
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deciphering the role of hydrothermal pretreatment in the conversion of biomass waste into hard carbon with superior electrochemical performance in sodium-ion batteries†
Wenxiu Li‡
ab,
Jian Cui‡b,
Wendong Yeb,
Panpan Su*b,
Xuefen Songb,
Tai Yang*a,
Yongguang Zhang*b and
Zhongwei Chen *b
*b
aSchool of Materials Science and Engineering, Hebei University of Technology, Tianjin, 300130, China. E-mail: yangtai@hebut.edu.cn
bPower Battery and Systems Research Center, State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: panpansu@dicp.ac.cn; ygzhang@dicp.ac.cn; zwchen@dicp.ac.cn
First published on 16th April 2025
Abstract
Due to the complex composition of biomass, the microstructure of derived hard carbon (HC) has a large number of defects and pores. This results in a low initial coulombic efficiency (ICE) and poor rate performance of sodium-ion batteries (SIBs), posing significant challenges to their practical application. This article proposes a hydrothermal pretreatment-assisted carbonization method to prepare almond shell-derived HC. Meanwhile, a comprehensive and systematic investigation was conducted to elucidate the impact of hydrothermal conditions (neutral and acidic conditions) on the crystallinity of the precursor and architecture of derived HC, as well as their correlation with sodium-ion storage performance. Hemicellulose in almond shells was selectively removed by the hydrothermal treatment in pure water, which effectively formed HC with high storage sites and regular morphology, thereby improving electrochemical performance. The optimized biomass-derived HC exhibited a remarkable reversible capacity of 317.1 mA h g−1 at 0.1 C, accompanied by an ICE of 85.9%. Even after 600 cycles at 1.0 C, an ultra-high capacity retention rate of 91.1% was achieved. Through in situ and ex situ characterization studies, it was demonstrated that the excellent sodium-ion storage capacity results from the adsorption on the surface and filling in closed pores of HC. This work provides important guidance for the design of biomass-derived HC as an anode material for SIBs.
Introduction
Due to the uneven distribution and limitations of lithium resources, sodium-ion batteries (SIBs) have been considered as a supplement to lithium-ion batteries (LIBs).1–3 However, compared with lithium ions, the larger radius of sodium ions renders traditional negative electrode materials of LIBs unsuitable for SIBs.4,5 Developing new electrode materials is the key to realizing the commercialization of SIBs.6 The negative electrode materials of SIBs include metal oxides,7 alloys,8 organic frameworks,9,10 and carbon-based materials.11,12 Among these, hard carbon (HC) has become the most promising negative electrode material for SIBs due to its merits of abundant resources, low operating voltage, and high theoretical specific capacity.13,14 Unlike the long-range ordered structure of graphite, HC is composed of randomly oriented and twisted graphite nanodomains with larger interlayer spaces (0.36–0.4 nm). It requires the precursors, which have branched structures and contain functional groups (e.g. oxygen, sulfur, nitrogen, etc.), in order to prohibit the rearrangement of large graphite layers during carbonization.15–17 Biomass perfectly meets the above demands and has become a promising precursor for HC.18,19Due to the complex composition, biomass-derived HC has high ash content, and the microstructure is rich in defects and open pores on the surface, resulting in low initial coulombic efficiency (ICE) and poor rate performance in SIBs.20 Pretreatment was widely adopted to tailor the composition of biomass to manipulate the microstructure of derived HC, thereby boosting the performance of SIBs.21,22 For instance, Kim et al.23 utilized ash-removal techniques to enhance the electrochemical performance of cocoa pod husk derived HC. A positive correlation between the acid treatment of peanut shells and their performance for SIBs was revealed by Dou et al.24 Additionally, hazelnut shell-derived HC with the aid of HCl treatment to remove the inorganic components exhibits high sodium-ion storage capacity.25 Zhang et al.26 effectively regulated the microstructure of tea stem-derived HC materials through simple acid treatment. Obviously, acid or alkali treatments can effectively remove inorganic impurities from biomass to improve the capacity of HC, while they may introduce pores and defects, which undermine the ICE and stability of SIBs.27,28 In contrast, the hydrothermal method is a more effective and environmentally friendly pretreatment to remove impurities from biomass. Combined with hydrothermal treatment, biomass shell-derived HC achieved a significant improvement in electrochemical performance.29 Previous studies have predominantly focused on the pretreatment of biomass to remove inorganic impurities, while neglecting its regulation on the microstructure of HC, which is closely related to their performance.30 Nevertheless, the cellulose structure will be disrupted, along with the removal of impurities under different hydrothermal conditions, which led to the formation of a large-scale graphitized structure and is not conducive to the sodium-ion storage behavior for derived HC. Therefore, a systematic study on hydrothermal pretreatment is needed to optimize the electrochemical performance of HC.
