
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAchieving Pt-coating-free anodes using double-layered catalyst layer structure for polymer electrolyte membrane water electrolysis†
Daehee Kimab,
Gi Hong Junga,
Young Hwa Yuna,
Jeesoo Parkc,
Sieon Ana,
Changsoo Leea,
Sechan Lee de,
Hyeonjung Parka,
Sang-Kyung Kima,
Hyun-Seok Chof,
Hee-Tak Kimc,
Namgee Jung*b,
Gisu Doo*a and
MinJoong Kim*g
de,
Hyeonjung Parka,
Sang-Kyung Kima,
Hyun-Seok Chof,
Hee-Tak Kimc,
Namgee Jung*b,
Gisu Doo*a and
MinJoong Kim*g
aHydrogen Research Department, Korea Institute of Energy Research (KIER), 152 Gajeong-ro, Yuseong-gu, Daejeon 34129, Republic of Korea. E-mail: dooanything@kier.re.kr
bGraduate School of Energy Science and Technology (GEST), Chungnam National University, 99 Daehak-ro, Yuseong-gu, Daejeon 34134, Republic of Korea. E-mail: njung@cnu.ac.kr
cDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea
dDepartment of Chemistry, Kookmin University, 77 Jeongneung-ro, Seongbuk-gu, Seoul 02707, Republic of Korea
eDepartment of Applied Chemistry, Kookmin University, 77 Jeongneung-ro, Seongbuk-gu, Seoul 02707, Republic of Korea
fDepartment of Chemical and Biomolecular Engineering, Sogang University, 35 Baekbeom-ro, Mapo-gu, Seoul 04107, Republic of Korea
gDepartment of Materials Science and Engineering, Kyung Hee University, 1732 Deogyeong-daero, Giheung-gu, Yongin, Gyeonggi-do 17104, Republic of Korea. E-mail: mj.kim@khu.ac.kr
First published on 17th May 2025
Abstract
Achieving high-performance and cost-effective proton exchange membrane water electrolysis (PEMWE) necessitates a reduction in the use of precious group metals (PGMs) while maintaining electrolysis efficiency. However, commercial high-activity IrOx catalysts often fail to deliver optimal performance at low loadings because of electron transport problems caused by the natural oxide (TiOx) on the Ti porous transport layer (PTL) surface. This is because the high work functions of IrOx catalysts create Schottky barriers in the low-work-function TiOx, thereby deteriorating the electrical conductivity at the interface. The Pt coating is commonly employed as a protective layer; however, hundreds of nanometers of coating significantly increase the cost of the electrolyzer. To address this challenge, we propose a double-layered catalyst layer (DL-CL) structure that enables the fabrication of Pt-coating-free anodes, with crystalline, low-work-function rutile IrO2 (R-IrO2) positioned at the PTL interface and highly active IrOx (HA-IrOx) positioned on the membrane side. The key advantage of the R-IrO2 layer over the Pt coating is its porous structure, which effectively prevents contact between HA-IrOx and TiOx with much lower loading (0.05–0.2 mg cm−2). The DL-CL minimizes the influence of TiOx on both performance and durability while enabling all PGMs to actively participate in the oxygen evolution reaction.
1 Introduction
Hydrogen is a key component of next-generation energy systems and plays a crucial role in achieving carbon neutrality.1–3 However, most hydrogen is still produced from fossil fuels, highlighting the need for efficient hydrogen production technologies linked to renewable energy sources.4,5 Given this, proton exchange membrane water electrolysis (PEMWE) has attracted considerable attention owing to its numerous advantages, including high current density, high hydrogen purity, and excellent responsiveness to power fluctuations.6,7 Nonetheless, there is a fatal drawback of high material cost, mainly due to platinum-group metals (PGMs) or titanium components that can withstand acidic environments and high anode potentials.8–10One of the biggest contributions to the PEMWE cost is Pt coatings, which are currently employed to conceal the native oxide (TiOx) of the titanium components.11,12 For industrial uses, hundreds of nanometers of Pt, which is equivalent to 0.5–1 mg cm−2, are coated on the Ti porous transport layer (PTL) and bipolar plate to enhance conductivity and corrosion resistance.13–15 However, to achieve the ultimate hydrogen production cost target of 1 USD per kg, reducing PGM usage is essential, and the U.S. Department of Energy (DOE) has proposed an ultimate goal of 0.125 mgPGM cm−2.16–18 This highlights the need to minimize or eliminate the Pt coatings, but significant challenges remain.19,20
Previous studies have pointed out the detrimental effects of TiOx on both electrolyzer performance and durability.13,21,22 In addition, a lower IrOx loading in the anode intensifies the problems caused by TiOx, that is, retardation of electron transport at the interface and deterioration of single-cell performance.12,23,24 Many researchers have observed abnormally poor single-cell performance of low-IrOx-loading anodes, mostly attributable to large ohmic polarization.19,25,26 Our recent publications have ascribed these electron-transport problems to the semiconducting properties of TiOx.27 Specifically, the high work functions of the IrOx catalysts and electron-withdrawing fluorinated ionomers result in electron depletion and upward band bending in the TiOx, lowering the electrical conductivity at the interface.27,28 Moreover, IrOx catalysts with higher activities tend to have higher work functions, promoting band bending and increasing the interfacial resistance.27,29,30 Because the low loading and high activity of IrOx catalysts are some of the biggest challenges in achieving high-performance and cost-effective anodes, the problems caused by TiOx are of great interest in PEMWE development.
Recently, our group focused on designing catalyst layer (CL) structures that can mitigate the abovementioned problems caused by TiOx and realize a Pt-coating-free anode. Our previous studies concluded that porous and low-work-function CLs are necessary to prevent band bending at the TiOx interface.27 Based on this conclusion, we suggested that an Ir metal nanofiber is the ideal anode catalyst structure.30 Owing to its high porosity and low work function, the Ir nanofiber CL effectively suppressed the problems caused by TiOx, exhibiting the same performance regardless of the Pt coating. However, the nanofiber catalyst has limitations in the complex manufacturing process and relatively low catalytic activity, requiring additional strategies for practical industrial applications.30,31
Herein, we propose a double-layered CL (DL-CL) structure that enables the fabrication of Pt-coating-free anodes with high activity and a simple manufacturing process.32 The DL-CL consists of two distinct layers of rutile IrO2 (R-IrO2) and highly amorphous IrOx (HA-IrOx) that face the Ti PTL and membrane, respectively. Because of its high crystallinity, low work function, and excellent electrochemical stability, thermally treated R-IrO2 is introduced at the TiOx interface to alleviate band bending in TiOx.21,33 However, since the R-IrO2 catalyst inherently possesses low activities for oxygen evolution reaction (OER), its loading is minimized to 0.05–0.2 mg cm−2, while the HA-IrOx catalyst is employed to reduce activation polarization.
In this regard, the DL-CL structure offers two significant benefits: (1) the minimal layer of low-work-function R-IrO2 effectively addresses interface electron transport issues, and (2) the highly active HA-IrOx CL in direct contact with the membrane maximizes the OER performance.34–37 Electrochemical analyses verify the outstanding single-cell performance of the DL-CL anodes, which retain both high activity and electrical conductivity regardless of the Pt coating on the PTL.38 Additionally, the longevity of the excellent interface properties is demonstrated in a constant-current durability test, in which TiOx does not cause an increase in ohmic resistance over 1000 h. We believe that this double-layer strategy represents a breakthrough in the development of high-performance and cost-effective PEMWE anodes.
2 Experimental section
2.1. Fabrication of the single-layered catalyst layer
All the CLs were prepared using a roll mill for slurry dispersion and subsequent blade casting. A commercial IrOx catalyst from Tanaka Kikinzoku Kogyo (TKK) was used as the HA-IrOx catalyst. For the R-IrO2 catalyst, another commercial iridium oxide black (Alfa Aesar, Premion) was annealed to obtain a high-crystalline IrO2 structure. The annealing was conducted at 500 °C in ambient air for 1 h, with a ramp rate of 5 °C min−1.In the case of cathode CLs, Pt/C (TEC10E50E, 46.9 wt% Pt, Tanaka Kikinzoku Kogyo) were utilized. All the CLs adopted a commercial Nafion ionomer solution (D2020, IEC = 1.05 mequiv. per g, Dupont) as a binder. The ionomer-to-carbon ratio of the cathode CLs was set to 1.0, whereas the ionomer content of the anode CLs was adjusted to 10 wt% of the total solid content. The solid components were dispersed in a mixed solution of n-propanol (NPA; Junsei Chemical) and deionized (DI) water using a roll mill with zirconia balls for 72 h. The uniformly dispersed slurry was coated on a polyimide (Kapton) film using a doctor blade and subsequently dried at a 50 °C hot plate. The Ir and Pt loadings were controlled to 0.50 mgIr cm−2 and 0.20 mgPt cm−2, respectively.
2.2. Fabrication of catalyst-coated membranes (CCMs)
Catalyst-coated membranes (CCMs) were fabricated by transferring the anode and cathode CLs of 9 cm2 onto a Nafion membrane (NRE 212, 50 μm, Dupont), followed by decal-transfer at 125 °C with 20 kgf cm−2 for 6 min. The CCMs were then assembled in a single-cell hardware with single-parallel-patterned Ti flow fields. Titanium-based materials were used for the anode PTL and bipolar plates (BPP), whereas carbon-based materials were employed for the cathode PTL. A titanium PTL (2GDL06N-025, 250 μm, Bekaert) and a carbon PTL with a microporous layer (JNTG30A3, 320 μm, JNT) were used as the anode and cathode, respectively. In addition, Pt-coated Ti PTL (2GDL06-025-BS02PT, 250 μm with a 200 nm Pt coating, Bekaert) was used to eliminate electron transport problems caused by the native TiOx.2.3. Double-layered CL (DL-CL) fabrication
To fabricate the DL-CL, the R-IrO2 catalyst was spray-coated onto an HA-IrOx CCM fabricated by the decal transfer process described in Section 2.2. The iridium loading of the HA-IrOx CL was 0.30 mgIr cm−2, while the R-IrO2 coating was controlled to 0.2 mgIr cm−2, resulting in a total iridium loading of 0.5 mgIr cm−2 for DL-CL. The R-IrO2 slurry for spray coating was prepared by dispersing R-IrO2 via bath sonication for 1 h. After the uniform dispersion slurry was loaded into an injector and coated onto the 80 °C-heated CCM using a spray coater. The loading ratios of the two catalysts were varied to determine the minimum amount of R-IrO2 required for fabricating high-performance DL-CL. The total catalyst loading was set to 0.50 mgIr cm−2, whereas the spray-coated R-IrO2 content was changed from 0.20 to 0.05 mgIr cm−2. Two CCMs with single-layered HA-IrOx and R-IrOx CLs directly spray-coated on the Nafion membranes were also fabricated for comparison with the DL-CL.2.4. Electrochemical single-cell analysis
Electrochemical analysis for the single cells was evaluated at 80 °C with the deionized (DI) water flow of 15 mL min−1. All single cells were activated 20 times by cycling chronopotentiometry (CP) and linear sweep voltammetry (LSV) over 20 times. For the CP step, the current was held at 1 A cm−2 for 1 min, and the LSV step was conducted from the open-circuit voltage (OCV) to 1.8 V with a scan rate of 10 mV s−1. After the activation, IV polarization curves were obtained at a current density range of 0.01–5.0 A cm−2 by maintaining each current step for 2 min and recording the steady-state voltage stability. Simultaneously, electrochemical impedance data were collected in the frequency range of 100 kHz to 100 mHz to break down the resistance components. To evaluate the durability of the anode, a DL-CL CCM using a thicker membrane (N115, 125 μm, Dupont) was prepared, and its anode, coupled with the bare Ti PTL, was subjected to a 1000 h durability test. A constant current density operation at 1 A cm−2 was applied throughout the operation. IV polarization and impedance analyses were conducted at six points of 70 h, 200 h, 350 h, 500 h, 750 h, and 1000 h to monitor degradation phenomena. Identical experiments were also performed using HA-IrOx anodes with and without the Pt coating for comparison. All the electrochemical measurements were performed using a potentiostat (HCP-803; BioLogic Science Instruments). For the overpotential breakdown analysis, three overpotential components (kinetic, ohmic, and other) were extracted from the polarization curves based on Tafel plots and ohmic resistance. Detailed procedures for deconvoluting the voltage losses into kinetic, ohmic, and other-related components are provided in ESI Note 1.†2.5. Characterization
The particle size and morphology of each catalyst were analyzed using field-emission transmission electron microscopy (FE-TEM) (F200, JEOL) at 200 kV. The geometric surface area and pore distribution were measured using Brunauer–Emmett–Teller (BET) analysis (3Flex, Micromeritics). The crystalline structure of each IrOx catalyst was determined using X-ray diffraction (XRD) (SmartLab, RIGAKU), and the surface oxidation states were confirmed using X-ray photoelectron spectroscopy (XPS; K-alpha, Thermo VG Scientific). All the XPS spectra were calibrated to the sp3 carbon peak of adventitious carbon (C 1s, 284.5 eV).39,40 The interface properties within the anode components were investigated using a work function analysis obtained from ultraviolet photoelectron spectroscopy (UPS; Axis-Supra, Kratos). The structures of the CLs were examined using scanning electron microscopy (SEM) (Regulus 8220, Hitachi) at an accelerating voltage of 10.0 kV. All characterizations were conducted at the Analysis Center for Energy Research (ACER) at the Korea Institute of Energy Research (KIER).For the in situ characterizations of the catalysts, X-ray absorption spectroscopy (XAS) of the Ir L3-edge (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 215 eV) was collected at the 7D beamlines of the Pohang Accelerator Laboratory (PAL) with a flux of 5 × 1012 photons per second at 300 mA and 2.5 GeV. The catalyst and Nafion dispersions were drop-coated onto MPL-coated carbon papers (JNTG30A3, JNT), which were subjected to the in situ analysis in a 0.5 M H2SO4 solution. The applied potentials were controlled at 1.0, 1.2, 1.4, 1.6, and 1.8 V versus a reversible hydrogen electrode (RHE). X-ray absorption near-edge structure (XANES) signals were measured in the fluorescence mode. Data fitting and analyses were conducted using the Athena and Artemis software.
215 eV) was collected at the 7D beamlines of the Pohang Accelerator Laboratory (PAL) with a flux of 5 × 1012 photons per second at 300 mA and 2.5 GeV. The catalyst and Nafion dispersions were drop-coated onto MPL-coated carbon papers (JNTG30A3, JNT), which were subjected to the in situ analysis in a 0.5 M H2SO4 solution. The applied potentials were controlled at 1.0, 1.2, 1.4, 1.6, and 1.8 V versus a reversible hydrogen electrode (RHE). X-ray absorption near-edge structure (XANES) signals were measured in the fluorescence mode. Data fitting and analyses were conducted using the Athena and Artemis software.
3 Results and discussion
3.1. Catalyst properties and TiOx effects
The single-cell performance of the PEMWE was significantly affected by the nanometer-thick native oxide (TiOx) on the Ti PTL. This phenomenon has been reported in previous studies, revealing that the electrical properties of TiOx depend significantly on the structural and material properties of the CL in contact.27,30 Hence, the single-cell performance is not solely determined by the intrinsic activity of the catalysts but more considerably by the physical properties of the catalysts and/or CLs that govern the electron transport resistance at the TiOx interface. However, because of the limited understanding of the TiOx phenomena and the lack of strategies for overcoming them, this study aimed to deepen the insights into the TiOx interface, design an improved electrode structure, and develop a durable, high-performance, Pt-coating-free anode.First, to explore the TiOx phenomenon, two iridium oxide catalysts with different physical properties were selected: a commercial amorphous catalyst with a high specific surface area (HA-IrOx) and a heat-treated catalyst with high crystallinity (R-IrO2). For the latter, another commercial amorphous iridium oxide (a-IrOx) was annealed at 500 °C in the air condition (Fig. S1†). The morphologies and structures of the catalysts were compared by TEM, as shown in Fig. 1a and b. HA-IrOx, with particle sizes ranging from 1 to 10 nm, exhibited a uniform contrast without any discernible crystalline pattern, demonstrating its amorphous structure. By contrast, R-IrO2 exhibited much larger particle sizes, ranging from 10 to 100 nm, along with clear crystalline patterns. Comparison with a-IrOx (Fig. S1a†) revealed that the annealing process reduced the surface roughness and clarified the interface between the particles, while increasing the overall particle size. Additionally, the crystalline patterns became evident after annealing, confirming the development of high crystallinity.
 | ||
| Fig. 1 Structural characterizations of the HA-IrOx and R-IrO2 catalysts and their effects on the single-cell anode interface (a and b) TEM images of the (a) HA-IrOx and (b) R-IrO2 catalysts with the insets of SAED pattern images. (c) Microstructure comparison of the catalysts using BET methods. (d) UPS spectra of the catalysts and bare Ti PTL and their work functions. (e and f) Single-cell analysis of the HA-IrOx and R-IrO2 anodes depending on the Pt coating. (e) IV polarization curves and (f) Nyquist plots of electrochemical impedance at 0.04 A cm−2. (g and h) Schematic representations illustrating the TiOx interfaces in the (g) HA-IrOx and (h) R-IrO2 anodes. | ||
Material characterizations with BET, XPS, and XRD were in good agreement with the TEM images.9,35,36 BET analysis revealed that the HA-IrOx catalyst exhibits a high specific surface area of 97.3 m2 g−1 (Fig. 1c), owing to its small particle size and rough surface structure. The value is five times larger than that of R-IrO2 (20.8 m2 g−1) and three times larger than that of another amorphous catalyst, a-IrOx (31.3 m2 g−1) (Fig. S1b†), implying the superior kinetic properties of HA-IrOx. In electronic analysis using XPS, the Ir 4f spectra (Fig. S1c and S2a†) indicated that the heat treatment stabilized the Ir3+ state of a-IrOx to Ir4+; therefore, R-IrO2 predominantly existed in the IrO2 state with Ir4+ signals at 61.9 and 64.9 eV. The HA-IrOx spectrum showed Ir3+ peaks at 62.3 and 65.3 eV, reflecting its amorphous nature. The XRD patterns displayed in Fig. S1d and S2b† show sharp peaks for R-IrO2 and broad peaks for HA-IrOx, highlighting the highly crystalline R-IrO2 and amorphous HA-IrOx structures.
The catalysts were further subjected to UPS analysis to estimate their interfacial properties with TiOx. The UPS plots in Fig. 1d and S1e† revealed that HA-IrOx has a work function of 4.90 eV, which decreases to 4.62 eV for a-IrOx and 4.27 eV for R-IrO2. These results suggest that a higher degree of amorphousness of iridium oxides is correlated with an increased work function. Because a larger gap in the work functions induces greater band bending at the semiconductor interface, the HA-IrOx/TiOx contact is likely to be the most susceptible to the electron transport problem. Notably, the TiOx layer of the Ti PTL in our study exhibited a work function of 4.20 eV, which was much smaller than those of the HA-IrOx and a-IrOx catalysts and comparable to that of R-IrO2.41 Therefore, the greater amorphousness or higher catalytic activity of IrOx is expected to impede the electron transport at the TiOx interface.
The interface properties of the catalysts were evaluated using single-cell analysis.42,43 Two catalysts, HA-IrOx and R-IrO2, were prepared as anode CLs at loading of 0.50 mgIr cm−2. By comparing the electrochemical data with and without a 200 nm Pt coating on the PTL, TiOx effects at each catalyst interface were investigated. As shown in the IV polarization curves of Fig. 1e, the HA-IrOx anodes manifested a considerable difference between the two polarization curves; the Pt coating led to a remarkable performance improvement, quadrupling the current density from 0.65 to 2.70 A cm−2 at 1.8 V. This is in sharp contrast to the R-IrO2 anodes, which showed nearly identical single-cell performances regardless of the Pt coating. These results imply that a severe problem occurred in the HA-IrOx anode because of TiOx, whereas it disappeared in the R-IrO2 case.
The different TiOx properties are also evident in the Nyquist plots (Fig. 1f) of the electrochemical impedance spectrum (EIS) at 0.04 A cm−2. The Nyquist plots were also fitted using equivalent circuits, as shown in Fig. S3,† to further analyze the impedance components. The specific equivalent circuit models used for each case are displayed in the corresponding insets of the plots. The HA-IrOx anodes exhibited substantial differences in the high-frequency impedances depending on the PTL type. For the Pt-coated anode, the high-frequency impedance showed a reasonable x-axis intercept (Rohm) at 0.088 ohm cm2. However, for the bare Ti PTL case, the intercept value markedly increased to 0.528 ohm cm2, and an additional semicircle (Rcontact) appeared at 15 kHz, indicating the severely retarded electron transport. Conversely, the R-IrO2 anodes made consistent impedance signals regardless of the PTL type, and their ohmic resistances were almost identical to 0.094–0.097 ohm cm2, implying excellent electron transport not affected by TiOx.
Notably, the intrinsic electrical conductivity of the catalysts was not a key contributor to the ohmic resistance. This is evident from the impedance comparison of the two Pt-coated anodes (Fig. 1f), where Rohm of HA-IrOx was slightly smaller than that of R-IrO2. This is contrary to the general assumption that the crystalline rutile phases of iridium oxide have higher conductivities than their amorphous phases. However, the different intrinsic conductivities are dictated by the CL resistance (RCL), as shown by the EIS analysis at a non-faradaic voltage of 1.35 V (Fig. S4a†).44,45 The HA-IrOx anode formed a constant phase element (CPE) in the frequency range of 4600–68 Hz, which stemmed from inefficient charge transport through the nonuniform and tortuous CL structure. By contrast, the CPE signal was absent, and nearly absolute capacitance signals appeared in the R-IrO2 case, reflecting its high electrical conductivity. In this regard, we believe that the interfacial properties of TiOx are a critical reason for the different ohmic resistances.
As mentioned above, the work functions of the catalysts are key factors in determining the TiOx interface properties. This is illustrated schematically in Fig. 1g and h. In the case of HA-IrOx (Fig. 1g), the work function mismatch at the interface may lead to a Schottky contact and severe band bending in TiOx. Considering the work function difference between HA-IrOx and TiOx, the electron transport barrier was expected to reach 0.6 eV. However, because the difference is negligible for the R-IrO2/TiOx interface (∼0.07 eV), it is believed that the R-IrO2 anode does not develop a critical Schottky contact at the interface, enabling efficient electron transport through TiOx (Fig. 1h).
Another speculation is associated with the amorphousness of the iridium oxides. The Ir L-edge spectra of in situ X-ray absorption near-edge spectroscopy (XANES) experiments in Fig. S5† show that the white-line peaks of the two catalysts behave differently with the applied potential.36 During a potential increase from 1.0 to 1.8 V, the white lines of R-IrO2 did not change at all (Fig. S5a†). By contrast, the white lines of HA-IrOx show a positive shift from 11219 to 11220 eV (Fig. S5b†), which is close to the increase in one d-band hole of Ir reported in previous studies. We inferred that positive charges easily accumulated in the HA-IrOx catalysts because of the unclear boundaries between the particles. In such cases, the accumulated positive charges may spread to neighboring TiOx, leading to electron depletion and deteriorated electrical conductivity.
Despite its excellent electron transport properties, the R-IrO2 catalyst exhibited poor single-cell performance owing to its inherently low catalytic activity compared to HA-IrOx as predicted by TEM, BET, and XPS analyses. The Tafel plots in Fig. S4b,† extracted from the single-cell polarization data, show that the HA-IrOx anode has a lower onset potential than R-IrO2. Moreover, the Tafel slopes of the Pt-coated anodes were 38.0 and 53.5 mV dec−1 for HA-IrOx and R-IrO2, respectively, probably owing to different reaction mechanisms for the two catalysts (i.e., lattice oxygen evolution mechanism versus adsorbate evolution mechanism). The crystalline rutile structure and large particle size are the main reasons for the large activation polarization for the R-IrO2 anode, and the comparison of the two catalyst anodes coupled with the Pt-coated PTL presents that the current density for R-IrO2, which is 2.0 A cm−2 at 1.8 V, is 25% smaller than that for the HA-IrOx anode (2.7 A cm−2).
The overall single-cell characteristics with varying catalysts and PTL types are summarized in the overpotential breakdown results in Fig. S6.† These results indicate that the physical properties of the iridium oxide catalyst play an important role in controlling the TiOx interface, but this results in a trade-off with the catalytic activity. Notwithstanding its high catalytic activity, HA-IrOx suffered significant performance problems when coupled with a bare Ti PTL. However, despite their high electrical conductivity, the R-IrO2 anodes exhibited comparatively low performance owing to their large activation polarization. This indicates that achieving high catalytic activity along with outstanding electrical conductivity at the CL/PTL interface is crucial for maximizing PEMWE single-cell performance.
3.2. Fabrication and analysis of double-layered catalyst layer
Industry and researchers currently employ Pt-coated PTLs to overcome the limitations of electron transport in high-activity IrOx anodes; however, the high material and manufacturing costs of Pt coatings pose a significant economic burden for commercialization. Specifically, the mostly used commercial 200 nm coating corresponds to 0.43 mgPt cm−2, which should be decreased or eliminated to reduce overall stack price. In this context, we propose a double-layered CL (DL-CL), which is a combination of the distinct layers of two catalysts (HA-IrOx and R-IrO2), as a simple and effective anode structure that exhibits both high performance and cost efficiency. In the DL-CL, the minimum amount of R-IrO2 was placed at the Ti PTL interface, whereas HA-IrOx with a dominant portion of the total iridium loading was positioned on the opposite side, as illustrated in Fig. 2a. This structure was intended to secure efficient interface electron transport by inserting the R-IrO2 catalyst at the CL/PTL interface while ensuring that the HA-IrOx catalysts actively participated in the OER near the membrane, not affected by TiOx. | ||
| Fig. 2 Superior single-cell performance of the double-layered catalyst layer (DL-CL). (a) Schematic of the DL-CL/TiOx interface, enabling efficient electron transport. (b) Cross-sectional SEM image of the DL-CL CCM. (c) and (d) Single-cell data comparisons of the DL-CL, HA-IrOx, and R-IrO2 anodes that were coupled with the bare Ti PTLs. (c) IV polarization curves and (d) Nyquist plots at 0.04 A cm−2. | ||
The DL-CL was fabricated in two steps. First, the HA-IrOx CL was prepared via ball milling and doctor blade coating. The prepared HA-IrOx CL, coupled with the cathode CL, was transferred to a membrane via decal transfer. Subsequently, the R-IrO2 slurry was spray-coated directly onto the HA-IrOx side of the CCM (Fig. S7†). The cross-sectional SEM image in Fig. 2b demonstrates a double-layered structure: a dense HA-IrOx CL and a comparatively porous R-IrO2 CL. Although the catalyst loadings of HA-IrOx and R-IrO2 were controlled to 0.30 and 0.20 mgIr cm−2, respectively, the two CLs featured similar thicknesses within the 1.17–1.18 μm range owing to the different porosities. This difference is corroborated by the high-magnification SEM images (Fig. S8†). Presumably, the smaller size of HA-IrOx forces more aggregation of the primary particles owing to stronger van der Waals interactions, resulting in smaller pores and lower porosity compared to R-IrO2. This is consistent with the Barrett–Joyner–Halenda analysis shown in Fig. S2c,† where the HA-IrOx particles possess only micropores with a size distribution of 2–20 nm, while R-IrO2 also had macropores larger than 100 nm.
The DL-CL was then applied to a single-cell anode, and its performance was compared with that of other CLs. When using the bare Ti PTLs, the DL-CL anode exhibited remarkably higher current densities compared to the single-layered CLs, as shown in Fig. 2c; the current density of DL-CL at 1.8 V was 340% higher than that of HA-IrOx CL and 55% higher than that of R-IrO2. The Nyquist plots in Fig. 2d and their equivalent circuit analysis in Fig. S9† show that the low ohmic resistance and small low-frequency semicircle (Rct) account for the superior performance of the DL-CL anode. Importantly, the ohmic resistance was as low as that of R-IrO2 at ∼0.08 ohm cm2, and the Rct was comparable to that of HA-IrOx, indicating that DL-CL combined the advantages of two catalyst anodes. In addition, the single-cell performance of the DL-CL anode did not change even without the Pt coating, as shown in the IV polarization curves in Fig. S10a.† The difference between the two DL-CL anodes was also negligible in the Nyquist plots (Fig. S10b†), highlighting the role of the R-IrO2 layer in stabilizing the TiOx interface. To clarify whether the fabrication method contributes to the improvement or not, we also characterized the single-layered CCMs of HA-IrOx and R-IrO2 prepared via the spray-coating method. The IV polarization and EIS results in Fig. S11† demonstrated that the spray-coating method did not notably enhance the performance compared to the decal-transferred method. Rather, the HA-IrOx sample showed a hugely deteriorated performance for the spray-coated case. These data indicate that the fabrication methods are not the critical contributors determining the DL-CL performance.
Interestingly, the DL-CL performed better than the HA-IrOx anode with the Pt-coated PTL (Fig. S10a†). Considering the similar ohmic and kinetic resistances (Fig. S10b†), we ascribed this enhancement to efficient charge transport through the DL-CL. This was supported by the non-faradaic impedance analysis results shown in Fig. S10c,† where the RCL of DL-CL was nearly half that of HA-IrOx. The highly conductive R-IrO2 layer appeared to increase the overall electrical conductivity, leading to performance enhancement, especially in the high-current-density region.
The Tafel plots in Fig. S10d† compare the kinetic performance of DL-CL with the HA-IrOx anodes of two different loadings (0.30 and 0.50 mgIr cm−2). The DL–CL plot is located in the middle of the two HA-IrOx anodes because of the presence of low-activity R-IrO2 catalysts. However, because the R-IrO2 fraction of the total 0.50 mgIr cm−2 was relatively small, the loss in kinetic performance was also not significant, and the onset potential and Tafel slope of the DL-CL were comparable to those of the HA-IrOx anodes with 0.50 mgIr cm−2 loading. These results demonstrated that the double-layered structure effectively retained the high activity of the HA-IrOx catalyst.
Notably, the R-IrO2 layer was significantly less compact than the Pt coating. According to the SEM image, an R-IrO2 layer with 1 mgIr cm−2 exhibits a thickness of 5.9 μm, which is 12 times larger than the theoretical thickness (0.47 μm (mgPt cm−2)−1) of the dense Pt coating. Therefore, the R-IrO2 layer was much thicker at the same PGM loading, making it more effective at separating the IrOx catalysts from the TiOx interface. This was further verified through single-cell experiments with varying R-IrO2 content in the DL-CL (Fig. S12†), where reducing the loading to 0.05 mgIr cm−2 in the DL-CL did not cause a performance drop or an increase in the ohmic resistance. The sustained high performance at such a low loading highlights the effectiveness of the R-IrO2 layer, given that the PGM content is only one-eighth that of the commercial Pt coating (0.05 mgIr cm−2 vs. 0.43 mgPt cm−2). The 0.05 mgIr cm−2 loading is even lower than the minimum Pt-coating loading (0.10 mgPt cm−2) required to ensure high IrOx anode performance, as reported by Geuß et al.46 In addition, the high porosity of the R-IrO2 layer is believed to facilitate oxygen, water, and electron transport through the CL (Fig. S10c†), which functions similarly to a microporous layer.
3.3. Structure effects of double-layered catalyst layer
To further identify the origin of the superior electrochemical performance of the DL-CL anodes, another double-layered CL with an inverted arrangement of the catalysts was fabricated in the same manner as the DL-CL. The inverted DL-CL (IDL-CL) structure is illustrated in Fig. 3a, and the HA-IrOx CL is placed at the PTL interface with the R-IrO2 CL on the PEM side. The catalyst loading and composition were the same as those of the DL-CL, and only the structure was reversed. The cross-sectional and surface SEM images shown in Fig. S13† demonstrate an inverted arrangement with a dense HA-IrOx CL on top and a porous R-IrO2 CL adjacent to the membrane. If the double-layered structure is the primary reason for the outstanding performance of the original DL-CL, the IDL-CL anode should exhibit an equally high performance. Otherwise, the catalyst arrangement was the dominant factor. | ||
| Fig. 3 Catalyst arrangement in DL-CL. (a) Schematic of the inverted DL-CL structure and its interface with TiOx. (b) IV polarization curves of the DL-CL and IDL-CL anodes depending on the Pt coating. (c) Schematic of the M-CL anode. (d) IV polarization curves of the DL-CL and M-CL anodes depending on the Pt coating. | ||
The single-cell analyses shown in Fig. 3b and S14a–c† verified that the latter statement is correct. The IDL-CL anode exhibited extremely poor performance when coupled with the bare Ti PTL, and its performance was significantly dependent on the Pt coating. Unlike the decent cell performance with the Pt-coated PTL (2.62 A cm−2 at 1.8 V), the uncoated anode did not perform properly and showed abnormal voltage losses at low current densities (0.15 A cm−2 at 1.8 V) (Fig. 3b). The impedance analysis at 0.04 A cm−2 (Fig. S14a†) demonstrated that a large semicircle in the high-frequency region (63–40 Hz) accounted for the abnormal voltage loss. Because the IDL-CL anode with the Pt-coated PTL exhibited outstanding kinetic performance (Fig. S14b†) and small overpotentials (Fig. S14c†), which was comparable to that of the DL-CL anodes, the TiOx interface was responsible for the performance deterioration of the uncoated anode. These results are analogous to the single-cell data of HA-IrOx and contrary to the DL-CL anodes, indicating that the superior performance of the DL-CL is not owing to its double-layered structure, but is mostly attributable to the optimized catalyst arrangement.
To further understand the structural effect of the CL on the TiOx interface, a mixed CL (M-CL) was prepared by homogeneously dispersing the HA-IrOx and R-IrO2 catalysts and then casting them into a single layer. The HA-IrOx catalyst was used at 1.5 times the amount of R-IrO2 to ensure the same catalyst ratio as that of DL-CL. Fig. 3c shows schematics of the M-CL structure and its interface with TiOx. Assuming a uniform catalyst distribution in the M-CL, TiOx was considered to form a mixed interface of the two catalysts, and single-cell analysis could reveal how the interface structure affects electron transport in TiOx (Fig. 3d and S15†). When paired with the bare Ti PTL, the M-CL anode exhibited reasonable performance without the severe polarization observed in the HA-IrOx and IDL-CL anodes. Electrochemical impedance analysis indicated that M-CL did not create a large high-frequency semicircle (Fig. S15a†). However, the overall performance was inferior to that of the DL-CL anode, and the ohmic resistance of the M-CL was 21% higher, implying contact resistance. An enlarged ohmic polarization of the M-CL anode was also observed in the overpotential breakdown analysis (Fig. S15b†), while the other polarizations were nearly the same as the DL-CL anode. In contrast, the M-CL anode exhibited a comparable performance and overpotentials to the DL-CL when the Pt-coated PTL was employed, suggesting that the M-CL was also affected by the presence of TiOx. We ascribed this to the HA-IrOx/TiOx interface, partially formed at the CL/PTL contact, which caused partial band bending in TiOx and reduced electrical conductivity. These investigations of the M-CL support the conclusion that setting HA-IrOx apart from the TiOx surface is crucial for interface engineering in Pt-coating-free anodes. The presence of R-IrO2 in the anode CL was not sufficient to mitigate the interfacial resistance; however, the detrimental components should be completely removed from TiOx to attain excellent electron transport.
3.4. Long-term durability test of the DL-CL/TiOx interface
However, the eliminated TiOx effect by the DL-CL has practical meaning only when this phenomenon persists for a long period of water electrolysis operation. Therefore, we conducted the long-term operation test of the DL-CL anode coupled with the bare Ti PTL at a constant current of 1 A cm−2 over 1000 h. HA-IrOx anodes with and without the Pt coating were also tested for comparison. The catalyst loading and composition were controlled as described above for the single-cell analysis, whereas the NR212 membranes were changed to thicker N115 membranes to exclude membrane deformation during the test.The chronological voltage profiles in Fig. 4a compare the three samples during constant-current operation. The HA-IrOx anode with the bare Ti PTL made a rapid voltage increase of 1.43 mV h−1 and jumped above the safety voltage limit (2.15 V) within 80 h. A performance drop is also observed in the IV polarization curves (Fig. 4b), and the Nyquist plots of the impedance signals (Fig. 4c and S16†) indicate that the enlarged high-frequency semicircle is responsible for the rapid voltage increase. These results indicate that the insulation by TiOx worsened during single-cell operation. We hypothesized that the thickening and/or increased amorphousness of the TiOx layer would lead to a significant decrease in the electrical conductivity drop at the interface.
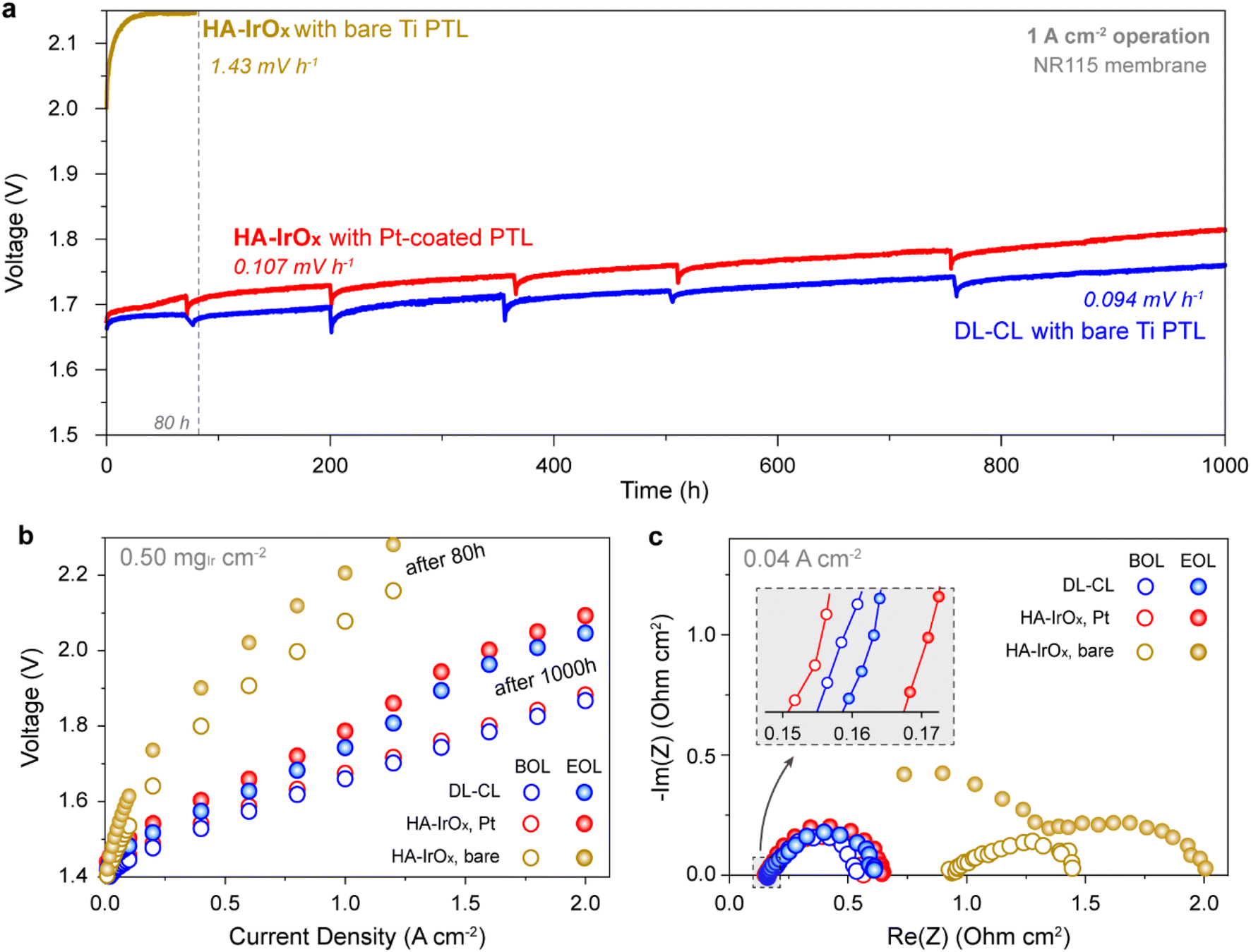 | ||
| Fig. 4 Durability test of the DL-CL anode with the bare Ti PTL. (a) Voltage profiles of the constant current operation at 1 A cm−2 for the HA-IrOx and DL-CL anodes. The bare Ti PTL was adopted for the DL-CL anode, and two HA-IrOx anodes with and without the Pt coating were tested for comparison. (b and c) Single-cell characterizations before and after the operation. (b) IV polarization curves. (c) Nyquist plots of impedance signals at 0.04 A cm−2 with a magnified view in the inset. | ||
Conversely, the HA-IrOx with the Pt-coated PTL underwent a relatively stable operation for 1000 h. A gradual voltage increase with a rate of 0.107 mV h−1 was observed, and the polarization curves and Nyquist plots showed that the degradation was associated with the decline in kinetic and mass transport properties. In particular, the change in Rct at 0.04 A cm−2 was more noticeable than that in the ohmic resistance in the Nyquist plots (Fig. 4c). This implies that high electron conductivity was retained in the anode owing to the Pt coating, but there was considerable degradation in the catalyst and/or CL, leading to decreased kinetic properties and further increased transport polarization. These results are summarized in the overpotential analysis shown in Fig. S17.†
In the case of the DL-CL coupled with the bare Ti PTL, the voltage profile was stable over 1000 h of operation. Despite the absence of the Pt coating, the voltage degradation rate (0.097 mV h−1) was even smaller than that of the Pt-coated HA-IrOx anode. Surprisingly, the ohmic resistance in the Nyquist plot did not increase at all (Fig. 4c), indicating invariant TiOx interface properties. Based on the breakdown of overpotentials shown in Fig. S17,† the kinetic and mass transport overpotentials were the primary reasons for the degradation. Given the similar degradation phenomenon of the Pt-coated HA-IrOx anode, it was thought that the dissolution and/or destruction of the HA-IrOx layer was the main contributor to the degradation of the two anodes. These align well with the previous publications that demonstrated the severe dissolution and/or agglomeration of the IrOx catalyst, playing a critical role in the PEMWE degradation.47–49 Although the degradation rate of the DL-CL was not impressive, when compared with that in publications, we would like to emphasize that the high retention ability of the excellent electron transport in the non-Pt-coated condition has not been presented previously; all the bare Ti PTL anodes, especially at low iridium loadings, resulted in huge ohmic resistance growth after the long-term operation, as shown in the HA-IrOx data and results in numerous publications.13,20,49,50 Although further investigation is required, R-IrO2 at the PTL interface is also beneficial for preventing the structural deformation of TiOx during the OER.
Taken together, the DL-CL anode exhibited excellent interface properties without any protective coating, lasting for a long-term operation of 1000 h. We believe that this strategy can pave the way for achieving high-performance and cost-effective PEMWE by realizing a Pt-coating-free anode. Possible following works can be optimizing the DL-CL materials to achieve a lower degradation rate, exploring the reason for the high stability of the TiOx interface, and suggesting practical methods for fabricating DL-CL anodes.
4 Conclusion
In summary, this study introduced a double-layered catalyst layer (DL-CL) that can effectively overcome electron transport limitations caused by TiOx at the anode interface. Two catalyst layers were strategically combined, one with a low work function (R-IrO2) and the other with a high-activity catalyst (HA-IrOx), to fabricate a DL-CL anode with excellent electron conductivity without the need for the Pt coating. Compared to single-layered CLs, the DL-CL anode exhibited superior single-cell performance, even with a bare Ti PTL, owing to the significantly lower kinetic and ohmic overpotentials, demonstrating the synergistic effect of the R-IrO2 and HA-IrOx catalysts. Further investigations by varying the CL structures revealed that the key factor for performance improvement was the catalyst arrangement, highlighting the importance of positioning a high-work-function catalyst away from the TiOx interface. A durability test further demonstrated that the DL-CL maintained a highly conductive TiOx interface over 1000 h of operation, underlining its practical feasibility. Given the high cost and material demand of conventional Pt coatings (∼200 nm or ∼0.43 mgPt cm−2), the DL-CL anode provides a promising alternative, replacing the Pt coating with a ultralow R-IrO2 loading (∼0.05 mgIr cm−2). This study establishes DL-CL as a viable strategy for cost-effective and durable anode designs in PEMWE.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
D. Kim and G. Doo conceptualized the experiments and drafted the manuscript. D. Kim, G. H. Jung, Y. H. Yun, and S. An conducted the experimental work. J. Park, C. Lee, S. Lee, H. Park, S.-K. Kim, H.-S. Cho, and H.-T. Kim performed analyses and characterizations. N. Jung, G. Doo, and M. J. Kim supervised the project and provided guidance through regular discussions. All authors reviewed the results and contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korea Government (MSIT) (No. 2022M3H4A3A01083536) and the Technology Innovation Program (1415188335, Development of Membrane Electrode Assembly and stack components for PEM water electrolysis) funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea). This work was also conducted under framework of the research and development program of the Korea Institute of Energy Research (C5-2407).References
- C. V. Pham, D. Escalera-López, K. Mayrhofer, S. Cherevko and S. Thiele, Adv. Energy Mater., 2021, 11, 2101998 CrossRef CAS.
- P. Shirvanian and F. van Berkel, Electrochem. Commun., 2020, 114, 106704 CrossRef CAS.
- W. H. Lee, Y.-J. Ko, J. H. Kim, C. H. Choi, K. H. Chae, H. Kim, Y. J. Hwang, B. K. Min, P. Strasser and H.-S. Oh, Nat. Commun., 2021, 12, 4271 CrossRef CAS PubMed.
- C. Van Pham, M. Bühler, J. Knöppel, M. Bierling, D. Seeberger, D. Escalera-López, K. J. Mayrhofer, S. Cherevko and S. Thiele, Appl. Catal. B Environ., 2020, 269, 118762 CrossRef.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- U. Babic, M. Suermann, F. N. Büchi, L. Gubler and T. J. Schmidt, J. Electrochem. Soc., 2017, 164, F387 CrossRef CAS.
- M. Schalenbach, G. Tjarks, M. Carmo, W. Lueke, M. Mueller and D. Stolten, J. Electrochem. Soc., 2016, 163, F3197 CrossRef CAS.
- N. Diklic, A. H. Clark, J. Herranz, J. S. Diercks, D. Aegerter, M. Nachtegaal, A. Beard and T. J. Schmidt, ACS Energy Lett., 2022, 7, 1735–1740 CrossRef CAS.
- S. Geiger, O. Kasian, B. R. Shrestha, A. M. Mingers, K. J. Mayrhofer and S. Cherevko, J. Electrochem. Soc., 2016, 163, F3132 CrossRef CAS.
- T. Lazaridis, B. M. Stühmeier, H. A. Gasteiger and H. A. El-Sayed, Nat. Catal., 2022, 5, 363–373 CrossRef CAS.
- C. Minke, M. Suermann, B. Bensmann and R. Hanke-Rauschenbach, Int. J. Hydrogen Energy, 2021, 46, 23581–23590 CrossRef CAS.
- M. Bernt, A. Siebel and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, F305–F314 CrossRef CAS.
- C. Liu, M. Shviro, A. S. Gago, S. F. Zaccarine, G. Bender, P. Gazdzicki, T. Morawietz, I. Biswas, M. Rasinski and A. Everwand, Adv. Energy Mater., 2021, 11, 2002926 CrossRef CAS.
- Z. Kang, J. Mo, G. Yang, Y. Li, D. A. Talley, S. T. Retterer, D. A. Cullen, T. J. Toops, M. P. Brady and G. Bender, Appl. Energy, 2017, 206, 983–990 CrossRef CAS.
- C. Rakousky, U. Reimer, K. Wippermann, M. Carmo, W. Lueke and D. Stolten, J. Power Sources, 2016, 326, 120–128 CrossRef CAS.
- C. Liu, J. A. Wrubel, E. Padgett and G. Bender, Appl. Energy, 2024, 356, 122274 CrossRef CAS.
- C. Liu, K. Wippermann, M. Rasinski, Y. Suo, M. Shviro, M. Carmo and W. Lehnert, ACS Appl. Mater. Interfaces, 2021, 13, 16182–16196 CrossRef CAS.
- A. T. Mayyas, M. F. Ruth, B. S. Pivovar, G. Bender, and K. B. Wipke, Manufacturing Cost Analysis for Proton Exchange Membrane Water Electrolyzers, National Renewable Energy Laboratory (NREL), Golden, CO (United States), 2019 Search PubMed.
- J. Lopata, Z. Kang, J. Young, G. Bender, J. Weidner and S. Shimpalee, J. Electrochem. Soc., 2020, 167, 064507 CrossRef CAS.
- C. Liu, M. Shviro, G. Bender, A. S. Gago, T. Morawietz, M. J. Dzara, I. Biswas, P. Gazdzicki, Z. Kang and S. F. Zaccarine, J. Electrochem. Soc., 2023, 170, 034508 CrossRef CAS.
- M. Bernt, C. Schramm, J. Schröter, C. Gebauer, J. Byrknes, C. Eickes and H. Gasteiger, J. Electrochem. Soc., 2021, 168, 084513 CrossRef CAS.
- A. Hartig-Weiß, M. Bernt, A. Siebel and H. A. Gasteiger, J. Electrochem. Soc., 2021, 168, 114511 CrossRef.
- U. Babic, E. Nilsson, A. Pătru, T. J. Schmidt and L. Gubler, J. Electrochem. Soc., 2019, 166, F214 CrossRef CAS.
- C. Rozain, E. Mayousse, N. Guillet and P. Millet, Appl. Catal. B Environ., 2016, 182, 123–131 CrossRef CAS.
- J. Mo, Z. Kang, S. T. Retterer, D. A. Cullen, T. J. Toops, J. B. Green Jr, M. M. Mench and F.-Y. Zhang, Sci. Adv., 2016, 2, e1600690 CrossRef PubMed.
- T. Schuler, J. M. Ciccone, B. Krentscher, F. Marone, C. Peter, T. J. Schmidt and F. N. Büchi, Adv. Energy Mater., 2020, 10, 1903216 CrossRef CAS.
- G. Doo, J. Park, J. Park, J. Heo, J. Jung, D. W. Lee, H. Bae, J. Hyun, E. Oh and J. Kwen, ACS Energy Lett., 2023, 8, 2214–2220 CrossRef CAS.
- W. Yang, T. Moehl, E. Service and S. D. Tilley, Adv. Energy Mater., 2021, 11, 2003569 CrossRef CAS.
- A. G. Scheuermann, J. P. Lawrence, K. W. Kemp, T. Ito, A. Walsh, C. E. Chidsey, P. K. Hurley and P. C. McIntyre, Nat. Mater., 2016, 15, 99–105 CrossRef CAS.
- G. Doo, H. Bae, J. Park, J. Hyun, I. Kim, D. W. Lee, E. Oh and H.-T. Kim, ACS Nano, 2024, 18, 23331–23340 CrossRef CAS.
- F. Hegge, F. Lombeck, E. Cruz Ortiz, L. Bohn, M. von Holst, M. Kroschel, J. Hübner, M. Breitwieser, P. Strasser and S. Vierrath, ACS Appl. Energy Mater., 2020, 3, 8276–8284 CrossRef CAS.
- Y. Qiu, H. Zhang, H. Zhong and F. Zhang, Int. J. Hydrogen Energy, 2013, 38, 5836–5844 CrossRef CAS.
- M. Povia, D. F. Abbott, J. Herranz, A. Heinritz, D. Lebedev, B.-J. Kim, E. Fabbri, A. Patru, J. Kohlbrecher and R. Schäublin, Energy Environ. Sci., 2019, 12, 3038–3052 RSC.
- J. Gao, C.-Q. Xu, S.-F. Hung, W. Liu, W. Cai, Z. Zeng, C. Jia, H. M. Chen, H. Xiao and J. Li, J. Am. Chem. Soc., 2019, 141, 3014–3023 CrossRef CAS.
- V. Pfeifer, T. Jones, J. V. Vélez, C. Massué, M. Greiner, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer and J. Allan, Phys. Chem. Chem. Phys., 2016, 18, 2292–2296 RSC.
- D. F. Abbott, D. Lebedev, K. Waltar, M. Povia, M. Nachtegaal, E. Fabbri, C. Copéret and T. J. Schmidt, Chem. Mater., 2016, 28, 6591–6604 CrossRef CAS.
- F. Karimi, A. Bazylak and B. A. Peppley, J. Electrochem. Soc., 2017, 164, F464 CrossRef CAS.
- B. Hasa, U. R. Aryal, S. Higashi, N. E. Tolouei, J. T. Lang, B. Erb, A. Smeltz, I. V. Zenyuk and G. Zhu, Appl. Catal. B Environ. Energy, 2025, 361, 124616 CrossRef CAS.
- J. P. H. Li, X. Zhou, Y. Pang, L. Zhu, E. I. Vovk, L. Cong, A. P. van Bavel, S. Li and Y. Yang, Phys. Chem. Chem. Phys., 2019, 21, 22351–22358 RSC.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Perkin-Elmer Corporation, Eden Prairie, 1992 Search PubMed.
- S. Kashiwaya, J. Morasch, V. Streibel, T. Toupance, W. Jaegermann and A. Klein, Surfaces, 2018, 1, 73–89 CrossRef.
- S. Chatterjee, X. Peng, S. Intikhab, G. Zeng, N. N. Kariuki, D. J. Myers, N. Danilovic and J. Snyder, Adv. Energy Mater., 2021, 11, 2101438 CrossRef CAS.
- M. Bernt, J. Schröter, M. Möckl and H. Gasteiger, J. Electrochem. Soc., 2020, 167, 124502 CrossRef CAS.
- U. Babic, T. J. Schmidt and L. Gubler, J. Electrochem. Soc., 2018, 165, J3016 CrossRef CAS.
- E. Padgett, G. Bender, A. Haug, K. Lewinski, F. Sun, H. Yu, D. A. Cullen, A. J. Steinbach and S. M. Alia, J. Electrochem. Soc., 2023, 170, 084512 CrossRef CAS.
- M. Geuß, L. Löttert, A. Hutzler, J. Schwarz, J. Nováková, I. Khalakhan, M. Gaberšček, K. J. Mayrhofer, S. Thiele and S. Cherevko, Chem. Eng. J., 2025, 162887 CrossRef.
- Z. Zeng, R. Ouimet, L. Bonville, A. Niedzwiecki, C. Capuano, K. Ayers, A. P. Soleymani, J. Jankovic, H. Yu and G. Mirshekari, J. Electrochem. Soc., 2022, 169, 054536 CrossRef CAS.
- M. Möckl, M. F. Ernst, M. Kornherr, F. Allebrod, M. Bernt, J. Byrknes, C. Eickes, C. Gebauer, A. Moskovtseva and H. A. Gasteiger, J. Electrochem. Soc., 2022, 169, 064505 CrossRef.
- A. Voronova, S. Kim, D. Kim, H.-Y. Park, J. H. Jang and B. Seo, Energy Environ. Sci., 2023, 16, 5170–5184 RSC.
- A. J. McLeod, L. V. Bühre, B. Bensmann, O. E. Herrera and W. Mérida, J. Power Sources, 2024, 589, 233750 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta01688f |
| This journal is © The Royal Society of Chemistry 2025 |