero-hydrogen-economy/, accessed 23 August 2024.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. Ferrero Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- M. El-Shafie and S. Kambara, Int. J. Hydrogen Energy, 2023, 48, 11237–11273 CrossRef CAS.
- O. Herbinet, P. Bartocci and A. Grinberg Dana, Fuel Commun., 2022, 11, 100064 CrossRef.
- X. Xu, E. Liu, N. Zhu, F. Liu, F. Qian, X. Xu, E. Liu, N. Zhu, F. Liu and F. Qian, Energies, 2022, 15, 1023 CrossRef CAS.
- J. Li, R. Zhang, J. Pan, H. Wei, G. Shu and L. Chen, Energy Convers. Manage., 2023, 280, 116827 CrossRef CAS.
- D. Erdemir and I. Dincer, Int. J. Energy Res., 2021, 45, 4827–4834 CrossRef.
- G. Langella, M. de Joannon, P. Sabia, P. Iodice and A. Amoresano, J. Phys.:Conf. Ser., 2022, 2385, 012036 CrossRef CAS.
- A. Wojcik, H. Middleton, I. Damopoulos and J. Van Herle, J. Power Sources, 2003, 118, 342–348 CrossRef CAS.
- G. Cinti, L. Barelli and G. Bidini, AIP Conf. Proc., 2019, 2191, 020048 CrossRef.
- S. S. Rathore, S. Biswas, D. Fini, A. P. Kulkarni and S. Giddey, Int. J. Hydrogen Energy, 2021, 46, 35365–35384 CrossRef CAS.
- M. Otto, L. Vesely, J. Kapat, M. Stoia, N. D. Applegate, G. Natsui, M. Asme, G. E. Aerospace and G. E. Research, ASME Open J. Eng., 2023, 2, 021033 CrossRef.
- H. Cao, J. Guo, F. Chang, C. Pistidda, W. Zhou, X. Zhang, A. Santoru, H. Wu, N. Schell, R. Niewa, P. Chen, T. Klassen and M. Dornheim, Chem. – Eur. J., 2017, 23, 9766–9771 CrossRef CAS PubMed.
- J. Humphreys, R. Lan and S. Tao, Adv. Energy Sustainability Res., 2021, 2, 2000043 CrossRef CAS.
- M. Hattori, S. Iijima, T. Nakao, H. Hosono and M. Hara, Nat. Commun., 2020, 111, 1–8 Search PubMed.
- M. Hattori, N. Okuyama, H. Kurosawa and M. Hara, J. Am. Chem. Soc., 2023, 145, 7888–7897 CrossRef CAS PubMed.
- H. D. Vandervell and K. C. Waugh, Chem. Phys. Lett., 1990, 171, 462–468 CrossRef CAS.
- G. Ertl, S. B. Lee and M. Weiss, Surf. Sci., 1982, 114, 527–545 CrossRef CAS.
- C. T. Rettner and H. Stein, Phys. Rev. Lett., 1987, 59, 2768–2771 CrossRef CAS PubMed.
- T. Oguri, J. Phys. Soc. Jpn., 1964, 19, 77–83 CrossRef CAS.
- P. K. Roy and S. Kumar, J. Environ. Chem. Eng., 2023, 11, 109097 CrossRef CAS.
- M. Kitano, Y. Inoue, M. Sasase, K. Kishida, Y. Kobayashi, K. Nishiyama, T. Tada, S. Kawamura, T. Yokoyama, M. Hara and H. Hosono, Angew. Chem., Int. Ed., 2018, 57, 2648–2652 CrossRef CAS PubMed.
- Y. Kobayashi, M. Kitano, S. Kawamura, T. Yokoyama and H. Hosono, Catal. Sci. Technol., 2017, 7, 47–50 RSC.
- S. Siporin, J. Catal., 2004, 225, 359–368 CrossRef CAS.
- A. Borodziński and M. Bonarowska, Langmuir, 1997, 13, 5613–5620 CrossRef.
- H. Liu and W. Han, Catal. Today, 2017, 297, 276–291 CrossRef CAS.
- R. Kojima and K. Aika, Appl. Catal., A, 2001, 218, 121–128 CrossRef CAS.
- Ammonia Production|Encyclopedia MDPI, https://encyclopedia.pub/entry/1129, accessed 23 August 2024.
- T. Wang and F. Abild-Pedersen, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2106527118 CrossRef CAS PubMed.
- International Energy Agency (IEA), Ammonia technology roadmap, 2021, DOI:10.1787/f6daa4a0-en.
- K. Smart, Johnson Matthey Technol. Rev., 2022, 66, 230–244 CrossRef CAS.
- M. Malmali, Y. Wei, A. McCormick and E. L. Cussler, Ind. Eng. Chem. Res., 2016, 55, 8922–8932 CrossRef CAS.
- M. Malmali, G. Le, J. Hendrickson, J. Prince, A. V. McCormick and E. L. Cussler, ACS Sustain. Chem. Eng., 2018, 6, 6536–6546 CrossRef CAS.
- M. Malmali, M. Reese, A. V. McCormick and E. L. Cussler, ACS Sustain. Chem. Eng., 2018, 6, 827–834 CrossRef CAS.
- F. Tian, J. Li, W. Chen, L. Tang and M. Wu, Int. J. Hydrogen Energy, 2024, 78, 92–122 CrossRef CAS.
- F. Tian, N. Zhou, W. Chen, J. Zhan, L. Tang and M. Wu, Adv. Sustainable Syst., 2024, 8, 2300618 CrossRef CAS.
- V. Shahed Gharahshiran and Y. Zheng, J. Energy Chem., 2024, 96, 1–38 CrossRef CAS.
- J. Mu, X. Gao, T. Yu, L. Zhao, W. Luo, H. Yang, Z. Liu, Z. Sun, Q. Gu and F. Li, Adv. Sci., 2024, 11, 2308979 CrossRef CAS PubMed.
- X. Long, F. Huang, Z. Yao, P. Li, T. Zhong, H. Zhao, S. Tian, D. Shu and C. He, Small, 2024, 20, 2400551 CrossRef CAS PubMed.
- H. Wan, A. Bagger and J. Rossmeisl, Angew. Chem., Int. Ed., 2021, 60, 21966–21972 CrossRef CAS PubMed.
- N. Segal, J. Catal., 1967, 8, 113–119 CrossRef CAS.
- D. King, J. Catal., 1965, 4, 253–259 CrossRef CAS.
- M. R. Hillis, C. Kemball and M. W. Roberts, Trans. Faraday Soc., 1966, 62, 3570 RSC.
- R. Kojima and K. Aika, Appl. Catal., A, 2001, 209, 317–325 CrossRef CAS.
- D. Mckay, J. S. J. S. J. Hargreaves, J. L. L. Rico, J. L. L. Rivera and X.-L. L. Sun, J. Solid State Chem., 2008, 181, 325–333 CrossRef CAS.
- R. Kojima and K. Aika, Appl. Catal., A, 2001, 215, 149–160 CrossRef CAS.
- N. Bion, F. Can, J. Cook, J. S. J. S. J. Hargreaves, A. L. L. Hector, W. Levason, A. R. R. McFarlane, M. Richard and K. Sardar, Appl. Catal., A, 2015, 504, 44–50 CrossRef CAS.