Almond shell, as a kind of waste fruit shell biomass material, is composed of cellulose, hemicellulose and lignin, which is a promising precursor for HC. Herein, we provide a hydrothermal pretreatment-assisted carbonization method to convert almond shells into highly efficient HC. In particular, the crystallinity of cellulose was selectively regulated by neutral (pure water) hydrothermal pretreatment, and the microstructure and the closed pore structure in derived HC were adjusted, which is conducive to enhancing sodium-ion storage capacity. The optimized biomass-derived HC exhibits a high reversible capacity of 317.1 mA h g−1 at 0.1 C with an ICE of 85.9%, and the capacity could be retained at 91.1% even after 600 cycles. This work provides significant guidance for the fabrication of biomass-derived HC as anode materials for commercial SIBs.
Experimental section
Synthetic methods
Almond shells were smashed to obtain the precursor powder (almond shells originated from Hebei province). The pretreatment was conducted via a hydrothermal method. 20.0 g of the precursor was dispersed in 100.0 mL of pure water and then transferred into a 150.0 mL Teflon-lined stainless-steel autoclave. Subsequently, the reactor was heated at a constant temperature of 180 °C for 24 h to conduct the hydrothermal reaction, then washed with deionized water until it became neutral and dried overnight in a vacuum oven at 60 °C. The obtained sample was named HY-W. The carbonization process was performed with a heating rate of 5 °C min−1 until 1100 °C and maintained for 5 h under an argon atmosphere. The obtained HC sample was named HC-W. HY-A and HC-A were prepared using the same method, except that the hydrothermal solvent was 1.0 M sulfuric acid aqueous solution (the sulfuric acid solution was purchased from China National Medicines Corporation Ltd). In contrast, the precursor without pretreatment was named HY-P, and HC-P was directly carbonized without undergoing hydrothermal treatment.Materials characterization
The morphological characteristics of the samples were measured through the application of scanning electron microscopy (SEM, JSM-7800F, Electronics Corporation, Japan) and transmission electron microscopy (TEM, JEM2100, Electronics Corporation, Japan). The thermal decomposition behavior of the precursor was determined by thermogravimetric analysis (TGA). The testing temperature range was set from room temperature to 1000 °C, with a heating rate of 10 °C min−1. The crystal structure of the HC materials was analyzed by X-ray diffraction (XRD, Empyrean X-ray diffractometer). The testing was performed using Cu Kα radiation within the range of 10° to 90°. The functional groups of the samples were tested by Fourier transform infrared spectroscopy (FTIR, Invenio, Bruker). The Brunauer–Emmett–Teller (BET) method was utilized to characterize the surface area and pore size distribution under the conditions of 77 K and nitrogen atmosphere. Small-Angle X-ray Scattering (SAXS) was conducted using an Anton Paar SAXSess mc2 to test the gyration radius of the samples. The molecular structure of the samples was analyzed by Raman spectroscopy (Nanowizard) with a wavelength of 532 nm. The molecular structure and valence state were analyzed by X-ray photoelectron spectroscopy (XPS, Thermo Fisher Escalab 250 Xi+).Electrochemical measurements
For electrode preparation, 92% HC material, 3% Super P, and 5% polyvinylidene fluoride were weighed and then thoroughly mixed with a suitable quantity of N-methylpyrrolidone to form a homogeneous slurry. Subsequently, the slurry was evenly coated onto double-sided carbon-coated aluminum foil, ensuring a uniform thickness. Next, the coated foil was placed in a vacuum environment at 100 °C and dried for 12 h to completely remove the solvent. After drying, the electrode was cut into circular pieces with a diameter of 12.0 mm, and the working electrode had a similar mass loading of 2.0–2.3 mg cm−2. For the assembly and disassembly of CR2032 coin cells, all operations were carried out in a glove box filled with argon. The water and oxygen content inside the glove box was strictly controlled to be less than 0.1 ppm. In the assembly, metallic sodium was selected as the counter electrode. The electrolyte consisted of 1.0 M sodium hexafluorophosphate dissolved in diglyme. Additionally, GF/A glass fiber with a diameter of 19.0 mm was used as the separator.The galvanostatic charge–discharge experimental tests were conducted using a Neware battery testing system, which was procured from Shenzhen. Cyclic voltammetry (CV) was conducted via an electrochemical workstation, with the scanning rates adjusted to range from 0.1 to 2.0 mV s−1. On the same electrochemical workstation, electrochemical impedance spectroscopy (EIS) was performed with an amplitude set to 5.0 mV and a frequency range from 10−2 to 105 Hz. For the galvanostatic intermittent titration technique (GITT), the test current was maintained at 20.0 mA g−1, with the pulse current lasting 10 minutes and the relaxation interval was set to 3 h. The scanning range of the in situ XRD was configured to be between 10° and 90°, with each scanning cycle completed within 10 minutes. The in situ XRD measurements were carried out during the charge and discharge processes at a current density of 0.15C.
Results and discussion
The synthesis process of almond shell-derived HC is shown in Fig. 1. The preparation method consists of two steps: hydrothermal treatment and carbonization. Under different hydrothermal conditions, cellulose with different degrees of crystallinity is formed. Subsequently, during the carbonization process, HC products are gradually generated, resulting in different pseudo graphitic microstructures. | ||
| Fig. 1 Schematic diagram of preparation of almond shell-derived HC. | ||
Thermogravimetric analysis (TGA) was employed to analyze the thermal decomposition behavior of almond shells. The curve (Fig. S1†) showed two main regions of mass loss for the precursors: a small peak corresponding to the release of inherent water at 100 °C and a weight loss occurring from 200 to 500 °C, which was due to the devolatilization of biomass. The main component of lignocellulose and most impurities could be decomposed in this region simultaneously.31 It is worth noting that the presence of these impurities disturbs the generation of highly efficient HC. The structure of hydrothermal pretreated precursors was determined using X-ray diffraction (XRD), which offers overall information about the cellulose crystal phase. The main peaks observed at approximately 15° and 22° corresponded to the (110) and (002) reflection planes of cellulose. The height of the (002) peak represents the sum of the crystalline and amorphous components. In this empirical method, the amorphous-only component is represented by the intensity at the minimum of about 18° between the (110) and (002) peaks. In addition, the crystallinity of cellulose could be calculated according to the formula:
| CrI = (I002 − IAM)/I002 | (1) |
Furthermore, Fourier transform infrared (FTIR) spectra were collected to comprehensively elucidate the chemical structure of the precursors under different hydrothermal conditions. As shown in Fig. S3,† there is a broad peak at approximately 1100 cm−1, corresponding to the complex aromatic structure of HY-P lignocellulose. After hydrothermal treatment in pure water medium, the C–O vibration peaks were enhanced at 1110 cm−1 and 1350 cm−1, indicating that HY-W mainly contains cellulose based structures. In contrast, these peaks for HY-A were relatively weak, indicating that fewer oxygen-containing functional groups were retained. This might be due to more polycondensation reactions occurring during the hydrothermal process, resulting in a lower cellulose content in the precursor. In summary, the HY-W sample exhibited a relatively high cellulose content, which was consistent with the structure evaluated by XRD.
In order to reveal the influences on derived HC from different degrees of crystallinity of cellulose, three types of samples (HY-P, HY-A and HY-W) were carbonized to obtain HCs. The micromorphology of the prepared HCs (HC-P, HC-A and HC-W) was characterized using a scanning electron microscope (SEM) and a high-resolution transmission electron microscope (HRTEM). As shown in Fig. 2a–c, the particles of the HC-P material were found to have an irregular morphology with abundant open porous structure. In contrast, both HC-A and HC-W exhibited regular morphology characteristics. Among them, HC-A showed a smooth surface morphology, but some flaky crystalline solids appeared on the surface of HC-W. HRTEM analysis was further used to elucidate the local structure of the prepared materials, as shown in Fig. 2d–f and S4.† The typical microstructure of HC with chaotic stacking graphite domains of short-range carbon layers could be observed in all images, and the interlayer spacing was measured to be about 0.4 nm. All the images indicated that the HC subjected to hydrothermal treatment presented a spherical morphology. Specifically, the size of HC obtained in pure water was relatively large, while that obtained under acidic conditions was relatively small. Carbon spheres of different sizes have different microstructures, which affect their electrochemical properties.
 | ||
| Fig. 2 SEM images of (a) HC-P, (b) HC-A, and (c) HC-W. TEM images of (d) HC-P, (e) HC-A, and (f) HC-W. (g) N2 adsorption–desorption isotherms and (h) SAXS patterns of HC-P, HC-A and HC-W. (i) Fitted SAXS pattern of HC-W. | ||
In order to determine the influences on the surface porous structure by hydrothermal pretreatment, the open porosity of HCs was characterized using N2 adsorption–desorption isotherms. It was observed that all the materials showed type IV curves with hysteresis loops (Fig. 2g), implying the generation of mesoporous structures.33 While HC-A exhibited a much larger gas adsorption capacity than HC-P and HC-W, it indicated that HC-A prepared from a lower crystallinity precursor would possess more exposed sites. In contrast, the smallest gas adsorption capacity was obtained from HC-W, which indicated that the high crystallinity was conducive to the formation of a denser surface structure. The surface structure is closely related to the electrochemical performance of ICE, which has an effect on the decomposition of electrolyte and the formation of the solid electrolyte interphase (SEI) film.34 In addition to the open porous structure, the closed pore was characterized using small-angle X-ray scattering (SAXS). As shown in Fig. 2h, all the samples exhibited a high scattering intensity from the closed pores. Compared with HC-A and HC-P, HC-W exhibited the highest scattering intensity, suggesting that HC-W formed the most closed pores.35 The plots fitted by three main contributions to the scattered intensity are shown in Fig. 2i, which include a power law at low Q, arising from scattering of the whole HC particles (Iparticles); a shoulder region at about 0.3–5 Å−1 attributed to the scattering from closed pores (Ipores) and a background term (Ibckg) coming from the tail of interatomic scattering:
| ISAXS(Q) = Iparticles(Q) + Ipores(Q) + Ibckg | (2) |
| Ibckg = C | (3) |
| Iparticles(Q) = A/QK | (4) |
Furthermore, the surface chemical composition and chemical states of C and O were revealed by X-ray photoelectron spectroscopy (XPS). Fig. 3a–f show that the high-resolution XPS spectrum of C 1s for HCs was deconvoluted into three distinct peaks with binding energies of 284.1, 284.8 and 288.4 eV, respectively, corresponding to sp2 C, C–C and COOR. For detailed results, the high-resolution O 1s spectra of HCs displayed distinct peaks for O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (533.0 eV), O–C (531.6 eV), and O (Arc) (530.0 eV). The presence of OC and O–C bonds in HC-A and HC-W moved towards lower bonding energy compared with HC-P, indicating that the hydrothermal pretreatment is conducive to the increase in electron density and the formation of an electron-rich surface.
C (533.0 eV), O–C (531.6 eV), and O (Arc) (530.0 eV). The presence of OC and O–C bonds in HC-A and HC-W moved towards lower bonding energy compared with HC-P, indicating that the hydrothermal pretreatment is conducive to the increase in electron density and the formation of an electron-rich surface.
 | ||
| Fig. 3 High-resolution (a) C 1s and (b) O 1s spectra of HC-P. High-resolution (c) C 1s and (d) O 1s spectra of HC-A. High-resolution (e) C 1s and (f) O 1s spectra of HC-W. Raman spectra of (g) HC-P, (h) HC-A, and (i) HC-W. | ||
To reveal the effects of hydrothermal pretreatment, the microstructure of prepared HCs was determined using XRD. Fig. S6† shows that the patterns of HC-P, HC-A, and HC-W presented two broad diffraction peaks at 2θ of 22° and 43°, which corresponded to the (002) and (100) crystal planes, respectively. Typically, the fact that all the peaks were broad indicated the disordered pseudo graphitic domains rather than long-range graphitization.36 According to the Bragg equation, the average interlayer spacing of the (002) plane was determined to be 0.4 nm in all HCs. However, the full width at half maximum (FWHM) of the (002) peak was different in the three samples. Among them, HC-A showed the smallest FWHM, indicating the highest degree of graphitization, and HC-W presented the largest FWHM, suggesting that it had the lowest degree of graphitization. These results demonstrated that the degree of graphitization in almond shell-derived HCs was related to the crystallinity of cellulose in the precursor.37 The crystal size was quantified by calculating La and Lc (Table S1†). The calculations showed that the parameters were not significantly different, which indicated that the changes in crystallinity brought by hydrothermal pretreatment could only change the degree of graphitic order rather than promote graphitic growth.
Raman spectroscopy further provided detailed crystallinity information. As shown in Fig. 3g–i and Table S2,† the D and G bands were located at 1350 and 1590 cm−1, respectively, which could be further divided into five sub-peaks at 1590, 1350, 1610, 1480 and 1240 cm−1, namely G, D1, D2, D3, and D4. The ratio of the D1 and G peaks was calculated based on the integral area (AD1/AG) to evaluate the disorder degree of the carbon. The AD1/AG values of HC-P, HC-A and HC-W were calculated to be 1.37, 1.31 and 1.42, respectively. Moreover, the results also showed that the HC-A had the highest degree of graphitization, even to an excessive degree; such a large-scale graphitized structure is not conducive to the formation of chaotically stacked graphitic planes in HC.38
The electrochemical performance of HCs was preliminarily evaluated using a coin-type half-cell system, with metallic Na as the counter electrode. Fig. 4a displays the galvanostatic charge–discharge curves at a current density of 0.1C (1.0C = 372.0 mA g−1) within the voltage range of 0–2.0 V. For all samples, the high reversible capacities were determined to be 263.4, 290.1 and 317.1 mA h g−1 for HC-P, HC-A and HC-W, respectively. The HC-W electrode delivered a higher ICE of 85.9% than HC-P (80.0%) and HC-A (80.1%). The loss in capacity during the first discharge/charge curves is mainly associated with the formation of the SEI due to the decomposition and irreversible ion trapping on the surface of HC. Obviously, HC-W possessed a denser surface structure, lower defects and open pores, which was propitious to form a more regular SEI.39 Considering the reversible capacity contributions in different regions, as shown in Fig. 4b: (1) both slope region reversible capacities of HC-A and HC-W were higher than those of HC-P, which was mainly from the sodium-ion adsorption; (2) HC-W exhibited a higher plateau region capacity than HC-A, which was possibly from the sodium-ion filling in closed pores.40 This indicates that hydrothermal pretreatment is helpful to improve the slope reversible capacity, and neutral hydrothermal conditions are more conducive to improving the plateau capacity than acidic hydrothermal conditions. Compared with HC-W, HC-A has a higher degree of graphitization. However, an excessively high degree of graphitization is not conducive to the adsorption and storage of sodium ions. Thus, the hydrothermal pretreatment of biomass in pure water can effectively manipulate the microstructure of derived HC, which leads to superior sodium-ion storage behavior both in the slope and plateau regions.
 | ||
| Fig. 4 (a) The initial discharge/charge curves of HC-P, HC-A and HC-W at 0.1C. (b) Discharge capacity contributions from the slope region and plateau region. (c) Rate performance of HC-P, HC-A and HC-W. (d) Cycling stability of HC-P, HC-A and HC-W at 1.0C for 600 cycles. (e) CV curves at different scan rates from 0.1 to 2.0 mV s−1 and (f) b-values of HC-W. (g) Capacitive contributions at different scan rates of HC-W. (h) Nyquist plots in the initial state. | ||
The rate performance was evaluated to determine the effect of the hydrothermal process on sodium-ion storage kinetic characteristics. As shown in Fig. 4c, HC-W exhibited the best rate performance, with reversible capacities of 306.0, 300.9, 293.1, 286.7, 280.3 and 272.5 mA h g−1 at various current densities, respectively. In particular, even at a current density of 10.0C, the reversible capacity could remain at 266.8 mA h g−1. When the current density was restored to 0.1C, the specific capacity of HC-W still remained at 300.7 mA h g−1. It is apparent that the rate performance of HC-W is better than others. The results indicate that an excessively high degree of graphitization is unfavorable for sodium-ion storage, resulting in a reduction in rate performance.41 The cycling stability of the prepared HCs at a current density of 1.0C is shown in Fig. 4d. After 600 cycles, the capacity retention rate of HC-W was 91.1%, which was higher than that of HC-P (80.7%) and HC-A (90.6%). It can be concluded that the stable carbon structure from biomass subjected to hydrothermal pretreatment in pure water can withstand the volume changes during the repeated sodiation/desodiation processes. During the cycling process, the structure is not prone to collapse and can retain relatively good cycling stability. Compared with reported studies on biomass-derived HC as anodes in SIBs, HC-W demonstrates much superior electrochemical performance (Table S3†).
Furthermore, the kinetic behavior of sodium-ion storage in HCs was investigated using cyclic voltammetry (CV) curves. As shown in Fig. 4e, S7 and S8,† there were sharp reduction/oxidation peaks at 0.01/0.1 V. Meanwhile, a relatively small reduction peak appeared at approximately 0.6 V in the first scanning cycle, which was attributed to the irreversible reaction between the electrolyte and the surface functional groups of HC and the formation of the SEI. The diffusion-controlled process and the surface-controlled process can be represented using the equation:
| i = aνb | (5) |
In addition, the sodium-ion storage and transport behaviors were analyzed through the electrochemical impedance spectroscopy (EIS) measurement. As shown in Fig. 4h, the Nyquist plot could be divided into two parts. A semicircle appeared in the high-frequency region related to the charge transfer resistance (Rct), and a sloping line appeared in the low-frequency region related to the Warburg impedance (Zw).21 In the initial state, the Rct of HC-W was 0.7 Ω, which was lower than that of HC-P (73.5 Ω) and HC-A (36.8 Ω). The lower Rct value indicates faster transport of sodium ions in negative electrodes, which is responsible for the improved rate performance. As an ideal sodium-ion storage structure, closed pores have faster electron and sodium-ion transport, which is kinetically advantageous for electrochemical processes.42,43 Therefore, the Na+ diffusion in HY-W is more convenient due to the highly disordered graphitic structures and abundant active closed pores. Moreover, the HY-W sample featured a relatively high proportion of closed pore volume. These structural properties collectively create a more favorable environment for the charge transfer process, effectively reducing the impedance encountered during the sodium-ion storage.
To gain a more comprehensive understanding of the kinetic characteristics of Na+ storage in HC, the galvanostatic intermittent titration technique (GITT) was employed to calculate the diffusion coefficient of Na+ (DNa+) of the anodes. The DNa+ can be obtained through the following formula:
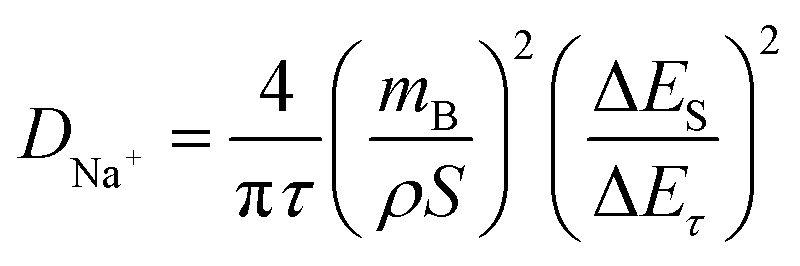 | (6) |
 | ||
| Fig. 5 (a) The diffusion coefficients of the discharge/charge process are calculated based on the GITT curves for HC-P, HC-A and HC-W. (b) The in situ XRD pattern of HC-W at 0.15C. (c) Ex situ SAXS and (d) closed pore size distribution of HC-W at 1.0 V, 0.1 V, and 0.01 V. (e) The illustration of the sodiation mechanism. | ||
Conclusions
In summary, we employed different conditions of hydrothermal treatment (neutral and acidic conditions) to pretreat the shell biomass materials and optimize derived HC. The results showed that the degree of crystallinity of cellulose could be improved by neutral hydrothermal pretreatment, which facilitated the formation of closed pores to enhance the sodium-ion storage capacity and regulated the surface defects to improve the ICE. In contrast, the large-scale graphitized structure would be generated by the hydrothermal treatment under acidic conditions, which is not conducive to the sodium-ion storage behavior of HC. As a result, HC derived from almond shells after neutral hydrothermal pretreatment delivers a high reversible capacity of 317.1 mA h g−1 with an ICE of 85.9% and excellent rate performance. This work provides a profound understanding of the influence of hydrothermal conditions (neutral and acidic conditions) on the chemical structure of the precursor and closed pores in derived HC, as well as their correlation with the sodium-ion storage microstructure in derived HC, thereby offering theoretical guidance for the structural design of effective biomass-derived HC materials.Data availability
Any data that support the findings of this study are included within the article, and all the data used in the manuscript are available on request from the corresponding author.Author contributions
Wenxiu Li and Jian Cui: investigation, methodology, formal analysis, writing – original draft. Wendong Ye and Xuefen Song: investigation, methodology. Panpan Su: investigation, writing – review & editing, supervision. Tai Yang: investigation, methodology, supervision. Yongguang Zhang and Zhongwei Chen: conceptualization, writing – review & editing, project administration, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial support from the Natural Science Foundation of Hebei Province, China (B2021202028 and B2020202052), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0600200), Dalian Revitalization Talents Program (No. 2022RG01), and the National Key Research and Development Program of China (2021YFA1501900).References
- Z. Tang, H. Wang, P. F. Wu, S. Y. Zhou, Y. C. Huang, R. Zhang, D. Sun, Y. G. Tang and H. Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202200475 CrossRef CAS PubMed.
- J. Q. Wang, Y. F. Zhu, Y. Su, J. X. Guo, S. Q. Chen, H. K. Liu, S. X. Dou, S. L. Chou and Y. Xiao, Chem. Soc. Rev., 2024, 53, 4230–4301 RSC.
- K. Sada, J. Darga and A. Manthiram, Adv. Energy Mater., 2023, 13, 2302321 CrossRef CAS.
- R. W. Shao, Z. F. Sun, L. Wang, J. H. Pan, L. C. Yi, Y. G. Zhang, J. J. Han, Z. P. Yao, J. Li, Z. H. Wen, S. Q. Chen, S. L. Chou, D. L. Peng and Q. B. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202320183 CrossRef CAS PubMed.
- Z. B. Zhang, R. Z. Wang, J. Q. Zeng, K. Y. Shi, C. B. Zhu and X. B. Yan, Adv. Funct. Mater., 2021, 31, 2106047 CrossRef CAS.
- Z. X. Guo, G. D. Qian, C. Y. Wang, G. Zhang, R. F. Yin, W. D. Liu, R. Liu and Y. N. Chen, Prog. Nat. Sci.: Mater. Int., 2023, 33, 1–7 CrossRef CAS.
- Y. M. Jiang, Z. Zhang, H. Y. Liao, Y. F. Zheng, X. T. Fu, J. N. Lu, S. Y. Cheng and Y. H. Gao, ACS Nano, 2024, 18, 7796–7824 CrossRef CAS.
- S. Jena, L. Sathishkumar, D. T. Tran, K. U. Jeong, N. H. Kim and J. H. Lee, Adv. Funct. Mater., 2024, 34, 2314147 CrossRef CAS.
- S. Y. Qiao, Q. W. Zhou, M. Ma, H. K. Liu, S. X. Dou and S. K. Chong, ACS Nano, 2023, 17, 11220–11252 CrossRef CAS PubMed.
- Z. Li, Y. Wei, K. Zhou, X. Huang, X. Zhou, J. Xu, T. Kong, J. W. Lucas Bao, X. L. Dong and Y. G. Wang, J. Energy Chem., 2025, 100, 557–564 CrossRef CAS.
- X. Y. Fan, X. R. Kong, P. T. Zhang and J. L. Wang, Energy Storage Mater., 2024, 69, 103386 CrossRef.
- S. A. Riza, R. G. Xu, Q. Liu, M. Hassan, Q. Yang, D. B. Mu, L. Li, F. Wu and R. J. Chen, New Carbon Mater., 2024, 39, 743–769 CrossRef CAS.
- J. R. Wang, L. Xi, C. X. Peng, X. Song, X. H. Wan, L. Y. Sun, M. N. Liu and J. Liu, Adv. Eng. Mater., 2024, 26, 2302063 CrossRef CAS.
- N. LeGe, X. X. He, Y. X. Wang, Y. J. Lei, Y. X. Yang, J. T. Xu, M. Liu, X. Q. Wu, W. H. Lai and S. L. Chou, Energy Environ. Sci., 2023, 16, 5688–5720 RSC.
- H. B. Zheng, J. Zeng, X. H. Wan, X. Song, C. X. Peng, J. R. Wang, L. Y. Sun, H. Wang, M. Zhu and J. Liu, Mater. Futures, 2024, 3, 032102 CrossRef CAS.
- X. W. Dou, I. Hasa, D. Saurel, C. Vaalma, L. M. Wu, D. Buchholz, D. Bresser, S. Komaba and S. Passerini, Mater. Today, 2019, 23, 87–104 CrossRef CAS.
- d. M. S. R. Carolina, L. Simonin, C. M. Ghimbeu, C. Vaulot, d. S. P. Denilson and D. Capucine, Fuel Process. Technol., 2022, 231, 107223 CrossRef.
- X. Y. Li, S. J. Zhang, J. J. Tang, J. Yang, K. Wen, J. Wang, P. Wang, X. Y. Zhou and Y. G. Zhang, J. Mater. Chem. A, 2024, 12, 21176–21189 RSC.
- P. Z. Wang, X. S. Zhu, Q. Q. Wang, X. Xu, X. S. Zhou and J. C. Bao, J. Mater. Chem. A, 2017, 5, 5761–5769 RSC.
- X. Zhao, Y. Ding, Q. Xu, X. Yu, Y. Liu and H. Shen, Adv. Energy Mater., 2019, 9, 1803648 CrossRef.
- F. Li, R. G. Mei, N. Wang, X. D. Lin, F. N. Mo, Y. C. Chen, C. H. Zhong, H. H. Chen, X. S. Dong and Q. X. Liu, Carbon, 2024, 230, 119556 CrossRef CAS.
- T. J. Tang, W. L. Zhu, P. P. Lan, X. X. Lan, H. R. Xie, P. K. Shen and Z. Q. Tian, Chem. Eng. J., 2023, 475, 146212 CrossRef CAS.
- R. F. Susanti, S. Alvin and J. Kim, J. Ind. Eng. Chem., 2020, 91, 317–329 CrossRef CAS.
- X. W. Dou, I. Hasa, D. Saurel, M. Jauregui, D. Buchholz, T. Rojo and S. Passerini, ChemSusChem, 2018, 11, 3276–3285 CrossRef CAS PubMed.
- J. C. Wang, J. H. Zhao, X. X. He, Y. Qiao, L. Li and S. L. Chou, Sustainable Mater. Technol., 2022, 33, e00446 CrossRef CAS.
- Q. S. He, H. X. Chen, X. Chen, J. J. Zheng, L. F. Que, F. D. Yu, J. H. Zhao, Y. M. Xie, M. L. Huang, C. Z. Lu, J. S. Meng and X. C. Zhang, Adv. Funct. Mater., 2024, 34, 2310226 CrossRef CAS.
- L. F. Zhao, Z. Hu, W. H. Lai, Y. Tao, J. Peng, Z. C. Miao, Y. X. Wang, S. L. Chou, H. K. Liu and S. X. Dou, Adv. Energy Mater., 2020, 11, 2002704 CrossRef.
- K. L. Hong, L. Qie, R. Zeng, Z. Q. Yi, W. Zhang, D. Wang, W. Yin, C. Wu, Q. J. Fan, W. X. Zhang and Y. H. Huang, J. Mater. Chem. A, 2014, 2, 12733–12738 RSC.
- N. Nieto, J. Porte, D. Saurel, L. Djuandhi, N. Sharma, A. Lopez-Urionabarrenechea, V. Palomares and T. Rojo, ChemSusChem, 2023, 16, e202301053 CrossRef CAS.
- C. Padwal, H. D. Pham, L. T. M. Hoang, S. Mundree and D. Dubal, Sustainable Mater. Technol., 2023, 35, e00547 CrossRef CAS.
- J. B. Liu, X. Y. Jiang, Z. H. Li, N. Li and T. Li, Ind. Crops Prod., 2024, 208, 117850 CrossRef CAS.
- G. Q. Chen, F. F. Hong, J. Y. Yuan, L. X. Li, M. Fang, W. J. Wei, X. H. Wang and Y. Wei, Carbohydr. Polym., 2022, 296, 119917 CrossRef CAS PubMed.
- M. Y. Su, K. Y. Zhang, E. H. Ang, X. L. Zhang, Y. N. Liu, J. L. Yang, Z. Y. Gu, F. A. Butt and X. L. Wu, Rare Met., 2024, 43, 2585–2596 CrossRef CAS.
- C. Matei Ghimbeu, A. Beda, B. Réty, H. El Marouazi, A. Vizintin, B. Tratnik, L. Simonin, J. Michel, J. Abou-Rjeily and R. Dominko, Adv. Energy Mater., 2024, 14, 2303833 CrossRef CAS.
- D. X. Xu, Y. M. Zhao, H. X. Chen, Z. Y. Lu, Y. F. Tian, S. Xin, G. Li and Y. G. Guo, Angew. Chem., Int. Ed., 2024, 63, e202401973 CrossRef CAS.
- Y. Chu, J. Zhang, Y. B. Zhang, Q. Li, Y. R. Jia, X. M. Dong, J. Xiao, Y. Tao and Q. H. Yang, Adv. Mater., 2023, 35, 2212186 CrossRef CAS.
- Z. Tang, R. Zhang, H. Y. Wang, S. Y. Zhou, Z. Y. Pan, Y. C. Huang, D. Sun, Y. G. Tang, X. B. Ji, K. Amine and M. H. Shao, Nat. Commun., 2023, 14, 6024 CrossRef CAS PubMed.
- R. Q. Dong, F. Wu, Y. Bai, Q. H. Li, X. Q. Yu, Y. Li, Q. Ni and C. Wu, Energy Mater. Adv., 2022, 2022, 9896218 Search PubMed.
- Z. Y. Guo, M. Yang, Q. Fan, Y. T. Chen, T. Xu, C. Y. Li, Z. Y. Li, Z. Y. Li, Q. Sun and H. Xia, Small, 2024, 20, 2407425 CrossRef CAS PubMed.
- G. Huang, H. Zhang, F. Gao, D. Y. Zhang, Z. Q. Zhang, Y. Liu, Z. T. Shang, C. Q. Gao, L. B. Luo, M. Terrones and Y. Q. Wang, Carbon, 2024, 228, 119354 CrossRef CAS.
- Z. Y. Huang, J. H. Huang, L. Zhong, W. L. Zhang and X. Q. Qiu, Small, 2024, 20, 2405632 CrossRef CAS.
- S. Zhou, Z. Tang, Z. Pan, Y. Huang, L. Zhao, X. Zhang, D. Sun, Y. Tang, A. S. Dhmees and H. Wang, SusMat, 2022, 2, 357–367 CrossRef CAS.
- J. Huang, S. Liu, E. Li, B. Dai, T. Lu, J. Teng, Y. Wu, X. Tang, L. Lei, D. Yin, K. Zhang and J. Li, Carbon, 2025, 234, 119981 CrossRef CAS.
- Z. Y. Guo, Z. Xu, F. Xie, J. L. Jiang, K. Zheng, S. Alabidun, M. Crespo-Ribadeneyra, Y. S. Hu, H. Au and M. M. Titirici, Adv. Mater., 2023, 35, 2304091 CrossRef CAS PubMed.
- X. Zhang, Y. J. Cao, G. D. Li, G. P. Liu, X. L. Dong, Y. G. Wang, X. L. Jiang, X. Zhang and Y. Y. Xia, Small, 2024, 20, 2311197 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta01104c |
| ‡ Wenxiu Li and Jian Cui contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |