
X-ray crystallographic evidence for the simultaneous presence of axial and rhombic sites in cupredoxins: atomic resolution X-ray crystal structure analysis of pseudoazurin and DFT modelling†
T. Yamaguchia,
K. Akaoa,
A. Takashinaa,
S. Asamuraa,
M. Unnoab,
R. K. Szilagyi*cd and
T. Kohzuma*ab
aInstitute of Quantum Beam Science, Ibaraki University, Bunkyo 2-1-1, Mito, Ibaraki 310-8512, Japan. E-mail: takamitsu.kohzuma.qbs@vc.ibaraki.ac.jp
bFrontier Research Center for Applied Atomic Sciences, Ibaraki University, Shirakata 162-1, Tokai, Ibaraki 319-1106, Japan
cDepartment of Chemistry and Biochemistry, Montana State University, Bozeman, MT 59717, USA. E-mail: szilagyi@montana.edu
dMTA-ELTE “Momentum” Chemical Structure/Function Laboratory, Budapest, 1117, Hungary
First published on 9th September 2016
Abstract
Crystal structure refinement of pseudoazurin from Achromobacter cycloclastes (AcPAz) was carried out at atomic (1.10 Å) resolution. The copper ion was localized in two positions at the metal binding site of AcPAz. The occupancies of the copper sites are consistent with the ratio of axial and rhombic signals from EPR spectra and the intensity ratios for blue (axial) and green (rhombic) copper sites from UV-VIS spectroscopy. Computational modelling using an approximately 6 Å protein environment around the Cu site for both the oxidized and reduced forms showed that a small scale inner sphere rearrangement can account for the co-existence of two different redox active sites for a mononuclear cupredoxin independently from the employed density functionals.
1. Introduction
Type 1 copper proteins function as electron carriers in many biological electron-transfer systems.1 Therefore, the electronic structure of the copper site has been studied extensively to elucidate the correlation between the structure, unique spectroscopic properties, and the Cu2+/Cu+ reduction potential.2–7 Electron paramagnetic resonance (EPR) spectroscopic features of the oxidized metalloprotein with an unusual small hyperfine coupling (A∥) value and unique electronic absorption spectra from ultraviolet-visible absorption spectroscopy (UV-VIS) were attributed to trigonally (axial) or tetragonally (rhombic) distorted tetrahedral Cu coordination environments. The two different coordination geometries eliminate the Jahn–Teller distortion force for a four-coordinate d9 metal site. These axial and rhombic sites correspond to optically different blue and green Cu proteins, respectively. In addition to the Jahn–Teller effect, the geometric distortion can be rationalized by the entatic state or rack-induced bonding from the protein environment.3,4 To date, the distortion model describing the presence of axial or rhombic sites invoked a single copper site.3 However, the possibility for the simultaneous presence of different copper sites in the same protein environment has already been suggested by early resonance Raman measurements.8 Furthermore, a flat potential surface along the Cu–S(Met) and Cu–S(Cys) distortion coordinates was demonstrated by computational modelling. Density functional theory (DFT) results show that large-scale structural distortions can take place involving the Cu site at negligible energetic costs.3,9,10 Importantly, the presence of a dual or a single Cu site as a stationary structure was dependent on the employed level of theory. Pure density functionals with the Generalized Gradient Approximation (GGA) for computational models containing the inner sphere environment9 localized two minima on the shallow potential energy surface. These correspond to axial (blue) and rhombic (green) Cu2+ sites. However, hybrid density functional studies with more complete basis sets consistently resulted in a single Cu site model.3,11 In addition to the employed functional, it has been shown that the completeness of the computational model can be equally critical as much as the level of theory due to the necessary treatment of the network of weak interactions that involve the metal site and its inner-sphere protein environment.12–14Pseudoazurin from Achromobacter cycloclastes (AcPAz) is an electron donor to nitrite reductase and nitrous oxide reductase in the denitrification process.15–17 In a previous study,18 the crystal structure of AcPAz was refined to 1.35 Å resolution (1BQK) with a single Cu site. The dual Cu positions could not be resolved from the electron density map. However, previous comprehensive spectroscopic observations indicated the simultaneous presence of two different Cu2+ sites.2,19,20 Improvement in crystallization conditions, thus in crystal quality, and developments of synchrotron beamlines and software resulted in identifying two positions for the Cu site in AcPAz at the resolution of 1.10 Å.
The X-band EPR spectrum of oxidized AcPAz showed the presence of two different spectral components as described in our previous studies.2 These components have characteristic g-values for an axial site (g⊥ = 2.05, g∥ = 2.23, A∥ = 5.2 mT) and simultaneously for a rhombic site (gx = 2.02, gy = 2.09, gz = 2.20, A∥ = 4.7 mT).2 The co-existence of the axial and rhombic EPR components were further elaborated by a high-resolution J-band EPR study,20 UV-VIS,19 resonance Raman,19 and X-ray absorption spectroscopic (XAS)21 measurements. Here we report the first direct structural evidence for the co-existence of axial and rhombic copper sites in AcPAz from an atomic (1.10 Å) resolution X-ray crystallographic analysis.
2. Experimental and modelling
2.1. AcPAz preparation and crystallization
The protein sample of AcPAz for crystallization was expressed by the cultivation of recombinant E. coli with IPTG induction. AcPAz was isolated from soluble fraction cell breakage using ion-exchange chromatography (CM-sephadex C-50 from GE Healthcare) as described earlier.2 The Cu/AcPAz molar ratio was measured to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by induced coupled plasma atomic emission spectroscopy (ICP-AES). UV-VIS characterization of the protein sample revealed an absorbance ratio at 280 and 594 nm (A280/A594) to be 1.4. Crystallization of AcPAz was performed by the hanging drop vapour diffusion method using 30% (w/v) polyethylene glycol (PEG 4000) as precipitant. The reservoir solution contained 0.1 M Tris–HCl (pH 8.5) buffer, 0.1 M MgCl2, and 5% (v/v) glycerol. The initial concentration of AcPAz in the drop was 48 mg cm−3.
:1 by induced coupled plasma atomic emission spectroscopy (ICP-AES). UV-VIS characterization of the protein sample revealed an absorbance ratio at 280 and 594 nm (A280/A594) to be 1.4. Crystallization of AcPAz was performed by the hanging drop vapour diffusion method using 30% (w/v) polyethylene glycol (PEG 4000) as precipitant. The reservoir solution contained 0.1 M Tris–HCl (pH 8.5) buffer, 0.1 M MgCl2, and 5% (v/v) glycerol. The initial concentration of AcPAz in the drop was 48 mg cm−3.
2.2. X-ray crystallography
Single crystal X-ray diffraction (XRD) data for the AcPAz crystal were collected by an ADSC Quantum 210r CCD detector in conjunction with a 1.0 Å X-ray radiation at beam line NW12A, Photon Factory Advanced Ring (PF-AR). The crystal temperature was maintained at 100 K by employing liquid nitrogen cryo-stream. Data were integrated and processed with HKL2000.22 The phase information was obtained by molecular replacement using oxidized AcPAz structure reported by Inoue, et al. (PDB ID: 1BQK).18 Further refinement was conducted using REFMAC5.23 COOT24 was used for visualization, model building, and correction during refinement cycles. The optimization of the positions and the occupancies of the disordered Cu sites in step size of 10% was carried out by visual minimization of residual Fo − Fc electron density map at 4σ contour level. See Fig. S1 in ESI† for representative examples of Fo − Fc map as a function of occupancies.2.3. Theoretical calculations
We carried out a concomitant computational investigation of the Cu positions, arrangement of the side chains from inner sphere coordination environment using a spectroscopically calibrated density functional and a saturated basis set (B38HFP86/def2TZVP).25–28 This specific hybrid functional was developed for modelling spectroscopic features of blue and green Cu sites.3 It has been successfully employed for other Cu-containing metalloprotein, such as galactose oxidase.29 For comparative purposes, we also report results obtained using a pure GGA density functional (BP86 (ref. 26 and 27)) and a popular hybrid GGA functional (B3LYP (ref. 30 and 31)). In contrast to past computational studies, the main conclusion of the given study is independent of the selected functional. The bulk of the protein environment was modelled with a polarizable continuum model32 employing an intermediate dielectric constant of 10. This value was selected on the basis of previous studies on cupredoxins by us3,7,11,21 and by others.9,10 The computational model contained 69 atoms that included the inner sphere coordination environment of the Cu site and the first outer sphere residues out to approximately 6 Å. The employed computational model encompassed all significant effects from covalent and ionic interactions including the H-bonding between the His amide groups and the S(Cys), and the dipole interactions from an amide trans to the S(Met) ligand.3. Results and discussion
3.1. Atomic resolution X-ray crystal structure analysis
Table 1 summarizes the data collection and refinement parameters for the structure of AcPAz. The crystals of AcPAz were grown under a newly developed crystallization condition as described in Experimental and modelling section. The AcPAz crystals belong to the I222 space group with cell parameters a = 56.1 Å, b = 63.4 Å, c = 67.2 Å. The asymmetric unit contains a single AcPAz molecule. The structure of AcPAz was refined at 1.10 Å resolution with R and Rfree values of 14.8% and 18.6%, respectively. The electron density maps at the Cu site in the refined structure models (PDB ID: 4YL4) are shown in Fig. 1. The residual Fo − Fc electron density map is unreasonable when the ellipsoidal electron density was modelled by a single Cu position (Fig. 1A). However, this residual density disappeared when the disordering of Cu atoms was refined to “position 1” with the occupancy of 40% and “position 2” with the occupancy of 60%. The percentage values were determined on the basis of fitting the ellipsoidal density map at the Cu site. It is important to emphasize that occupancies of Cu positions well approximate the independent axial and rhombic populations from EPR measurements (23%/77% from X-band EPR;2 37%/63% from J-band EPR20).| a Numbers in parentheses are for the highest-resolution shell. | |
|---|---|
| Beam line | PF-AR NW12A |
| Wavelength/Å | 1.00000 |
| Resolution range/Å | 50.0–1.10 (1.14–1.10)a |
| Space group | I222 |
| Cell parameter/Å | a = 56.1 |
| b = 63.4 | |
| c = 67.2 | |
| Mosaisity/° | 0.428 |
| Reflections measured | 329789 |
| Reflection unique | 47290 |
| R merge, % | 6.0 (34.3)a |
| I/σ | 34.3 (3.96)a |
| Completeness, % | 97.1 (99.6)a |
| Refinement range/Å | 13.0–1.10 |
| R factor, % | 14.8 |
| Rfree, % | 18.6 |
| RMSD bond length/Å | 0.025 |
| RMSD bond angles/° | 2.42 |
| Number of non-hydrogen atoms | 1214 |
| Number of water atoms | 142 |
| Average B-factor (protein per Å2) | 18.8 |
| PDB entry | 4YL4 |
 | ||
| Fig. 1 The stereo view of 2Fo − Fc (grey at 1.5σ level) and Fo − Fc (green at 4.0σ and red, −4.0σ levels) maps around the Cu site of AcPAz modelled with a single Cu position (A) and dual Cu positions (B). Positions 1 and 2 are labelled as ‘axial’ and ‘rhombic’ in Fig. 1B, respectively. | ||
In order to further substantiate the existence of dual Cu positions, Type 1 copper sites from Paracoccus denitrificans amicyanin (PdAmi),33 AcNiR,34 Populus nigra plastocyanin (PnPc),35 and Hyphomicrobium denitrificans pseudoazurin (HdPAz)36 determined at resolutions of 0.75, 0.90, 1.00, and 1.18 Å, respectively, were compared. Fig. 2 shows the structures with 2Fo − Fc and Fo − Fc electron density maps from the Protein Data Bank. During the refinements by others, the Cu site was modelled with a single position; however, significant residual electron densities were observed in the Fo − Fc maps. Remarkably, the 2Fo − Fc and Fo − Fc electron density maps of these cupredoxins are highly similar to that of the AcPAz structure (Fig. 1A) refined with a single Cu site. Our given extended crystallographic analysis suggests that considering an additional position for the copper site can result in the elimination of the Fo − Fc map around the Cu. This would indicate the co-existence of axial and rhombic sites in other Type 1 cupredoxins than AcPAz.
 | ||
| Fig. 2 2Fo − Fc (grey at 1.5σ level) and Fo − Fc (green at 4.0σ and red −4.0σ levels) maps at the Cu binding site of PdAmi (A, 2OV0), AcNiR (B, 2BW4), PnPc (C, 4DP9), and HdPAz (D, 3EF4). Maps were generated from structures and electron density maps deposited in the Protein Data Bank. | ||
The two positions for the Cu atoms in AcPAz can be unambiguously assigned to the axial (minor component, ∼40%) and rhombic (major component, ∼60%) sites, when the Cu–ligand distances and angles in Table 2 are compared. The characteristically long Cu–S(Met) distance and close to 120° basal L–Cu–L′ angles correspond to the axial site in PnPc.35 Contrary, the short Cu–S(Met) distance and close to perpendicular basal L–Cu–L′ angles correspond to the rhombic Cu site in AcNiR.34 The axial site in position 1 can be clearly identified by an approximately 0.5 Å longer Cu–S(Met86) distance in comparison to the rhombic site in position 2 (3.10 Å vs. 2.60 Å, respectively). Interestingly, the Cu–S(Cys78) distances in AcPAz for both Cu positions were found to be identical (2.21 Å). Generally, the Cu–S(Cys) distance in the axial site is expected to be shorter than the rhombic site from the higher Cu–S(Cys) frequency in resonance Raman spectra,19 and the increased Cu–S bond covalency from S K-edge XAS.21 Highly similar bond lengths for the oxidized axial and rhombic Cu sites in AcPAz were also observed by Cu K-edge extended X-ray absorption fine structure (EXAFS) analysis.21 The inconsistencies between the resonance Raman results and the XRD/XAS analyses can be resolved by considering a balance between covalent and ionic Cu–ligand interactions in determining the bond strength. Ionic interactions at short distances are of comparable strength to covalent interactions with good orbital overlap.21 The shorter Cu–N(His81) bond can be rationalized by the longer Cu–S(Met86) distance in the axial site in comparison to the rhombic site. The Cu–N bond lengths for the axial (1.86–2.03 Å) and rhombic (1.96–2.21 Å) sites of AcPAz show a significantly larger range than the corresponding distances in PnPc (1.94–1.99 Å) and AcNiR (2.03–2.04 Å). The Cu positions in PnPc and AcNiR were modelled by a single position despite the indication for the simultaneous presence of axial and rhombic sites by J-band EPR for plastocyanin20 and resonance Raman for nitrite reductase.8 The reported Cu–L distances in PnPc and AcNiR may well be considered as the population weighted average of shorter and longer Cu–N(His) distances in the axial and the rhombic sites, respectively. The EXAFS analyses of the wild type and several variants of AcPAz could not differentiate between the two Cu–N(His) distances either,21 since they show an average value of 1.94–1.96 Å similarly to those in PnPc and AcNiR XRD structures.
| Internal coordinates | AcPAz position 1 | AcPAz position 2 | Internal coordinates | Poplar nigra plastocyanin | Internal coordinates | Ac nitrite reductase |
|---|---|---|---|---|---|---|
| Cu–N(His40) | 2.03 | 1.96 | Cu–N(His37) | 1.94 | Cu–N(His95) | 2.04 |
| Cu–S(Cys78) | 2.21 | 2.21 | Cu–S(Cys84) | 2.16 | Cu–S(Cys126) | 2.23 |
| Cu–N(His81) | 1.86 | 2.21 | Cu–N(His87) | 1.99 | Cu–N(His145) | 2.03 |
| Cu–S(Met86) | 3.10 | 2.60 | Cu–S(Met92) | 2.78 | Cu–S(Met150) | 2.49 |
| N(His40)–Cu–S(Cys78) | 132 | 136 | N(His37)–Cu–S(Cys84) | 131 | N(His95)–Cu–S(Cys126) | 127 |
| N(His40)–Cu–N(His81) | 106 | 96 | N(His37)–Cu–N(His87) | 100 | N(His95)–Cu–N(His145) | 101 |
| N(His40)–Cu–S(Met86) | 77 | 91 | N(His37)–Cu–S(Met92) | 88 | N(His95)–Cu–S(Met150) | 86 |
| S(Cys78)–Cu–N(His81) | 121 | 107 | S(Cys84)–Cu–N(His87) | 119 | S(Cys126)–Cu–N(His145) | 108 |
| S(Cys78)–Cu–S(Met86) | 99 | 115 | S(Cys84)–Cu–S(Met92) | 109 | S(Cys126)–Cu–S(Met150) | 108 |
| N(His81)–Cu–S(Met86) | 100 | 107 | N(His87)–Cu–S(Met92) | 104 | N(His145)–Cu–S(Met150) | 127 |
The comparison of the two Cu–N(His) distances among the four crystallographic structures in Table 2 reveals a notable relationship. The Cu–N(His40) distance of position 1 (axial) is longer than position 2 (rhombic) by 0.07 Å for AcPAz. However, PnPc as the reference for axial Cu site has a 0.10 Å shorter bond length than rhombic site of AcNiR. Furthermore, in rhombic AcPAz the short Cu–N(His40) bond correlates with the long Cu–N(His81) bond (0.17 Å deviation), and vice versa for the axial AcPAz (0.25 Å deviation). In PnPc and AcNiR, these deviations are however only less than 0.05 Å. Given the high resolution of the crystal structures in Table 2, the origin of the peculiar trend in Cu–N ligand distances may lie in the second sphere or perhaps further outer sphere protein environment effects. The L–Cu–L′ angles for the axial and rhombic sites also differ significantly from each other.
Moreover, we carefully considered whether the presence of the two different Cu positions was due to disorder in the coordinating amino acid (His40, Cys78, His81, and Met86). This was observed by others during the refinement of Cu positions of Met98Gln variant of amicyanin that showed disorder of Cu and Cu-coordinating Gln98 residue.33 The refinement of multiple Cu sites in the amicyanin structure necessitates the evaluation of the structural differences between oxidized and reduced forms. As we have seen in the EXAFS study,21 during X-ray irradiation the samples can undergo radiation damage, particularly photoreduction.
The Cu–ligand distances in Type 1 copper site is expected to elongate upon Cu reduction due to an increased electrostatic repulsion between metal and ligands in addition to the loss of covalent interactions between Cu 3d and ligand 2p/3p orbitals. Therefore, the simultaneous presence of Cu2+ and Cu+ ions may provide an explanation for the dual Cu positions alternative to the presence of both axial and rhombic oxidized Cu sites. In order to evaluate this possibility, we examined the structural effects of reduction on the Cu coordination geometry in Type 1 copper proteins with [N2S2] coordination. The considered crystal structures were obtained under similar pH conditions for the same Type 1 copper proteins.18,35,37–41 Cu–ligand distances for all structures are summarized in Table S1 of ESI.† Surprisingly, some of the structures (#1–4 and #11–13 in Table S1†) show the shortening of Cu–ligand distances upon Cu reduction. The average elongation of Cu–S(Met) for all the Type 1 copper proteins was 0.05 ± 0.10 Å between the reduced and oxidized sites, which is inconsistent with the observed difference of 0.50 Å for the two Cu positions of AcPAz. By focusing on the AcPAz structures, the average elongation of Cu–S(Met) (#5–8 in Table S1†) upon reduction (0.11 ± 0.05 Å) was found to be still too small. The corresponding changes in the Cu–S(Cys) and Cu–N(His) distances for all entries in Table S1† (−0.01 ± 0.08 Å and 0.05 ± 0.11 Å) or for the AcPAz structures only (0.05 ± 0.04 Å and 0.12 ± 0.15 Å) also do not support the alternative explanation of a redox dependent presence of the two Cu positions. Thus, the rationale for the presence of partially reduced Cu site can be excluded on the basis of available crystal structures.
3.2. Theoretical calculations
The flexibility of the inner sphere coordination environment was evaluated for accommodating the two Cu sites within the same protein matrix. A systematic analysis was carried out for fully and partially optimized inner sphere coordination environments around the Cu site in the oxidized and reduced states using the given atomic resolution crystal structure of AcPAz. The full structural optimizations employing the saturated basis set for all atoms, pure GGA (BP86) and hybrid GGA (B3LYP and B38HFP86) functionals converge to a single Cu site structure with Cu–S(Met) distances of 2.53, 2.63, and 2.61 Å for the oxidized, and 2.58, 2.99, and 2.72 Å for the reduced forms (Fig. 3). These results suggest that considering inner coordination sphere interactions alone, there is a single metal site geometry for the Type 1 copper proteins in agreement with the prediction from the previously defined coupled distortion coordinate model.3 The above structural optimization results show a notable repulsive nature of the popular B3LYP potential, where basis set saturation results in elongated metal–ligand distances.42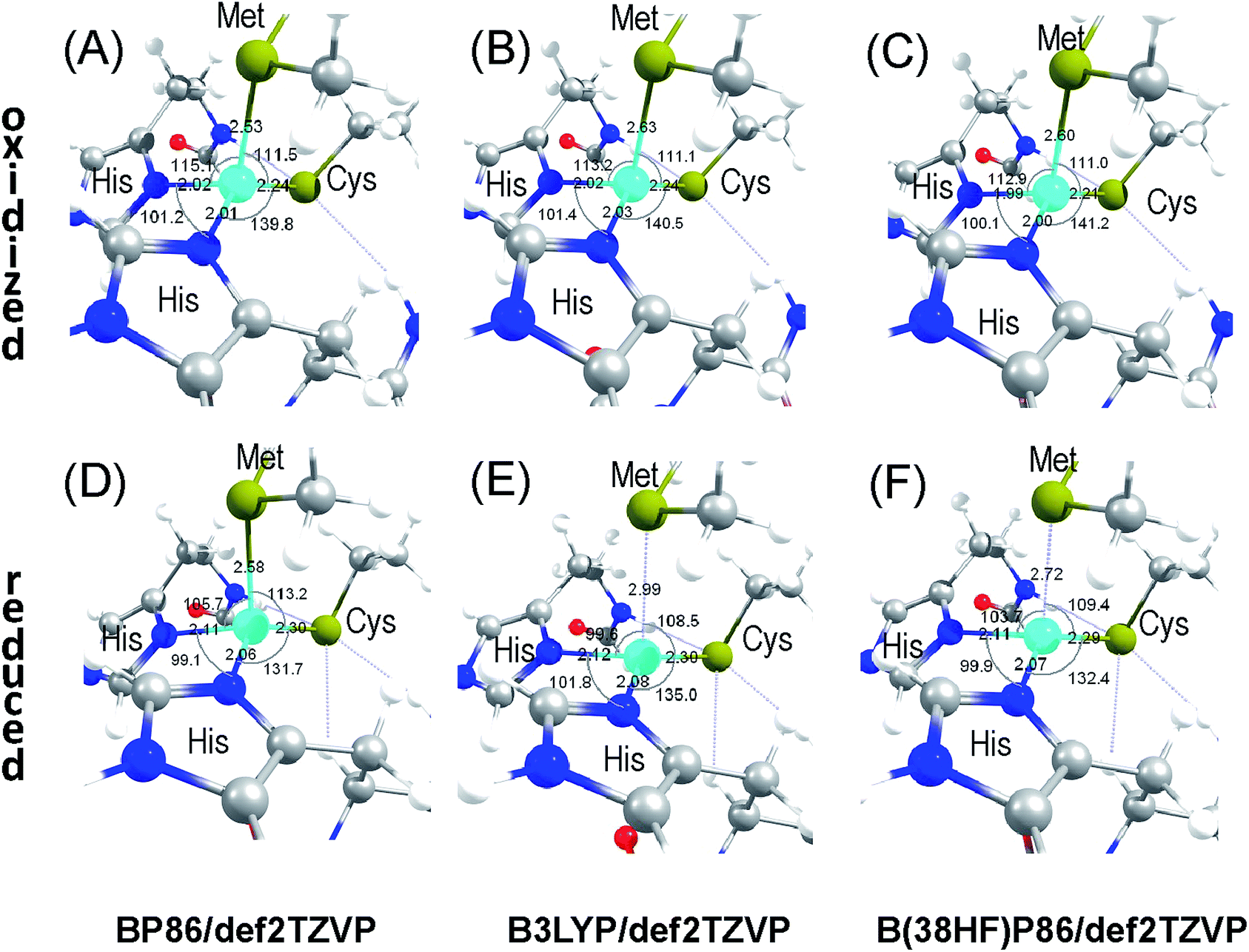 | ||
| Fig. 3 Optimized inner sphere environment for the oxidized (A–C) and reduced (D–F) Cu sites in AcPAz starting from separate axial and rhombic sites, but converging to a single site geometry using BP86 (A and D), B3LYP (B and E), and B38HFP86 (C and F) functionals with def2TZVP basis sets with all α- and β-carbon atoms fixed at their crystallographic positions. | ||
Although the computational results in Fig. 3 only support a single Cu geometry, the crystal structure shows the presence of two different Cu positions. This discrepancy can be resolved by considering a more dominant role for the complex network of weak interactions from outer spheres than the Cu–ligand inner sphere itself. These outer sphere effects cannot be captured even with the employed, fully optimized extended inner sphere model.
Accepting the experimental Cu positions, we carried out partial structural optimizations with constrained Cu2+, α- and β-carbon positions fixed at their crystallographic values. These calculations allowed us to refine the ligand positions that are the most suitable to maintain an axial or a rhombic environment. Fig. 4 compares the partially optimized structures for the oxidized form. The first important observation from Fig. 4C is that both axial and rhombic ligand environments highly resemble each other. Thus, their individual crystallographic electron densities are also expected to be highly similar. This similarity poses a challenge to the refinement of two different ligand environments corresponding to the different Cu positions. The axial site (Fig. 4A) has a long (calc.: 3.17 Å, exp.: 3.10 Å) while the rhombic site (Fig. 4B) has the short Cu–S(Met) distance (calc.: 2.61 Å, exp.: 2.60 Å). Importantly, the BP86 (axial: 3.21 vs. rhombic: 2.59 Å) and the B3LYP (axial: 3.27 vs. rhombic: 2.65 Å) calculations give comparable results to those shown in Fig. 4 with the same structural constraints imposed as discussed above. The asymmetry between the two Cu–N(His) distances is eliminated for the rhombic site; however, it is maintained reasonably well for the axial site. The L–Cu–L′ angles are closer to the ideal 120° value for the axial site with a trigonal basal plane. They are closer to 90° for the rhombic site with a tetragonal geometry. The root mean square deviation between the positions of the 14 optimized atoms in the models shown in Fig. 4C is 0.22 Å. With the exception of the displacement of the S(Cys) ligand, all the other ligand atoms remain practically unchanged. This observation serves as a justification for the little to no disorder in the inner sphere side chain atomic positions in AcPAz. It is also important to note that the structural optimizations resulted in an energetically less favourable axial site (Fig. 3A) by 20 kJ mol−1 than the rhombic (Fig. 3B), which is in qualitative agreement with the axial site being the minor and the rhombic site being the major component.
 | ||
| Fig. 4 Partially optimized structures for the oxidized (A) axial and (B) rhombic Cu sites of AcPAz at B38HFP86/def2TZVP level with Cartesian coordinates of the Cu2+ ion, all α- and β-carbon atoms kept fixed at their crystallographic values. Panel (C) shows the overlay of the optimized atomic positions for the inner sphere ligands. | ||
As an independent evaluation for the energetic consequence of the presence of two Cu2+ positions within the same protein environment, a molecular field analysis (MFA) was carried out using the Cu2+ ion as a probe (Fig. 5A). Using the B38HFP86 hybrid GGA functional with saturated basis set, the global minima of the MFA showed a single Cu site with Cu–L distances and L–Cu–L′ angles that are closer to the rhombic than to the axial site (Fig. 5B). Considering an approximately 20 kJ mol−1 energy range as determined from the difference between the partially optimized axial and rhombic sites with frozen Cu2+ positions (see above), the MFA calculations indicate an elongated potential energy well for the most likely Cu positions as shown in Fig. 5C. The shape and orientation of the Cu positions in the energy well approximate the elongated 2Fo − Fc electron density maps of Fig. 1 and 2.
 | ||
| Fig. 5 Molecular field analysis of the inner sphere environment at the metal binding site of AcPAz using Cu2+ ion as probe ((A) Cu positions evaluated in MFA, (B) the lowest energy structure, (C) range of low energy structures within 20 kJ mol−1). The blue dots correspond to the position of Cu2+ tested in MFA. | ||
Given that multiple Cu sites have already been refined from X-ray crystallographic data for the related amicyanine copper protein,33 the variation in the reduced Cu site geometry was also evaluated as a function of crystallographic Cu positions. The axial and rhombic reduced forms of the AcPAz inner sphere environments are compared in Fig. 6A and B. Unexpectedly, the deviation of the calculated Cu–S(Met) bond lengths between axial and rhombic sites in the Cu+ oxidation state (0.57 Å) is identical to that in the Cu2+ state (0.56 Å) (Fig. 3). This comparison demonstrates that the Cu position for axial and rhombic sites can be separated by approximately 0.6 Å along the Cu–S(Met) bond, independently from the oxidation state for the AcPAz metal site. Although it is not possible to rule out unambiguously the possibility of the presence of some amount of reduced Cu site as an artefact due to radiation damage/photoreduction, the assignment of ‘axial’ and ‘rhombic’ geometries for the positions 1 and 2 can be computationally confirmed by comparing crystallographic and optimized Cu–S(Met) distances. Moreover, it is notable that the Cu–N(His) distances in the reduced structures remain asymmetric. While this is true for the rhombic site, a reverse relationship was observed in calculations than in the crystal structure. The difference in the Cu–S(Cys) distances between the two sites is negligible, which is consistent with the identical bond length in the crystal structure (see also Table S1† for other relevant structures).
 | ||
| Fig. 6 Partially optimized structures for the reduced (A) axial and (B) rhombic Cu sites of AcPAz at B38HFP86/def2TZVP level with Cartesian coordinates of the Cu2+ ion, all α- and β-carbon kept fixed at their crystallographic values. | ||
In the light of the above discussion for the crystallographic results and computational modelling, the dual positions of Cu atom in AcPAz can be explained by the presence of two energetically comparable minima on the potential energy surface within a highly similar ligand environment. The existence of these minima can be facilitated by outer sphere protein environmental effects, since a single equilibrium structure is localized in all computational models without a constrain on the Cu position regardless of the level of theory. This conclusion when considering only the inner sphere ligands challenges the concept of a potential energy surface for the Cu binding with a single stationary structure at the rhombic limit due to Jahn–Teller distortion.3,4 An outer sphere network of interactions from the protein matrix, which has already been shown to be important for the regulation of Cu site structure and reactivity,19,43–47 can provide comparable energetic strain to the Jahn–Teller distortion that moves the Cu site away from its lowest energy rhombic structure and trap it in a higher energy axial structure. The structural basis of a single minimum on a coupled geometric distortion coordinate between the apical Cu–S(Met) and the basal Cu–S(Cys) distances was previously developed using low resolution crystal structures (1.90–2.70 Å for NiR,48 1.33 Å for Pc,49 1.80 Å for cucumber basic protein50). Now with the availability of atomic resolution crystal structures and careful consideration of the electron density around the Cu site, the simultaneous presence of axial and rhombic sites can be realized within the same protein crystal. This confirmation is in agreement with the interpretation of a broad range of spectroscopic results and computational analyses.
4. Conclusions
The structure of AcPAz at 1.10 Å resolution demonstrated the co-existence of axial and rhombic Cu sites in the same crystal. The occupation of Cu positions in a Type 1 site was found to be consistent with their population from independent EPR spectroscopic measurements. It is important to highlight that the independent XRD, EPR,2,20 and XAS21 measurements were performed at approximately same temperatures. In our past experience, the AcPAz does not show thermochromic behaviour.2,20 These results strongly suggest the possibility for the presence of dual Cu geometry in Type 1 copper proteins. The presence of two Cu sites was already proposed earlier on the basis of spectroscopic studies of AcPAz,2,19,20 AcNiR,8 and Pc20 metalloproteins. The characteristically elongated electron density maps are commonly found in other blue and green copper proteins. The structural and spectroscopic observations suggest that the dual Cu positions can be a common feature for all Type 1 copper proteins. The chemical functional relevance of the dual Cu sites may include versatility through allosteric interactions in electron-transfer processes at reduction potential that is adjustable on-demand. Furthermore, the presence of dual Cu sites may contribute to accommodating various redox partners as required in nitrite reductase versus nitrous oxide reductase. Kinetic experiments aimed at biological functional relevance are currently underway. The origin of the presence of two Cu positions cannot be correlated with a mixture of reduced and oxidized forms as described for amicyanine.33 It is more likely that protein dynamics is responsible for the dual Cu sites through a fascinating network of weak interactions. This is in contrast to previous models, where the Cu site moves its ligand environment due to Jahn–Teller distortion force.Acknowledgements
We are grateful to Professor Yasuhito Shomura of Ibaraki University for his insightful discussions. This work was supported by Grant-in-Aid for Scientific Research from JSPS No. 22550145 (TK) and the Sekisho Scholar Award (AT & TY). This work was also partly supported by the Reimei Kenkyu Award from Japan Atomic Energy Agency, JAEA (MU). The MTA-ELTE Chemical Structure & Function “Momentum” Laboratory (ID 96122) is supported by the Hungarian Academy of Sciences, Budapest, Hungary (Contract No. LP2015-10/2015). Portion of this research was conducted at Photon Factory (PF) BL NW12A as part of proposals 2009G578, 2011G519, and 2013G504.Notes and references
- C. Dennison, Coord. Chem. Rev., 2005, 249, 3025–3054 CrossRef CAS
.
- R. F. Abdelhamid, Y. Obara, Y. Uchida, T. Kohzuma, D. M. Dooley, D. E. Brown and H. Hori, JBIC, J. Biol. Inorg. Chem., 2007, 12, 165–173 CrossRef CAS PubMed
- E. I. Solomon, R. K. Szilagyi, S. D. George and L. Basumallick, Chem. Rev., 2004, 104, 419–458 CrossRef CAS PubMed
- L. B. LaCroix, S. E. Shadle, Y. N. Wang, B. A. Averill, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1996, 118, 7755–7768 CrossRef CAS
- S. DeBeer, D. W. Randall, A. M. Nersissian, J. S. Valentine, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Phys. Chem. B, 2000, 104, 10814–10819 CrossRef CAS
- S. E. Shadle, J. E. Pennerhahn, H. J. Schugar, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1993, 115, 767–776 CrossRef CAS
- S. D. George, L. Basumallick, R. K. Szilagyi, D. W. Randall, M. G. Hill, A. M. Nersissian, J. S. Valentine, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2003, 125, 11314–11328 CrossRef PubMed
- D. M. Dooley, R. S. Moog, M. Y. Liu, W. J. Payne and J. LeGall, J. Biol. Chem., 1988, 263, 14625–14628 CAS
- K. Pierloot, J. O. A. De Kerpel, U. Ryde, M. H. M. Olsson and B. O. Roos, J. Am. Chem. Soc., 1998, 120, 13156–13166 CrossRef CAS
- U. Ryde, M. H. M. Olsson, K. Pierloot and B. O. Roos, J. Mol. Biol., 1996, 261, 586–596 CrossRef CAS PubMed
- R. K. Szilagyi and E. I. Solomon, Curr. Opin. Chem. Biol., 2002, 6, 250–258 CrossRef CAS PubMed
- D. F. Hansen, S. I. Gorelsky, R. Sarangi, K. O. Hodgson, B. Hedman, H. E. M. Christensen, E. I. Solomon and J. J. Led, JBIC, J. Biol. Inorg. Chem., 2006, 11, 277–285 CrossRef PubMed
- R. G. Hadt, S. I. Gorelsky and E. I. Solomon, J. Am. Chem. Soc., 2014, 136, 15034–15045 CrossRef CAS PubMed
- T. V. Harris and R. K. Szilagyi, J. Comput. Chem., 2016, 37, 1681–1696 CrossRef CAS PubMed
- M. Y. Liu, M. C. Liu, W. J. Payne and J. Legall, J. Bacteriol., 1986, 166, 604–608 CAS
- T. Kohzuma, S. Takase, S. Shidara and S. Suzuki, Chem. Lett., 1993, 22, 149–152 CrossRef
- K. Fujita, M. Hirasawa-Fujita, D. E. Brown, Y. Obara, F. Ijima, T. Kohzuma and D. M. Dooley, J. Inorg. Biochem., 2012, 115, 163–173 CrossRef CAS PubMed
- T. Inoue, N. Nishio, S. Suzuki, K. Kataoka, T. Kohzuma and Y. Kai, J. Biol. Chem., 1999, 274, 17845–17852 CrossRef CAS PubMed
- M. B. Fitzpatrick, Y. Obara, K. Fujita, D. E. Brown, D. M. Dooley, T. Kohzuma and R. S. Czernuszewicz, J. Inorg. Biochem., 2010, 104, 250–260 CrossRef CAS PubMed
- P. Gast, F. G. J. Broeren, S. Sottini, R. Aoki, A. Takashina, T. Yamaguchi, T. Kohzuma and E. J. J. Groenen, J. Inorg. Biochem., 2014, 137, 57–63 CrossRef CAS PubMed
- T. Yamaguchi, J. Yano, V. K. Yachandra, Y. Nihei, H. Togashi, R. K. Szilagyi and T. Kohzuma, Bull. Chem. Soc. Jpn., 2015, 88, 1642–1652 CrossRef CAS
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS
- A. A. Vagin, R. A. Steiner, A. A. Lebedev, L. Potterton, S. McNicholas, F. Long and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2184–2195 Search PubMed
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef PubMed
- R. K. Szilagyi, M. Metz and E. I. Solomon, J. Phys. Chem. A, 2002, 106, 2994–3007 CrossRef CAS
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef
- D. Rokhsana, A. E. Howells, D. M. Dooley and R. K. Szilagyi, Inorg. Chem., 2012, 51, 3513–3524 CrossRef CAS PubMed
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed
- C. J. Carrell, J. K. Ma, W. E. Antholine, J. P. Hosler, F. S. Mathews and V. L. Davidson, Biochemistry, 2007, 46, 1900–1912 CrossRef CAS PubMed
- S. V. Antonyuk, R. W. Strange, G. Sawers, R. R. Eady and S. S. Hasnain, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12041–12046 CrossRef CAS PubMed
- G. S. Kachalova, A. C. Shosheva, G. P. Bourenkov, A. A. Donchev, M. I. Dimitrov and H. D. Bartunik, J. Inorg. Biochem., 2012, 115, 174–181 CrossRef CAS PubMed
- D. Hira, M. Nojiri and S. Suzuki, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2008, 65, 85–92 CrossRef PubMed
- T. Inoue, N. Nishio, Y. Kai, S. Harada, Y. Ohshiro, S. Suzuki, T. Kohzuma, S. Shidara and H. Iwasaki, J. Biochem., 1993, 114, 761–762 CAS
- C. A. P. Libeu, M. Kukimoto, M. Nishiyama, S. Horinouchi and E. T. Adman, Biochemistry, 1997, 36, 13160–13179 CrossRef CAS PubMed
- K. Petratos, Z. Dauter and K. S. Wilson, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1988, 44, 628–636 CrossRef
- E. Vakoufari, K. S. Wilson and K. Petratos, FEBS Lett., 1994, 347, 203–206 CrossRef CAS PubMed
- M. Velarde, R. Huber, S. Yanagisawa, C. Dennison and A. Messerschmidt, Biochemistry, 2007, 46, 9981–9991 CrossRef CAS PubMed
- R. K. Szilagyi and M. A. Winslow, J. Comput. Chem., 2006, 27, 1385–1397 CrossRef CAS PubMed
- K. M. Lancester, in Structure and Bonding: Molecular Electronic Structures of Transition Metal Complexes I, ed. D. M. P. Mingos, P. Day and J. P. Dahl, 2012, vol. 142, pp. 119–153 Search PubMed
- J. J. Warren, K. M. Lancaster, J. H. Richards and H. B. Gray, J. Inorg. Biochem., 2012, 115, 119–126 CrossRef CAS PubMed
- N. M. Marshall, D. K. Garner, T. D. Wilson, Y. G. Gao, H. Robinson, M. J. Nilges and Y. Lu, Nature, 2009, 462, 113–116 CrossRef CAS PubMed
- Y. Lu, N. Sieracki and N. M. Marshall, Nature, 2009, 460, 855–862 CrossRef CAS PubMed
- M. C. Machczynski, H. B. Gray and J. H. Richards, J. Inorg. Biochem., 2002, 88, 375–380 CrossRef CAS PubMed
- E. T. Adman, J. W. Godden and S. Turley, J. Biol. Chem., 1995, 270, 27458–27474 CrossRef CAS PubMed
- J. Guss, H. Bartunik and H. Freeman, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1992, 48, 790–811 CrossRef
- J. M. Guss, E. A. Merritt, R. P. Phizackerley and H. C. Freeman, J. Mol. Biol., 1996, 262, 686–705 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Montana State University Scholarworks: http://doi.org/10.15788/M2PP4F. See DOI: 10.1039/c6ra19282c |
| This journal is © The Royal Society of Chemistry 2016 |