- C. J. H. H. Jacobsen, Chem. Commun., 2000, 1057–1058 RSC.
- C. J. H. H. Jacobsen, S. Dahl, B. S. G. S. Clausen, S. Bahn, A. Logadottir and J. K. Nørskov, J. Am. Chem. Soc., 2001, 123, 8404–8405 CrossRef CAS PubMed.
- A. Logadottir, T. Rod, J. Nørskov, B. Hammer, S. Dahl and C. J. Jacobsen, J. Catal., 2001, 197, 229–231 CrossRef CAS.
- Y. Cao, E. Toshcheva, W. Almaksoud, R. Ahmad, T. Tsumori, R. Rai, Y. Tang, L. Cavallo, H. Kageyama and Y. Kobayashi, ChemSusChem, 2023, 16, e202300234 CrossRef CAS PubMed.
- C. Zhang, S. Shi, B. Fang, J. Ni, J. Lin, X. Wang, B. Lin and L. Jiang, Chin. Chem. Lett., 2023, 34, 107237 CrossRef CAS.
- M. Miyazaki, K. Ikejima, K. Ogasawara, M. Kitano and H. Hosono, ChemSusChem, 2023, 16, e202300551 CrossRef CAS PubMed.
- S. Al Sobhi, J. S. J. Hargreaves, A. L. Hector and S. Laassiri, Dalton Trans., 2019, 48, 16786–16792 RSC.
- Y. Zhou, X. Peng, T. Zhang, H. Cai, B. Lin, L. Zheng, X. Wang and L. Jiang, ACS Catal., 2022, 12, 7633–7642 CrossRef CAS.
- S. Al Sobhi, N. Bion, J. S. J. Hargreaves, A. L. Hector, S. Laassiri, W. Levason, A. W. Lodge, A. R. McFarlane and C. Ritter, Mater. Res. Bull., 2019, 118, 110519 CrossRef CAS.
- X. Peng, H. X. Liu, Y. Zhang, Z. Q. Huang, L. Yang, Y. Jiang, X. Wang, L. Zheng, C. Chang, C. T. Au, L. Jiang and J. Li, Chem. Sci., 2021, 12, 7125–7137 RSC.
- Y. Zhou, C. Wang, X. Peng, T. Zhang, X. Wang, Y. Jiang, H. Qi, L. Zheng, J. Lin and L. Jiang, CCS Chem., 2022, 4, 1758–1769 CrossRef CAS.
- R. Kojima and K. I. Aika, Appl. Catal., A, 2001, 215, 149–160 CrossRef CAS.
- T.-N. N. Ye, S.-W. W. Park, Y. Lu, J. Li, M. Sasase, M. Kitano, T. Tada and H. Hosono, Nature, 2020, 583, 391–395 CrossRef CAS PubMed.
- T.-N. N. Ye, S.-W. W. Park, Y. Lu, J. Li, M. Sasase, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2020, 142, 14374–14383 CrossRef CAS PubMed.
- Y. Zhou, C.-Q. Q. Xu, Z. Tan, H. Cai, X. Wang, J. J. Li, L. Zheng, C. T. Au, J. J. Li and L. Jiang, ACS Catal., 2022, 12, 2651–2660 CrossRef CAS.
- J. Humphreys, R. Lan, S. Chen, M. Walker, Y. Han and S. Tao, Appl. Catal., B, 2021, 285, 119843 CrossRef CAS.
- Z. Chen, Y. Ye, T. Peng, C. Wu, H. Li, X. Pan and X. Bao, ACS Catal., 2023, 13, 14385–14394 CrossRef CAS.
- S. Ghosh, S. M. Jeong and S. R. Polaki, Korean J. Chem. Eng., 2018, 35, 1389–1408 CrossRef CAS.
- Y. Zhang, J. Zhao, D. Yang, B. Wang, Y. Zhou, J. Wang, H. Chen, T. Mei, S. Ye and J. Qu, Nat. Chem., 2022, 14, 46–52 CrossRef CAS PubMed.
- R. Morimoto, T. Ogawa, K. Torii, T. Seno, H. Okumura and K. N. Ishihara, ChemRxiv, 2023, DOI:10.26434/chemrxiv-2023-ng4n7-v2.
- Y. Cao, A. Saito, Y. Kobayashi, H. Ubukata, Y. Tang and H. Kageyama, ChemCatChem, 2021, 13, 191–195 CrossRef CAS.
- Q. Wang, H. Wen, Y. Guan, S. Zhang, W. Gao, J. Guo and P. Chen, ACS Catal., 2023, 13, 9882–9890 CrossRef CAS.
- H. Yan, W. Gao, Q. Wang, J. Guo and P. Chen, Faraday Discuss., 2023, 243, 55–64 RSC.
- C. Liu, Q. Wang, J. Guo, T. Vegge, P. Chen and H. A. Hansen, ACS Catal., 2022, 12, 4194–4202 CrossRef CAS.
- W. Gao, Q. Wang, Y. Guan, H. Yan, J. Guo and P. Chen, Faraday Discuss., 2023, 243, 27–37 RSC.
- Y. Goto, M. Kikugawa, K. Kobayashi, Y. Manaka, T. Nanba, H. Matsumoto, M. Matsumoto, M. Aoki and H. Imagawa, RSC Adv., 2023, 13, 15410–15415 RSC.
- M. Hara, M. Hattori and H. Kurosawa, Iron to catalyze ammonia synthesis at low temperature, 2022, DOI:10.21203/RS.3.RS-1426351/V2.
- P. Wang, F. Chang, W. Gao, J. Guo, G. Wu, T. He and P. Chen, Nat. Chem., 2017, 9, 64–70 CrossRef CAS PubMed.
- H. Yan, W. Gao, Q. Wang, J. Guo and P. Chen, Faraday Discuss., 2023, 243, 55–64 RSC.
- Y. Inoue, M. Kitano, K. Kishida, H. Abe, Y. Niwa, M. Sasase, Y. Fujita, H. Ishikawa, T. Yokoyama, M. Hara and H. Hosono, ACS Catal., 2016, 6, 7577–7584 CrossRef CAS.
- P. K. Roy and S. Kumar, Int. J. Hydrogen Energy, 2024, 64, 497–506 CrossRef CAS.
- Q. Wang, J. Pan, J. Guo, H. A. Hansen, H. Xie, L. Jiang, L. Hua, H. Li, Y. Guan, P. Wang, W. Gao, L. Liu, H. Cao, Z. Xiong, T. Vegge and P. Chen, Nat. Catal., 2021, 411(4), 959–967 CrossRef.
- P. K. Roy and S. Kumar, ACS Appl. Energy Mater., 2020, 3, 7167–7179 CrossRef CAS.
- M. Kitano, S. Kanbara, Y. Inoue, N. Kuganathan, P. V. Sushko, T. Yokoyama, M. Hara and H. Hosono, Nat. Commun., 2015, 61(6), 1–9 Search PubMed.
- S. Kanbara, M. Kitano, Y. Inoue, T. Yokoyama, M. Hara and H. Hosono, J. Am. Chem. Soc., 2015, 137, 14517–14524 CrossRef CAS PubMed.
- Y. Gong, J. Wu, M. Kitano, J. Wang, T. N. Ye, J. Li, Y. Kobayashi, K. Kishida, H. Abe, Y. Niwa, H. Yang, T. Tada and H. Hosono, Nat. Catal., 2018, 13(1), 178–185 CrossRef.
- Y. Lu, J. Li, T. Tada, Y. Toda, S. Ueda, T. Yokoyama, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2016, 138, 3970–3973 CrossRef CAS PubMed.
- T. N. Ye, Y. Lu, J. Li, T. Nakao, H. Yang, T. Tada, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2017, 139, 17089–17097 CrossRef CAS PubMed.
- C. Li, Z. Zhang, Y. Zheng, B. Fang, J. Ni, J. Lin, B. Lin, X. Wang and L. Jiang, Chem. Eng. Sci., 2022, 251, 117434 CrossRef CAS.
- T.-N. N. Ye, Y. Lu, Y. Kobayashi, J. Li, S.-W. W. Park, M. Sasase, M. Kitano and H. Hosono, J. Phys. Chem. C, 2020, 124, 28589–28595 CrossRef CAS.
- Y. Gong, H. Li, J. Wu, X. Song, X. Yang, X. Bao, X. Han, M. Kitano, J. Wang and H. Hosono, J. Am. Chem. Soc., 2022, 144, 8683–8692 CrossRef CAS PubMed.
- C. Croisé, K. Alabd, S. Tencé, E. Gaudin, A. Villesuzanne, X. Courtois, N. Bion and F. Can, ChemCatChem, 2023, 15, e202201172 CrossRef.
- G. Liang, J. Huot and R. Schulz, J. Alloys Compd., 2001, 320, 133–139 CrossRef CAS.
- H. J. Ahn and J. Y. Lee, Int. J. Hydrogen Energy, 1991, 16, 93–99 CrossRef CAS.
- R. L. Cohen and K. W. West, J. Less-Common Met., 1983, 95, 17–23 CrossRef CAS.
- Y. Nakamura, K. Sato, S. Fujitani, K. Nishio, K. Oguro and I. Uehara, J. Alloys Compd., 1998, 267, 205–210 CrossRef CAS.
- H. Li, Y. Gong, H. Yang, X. Yang, K. Li, J. Wang and H. Hosono, ChemSusChem, 2023, 16, e202301016 CrossRef CAS PubMed.
- C. Croisé, K. Alabd, A. Villesuzanne, F. Can, X. Courtois, E. Gaudin, S. Tencé and N. Bion, Catal. Commun., 2023, 179, 106689 CrossRef.
- H. Hosono and M. Kitano, Chem. Rev., 2021, 121, 3121–3185 CrossRef CAS PubMed.
- J. L. Dye, Acc. Chem. Res., 2009, 42, 1564–1572 CrossRef CAS PubMed.
- Y. Jiang, R. Takashima, T. Nakao, M. Miyazaki, Y. Lu, M. Sasase, Y. Niwa, H. Abe, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2023, 145, 10669–10680 CrossRef CAS PubMed.
- Y. Gong, H. Li, C. Li, X. Yang, J. Wang and H. Hosono, Chem. Mater., 2022, 34, 1677–1685 CrossRef CAS.
- J. Kammert, J. Moon, Y. Cheng, L. Daemen, S. Irle, V. Fung, J. Liu, K. Page, X. Ma, V. Phaneuf, J. Tong, A. J. Ramirez-Cuesta and Z. Wu, J. Am. Chem. Soc., 2020, 142, 7655–7667 CrossRef CAS PubMed.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S. W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 411(4), 934–940 CrossRef PubMed.
- Z. Kowalczyk, S. Jodzis, W. Raróg, J. Zieliński, J. Pielaszek and A. Presz, Appl. Catal., A, 1999, 184, 95–102 CrossRef CAS.
- W. Raróg-Pilecka, E. Miśkiewicz, S. Jodzis, J. Petryk, D. Łomot, Z. Kaszkur, Z. Karpiński and Z. Kowalczyk, J. Catal., 2006, 239, 313–325 CrossRef.
- Y. Kadowaki and K. I. Aika, J. Catal., 1996, 161, 178–185 CrossRef CAS.
- X. Zhang, L. Liu, J. Wang, X. Ju, R. Si, J. Feng, J. Guo and P. Chen, J. Catal., 2023, 417, 382–395 CrossRef CAS.
- H. Ronduda, M. Zybert, A. Dziewulska, W. Patkowski, K. Sobczak, A. Ostrowski and W. Raróg-Pilecka, Surf. Interfaces, 2023, 36, 102530 CrossRef CAS.
- J. Feng, L. Liu, X. Ju, Q. Jiang, J. Wang, J. Guo, T. He and P. Chen, Catal. Sci. Technol., 2023, 13, 4156–4167 RSC.
- W. J. Movick, F. Kishimoto and K. Takanabe, Chem. Eng. J., 2023, 452, 139525 CrossRef CAS.
- C. Li, M. Li, Y. Zheng, B. Fang, J. Lin, J. Ni, B. Lin and L. Jiang, Appl. Catal., B, 2023, 320, 121982 CrossRef CAS.
- Y. Baik, M. Kwen, K. Lee, S. Chi, S. Lee, K. Cho, H. Kim and M. Choi, J. Am. Chem. Soc., 2023, 145, 11364–11374 CrossRef CAS PubMed.
- W. Patkowski, M. Zybert, H. Ronduda, G. Gawrońska, A. Albrecht, D. Moszyński, A. Fidler, P. Dłużewski and W. Raróg-Pilecka, Catal, 2023, 13, 405 CAS.
- R. Javaid and T. Nanba, Int. J. Hydrogen Energy, 2023, 48, 11214–11224 CrossRef CAS.
- R. Javaid and T. Nanba, Top. Catal., 2023, 66, 452–460 CrossRef CAS.
- J. Feng, L. Liu, X. Zhang, J. Wang, X. Ju, R. Li, J. Guo, T. He and P. Chen, Catal. Sci. Technol., 2023, 13, 844–853 RSC.
- H. M. Vieri, A. Badakhsh and S. H. Choi, Int. J. Energy Res., 2023, 2023, 2072245 CrossRef.
- X. Zhang, L. Liu, A. Wu, J. Zhu, R. Si, J. Guo, R. Chen, Q. Jiang, X. Ju, J. Feng, Z. Xiong, T. He and P. Chen, ACS Catal., 2022, 12, 2178–2190 CrossRef CAS.
- M. Osozawa, A. Hori, K. Fukai, T. Honma, K. Oshima and S. Satokawa, Int. J. Hydrogen Energy, 2022, 47, 2433–2441 CrossRef CAS.
- B. Fang, C. Zhang, Z. Qi, C. Li, J. Ni, X. Wang, J. Lin, C. T. Au, B. Lin and L. Jiang, AIChE J., 2022, 68, e17849 CrossRef CAS.
- S. I. Miyahara, K. Sato, K. Tsujimaru, Y. Wada, Y. Ogura, T. Toriyama, T. Yamamoto, S. Matsumura, K. Inazu and K. Nagaoka, ACS Omega, 2022, 7, 24452–24460 CrossRef CAS PubMed.
- S. E. Sivan, K. H. Kang, S. J. Han, O. Francis Ngome Okello, S. Y. Choi, V. Sudheeshkumar, R. W. J. Scott, H. J. Chae, S. Park and U. H. Lee, J. Catal., 2022, 408, 316–328 CrossRef CAS.
- J. Wang, L. Liu, X. Zhang, J. Yu, X. Ju, J. Feng, J. Guo, T. He and P. Chen, Catal. Sci. Technol., 2022, 12, 7501–7509 RSC.
- S. ichiro Miyahara, K. Sato, Y. Kawano, K. Imamura, Y. Ogura, K. Tsujimaru and K. Nagaoka, Catal. Today, 2021, 376, 36–40 CrossRef.
- H. Ronduda, M. Zybert, W. Patkowski, K. Sobczak, D. Moszyński, A. Albrecht, A. Sarnecki and W. Raróg-Pilecka, RSC Adv., 2022, 12, 33876–33888 RSC.
- C. Li, S. Yu, Y. Shi, M. Li, B. Fang, J. Lin, J. Ni, X. Wang, B. Lin and L. Jiang, Chem. Eng. Sci., 2022, 262, 118045 CrossRef CAS.
- B. Lin, B. Fang, Y. Wu, C. Li, J. Ni, X. Wang, J. Lin, C. T. Au and L. Jiang, ACS Catal., 2021, 11, 1331–1339 CrossRef CAS.
- J. Huang, J. Pan, Z. You and X. Jiang, Int. J. Hydrogen Energy, 2022, 47, 28019–28024 CrossRef CAS.
- R. Woo, K. Lee, B. S. An, S. H. Kim, H. K. Ju, J. H. Kim, J. Shim, H. T. Beum, K. Cho, Y. S. Bae and H. C. Yoon, Chem. Eng. J., 2023, 475, 146354 CrossRef CAS.
- Y. Zhao, J. Huang, M. Yuan, X. Li, Y. Wang, M. Li, J. Li and Z. You, Catal. Lett., 2024, 154, 1715–1729 CrossRef CAS.
- Y. Zhou, J. Wang, L. Liang, Q. Sai, J. Ni, C. tong Au, X. Lin, X. Wang, Y. Zheng, L. Zheng and L. Jiang, J. Catal., 2021, 404, 501–511 CrossRef CAS.
- H. Kim, A. Jan, D. H. Kwon, H. Il Ji, K. J. Yoon, J. H. Lee, Y. Jun, J. W. Son and S. Yang, Small, 2023, 19, 2205424 CrossRef CAS PubMed.
- K. Sato and K. Nagaoka, Chem. Lett., 2021, 50, 687–696 CrossRef CAS.
- W. Rarogpilecka, E. Miskiewicz, L. Kepinski, Z. Kaszkur, K. Kielar and Z. Kowalczyk, J. Catal., 2007, 249, 24–33 CrossRef CAS.
- M. Karolewska, E. Truszkiewicz, M. Wściseł, B. Mierzwa, L. Kȩpiński and W. Raróg-Pilecka, J. Catal., 2013, 303, 130–134 CrossRef CAS.
- B. Lin, Y. Qi, K. Wei and J. Lin, RSC Adv., 2014, 4, 38093 RSC.
- K. Sato, K. Nagaoka, S. I. Miyahara, K. Tsujimaru, Y. Wada, T. Toriyama, T. Yamamoto, S. Matsumura, K. Inazu, H. Mohri, T. Iwasa and T. Taketsugu, ACS Catal., 2021, 11, 13050–13061 CrossRef CAS.
- H. Ronduda, M. Zybert, W. Patkowski, A. Ostrowski, P. Jodłowski, D. Szymański and W. Raróg-Pilecka, Int. J. Hydrogen Energy, 2022, 47, 35689–35700 CrossRef CAS.
- H. Ronduda, M. Zybert, W. Patkowski, A. Ostrowski, P. Jodłowski, D. Szymański, L. Kępiński and W. Raróg-Pilecka, RSC Adv., 2021, 11, 14218–14228 RSC.
- H. Ronduda, M. Zybert, W. Patkowski, A. Ostrowski, P. Jodłowski, D. Szymański, L. Kępiński and W. Raróg-Pilecka, Int. J. Hydrogen Energy, 2022, 47, 6666–6678 CrossRef CAS.
- E. Apen and J. L. Gland, Surf. Sci., 1994, 321, 308–317 CrossRef CAS.
- M. K. Agusta and H. Kasai, Surf. Sci., 2012, 606, 766–771 CrossRef CAS.
- X. Lu, S. Francis, D. Motta, N. Dimitratos and A. Roldan, Phys. Chem. Chem. Phys., 2020, 22, 3883–3896 RSC.
- M. Chen, M. Yuan, J. Li and Z. You, Appl. Catal., A, 2018, 554, 1–9 CrossRef CAS.
- J. Huang, M. Yuan, X. Li, Y. Wang, M. Li, J. Li and Z. You, J. Catal., 2020, 389, 556–565 CrossRef CAS.
- J. Huang, M. Yuan, X. Li, Y. Wang, M. Li, J. Li and Z. You, Appl. Catal., A, 2021, 615, 118058 CrossRef CAS.
- K. ichi Aika, T. Takano and S. Murata, J. Catal., 1992, 136, 126–140 CrossRef.
- D. Szmigiel, W. Raróg-Pilecka, E. Miśkiewicz, M. Gliński, M. Kielak, Z. Kaszkur and Z. Kowalczyk, Appl. Catal., A, 2004, 273, 105–112 CrossRef CAS.
- D. Szmigiel, H. Bielawa, M. Kurtz, O. Hinrichsen, M. Muhler, W. Raróg, S. Jodzis, Z. Kowalczyk, L. Znak and J. Zieliński, J. Catal., 2002, 205, 205–212 CrossRef CAS.
- S. E. Siporin and R. J. Davis, J. Catal., 2004, 222, 315–322 CrossRef CAS.
- Y. Baik, M. Kwen, K. Lee, S. Chi, S. Lee, K. Cho, H. Kim and M. Choi, J. Am. Chem. Soc., 2023, 145, 11364–11374 CrossRef CAS PubMed.
- K. Lee, R. Woo, H. C. Woo, G. Ko, K. Cho, Y. Park, M. Choi and H. C. Yoon, J. Catal., 2024, 434, 115530 CrossRef CAS.
- S. Murata and K. I. Aika, J. Catal., 1992, 136, 110–117 CrossRef CAS.
- C. Fernández, N. Bion, E. M. Gaigneaux, D. Duprez and P. Ruiz, J. Catal., 2016, 344, 16–28 CrossRef.
- B. Lin, L. Heng, B. Fang, H. Yin, J. Ni, X. Wang, J. Lin and L. Jiang, ACS Catal., 2019, 9, 1635–1644 CrossRef CAS.
- S. Wu, Y.-K. Peng, T.-Y. Chen, J. Mo, A. Large, I. McPherson, H.-L. Chou, I. Wilkinson, F. Venturini, D. Grinter, P. Ferrer Escorihuela, G. Held and S. C. E. Tsang, ACS Catal., 2020, 10, 5614–5622 CrossRef CAS.
- The Ammonia-Synthesis Catalyst of the Next Generation: Barium-Promoted Oxide-Supported Ruthenium – Bielawa – 2001 – Angewandte Chemie International Edition – Wiley Online Library, https://onlinelibrary.wiley.com/doi/abs/10.1002/1521-3773(20010316)40:6%3C1061::AID-ANIE10610%3E3.0.CO;2-B, accessed 24 August 2024.
- C. J. H. Jacobsen, S. Dahl, P. L. Hansen, E. Törnqvist, L. Jensen, H. Topsøe, D. V. Prip, P. B. Møenshaug and I. Chorkendorff, J. Mol. Catal. A:Chem., 2000, 163, 19–26 CrossRef CAS.
- K. Sato, K. Imamura, Y. Kawano, S. ichiro Miyahara, T. Yamamoto, S. Matsumura and K. Nagaoka, Chem. Sci., 2016, 8, 674–679 RSC.
- Y. Niwa and K. I. Aika, J. Catal., 1996, 162, 138–142 CrossRef CAS.
- Y. Ogura, K. Sato, S. I. Miyahara, Y. Kawano, T. Toriyama, T. Yamamoto, S. Matsumura, S. Hosokawa and K. Nagaoka, Chem. Sci., 2018, 9, 2230–2237 RSC.
- Y. Ogura, K. Tsujimaru, K. Sato, S. Miyahara, T. Toriyama, T. Yamamoto, S. Matsumura and K. Nagaoka, ACS Sustain. Chem. Eng., 2018, 6, 17258–17266 CrossRef CAS.
- Z. Wang, Z. Cai and Z. Wei, ACS Sustain. Chem. Eng., 2019, 7, 8226–8235 CrossRef CAS.
- Z. You, K. Inazu, K. ichi Aika and T. Baba, J. Catal., 2007, 251, 321–331 CrossRef CAS.
- Y. Wu, C. Li, B. Fang, X. Wang, J. Ni, B. Lin, J. Lin and L. Jiang, Chem. Commun., 2020, 56, 1141–1144 RSC.
- K. Sato, S. Miyahara, Y. Ogura, K. Tsujimaru, Y. Wada, T. Toriyama, T. Yamamoto, S. Matsumura and K. Nagaoka, ACS Sustain. Chem. Eng., 2020, 8, 2726–2734 CrossRef CAS.
- K. Sato, S. Miyahara, K. Tsujimaru, Y. Wada, T. Toriyama, T. Yamamoto, S. Matsumura, K. Inazu, H. Mohri, T. Iwasa, T. Taketsugu and K. Nagaoka, ACS Catal., 2021, 11, 13050–13061 CrossRef CAS.
- J. P. Lange, Angew. Chem., Int. Ed., 2015, 54, 13186–13197 CrossRef CAS PubMed.
- J. P. Lange, Catal. Sci. Technol., 2016, 6, 4759–4767 RSC.
- J. Zhang, X. Li, J. Zheng, M. Du, X. Wu, J. Song, C. Cheng, T. Li and W. Yang, Energy Convers. Manage., 2023, 293, 117482 CrossRef CAS.
- M. L. Carreon, J. Phys. D Appl. Phys., 2019, 52, 483001 CrossRef CAS.
- P. Shrestha, D. P. Subedi and U. M. Joshi, Electrical characterization of atmospheric pressure dielectric barrier discharge in air, IAEA-TECDOC-CD-1713, International Atomic Energy Agency, Division of Physical and Chemical Sciences, Vienna, Austria, 2013 Search PubMed.
- U. N. Pal, M. Kumar, H. Khatun and A. K. Sharma, J. Phys.:Conf. Ser., 2008, 114, 012065 CrossRef.
- K. Ollegott, P. Wirth, C. Oberste-Beulmann, P. Awakowicz and M. Muhler, Chem. Ing. Tech., 2020, 92, 1542–1558 CrossRef CAS.
- Dielectric barrier discharge (DBD)|Institute of Interfacial Process Engineering and Plasma Technology|University of Stuttgart, https://www.igvp.uni-stuttgart.de/en/research/plasma-technology/sources/barrier/, accessed 25 August 2024.
- J. R. Roberts, Vac. Ultrav. Spectrosc., 2000, 37–63 CAS.
- E. N. Eremin, A. N. Maltsev and V. L. Syaduk, Russ. J. Phys. Chem., 1971, 45(5), 635 Search PubMed.
- L. R. Winter, B. Ashford, J. Hong, A. B. Murphy and J. G. Chen, ACS Catal., 2020, 10, 14763–14774 CrossRef CAS.
- Y. Wang, W. Yang, S. Xu, S. Zhao, G. Chen, A. Weidenkaff, C. Hardacre, X. Fan, J. Huang and X. Tu, J. Am. Chem. Soc., 2022, 144, 12020–12031 CrossRef CAS PubMed.
- Q. Xie, S. Zhuge, X. Song, M. Lu, F. Yu, R. Ruan and Y. Nie, J. Phys. D Appl. Phys., 2019, 53, 064002 CrossRef.
- F. Gorky, S. R. Guthrie, C. S. Smoljan, J. M. Crawford, M. A. Carreon and M. L. Carreon, J. Phys. D Appl. Phys., 2021, 54, 264003 CrossRef CAS.
- T. Mizushima, K. Matsumoto, H. Ohkita and N. Kakuta, Plasma Chem. Plasma Process., 2007, 27, 1–11 CrossRef CAS.
- K. Li, S. Chen, H. Wang and F. Wang, Appl. Catal., A, 2023, 650, 118983 CrossRef CAS.
- S. Li, Y. Shao, H. Chen and X. Fan, Ind. Eng. Chem. Res., 2022, 61, 3292–3302 CrossRef CAS.
- Y. Wang, M. Craven, X. Yu, J. Ding, P. Bryant, J. Huang and X. Tu, ACS Catal., 2019, 9, 10780–10793 CrossRef CAS PubMed.
- X. Zhu, X. Hu, X. Wu, Y. Cai, H. Zhang and X. Tu, J. Phys. D Appl. Phys., 2020, 53, 164002 CrossRef CAS.
- M. Iwamoto, M. Horikoshi, R. Hashimoto, K. Shimano, T. Sawaguchi, H. Teduka and M. Matsukata, Catal, 2020, 10, 590 CAS.
- Y. Zhang, J. Niu, S. Chen, J. Sun, Q. Chen, X. Zhao, B. S. Patil, A. S. R. van Kaathoven, F. J. J. Peeters, N. Cherkasov, J. Lang, Q. Wang and V. Hessel, J. Phys. D Appl. Phys., 2020, 53, 144003 CrossRef.
- J. R. Shah, F. Gorky, J. Lucero, M. A. Carreon and M. L. Carreon, Ind. Eng. Chem. Res., 2020, 59, 5167–5176 CrossRef CAS.
- A. Gómez-Ramŕez, J. Cotrino, R. M. Lambert and A. R. González-Elipe, Plasma Sources Sci. Technol., 2015, 24, 065011 CrossRef.
- A. Gómez-Ramírez, A. M. Montoro-Damas, J. Cotrino, R. M. Lambert and A. R. González-Elipe, Plasma Processes Polym., 2017, 14, 1600081 CrossRef.
- K. Aihara, M. Akiyama, T. Deguchi, M. Tanaka, R. Hagiwara and M. Iwamoto, Chem. Commun., 2016, 52, 13560–13563 RSC.
- M. Iwamoto, M. Akiyama, K. Aihara and T. Deguchi, ACS Catal., 2017, 7, 6924–6929 CrossRef CAS.
- C. Ndayirinde, Y. Gorbanev, R. G. Ciocarlan, R. De Meyer, A. Smets, E. Vlasov, S. Bals, P. Cool and A. Bogaerts, Catal. Today, 2023, 419, 114156 CrossRef CAS.
- B. S. Patil, N. Cherkasov, N. V. Srinath, J. Lang, A. O. Ibhadon, Q. Wang and V. Hessel, Catal. Today, 2021, 362, 2–10 CrossRef CAS.
- T. Mizushima, K. Matsumoto, J. I. Sugoh, H. Ohkita and N. Kakuta, Appl. Catal., A, 2004, 265, 53–59 CrossRef CAS.
- X. Hu, X. Zhu, X. Wu, Y. Cai and X. Tu, Plasma Processes Polym., 2020, 17, 2000072 CrossRef CAS.
- P. Peng, Y. Li, Y. Cheng, S. Deng, P. Chen and R. Ruan, Plasma Chem. Plasma Process., 2016, 36, 1201–1210 CrossRef CAS.
- H. H. Kim, Y. Teramoto, A. Ogata, H. Takagi and T. Nanba, Plasma Processes Polym., 2017, 14, 1600157 CrossRef.
- P. Peng, Y. Cheng, R. Hatzenbeller, M. Addy, N. Zhou, C. Schiappacasse, D. Chen, Y. Zhang, E. Anderson, Y. Liu, P. Chen and R. Ruan, Int. J. Hydrogen Energy, 2017, 42, 19056–19066 CrossRef CAS.
- Y. Wang, M. Craven, X. Yu, J. Ding, P. Bryant, J. Huang and X. Tu, ACS Catal., 2019, 9, 10780–10793 CrossRef CAS PubMed.
- P. Mehta, P. Barboun, F. A. Herrera, J. Kim, P. Rumbach, D. B. Go, J. C. Hicks and W. F. Schneider, Nat. Catal., 2018, 14(1), 269–275 CrossRef.
- Y. Liu, C.-W. Wang, X.-F. Xu, B.-W. Liu, G.-M. Zhang, Z.-W. Liu, Q. Chen and H.-B. Zhang, Plasma Chem. Plasma Process., 2022, 42, 267–282 CrossRef CAS.
- F. Gorky, A. Best, J. Jasinski, B. J. Allen, A. C. Alba-Rubio and M. L. Carreon, J. Catal., 2021, 393, 369–380 CrossRef CAS.
- A. Bajpai and S. Kumar, Mol. Catal., 2024, 557, 113961 CrossRef CAS.
- H. Shen, C. Choi, J. Masa, X. Li, J. Qiu, Y. Jung and Z. Sun, Chem, 2021, 7, 1708–1754 CAS.
- Who really discovered the Haber process?|Feature|RSC Education, https://edu.rsc.org/feature/who-really-discovered-the-haber-process/2020277.article, accessed 25 August 2024.
- A. U. Shetty and R. Sankannavar, J. Energy Chem., 2024, 92, 681–697 CrossRef CAS.
- P. A. Kempler and A. C. Nielander, Nat. Commun., 2023, 141, 1–4 Search PubMed.
- W. Guo, K. Zhang, Z. Liang, R. Zou and Q. Xu, Chem. Soc. Rev., 2019, 48, 5658–5716 RSC.
- M. Zhang, X. Ai, X. Liang, H. Chen and X. Zou, Adv. Funct. Mater., 2023, 33, 2306358 CrossRef CAS.
- X. Yang, F. Ling, J. Su, X. Zi, H. Zhang, H. Zhang, J. Li, M. Zhou and Y. Wang, Appl. Catal., B, 2020, 264, 118477 CrossRef CAS.
- Q. Li, L. He, C. Sun and X. Zhang, J. Phys. Chem. C, 2017, 122(121), 27563–27568 CrossRef.
- D. Krishnamurthy, N. Lazouski, M. L. Gala, K. Manthiram and V. Viswanathan, ACS Cent. Sci., 2021, 7, 2073–2082 CrossRef CAS PubMed.
- M. M. M. Kuypers, H. K. Marchant and B. Kartal, Nat. Rev. Microbiol., 2018, 165(16), 263–276 CrossRef PubMed.
- J. Mu, X. W. Gao, T. Yu, L. K. Zhao, W. Bin Luo, H. Yang, Z. M. Liu, Z. Sun, Q. F. Gu and F. Li, Adv. Sci., 2024, 11, 2308979 CrossRef CAS PubMed.
- X. Zhang, Y. Wang, C. Liu, Y. Yu, S. Lu and B. Zhang, Chem. Eng. J., 2021, 403, 126269 CrossRef CAS.
- S. Niu, J. Energy Chem., 2023, 86, 69–83 CrossRef CAS.
- J. Long, S. Chen, Y. Zhang, C. Guo, X. Fu, D. Deng and J. Xiao, Angew. Chem., Int. Ed., 2020, 59, 9711–9718 CrossRef CAS PubMed.
- H. Niu, Z. Zhang, X. Wang, X. Wan, C. Kuai, Y. Guo, H. Niu, X. Wang, X. Wan, C. Kuai, Y. Guo and Z. Zhang, Small, 2021, 17, 2102396 CrossRef CAS PubMed.
- L. Chen, D. Shen, B. Li, Z. Xiao, W. Sun, X. Liu, J. Ma, C. Li and W. Wang, Ceram. Int., 2023, 49, 23129–23139 CrossRef CAS.
- Q. Wu, H. Wang, S. Shen, B. Huang, Y. Dai and Y. Ma, J. Mater. Chem. A, 2021, 9, 5434–5441 RSC.
- Y. Li, C. Cheng, S. Han, Y. Huang, X. Du, B. Zhang and Y. Yu, ACS Energy Lett., 2022, 7, 1187–1194 CrossRef CAS.
- C. He, H. Yang, M. Xi, L. Fu, J. Huo and C. Zhao, Environ. Res., 2022, 212, 113479 CrossRef CAS PubMed.
- J. Wu and Y. X. Yu, Int. J. Hydrogen Energy, 2023, 48, 5961–5975 CrossRef CAS.
- X. Qian, Y. Wei, M. Sun, Y. Han, X. Zhang, J. Tian and M. Shao, Chin. J. Catal., 2022, 43, 1937–1944 CrossRef CAS.
- Y. Cui, A. Dong, Y. Qu, J. Zhang, M. Zhao, Z. Wang and Q. Jiang, Chem. Eng. J., 2021, 426, 131843 CrossRef CAS.
- H. Chen, T. Wu, X. Li, S. Lu, F. Zhang, Y. Wang, H. Zhao, Q. Liu, Y. Luo, A. M. Asiri, Z. Feng, Y. Zhang and X. Sun, ACS Sustain. Chem. Eng., 2021, 9, 1512–1517 CrossRef CAS.
- H. Chen, J. Liang, K. Dong, L. Yue, T. Li, Y. Luo, Z. Feng, N. Li, M. S. Hamdy, A. A. Alshehri, Y. Wang, X. Sun and Q. Liu, Inorg. Chem. Front., 2022, 9, 1514–1519 RSC.
- G. Lin, Q. Ju, X. Guo, W. Zhao, S. Adimi, J. Ye, Q. Bi, J. Wang, M. Yang and F. Huang, Adv. Mater., 2021, 33, 2007509 CrossRef CAS PubMed.
- Z. Du, J. Liang, S. Li, Z. Xu, T. Li, Q. Liu, Y. Luo, F. Zhang, Y. Liu, Q. Kong, X. Shi, B. Tang, A. M. Asiri, B. Li and X. Sun, J. Mater. Chem. A, 2021, 9, 13861–13866 RSC.
- T. Xu, J. Liang, Y. Wang, S. Li, Z. Du, T. Li, Q. Liu, Y. Luo, F. Zhang, X. Shi, B. Tang, Q. Kong, A. M. Asiri, C. Yang, D. Ma and X. Sun, Nano Res., 2022, 15, 1039–1046 CrossRef CAS.
- Q. Liu, Y. Lin, S. Gu, Z. Cheng, L. Xie, S. Sun, L. Zhang, Y. Luo, A. A. Alshehri, M. S. Hamdy, Q. Kong, J. Wang and X. Sun, Nano Res., 2022, 15, 7134–7138 CrossRef CAS.
- S. Li, Y. Wu, Q. Liu, B. Li, T. Li, H. Zhao, A. A. Alshehri, K. A. Alzahrani, Y. Luo, L. Li and X. Sun, Inorg. Chem. Front., 2021, 8, 3105–3110 RSC.
- J. Mu, Z. Zhao, X. W. Gao, Z. M. Liu, W. Bin Luo, Z. Sun, Q. F. Gu and F. Li, Adv. Energy Mater., 2024, 14, 2303558 CrossRef CAS.
- K. Chen, R. Wang, Q. Mei, F. Ding, H. Liu, G. Yang, B. Bai and Q. Wang, Appl. Catal., B, 2024, 344, 123670 CrossRef CAS.
- A. Rasyotra, A. Thakur, B. Gaykwad, R. Mandalia, R. Ranganathan and K. Jasuja, ACS Appl. Mater. Interfaces, 2024, 16, 24473–24482 CrossRef CAS PubMed.
- J. Xia, H. Guo, M. Cheng, C. Chen, M. Wang, Y. Xiang, T. Li and E. Traversa, J. Mater. Chem. A, 2021, 9, 2145–2151 RSC.
- Z. Wang, Z. Dai, S. Wang, H. Zhang, W. Tian, Y. Xu, X. Li, L. Wang and H. Wang, Chem. Eng. J., 2021, 416, 129105 CrossRef CAS.
- S. Murmu, S. Paul, S. Kapse, R. Thapa, S. Chattopadhyay, A. N., S. N. Jha, D. Bhattacharyya and U. K. Ghorai, J. Mater. Chem. A, 2021, 9, 14477–14484 RSC.
- Y. Wang, M. Batmunkh, H. Mao, H. Li, B. Jia, S. Wu, D. Liu, X. Song, Y. Sun and T. Ma, Chin. Chem. Lett., 2022, 33, 394–398 CrossRef CAS.
- M. Liu, S. Yin, T. Ren, Y. Xu, Z. Wang, X. Li, L. Wang and H. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 47458–47464 CrossRef CAS PubMed.
- J. Liang, H. Chen, T. Mou, L. Zhang, Y. Lin, L. Yue, Y. Luo, Q. Liu, N. Li, A. A. Alshehri, I. Shakir, P. O. Agboola, Y. Wang, B. Tang, D. Ma and X. Sun, J. Mater. Chem. A, 2022, 10, 6454–6462 RSC.
- L. Zhang, X. Ji, X. Ren, Y. Ma, X. Shi, Z. Tian, A. M. Asiri, L. Chen, B. Tang, X. Sun, L. Zhang, X. Ji, X. Ren, X. Sun, Y. Ma, X. Shi, B. Tang, Z. Tian, L. Chen and A. M. Asiri, Adv. Mater., 2018, 30, 1800191 CrossRef PubMed.
- J. Kong, M.-S. S. Kim, R. Akbar, H. S. H. Y. Park, J. H. Jang, H. Kim, K. Hur and H. S. H. Y. Park, ACS Appl. Mater. Interfaces, 2021, 13, 24593–24603 CrossRef CAS PubMed.
- U. K. Ghorai, S. Paul, B. Ghorai, A. Adalder, S. Kapse, R. Thapa, A. Nagendra and A. Gain, ACS Nano, 2021, 15, 5230–5239 CrossRef CAS PubMed.
- H. Y. Zhou, Y. Bin Qu, J. C. Li, Z. L. Wang, C. C. Yang and Q. Jiang, Appl. Catal., B, 2022, 305, 121023 CrossRef CAS.
- J. Liang, W. F. Hu, B. Song, T. Mou, L. Zhang, Y. Luo, Q. Liu, A. A. Alshehri, M. S. Hamdy, L. M. Yang and X. Sun, Inorg. Chem. Front., 2022, 9, 1366–1372 RSC.
- Y. Luo, Q. Li, Y. Tian, Y. Liu and K. Chu, J. Mater. Chem. A, 2022, 10, 1742–1749 RSC.
- X. Zhu, J. Zhao, L. Ji, T. Wu, T. Wang, S. Gao, A. A. Alshehri, K. A. Alzahrani, Y. Luo, Y. Xiang, B. Zheng and X. Sun, Nano Res., 2020, 13, 209–214 CrossRef CAS.
- K. Chu, Y. Liu, Y. Li, Y. Guo and Y. Tian, ACS Appl. Mater. Interfaces, 2020, 12, 7081–7090 CrossRef CAS PubMed.
- Y. Gu, B. Xi, W. Tian, H. Zhang, Q. Fu, S. Xiong, Y. Gu, B. J. Xi, W. Z. Tian, H. Zhang, Q. Fu and S. L. Xiong, Adv. Mater., 2021, 33, 2100429 CrossRef CAS PubMed.
- N. Mukherjee, A. Adalder, N. Barman, R. Thapa, R. Urkude, B. Ghosh and U. K. Ghorai, J. Mater. Chem. A, 2024, 12, 3352–3361 RSC.
- S. Ye, Z. Chen, G. Zhang, W. Chen, C. Peng, X. Yang, L. Zheng, Y. Li, X. Ren, H. Cao, D. Xue, J. Qiu, Q. Zhang and J. Liu, Energy Environ. Sci., 2022, 15, 760–770 RSC.
- Z. Song, Y. Liu, Y. Zhong, Q. Guo, J. Zeng, Z. Geng, Z. Song, Y. Liu, Y. Zhong, J. Zeng, Z. Geng and Q. Guo, Adv. Mater., 2022, 34, 2204306 CrossRef CAS PubMed.
- X. Li, X. Zhao, Y. Zhou, J. Hu, H. Zhang, X. Hu and G. Hu, Appl. Surf. Sci., 2022, 584, 152556 CrossRef CAS.
- M. Jiang, A. Tao, Y. Hu, L. Wang, K. Zhang, X. Song, W. Yan, Z. Tie and Z. Jin, ACS Appl. Mater. Interfaces, 2022, 14, 17470–17478 CrossRef CAS PubMed.
- Y. Xu, K. Ren, T. Ren, M. Wang, Z. Wang, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2022, 306, 121094 CrossRef CAS.
- M. Liu, Q. Mao, K. Shi, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 13169–13176 CrossRef CAS PubMed.
- Y. Xu, K. Ren, T. Ren, M. Wang, M. Liu, Z. Wang, X. Li, L. Wang and H. Wang, Chem. Commun., 2021, 57, 7525–7528 RSC.
- Y. Xu, Y. Sheng, M. Wang, T. Liu, H. Yu, K. Deng, Z. Wang, L. Wang and H. Wang, J. Mater. Chem. A, 2022, 10, 16290–16296 RSC.
- Q. Liu, L. Xie, J. Liang, Y. Ren, Y. Wang, L. Zhang, L. Yue, T. Li, Y. Luo, N. Li, B. Tang, Y. Liu, S. Gao, A. A. Alshehri, I. Shakir, P. O. Agboola, Q. Kong, Q. Wang, D. Ma, X. Sun, Q. Liu, L. Xie, Q. Kong, Q. Wang, J. Liang, Y. Ren, L. Zhang, L. Yue, T. Li, Y. Luo, X. Sun, Y. Wang, D. Ma, N. Li, B. Tang, Y. Liu, S. Gao, A. A. Alshehri, I. Shakir and P. O. Agboola, Small, 2022, 18, 2106961 CrossRef CAS PubMed.
- Z. Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F. Y. Chen, J. Y. (Timothy) Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 121, 1–10 Search PubMed.
- Y. Zhao, Y. Liu, Z. Zhang, Z. Mo, C. Wang and S. Gao, Nano Energy, 2022, 97, 107124 CrossRef CAS.
- R. Zhang, C. Li, H. Cui, Y. Wang, S. Zhang, P. Li, Y. Hou, Y. Guo, G. Liang, Z. Huang, C. Peng and C. Zhi, Nat. Commun., 2023, 141, 1–11 Search PubMed.
- Y. Hai, X. Li, Y. Cao, X. Wang, L. Meng, Y. Yang and M. Luo, ACS Appl. Mater. Interfaces, 2024, 16, 11431–11439 CrossRef CAS PubMed.
- S. Luo, H. Guo, T. Li, H. Wu, F. Zhang, C. Tang, G. Chen, G. Yang and Y. Zhou, Appl. Catal., B, 2024, 351, 123967 CrossRef CAS.
- Y. Luo, K. Chen, P. Shen, X. Li, X. Li, Y. Li and K. Chu, J. Colloid Interface Sci., 2023, 629, 950–957 CrossRef CAS PubMed.
- Z. Li, J. Liang, Q. Liu, L. Xie, L. Zhang, Y. Ren, L. Yue, N. Li, B. Tang, A. A. Alshehri, M. S. Hamdy, Y. Luo, Q. Kong and X. Sun, Mater. Today Phys., 2022, 23, 100619 CrossRef CAS.
- X. Lu, J. Yu, J. Cai, Q. Zhang, S. Yang, L. Gu, G. I. N. Waterhouse, S. Q. Zang, B. Yang and S. Lu, Cell Rep. Phys. Sci., 2022, 3, 100961 CrossRef CAS.
- S. Zhang, M. Li, J. Li, Q. Song and X. Liu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2115504119 CrossRef CAS PubMed.
- X. Fan, J. Liang, L. Zhang, D. Zhao, L. Yue, Y. Luo, Q. Liu, L. Xie, N. Li, B. Tang, Q. Kong and X. Sun, Carbon Neutralization, 2022, 1, 6–13 CrossRef.
- W. Qiu, X. Chen, Y. Liu, D. Xiao, P. Wang, R. Li, K. Liu, Z. Jin and P. Li, Appl. Catal., B, 2022, 315, 121548 CrossRef CAS.
- J. Ni, J. Yan, F. Li, H. Qi, Q. Xu, C. Su, L. Sun, H. Sun, J. Ding and B. Liu, Adv. Energy Mater., 2024, 14, 2400065 CrossRef CAS.
- T. Ren, K. Ren, M. Wang, M. Liu, Z. Wang, H. Wang, X. Li, L. Wang and Y. Xu, Chem. Eng. J., 2021, 426, 130759 CrossRef CAS.
- D. Zhao, C. Ma, J. Li, R. Li, X. Fan, L. Zhang, K. Dong, Y. Luo, D. Zheng, S. Sun, Q. Liu, Q. Li, Q. Lu and X. Sun, Inorg. Chem. Front., 2022, 9, 6412–6417 RSC.
- X. Fan, D. Zhao, Z. Deng, L. Zhang, J. Li, Z. Li, S. Sun, Y. Luo, D. Zheng, Y. Wang, B. Ying, J. Zhang, A. A. Alshehri, Y. Lin, C. Tang, X. Sun and Y. Zheng, Small, 2023, 19, 2208036 CrossRef CAS PubMed.
- H. Wang, D. Zhao, C. Liu, X. Fan, Z. Li, Y. Luo, D. Zheng, S. Sun, J. Chen, J. Zhang, Y. Liu, S. Gao, F. Gong and X. Sun, J. Mater. Chem. A, 2022, 10, 24462–24467 RSC.
- Z. Niu, S. Fan, X. Li, Z. Liu, J. Wang, J. Duan, M. O. Tadé and S. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 35477–35484 CrossRef CAS PubMed.
- Z. Deng, C. Ma, Z. Li, Y. Luo, L. Zhang, S. Sun, Q. Liu, J. Du, Q. Lu, B. Zheng and X. Sun, ACS Appl. Mater. Interfaces, 2022, 14, 46595–46602 CrossRef CAS PubMed.
- T. H. Jeon, Z.-Y. Wu, F.-Y. Chen, W. Choi, P. J. J. Alvarez and H. Wang, J. Phys. Chem. C, 2022, 126, 6982–6989 CrossRef CAS.
- Q. Liu, G. Wen, D. Zhao, L. Xie, S. Sun, L. Zhang, Y. Luo, A. Ali Alshehri, M. S. Hamdy, Q. Kong and X. Sun, J. Colloid Interface Sci., 2022, 623, 513–519 CrossRef CAS PubMed.
- Y. Guo, R. Zhang, S. Zhang, Y. Zhao, Q. Yang, Z. Huang, B. Dong and C. Zhi, Energy Environ. Sci., 2021, 14, 3938–3944 RSC.
- X. Li, P. Shen, X. Li, D. Ma and K. Chu, ACS Nano, 2023, 17, 1081–1090 CrossRef CAS PubMed.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |